
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis of oligomers containing alternating C-glycosyl α-amino acids and proteinogenic α-amino acids†
Ivana
Colić
,
Barbara
Bogović
and
Ivanka
Jerić
 *
*
Division of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička cesta 54, 10 000 Zagreb, Croatia. E-mail: ijeric@irb.hr
First published on 26th June 2024
Abstract
C-Glycosyl amino acids are a group of C-glycosides in which a carbohydrate molecule is attached to the side chain or backbone of the amino acid via a C–C bond. Despite the numerous methods that have been developed for their synthesis, the C-glycosyl α-amino acids and their oligomers are relatively unexplored. In this work, we presented a protocol for the synthesis of oligomers containing alternating C-glycosyl α-amino acids and proteinogenic α-amino acids. The methodology is based on the modification of α-acyloxyamides obtained from the Passerini reaction using α-D-galactopyranose- and α-L-sorbofuranose-derived aldehydes as C-glycosyl donors. The protocol enabled the synthesis of homo- and heterochiral tetramers, and homo- and heterovalent tetramers in very good yields.
Introduction
It is estimated that about 800 non-proteinogenic amino acids (NPAAs) have been identified in bacteria, fungi, plants and marine organisms, leaving the 20 proteinogenic amino acids in the minority at 4%.1 The existence of many hundreds of biologically active natural peptides with one or more NPAAs, reflects the ability of these particular monomers to confer some useful functional properties, not available in the basic set of 20 amino acids. The incorporation of NPAAs into peptides leads to modifications in the secondary structure that often results in improved selectivity, bioavailability, and stability.2 Moreover, they are extensively used in the synthesis of peptide catalysts,3 and functional materials.4 Finally, NPAAs are versatile intermediates in organic synthesis, especially in drug discovery. The broad utility of NPAAs therefore makes them one of the most important classes of organic molecules.Given the importance and application of NPAAs, there is a strong impetus to access various collections of NPAAs and understand their influence on the structural properties of the molecule into which they are incorporated. The focus of our research is on C-glycosyl amino acids, a group of C-glycosides in which a carbohydrate molecule is linked to the side chain or backbone of the amino acid by a C–C bond. C-Glycosyl amino acids are found in nature, mostly as bacterial secondary metabolites, for example peptidyl nucleoside antibiotic amipurimycin, neodysiherbaine with powerful neuropharmacological activity, and nikkomycin, which have potent antimycotic activities against various human pathogenic fungi and bacteria.5 However, compared to O- and N-glycosylation, installation of C-glycosyl units is much more difficult and has lagged far behind. Methodologies developed for the synthesis of C-glycosyl α-amino acids include alkylation of α-amino acid equivalents, Strecker reactions, hydrogenation of dehydroamino acids, multicomponent reactions with sugar derivatives, and de novo synthesis of C-glycosyl amino acids.6–10 Also, olefin cross-metathesis/cyclization strategy,11 condensation of barbituric acid with unprotected carbohydrates followed by subsequent barbiturate oxidative cleavage12 and Ni-catalysed reductive hydroglycosylation of alkynes13 were employed. Although often reliable, these methods suffer from multistep synthetic procedures, harsh reaction conditions, and limited substrate scope, accompanied with poor control of stereoselectivity. Novel, mild alternatives include photo-catalysed addition of glycosyl radicals to α-imino esters14 and photo-induced Cu-catalysed asymmetric C(sp3)–H alkylation of glycine derivatives,15 but purification of such radical reactions could be difficult and often hinder scale-up. Despite developed methodologies, most of published data refer to amino acids with carbohydrate unit placed at the side-chain,10,13,16 or C-glycosyl β-amino acids,8,9,11,16b while examples with C-glycosyl α-amino acids with carbohydrate unit placed directly to Cα atom,12,14,15 or C-glycosyl γ-amino acids are scarce17 (Fig. 1A).
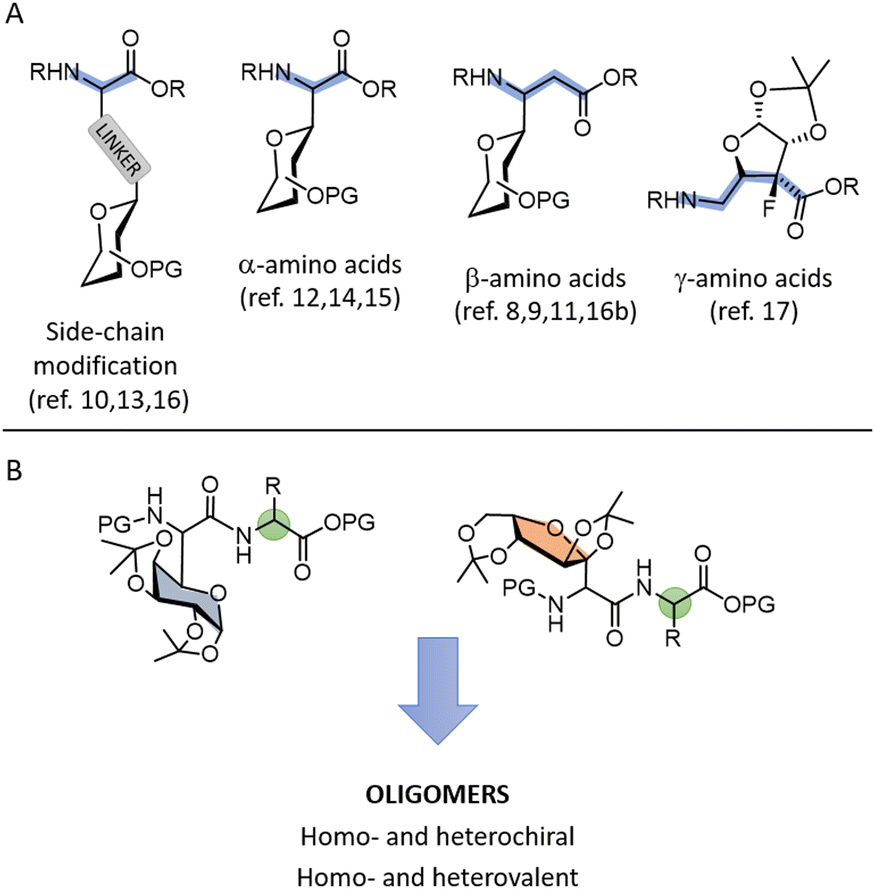 | ||
| Fig. 1 (A) Types of C-glycosyl amino acids. (B) Two types of dipeptides with embedded C-glycosyl α-amino acids prepared in this work. | ||
Site-specific modification of peptides with C-glycosyl amino acid(s) has been extensively studied and has provided significant knowledge about the impact of such modification on stability or affinity for biomolecules.18,19 In contrast, methods for peptide synthesis with embedded C-glycosyl amino acids have been almost neglected, so that knowledge about the effects of C-glycosyl amino acid(s) incorporated into peptides on their structural and functional properties is very limited. Therefore, for a broader and more efficient utilisation of C-glycosyl amino acids, a reliable synthetic protocol is required not only for their synthesis but also for their oligomers.
Addressing this gap, we aimed to explore the scope and limitations of peptide synthesis with proteinogenic amino acids and two types of C-glycosyl α-amino acids. In our previous work, we used mono- and bis-isopropylidene-protected carbohydrate aldehydes in the Passerini reaction to obtain α-acyloxyamides with C-glycosyl moiety. The reaction is robust and highly efficient with various carbohydrates, while the diastereoselectivity is affected by the structure of the carbohydrate aldehyde and the best results gave 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 d.r.20 The S-configuration of the newly formed chiral centre in the main diastereoisomer was confirmed. In this work, we report the post-Passerini modification to obtain C-glycosyl α-amino acids. The use of α-amino acid-derived isocyanides in the Passerini reaction allows the formation of hybrid dipeptide structures containing C-glycosyl α-amino acid and proteinogenic amino acid (Fig. 1B). Such building blocks enable access to oligomers with an alternating distribution of C-glycosyl amino acid and proteinogenic amino acid and provide insight into the role of monomer structure and chirality on the formation of different oligomeric structures.
:10 d.r.20 The S-configuration of the newly formed chiral centre in the main diastereoisomer was confirmed. In this work, we report the post-Passerini modification to obtain C-glycosyl α-amino acids. The use of α-amino acid-derived isocyanides in the Passerini reaction allows the formation of hybrid dipeptide structures containing C-glycosyl α-amino acid and proteinogenic amino acid (Fig. 1B). Such building blocks enable access to oligomers with an alternating distribution of C-glycosyl amino acid and proteinogenic amino acid and provide insight into the role of monomer structure and chirality on the formation of different oligomeric structures.
Results and discussion
The anticipated methodology for the synthesis of C-glycosyl α-amino acids is based on the modification of the Passerini product; hydrolysis followed by the OH → NH2 transformation (with inversion of the configuration) to obtain C-glycosyl α-amino acid (Scheme 1). The main advantages of C-glycosyl amino acids are their tuneable properties in terms of ring size, stereochemistry, substituents, conformation and adjustable hydrophilic/hydrophobic character as a function of the free/protected nature of their OH groups. For this study we selected two types of glycosyl donors: 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose-derived aldehyde (D-Gal) to access the C-glycosyl α-amino acid in pyranose form and with tertiary Cβ atom, while 2,3:4,6-di-O-isopropylidene-α-L-sorbofuranose-derived aldehyde (L-Sor) enabled access to C-glycosyl α-amino acid in furanose form and with tetrasubstituted Cβ atom (Scheme 1). As isocyanide sources we chose the hydrophobic leucine, valine and phenylalanine in both chiral forms to obtain homo- and heterochiral dipeptides, and additionally the non-chiral amino acid glycine (Scheme 1). | ||
| Scheme 1 General approach towards dipeptides comprising C-glycosyl α-amino acid by modification of Passerini products. Green circle in glycosyl donors represents Cβ atom in C-glycosyl α-amino acids (tertiary for D-Gal vs. tetrasubstituted for L-Sor). | ||
Carbohydrate aldehydes were obtained by oxidation of the corresponding bis-isopropylidene-protected α-D-galactopyranose and α-L-sorbofuranose with Dess–Martin periodinane, while isocyanides were obtained by dehydration of the amino acid-derived N-formamides. The Passerini products Gal-1a–Gal-1g were obtained from galactose-derived aldehyde, acetic acid and selected amino acid-derived isocyanide according to published methodology.20 All products were isolated in excellent yield (80–95%) as an inseparable mixture of two diastereoisomers (Scheme 2A). Diastereoselectivity was determined from the 1H NMR spectra of the isolated products and ranged from 65:35 d.r. found in product Gal-1g to 80:20 d.r. found in products Gal-1d, Gal-1e and Gal-1f. Therefore, any specific sterical or stereochemical influence of the amino acid isocyanide was not observed and homochiral and heterochiral Passerini products were obtained in comparable yields and diastereoselectivity. Two diastereoisomers of Gal-1b were separated by column chromatography, however, separation was slow and incomplete and resulted in the loss of material, so we proceeded without separation of diastereoisomers for all Passerini products.
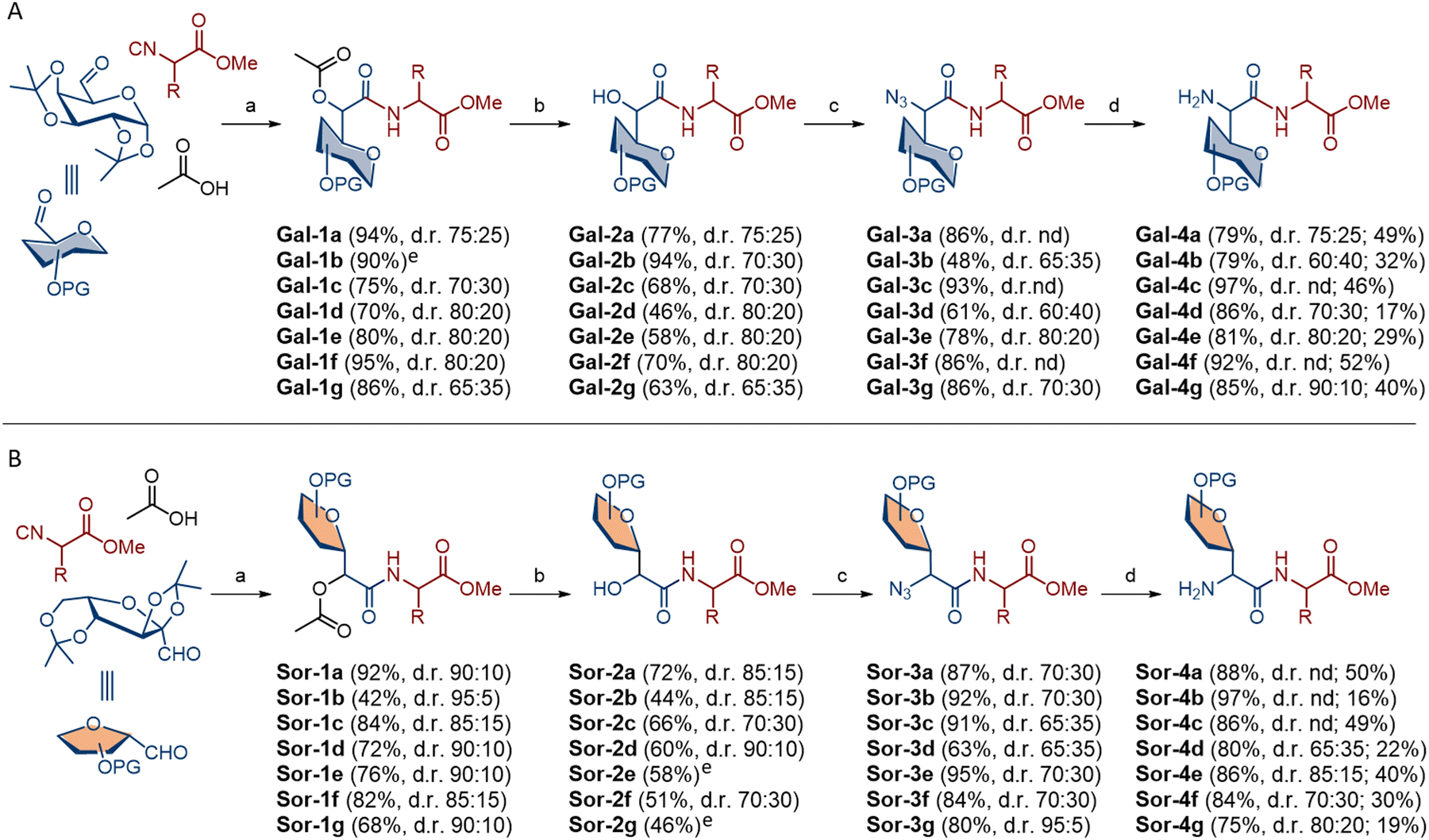 | ||
| Scheme 2 Synthesis of homo- and heterochiral dipeptides containing C-glycosyl α-amino acid derived from D-galactose (A) and L-sorbose (B). Reagent and conditions: (a) 0.1 mmol of all components in DCM, at RT, overnight; (b) NaOH (5 eq.) in MeOH (0.1 M), RT, 5 h; then K2CO3 (1.5 eq.) and MeI (3 eq.) in DMF (0.2 M) at 75 °C, overnight; (c) pyridine (10 eq.) and Tf2O (3 eq.) in DCM (0.04 M) at −20 °C, 3 h; then NaN3 (5 eq.) in DMF (0.2 M) at −20 °C, 24 h; (d) NaBH4 (1.5 eq.) and NiCl2·x6H2O (0.01 eq.) in DCM–EtOH (1:5, v/v, 0.02 M), at RT, 12 h; (e) main diastereoisomer was isolated by column chromatography; nd – not determined. Overall yields of Gal-4 and Sor-4 are also given (second number in parenthesis). | ||
Hydrolysis of the Passerini products Gal-1a–Gal-1g under basic conditions gave α-hydroxy C-glycosyl dipeptides as free acids, which were converted to methyl esters Gal-2a–Gal-2g with MeI and K2CO3 in DMF at 46–94% yield over two reaction steps. Next step was α-hydroxyl group activation with triflic anhydride, and the subsequent nucleophilic substitution with sodium azide afforded azido dipeptides Gal-3a–Gal-3g in 48–93% yield over two reaction steps. Finally, azide reduction to amine was performed with NaBH4 and dipeptide esters Gal-4a–Gal-4g were obtained in 85–97% yield (Scheme 2A). Following the same approach, sorbose-derived Passerini products Sor-1a–Sor-1g were isolated in 42–92% yield with diastereoselectivity from 85:15 d.r. to 95:5 d.r. (Scheme 2B). Unfortunately, as with galactose-related Passerini products, separation of two diastereoisomers failed. Hydrolysis of Passerini products followed by the C-carboxyl group protection afforded methyl esters Sor-2a–Sor-2g at 44–72% yield, while azides Sor-3a–Sor-3g were obtained in 63–95% yield over two reaction steps. At the final step, azide reduction gave dipeptide esters Sor-4a–Sor-4g in 75–97% yield.
Comparison of two glycosyl donors revealed very good to excellent reactivity with one or two exceptions in each reaction step, and dipeptides were obtained in 16–52% overall yield. We also tested the robustness of each reaction step. Most reactions were performed at the 1–3 mmol scale, but they were also tolerated at the 5 mmol scale. However, the best yields for the Passerini products and amines were obtained at the 2 mmol scale. The main limitation of the method is the large-scale synthesis of isocyanides from amino acids, where the dehydration of the N-formamides was accompanied by a loss of enantiomeric purity. In addition, isocyanides stored at −20 °C for a month or longer undergo slow racemisation. These limitations should be considered when working with amino acid-derived isocyanides. In addition, purification of sorbose-related dimers in each reaction step were generally more challenging, resulting in somewhat lower yields and disturbance of the diastereomeric ratio. Despite the complexity caused by the presence of diastereoisomers, the NMR spectra of the individual compound classes show some common features. The formation of a new stereogenic centre in the Passerini reaction is evident by the appearance of a signal at ∼5.10 ppm for the major S isomer and ∼5.30 ppm for the minor R isomer in the Gal series. In the Sor series, the difference in the chemical environment of the new stereogenic centre in two diastereoisomers is less pronounced and the major S isomer is found ∼5.30 ppm, while the minor R isomer is found slightly further down (∼5.40 ppm). The removal of the acetyl group in the Passerini compounds resulted in the stereogenic proton being found in a higher field (4.5–5 ppm) in both the Gal and Sor series. The NMR spectra of the dipeptides Gal-3 and Sor-3 as well as the dipeptides Gal-4 and Sor-4 are characterised by minor changes compared to the α-hydroxyl esters Gal-2 and Sor-2.
With a library of dipeptides bearing two types of C-glycosyl amino acids, we embarked on the synthesis of oligomers. We opted for the solution-phase [2+2] condensation to obtain corresponding tetramers. Amine group protection was carried out with benzyl chloroformate followed by the methyl ester removal. Cbz-protected dipeptides Gal-5a–Gal-5g were obtained in 37–79% yield over two reaction steps. Quite unexpectedly, we experienced difficulties with Cbz-protection of sorbose-related dipeptides. Reaction was incomplete, and purification of products from the reaction mixture was unsuccessful. We tried to optimize the reaction conditions by replacing benzyl chloroformate with dibenzyl dicarbonate and performing methyl ester removal before Cbz protection, but without improvements. We managed to isolate only Sor-5g in 39% yield. Nevertheless, sorbose-related dipeptides we utilized as amine components in coupling with Cbz-protected galactose-related dipeptides (below).
The formation of amide bonds with sterically hindered amino acids is often challenging and requires modification of traditional amide synthesis mediated by coupling reagents. In our group of dipeptides, the presence of a bulky, isopropylidene-protected carbohydrate motif near the peptide backbone could hinder nucleophilic access to the activated carboxyl group, especially for branched amino acids such as leucine and valine. So, we tested dipeptide couplings with the uronium-type HATU reagent in DCM at room temperature and were pleased with outcomes. In galactose series, six homo-valent tetrapeptides were obtained in very good yield (60–81%, Scheme 3), while the formation of Gal-6g was confirmed with LC-MS, but its purification was difficult and incomplete. Sorbose-related tetrapeptide Sor-6g was successfully obtained in 55% yield (Scheme 3). In addition, the protocol can be applied for further peptide elongation. Coupling of Gal-6d with Gal-4d ([4+2] condensation) was as efficient as coupling of Gal-5d with Cbz deprotected Gal-5d ([2+4] condensation) and afforded Gal-7d in 95% and 88% yield, respectively (Scheme 3).
 | ||
| Scheme 3 Synthesis of tetramers. Reagent and conditions: (a) Z-Cl (1.5 eq.) and DIPEA (3 eq.) in DCM, at RT, overnight; then NaOH (5 eq.) in MeOH, at RT, 5 h; (b) HATU (1.1 eq.) NMM (1.1 eq.), corresponding Gal-4 or Sor-4g (1 eq.) in DCM, RT, overnight. Structure of hexapeptide Gal-7d is given in the rectangle. | ||
Although coupling reaction were performed with diastereomeric mixtures of dipeptides Gal-5 and Gal-4, all tetramers, homo- and heterochiral, were obtained in good to very good yields. This implies that the coupling reaction is quite robust and the chirality of the C-terminal amino acid in Gal-5 has no significant influence on the nucleophilic attack of the amino components Gal-4.
Next, we aimed to synthesize heterovalent tetrapeptides by coupling selected galactose- and sorbose-related dipeptides. Although the protection of sorbose-related dipeptides proved to be problematic, the coupling of Gal-5a and Sor-4a gave, to our delight, tetramer 8 in 88% yield, while the coupling of Gal-5f and Sor-4f gave tetramer 9 in 91% yield (Scheme 4). Finally, coupling of the homochiral galactose-related dipeptide Gal-5f with the heterochiral sorbose-related dipeptide Sor-4a gave the heterochiral and heterovalent tetramer 10 in 94% yield. The method is therefore flexible and can also be used for the formation of “mixed” tetrapeptides.
 | ||
| Scheme 4 Heterovalent tetrapeptides. | ||
Finally, we tested isopropylidene groups deprotection under acidic conditions. Treatment of Gal-6c and Gal-6g with TFA/H2O (9:1) at room temperature resulted in removal of isopropylidene groups after 5 h giving a mixture of multiple anomeric forms.
Conclusions
In conclusion, we have presented a protocol for the synthesis of oligomers containing alternating C-glycosyl α-amino acids and proteinogenic α-amino acids. The protocol is based on the coupling of dipeptide building blocks obtained by the modification of Passerini products. α-D-Galactopyranose- and α-L-sorbofuranose-derived aldehydes along with leucine, valine and phenylalanine isocyanides in both chiral forms, as well as glycine isocyanide, were used for the formation of dipeptide building blocks. The protocol enabled the synthesis of homo- and heterochiral tetramers in very good yields. Furthermore, the synthetic utility of the protocol was demonstrated by the synthesis of homo- and heterovalent tetramers, and even hexamers are accessible with this approach.Non-proteinogenic amino acids have wide application in medicinal chemistry, development of catalyst and functional materials. However, many classes of non-proteinogenic amino acids remain unexploited due to a lack of a synthetic protocol and/or sufficient knowledge about their influence on the structural properties of the molecule into which they are incorporated. Our next step will therefore be the systematic replacement of proteinogenic amino acid(s) in selected peptides (e.g. β sheet-forming peptides) with specific C-glycosyl α-amino acid(s) to determine how their number and distribution within the peptide chain (alternating, consecutive, site-specific) modulate peptide conformation.
Experimental
General procedures for all reaction steps are given below, together with characterization data for one representative compounds, while characterization data and NMR spectra for all compounds are provided in the ESI† file. (3aR,5S,5aR,8aS,8bR)-2,2,7,7-tetramethyltetra-hydro-5H-bis([1,3]dioxolo)[4,5-b:4′,5′-d]pyran-5-carbaldehyde (Gal-related aldehyde) and (3aS,3bR,7aS,8aR)-2,2,5,5-tetramethyltetrahydro-8aH-[1,3]dioxolo[4′,5′:4,5]furo[3,2-d][1,3]dioxine-8a-carbaldehyde (Sor-related aldehyde) were prepared following published procedure.21General procedure for the synthesis of amino acid-derived isocyanides
Amino acid methyl ester (1 equiv.) was dissolved in DCM and solution was cooled to 0 °C. Formic acid was added dropwise followed by the addition of N,N′-dicyclohexylcarbodiimide (DCC, 1.3 equiv.) and 4-dimethylaminopyridine (DMAP, 0.2 equiv.). The reaction mixture was stirred overnight at room temperature. The reaction was terminated by addition of saturated NaHCO3 solution, extracted with DCM and washed with saturated NaCl solution. The organic layer was dried over anhydrous Na2SO4. The product was purified by flash chromatography on a silica gel column in a solvent system: petrol ether/EtOAc (1:1). Isolated product was dissolved in dry THF and cooled to −78 °C. NMM (4 equiv.) was added dropwise followed by the addition of triphosgene (0.5 equiv.) dissolved in dry THF. Reaction mixture was stirred for 3 hours at −78 °C, and then terminated by addition of saturated NaHCO3 solution and extracted with DCM. The organic layer was dried over anhydrous Na2SO4, solvent evaporated and the residue purified by flash chromatography on a silica gel column in a solvent system: petrol ether/EtOAc (1:1).
:1, v/v); C8H13NO2). 1H NMR (600 MHz, CDCl3) δ 4.28 (dd, J = 9.9, 4.6 Hz, αLeu, 1H), 3.82 (s, OMe, 3H), 1.96–1.77 (m, ββ′Leu, 2H), 1.75–1.63 (m, γLeu, 1H), 0.98 (dd, J = 13.7, 6.6 Hz, δδ′Leu, 6H). 13C NMR (151 MHz, CDCl3) δ 167.5, 160.0, 54.9, 53.2, 41.2, 24.7, 22.5, 20.8.
General procedure for the synthesis of Passerini products
To a flask containing 0.5 M solution of galactose- or sorbose-derived aldehyde (1 equiv.) in dry DCM were added acetic acid (1 equiv.) and the isocyanide component (1 equiv.) dissolved in 200 μL of dry DCM. With all reactants added, the solution was allowed to stir overnight at room temperature. The reactions were concentrated under reduced pressure, and the residue was purified by flash column chromatography on a silica gel column in a solvent system: petrol ether/EtOAc (1:1).
:1, v/v); C22H35NO10; mixture of two diastereoisomers, d.r. 75:25. Chemical shifts are given for both diastereoisomers. 1H NMR (300 MHz, CDCl3): δ 6.69 (d, J = 7.9 Hz, NHLeu, 1H), 5.55–5.48 (m, H1Gal, 1H), 5.35 (d, J = 7.2 Hz, αGal, 0.25H), 5.08 (d, J = 9.6 Hz, αGal, 0.75H), 4.68–4.54 (m, αLeu, Gal, 2H), 4.35–4.26 (m, Gal, 2H), 4.24–4.17 (m, Gal, 1H), 3.72, 3.70 (s, OMe, 3H), 2.15, 2.14 (s, OAc, 3H), 1.72–1.60 (m, ββ′Leu, 2H), 1.57–1.53 (m, γLeu, CH3, 4H), 1.46, 1.44 (s, CH3, 3H), 1.33–1.29 (m, CH3, 6H), 0.96–0.89 (m, δδ′Leu, 6H). 13C NMR (151 MHz, CDCl3) δ 172.9, 172.7, 171.3, 170.5, 169.6, 167.9, 166.9, 109.8, 109.7, 109.6, 109.2, 96.4, 96.3, 73.0, 71.3, 70.9, 70.8, 70.6, 70.5, 70.4, 67.2, 66.9, 60.2, 52.5, 52.2, 51.1, 50.9, 41.5, 41.4, 29.8, 26.2, 26.1, 25.14, 25.10, 24.9, 24.9, 24.5, 24.3, 22.9, 22.8, 22.2, 22.1, 21.2, 21.0, 20.8.
General procedure for the synthesis of α-hydroxy dipeptide esters
Passerini product was dissolved in methanol (c = 0.1 M) and solid NaOH was added (5 equiv.) Reaction mixture was stirred at room temperature for 5 hours. Solvent was evaporated and the residue purified by flash column chromatography on a silica gel column in a solvent system: petrol ether/EtOAc (1:1) and EtOAc/EtOH/AcOH/H2O (70:10:2:2). Isolated hydroxy acid was dissolved in dry DMF (c = 0.2 M), K2CO3 (1.5 equiv.) and MeI (3 equiv.) were added, and the reaction mixture was stirred overnight at 75 °C. The reactions were concentrated under reduced pressure, and the residue was dissolved in DCM and extracted with saturated NaHCO3 solution. Organic layer was washed with saturated NaCl solution, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column in a solvent system: petrol ether/EtOAc (1:1).
:1, v/v); C20H33NO9; mixture of two diastereoisomers d.r. 75:25. Chemical shifts are given for both diastereoisomers. 1H NMR (600 MHz, CDCl3) δ 7.25–7.08 (m, NHLeu, 1H), 5.61–5.46 (m, H1Gal, 1H), 4.67–4.55 (m, αLeu, Gal, 2H), 4.53–4.44 (m, Gal, 1H), 4.42–4.19 (m, Gal, 2H), 4.05 (t, J = 6.9 Hz, Gal, 1H), 3.72 (s, OMe, 3H), 1.72–1.57 (m, ββ′γLeu, 3H), 1.54–1.44 (m, CH3, 6H), 1.42–1.27 (m, CH3, 6H), 1.01–0.89 (m, δδ′Leu, 6H). 13C NMR (151 MHz, CDCl3) δ 172.9, 172.6, 172.3, 171.3, 109.9, 109.7, 109.4, 109.4, 96.5, 96.4, 73.5, 72.7, 71.4, 71.0, 70.8, 70.7, 70.6, 70.4, 67.3, 66.7, 60.5, 52.4, 51.1, 51.0, 50.8, 41.4, 41.2, 29.8, 26.2, 25.9, 25.3, 25.1, 24.9, 24.1, 24.0, 22.9, 22.1, 21.9, 14.3.
General procedure for the synthesis of azido dipeptide esters
α-hydroxy derivative was dissolved in dry DCM (c = 0.04 M) and pyridine (10 equiv.) and trifluoromethanesulfonic anhydride (3 equiv.) were added dropwise to precooled solution (−20 °C). The reaction mixture was stirred under argon for 3 hours, and then extracted with water. The organic layer was washed with 10% solution of KHSO4, saturated NaHCO3 solution and saturated NaCl solution, and then dried over Na2SO4. The solvent was evaporated, and the crude product was used in the next step.Triflate derivative was dissolved in dry DMF (c = 0.2 M), cooled to 0 °C and NaN3 (5 equiv.) was added. The reaction was stirred at room temperature for 12 hours. Solvent was evaporated, residue dissolved in EtOAc and extracted with saturated NaHCO3 solution. The organic layer was dried over anhydrous Na2SO4, solvent evaporated and the residue purified by flash chromatography on a silica gel column in a solvent system: petrol ether/EtOAc (1:1).
:1, v/v); C20H32N4O8; mixture of two diastereoisomers, d.r. nd. Chemical shifts are given for both diastereoisomers. 1H NMR (300 MHz, CDCl3) δ 6.90–6.56 (m, NHLeu, 1H), 5.62–5.51 (m, H1Gal, 1H), 4.71–4.55 (m, αGal, αLeu, 2H), 4.48–4.25 (m, Gal, 2H), 4.21–3.94 (m, Gal, 2H), 3.73 (br s, OMe, 3H), 1.72–1.63 (m, ββ′γLeu, 3H), 1.56–1.46 (m, CH3, 6H), 1.39–1.17 (m, CH3, 6H), 0.94 (d, J = 5.8 Hz, δδ′Leu, 6H). 13C NMR (151 MHz, CDCl3) δ 172.9, 172.7, 171.3, 168.2, 167.1, 109.9, 109.8, 109.6, 109.4, 96.6, 96.5, 71.475, 71.1, 70.7, 70.7, 70.6, 70.5, 68.5, 68.3, 67.3, 63.3, 62.9, 61.6, 60.5, 52.6, 52.4, 51.1, 51.0, 41.3, 29.8, 26.1, 26.1, 26.0, 26.0, 25.1, 25.0, 24.9, 24.9, 24.4, 24.4, 22.9, 22.9, 22.0, 21.2.
General procedure for the synthesis of C-glycosyl dipeptides
Azide derivative was dissolved in DCM–MeOH mixture (1:5, v/v, 0.02 M) and NaBH4 (1.5 equiv.) and NiCl2·6H2O (0.01 equiv.) were added. The reaction mixture was stirred at room temperature until the consumption of the starting compound (typically 12 h). The solvent was evaporated, and the residue was extracted with DCM and a saturated NaHCO3 solution. The organic layer was dried over Na2SO4 and evaporated.
:1, v/v); C20H34N2O8; mixture of diastereoisomers d.r. 75:25. Chemical shifts are given for both diastereoisomers. 1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 8.3 Hz, NHLeu, 0.25H), 7.67 (br d, NHLeu, 0.75H), 5.56 (br d, H1Gal, 0.25H), 5.52 (br d, H1Gal, 0.75H), 4.75–4.51 (m, αGal, αLeu, 2H), 4.49–4.23 (m, Gal, 2H), 4.16 (br d, Gal, 0.25H), 4.12 (br d, Gal, 0.75H), 3.71 (br s, OMe, 3H), 3.68–3.56 (m, Gal, 1H), 1.93–1.76 (m, βLeu, 1H), 1.73–1.59 (m, β′γLeu, 2H), 1.58–1.52 (m, CH3, 3H), 1.46 (s, CH3, 3H), 1.35–1.27 (m, CH3, 6H), 1.09–0.80 (m, δδ′Leu, 6H). 13C NMR (151 MHz, CDCl3) δ 173.6, 173.2, 172.5, 172.0, 109.43, 109.41, 109.24, 109.18, 96.6, 96.5, 73.3, 73.1, 71.7, 71.1, 70.9, 70.8, 68.2, 67.8, 67.5, 66.9, 56.1, 55.9, 52.3, 52.3, 50.8, 50.7, 41.6, 41.3, 26.2, 26.1, 26.0, 25.3, 25.1, 25.1, 24.9, 24.3, 24.0, 23.1, 22.9, 22.1, 21.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H34N2O8 431.2393; found 431.2389.
General procedure for the synthesis of Cbz-protected dipeptides
C-Glycosyl α-amino acid containing dipeptide was dissolved in dry DCM (c = 0.1 M) and N,N-diisopropylethylamine (DIPEA, 3.0 equiv.) was added. The reaction mixture was stirred at room temperature under argon for 15 minutes, and then benzyl chloroformate (1.5 equiv.) was added dropwise. With all reactants added, the solution was allowed to stir under argon overnight at room temperature. The reaction was terminated by addition of saturated NaHCO3, extracted with DCM and washed with saturated NaCl solution. The organic layer was dried over anhydrous Na2SO4 and evaporated. The product was purified by flash chromatography on a silica gel column in a solvent system: DCM/MeOH (10:1). The isolated Cbz-protected product was dissolved in methanol (c = 0.1 M) and solid NaOH was added (5 equiv.) Reaction mixture was stirred at room temperature for 5 hours. The reaction was quenched by adding a 10% citric acid solution to adjust the pH of the solution to 4. Then, it was extracted with DCM and washed with saturated NaCl solution. The organic layer was dried over anhydrous Na2SO4, the solvent was evaporated, and the crude product was used in the next step.
:1, v/v); C27H38N2O10; mixture of diastereoisomers d.r. 70:30. Chemical shifts are given for both diastereoisomers. 1H NMR (600 MHz, CDCl3) δ 7.49–7.21 (m, ArCbz, NH, 6H), 7.19–6.81 (m, NH, 1H), 5.73–5.41 (m, Gal, 2H), 5.27–4.99 (m, CH2Cbz, 2H), 4.63–4.53 (m, Gal, αLeu, 2.3H), 4.48–4.44 (m, αLeu, 0.7H), 4.31–4.25 (m, Gal, 2H), 1.70–1.40 (m, ββ′γLeu, CH3, 6H), 1.40–1.22 (m, CH3, 9H), 1.08–0.80 (m, δδ′Leu, 6H). 13C NMR (151 MHz, CDCl3) δ 175.7, 175.1, 169.8, 169.7, 157.0, 136.1, 136.0, 128.7, 128.6, 128.5, 128.4, 128.2, 127.9, 127.8, 127.7, 127.0, 109.6, 109.3, 96.5, 96.2, 71.6, 70.9, 70.8, 70.7, 70.4, 67.4, 66.7, 66.5, 65.8, 65.3, 56.5, 56.1, 51.1, 50.9, 47.5, 46.3, 41.3, 40.8, 29.7, 26.1, 26.0, 25.8, 25.8, 25.0, 24.9, 24.9, 24.7, 24.0, 23.9, 22.9, 22.7, 21.9, 21.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H39N2O10 551.26047; found 551.26044.
General procedure for the synthesis of oligomers
Cbz-protected dipeptide was dissolved in dry DCM (c = 0.05 M). N-Methylmorpholine (NMM, 1.1 equiv.) was added dropwise followed by the addition of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, 1.1 equiv.). The reaction mixture was stirred at room temperature for 30 minutes, and then the amino component (1 equiv.) dissolved in 500 μL of dry DCM, was added dropwise. Reaction mixture was allowed to stir overnight at room temperature. The reaction was quenched by adding a saturated NH4Cl solution, then extracted with DCM and washed with saturated NaCl solution. The organic layer was dried over anhydrous Na2SO4, the solvent evaporated, and the residue purified by flash chromatography on a silica gel column using a solvent system: DCM/MeOH (10:1, v/v).
Tetramer Gal-6a
Yield: 75% (79 mg); yellow oil; Rf = 0.48 (DCM/MeOH 10:1, v/v); C47H70N4O17.1H NMR (600 MHz, CDCl3) δ 7.48–6.77 (m, 9H), 5.63–5.38 (m, 2H), 5.31–4.98 (m, 2H), 4.60–4.22 (m, 12H), 3.82–3.58 (m, 3H), 1.71–1.57 (m, 6H), 1.55–1.41 (m, 12H), 1.32–1.27 (m, 12H), 1.02–0.78 (m, 12H). 13C NMR (151 MHz, CDCl3) δ 173.0, 172.9, 172.1, 172.0, 170.1, 169.7, 169.6, 169.3, 169.0, 168.9, 157.1, 156.8, 136.3, 136.1, 128.9, 128.7, 128.6, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.8, 109.5, 109.4, 109.3, 96.7, 96.6, 96.4, 96.3, 96.2, 72.0, 71.9, 71.8, 71.1, 71.0, 70.98, 70.87, 70.8, 67.4, 67.2, 66.8, 66.65, 66.60, 66.24, 66.18, 54.2, 54.1, 52.32, 52.26, 51.3, 51.2, 41.1, 41.0, 40.9, 40.6, 40.4, 26.2, 26.10, 26.07, 26.0, 25.95, 25.90, 25.21, 25.19, 25.1, 24.8, 24.7, 24.6, 24.2, 24.1, 24.04 23.2, 23.1, 23.0, 22.9, 22.9, 22.8, 22.2, 22.14, 22.12, 21.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C47H71N4O17 963.4814; found 963.4809.
Author contributions
Ivana Colić: conceptualization, methodology, investigation; Barbara Bogović: investigation; Ivanka Jerić: writing–original draft, review and editing, supervision, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Croatian Science Foundation, Grant Number 9617.References
- Y. Ding, J. P. Ting, J. Liu, S. Al-Azzam, P. Pandya and S. Afshar, Amino Acids, 2020, 52, 1207 CrossRef CAS PubMed.
- (a) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807 CrossRef CAS PubMed; (b) A. Wester, M. Devocelle, E. A. Tallant, M. C. Chappell, P. E. Gallagher and F. Paradisi, Amino Acids, 2017, 49, 1733 CrossRef CAS PubMed; (c) J. S. M. Svendsen, T. M. Grant, D. Rennison, M. A. Brimble and J. Svenson, Acc. Chem. Res., 2019, 52, 749 CrossRef CAS PubMed.
- (a) A. J. Metrano, A. J. Chinn, C. R. Shugrue, E. A. Stone, B. Kim and S. J. Miller, Chem. Rev., 2020, 120, 11479 CrossRef CAS PubMed; (b) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed; (c) A. E. Wendlandt, P. Vangal and E. N. Jacobsen, Nature, 2018, 556, 447 CrossRef CAS PubMed; (d) Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Science, 2017, 355, 162 CrossRef CAS PubMed; (e) Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2020, 142, 17211 CrossRef CAS PubMed.
- A. Levin, T. Hakala, L. Schnaider, G. Bernardes, E. Gazit and T. Knowles, Nat. Rev. Chem., 2020, 4, 615 CrossRef CAS.
- S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591 RSC.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281 CrossRef CAS PubMed.
- H. Liao, J. Ma, H. Yao and X.-W. Liu, Org. Biomol. Chem., 2018, 16, 1791 RSC.
- A. Dondoni and A. Massi, Acc. Chem. Res., 2006, 39, 451 CrossRef CAS PubMed.
- A. Dondoni, A. Massi and S. Sabbatini, Chem. – Eur. J., 2005, 11, 7110 CrossRef CAS PubMed.
- S. Koch, D. Schollmeyer, H. Löwe and H. Kunz, Chem. – Eur. J., 2013, 19, 7020 CrossRef CAS PubMed.
- E. G. Nolen, A. J. Kurish, J. M. Potter, L. A. Donahue and M. D. Orlando, Org. Lett., 2005, 7, 3383 CrossRef CAS PubMed.
- M.-C. Frantz, S. Dropsit-Montovert, F. Pic, A. Prévot-Guéguiniat, C. Aracil, Y. Ding, M. Lima, F. Alvarez, S. Ramos, L. Mao, L. Lu, J. Xu, X. Marat and M. Dalko-Csiba, Org. Lett., 2019, 21, 2684 CrossRef CAS PubMed.
- Y.-H. Liu, Y.-N. Xia, T. Gulzar, B. Wei, H. Li, D. Zhu, Z. Hu, P. Xu and B. Yu, Nat. Commun., 2021, 12, 4924 CrossRef CAS PubMed.
- P. Ji, Y. Zhang, Y. Wei, H. Huang, W. Hu, P. A. Mariano and W. Wang, Org. Lett., 2019, 21, 3086 CrossRef CAS PubMed.
- R. Qi, C. Wang, Z. Ma, H. Wang, Q. Chen, L. Liu, D. Pan, X. Ren, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2022, 61, e2022008 Search PubMed.
- (a) N. R. M. Reintjens, T. S. Koemans, N. Zilverschoon, R. Castelli, R. A. Cordfunke, J. Wouter Drijfhout, N. J. Meeuwenoord, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel and J. D. C. Codée, Eur. J. Org. Chem., 2020, 5126 CrossRef CAS; (b) K. Jayakantha and Y. D. Vankar, Org. Lett., 2005, 7, 5441 CrossRef PubMed.
- S. S. Burade, T. Saha, N. Bhuma, N. Kumbhar, A. Kotmale, P. R. Rajamohanan, R. G. Gonnade, P. Talukdar and D. D. Dhavale, Org. Lett., 2017, 19, 5948 CrossRef CAS PubMed.
- S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Macromol. Biosci., 2019, 19, 1800425 CrossRef PubMed.
- K. S. Bücher, P. B. Konietzny, N. L. Snyder and L. Hartmann, Chem. – Eur. J., 2019, 25, 3301 CrossRef PubMed.
- K. Vlahoviček-Kahlina, M. Vazdar, A. Jakas, V. Smrečki and I. Jerić, J. Org. Chem., 2018, 83, 13146 CrossRef PubMed.
- A. Jakas, A. Višnjevac and I. Jeric, J. Org. Chem., 2020, 85, 3766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and NMR spectra of all compounds. See DOI: https://doi.org/10.1039/d4nj02059f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |