
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liposome biodistribution mapping with in vivo X-ray fluorescence imaging†
Giovanni Marco
Saladino
 *ab,
Po-Han
Chao
b,
Bertha
Brodin
a,
Shyh-Dar
Li
b and
Hans Martin
Hertz
a
*ab,
Po-Han
Chao
b,
Bertha
Brodin
a,
Shyh-Dar
Li
b and
Hans Martin
Hertz
a
aDepartment of Applied Physics, Bio-Opto-Nano Physics, KTH Royal Institute of Technology, SE 10691, Stockholm, Sweden. E-mail: saladino@kth.se
bFaculty of Pharmaceutical Sciences, University of British Columbia, V6T 1Z3, Vancouver, British Columbia, Canada
First published on 27th August 2024
Abstract
Lipid-based nanoparticles are organic nanostructures constituted of phospholipids and cholesterol, displaying high in vivo biocompatibility. They have been demonstrated as effective nanocarriers for drug delivery and targeting. Mapping liposome distribution is crucial as it enables a precise understanding of delivery kinetics, tissue targeting efficiency, and potential off-target effects. Recently, ruthenium-encapsulated liposomes have shown potential for targeted drug delivery, photodynamic therapy, and optical fluorescence imaging. In the present work, we design Ru(bpy)3-encapsulated liposomes (Ru-Lipo) empowering optical and X-ray fluorescence (XRF) properties for dual mode imaging and demonstrate the passivation role of liposomes over the free Ru(bpy)3 compound. We employ whole-body XRF imaging to map the in vivo biodistribution of Ru-Lipo in mice, enabling tumor detection and longitudinal studies with elemental specificity and resolution down to the sub-millimeter scale. Quantitative XRF computed tomography on extracted organs permits targeting efficiency evaluations. These findings highlight the promising role of XRF imaging in pharmacokinetic studies and theranostic applications for the rapid optimization of drug delivery and assessment of targeting efficiency.
Introduction
Liposomes have been demonstrated to serve as efficient nanocarriers for drug delivery and targeting, with many products approved by the European Medicines Agency (EMA) and Food and Drug Administration (FDA).1,2 The lipid bilayer structure of these nanoparticles mimics the natural cell membrane, facilitating interactions with biological systems. This feature enables encapsulation of a wide range of therapeutic agents, including hydrophobic and hydrophilic drugs, peptides, proteins, and nucleic acids.3,4 Additionally, lipid-based nanoparticles can be modified with targeting ligands or surface modifications to enhance specific accumulation at desired sites, such as tumors or inflamed tissues, while minimizing off-target effects.5 Furthermore, they can encapsulate various types of imaging agents, such as fluorescent dyes for optical imaging,6–8 radionuclides for nuclear imaging,9 or contrast agents for magnetic resonance imaging (MRI).10,11 By loading the liposomes with two different types of imaging agents, they can enable dual mode imaging.12,13Light-sensitive ruthenium(II) polypyridyl compounds represent powerful tools in photochemistry, recently emerging as potential prodrugs for photodynamic therapy (PDT),14–16 which employs visible and near-infrared light for cancer treatment enabling precise spatial and temporal control over the cytotoxicity, thus reducing adverse effects in cancer patients.17 In this context, sterically hindered liposomes, including a polyethylene glycol (PEG) capping, can be employed as versatile and biocompatible contrast agents and drug carriers for treating various diseases owing to their prolonged circulation time in the bloodstream.18,19
Recently, X-ray fluorescence (XRF) has been applied for in vivo imaging of small animals with high resolution and elemental specificity, using a laboratory setup.20 This has been achieved through the design of inorganic nanoparticles (NPs) as contrast agents, consisting of elements such as molybdenum (Mo), ruthenium (Ru), or rhodium (Rh), whose absorption edge aligns with the energy of the X-ray source (24 keV).21–23 Nevertheless, the highly crystalline nature of inorganic NPs has recently brought concerns over long-term toxicity, due to low dissolution rates, high residence times, and undesired accumulations.24,25
In the present work, we design liposomes encapsulating a ruthenium(II) polypyridyl compound, which can function alone as a dual mode imaging agent, enabling both XRF and optical fluorescence imaging. Owing to its properties, we map the liposome biodistribution in vivo with longitudinal studies using both the imaging modalities, achieving passive tumor targeting. The results evidence that the high penetration depth, high resolution, and elemental specificity of XRF imaging enable the rapid and accurate evaluation of the Ru-Lipo biodistribution in vivo and their quantification in extracted organs. The generality of the synthesis method and the ruthenium-specific detectability with XRF pave the way to theranostic application using PDT-specific ruthenium(II) polypyridyl compounds.
Results and discussion
Methodology
We conceptually illustrated the enabled dual-mode in vivo imaging methodology with optical fluorescence and XRF (Fig. 1). PEGylated Ru-Lipo were designed to empower both optical fluorescence and XRF properties, exploiting the intrinsic adsorption edge of ruthenium as the XRF contrast agent and the optical fluorescence properties of the organic dye, Ru(bpy)3 (Fig. 1a). After assessing the viability in vitro, Ru-Lipo could be employed as dual-mode contrast agents, intravenously injected into mice. Biodistribution and pharmacokinetics or Ru-Lipo could be investigated with projection images obtained via optical fluorescence and XRF imaging. Optical fluorescence imaging was achieved by employing a commercial in vivo imaging system (IVIS), by matching the excitation (465 nm) and emission (620 nm) windows with the optical fluorescence characteristics of Ru(bpy)3 (Fig. 1b).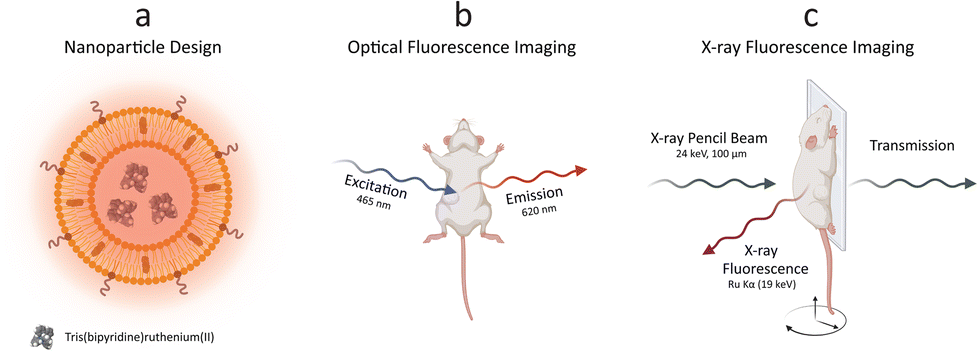 | ||
| Fig. 1 Dual mode imaging with ruthenium-encapsulated liposomes. Schematic illustration of (a) the nanoparticle design, (b) optical fluorescence imaging, and (c) X-ray fluorescence imaging. PEGylated liposomes were synthesized with the thin-film hydration method, encapsulating a hydrophilic ruthenium dye. Optical fluorescence imaging was performed with a commercial in vivo imaging system (IVIS), while X-ray fluorescence imaging was achieved with a laboratory liquid-metal jet X-ray source. | ||
A schematic representation of the in vivo XRF imaging arrangement (Fig. 1c) highlighted the employment of a monochromatic X-ray pencil beam (24 keV), generating XRF radiation when exciting a contrast agent constituted of an element (Ru) with its adsorption edge (KRu = 22 keV) matching the X-ray source energy. The transmitted radiation could be employed for conventional X-ray (transmission) imaging to be overlaid by the XRF signal. A movable stage, monitoring and anesthesia equipment enabled in vivo imaging of mice in scanning mode, with a resolution of down to 100 μm (spot size).20
Contrast agent design
The Ru-Lipo formulation was prepared through hydration of lipid thin-films, followed by membrane extrusion to control the size, dialysis against saline to remove the unencapsulated contrast agent, and concentrated with centrifuge filters (Fig. S1†). The physical characteristics of Ru-Lipo were studied, including morphological analysis, hydrodynamic size and ζ-potential (Fig. 2). Cryo-TEM micrographs of Ru-Lipo evidenced a uniform spherical morphology (Fig. 2a) and an intact lipidic bilayer membrane (Fig. S2a†) with an estimated diameter of 88 ± 29 nm (Fig. S2b†). | ||
| Fig. 2 Nanoparticle characterization. (a) Cryo-TEM micrograph of ruthenium-encapsulated liposomes, Ru-Lipo. Scale bar, 200 nm. (b) Average hydrodynamic size values in saline solution (0.9%) after each synthesis step. (c) ζ-Potential of Ru-Lipo in water (pH 6.5). | ||
The hydrodynamic size distribution highlighted a nearly monodisperse sample with an intensity peak at 87 nm, with a polydispersity index (PDI) of 0.03, indicating a uniform particle population (Fig. S3a†), consistent with the results obtained by electron microscopy. A longitudinal study conducted at 37 °C over a period of 3 days highlighted the stability of the synthesized Ru-Lipo, with negligible variations of the hydrodynamic size (Fig. S3b†). After 72 h, the release of Ru(bpy)3 was estimated to be 6.4 ± 0.1%, as determined by optical spectroscopy. The hydrodynamic size was followed over the three synthesis steps, showing no significant changes between extrusion and ultrafiltration steps (Fig. 2b). The ζ-potential of Ru-Lipo, indicating their surface charge, was estimated as −29 ± 9 mV, indicating a negative charge in water, at pH 6.5 (Fig. 2c), typical for liposomes PEGylated with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG), which do not shield the entire surface charge, thus leading to negatively charged sterically stabilized liposomes.26
Owing to its dual-mode properties, the Ru-Lipo formulation was also measured for Ru content by means of optical and X-ray fluorescence (Fig. 3). By exposing several Ru-Lipo dilutions to the X-ray pencil beam of a liquid metal-jet X-ray source (24 keV),20 the XRF signal from Ru Kα and Ru Kβ emission peaks could be recorded and collected by photon-counting silicon-drift detectors (Fig. 3a). Furthermore, Thomson scattering (λT) and Compton scattering (λC) could be identified, and the latter's contributions subtracted as the background from the XRF signal (Fig. S4a†). The Ru Kα emission peaks were employed for quantitative estimations (Fig. S4b†) and subsequent XRF imaging. Besides the XRF properties, Ru-Lipo exhibited the same optical fluorescence emission profile as the free Ru(bpy)3 (Fig. S5a†). The excitation and emission spectra highlighted intensity peaks in the visible range, at 450 nm and 630 nm, respectively (Fig. 3b). Furthermore, the addition of Triton X 10% to a diluted dispersion of Ru-Lipo was used for liposome's membrane disruption27 and led to a variation in the sample's optical fluorescence of 2%, which was ascribed to random errors and solvent evaporation. Hence, the encapsulation of Ru(bpy)3 in the liposome core did not lead to fluorescence quenching effects due to potentially limited hydrated environment. For this reason, optical fluorescence could be used for the quantitative estimations of encapsulation efficiency (EE) and Ru(bpy)3 concentration.
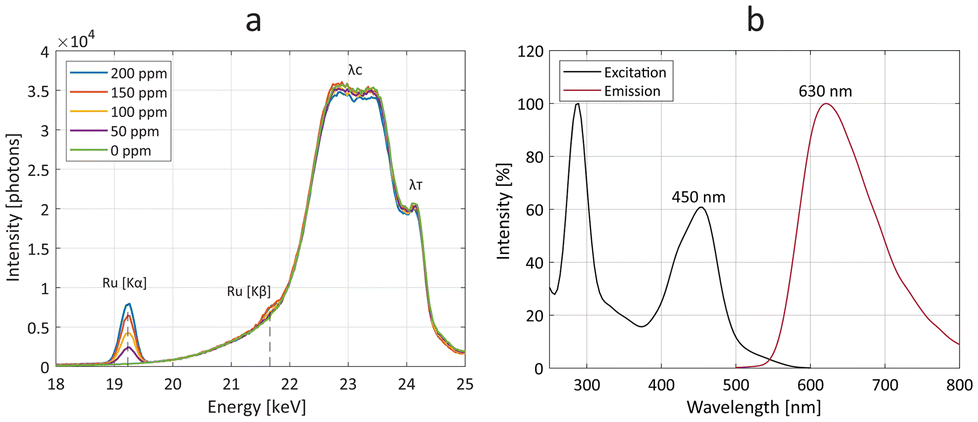 | ||
| Fig. 3 Optical and X-ray fluorescence properties. (a) X-ray fluorescence spectra of stock solutions with several concentrations of ruthenium-encapsulated liposomes (Ru-Lipo), highlighting the Ru Kα and Ru Kβ emission peaks. Compton (λC) and Thomson (λT) scattering are also indicated. (b) Excitation (black) and emission (red) optical fluorescence spectra of Ru-Lipo, evidencing the excitation and emission peaks in the visible range, at 450 nm and 630 nm, respectively. | ||
A calibration curve was obtained for Ru(bpy)3 concentration values of up to 40 μM, by a linear fit (Fig. S5b†). The EE and Ru-to-lipid weight ratio, 10% and 1.8% respectively (Table S1†), were in line with other formulations prepared with 1,2-distearoyl-sn-glycero-3-phosphatidylcholine (DSPC) and hydrophilic loads.28 DSPC was chosen as the main lipid component for yielding liposomes empowering slow load release, long circulation times, and high tumoral delivery, compared to other phospholipids.28 The PEGylation introduced through DSPE-PEG enhances liposome stability in vivo, by limiting opsonization and absorption by the reticuloendothelial system.29 The constituents of the Ru-Lipo formulation were investigated with Fourier-transform infrared spectroscopy (FT-IR) and ultra-performance liquid chromatography (UPLC). All the FT-IR spectra (Fig. S6a†) of the lipidic components exhibited the typical lipidic backbone bands, including δs(CH2) at 1468 cm−1, νs(CH2) at 2850 cm−1, and νas(CH2) at 2916 cm−1.30 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration for DSPC was associated with the band at 1728 cm−1,31 while the band at 772 cm−1 was ascribed to the bipyridine ring breathing of Ru(bpy)3.32 The UPLC analysis (Fig. S6b†) evidenced the presence of all the three lipidic components in Ru-Lipo, DSPC, cholesterol, and DSPE-PEG. Furthermore, it permitted to estimate the effective lipid ratio (Table S1†), highlighting DSPC as the limiting lipidic component for the final formulation. The Ru-to-lipid weight ratio was hereby limited by the solubility of the compound. In future endeavors, the Ru-to-lipid weight ratio could be increased through the implementation of solvent-assisted active loading mechanisms.33 This approach would enhance the relative concentration of the XRF-active element (Ru). Consequently, it would be possible to achieve higher sensitivity with XRF imaging. Furthermore, the replacement of Ru(bpy)3 with Ru-containing photosensitizers for phototherapy applications, including compounds currently under evaluation in clinical trials,34–36 can enable the development of theranostic nanoparticles.
O stretching vibration for DSPC was associated with the band at 1728 cm−1,31 while the band at 772 cm−1 was ascribed to the bipyridine ring breathing of Ru(bpy)3.32 The UPLC analysis (Fig. S6b†) evidenced the presence of all the three lipidic components in Ru-Lipo, DSPC, cholesterol, and DSPE-PEG. Furthermore, it permitted to estimate the effective lipid ratio (Table S1†), highlighting DSPC as the limiting lipidic component for the final formulation. The Ru-to-lipid weight ratio was hereby limited by the solubility of the compound. In future endeavors, the Ru-to-lipid weight ratio could be increased through the implementation of solvent-assisted active loading mechanisms.33 This approach would enhance the relative concentration of the XRF-active element (Ru). Consequently, it would be possible to achieve higher sensitivity with XRF imaging. Furthermore, the replacement of Ru(bpy)3 with Ru-containing photosensitizers for phototherapy applications, including compounds currently under evaluation in clinical trials,34–36 can enable the development of theranostic nanoparticles.
Biocompatibility
The synthesized Ru-Lipo were tested in vitro using a real-time cell analysis (RTCA) assay to assess their role in ameliorating the toxicity of the free dye, Ru(bpy)3. RTCA enabled longitudinal studies, by measuring the cell proliferation over time of murine macrophages (Fig. 4). This cell line (RAW 264.7) was selected for in vitro toxicity evaluations owing to the macrophage's role in the immune response of external stimuli, including nanoparticles, leading to nanoparticle uptake and sequestration in liver and spleen.37–39 Furthermore, in our previous studies, macrophages consistently exhibited a strong concentration-dependent response,22,40 making them the preferred choice for assessing nanoparticle cytotoxicity prior to in vivo studies.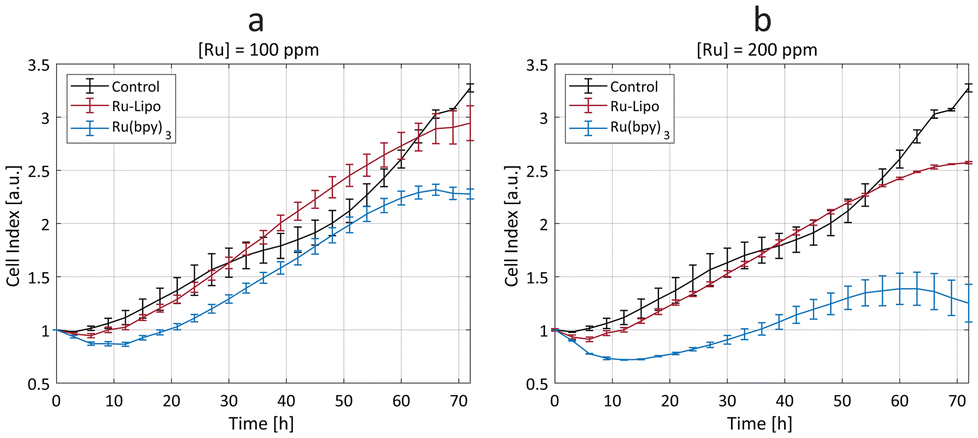 | ||
| Fig. 4 Cytotoxicity study. Real-time cell analysis (RTCA) assay on macrophages (RAW 264.7), after exposure to ruthenium-encapsulated liposomes (Ru-Lipo, in red) and free ruthenium dye, Ru(bpy)3 (in blue) with two ruthenium concentrations, (a) 100 ppm and (b) 200 ppm. The cell index values are compared to unexposed (negative) control cells (black). Measurements were made in triplicates (±SD). | ||
Cells exposed to a low concentration (50 ppm) did not evidence any significant difference between Ru-Lipo and the free dye, Ru(bpy)3 (Fig. S7a†). Both the higher tested ruthenium concentrations (100 and 200 ppm) highlighted a significant passivation role of the liposomes (Fig. 4), with Ru-Lipo and Ru(bpy)3 leading to a concentration- and time-dependent viability. The free dye exhibited a half-maximal inhibitory concentration (IC50) of 200 ppm at 48 h, while Ru-Lipo did not show any significant difference in cell index compared to unexposed control cells, during the proliferation phase (t < 60 h), at the tested concentrations and exposure times. By comparing the same ruthenium concentrations in form of free and encapsulated dye, it was possible to validate the beneficial role of employing liposomes as the carriers for XRF contrast agents, as a possible alternative to inorganic passivation strategies, such as silica shell coating or conjugation with carbon quantum dots.21,41 Finally, live imaging of cells exposed to Ru-Lipo demonstrated the possibility to track and visualize the liposomes in vitro (Fig. S7b†), by means of their optical fluorescence properties (Fig. S5a†).
Preclinical imaging
Owing to the dual fluorescence properties of the designed contrast agents, Ru-Lipo could be employed both for XRF and optical fluorescence imaging. Mice injected with Ru-Lipo were imaged in vivo employing the laboratory liquid-metal jet X-ray source or a commercial optical fluorescence imaging system (Fig. 5). In vivo XRF projection images highlighted a specific biodistribution pattern, by detecting Ru Kα emission photons and overlaying them on the simultaneously acquired X-ray transmission image (Fig. 5a).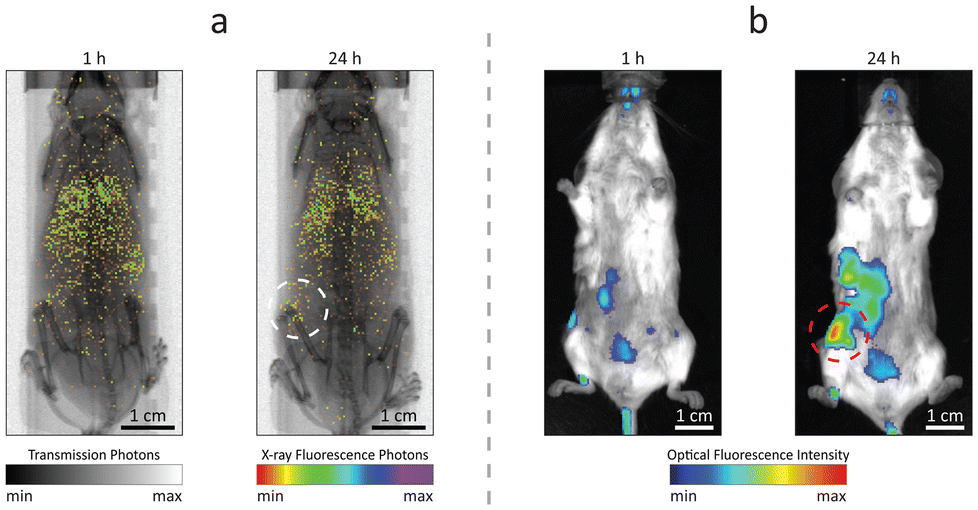 | ||
| Fig. 5 Dual mode imaging. In vivo (a) X-ray fluorescence projection images and (b) optical fluorescence imaging of mice, 1 h and 24 h after injection with ruthenium-encapsulated liposomes (Ru-Lipo). The tumor area is highlighted with dashed circles. Scale bars, 1 cm. | ||
Longitudinal scans on the same mice at several time points (1 h, 5 h, and 24 h) permitted to identify the optimal time interval between injection and imaging for tumor detection (Fig. S8†). At early stages (1 h and 5 h), Ru-Lipo exhibited a scattered biodistribution, including detection from the lungs, liver, and spleen, indicating long circulation times. Finally, Ru-Lipo accumulated mainly in spleen, liver, and xenografted tumor, 24 h after injection.
The passive tumor targeting was confirmed with the IVIS system, exploiting the optical fluorescence properties of Ru-Lipo (Fig. 5b). The organ distribution study with optical fluorescence was hindered by the limited penetration depth and tissue autofluorescence in the optical spectrum (Fig. S9†), which only permits the signal detection up to a few millimeters beneath the skin surface.42,43 These results supported the possibility to perform dual-mode imaging solely employing the intrinsic properties of Ru(bpy)3 together with the passivating and capping functions of liposomes to achieve biocompatible and sufficiently confined contrast agents. Liver and spleen retention of intravenously injected nanoparticles by resident macrophages is a common and undesired outcome.44,45 Nanoparticle sequestration can be limited by inducing a temporary macrophage depletion or by introducing specific surface functionalization to minimize opsonization.46 In the present work, PEGylation likely shielded the liposome surface from fast opsonization and phagocytosis by the mononuclear phagocytic system. For this reason, Ru-Lipo reduced the previously encountered undesired accumulations in the liver, when employing inorganic contrast agents, enabling tumor detection without macrophage depletion.40,47 In the future, improved tumor detection could be achieved by refining the Ru-Lipo design aiming at increasing the Ru-to-Lipid ratio, thus enabling higher injection doses.
Inorganic Ru NPs are known to have slow dissolution rates even in acids, often requiring microwave digestion processing.48 Furthermore, recent in vivo studies employing Ru NPs as XRF contrast agents highlighted long residence times with accumulations in liver, spleen, and skin persisting for over two weeks, after intravenous administration.25 On the other hand, PEGylated liposomes possess a half-life of up to 45 h,49 and their cargo loading undergoes rapid renal excretion after liposome degradation.29,50 For this reason, the designed Ru-Lipo constitute a new generation of XRF contrast agents.
The efficacy, safety, and overall therapeutic potential of nanomedicines are largely determined by their biodistribution profiles.51,52 Therefore, quantitative analysis of nanoparticle distribution in organs is crucial for the design and development of effective nanomedicines. The main organs affected by liposome accumulations and tumor were excised, extracted, and scanned ex vivo with the XRF imaging setup (Fig. 6).
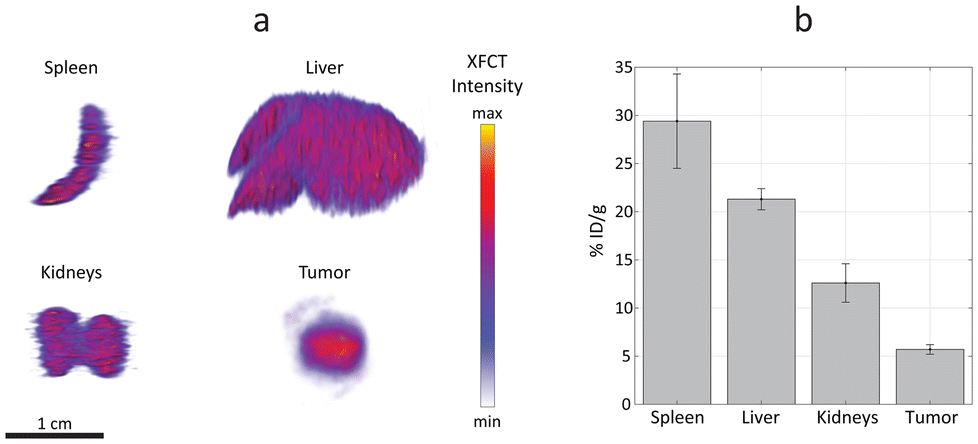 | ||
| Fig. 6 Quantitative accumulation estimation. (a) X-ray fluorescence computed tomography (XFCT) of spleen, liver, kidneys, and tumors, extracted from mice injected with ruthenium-encapsulated liposomes (Ru-Lipo). (b) Quantitative estimation of Ru-Lipo uptake in specific organs and tumors via XFCT, normalized to tissue weight and injected dose (% ID per g, ±SD). Scale bar, 1 cm. | ||
XRF computed tomography (XFCT) has been demonstrated to provide reliable quantitative estimations, previously validated with inductively coupled plasma (ICP) mass spectrometry.53 In the present work, the tomographic reconstructions highlight a homogenous distribution of Ru-Lipo in liver, spleen, and kidneys (Fig. 6a). The successful passive targeting of Ru-Lipo to xenografted tumors was confirmed with XFCT, evidencing the highest local Ru concentration in the tumor core.
Typically, ICP techniques are employed for quantitative studies in nanomedicine, particularly for determining nanoparticle distribution in organs.54,55 However, these methods often lead to extended processing times, which can be a significant drawback in time-sensitive research and applications. Furthermore, the ICP-analyzed sample usually consists of only a small acid-digested portion of the overall organ, which may result in partial or biased information about the nanoparticle distribution. XFCT allowed to obtain a 3D distribution map leading to an average estimation of the overall Ru-Lipo accumulation in the organs and tumor (Fig. 6b), providing a more comprehensive and representative analysis of nanoparticle distribution.
The quantitative nanoparticle distribution analysis demonstrated that the spleen was responsible for the highest liposome sequestration normalized by organ weight (29 ± 5% ID per g), followed by liver (21 ± 1% ID per g) and kidneys (13 ± 2% ID per g). In fact, nanoparticle sequestration by the liver resident macrophages (Kupffer cells) and splenic macrophages is well documented.37,38 The minimal accumulations in kidneys were attributed to the choice of DSPC, as the main lipidic component of Ru-Lipo, which can lower the risk of nephrotoxicity.28 The limited variability among different samples highlights the reliability of this methodology for quantitatively estimating nanoparticle distribution in organs. We speculate that undetected Ru-Lipo were either excreted within the first 24 hours or were still in circulation, below the detection limit.22 Overall, this strategy for liposome design enabled in vivo tumor detection via XRF imaging with a single administration of Ru-Lipo, registering a tumor delivery efficiency of 5.7 ± 0.5% ID per g, consistent with existing literature.55
Conclusions
In the present work, we introduced a methodology for biodistribution studies of ruthenium-encapsulated liposomes using XRF imaging. The same XRF imaging setup used for in vivo image acquisition was also used for the quantitative analysis of nanoparticles in organs. This not only streamlined the process, but also ensured consistency between the imaging and analysis stages, leading to more rapid and reliable results. Dual mode imaging with optical and XRF fluorescence validated the passive targeting of tumors. Moreover, the transition from inorganic to organic nanoparticles as the XRF contrast agents enabled tumor detection without the need of macrophage depletion. Furthermore, the wider availability of FDA-approved liposomal formulations enhances the possible applications of XRF imaging in pharmaceutical and medical research. In the future, the careful choice of specific ruthenium(II) polypyridyl compounds with therapeutic action will confer the possibility to develop theranostic nanoparticles, owing to the elemental specificity of XRF imaging for ruthenium.Experimental
Materials
1,2-Distearoyl-sn-glycero-3-phosphatidylcholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (CH), tris(2,2-bipyridyl)dichlororuthenium(II) hexahydrate (Ru(bpy)3), and Triton X 10% were obtained from Sigma Aldrich. All other reagents were of analytical grade.Liposome preparation
The lipid components (DSPC, CH and DSPE-PEG) were mixed in chloroform at a mole ratio of 56.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38.3:5.3, for a total of 60 mg. Solvent was evaporated at 50 °C with Rotavapor. The thin film was then hydrated with an aqueous solution of Ru(bpy)3 (1 mL, 0.1 mmol in saline solution 0.9%) at 50 °C, to form multi-lamellar vesicles (MLVs). MLVs were extruded 21 times through polycarbonate filters (Nuclepore Track-Etch Membranes, Whatman) of 0.2 μm pore size first, and then repeated with 0.1 μm pore size, each at 65 °C, to adjust liposome size to around 100 nm. Formulations were then transferred to a dialysis tube (Biosciences Pur-A-Lyzer Midi Dialysis Kit 12 kDa MWCO, Sigma Aldrich, SE) and dialyzed against saline (500 mL) for 0.5 h. Saline was changed and dialyzed again for 1 h, and then overnight. Finally, the formulation was concentrated with centrifuge filter units (Amicon® Ultra Centrifugal Filter, 30 kDa MWCO) (Fig. S1†). Empty liposomes were synthesized following the same steps, without the addition of Ru(bpy)3.
:38.3:5.3, for a total of 60 mg. Solvent was evaporated at 50 °C with Rotavapor. The thin film was then hydrated with an aqueous solution of Ru(bpy)3 (1 mL, 0.1 mmol in saline solution 0.9%) at 50 °C, to form multi-lamellar vesicles (MLVs). MLVs were extruded 21 times through polycarbonate filters (Nuclepore Track-Etch Membranes, Whatman) of 0.2 μm pore size first, and then repeated with 0.1 μm pore size, each at 65 °C, to adjust liposome size to around 100 nm. Formulations were then transferred to a dialysis tube (Biosciences Pur-A-Lyzer Midi Dialysis Kit 12 kDa MWCO, Sigma Aldrich, SE) and dialyzed against saline (500 mL) for 0.5 h. Saline was changed and dialyzed again for 1 h, and then overnight. Finally, the formulation was concentrated with centrifuge filter units (Amicon® Ultra Centrifugal Filter, 30 kDa MWCO) (Fig. S1†). Empty liposomes were synthesized following the same steps, without the addition of Ru(bpy)3.
Liposome characterization
The size and zeta potential of the liposomes were measured by dynamic light scattering (DLS) with a Zetasizer Nano (Malvern Instruments Ltd, United Kingdom). Cryogenic transmission electron microscopy (Krios G3i Cryo-TEM, Thermo Fisher Scientific, Sweden) was employed for the morphological characterization of Ru-Lipo. Ultraviolet-visible spectrophotometry (UV-vis, NP80, Implen) was employed to estimate the load release percentage after incubation in saline solution (0.9%) at 37 °C for 72 h. The filtrate was collected using a centrifuge filter unit and the optical absorption at 430 nm was compared to known amounts of Ru(bpy)3. Liposome disruption studies were performed with Triton X 10%.27 The structural characterization was investigated with FT-IR (PerkinElmer, USA) and Acquity UPLC chromatography system (Waters, USA). The concentration was confirmed with XRF spectrum acquisitions of the Ru-Lipo stocks by obtaining a calibration curve using several dilutions of Ru standards (Fig. S4b†).20 The X-ray spectra were acquired with scans of 180 s. Optical fluorescence measurements were obtained with the Synergy Mx Monochromator-Based Multi-Mode Microplate Reader (BioTek, VT, USA). The encapsulation efficiency (EE) was determined as [Ru]L/[Ru]0 × 100%, where [Ru]L is the concentration of Ru in the liposome, and [Ru]0 is the concentration of the hydration solution.Cell studies
The real-time cell analysis assay (xCELLigence Agilent, St Clara, USA) was used to demonstrate the passivation role of the encapsulating liposomes (Ru-Lipo) over the free dye – Ru(bpy)3. The assay was performed on RAW 264.7 (ATCC-TIB-71) cell line, at two Ru concentrations (200 and 100 ppm) in triplicates (96-well plate, biological replicates, ±SD). Untreated cells were the negative control. The estimated viability was based on the quantification of the impedance, an indicator of cell proliferation. The cells were allowed to adhere to the plate surface for 24 hours before exposure to the compound (time = 0). Live images of the cells were obtained using an EVOS 5000 Imaging System (Thermo Fisher Scientific, MA, USA).Animal studies
Experiments with mice were approved by the regional animal ethics committee of Northern Stockholm, Sweden (ethical permit number 13156-2022), according to institutional, national, and European guidelines for animal handling and research (L150/SJVFS 2019:9 and 2010/63/EU) or following the protocol (A22-0141) approved by the Animal Care Committee of the University of British Columbia (Vancouver, BC, Canada). Eight-week-old female albino mice (BALB/c) were obtained from Janvier Labs (France) or Charles River Laboratories (Wilmington, USA) and housed under controlled temperature (21 ± 1 °C) and humidity (55 ± 5%) conditions, with light–dark cycle and ad libitum feeding. The general conditions of the mice were assessed before and during the study, checking for possible onsets of behavioural and/or morphological changes. 6 mice were xenografted with the syngeneic breast adenocarcinoma cell line 4T1 by subcutaneously injecting ≈106 cells suspended in phosphate-buffered saline (PBS, 100 μL). After 1 week, the xenografted mice were intravenously injected with Ru-Lipo formulations (100 μL, 10 mgRu kg−1). Mice were imaged with either optical fluorescence or XRF imaging arrangements.X-ray fluorescence imaging
Whole-body XRF projection images were acquired in vivo with our laboratory liquid-metal jet X-ray source for XRF imaging.56 XRF scans were performed under anesthesia with isoflurane (Abbott, Sweden) at several time points (1 h, 5 h, and 24 h). During the imaging sessions, ophthalmic ointment (Oculentum simplex, APL, Sweden) was applied to the eyes for cornea protection; temperature and respiration were also monitored. A step size of 200 μm and exposure time of 10 ms per step were chosen, resulting in a 15 min scanning time. A binning factor of 2 was used for XRF imaging. The average radiation dose was equal to 1 mGy for a whole-body projection image.57 XFCT was acquired for extracted and fixed tissues using a voxel size of 200 × 200 × 200 μm3, acquiring 30 projections over 180°, with an exposure time of 25 ms per step, resulting into scanning times ranging from about 1 h for spleen, tumor, and kidneys to up to 3.5 h for the liver.Optical fluorescence imaging
Anesthetized mice were imaged using an IVIS® Imaging System (Caliper Life Sciences, Waltham, MA, USA) at several time points (1 h, 5 h, and 24 h). For the acquisition, the exposure time was set to 30 s and a binning factor of 4 was used. The employed excitation and emission filters were 465 nm and 620 nm, respectively, to match the optical fluorescence properties of Ru(bpy)3.Author contributions
Conceptualization, methodology, formal analysis, data curation, software, writing—original draft, and visualization: G. M. S. Investigation: G. M. S., P. C., and B. B. Writing—review and editing: G. M. S., P. C., B. B., S. L., and H. M. H. Supervision and resources: S. L. and H. M. H.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank K. Andersson for valuable expertise in laboratory animal science and excellent control of animal welfare and Prof. M. Toprak for the fruitful discussions. GMS enthusiastically acknowledges the Olle Eriksson Foundation for Materials Engineering (VT-2022-0002) for funding his research stay at the University of British Columbia. This work was supported by the Knut and Alice Wallenberg Foundation (KAW 2016.0057), the Appendix Cancer/Pseudomyxoma Peritonei Research Foundation (18004), the Canadian Cancer Society (706231), and the NanoMedicines Innovation Network (2020-T1-02). The Li lab has been supported by the Canadian Institutes of Health Research (CIHR, PJT-168861), and the Natural Science and Engineering Research Council in Canada (NSERC, RGPIN-2023-04020). The electron micrograph acquisition was performed by the 3D-EM Core Facility at the Karolinska Institute. Fig. 1, Fig. S1,† and ToC figure were created using BioRender.com.References
- R. K. Thapa and J. O. Kim, J. Pharm. Invest., 2023, 53, 19 CrossRef CAS PubMed.
- S. Giordani, V. Marassi, A. Zattoni, B. Roda and P. Reschiglian, J. Pharm. Biomed. Anal., 2023, 236, 115751 CrossRef CAS PubMed.
- S. H. Crayton, A. K. Chen, J. F. Liu, E. M. Higbee-Dempsey, C. H. Huang, A. Tsourkas and Z. Cheng, Comprehensive Biomaterials II, 2017, p. 424 Search PubMed.
- C. Wang, X. Lan, L. Zhu, Y. Wang, X. Gao, J. Li, H. Tian, Z. Liang and W. Xu, Small, 2024, 2309031 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2020, 20, 101 CrossRef PubMed.
- V. Deissler, R. Rüger, W. Frank, A. Fahr, W. A. Kaiser and I. Hilger, Small, 2008, 4, 1240 CrossRef CAS PubMed.
- K. N. Tasnim, A. Rahman, S. M. Newaj, O. Mahmud, S. Monira, T. Z. Khan, H. M. Reza, M. Shin and S. Md. Sharker, ACS Appl. Bio Mater., 2024, 7(5), 3190 CrossRef CAS PubMed.
- Y. Bian, Y. Zhang, B. Hu, Y. Huang, W. Liang, Q. Yuan, J. Zhang, X. Gao, D. Su, Y. Bian, W. Liang, Q. Yuan, X. Gao, D. Su, Y. Zhang and J. Zhang, Small, 2024, 2401282 CrossRef PubMed.
- F. Man, P. J. Gawne and R. T. M. de Rosales, Adv. Drug Delivery Rev., 2019, 143, 134 CrossRef CAS PubMed.
- W. J. M. Mulder, G. J. Strijkers, A. W. Griffioen, L. Van Bloois, G. Molema, G. Storm, G. A. Koning and K. Nicolay, Bioconjugate Chem., 2004, 15, 799 CrossRef CAS PubMed.
- P. Šimečková, F. Hubatka, J. Kotouček, P. Turánek Knötigová, J. Mašek, J. Slavík, O. Kováč, J. Neča, P. Kulich, D. Hrebík, J. Stráská, K. Pěnčíková, J. Procházková, P. Diviš, S. Macaulay, R. Mikulík, M. Raška, M. Machala and J. Turánek, Sci. Rep., 2020, 10, 1 CrossRef PubMed.
- A. de Vries, M. B. Kok, H. M. H. F. Sanders, K. Nicolay, G. J. Strijkers and H. Grüll, Contrast Media Mol. Imaging, 2012, 7, 68 CrossRef CAS PubMed.
- M. K. Ravoori, S. Singh, R. Bhavane, A. K. Sood, B. Anvari, J. Bankson, A. Annapragada and V. Kundra, Sci. Rep., 2016, 6, 1 CrossRef PubMed.
- J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini, H. Chao and G. Gasser, Nat. Commun., 2020, 11, 1 CrossRef PubMed.
- L. Conti, E. Macedi, C. Giorgi, B. Valtancoli and V. Fusi, Coord. Chem. Rev., 2022, 469, 214656 CrossRef CAS.
- C. Parella, A. Blanquer, S. Sinha, E. Hümpfner, J. Hernando, E. Mora, X. Fontrodona, Z. Kelemen, C. Nogués, R. Núñez and I. Romero, Dyes Pigm., 2024, 224, 111985 CrossRef CAS.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS PubMed.
- D. Gao, Z. Luo, Y. He, L. Yang, D. Hu, Y. Liang, H. Zheng, X. Liu and Z. Sheng, Small, 2023, 19, 2206544 CrossRef CAS PubMed.
- K. G. Y. Arsana, G. M. Saladino, B. Brodin, M. S. Toprak and H. M. Hertz, Int. J. Mol. Sci., 2024, 25, 920 CrossRef CAS PubMed.
- G. M. Saladino, C. Vogt, Y. Li, K. Shaker, B. Brodin, M. Svenda, H. M. Hertz and M. S. Toprak, ACS Nano, 2021, 15(3), 5077 CrossRef CAS PubMed.
- G. M. Saladino, C. Vogt, B. Brodin, K. Shaker, N. I. Kilic, K. Andersson, M. Arsenian-Henriksson, M. S. Toprak and H. M. Hertz, Nanoscale, 2022, 15, 2214 RSC.
- N. I. Kilic, G. M. Saladino, S. Johansson, R. Shen, C. McDorman, M. S. Toprak and S. Johansson, ACS Appl. Mater. Interfaces, 2023, 15, 49794 CrossRef CAS PubMed.
- R. Mohammapdour and H. Ghandehari, Adv. Drug Delivery Rev., 2022, 180, 114022 CrossRef CAS PubMed.
- C. Vogt, G. M. Saladino, K. Shaker, M. Arsenian-Henriksson, H. M. Hertz, M. S. Toprak and B. A. Brodin, Nanomedicine, 2023, 18(18), 1161 CrossRef CAS PubMed.
- M. Petrini, W. J. M. Lokerse, A. Mach, M. Hossann, O. M. Merkel and L. H. Lindner, Int. J. Nanomed., 2021, 16, 4045 CrossRef PubMed.
- J. R. Jimah, P. H. Schlesinger and N. H. Tolia, Bio-Protoc., 2017, 7(15), e2433 Search PubMed.
- Y. Zhao, J. P. May, I. W. Chen, E. Undzys and S. D. Li, Pharm. Res., 2015, 32, 3261 CrossRef CAS PubMed.
- S. Paramshetti, M. Angolkar, S. Talath, R. A. M. Osmani, A. Spandana, A. Al Fatease, U. Hani, K. V. R. N. S. Ramesh and E. Singh, Life Sci., 2024, 346, 122616 CrossRef CAS PubMed.
- A. R. Caldas, J. Catita, R. Machado, A. Ribeiro, F. Cerqueira, B. Horta, R. Medeiros, M. Lúcio and C. M. Lopes, Pharmaceutics, 2021, 13, 1202 CrossRef CAS PubMed.
- B. Kästner, C. M. Johnson, P. Hermann, M. Kruskopf, K. Pierz, A. Hoehl, A. Hornemann, G. Ulrich, J. Fehmel, P. Patoka, E. Rühl and G. Ulm, ACS Omega, 2018, 3, 4141 CrossRef PubMed.
- M. U. Munshi, J. Martens, G. Berden and J. Oomens, J. Phys. Chem. A, 2020, 124, 2449 CrossRef CAS PubMed.
- W. L. Tang, W. H. Tang, A. Szeitz, J. Kulkarni, P. Cullis and S. D. Li, Biomaterials, 2018, 166, 13 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797 CrossRef CAS PubMed.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23 CrossRef CAS PubMed.
- M. Ankathatti Munegowda, A. Manalac, M. Weersink, S. A. McFarland and L. Lilge, Coord. Chem. Rev., 2022, 470, 214712 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- W. Ngo, S. Ahmed, C. Blackadar, B. Bussin, Q. Ji, S. M. Mladjenovic, Z. Sepahi and W. C. W. Chan, Adv. Drug Delivery Rev., 2022, 185, 114238 CrossRef CAS PubMed.
- T. Anchordoquy, N. Artzi, I. V. Balyasnikova, Y. Barenholz, N. M. La-Beck, J. S. Brenner, W. C. W. Chan, P. Decuzzi, A. A. Exner, A. Gabizon, B. Godin, S. K. Lai, T. Lammers, M. J. Mitchell, S. M. Moghimi, V. R. Muzykantov, D. Peer, J. Nguyen, R. Popovtzer, M. Ricco, N. J. Serkova, R. Singh, A. Schroeder, A. A. Schwendeman, J. P. Straehla, T. Teesalu, S. Tilden and D. Simberg, ACS Nano, 2024, 18, 13983 CrossRef CAS PubMed.
- G. M. Saladino, B. Brodin, R. Kakadiya, M. S. Toprak and H. M. Hertz, Sci. Adv., 2024, 10, eadl2267 CrossRef CAS PubMed.
- G. M. Saladino, N. I. Kilic, B. Brodin, B. Hamawandi, I. Yazgan, H. M. Hertz and M. S. Toprak, Nanomaterials, 2021, 11(9), 2165 CrossRef CAS PubMed.
- X. Meng, F. Yang, H. Dong, L. Dou and X. Zhang, Nano Today, 2021, 38, 101156 CrossRef CAS.
- L. Finlayson, I. R. M. Barnard, L. McMillan, S. H. Ibbotson, C. T. A. Brown, E. Eadie and K. Wood, Photochem. Photobiol., 2022, 98, 974 CrossRef CAS PubMed.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, J. Controlled Release, 2016, 240, 332 CrossRef CAS PubMed.
- W. Poon, Y. N. Zhang, B. Ouyang, B. R. Kingston, J. L. Y. Wu, S. Wilhelm and W. C. W. Chan, ACS Nano, 2019, 13, 5785 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28 CrossRef CAS PubMed.
- E. Samuelsson, H. Shen, E. Blanco, M. Ferrari and J. Wolfram, Colloids Surf., B, 2017, 158, 356 CrossRef CAS PubMed.
- Y. Yang, C. Sun, Y. Ren, S. Hao and D. Jiang, Sci. Rep., 2014, 4, 4540 CrossRef PubMed.
- J. Heyes, K. Hall, V. Tailor, R. Lenz and I. MacLachlan, J. Controlled Release, 2006, 112, 280 CrossRef CAS PubMed.
- S. Ait-Oudhia, D. E. Mager and R. M. Straubinger, Pharmaceutics, 2014, 6, 137 CrossRef PubMed.
- J. M. Metselaar and T. Lammers, Drug Delivery Transl. Res., 2020, 10, 721 CrossRef PubMed.
- E. K. H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216 Search PubMed.
- J. C. Larsson, C. Vogt, W. Vågberg, M. S. Toprak, J. Dzieran, M. Arsenian-Henriksson and H. M. Hertz, Phys. Med. Biol., 2018, 63, 164001 CrossRef PubMed.
- D. Clases and R. Gonzalez de Vega, Anal. Bioanal. Chem., 2022, 414, 7363 CrossRef CAS PubMed.
- Q. Chen, L. Yuan, W.-C. Chou, Y.-H. Cheng, C. He, N. A. Monteiro-Riviere, J. E. Riviere and Z. Lin, ACS Nano, 2023, 17(20), 19810 CrossRef CAS PubMed.
- H. M. Hertz, J. C. Larsson, U. Lundström, D. H. Larsson and C. Vogt, Opt. Lett., 2014, 39, 2790 CrossRef CAS PubMed.
- K. Shaker, C. Vogt, Y. Katsu-Jimenez, R. V. Kuiper, K. Andersson, Y. Li, J. C. Larsson, A. Rodriguez-Garcia, M. S. Toprak, M. Arsenian-Henriksson and H. M. Hertz, IEEE Trans. Med. Imaging, 2020, 39, 3910 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02793k |
| This journal is © The Royal Society of Chemistry 2024 |