
Integrative studies of ionic liquid interface layers: bridging experiments, theoretical models and simulations
Rong
An†
 *a,
Nanhua
Wu†
b,
Qingwei
Gao
c,
Yihui
Dong
d,
Aatto
Laaksonen
*efgh,
Faiz Ullah
Shah
i,
Xiaoyan
Ji
*e and
Harald
Fuchs
*aj
*a,
Nanhua
Wu†
b,
Qingwei
Gao
c,
Yihui
Dong
d,
Aatto
Laaksonen
*efgh,
Faiz Ullah
Shah
i,
Xiaoyan
Ji
*e and
Harald
Fuchs
*aj
aHerbert Gleiter Institute of Nanoscience, School of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: ran@njust.edu.cn
bKey Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering, Changzhou University, Changzhou 213164, China
cCollege of Environmental and Chemical Engineering, Shanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, Shanghai University of Electric Power, Shanghai 200090, China
dDepartment of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot, 76100, Israel
eEnergy Engineering, Division of Energy Science, Luleå University of Technology, 97187 Luleå, Sweden. E-mail: xiaoyan.ji@ltu.se
fDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, SE-10691 Stockholm, Sweden. E-mail: aatto@mmk.su.se
gCenter of Advanced Research in Bionanoconjugates and Biopolymers, ‘‘Petru Poni” Institute of Macromolecular Chemistry, Iasi 700469, Romania
hState Key Laboratory of Materials-Oriented and Chemical Engineering, Nanjing Tech University, Nanjing 211816, China
iChemistry of Interfaces, Luleå University of Technology, 97187 Luleå, Sweden
jCenter for Nanotechnology (CeNTech), Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany. E-mail: fuchsh@uni-muenster.de
First published on 10th February 2024
Abstract
Ionic liquids (ILs) are a class of salts existing in the liquid state below 100 °C, possessing low volatility, high thermal stability as well as many highly attractive solvent and electrochemical capabilities, etc., making them highly tunable for a great variety of applications, such as lubricants, electrolytes, and soft functional materials. In many applications, ILs are first either physi- or chemisorbed on a solid surface to successively create more functional materials. The functions of ILs at solid surfaces can differ considerably from those of bulk ILs, mainly due to distinct interfacial layers with tunable structures resulting in new ionic liquid interface layer properties and enhanced performance. Due to an almost infinite number of possible combinations among the cations and anions to form ILs, the diversity of various solid surfaces, as well as different external conditions and stimuli, a detailed molecular-level understanding of their structure–property relationship is of utmost significance for a judicious design of IL–solid interfaces with appropriate properties for task-specific applications. Many experimental techniques, such as atomic force microscopy, surface force apparatus, and so on, have been used for studying the ion structuring of the IL interface layer. Molecular Dynamics simulations have been widely used to investigate the microscopic behavior of the IL interface layer. To interpret and clarify the IL structure and dynamics as well as to predict their properties, it is always beneficial to combine both experiments and simulations as close as possible. In another theoretical model development to bridge the structure and properties of the IL interface layer with performance, thermodynamic prediction & property modeling has been demonstrated as an effective tool to add the properties and function of the studied nanomaterials. Herein, we present recent findings from applying the multiscale triangle “experiment–simulation–thermodynamic modeling” in the studies of ion structuring of ILs in the vicinity of solid surfaces, as well as how it qualitatively and quantitatively correlates to the overall ILs properties, performance, and function. We introduce the most common techniques behind “experiment–simulation–thermodynamic modeling” and how they are applied for studying the IL interface layer structuring, and we highlight the possibilities of the IL interface layer structuring in applications such as lubrication and energy storage.
 Rong An | Rong An received her PhD degree in Chemical Engineering from Nanjing Tech University, China, in 2013. She then worked as a postdoc researcher in Chemical and Biomolecular Engineering at North Carolina State University from 2013 to 2015. She joined School of Materials Science and Engineering/Herbert Gleiter Institute of Nanoscience, at Nanjing University of Science and Technology since 2015. Her current position is an Associate Professor. Her research interests include ionic liquid-solid interations, interface and surface science, liquid-solid interfacial phenomena, ionic liquid-based lubrication and energy storage, etc. |
 Nanhua Wu | Nanhua Wu received her Dual-PhD degree in Chemical Engineering from Nanjing Tech University, China, in 2019, and Luleå University of Technology, Sweden in 2020, respectively. She joined School of Petrochemical Engineering at Changzhou University since 2020. Her current position is an Associate Professor. Her research interests focus on the modeling of interfacial molecular thermodynamics in the area of catalysis, ionic liquids, membrane, etc. |
 Aatto Laaksonen | Aatto Laaksonen is a Professor of Physical Chemistry at Stockholm University (SU), PhD in 1981 (SU), PDF Daresbury Laboratory (UK) in 1982 with Vic Saunders and at IBM Poughkeepsie/Kingston (USA) from 1983 to 1985 with Enrico Clementi, Sabbatical at Dalhousie University 1993–1995 (Canada) with Rod Wasylishen. He is a visiting professor at JAERI 2002, 2005 (Japan), at Cagliari University 2008, 2009, 2011, 2015 (Italy), at Nanjing Tech University 2018-present (China), Petru Poni Institute 2018-present (Romania) and Luleå University of Technology 2020-present. His research focuses on multiscale modelling/simulations, model development in materials sciences and green chemical engineering. |
 Faiz Ullah Shah | Faiz Ullah Shah is an Associate Professor in Chemistry of Interfaces at Luleå University of Technology, Sweden. In 2011, he obtained his PhD degree in Chemistry from Luleå University of Technology, Sweden. Since 2013, he is leading a research group of PhD students, postdoctoral researchers and master students. His research is focused on the development of halogen-free ionic liquids and electrolytes for various energy related applications including batteries and supercapacitors. He has co-authored more than 80 research articles and is a co-inventor of eight patents. |
 Xiaoyan Ji | Xiaoyan Ji is a full Professor in Division of Energy Science at Luleå University of Technology, Sweden. She received her PhD degree in Chemical Engineering from Nanjing Tech University, China, in 2000 with 20+ years experience of research and development in the area of Chemical Engineering and Energy Engineering. Her research work is from theoretical modeling to technology developing, process simulating and integrating. Up to now, as a first author or main contributor, she has published 200+ journal articles. |
 Harald Fuchs | Harald Fuchs is a Chair-Professor of Experimental Physics at the University of Münster, Germany. He spent from 1984 to 1985 a post-doctoral year with IBM Research Laboratory Zurich in the group of G. Binnig and H. Rohrer. He is the founder of the Center for nanotechnology in Münster and its scientific director. His research focuses on nanoscale science and nanotechnology, ranging from scanning probe microscopy to self-organized nanostructure fabrication, on-surface chemical reactions, and nano-bio systems. He is an elected member of the German National Academy of Science ‘Leopoldina’, the German National Academy of Science and Engineering ‘acatech’, and the ‘TWAS’. |
1. Introduction
Ionic liquids (ILs) are a class of salts existing in the liquid state below 100 °C, while many of them are liquids already at room temperature. As salts, their ions (organic or inorganic) form relatively short-lived ion pairs. They possess unique properties of great technological interest, such as negligible vapor pressure, nonflammability, high thermal and chemical stability, wide liquid range and electrochemical window, high ionic conductivity, and more, all with a rational molecular designability for a great variety of applications due to their rich diversity of physical and chemical properties.1–3 Although the first IL, ethylammonium nitrate (EAN), was discovered more than a century ago, they have caught much attention from both the academic and industrial communities during the last two decades. The potential energy-related applications are solar and fuel cells, high-temperature heat transfer fluids, carbon capture and utilization, and biomass processing. In addition, the particularly interesting is their tribological applications as electrically conductive lubricants for modern electric vehicles4,5 and electrolytes for energy storage devices such as batteries and supercapacitors, due to their excellent electrochemical performance in general.6–14ILs in a variety of recent applications are often used in direct contact with solid surfaces or embedded in pores and constraint spaces,2,3 as the function of ILs at surfaces is considerably different and improved from that of bulk ILs. This is due to the altered property of ILs by forming well-defined interfacial layers with well-tunable structures.2,3,6,7,15 The ion structuring of the near-(solid) surface ILs is determined by a variety of attractive interactions ranging from short-range van der Waals forces to long-range electrostatic forces, and hydrogen bonding, etc.,2,16,17 depending on the surface properties (material, chemistry, roughness, curvature, etc.),3,16,18,19 the nature of ILs,6,20,21 as well as external conditions.7,22,23 Ion arrangements of the ILs in direct contact or close vicinity can be precisely controlled by fine-tuning the ILs and adapting the solid materials at the interfaces. For example, imidazolium-based ILs at the surface exhibit a dramatic shift towards a more ordered solid-like phase below a threshold thickness. This is related to the metallic nature of the underlying materials, i.e., more metallic surfaces facilitating the phase shift.18 The ILs with cations/anions having a longer alkyl chain exhibit well-pronounced “…cation–cation–anion–anion…” bilayer structures induced by van der Waals forces, rather than alternating cation–anion monolayer structures by electrostatic forces in shorter-chained ILs.6,21,24 External stimuli can play a critical role in the ion structuring of the IL interface layer. For example, charged surfaces1,25 can create fairly dense ion layers next to the surface, while decaying exponentially into the bulk IL.26 In this way, the tailorable ion structures of the IL interface layer largely determine their properties and performance.27,28 Considering the excessively large number of possible ILs (estimated to ∼1018) as well as the great diversity of available solid surfaces and external conditions and stimuli, theoretical modeling is critically important to bridge the interfacial structure and properties/performance of the IL interface layer and to provide a quantitative understanding of the structure–property relationships, which is a prerequisite for the design of ILs with properties tailored for targeted applications.
Many experimental techniques have been used to study the ion structuring of the IL interface layer.2,23,29 Among them, surface force apparatus (SFA)30–34 and atomic force microscopy (AFM)17,25,33,35,36 have emerged as more important experimental tools for detailed investigations due to their accuracy and atomic resolution. In parallel, molecular simulations, such as molecular dynamics (MD) and Monte Carlo (MC), have been widely used to investigate the structures of the IL interface layer.23,37 There is no doubt that it is important to combine simulation and modeling with advanced experimental techniques to interpret and clarify the IL structure at the microscopic level, as well as to predict the properties. But even further, to better bridge the experiments and simulations is to include modern thermodynamic property & prediction modeling in the constellation to create a powerful multiscale triangle. We will call it here simply for “thermodynamic modeling” (TDM) although it may mean different things depending on the subfield of research. We intend to stress that during the last decades, and thermodynamic modeling has continuously become more accurate and predictive but also contains more molecular features. Thermodynamic models have been traditionally based on their fundamental principles and mathematical rules with collected reference data of thermodynamic and transport properties to describe the behavior of substances and systems. It has been always used exclusively in chemical and process engineering. Thermodynamic models rely much on the use of a large variety of equations of state (EOS) starting from the van der Waals equation. Classical density functional theory (DFT) is a highly powerful TDM method for equilibrium properties (free energy, phase behavior, etc), suitable for inhomogeneous systems and phase transitions. It can predict TM properties for complex chain-like fluids like polymers and ILs. Many TDM methods can also take molecular information from both experiments and molecular modeling and simulations, which has become a powerful and general tool alongside molecular simulations. At its best, it provides the means to couple the microscopic description of matter with macroscopic properties and function of the real world. Molecular simulations (MD & MC) as such can give thermodynamic quantities after applying statistical mechanics in the analysis, but the particle-based simulations can only take us to the mesoscale from where many orders of magnitude remain to the macroscale, and to get there continuum models are needed, or alternatively thermodynamic modeling. Therefore, the combination of experiments, molecular simulations, and thermodynamic modeling as a “tripod” is an ideal multi-scale tool. This integrated bottom-up/top-down approach has been used for homogeneous and bulk systems, but as a general tool, it can be applied to study the heterogeneous systems and interfaces between bulk and surfaces. The use of the triangle can provide a more comprehensive understanding. It is a way to perform verification and validation of the findings and test hypotheses. But after all, it gives a good insight into these complex systems of IL interface layers and allows predictive capabilities. It is an ultimate synergetic approach. We have collected the recent advances from literature where this approach, as a whole or partially, is used for IL interface layers to find how their nanostructure affects their properties and then makes use in different applications.
Excellent reviews have already been published from the aspects of experiments and/or molecular simulations, to reveal the IL interface layer structuring and their properties.1–4,23,25,29,38 However, molecular thermodynamic models have been developed to describe melting temperature,39–41 gas adsorption42 of confined nano-phases, surface wetting,39,43,44etc., but for ILs, most of them are used for predicting the bulk characteristics.45–50 To the best of our knowledge, no reviews have been reported to include even the research on thermodynamic models in the studies of IL interface layers. Not to mention a systematic survey on the ion structuring and the associating properties of the IL interface layer as well as their structure–property relationships. The main motivation behind this review is to introduce and highlight the development and application of multiscale triangle “experiment–simulation–thermodynamic modeling” to study the IL interface layer structures and properties, representing many exciting challenges while being of paramount2,3,29 importance from both the scientific and applied perspectives.
This review is organized as follows. We introduce the multi-scale the “experiment–simulation–thermodynamic modeling” triangle and how it can be applied on the studies of IL interface layers in Section 2. In Section 3 we detail the studies of mono- and bilayer structuring of ILs on surfaces using the triangle. Structure–property relationships of IL interface layers are collected in Section 4. Among many applications of IL interface layers, we choose to present those connected to tribology and lubrication as well as energy storage in Section 5. Summary and future outlook are given finally in Section 6.
2. Structure–property relationships of ionic liquids interface layer by “experiment–simulation–thermodynamic modeling” tool
Several findings in literature have demonstrated a clear effect of the ion structuring of IL interface layers on the overall properties and performance in different applications, and for the IL-based lubricants and electrolytes in particular. For example, more ordered IL layers in the vicinity of a solid surface can both increase the friction51–53 and reduce it.52,54,55 In the case of electrolytes, the ordered ion packing of ILs can enhance the charge storage performance,6 while the structuring of ILs can slow down the capacitive process.56 These different responses observed in these properties due to the structures of ILs can be given completely by different interpretations.2.1 Overview of various techniques to study interface layer ionic liquids and their applications
Relevant experimental investigations, such as sum frequency generation vibrational spectroscopy (SFG), high-energy reflection and scattering using X-rays and neutrons, nuclear magnetic resonance (NMR), X-ray photoelectron spectroscopy (XPS), quartz crystal microbalance (QCM), surface-force apparatus (SFA), and atomic force microscopy (AFM), have been carried out to reveal detailed structures of IL interface layers. In studies of ion nano-structuring of ILs at solid interfaces, SFA (Fig. 1a, left) and AFM (Fig. 1b, left) are the most extensively utilized experimental techniques to unveil the intrinsic mechanism behind IL–solid interfacial phenomena at the nanoscale, as a layering structure of IL interface layers is resistive against an external pressing force to a certain extent.57–60 Thus, the force-sensitive techniques, i.e., SFA and/or AFM have been naturally employed to directly probe the layering structures of the interfaces, allowing for the ion structure to be visualized to obtain not only the number and thickness of layering structures but also the force that each layer can resist. Together with molecular simulation (Fig. 1c), these tools have provided molecular insights on the microstructural changes of ILs at solid interfaces, i.e., how the ion self-assembly underpins much of their complex chemical and physical behavior. It is crucial to note that the local structural features of ILs are correlated strongly with their properties,27 and a detailed understanding of how the ion structuring of the IL interface layer at the molecular level affects their properties is a prerequisite for the design of ILs with properties tailored for targeted applications.28 In this respect, thermodynamic modeling (Fig. 1d) plays an essential role in relating the structure–property of the IL interface layer, allowing the judicious design of IL–solid interfaces for task-specific applications.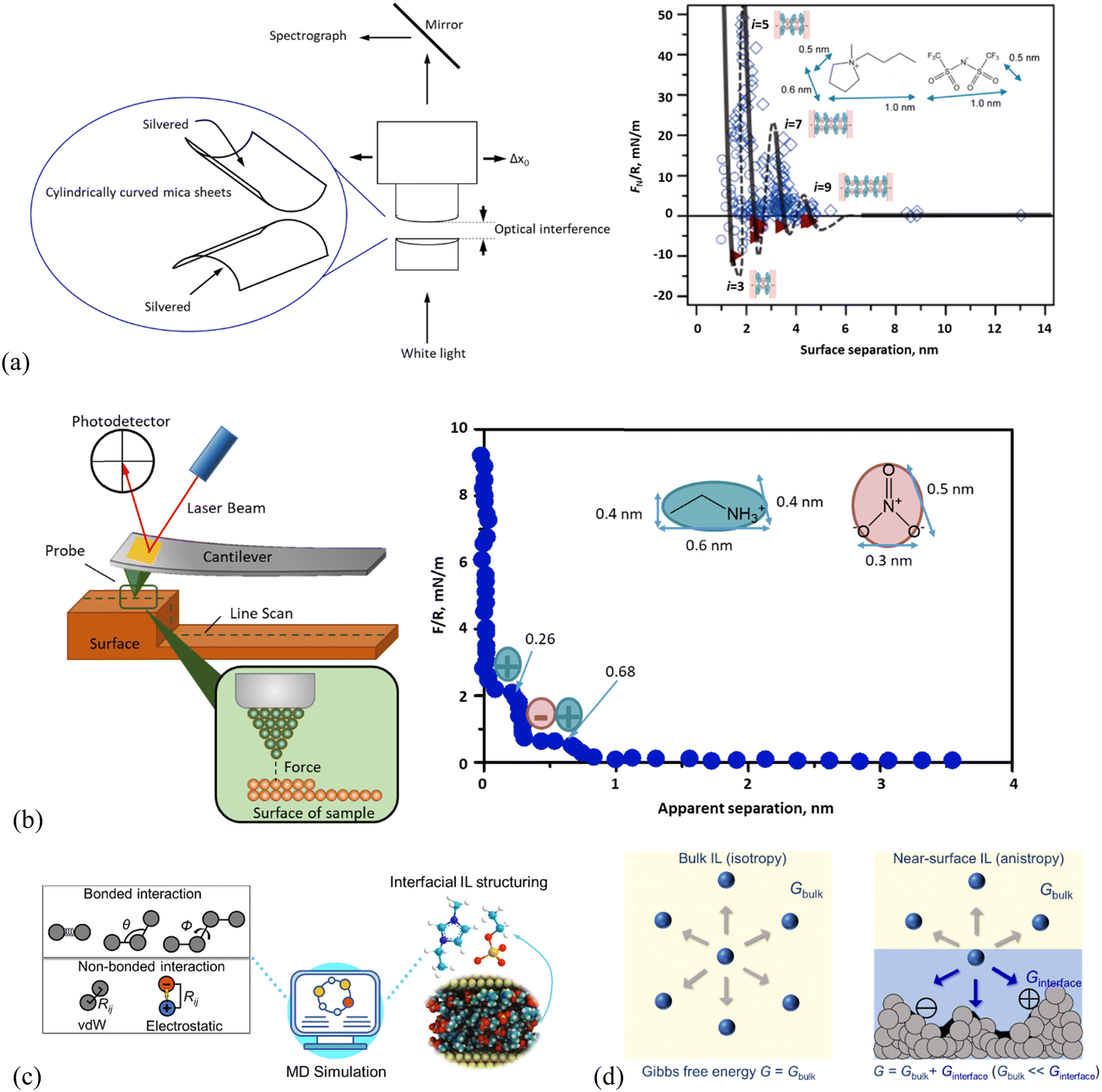 | ||
| Fig. 1 Schematic presentation of the working principle in (a) surface force apparatus (SFA) and a typical SFA-measured force curve for [C4C1Pyrr][NTf2] confined between mica sheets, adapted from ref. 51 with permission from RSC, copyright 2013, (b) atomic force microscopy (AFM) and AFM-measured normal force curve for EAN on mica with a 5 μm-radius silica colloid probe. Adapted from ref. 73 with permission from RSC, copyright 2016. In the right panel of (a), open diamonds and circles indicate points measured on approach and retraction of the surfaces, respectively. Filled triangles in (a) indicate points measured from the jump-apart of the surfaces from an adhesive minimum. i means the number of ion layers. The lines are a guide to the eye to show the oscillatory nature of the forces, with solid lines through measured regions and dashed lines through the regions inaccessible to measurement due to the jump-in from the previous energy barrier. (c) Molecular dynamics (MD) simulation method for detailing the structure and dynamics of the ionic liquid interface layer. Adapted from ref. 74 and 75 with permissions from Wiley, copyright 2022, and MDPI, copyright 2019. (d) Molecular thermodynamics modeling, here in quantitatively describing bulk and ionic liquid interface layer via Gibbs free energy (G). The solid surface in contact with the IL (right panel) shows varying nanostructured features, and/or chemical heterogeneities, and/or surface charges. | ||
DFT is another powerful and efficient theoretical tool to model the structuring layers of interfacial ILs and their properties95 by minimizing the grand potential.95,96 In general, DFT is developed based on any equation of state (EOS),97 while for ILs, owing to their complexicity, the EOS based on statistical associating fluid theory (SAFT) is preferred, which have been intensively developed to study the properties of ILs.98 To present the layering structures and properties of the interfacial ILs, an external potential is needed to consider the strongly assymetruic interactions between ILs and the solid surface.99 DFTs have been intensively developed to study the properties such as gas adsorption isotherms in metal organic frameworks,100,101 partition and selectivity of IL mixtures in porous electrodes,102 surfactant and polyelectrolyte on rock surface for selecting appropriate polyelectrolytes suitable for different reservoirs,103 interfacial layering of ILs,104 interfacial structure and differential capacitance of IL–graphite interface,105etc. Meanwhile, further development has been conducted and extended to the dimention more than one, as well as complex structures including the surface roughness and heterogeneity. In developing DFT, those based on third-order thermodynamic perturbation theory of Feynman-Hibbs-corrected Mie potentials and using the modified fundamental measure theory were recently reported.106 Perturbed chain-SAFT-based DFT was employed to study the selective adsorption of mixed ILs confined in rough slit pores of electrodes, where the Helmholtz energy functional of the DFT model accounts for the hard-sphere, the dispersive, and the electrostatic correlation interactions among ILs ions.102 To accelate the computation speed, different althorighms have been combined and integrated into DFT. For example, the Chebyshev pseudospectral collocation method was integrated into DFT to efficiently describe the density profile of the IL–CO2 system inside electroneutral nanopores.98 This Chebyshev pseudo-spectral collocation method was combined with Anderson mixing algorithm to accelerate the interative process, to study the effects of temperature, pressure, pore size, and pore shape on the gas solubility in the ILs confined inside silica nanopores.107 A two-dimensional DFT model was used to investigate the distribution and partition behavior of imidazolium-based Ls in slit nanopores with rough surfaces, where the Helmholtz free-energy functionals were used to account for the excluded-volume effect and chain formation.108 While it should be addressed that most of studies is limited to the systems other than ILs. In summary, DFT has become a fantastic tool for characterizing the ion distribution of the ILs at the interface,109,110 while obtaining the external function still remains a challenge.
2.2 Quantized friction and nano-structuring of interface layer ionic liquids seen in AFM and SFA
Studies of AFM nano-frictional response allow the variations in friction force with a normal load to be correlated with the interfacial IL structure.20,53,55,73,111–116 AFM-measured nanoscale friction force offers a unique insight into the nano-structures of ILs at the interface. Few studies are reported on the phenomenon of ‘quantized friction’, in which the friction coefficient can be indexed by the number of layers of the IL film at solid surfaces, providing valuable insight into the overall interface structure of ILs.65 For example, as shown in Fig. 2(a), for ethylammonium nitrate (EAN) on mica at room temperature, two distinct regimes in a quantized manner can be obtained. One is for loosely ordered ion layers obtained at smaller normal loads <20 nN and another for highly ordered ion layers at higher loads larger than 20 nN.73 While for 1-(4-hydroxybutyl)pyridinium bis(trifluoromethanesulfonyl)imide ([HOC4Py][NTf2]) at the mica surface, a three-regime quantized nanofriction feature with two critical normal forces (breakpoints) at 12 and 81 nN was detected (Fig. 2(b)).117 (1) The first breakpoint (12 nN) means when the normal force is larger than 12 nN, and the AFM probe should have reached the boundary regime, where a strongly adsorbed cation boundary layer resists getting “squeezed out” and the friction force increases monotonously with a normal load. (2) The second breakpoint is detected at a normal force of 81 nN, indicating a rupture of an additional layer. However, the adsorbed cation-enriched boundary layer should have not been completely squeezed out, resulting in direct contact between the AFM probe and mica. Further, the Infrared spectra can demonstrate that strong stabilizing H-bonding among the [HOC4Py]+ cations is formed in a bilayer cation structure. This structure is similar to that observed by Mao et al.,6 which is more difficult to squeeze out. (3) The first [HOC4Py]+ cation layer is shaved away when the normal load is >81 nN due to the destruction of the H-bonds between the two cation layers, and only the final cation layer remains to prevent a direct probe–mica surface contact (cf. the inset scheme in Fig. 2b).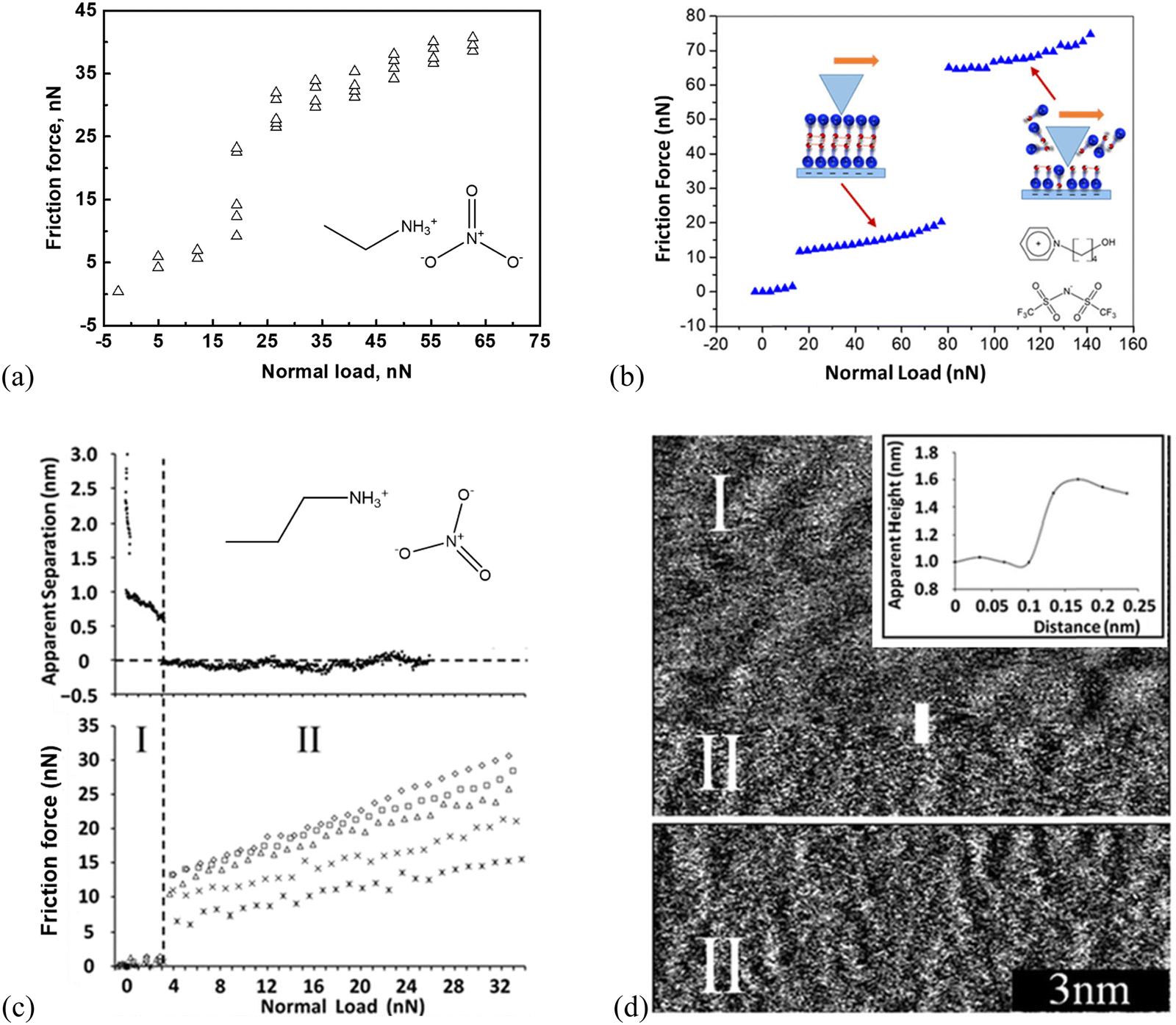 | ||
| Fig. 2 Friction-load plots obtained by a (a) colloid silica probe with a radius of 3.4 μm in EAN, adapted from ref. 73 with permission from RSC, copyright 2016, and (b) sharp AFM silicon probe in [HOC4Py][NTf2] supported by mica at room temperature. Reproduced from ref. 117 with permission from ACS, copyright 2020. (c) Top: Apparent separation between a mica surface and the silica colloid probe (diameter ∼5 μm) as a function of normal load for the mica–PAN system. Bottom: Friction force as a function of normal load for various sliding velocities using silica colloid probe. Stars: 5 μm s−1, crosses: 10 μm s−1, triangles: 20 μm s−1, squares: 30 μm s−1, diamonds: 40 μm s−1. Reproduced from ref. 111 with permission from RSC, copyright 2013. (d) Sharp AFM tip images of the PAN–mica interface. The inset is a section analysis for the sharp AFM probe moving from regime I to II. Reproduced from ref. 111 with permission from RSC, copyright 2013. | ||
Propylammonium nitrate (PAN) on a mica surface shows a two-distinct-layering frictional behavior (Fig. 2c, bottom), in which the silica colloid probe slides along either an upper layer (I) or the innermost layer (II).111 More specifically, as shown in Fig. 2c, the approaching force curve (top) demonstrates the quantized frictional regimes I and II, exhibiting two discrete jumps at large separations (II) and two subsequent ion layers (I). The friction force (bottom) jumps abruptly from ∼1 to 7–13 nN, revealing that the AFM colloid probe moves from the contact with the upper ionic liquid interface layer (I) to the innermost layer, closer to the surface (II). The corresponding topographic images further confirm the upper ion layer (I). It has a distinct, undulating structure with a periodicity of about 1 nm and is evocative of its bulk nanostructures (Fig. 2d). After the sharp AFM probe jumps inward to the innermost layer (II), a “worm-like” morphology is observed, and the height difference between the regimes I and II is around 0.5 nm (Fig. 2d). This is consistent with both the step size in the force curve (Fig. 2c, top) and the PAN ion pair diameter (∼0.56 nm).
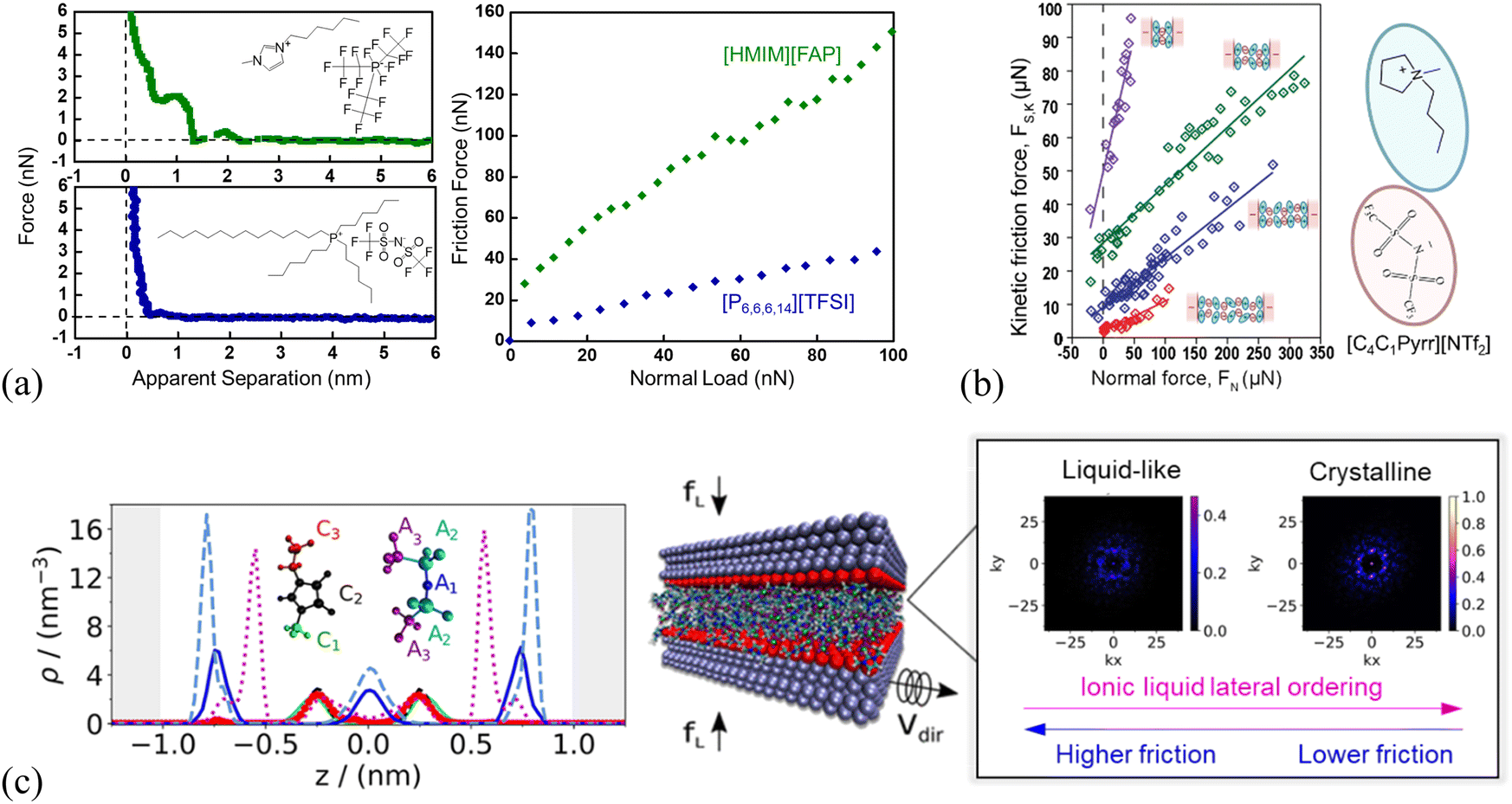
With SFA, a layered ion structuring and quantized friction behavior are simultaneously observed in the dicationic IL, 1,10-bis(3-methylimidazolium)decane di[bis(trifluoromethylsulfonyl)]-imide ([C10(C1Im)2][NTf2]2) confined by the mica surfaces (Fig. 3a). The measured friction FS appears extremely low when the film thickness (separation) D > 3.2 nm, while FS increases linearly with the normal force FN. This is because FN increases sufficiently to expel a further ion-pair layer of the IL, decreasing the separation D to 2.4 nm. When the normal force is increased further to squeeze another ion pair layer out with a separation D of 1.6 nm, the value of FS is larger and increases with a steeper gradient.
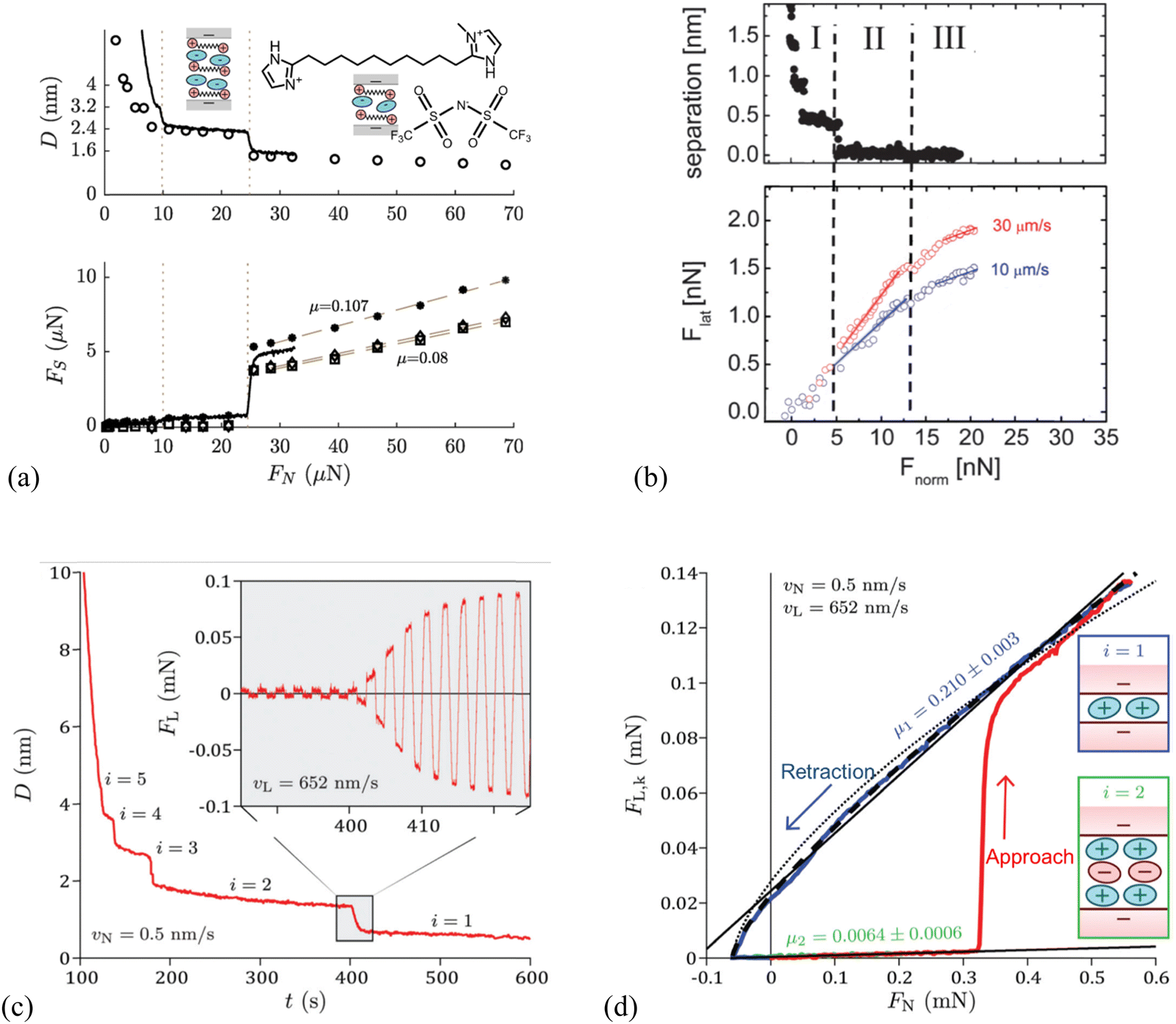 | ||
| Fig. 3 (a) Top: Surface separation D as a function of normal force FN during friction measurements by SFA for the dicationic IL, 1,10-bis(3-methylimidazolium)decane di[bis(trifluoromethylsulfonyl)]-imide, shortened as [C10(C1Im)2][NTf2]2. The continuous line corresponds to an approach performed while shearing continuously at a velocity of 470 nm s−1, whereas the circles represent the distances at which shear measurements were performed during a stepped approach. Bottom: Kinetic friction force FS as a function of FN, for a continuous approach while shearing at a velocity of 470 nm s−1 (solid line), and for measurements in the stepped approach (markers). The squares, diamonds, and asterisks represent measurements at a shearing velocity of 470, 105, and 52 nm s−1, respectively. Reproduced from ref. 118 with permission from ACS, copyright 2019. (b) Top: Separation D versus normal force Fnorm of EAN at mica surfaces measured by an AFM silica colloid probe with a diameter of 20.8 μm, and bottom: lateral force Flatversus normal force Fnorm at sliding velocities of 10 and 30 μm s−1. Dashed vertical lines denote the divisions between Intervals I, II, and III. Solid lines denote linear fits to increasing load data from which friction coefficients μ are obtained. Reproduced from ref. 119 with permission from RSC, copyright 2012. SFA measurements for 1-butyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide ([C4C1Pyrr][NTf2]), when moving the top surface with the piezoelectric tube at a normal velocity vN = 0.5 nm s−1 and a lateral velocity vL = 652 nm s−1: reproduced from ref. 65 with permission from RSC, copyright 2020. (c) Temporal evolution of the apical distance D on full approach. The inset shows the temporal evolution of the lateral friction force FL during the squeeze-out transition from layer i = 2 to layer i = 1. (d) Kinetic friction FL,k as a function of load FN on full approach (in red) then retraction from layer i = 1 (in blue). | ||
It should be mentioned that the magnitude and gradient of FS are weakly dependent on the shearing velocity, e.g., slow velocity results in large FS on the layer with a separation of 1.6 nm, while the friction coefficient μ shows a very weak velocity dependence. Apparently, Fig. 3(a) reveals a quantized friction behavior, in which discontinuities occur in friction as the molecular layers of the IL are squeezed out and the friction coefficient μ depends on the number of confined ion layers.118Fig. 3(b) shows a nano-frictional force response of EAN at mica surfaces with an AFM silica colloid probe, which can be decomposed into 3 linear regions, i.e., Intervals I, II and III, each having a distinct slope. As the applied normal load Fnorm is increased, the apparent separation decreases from >2 to 0.4 nm as 4 weak ion pair layers are ruptured, and then remains almost constant up to 5 nN load (Interval I). The friction force Flat increases linearly with the normal load over the same loading range and is independent of the sliding velocity. When the load exceeds 5 nN with the apparent separation decreasing to 0.1 nm (Interval II), only a single confined layer remains, consisting mostly of cations adsorbed to mica for neutralizing the surface charge. A break is observed in the friction data in Interval II, which is most noticeable at a higher sliding velocity. The resulting friction coefficient, which is defined by the gradient of the lateral vs. normal loads based on Amontons’ law, depends strongly on the velocity in Interval II, and is smaller than that obtained in Interval I for thicker confined films at lower normal load. A final transition to Interval III is observed due to the structural transition of the ion layers when the normal load is further increased above 13 nN, corresponding to a separation decreasing to zero, and the friction coefficient is further reduced compared with that in Interval II and independent of the velocity.119
Similar quantized friction phenomena have also been found in other investigations performed by AFM120 and SFA65 in which the friction coefficient is clearly indexed by the number of ions composing the IL film. Confining the 1-butyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide ([C4C1Pyrr][NTf2]) between mica surfaces forms a structured film responding to shear in a pronounced quantized way (Fig. 3c).65 This friction response has been determined by SFA more precisely, comprehensively and rapidly, based on the simultaneous application of normal and lateral motions to the surfaces. Fig. 3(c) shows temporal evolutions of the apical distance D and lateral friction force FL in which the apical distance varies by 0.64 nm in 10 s, associating with about four shearing cycles (inset). The amplitude of FL is observed to increase continuously during the squeeze-out transition from layer i = 2 to i = 1. The extracted average lateral friction force during the creep stage for each shearing period is identified as the kinetic friction force FL,k, and the resulting friction coefficient μ is clearly indexed by the number of ions of the IL film, which is the signature of the quantized friction phenomenon (Fig. 3d). In layer i = 2 (retraction branch in green) where the film is composed of two cation layers and one anion layer, the friction coefficient μ is 0.0064 ± 0.0006. While the value of friction coefficient is almost two orders of magnitude larger in layer i = 1, which is composed of only a monolayer of cations (μ = 0.210 ± 0.003, retraction branch in blue) than that in layer i = 2, and a transition happens at a load FN = 0.33 mN between the two regimes.
2.3 Electrically controlled friction and nano-structuring of interface layer ionic liquids
The IL lubrication at solid–liquid interfaces can be well-controlled via electrical polarization of the sliding surfaces to regulate the interface layer ion structural features.23 Urbakh's group elucidated the connection between the charge effect on the structure of the IL layers and their lubricating properties using MD simulation, in which, the friction force varies with the surface charge density through ordering the interface ions layer and switching between anion and cation layers in contact with substrate surfaces. They found that large surface charge densities are beneficial to the reduction of friction coefficient and strong interaction between the ionic layers and surfaces to prevent the ‘‘squeeze-out’’ of ILs under high pressure.81 In particular, the [C2MIM][NTf2] IL confined between charged surfaces has recently been found to be less compressible than that confined between neutral surfaces, favoring the use of charged surfaces over neutral ones for the lubrication by sustaining loads of several GPa.121Interestingly, the friction force of imidazolium ILs is found to feature a nonmonotonic behavior with surface charge at high normal loads of ∼200 and 300 MPa, showing a maximum at a specific charge density (Fig. 4a). This maximum results from the interplay between the increase of the roughness of the energy landscape with surface charge and the shift of the slippage plane from the solid–liquid interface to the interior of the lubricating IL film (cf. inset illustrations, Fig. 4a).122 A previous simulation by Urbakh's group123 also showed that the variation of the friction force with the surface charge density is determined by the regime of motion of the confined [BMIM][BF4] IL between the graphite substrate and tip, depending on the strengths of the ion–substrate and ion–tip interactions (Fig. 4b). In the case of the weak ion–substrate interaction (εis = 0.23 kJ mol−1, Fig. 4b, blue), the friction as a function of surface charge density has a minimum for the uncharged graphite surface and increases by an order of magnitude with the surface charging of either sign. This friction feature here is related to the shift of the slippage plane, where the frictional energy dissipation mainly occurs at the IL–graphite interface in the case of weak ion–substrate interaction. In comparison, most ions stick to the graphite substrate surface when the IL ions interact strongly with the graphite substrate, and the slippage occurs at the IL–tip interface. Thus, the friction has a maximum for the uncharged graphite substrate and decreases with increasing the charge of any sign, for the strong ion–substrate interaction (εis = 70 kJ mol−1, Fig. 4b, red).
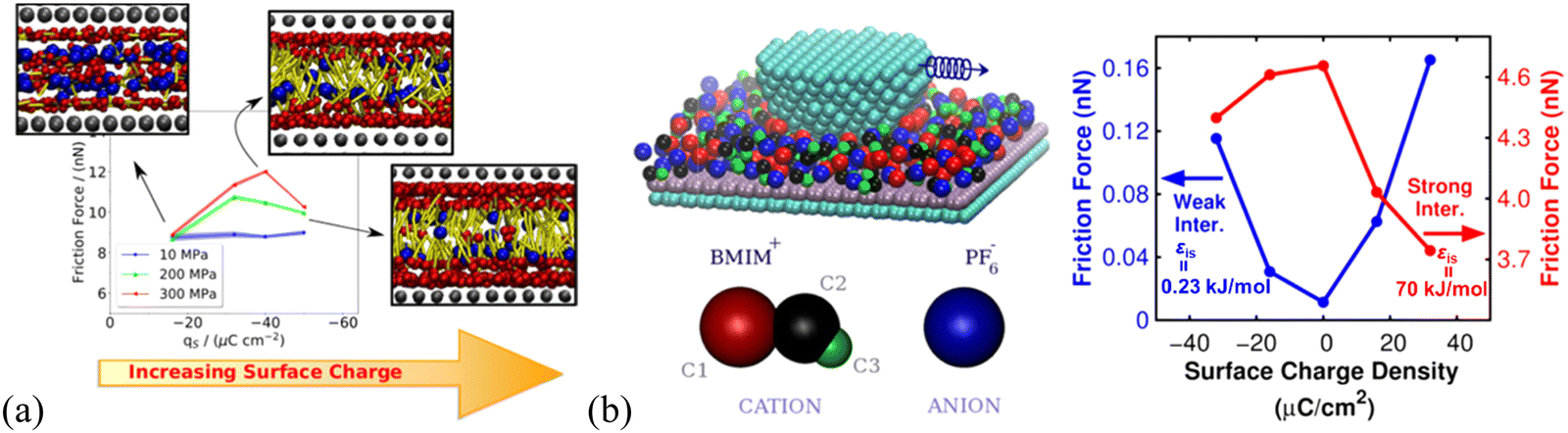 | ||
| Fig. 4 (a) Dependence of the friction force of [C6MIM][BF4] on the surface charge and normal load applied on the slabs, using non-equilibrium MD simulation. Insets are the front views of the confined ILs at 300 MPa next to the moving wall for different charge densities. The three beads in the charged head group of the cation are represented as red spheres. The blue spheres represent the anion and the cation aliphatic tails are colored in yellow. Reproduced from ref. 122 with permission from ACS, copyright 2020. (b) Friction force as a function of graphite surface charge density calculated for two ion–substrate interactions: εis = 0.23 kJ mol−1, where slippage occurs at the interface between the [BMIM][BF4] IL layer and the substrate, and εis = 70 kJ mol−1, where slippage takes place mainly at the interface between the tip and the IL layer. Here, the simulations are performed for the uncharged tip and similar results can also be obtained for a negatively charged tip. Reproduced from ref. 123 with permission from ACS, copyright 2018. | ||
The responses of 1-butyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate ([BMIM]FAP) and 1-hexyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate ([HMIM]FAP) to the variation of surface potentials at Au(111) are the same, i.e., all are more lubricating at negative potentials than neutral surfaces while poorly lubricating at positive surfaces (Fig. 5a, left and middle panels).124 This reveals that the imidazolium-enriched layer is more lubricating than the mixed layer, followed by the [FAP]− anion-enriched layer (inset illustration in Fig. 5a, left). The negative surface potential induces the cations of the ILs to orient with charged groups towards the surface such that the alkyl tails form a well-defined sliding plane.114 The poorest lubricity appears in the [FAP]− anion enriched layer at positive potentials because of the disordered coil morphology of the anions producing a rough sliding plane,125 and the weak interaction of the anion with the Au(111) surface due to the larger size and more delocalized charge.114,124 The mixed cation–anion layer formed at the neutral surface reveals a lubricity in between. It is found in Fig. 5(a) that the lubricity can be altered by changing the cation alkyl chain length at negative potentials and neutral surfaces, in which the long chain facilitates the interfacial lubricity. In comparison, the response of the friction force in 1-butyl-3-methylimidazolium iodide ([BMIM]I) to surface potential is completely different from that in the ILs with [FAP]− anion. A higher friction is observed in [BMIM]I at a more negatively charged Au(111) surface (Fig. 5a, right), because of the spherical shape and localized charge of I-anions, allowing the strong ion adsorption to form a smooth, well-ordered surface plane. Thus, the interfacial lubrication at IL–solid interfaces can be well-tuned by modifying the composition and structure of the interface layer ions through varying the applied voltages.
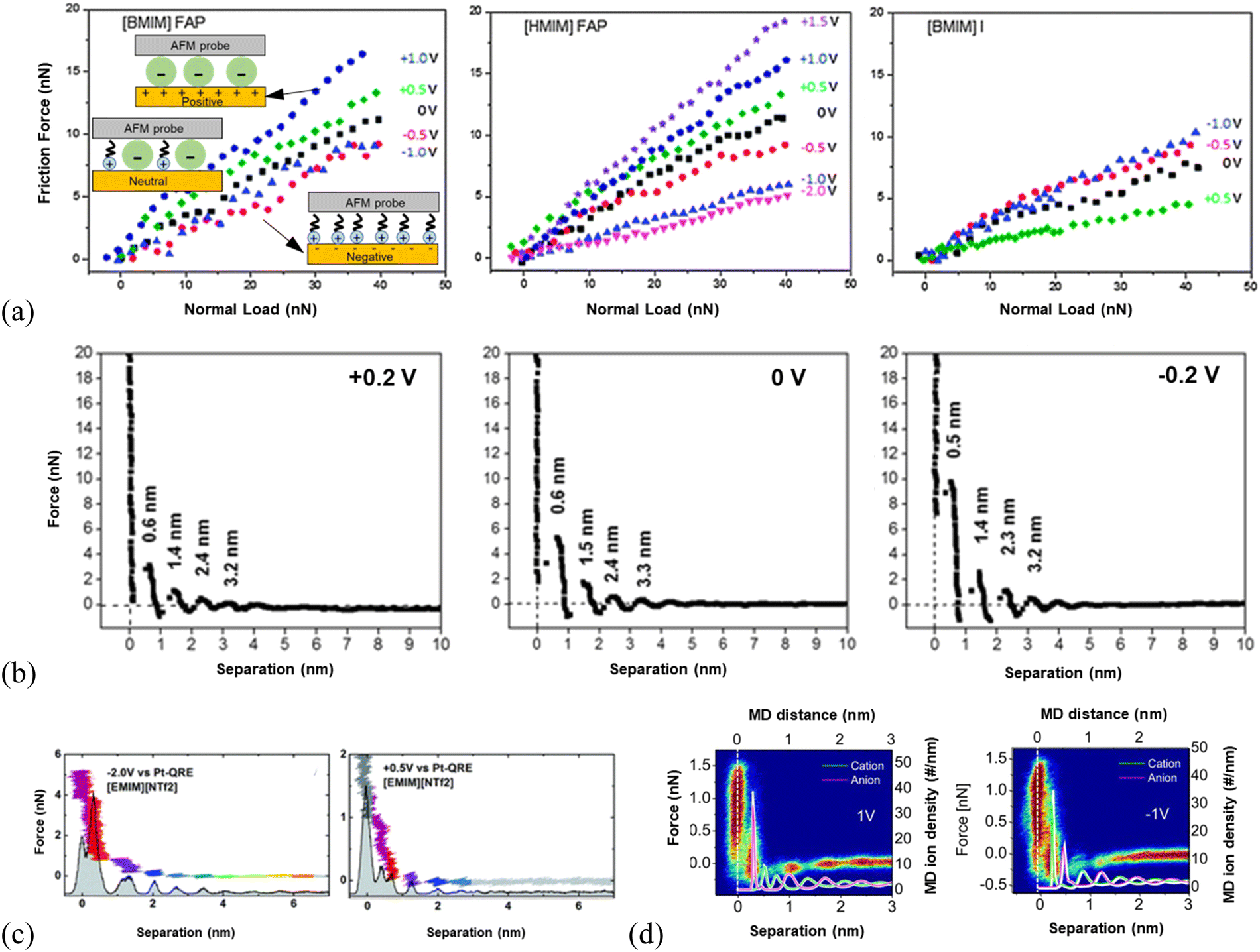 | ||
| Fig. 5 (a) Friction force versus normal load as a function of surface potential for [BMIM]FAP (left), [HMIM]FAP (middle) and [BMIM]I (right) at Au(111) surfaces, with silica AFM probes. Reproduced from ref. 54 with permission from RSC, copyright 2013. (b) A comparison of the AFM force-separation curves of [Py1,4][TFSA] at Au(111) under varying surface potentials. Reproduced from ref. 126 with permission from RSC, copyright 2018. (c) AFM normal force as a function of the tip-sample separation in [EMIM][NTf2] for electrical potentials of −2 V (left) and +0.5 V (right) at Au(100) surfaces. The histogram underlying the force curve represents the probability of finding the tip at a given distance to the sample during approach. The colors serve as a guide to the eye to compare equal molecular layers in the panels. Reproduced from ref. 127 with permission from RSC, copyright 2017. (d) Comparison of AFM force curves with MD simulations of [EMIM][NTf2] cation and anion positions at positively (left) and negatively (right) polarized graphite surfaces. The white line denotes the position of the graphite surface. Reproduced from ref. 72 with permission from ACS, copyright 2013. | ||
AFM approaching force curves have shown that the distinctiveness of the ion layering of interface layer ILs varies with the applied surface potential.2 For example, the force required to rupture the ion layer of [Py1,4][TFSA] (1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide) adjacent to Au(111) surface was found to vary with the applied surface potential. The rupturing force of the innermost ion layer is 10, 6, and 3 nN for the IL at −0.2, 0, and +0.2 V, respectively (Fig. 5b), indicating an increase in the strength of ion adsorption at more negative surface potentials, corresponding to a relatively strong near-surface structure.126 For the AFM probe approaching [EMIM][NTf2] (1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide) on a negatively polarized Au(100) surface (left, Fig. 5c), the force curves revealed a more pronounced layering of the IL compared to that at the positive surface (right, Fig. 5c). The layering structure of the ionic liquid interface can be further recognized in the underlying histogram of the probability of finding the tip at a given distance.127 A detailed picture of the molecular ordering of [EMIM][NTf2] at the charged graphite72 and [BMIM][NTf2] at the charged silica surface128 has also been demonstrated by both AFM experiments and MD simulations, which is comparable to the configuration of cations and anions at charged Au(100) surfaces.127 As shown in the comparison of AFM force curves overlaid with the ion density curves by simulation in the left panel of Fig. 5(d), the ion layer of [EMIM][NTf2] closest to the positively biased graphite surface matches the calculated position of the first anion layer. While at −1 V, the first layer is detected as the strongly bound cation layer, which balances the negative surface charge in accordance with the simulations.72 Thus, the charge at the surface induces the ordering of ILs into layers of alternating charge, which would favor the lubrication of ILs at solid surfaces.
To briefly summarize, the nano-frictional properties of ionic liquid interface layer, particularly, the ‘quantized friction’ phenomenon observed by either AFM or SFA does depend strongly on the ILs nano-structuring. The dependence of nanofriction on the ion nanostructures of interface layer ILs can be influenced by external stimuli, such as the electrical potential, but is mostly being studied qualitatively, and the quantitative description of the relationship between interfacial ILs nanostructure and properties has not yet been developed.
2.4 Structure–property relationships for interface layer ionic liquids
Thermodynamic modeling analysis reveals a wealth of information of the microscopic structure and mechanisms. It can be used to provide quantitative structure–property relationships (Fig. 6a), highly important in all design and engineering of ILs. Still, reliable interaction parameters for the molecular thermodynamic modeling is demanding (Fig. 6a) and requires reliable and accurate experimental data.129,130 Molecular simulations are the best tools to verify and validate the feasibility of the experimentally derived interaction parameters (Fig. 6a).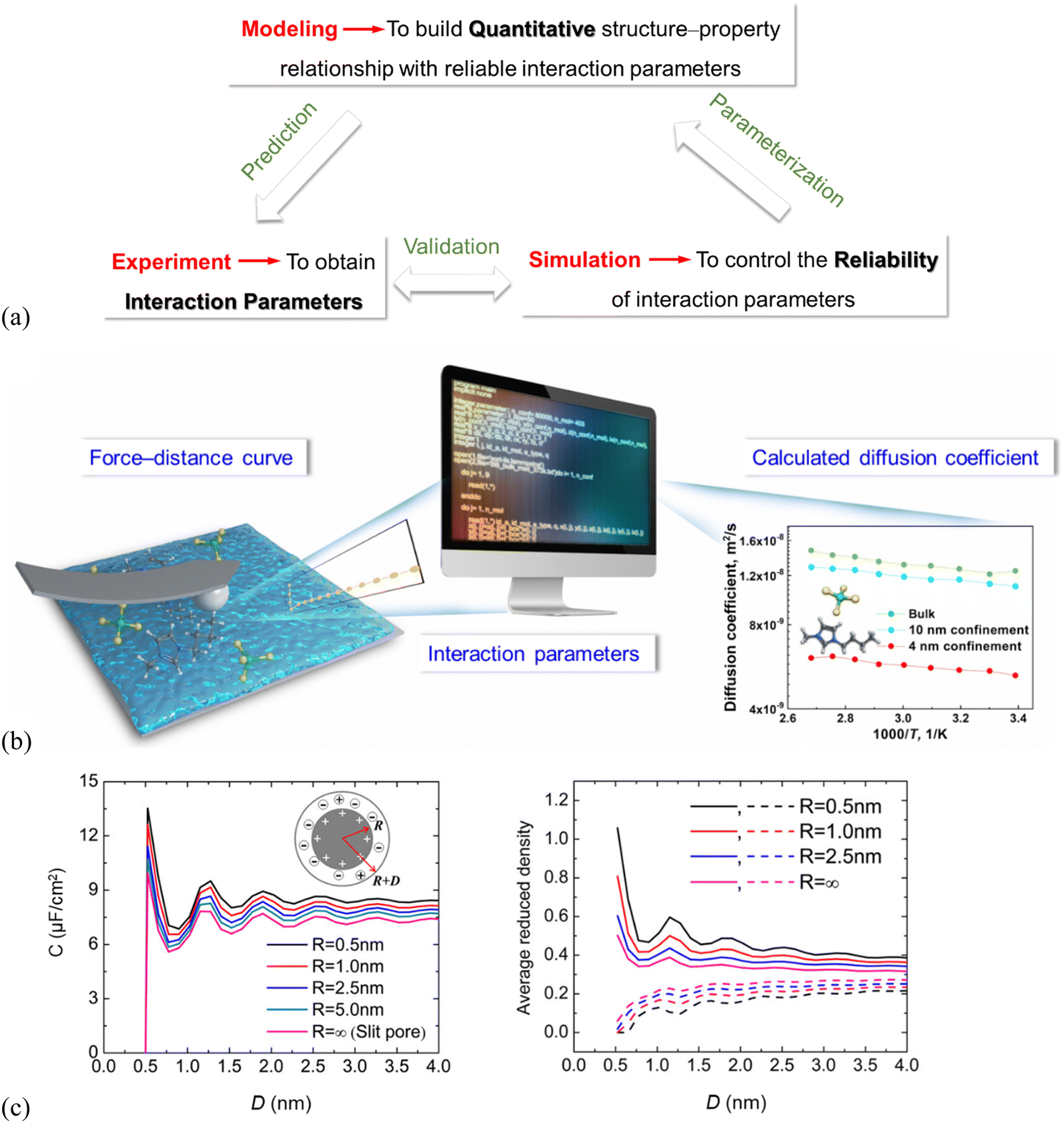 | ||
| Fig. 6 (a) Multiscale–triangle “experiment–simulation–thermodynamic modeling” technique enabling structure–property relationships. (b) Schematic illustration showing interaction parameters derived from CP–AFM-measured force–distance curves, which can be used in simulations to calculate temperature–dependent diffusion coefficients of the nano-porous SiO2 confined IL (4, 10 nm pore size) and for ILs in the bulk. Adapted from ref. 90 with permission from RSC, copyright 2022. (c) Left: DFT-predicted overall integral capacitance (C) of room-temperature ILs vs. thickness (D) for spherical shells of different inner radii (R), right: average densities of counterions (solid lines) and co-ions (dashed lines) inside the spherical shell at different thicknesses. The surface electrical potential is ψ0 = 1.5 V. Reproduced from ref. 109 with permission from ACS, copyright 2016. | ||
There are recently only a few studies to derive interaction parameters directly from experiments. The force curves of IL systems, measured by using SFA and AFM, show typically an oscillatory behavior, indirectly containing information about the number density distribution of the IL cations/anions on the solid substrate. Amano, et al.92 did transform their high-resolution AFM-measured force curves into the number density distributions to estimate the hydration structure of liquids at the muscovite mica (001) surface, but only for the molecular liquid region. Wei, et al.131,132 recently proposed an experimental approach with colloid probe AFM (CP–AFM) to quantify the molecular interactions of imidazolium-based ILs with charged SiO2 surfaces.90,91 The CP–AFM-measured adhesion force offers a direct measure of the IL interactions with SiO2, which can be used to fit the interaction parameters for the molecular simulations (Fig. 6b).90 The CP–AFM-derived interaction parameters are further used in MD simulations to compute the temperature–dependent diffusion coefficient of the ILs confined in cylindrically porous SiO2 with varying pore size (4, 10 nm, cf.Fig. 6b) and compared with those measured experimentally by Nuclear Magnetic Resonance (NMR) spectroscopy, confirming the reliability of the AFM-quantified molecular interactions. An enhanced diffusion rate in ILs at elevated temperatures was observed by simulations for the bulk ILs as well as the confined ones in cylindrically porous SiO2 with 4 and 10 nm pores, in agreement with that in NMR measurements. However, the diffusion coefficients in the bulk and confined ILs calculated in simulations based on these AFM-quantified interaction parameters are 1–3 orders of magnitude higher than those measured by NMR. The main reason for the much faster diffusion obtained in the simulation is the complete neglecting of the electrostatic interactions as the cation and anion are considered as an ion pair and the charges are simply switched off. Also, the surface topologies of the nano-porous SiO2 used for NMR measurements are complex in comparison with the SiO2 model in simulations. Thus, the MD simulation with CP–AFM-derived interaction parameters succeeds in predicting the trends of diffusion coefficients, but fails in reproducing the NMR-measured values. This demonstrates the need for considering the cation/anion charges and surface complexities in deriving the interaction parameters for both simulations and experiments.90 The lack of accurate interaction parameters is, due to the rather limited number of studies, still being reported in using thermodynamic modeling analysis to quantitatively predict the properties of the interface layer ILs, although the modeling is expected to greatly improve the quantification of the structure–property relationships.
The interaction parameters42 were also reported to be derived from molecular simulations and thermodynamic modeling41 by describing the substrate effect on the Gibbs free energy of the supported metal nanoparticles, to quantify CO2 absorption working capacity of the supported imidazolium-based ILs. The thermodynamic modeling was performed to describe the Gibbs free energy of CO2 in the supported ILs, so that the CO2 solubility could be quantitatively predicted in the supported ILs.42 The interface layer ILs increase the CO2 solubility compared to the bulk ILs. This can be explained by dividing the Gibbs free energy into three parts, i.e., the terms for bulk, IL–CO2 interface, and IL–substrate interface. The CO2 solubility increases in the IL–substrate interface. DFT studies show a qualitative correlation between the differential capacitance of electric double layers in ILs at electrode surfaces and the interfacial ion layering structure.95,109 The left panel in Fig. 6(c) shows the oscillatory dependence of overall capacitance (C) of room-temperature ILs for spherical shells of varied inner radii (i.e. pore size, R) and thickness (D), which is consistent with those in average densities of the IL-counterions inside the spherical shells (Fig. 6c, right). This can be explained because a smaller inner core radius results in more counterions inside the pore leading to a larger capacitance.109
Wang, et al. used the colloid probe AFM to demonstrate that the phosphonium-based IL (trihexyltetradecyl-phosphonium bis(salicylato)borate) ([P6,6,6,14][BScB]) with a long cation chain prefers to form stiffer ion layers at the charged gold surface with a “…cation–cation–anion–anion…” bilayer structure through van der Waals interactions among the nonpolar long alkyl tails of the cations. The formed bilayer structure of this IL exhibits an enhanced capacitance than the monolayer of the IL with a shorter chain, as the excess ions packed in the bilayer structure promote the IL to reach bulk electroneutrality over a shorter distance, enhancing the capacitance.21 Similar enhanced charge storage performance was reported earlier in the bilayer ILs with long amphiphilic chains at electrified surfaces by Yang et al.96 and Mao et al.6 For the latter, they found that a surface-active IL, 1-butyl-3-methylimidazolium 1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate([C4C1Im][AOT]) with a long anion chain exhibits significant and competing van der Waals interactions owing to the non-polar surfactant tails, leading to unusual bilayer ion structure and displaying optimum electrocapacitive performance. These but still qualitative findings on structure–property relationship of interface layer ILs further suggest that there is an urgent need to develop generalized thermodynamic models, based on accurate molecular parameters to quantitatively describe the interfacial ILs nanostructure and properties.
3. “Experiment–simulation–thermodynamic modeling” studies on nano-structuring of interface layer ionic liquids
3.1 Monolayer and bilayer structures of interface layer ionic liquids by experimental techniques
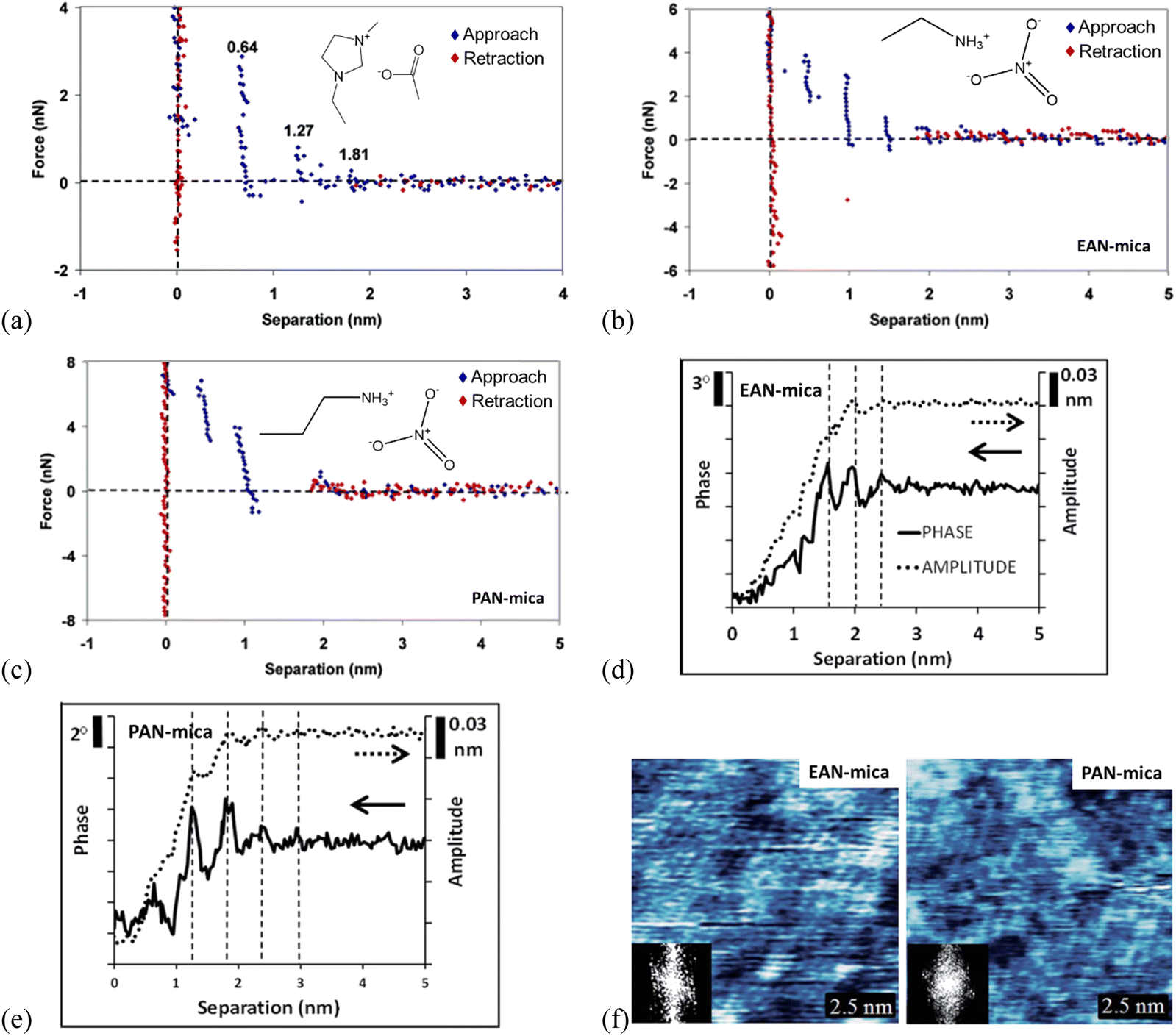 | ||
| Fig. 7 (a) Force curves for a Si3N4 AFM probe approaching and retracting from a mica surface in [C2mim]Ac. The separation distances for each ion layer prior to a push-through are given on the plot. Reproduced from ref. 71 with permission from ACS, copyright 2007. Force curves for an AFM Si3N4 probe approaching and retracting (b) EAN-, (c) PAN-mica interfaces. Reproduced from ref. 71 with permission from ACS, copyright 2007. Typical amplitude (dotted) and phase (solid) data recorded as an oscillating AFM probe approaches a mica substrate immersed in (d) EAN, and (e) PAN. Topographic images of (f) EAN and PAN innermost ion layers adsorbed to a mica surface and the inset show the 2D Fourier transforms by dynamic mode AFM. The x-axis in (d) and (e) measures the extension of the AFM Z piezo. Reproduced from ref. 35 with permission from RSC, copyright 2015. | ||
AFM force curves in Fig. 7(b) and (c) compare the ion layering in ILs of different sizes, in which the longer-chained propylammonium nitrate (PAN) exhibits fewer ion layers than the shorter ethylammonium nitrate (EAN) on mica, due to the higher molecular flexibility of PAN.71 Meanwhile, the force profiles oscillate periodically with the characteristic oscillation length corresponding to the physical dimension of the ion pair for the ILs confined between Si3N4 probes and mica. The magnitude of the force required to rupture each ion layer increases as the probe moves closer to the surface for both EAN and PAN, suggesting that the layers closer to the mica are more ordered, with decreasing ordering into layers further from the mica substrate. A stronger attractive interaction of the longer PAN with mica is detected, and the force steps in PAN are not vertical like those in EAN, suggesting the detected ion layers in PAN are more compressible.
Combining with the dynamic mode AFM, the 3-dimensional ion ordering of ILs at solid interfaces can be captured further.35 In the dynamic mode AFM, the cantilever can oscillate close to or at resonance frequency and the cantilever oscillation alters as the height changes, to adjust the probe-surface distance. This allows a simultaneous detection of the structure in both lateral and normal directions to obtain 3-dimensional information on the ion ordering at solid interfaces.136 With the dynamic mode AFM, the phase and amplitude of EAN and PAN at mica surfaces are measured as a function of the separation, revealing clearly layered ion structures in the normal direction (Fig. 7d and e), although the steps tend to be less distinct in the amplitude than in the phase data.35 For both EAN and PAN, the phase and amplitude are constant when the probe-surface separation is greater than 3 nm, indicating that the AFM probe is insensitive to the bulk ILs. While as the separation is decreased below 3 nm, the phase and amplitude decrease as a consequence of the confining effect of the probe and mica substrate. The obtained phase and amplitude signals exhibit periodic oscillations as the probe moves closer to the mica, and the oscillation periods in EAN and PAN in Fig. 7(d) and x1(e) are the same as the step sizes in AFM force curves (Fig. 7b and c), respectively. The magnitude of the oscillations increases closer to the surface due to a more pronounced ion ordering and enhanced confinement near the surface. The oscillations in PAN are more intense than EAN due to the longer alkyl chains of PAN, producing stronger solvophobic segregation. In the lateral direction, the topographic images of the IL-mica innermost layer obtained by the dynamic mode AFM (Fig. 7f) exhibit a regular lattice structure. The insets 2D Fourier transforms show a hexagonal symmetry of the lattice with a repeated spacing of 0.5 nm, which is in agreement with that of the mica lattice, as a consequence that the cations are electrostatically bound to mica-charged sites.
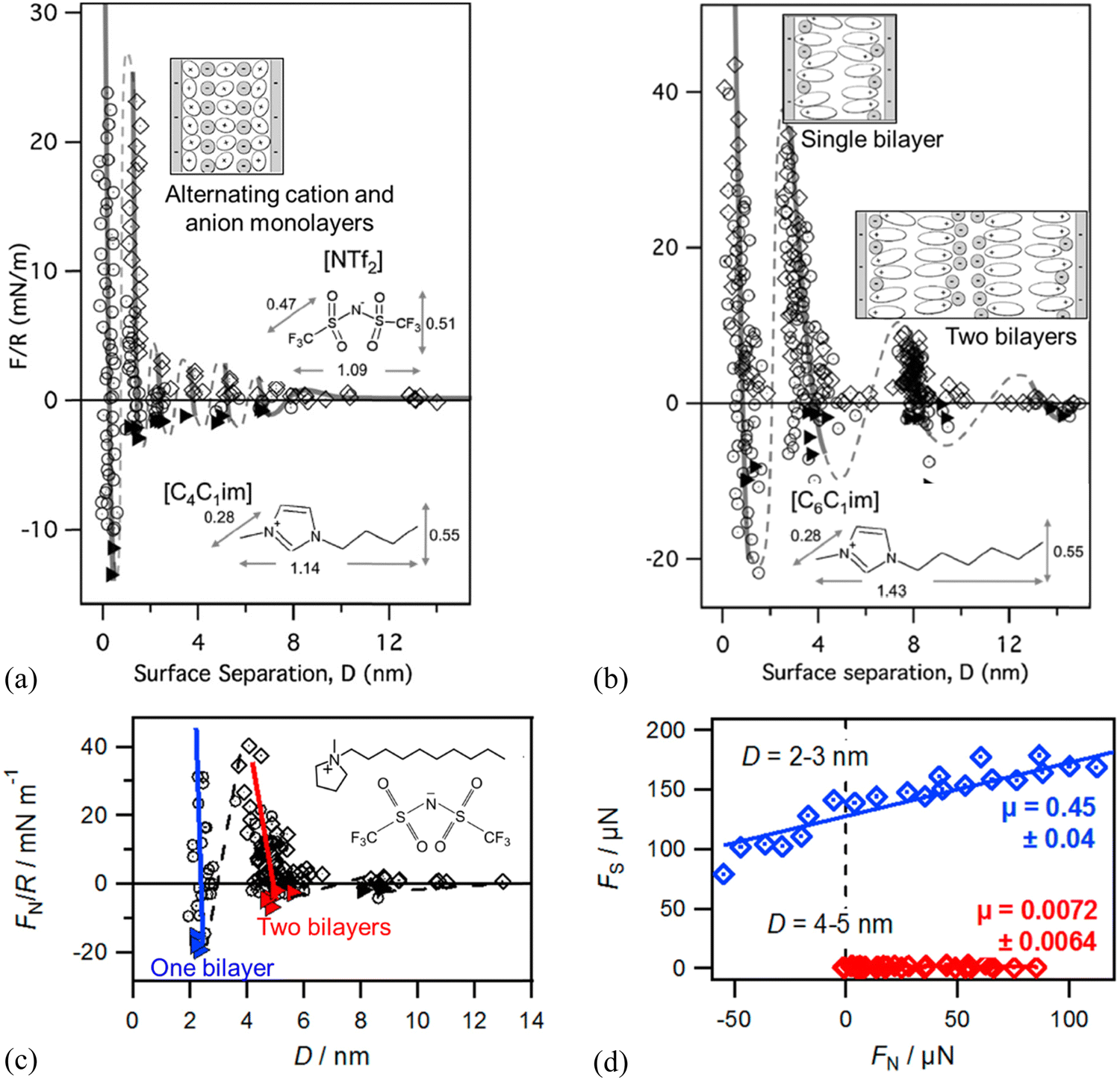 | ||
| Fig. 8 SFA-measured normal force (F) between two negatively charged mica surfaces (normalized by the mica curvature, R) versus film thickness (D) for (a) [C4C1im][NTf2] and (b) [C6C1im][NTf2]. The lines are a guide to the eye to show the oscillatory nature of the forces. The insets show ion orientations and molecular structure of differently lengthened cations and the anion. Reproduced from ref. 137 with permission from RSC, copyright 2011. (c) SFA-measured normal force FN/R versus D showing sequential squeeze-out of bilayers of [C10C1Pyrr][NTf2] between two mica sheets. Open diamonds and circles show data measured upon approach and retraction of the surfaces, respectively, and adhesive minima are marked as filled triangles. The inset shows the molecular structure of [C10C1Pyrr][NTf2]. Reproduced from ref. 138 with permission from ACS, copyright 2013. (d) SFA-measured friction force FSversus FN measured for one and two bilayers of [C10C1Pyrr][NTf2]. Solid lines are linear fits in (d) to obtain friction coefficient μ (slope) following Amontons’ law. Reproduced from ref. 142 with permission from ACS, copyright 2014. | ||
The dynamic friction force was also measured by SFA across one and two bilayers of 1-decyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide ([C10C1Pyrr][NTf2]) confined between two mica sheets (Fig. 8c and d).142 As shown in Fig. 8(d), the friction force versus normal load measured by SFA gives a low friction coefficient μ = 0.0072 ± 0.0064 for a film of 5.0 nm consisting of two bilayers, which corresponds to two bilayers in the red regime in Fig. 8(c). When the IL film is squeezed down to one bilayer with a film thickness of 2.4 nm corresponding to one bilayer (blue regime in Fig. 8c), it becomes very sticky with a concomitant abrupt increase in friction and a high friction coefficient of μ = 0.45 ± 0.04 (Fig. 8d). The friction for the two bilayers in [C10C1Pyrr][NTf2] is lower than that for any of those alternating cation–anion monolayers (9, 7, 5, and 3 ion monolayers)51 in shorter-chained [C4C1Pyrr][NTf2], whereas the friction for one bilayer is higher than that for any of the monolayer regimes.
3.2 Monolayer and bilayer structures – molecular simulations & thermodynamic modeling
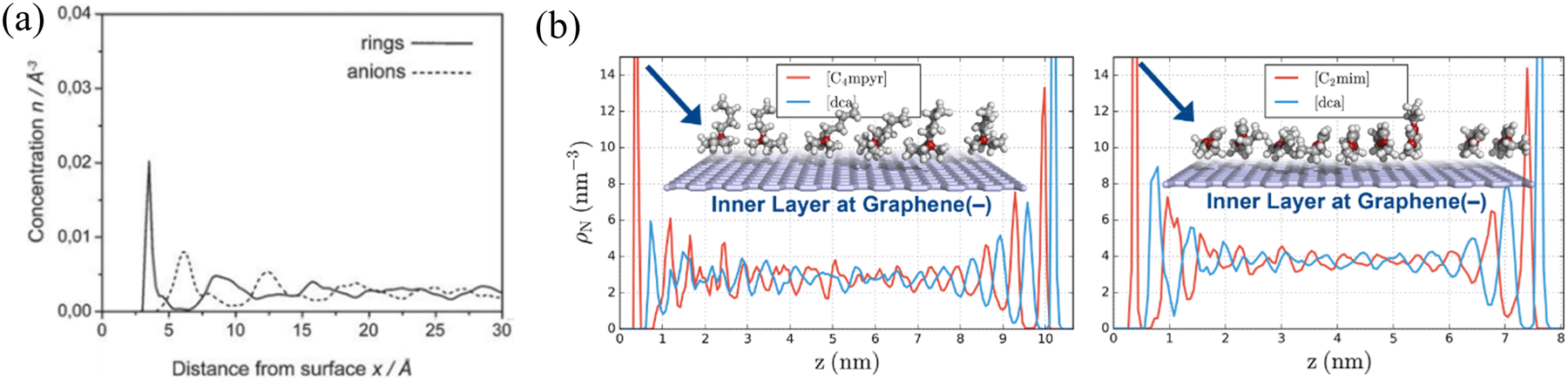 | ||
| Fig. 9 (a) The concentration profiles of imidazolium rings and anions of [BMIM][PF6] in the vicinity of the negatively charged graphite surface. Reproduced from ref. 160 with permission from RSC, copyright 2019. (b) Partial density profiles of 1-ethyl-3-methylimidazolium dicyanamide ([C2mim][dca]) and N-butyl-N-methylpyrrolidinium dicyanamide ([C4myr][dca]) at both charged graphene surfaces. Reproduced from ref. 149 with permission from RSC, copyright 2017. | ||
Classical density functional theory (DFT) has been employed to characterize and predict the IL interface layer nano-structuring.95,96,109,110Fig. 10 presents DFT predictions of the ionic distributions, the local mean electrical potential (φ(z)), and the integrated local charge density (q(z)) for three different ILs (symmetric IL with cation and anion of the same size in Fig. 10(a), asymmetric IL with a larger cation than the anion in Fig. 10(b), and asymmetric IL with a smaller cation than the anion in Fig. 10c) at charged surfaces.104 Distinctive multiple alternating layers of cations and anions were observed near the charged surface beyond 10 times the ionic diameter (Fig. 10a, top). The observed alternative monolayer ion structuring is concomitant with strong charge inversion (or ‘‘over screening’’) as shown by the oscillatory distributions of the local mean electric potential and the integrated charge density (Fig. 10a, bottom). Fig. 10(b) shows that an increase in the cation size results in a significant reduction of both the anion contact density and the magnitudes of charge oscillations, although the number of oscillatory layers remains largely unchanged. The reduced contact density and the magnitudes of charge oscillations are because of the weakened electrostatic effect by the larger cations near the surface due to the enhanced cation excluded volume. While the reduction of the cation size strengthens the electrostatic interactions, reduces the number of ionic layers at the surface, and makes the layering structure less distinctive (Fig. 10c). The less distinctive layering structure in the IL with a smaller cation calculated by DFT in Fig. 10(c) seems to contradict the SFA experimental results in Fig. 8(a) and (b) where 6 oscillatory layers were observed in the IL with the smaller [C4C1im]+ cation but only 4 oscillations for the larger [C6C1im]+ cation. However, it is noteworthy that the longer-chained [C6C1im][NTf2] exhibits a distinct amphiphilicity with the positive charge at the headgroup and a non-polar tail, which can induce a strong van der Waals force, resulting in a “…cation–cation–anion–anion…” bilayering structure. However, in the DFT calculations, the ILs were assumed to be spherical particles or segments, implying that they are non-amphiphilic. Therefore, in SFA measurements, the larger amphiphilic [C6C1im][NTf2] exhibits less oscillations than those of the smaller [C4C1im][NTf2], but still showing a more distinctive layering structure with a much greater repeat distance, in agreement with the DFT predictions.
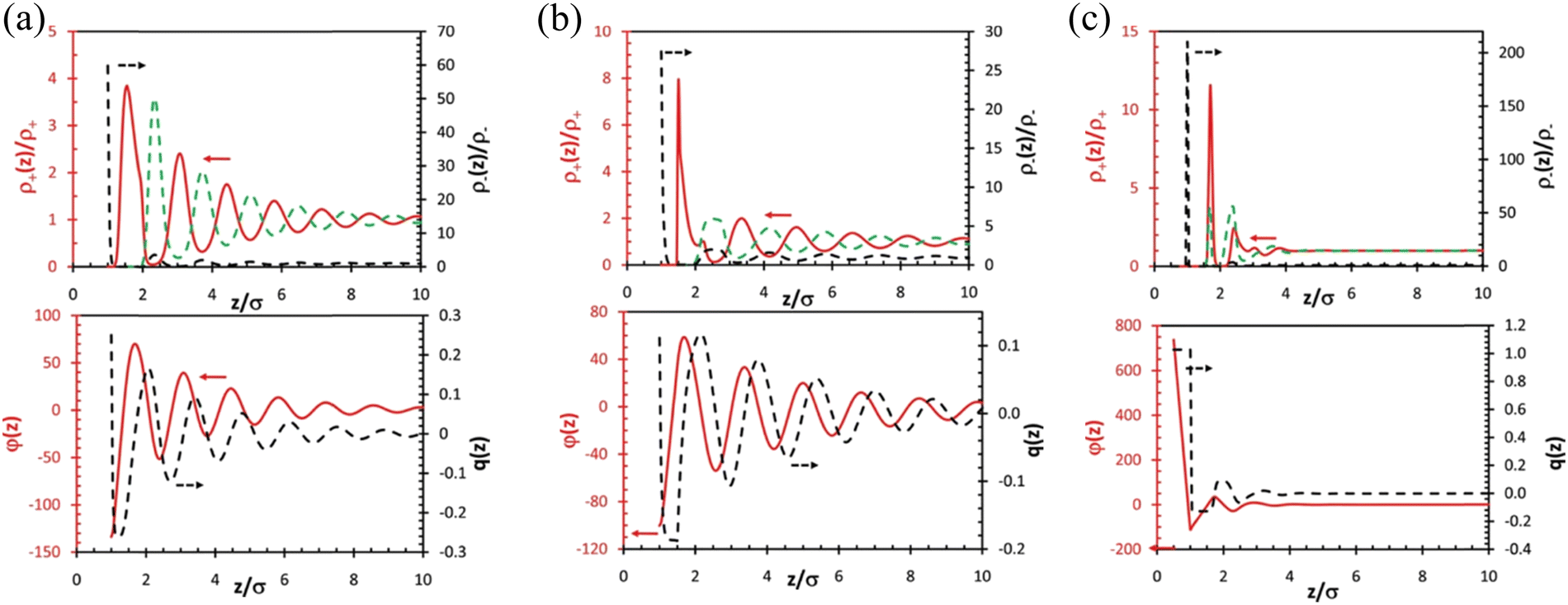 | ||
| Fig. 10 (a) Top: DFT-predicted distributions of cations (red solid line) and anions (black dashed line) for a symmetric IL (σ+ = σ–= σ) near a charged surface with reduced charge density Qσ2/e = 0.25. The green dashed line represents the anion density beyond the first layer plotted using the same scale of that for the cation density (left axis). Bottom: The reduced mean electric potential, φ(z), and the integrated charge distribution function, q(z), near the electrode. (b) The same as (a) but for an IL with asymmetric ionic size (σ+ = 1.5σ, σ–= σ). (c) The same as (a) but for an IL with asymmetric ionic size (σ+ = 0.5σ, σ–= σ). Reproduced from ref. 104 with permission from RSC, copyright 2011. σ+ and σ– represent the size of cation and anion, respectively. | ||
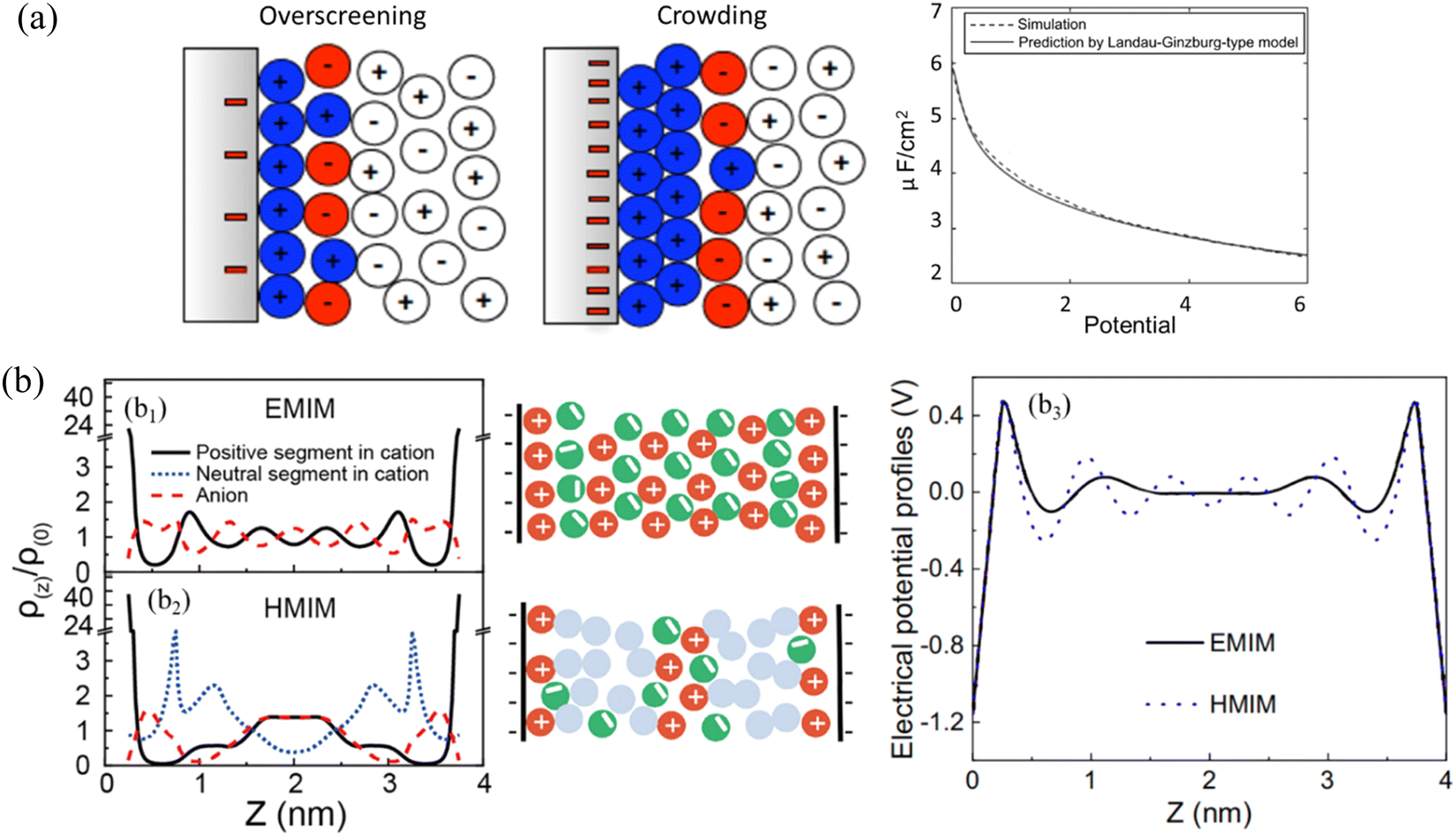 | ||
| Fig. 11 (a) Structure of the IL double layer (in color) predicted by Landau–Ginzburg-type continuum theory at a moderate voltage of 0.26 V (left), and a high voltage of 2.6 V (right). Right panel: Comparison of double-layer differential capacitance obtained from Landau–Ginzburg-type model (solid curve) and simulation (dashed curve). Reproduced from ref. 162 with permission from APS, copyright 2011. (b) b1 and b2: DFT-predicted density profiles (left) and schematics (middle) of anions (red dashed line), the positive (black solid line) and neutral (blue dotted line) segments of cations inside a negatively charged nanopore at the electrical surface potential of –ψ for ILs containing (b1) 1-methyl-3-ethylimidazolium ([EMIM]+) and (b2) 1-methyl-3-hexylimidazolium ([HMIM]+) cations. b3: Electrical potential profiles inside a negative nanopore at the electrical surface potential of –ψ for ILs containing [EMIM]+ (blue dotted line) and [HMIM]+ (black solid line). Reproduced from ref. 96 with permission from Elsevier, copyright 2020. | ||
Aided by DFT, the alternative cation–anion monolayer structure for the symmetrical IL containing 1-methyl-3-ethylimidazolium ([EMIM]+) within a negatively charged pore (–ψ) can be seen in Fig. 11(b1). While the addition of a long chain into the cation, i.e., the IL containing 1-methyl-3-hexylimidazolium ([HMIM]+, cf.Fig. 11b2), disrupts the mono-layering of ions that is normally seen for a symmetrical IL in Fig. 11(b1). In the IL with a long [HMIM]+ cation chain (Fig. 11b2), the van der Waals force will dictate the arrangement of ions, forming a new layer of neutral segments, so that bilayer IL structuring occurs. As a result, the electrical potential profiles of the first layer are more balanced in the IL with a longer [HMIM]+ cation than those of the symmetric IL system with a [EMIM]+ cation, and the oscillation of charge smooths for the longer IL (Fig. 11b3).96 Despite the years of active research and the significant efforts of theorists and simulators, the studies on the ILs interface layer with the combination of modeling and simulation are still fairly lacking.165 Vaganova et al.166 proposed an approach that combines DFT and MD simulations to study the behavior of pure methane, ethane and carbon dioxide and their binary mixtures in slit-like carbon nanopores, showing a good agreement between the structures obtained by theory and simulation. This approach consists of two principal steps: a DFT calculation of fluid composition and density distribution in a nanopore under specified thermodynamic conditions and an MD simulation of the confined system with obtained characteristics. Malpica-Morales et al.167 combined machine learning techniques with DFT to reconstruct the external potential which accounts for the interaction of the many-particle system with an external field and thus affects the density distribution. This combination accurately infers the density profile of the many-particle system, probably serve as a prototype for studying a wide variety of applications, especially for ILs interface layering behavior. Here are a few examples just for quanlitative validation between modeling and simulation: electrolyte perturbed-chain SAFT-DFT model was developed to describe the behavior of the imidazolium-based IL and CO2–IL mixture confined in silica nanopores, in which an ionic functional was proposed for electrostatic free-energy contribution. The DFT model predicting long-range oscillatory density profiles of ILs confined in silica slit and cylindrical pores agree well with those predicted by molecular simulation.168 The strong oscillations of ionic density profiles and the local electrostatic potential were predicted by DFT to describe the double layer structure of an IL at an electrode surface using a dimer model, which was confirmed further by simulation.169 DFT-based MD simulations were conducted to study the interface between graphene and 1-ethyl-3-methyl-imidazolium tetrafluoroborate IL to examine the ions arrangement at variable surface charge densities.170
3.3 Combined atomic force microscopy & simulations
AFM studies have been combined with MD simulations to examine the distinct effect of the addition of salts into ILs at solid interfaces on the cation–anion monolayers.7 In Fig. 12(a), neat IL, i.e., N-methyl-N-propylpyrrolidinium bis(fluorosulfonyl)imide ([C3mpyr][FSI]) gives rise to five oscillations at gold surfaces in the approaching force curve, with the step period of 0.60 nm, being consistent with the size of the cation–anion pair. The solvation force, required to squeeze-out a layer, increases closer to the surface as the ordering of the layered ions is enhanced. In contrast, with the addition of 50 mol% NaFSI in the [C3mpyr][FSI] IL (Fig. 12b), the ion structure is significantly changed stepwise. A considerable decrease is observed in the solvation force, indicating that the reduced force to rupture each ion layer, due to the weakening of the ion–ion association caused by NaFSI salt. The observed shift of the step position in Fig. 12(b) suggests that the dimensions of ion packing have been changed by NaFSI, attributed to the Na+ altering the chemical composition of the inner ion layers.7 The presented ion number density profiles show around five peaks for the [C3mpyr][FSI] IL system without NaFSI salt, agreeing well with the AFM results (Fig. 12a). With the addition of 50 mol% NaFSI salt, the number of peaks in the density profiles of both cations and anions reduces to three, again reproducing the change observed in the AFM measurements (Fig. 12b). The height of the first sharp peak for the IL cation reduces as the salt is added, whereas that of the [FSI]− increases, suggesting a growth in the number of anions in the innermost layer.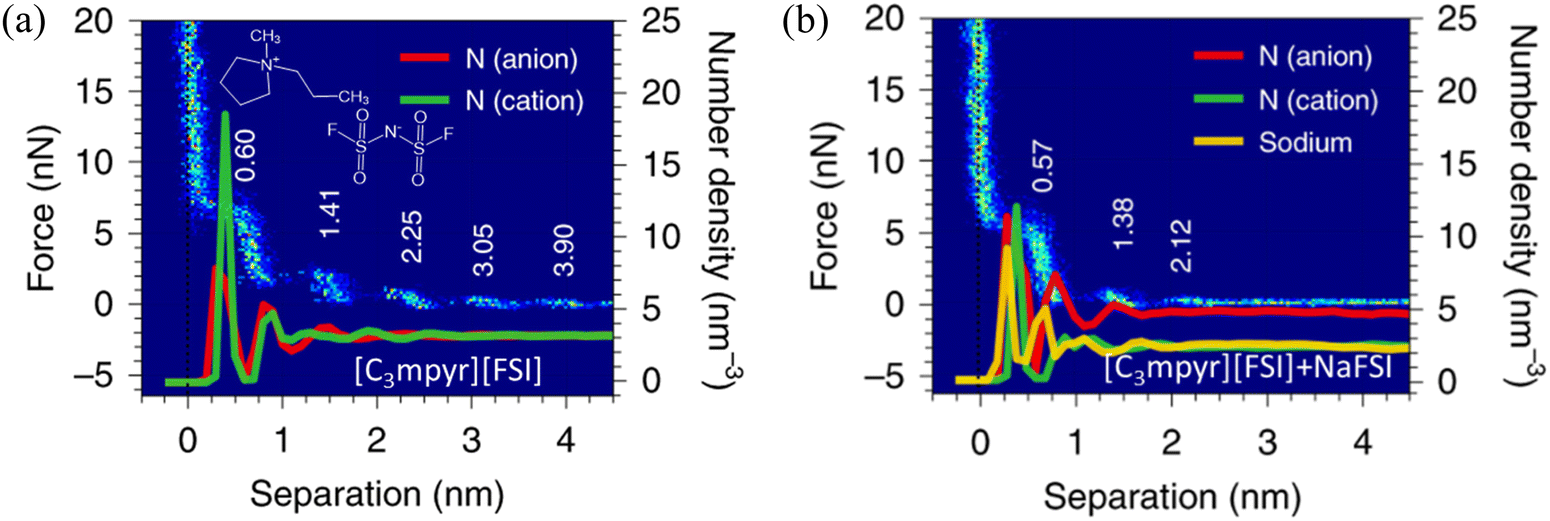 | ||
| Fig. 12 AFM-measured force curves and MD-calculated ion number density profiles of the [C3mpyr][FSI] IL (a) without and (b) with an addition of 50 mol% NaFSI on gold electrodes at open circuit potential (OCP). Reproduced from ref. 7 with permission from Springer, copyright 2020. | ||
To reveal the molecular origin of the experimentally detected ionic layers in the amphiphilic IL at solid surfaces, Mao et al.6 compared the different ion structures of non-amphiphilic (Fig. 13a) and amphiphilic (Fig. 13b) ILs at graphite surfaces through the combination of the atomistic molecular dynamics (MD) simulation and AFM force curves. The conventional non-amphiphilic 1-butyl-3-methylimidazolium tetrafluoroborate ([C4C1Im][BF4]) and amphiphilic 1-butyl-3-methylimidazolium 1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate ([C4C1Im][AOT]) are compared. The latter IL, i.e., [C4C1Im][AOT], is amphiphilic because it possesses a special anion [AOT]− with a negative charge located at one end of the ion (polar head) and a bulky branched di-chain hydrocarbon group at the other end (non-polar tail). The left panels of Fig. 13(a) and (b) show the ion number density (ρN) profiles of [C4C1Im][BF4] and [C4C1Im][AOT], respectively, obtained from MD simulations at charged graphite surfaces with applied potentials (U) of ±2 V. The non-amphiphilic [C4C1Im][BF4] IL exhibits a lamellar structure of alternating charges, persisting for around 8 ion layers (Fig. 13a). While for the amphiphilic [C4C1Im][AOT], it is interesting that the surface-induced charge-alternating structure only persists for around two ion layers, with the first peak showing a much larger ρN (Fig. 13b) relative to that of [C4C1Im][BF4].
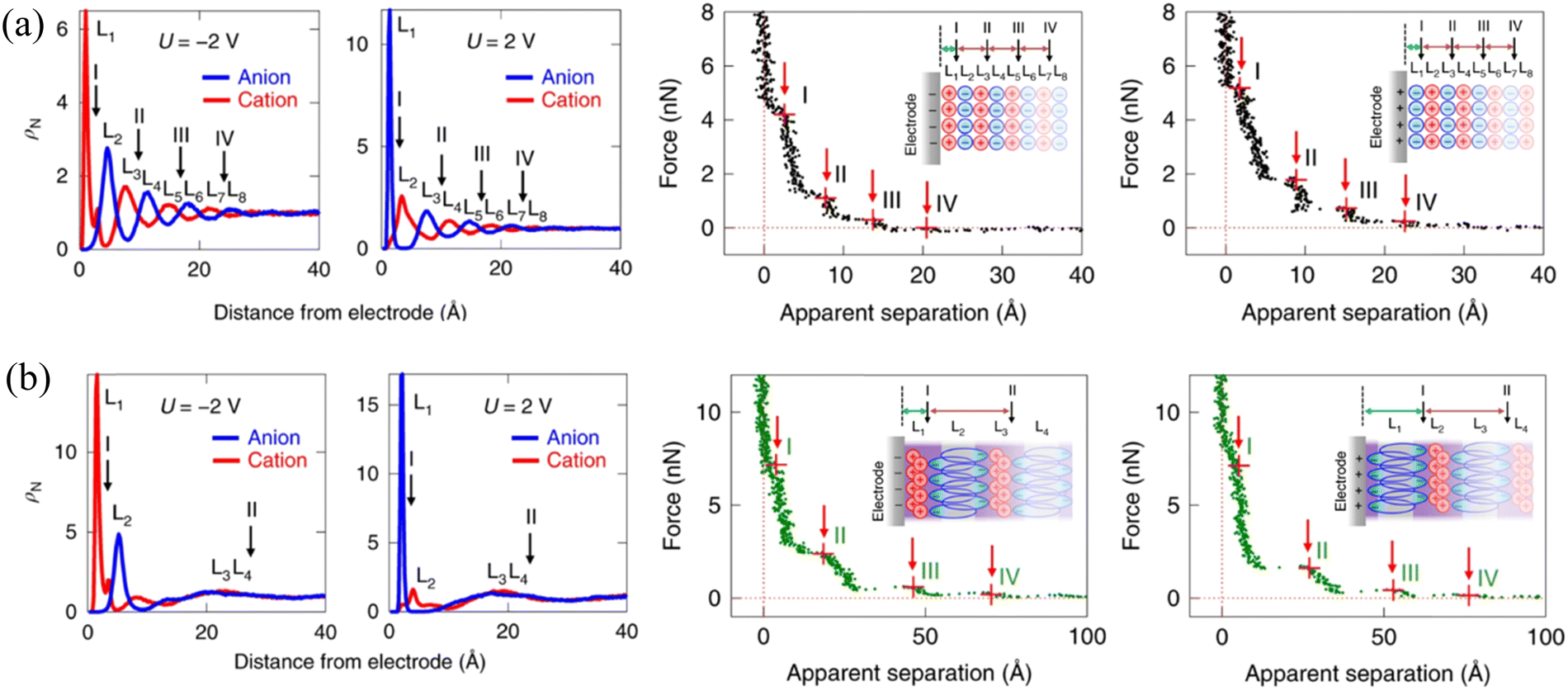 | ||
| Fig. 13 Ion number density profiles obtained from MD simulations (left) and typical AFM force curves (right) of (a) non-amphiphilic [C4C1Im][BF4] and (b) amphiphilic [C4C1Im][AOT] on negatively and positively charged graphite electrodes. Reproduced from ref. 6 with permission from Springer, copyright 2019. Insets in AFM force curves are schematics of hypothesized ion arrangements of the ILs at charged graphite surfaces. Red arrows with I, II, III and IV in the force curves indicate the possible positions corresponding to the AFM push-through locations, corresponding to the black arrows marked positions in ion number density profiles from MD. | ||
AFM force curves in the right panels of Fig. 13(a) and (b) demonstrate the alternating cation–anion molecular layering in [C4C1Im][BF4] and unusual interfacial ion distribution in the amphiphilic [C4C1Im][AOT] that have been observed in MD simulations. The separation between steps for [C4C1Im][AOT] is 22–24 Å (right, Fig. 13b), which is three to four times larger than those for [C4C1Im][BF4] (6–7 Å), corresponding to the physical dimension of an ion pair of [C4C1Im]+ and [BF4]− (right, Fig. 13a). The presented unusual interfacial ion distribution in the amphiphilic [C4C1Im][AOT] results from the significant van der Waals interactions among the non-polar surfactant tails in the anion. At a positively charged electrode surface, the polar heads of anions [AOT]− are electrostatically attracted to the interface, whereas the non-polar tails orientate outward, in close proximity to the non-polar tails of the next anion layer. A bilayer thus forms in parallel to the positively charged surface, with a neutralizing cation layer adjacent to the anionic polar heads. Near a negatively charged surface, the innermost ion layer is enriched in cations, fully neutralizing the adjacent anionic bilayer, and resulting in a larger separation (right, Fig. 13b, inset). In contrast, the distribution of most cations and anions in the non-amphiphilic [C4C1Im][BF4] is dominated by the Coulombic interactions, forming ‘neutral aggregates’ (right, Fig. 13a, inset) with the characteristic separation of an ion-pair dimension. Similar “…anion–anion–cation–cation…” bilayer structure has also been observed in the amphiphilic phosphonium-based IL trihexyltetradecyl-phosphonium bis(salicylato)borate ([P6,6,6,14][BScB]).21 The observed bilayer structure of the amphiphilic ILs, allowing the excess ions to pack at charged surfaces, facilitates the improvement of the capacitive energy storage capability.6,21,96
4. How interface layer ionic liquid nano-structuring enables lubrication and energy storage
ILs exhibit many attractive properties in proximity to solid surfaces, and can form well-defined ion layers at interfaces where the composition, structure, dynamics, etc. are tunable. A large volume of work has appeared over the past few years to witness the growth of ILs in the vicinity of solid surfaces for engineering and electrochemical applications, especially in lubrication and energy storage.4–7,12,15,29,38,1714.1 Lubrication and friction reduction
ILs can lead to a better friction reduction than conventional molecular lubricants, because they can form a robust boundary film better resisting the squeeze-out at heavy loads. ILs self-assemble to ordered ion layers near the surface exhibiting strong interactions with surfaces.30 The interface ion layering of ILs simply plays a pivotal role in improving the lubricating efficiency at IL–solid interfaces.2,171 A super-lubrication with a super-low friction coefficient (μ) of 0.001 at pressures up to 78 MPa has been observed at varying sliding velocities by AFM in imidazolium ILs at graphite surfaces even without electric field regulation (Fig. 14a).20 The ordered striped structure driven by the “atomic-locking” effect between carbon atoms on the alkyl chain of ILs and graphite unveils the hidden structural feature of IL ions dominating this extreme lubrication. Taking the imidazolium IL, [C18mim][NTf2] as an example, it is obvious that the IL self-assembles into smooth films (Fig. 14b) with a thickness of ∼0.5 nm corresponding to a monolayer IL with the imidazole ring lying flat on the surface. A well-ordered striped structure with a periodicity of about 6 nm has been observed in the high-resolution image (Fig. 14c), with bright striped regions representing the self-assembled ILs on the surface. In combination with simulation, the alternate ordered and disordered ionic regions are recognized, where a head-to-head structure pattern is shown with two different configurations of the imidazole ring arranged alternately in the same column (Fig. 14d). The cross-sectional profiles from AFM experiment and simulation in Fig. 14(e) indicate that the length of one period of this head-to-head ion structure can be divided into three regions, in which, regions I, II, and III correspond to imidazole rings, anions, and alkyl chains, respectively. The height difference between the experiment and simulation is attributed mainly to the out-of-plane flexibility of the warped imidazole rings, which are deformed and pushed closely to the graphite surface under the sliding AFM tip.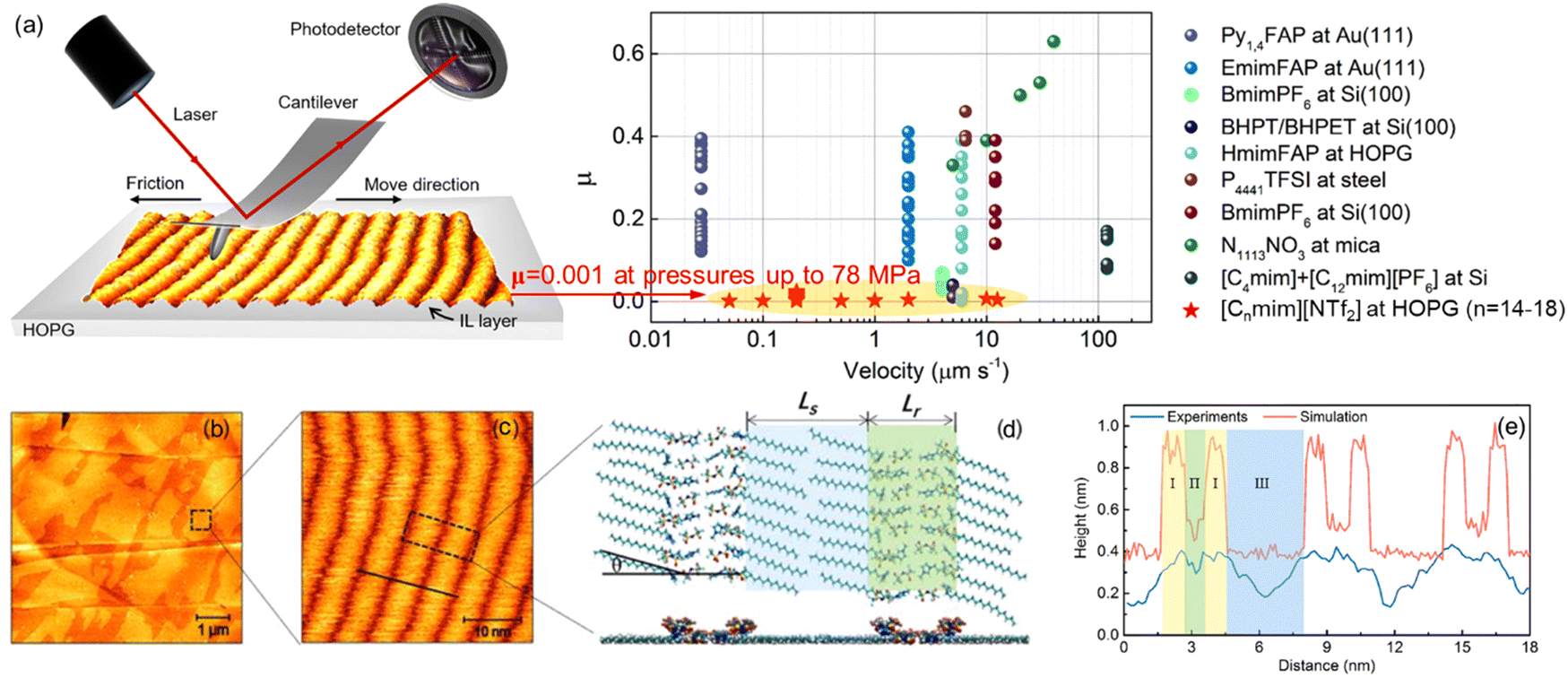 | ||
| Fig. 14 (a) Friction coefficient (μ) of various ILs obtained by AFM experiments. (b)–(e) Typical AFM images and simulation results of [C18mim][NTf2] on the HOPG surface: (b) topography image of [C18mim][NTf2] and (c) a close-up view of the fine structure. (d) Top and side views of arrangements for cations and anions obtained by simulation, Ls and Lr represent the lengths of the alkyl chain and anion region, respectively, and graphite surface is now shown here. (e) Comparison of cross-sectional profiles in the experiment (black line in (c)) and simulation. Reproduced from ref. 20 with permission from ACS, copyright 2022. | ||
Despite great progress in the fundamental understanding of the ion layering of ILs at solid surfaces and the friction through various advanced techniques, the influence of the layering structure of ILs on friction is still controversial. The layering structures of 1-hexyl-3-methylimidazolium tris(pentafluoroethyl) trifluorophosphate ([HMIM][FAP]) and trihexyl-(tetradecyl)phosphonium bis(trifluoromethane)sulfonamide ([P6,6,6,14][TFSI]) confined between a sharp AFM Si probe and a stainless steel surface are compared by AFM force curves together with the friction force (Fig. 15a).52 The force curves of [HMIM][FAP] (Fig. 15a, left) exhibit more ion layers near the surface than [P6,6,6,14][TFSI], because the bulkiness of the [P6,6,6,14]+ cation in [P6,6,6,14][TFSI] weakens the electrostatic interaction with the anion and results in the weaker structure. Therefore, the sliding AFM probe expels the weak ionic liquid interface layer structure of [P6,6,6,14][TFSI] with minimal energy loss and low friction (Fig. 15a, right). A quantized friction feature is observed in the IL, 1-butyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide ([C4C1Pyrr][NTf2]), confined between two mica sheets in a SFA, as shown in Fig. 15(b). Each of the quantized friction regimes follows an approximately linear friction vs. normal load relationship, and the resulting gradient, i.e., the friction coefficient μ increases as the layering ion pairs become thinner. This is attributed to the greater degree of interlocking of the layers closer to the surface, which are more ordered, with a greater ion excess concentration. These more ordered and densely packed ion layers result in a ‘stronger inter-layer attraction for the same value of contact area and a greater barrier for unlocking the surfaces to allow shear’.51 A higher friction force, as a result of a stronger layering structure of IL ions at the surface, has also been found by AFM in the imidazole-based ILs on the surfaces of mica and carbon.53,113,172 It is apparent that the strongly structured ion layers of the interface layer IL result in a higher friction, because of more dissipated energy, and a higher friction is required by breaking the stronger interlayer bonds.
 | ||
| Fig. 15 (a) AFM force curves (left) and friction force vs. normal load (right) of [HMIM][FAP] and [P6,6,6,14][TFSI] on the stainless steel surface. Reproduced from ref. 52 with permission from ACS, copyright 2017. (b) Kinetic friction force FS,kvs. the normal load FN measured for different numbers of ion layers of the IL 1-butyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide ([C4C1Pyrr][NTf2]) confined between mica surfaces in a SFA. Reproduced from ref. 51 with permission from RSC, copyright 2013. (c) Left: Number density profiles of the [C2MIM][NTf2] IL confined between two positively charged slabs (+32 μC cm−2) under a load of 400 MPa using the nonequilibrium MD simulations. The color of the line corresponds to that of each group in the IL (see legend). Middle: The slabs confining the IL, with a normal load, fL, and the top slab moving along the y direction at a constant velocity, Vdir, with a fixed lower slab. Right: 2D structure factor Sm(kx, ky) for the layer 2 and 3 in the first 3 ion layers of the confined [C2MIM][NTf2] nearest to the slab surface, showing the ion crystalline ordering. kx and ky represent the wave-vectors in x and y directions, and the bright spots indicate the formation of 2D crystalline structures. Layer 1 corresponds to the anion-rich layer next to the fixed wall, layer 2 to the adjacent cation-rich layer, and layer 3 to the anion-rich layer in the middle of the IL film. Reproduced from ref. 173 with permission from ACS, copyright 2020. | ||
However, Atkin's group54,55 found a different ion ordering dependence of the friction, where more ordered IL layers lead to a smoother sliding plane, resulting in a smaller friction, and this is further demonstrated by simulation findings.173 With atomistic nonequilibrium molecular dynamics (NEMD) simulations, the friction is observed to vary with the ion layering structure of imidazolium ILs at surfaces, in which these ILs feature the lower friction forces for the more ordered ion layers (Fig. 15c).23,173 As shown in Fig. 15c (left panel), the number density profiles of 1-ethyl-3-methyl-imidazolium bis-(trifluoromethylsulfonyl)imide ([C2MIM][NTf2]) between two positively charged slabs under a load of 400 MPa exhibit 5 ionic layers. The characteristic alternating charge structure of the IL under confinement is clearly observed. With the time-averaged 2D structure factor Sm(kx, ky),173 the ion crystalline ordering is observed for the first 3 ion layers of the confined [C2MIM][NTf2] nearest to the slab surface (right panel, Fig. 15c). Layer 1 corresponds to the anion-rich layer next to the fixed wall which is not shown in Fig. 15c, layer 2 to the adjacent cation-rich layer, and layer 3 to the anion-rich layer in the middle of the IL film. The appeared well-defined bright spots in layer 3 of [C2MIM][NTf2] (right panel, Fig. 15c), are indicative of crystalline order throughout the confined IL film induced by the positive charged slab surface, while the structure of layer 2 is clearly disordered liquid-like. The observed crystalline ordering of the IL ions is closely related to the friction forces, in which ordered ion structures are weakly commensurate or incommensurate with the slab structure facilitating the decrease of the friction. Thus, there is a controversy over the correlation between the ion ordering of ILs and the friction at IL–solid interfaces, which needs further investigation.
4.2 Making use of nano-structuring of interface layer ionic liquids in energy storage
Conventional organic solvent-based electrolytes have a number of limitations including flammability, unsatisfactory electrochemical stability, thermal instability, and decomposition of the salts that are adversely affecting the overall performance and service life of energy storage devices.174 IL-Based electrolytes are considered as promising alternative electrolytes with great potential to overcome the drawbacks associated with these conventional electrolytes.175 ILs have a combination of properties making them appropriate electrolytes for energy storage devices. Some of these properties include non-flammability, negligible vapor pressure, chemical, thermal and electrochemical stabilities, inherent ionic conductivity, and wide electrochemical stability windows up to 6.0 V.176–178 In the case of batteries, the energy storage is based on faradaic reactions and the voltage corresponds to the redox couple, however, energy is stored through compression of IL ions along the electrode surfaces in the case of an electric double-layer capacitor. In the latter, energy storage occurs via physical separation by the ions under the influence of applied electric potential, and no chemical reactions are involved.We focus on the interfacial structuring of ILs as electrolytes in electric double-layer capacitors, where an electric double layer (EDL) is formed in contact with electrode surfaces, i.e., the ILs screen charged surfaces by the formation of bound layers and diffuse electric double layers containing dissociated ions. As illustrated in Fig. 16(a),179 the dissociated IL ions of the opposite signs are accumulated on the surface of carbon electrodes as a result of the applied negative potentials and the amount of ions is proportional to the applied potential. The resulting local Coulombic rearrangements of the IL ions at negatively charged carbon electrode surfaces are responsible for the energy storage performance. Apparently, the nature of the electrode can affect the structure of the EDL and thus plays a key role in the differential capacitance of the supercapacitors.180 For example, Su et al.181 functionalized gold electrode surfaces by –COOH and –NH2 groups to adjust surface chemistry and control the ion structuring of [BMIM][PF6] IL, strongly influencing the capacitance process of the IL. With in situ colloid probe AFM detection of nano-friction and torsional resonance frequency at the IL–electrode interfaces, they observed stiffer ion layers on the Au–COOH surface than on Au–NH2, demonstrating more ordered ion layers on the –COOH-terminated Au surface. The strength of the ion layering on the Au–COOH surface is facilitated by the π–π+ stacking interaction between the delocalized π+ electrons from the imidazolium ring of the [BMIM]+ and the localized π electrons from the sp2 carbon on the –COOH group. On the contrary, the weaker IL–surface interactions on the Au–NH2 surface lead to aggregating IL droplets and are unable to induce a strong templating effect on the IL ions. The resulting uniform structuring ion layers of the IL at Au–COOH speed up the capacitive process and make the electrochemical response more sensitive (Fig. 16b). With MD simulations, Vatamanu et al. observed a noticeable improvement in the capacitive storage of the IL by increasing the curvature of the electrode, as an effectively larger volume of the ILs interface layer allows more counterions per surface area in contrast to a flat surface, leading to an increased capacitance.182
 | ||
| Fig. 16 (a) Schematic of the carbon electrode–IL interface in electric double-layer capacitors showing local ordering transitions of IL ions caused by varying surface potentials. Adapted from ref. 179 with permission from RSC, copyright 2018. (b) Cyclic voltammetry curves of [BMIM][PF6] on the surface of –COOH and –NH2 terminated gold electrodes. The inset circles showing the colloid probe AFM obtained ion structuring of the IL at the functionalized electrode surfaces. Reproduced from ref. 181 with permission from ACS, copyright 2023. | ||
Besides, the chemical structure of the IL has a direct influence on the ion distribution near the charged electrode to affect the differential capacitance of the supercapacitors. For example, the imidazolium cations of ILs, which get chemisorbed on metals such as Ag and Au, were found to result in voltage-mediated structural transitions and large peaks in capacitance profiles.183–185 MD simulations were performed to compare differential capacitance profiles of [BMIM][BF4] and [BMIM][TFSI] at graphene electrodes, which exhibit similar camel-shaped capacitance profiles with two peaks on either side of a minimum centered at the potential of zero charge (left panel, Fig. 17).186 However, their capacitance profiles at large absolute voltages are different, where the differential capacitance begins to decrease at both positive and negative polarizations for [BMIM][BF4], but only at positive polarization for [BMIM][TFSI]. The different capacitive behavior at the positively polarized carbon electrode surface is due to the different anions restructuring of [BMIM][BF4] and [BMIM][TFSI] within the inner layer adjacent to the electrode surface (cf. middle and right panels in Fig. 17).
 | ||
| Fig. 17 Left: Differential capacitance “CD” of [BMIM][BF4] and [BMIM][TFSI] electrolytes at graphene electrode as a function of electrode potential (ΔVelectrode), plotted relative to the potential of zero charge. Snapshots of [BMIM][BF4] (middle) and [BMIM][TFSI] (right) electrolytes near the electrode surface at approximately +1.6 V relative to PZC (potential of zero charge). Orange: [BMIM]+ cation; green: [BF4]− (middle panel) and [TFSI]− (right panel) anions. The color bar on the right indicates the value of image charges on each atom of the graphene electrode. Reproduced from ref. 186 with permission from ACS, copyright 2021. | ||
Recent studies have demonstrated that IL electrolytes comprising structural analogous anions have different modes of interactions with carbon electrode surfaces, leading to different capacitive performances on the same carbon electrode.175,187 For example, the IL containing non-aromatic tetrahydrofuran-2-carboxylate anion [HFuA]− with delocalized electrons has shown somewhat tilted orientation on the surface of graphene associated with the best capacitive performance as compared with the other two ILs containing aromatic anions, 2-furoate [FuA]− and thiophene-2-carboxylate [TpA]− anions. The configurations of [P4444]+ cation (Fig. 18a and e), [FuA]− (Fig. 18b and f), [HFuA]− (Fig. 18c and g), and [TpA]− (Fig. 18d and h) anions at electrified graphene electrodes with a surface charge density of 10 μC cm−2 are shown in Fig. 18. The tetra(n-butyl)phosphonium [P4444]+ cation exhibited random distributions near the surface with no specific coordination with the graphene electrode. The aromatic anions, [FuA]− and [TpA]−, exhibited perpendicular distributions to the positively charged graphene electrode resulting in very strong surface interactions that might need a large voltage to break the interfacial structures. This was the main reason that the [P4444][FuA] and [P4444][TpA] ILs have provided relatively lower capacitances than the [P4444][HFuA] IL.188
 | ||
| Fig. 18 Ion configures of [P4444]+ (a) and (e) cation in cation-cathode interfacial region, and of [FuA]− (b) and (f), [HFuA]− (c) and (g), and [TpA]− (d) and (h) anions in anion-anode interfacial region. The lower panels correspond to the side-views of the top-view configurations shown in upper panels. Adapted from ref. 188 with permission from Elsevier, copyright 2022. | ||
5. Summary and outlook
We have reviewed recent studies of the multifaceted layering behavior of ionic liquids at a variety of different surfaces, studied by experiments, molecular simulations, and thermodynamic modeling methods. Ionic liquid interface-layered nanostructures are important in numerous applications. The ordered surface structures after embedding them on surfaces and in constrained spaces greatly enhance many of IL properties, from those observed in bulk. They provide a convenient platform for novel innovations within sensor, electrode, battery, capacitor, and lubricant technologies. There are several research methods and strategies to study IL interface layers. We have chosen to survey more closely three highly powerful methodologies. First, advanced experimental techniques for surface analysis: surface force apparatus (SFA) and atomic force microscopy (AFM). SFA provides detailed and highly accurate information about interaction forces (attractive and repulsive) between surfaces in a controlled manner in a variety of mediums. AFM with its very sharp tip at the end of a cantilever scans the surface using different modes. It can visualize the topography and morphology of ILs on a solid surface at an atomistic resolution. It provides information about structure, arrangements, organization, thickness, elasticity, and friction, etc. Although fundamentally and operationally different, they can give similar and complementary information. Second, Molecular simulations, and in particular molecular dynamics (MD), which is invaluable to studying the structure, thermodynamics, and dynamics of any type of molecular system, from solid metals to large biological systems in realistic environments and conditions. MD can be performed from first-principles to classical atomistic and coarse-grained mesoscopic systems at and out of equilibrium, including all possible hybrid methods among them. It is an ideal tool for studying ILs on different surfaces of constrained spaces. Third, thermodynamics provides an incredible and complete toolbox to study matter in its all possible states both at equilibrium and non-equilibrium. Already van der Waals gave us the first thermodynamic model which inspired to a wealth of state functions. Although thermodynamics works well without molecular degrees of freedom and is best for macroscopic matter, it can take molecular information from both experiments and simulations. Thermodynamics is highly useful for surface studies providing quantities like adsorption isotherms, solvation-free energies, phase equilibria, activity coefficients, surface/interface tension to mention a few. Many of these quantities can also be calculated from MD simulations or obtained experimentally.As all methods operate on their specific resolutions (molecular or material) and scales (length, time, etc.) providing results and data on properties of what they are designed to do, it is preferable to combine several of them. This is also normally done in the characterization of newly synthesized materials. Designing new materials and products with specifically tailored properties starting from molecular building blocks requires connecting ten orders of magnitude in the length scale. This is impossible using any single method or methodology. It can only be done using different multi-scale strategies. Multi-scale modeling, both “from bottom-up” and “from top-down” has been developed during the last two decades based on several strategies to connect and couple the scales. By writing this review our idea was to propose a strategy for multi-scale studies by using a triangle made of (1) advanced experimental tools, (2) MD simulations, and (3) thermodynamic modeling. The latter is an essential tool in chemical engineering. However, it has developed rapidly during the last decades and can be connected as a complementary tool with both experiments and molecular simulations. Therefore, it works well as a bridge between (1) and (2). We will demonstrate it for interfacial ILs. Mainly, because it is our own area of research but also because they are among the most complicated molecular systems due to all molecular interactions are there in concert. However, the modeling for ILs interface layers is somewhat scarce, which inspires and promotes us to perform it now, especially to build a multi-scale methodology by integrating modeling with experiments and simulations. Therefore, there is an urgent need to bridge theoretical modeling, simulation and advanced experimental tools to qualitatively and quantitatively study the ILs interface layer to enable the judicious design of IL–solid interfaces with appropriate properties for task-specific applications.
Experimental, molecular simulation, and thermodynamic modeling studies of the IL interface layer have shown different subtle responses of the overall properties and performance to the ion structuring for the interface layer ILs, but being tied to completely different interpretations. The combination of experimental techniques and/or computational simulations and/or thermodynamic modeling analysis is one important route to build up the quantitative structure–property relationship of interface layer ILs to deeply understand the mechanism in the ILs-based applications, and identify the proper ILs for specific solid interfaces with targeted properties/performance, and is seen in just a few other scenarios, such as CO2 adsorption42 with supported ILs.
However, establishing any quantitative structure–property relationships of interface layer ILs requires more research. The property/performance of ILs at solid surfaces is a complex function of the characteristics of the contacting surfaces, the nature of the IL, and external stimuli. Thus, the picture emerging now is that a large number of potential IL cations and anions are available, which provides enormous scope for scientific innovations, meaning that the use of ILs often results in reduced efficiencies or deviations from expected behavior. Using solid surfaces with a wide variety of adjustable properties further imposes challenges in identifying an optimum IL for any given solid surface in any specific application. Therefore, a good understanding and control of the chosen ILs, their structuring and ion dynamics when interacting with the solid surfaces, and how they are affected by the external conditions are needed to create assemblies for applications like boundary lubrication and high-performance energy storage devices. It is highly important to achieve an atomic-scale understanding of the molecular origin of the interface layer ILs behavior, providing quantitative structure–property relationships for exquisite control of functional IL–solid interfaces.
Theoretical studies, including computational simulations and thermodynamic modeling, can help in identifying the conditions that lead to optimal performance by achieving quantitative structure–property relationships of interface layer ILs. Nevertheless, the key problem in these theoretical studies is that the interaction parameters must be accurately introduced as an input to the model, and acquiring these ‘inputs’ experimentally is challenging. A colloid probe AFM-based methodology was proposed to attempt to quantify the molecular interactions of imidazolium-based ILs with SiO2 surfaces, in which, the AFM-measured adhesion force offers a direct measure of the IL interactions with the solid SiO2, that can be used to fit interaction parameters for theoretical calculations.90,91 However, the applicability and reliability of these derived interaction parameters still need to be validated.
The translation of ILs interface layer structuring to applications would also be of interest. When considering IL-based applications such as lubrication and energy storage, the corresponding properties/performance, i.e., friction and capacitance, are expected to be controllable at will. Although some of the properties are imposed by external conditions (e.g., temperature and potential) and the specific properties of surfaces (e.g. nanostructure and chemical heterogeneity), ILs themselves offer a vast parameter space in terms of their chemical compositions, which can be further diversified using solvents and additives.23 Therefore, it will be necessary to fully characterize the role of surface characteristics, nature of ILs, and external stimuli to build up the quantitative structure–property relationship. Previous exploration in molecular liquids at solid surfaces, combined the AFM experiment and thermodynamics modeling to find that the nanofriction coefficient, μ, is a function of three dimensionless parameters reflecting the intermolecular forces involved and the structure of the solid substrate according to the following equation: μ = 0.0079·ρs·Δs·σls2, where ρs is the atomic density of the solid, Δs is the spacing between layers of solid atoms, and σls is the molecular diameter that characterizes the liquid–solid interaction.43 This quantitative description cannot be extended straightforwardly to ILs and complex solid interfaces, but, at least, it demonstrates the possibility of building up quantitative structure–property relationships.
Detailed scenarios for solving these problems lie beyond the scope of this review. The exploration of building up quantitative structure–property relationships of interface layer ILs is only in its infancy, and there is still a lot of work for physicists and chemists to do to realize this quantification and guide for screening the best ILs-based lubricants and electrolytes. We hope that this review will motivate further work and enable the multiscale–triangle “experiment–simulation–thermodynamic modeling” in building up quantitative structure–property relationships for guidance in the virtual design of IL-based applications.
Abbreviations
| [C4C1Pyrr][NTf2] | 1-Butyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide |
| [C10C1Pyrr][NTf2] | 1-Decyl-1-methylpyrrolidinium bis[(trifluoromethane)sulfonyl]imide |
| [C4C1im][NTf2] | 1-Butlyl-3-methylimidazoliym bis(trifluoronmenthylsuldonyl)imide |
| [C6C1im][NTf2] | 1-Hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| [C2mim]Ac | 1-Ethyl-3-methylimidazolium acetate |
| EAN | Ethylammonium nitrate |
| PAN | Propylammonium nitrate |
| [C4C1Im][BF4] | 1-Butyl-3-methylimidazolium tetrafluoroborate |
| [C4C1Im][AOT] | 1-Butyl-3-methylimidazolium 1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate |
| [C3mpyr][FSI] | N-Methyl-N-propylpyrrolidinium bis(fluorosulfonyl)imide |
| [HMIM][FAP] | 1-Hexyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate |
| [P6,6,6,14][TFSI] | Trihexyl-(tetradecyl)phosphonium bis(trifluoromethane)sulfonamide |
| [HOC4Py][NTf2] | 1-(4-Hydroxybutyl)pyridinium bis(trifluoromethanesulfonyl)imide |
| [P6,6,6,14][BScB] | Trihexyltetradecylphosphonium bis(salicylato)borate |
| [C2MIM][NTf2] | 1-Ethyl-3-methyl-imidazolium bis-(trifluoromethylsulfonyl)imide |
| [Py1,4][TFSA] | 1-Butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide |
| [BMIM]FAP | 1-Butyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate |
| [HMIM]FAP | 1-Hexyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate |
| [BMIM]I | 1-Butyl-3-methylimidazolium iodide |
| [EMIM][NTf2] | 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| [C2mim][dca] | 1-Ethyl-3-methylimidazolium dicyanamide |
| [C4myr][dca] | N-Butyl-N-methylpyrrolidinium dicyanamide |
| [P4444][FuA] | Tetra(n-butyl)phosphonium furan-2-carboxylate |
| [P4444][TpA] | Tetra(n-butyl)phosphonium thiophene-2-carboxylate |
| [P4444][HFuA] | Tetra(n-butyl)phosphonium tetrahydrofuran-2-carboxylate |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to the funding by the China Postdoctoral Science Foundation and the National Natural Science Foundation of China (Grant No. 21838004, 22108015, 21978134). X. J. thanks the financial support from the Swedish Research Council (Grant No. 2020-03899), STINT (Grant No. CH2019-8287), and Horizon-EIC, Pathfinder challenges (Grant No. 101070976). F. U. S gratefully acknowledges the financial support from the Swedish Research Council (Grant No. 2018-04133). A. L. acknowledges Kempe Foundation project (Grant No. SMK21-0011), Swedish Research Council (Grant No. 2019-03865) and European Union's Horizon Europe research and innovation programme under grant agreement No 101086667, project BioMat4CAST.References
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- S. Zhang, J. Zhang, Y. Zhang and Y. Deng, Chem. Rev., 2017, 117, 6755–6833 CrossRef CAS PubMed.
- M. Cai, Q. Yu, W. Liu and F. Zhou, Chem. Soc. Rev., 2020, 49, 7753–7818 RSC.
- F. Zhou, Y. Liang and W. Liu, Chem. Soc. Rev., 2009, 38, 2590–2599 RSC.
- X. Mao, P. Brown, C. Červinka, G. Hazell, H. Li, Y. Ren, D. Chen, R. Atkin, J. Eastoe, I. Grillo, A. A. H. Padua, M. F. Costa Gomes and T. A. Hatton, Nat. Mater., 2019, 18, 1350–1357 CrossRef CAS PubMed.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Nat. Mater., 2020, 19, 1096–1101 CrossRef CAS PubMed.
- A. Eftekhari, Energy Storage Mater., 2017, 9, 47–69 CrossRef.
- K. L. Van Aken, M. Beidaghi and Y. Gogotsi, Angew. Chem., Int. Ed., 2015, 54, 4806–4809 CrossRef CAS PubMed.
- P. F. R. Ortega, G. A. dos Santos Jr., J. P. C. Trigueiro, G. G. Silva, N. Quintanal, C. Blanco, R. L. Lavall and R. Santamaría, J. Phys. Chem. C, 2020, 124, 15818–15830 CrossRef CAS.
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Acc. Chem. Res., 2019, 52, 686–694 CrossRef CAS PubMed.
- S. Bi, H. Banda, M. Chen, L. Niu, M. Chen, T. Wu, J. Wang, R. Wang, J. Feng, T. Chen, M. Dincă, A. A. Kornyshev and G. Feng, Nat. Mater., 2020, 19, 552–558 CrossRef CAS PubMed.
- M. Chen, J. Wu, T. Ye, J. Ye, C. Zhao, S. Bi, J. Yan, B. Mao and G. Feng, Nat. Commun., 2020, 11, 5809 CrossRef CAS PubMed.
- X. Wang, M. Salari, D.-E. Jiang, J. Chapman Varela, B. Anasori, D. J. Wesolowski, S. Dai, M. W. Grinstaff and Y. Gogotsi, Nat. Rev. Mater., 2020, 5, 787–808 CrossRef CAS.
- M. Radiom, Curr. Opin. Colloid Interface Sci., 2019, 39, 148–161 CrossRef CAS.
- R. Futamura, T. Iiyama, Y. Takasaki, Y. Gogotsi, M. J. Biggs, M. Salanne, J. Ségalini, P. Simon and K. Kaneko, Nat. Mater., 2017, 16, 1225–1232 CrossRef CAS PubMed.
- R. An, A. Laaksonen, M. Wu, Y. Zhu, F. U. Shah, X. Lu and X. Ji, Nanoscale, 2022, 14, 11098–11128 RSC.
- J. Comtet, A. Niguès, V. Kaiser, B. Coasne, L. Bocquet and A. Siria, Nat. Mater., 2017, 16, 634–639 CrossRef CAS PubMed.
- A. Sheehan, L. A. Jurado, S. N. Ramakrishna, A. Arcifa, A. Rossi, N. D. Spencer and R. M. Espinosa-Marzal, Nanoscale, 2016, 8, 4094–4106 RSC.
- Y. Lu, Y. Wang, F. Huo, W. Chen, M. Ma, W.-L. Ding, H. He and S. Zhang, ACS Nano, 2022, 16, 16471–16480 CrossRef CAS PubMed.
- T. Wang, L. Li, F. Zhang, Z. Dai, F. U. Shah, W. Wang, F. Xu and R. An, Phys. Chem. Chem. Phys., 2022, 24, 25411–25419 RSC.
- T. Sun, Y. Lu, J. Lu, H. Dong, W. Ding, Y. Wang, X. Yang and H. He, J. Phys. Chem. Lett., 2022, 13, 7113–7120 CrossRef CAS PubMed.
- F. Bresme, A. A. Kornyshev, S. Perkin and M. Urbakh, Nat. Mater., 2022, 21, 848–858 CrossRef CAS PubMed.
- J. Yang, C. Lian and H. Liu, Chem. Eng. Sci., 2020, 227, 115927 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2010, 12, 1709–1723 RSC.
- M. Mezger, H. Schröder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schöder, V. Honkimäki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed.
- Y. Hu and X. Peng, Structure and Bonding, in Structures and Interactions of Ionic Liquids, ed. S. Zhang, J. Wang, X. Lu and Q. Zhou, Springer-Verlag Berlin, Heidelberg, 2014, vol. 151 Search PubMed.
- E. J. Maginn, Acc. Chem. Res., 2007, 40, 1200–1207 CrossRef CAS PubMed.
- Y.-L. Wang, B. Li, S. Sarman, F. Mocci, Z.-Y. Lu, J. Yuan, A. Laaksonen and M. D. Fayer, Chem. Rev., 2020, 120, 5798–5877 CrossRef CAS PubMed.
- S. Perkin, T. Albrecht and J. Klein, Phys. Chem. Chem. Phys., 2010, 12, 1243–1247 RSC.
- U. Raviv and J. Klein, Science, 2002, 297, 1540–1543 CrossRef CAS PubMed.
- A. M. Smith, A. A. Lee and S. Perkin, Phys. Rev. Lett., 2017, 118, 096002 CrossRef PubMed.
- Y. Wang and L. Li, Langmuir, 2020, 36, 2743–2756 CrossRef CAS PubMed.
- L. R. Griffin, K. L. Browning, S. M. Clarke, A. M. Smith, S. Perkin, M. W. A. Skoda and S. E. Norman, Phys. Chem. Chem. Phys., 2017, 19, 297–304 RSC.
- A. Elbourne, K. Voïtchovsky, G. G. Warr and R. Atkin, Chem. Sci., 2015, 6, 527–536 RSC.
- S. Zhou, K. S. Panse, M. H. Motevaselian, N. R. Aluru and Y. Zhang, ACS Nano, 2020, 14, 17515–17523 CrossRef CAS PubMed.
- J. P. de Souza, K. Pivnic, M. Z. Bazant, M. Urbakh and A. A. Kornyshev, J. Phys. Chem. B, 2022, 126, 1242–1253 CrossRef CAS PubMed.
- Y. Zhou and J. Qu, ACS Appl. Mater. Interfaces, 2017, 9, 3209–3222 CrossRef CAS PubMed.
- K. E. Gubbins, Y. Long and M. Śliwinska-Bartkowiak, J. Chem. Thermodyn., 2014, 74, 169–183 CrossRef CAS.
- N. Wu, X. Ji, L. Li, J. Zhu and X. Lu, Chem. Eng. Sci., 2021, 229, 116164 CrossRef CAS.
- N. Wu, X. Ji, R. An, C. Liu and X. Lu, AIChE J., 2017, 63, 4595–4603 CrossRef CAS.
- N. Wu, X. Ji, W. Xie, C. Liu, X. Feng and X. Lu, Langmuir, 2017, 33, 11719–11726 CrossRef CAS PubMed.
- R. An, L. Huang, Y. Long, B. Kalanyan, X. Lu and K. E. Gubbins, Langmuir, 2016, 32, 743–750 CrossRef CAS PubMed.
- R. An, L. Huang, K. P. Mineart, Y. Dong, R. J. Spontak and K. E. Gubbins, Soft Matter, 2017, 13, 3492–3505 RSC.
- G. Yu, Z. Wei, K. Chen, R. Guo and Z. Lei, AIChE J., 2022, 68, e17575 CrossRef CAS.
- K. Paduszyński and U. Domańska, J. Phys. Chem. B, 2012, 116, 5002–5018 CrossRef PubMed.
- T. Zhou, H. Shi, X. Ding and Y. Zhou, Chem. Eng. Sci., 2021, 229, 116076 CrossRef CAS.
- Y. Huang, X. Zhang, X. Zhang, H. Dong and S. Zhang, Ind. Eng. Chem. Res., 2014, 53, 11805–11817 CrossRef CAS.
- I. I. I. Alkhatib, D. Bahamon, A. Al Hajaj and L. F. Vega, Ind. Eng. Chem. Res., 2022, 61, 12190–12207 CrossRef CAS.
- X. Liu, Y. Chen, S. Zeng, X. Zhang, S. Zhang, X. Liang, R. Gani and G. M. Kontogeorgis, AIChE J., 2020, 66, e16794 CrossRef CAS.
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, T. Welton and S. Perkin, Phys. Chem. Chem. Phys., 2013, 15, 15317–15320 RSC.
- P. K. Cooper, C. J. Wear, H. Li and R. Atkin, ACS Sustainable Chem. Eng., 2017, 5, 11737–11743 CrossRef CAS.
- R. An, X. Qiu, F. U. Shah, K. Riehemann and H. Fuchs, Phys. Chem. Chem. Phys., 2020, 22, 14941–14952 RSC.
- H. Li, M. W. Rutland and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 14616–14623 RSC.
- H. Li, M. W. Rutland, M. Watanabe and R. Atkin, Faraday Discuss., 2017, 199, 311–322 RSC.
- R. Atkin, N. Borisenko, M. Drueschler, S. Z. El Abedin, F. Endres, R. Hayes, B. Huber and B. Roling, Phys. Chem. Chem. Phys., 2011, 13, 6849–6857 RSC.
- C. Rodenbücher, K. Wippermann and C. Korte, Appl. Sci., 2019, 9, 2207 CrossRef.
- Y.-X. Zhong, J.-W. Yan, M.-G. Li, X. Zhang, D.-W. He and B.-W. Mao, J. Am. Chem. Soc., 2014, 136, 14682–14685 CrossRef CAS.
- X. Zhang, Y.-X. Zhong, J.-W. Yan, Y.-Z. Su, M. Zhang and B.-W. Mao, Chem. Commun., 2012, 48, 582–584 RSC.
- J. M. Black, M. Zhu, P. Zhang, R. R. Unocic, D. Guo, M. B. Okatan, S. Dai, P. T. Cummings, S. V. Kalinin, G. Feng and N. Balke, Sci. Rep., 2016, 6, 32389 CrossRef CAS PubMed.
- J. Marra and J. J. B. Israelachvili, Biochemistry, 1985, 24, 4608–4618 CrossRef CAS PubMed.
- U. Raviv and J. Klein, Science, 2002, 297, 1540–1543 CrossRef CAS.
- J. Klein, Nature, 1980, 288, 248–250 CrossRef CAS.
- J. Klein and P. Luckham, Nature, 1982, 300, 429–431 CrossRef CAS.
- R. Lhermerout and S. Perkin, Phys. Chem. Chem. Phys., 2020, 22, 455–466 RSC.
- L. Xiang, J. Zhang, L. Gong and H. Zeng, Soft Matter, 2020, 16, 6697–6719 RSC.
- D. Alsteens, H. E. Gaub, R. Newton, M. Pfreundschuh, C. Gerber and D. J. Müller, Nat. Rev. Mater., 2017, 2, 17008 CrossRef CAS.
- R. García and R. Pérez, Surf. Sci. Rep., 2002, 47, 197–301 CrossRef.
- H.-J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855–6863 CrossRef CAS.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CrossRef CAS.
- J. M. Black, D. Walters, A. Labuda, G. Feng, P. C. Hillesheim, S. Dai, P. T. Cummings, S. V. Kalinin, R. Proksch and N. Balke, Nano Lett., 2013, 13, 5954–5960 CrossRef CAS PubMed.
- N. Hjalmarsson, R. Atkin and M. W. Rutland, Phys. Chem. Chem. Phys., 2016, 18, 9232–9239 RSC.
- M. Dong, K. Zhang, X. Wan, S. Wang, S. Fan, Z. Ye, Y. Wang, Y. Yan and X. Peng, Small, 2022, 18, 2108026 CrossRef CAS PubMed.
- M. Aminpour, C. Montemagno and J. A. Tuszynski, Molecules, 2019, 24, 1693 CrossRef CAS PubMed.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, 2017 Search PubMed.
- D. Bedrov, J.-P. Piquemal, O. Borodin, A. D. MacKerell, Jr., B. Roux and C. Schröder, Chem. Rev., 2019, 119, 7940–7995 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding Molecular Simulation: From Algorithms to Applications, Academic Press, 2002 Search PubMed.
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC.
- S. Baldelli, Acc. Chem. Res., 2008, 41, 421–431 CrossRef CAS PubMed.
- O. Y. Fajardo, F. Bresme, A. A. Kornyshev and M. Urbakh, Sci. Rep., 2015, 5, 7698 CrossRef CAS PubMed.
- Y. Wang, C. Wang, Y. Zhang, F. Huo, H. He and S. Zhang, Small, 2019, 15, 1804508 CrossRef PubMed.
- G. Saielli, J. Phys. Chem. B, 2016, 120, 2569–2577 CrossRef CAS PubMed.
- C. Qian, Y. Wang, H. He, F. Huo, N. Wei and S. Zhang, J. Phys. Chem. C, 2018, 122, 22194–22200 CrossRef CAS.
- G. Jin, X. Song, Q. Gao, Y. Zhang, Y. Chen, X. Lu and Y. Zhu, Ind. Eng. Chem. Res., 2022, 61, 3754–3765 CrossRef CAS.
- R. An, G. Zhou, Y. Zhu, W. Zhu, L. Huang and F. U. Shah, Adv. Mater. Interfaces, 2018, 5, 1800263 CrossRef.
- X. Zhong, Z. Fan, Z. Liu and D. Cao, J. Phys. Chem. B, 2012, 116, 3249–3263 CrossRef CAS PubMed.
- Y. Wang, J. Liu, X. Yuan, M. Wang, Y. Nie, S. Zhang and H. He, Chem. Eng. J., 2023, 455, 140370 CrossRef CAS.
- Y. Lu, Y. Wang, F. Huo, W. Chen, M. Ma, W.-L. Ding, H. He and S. Zhang, ACS Nano, 2022, 16, 16471–16480 CrossRef CAS PubMed.
- Y. Wei, Z. Dai, Y. Dong, A. Filippov, X. Ji, A. Laaksonen, F. U. Shah, R. An and H. Fuchs, Phys. Chem. Chem. Phys., 2022, 24, 12808–12815 RSC.
- Y. Wei, Y. Dong, X. Ji, F. Ullah Shah, A. Laaksonen, R. An and K. Riehemann, J. Mol. Liq., 2022, 350, 118506 CrossRef CAS.
- K.-i Amano, Y. Liang, K. Miyazawa, K. Kobayashi, K. Hashimoto, K. Fukami, N. Nishi, T. Sakka, H. Onishi and T. Fukuma, Phys. Chem. Chem. Phys., 2016, 18, 15534–15544 RSC.
- Y. Wang, C. Qian, F. Huo, B. Xu, H. He and S. Zhang, AIChE J., 2021, 67, e17060 CrossRef CAS.
- M. Wang, Y. Wang, T. Zhang, Y. Xia and H. He, AIChE J., 2023, 69, e18222 CrossRef CAS.
- D.-e Jiang, D. Meng and J. Wu, Chem. Phys. Lett., 2011, 504, 153–158 CrossRef CAS.
- J. Yang, C. Lian and H. Liu, Chem. Eng. Sci., 2020, 227, 115927 CrossRef CAS.
- P. Rehner, G. Bauer and J. Gross, Ind. Eng. Chem. Res., 2023, 62, 5347–5357 CrossRef CAS.
- Y. Sun, X. Lu, G. Shen and X. Ji, Ind. Eng. Chem. Res., 2021, 60, 9269–9285 CrossRef CAS.
- G. Shen, X. Lu and X. Ji, Fluid Phase Equilib., 2014, 382, 116–126 CrossRef CAS.
- M. Zhou, J. Wang, J. Garcia, Y. Liu and J. Wu, Ind. Eng. Chem. Res., 2021, 60, 17016–17025 CrossRef CAS.
- X. Wu, Z. Cao, X. Lu and W. Cai, Chem. Eng. J., 2023, 459, 141612 CrossRef CAS.
- G. Shen, H. Yang, Y. Hu, X. Zhang, F. Zhou, H. Li and K. Hong, Nanomaterials, 2022, 13, 1–13 CrossRef.
- J. Cheng, J. Xu, J. Yang, W. Lv, C. Lian and H. Liu, Chem. Eng. Sci., 2022, 261, 117957 CrossRef CAS.
- J. Wu, T. Jiang, D.-E. Jiang, Z. Jin and D. Henderson, Soft Matter, 2011, 7, 11222–11231 RSC.
- G. Shen, Y. Sun, Y. Wang, X. Lu and X. Ji, J. Mol. Liq., 2020, 310, 113199 CrossRef CAS.
- M. Hammer, G. Bauer, R. Stierle, J. Gross and Ø. Wilhelmsen, J. Chem. Phys., 2023, 158, 104107 CrossRef CAS PubMed.
- Y. Sun, Z. Dai, G. Shen, X. Lu, X. Ling and X. Ji, Front. Chem., 2022, 9, 801551 CrossRef PubMed.
- G. Shen, D. Zhang, Y. Hu, X. Zhang, F. Zhou, Y. Qian, X. Lu and X. Ji, J. Chem. Phys., 2022, 157, 014701 CrossRef CAS PubMed.
- C. Lian, D.-E. Jiang, H. Liu and J. Wu, J. Phys. Chem. C, 2016, 120, 8704–8710 CrossRef CAS.
- A. L. Frischknecht, D. O. Halligan and M. L. Parks, J. Chem. Phys., 2014, 141, 054708 CrossRef.
- A. Elbourne, J. Sweeney, G. B. Webber, E. J. Wanless, G. G. Warr, M. W. Rutland and R. Atkin, Chem. Commun., 2013, 49, 6797–6799 RSC.
- H. Li, Y. Zhang, S. Jones, R. Segalman, G. G. Warr and R. Atkin, J. Colloid Interface Sci., 2022, 606, 1170–1178 CrossRef CAS PubMed.
- R. An, Y. Wei, X. Qiu, Z. Dai, M. Wu, E. Gnecco, F. U. Shah and W. Zhang, Friction, 2022, 10, 1893–1912 CrossRef CAS.
- J. Sweeney, F. Hausen, R. Hayes, G. B. Webber, F. Endres, M. W. Rutland, R. Bennewitz and R. Atkin, Phys. Rev. Lett., 2012, 109, 155502 CrossRef PubMed.
- H. Li, R. J. Wood, M. W. Rutland and R. Atkin, Chem. Commun., 2014, 50, 4368–4370 RSC.
- X. Qiu, L. Lu, Z. Qu, J. Liao, Q. Fan, F. U. Shah, W. Zhang and R. An, Friction, 2022, 10, 268–281 CrossRef.
- H. Li, T. Niemann, R. Ludwig and R. Atkin, J. Phys. Chem. Lett., 2020, 11, 3905–3910 CrossRef CAS PubMed.
- C. S. Perez-Martinez and S. Perkin, Langmuir, 2019, 35, 15444–15450 CrossRef CAS PubMed.
- O. Werzer, E. D. Cranston, G. G. Warr, R. Atkin and M. W. Rutland, Phys. Chem. Chem. Phys., 2012, 14, 5147–5152 RSC.
- J. Hoth, F. Hausen, M. H. Müser and R. Bennewitz, J. Phys.: Condens. Matter, 2014, 26, 284110 CrossRef PubMed.
- S. Di Lecce, A. A. Kornyshev, M. Urbakh and F. Bresme, Phys. Chem. Chem. Phys., 2021, 23, 22174–22183 RSC.
- S. Di Lecce, A. A. Kornyshev, M. Urbakh and F. Bresme, ACS Appl. Mater. Interfaces, 2020, 12, 4105–4113 CrossRef CAS PubMed.
- K. Pivnic, O. Y. Fajardo, F. Bresme, A. A. Kornyshev and M. Urbakh, J. Phys. Chem. C, 2018, 122, 5004–5012 CrossRef CAS.
- R. Atkin, N. Borisenko, M. Drüschler, S. Z. El Abedin, F. Endres, R. Hayes, B. Huber and B. Roling, Phys. Chem. Chem. Phys., 2011, 13, 6849–6857 RSC.
- T. Waldmann, H.-H. Huang, H. E. Hoster, O. Höfft, F. Endres and R. J. Behm, ChemPhysChem, 2011, 12, 2565–2567 CrossRef CAS PubMed.
- V. Hoffmann, G. Pulletikurthi, T. Carstens, A. Lahiri, A. Borodin, M. Schammer, B. Horstmann, A. Latz and F. Endres, Phys. Chem. Chem. Phys., 2018, 20, 4760–4771 RSC.
- G. Krämer, F. Hausen and R. Bennewitz, Faraday Discuss., 2017, 199, 299–309 RSC.
- F. Federici Canova, M. Mizukami, T. Imamura, K. Kurihara and A. L. Shluger, Phys. Chem. Chem. Phys., 2015, 17, 17661–17669 RSC.
- A. Moradzadeh, M. H. Motevaselian, S. Y. Mashayak and N. R. Aluru, J. Chem. Theory Comput., 2018, 14, 3252–3261 CrossRef CAS PubMed.
- F. Uhlig, J. Zeman, J. Smiatek and C. Holm, J. Chem. Theory Comput., 2018, 14, 1471–1486 CrossRef CAS PubMed.
- S. Zimmermann, W. Klauser, J. Mead, S. Wang, H. Huang and S. Fatikow, Nano Res., 2019, 12, 389–396 CrossRef CAS.
- J. Erath, S. Schmidt and A. Fery, Soft Matter, 2010, 6, 1432–1437 RSC.
- R. Lhermerout and S. Perkin, Phys. Rev. Fluid, 2018, 3, 014201 CrossRef.
- M. A. Gebbie, M. Valtiner, X. Banquy, E. T. Fox, W. A. Henderson and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9674–9679 CrossRef CAS PubMed.
- M. A. Gebbie, A. M. Smith, H. A. Dobbs, A. A. Lee, G. G. Warr, X. Banquy, M. Valtiner, M. W. Rutland, J. N. Israelachvili, S. Perkin and R. Atkin, Chem. Commun., 2017, 53, 1214–1224 RSC.
- Y. F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber and D. J. Müller, Nat. Nanotechnol., 2017, 12, 295–307 CrossRef.
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Chem. Commun., 2011, 47, 6572–6574 RSC.
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, P. Licence, A. Dolan, T. Welton and S. Perkin, J. Phys. Chem. Lett., 2013, 4, 378–382 CrossRef CAS.
- A. M. Smith, K. R. J. Lovelock and S. Perkin, Faraday Discuss., 2013, 167, 279–292 RSC.
- B. Yoo, J. K. Shah, Y. Zhu and E. J. Maginn, Soft Matter, 2014, 10, 8641–8651 RSC.
- A. A. d Freitas, K. Shimizu, A. M. Smith, S. Perkin and J. N. C. Lopes, J. Chem. Phys., 2018, 148, 193808 CrossRef PubMed.
- A. M. Smith, M. A. Parkes and S. Perkin, J. Phys. Chem. Lett., 2014, 5, 4032–4037 CrossRef CAS PubMed.
- Y. Shen, X. X. He and F. R. Hung, J. Phys. Chem. C, 2015, 119, 24489–24500 CrossRef CAS.
- M. Rouha and P. T. Cummings, Phys. Chem. Chem. Phys., 2015, 17, 4152–4159 RSC.
- O. Y. Fajardo, F. Bresme, A. A. Kornyshev and M. Urbakh, J. Phys. Chem. Lett., 2015, 6, 3998–4004 CrossRef CAS PubMed.
- J. Jiang, D. P. Cao, D. E. Jiang and J. Z. Wu, J. Phys. Chem. Lett., 2014, 5, 2195–2200 CrossRef CAS PubMed.
- Y. L. Wang, A. Laaksonen and Z. Y. Lu, Phys. Chem. Chem. Phys., 2013, 15, 13559–13569 RSC.
- X. J. Zhong, Z. Fan, Z. P. Liu and D. P. Cao, J. Phys. Chem. B, 2012, 116, 3249–3263 CrossRef CAS PubMed.
- S. Begić, E. Jónsson, F. Chen and M. Forsyth, Phys. Chem. Chem. Phys., 2017, 19, 30010–30020 RSC.
- C. Wang, C. Qian, Z. Li, N. Wei, N. Zhang, Y. Wang and H. He, Ind. Eng. Chem. Res., 2020, 59, 8028–8036 CrossRef CAS.
- Y. Zhang and P. T. Cummings, ACS Appl. Mater. Interfaces, 2019, 11, 42680–42689 CrossRef CAS PubMed.
- N. Vucemilovic-Alagic, R. D. Banhatti, R. Stepic, C. R. Wick, D. Berger, M. U. Gaimann, A. Baer, J. Harting, D. M. Smith and A. S. Smith, J. Colloid Interface Sci., 2019, 553, 350–363 CrossRef CAS PubMed.
- C. Qian, B. Ding, Z. W. Wu, W. L. Ding, F. Huo, H. Y. He, N. Wei, Y. L. Wang and X. P. Zhang, Ind. Eng. Chem. Res., 2019, 58, 20109–20115 CrossRef CAS.
- Y. L. Wang, F. Huo, H. Y. He and S. J. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 17773–17780 RSC.
- A. Ruzanov, M. Lembinen, P. Jakovits, S. N. Srirama, I. V. Voroshylova, M. Cordeiro, C. M. Pereira, J. Rossmeisl and V. B. Ivanistsev, Phys. Chem. Chem. Phys., 2018, 20, 10275–10285 RSC.
- S. J. Zhang, Y. L. Wang, H. Y. He, F. Huo, Y. M. Lu, X. C. Zhang and K. Dong, Green Energy Environ., 2017, 2, 329–330 CrossRef.
- Y.-L. Wang, M. Golets, B. Li, S. Sarman and A. Laaksonen, ACS Appl. Mater. Interfaces, 2017, 9, 4976–4987 CrossRef CAS PubMed.
- J. N. Neal, D. J. Wesolowski, D. Henderson and J. Wu, J. Chem. Phys., 2017, 146, 174701 CrossRef PubMed.
- S. Maolin, Z. Fuchun, W. Guozhong, F. Haiping, W. Chunlei, C. Shimou, Z. Yi and H. Jun, J. Chem. Phys., 2008, 128, 134504 CrossRef PubMed.
- S. A. Kislenko, I. S. Samoylov and R. H. Amirov, Phys. Chem. Chem. Phys., 2009, 11, 5584–5590 RSC.
- Z. Dai, Y. You, Y. Zhu, S. Wang, W. Zhu and X. Lu, J. Phys. Chem. B, 2019, 123, 6857–6869 CrossRef CAS PubMed.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106, 046102 CrossRef PubMed.
- M. V. Fedorov and A. A. Kornyshev, Electrochim. Acta, 2008, 53, 6835–6840 CrossRef CAS.
- M. Chu, M. Miller and P. Dutta, ACS Cent. Sci., 2016, 2, 175–180 CrossRef CAS PubMed.
- S. Kondrat, G. Feng, F. Bresme, M. Urbakh and A. A. Kornyshev, Chem. Rev., 2023, 123, 6668–6715 CrossRef CAS PubMed.
- M. Vaganova, I. Nesterova, Y. Kanygin, A. Kazennov and A. Khlyupin, Chem. Eng. Sci., 2022, 250, 117383 CrossRef CAS.
- A. Malpica-Morales, P. Yatsyshin, M. A. Durán-Olivencia and S. Kalliadasis, J. Chem. Phys., 2023, 159, 104109 CrossRef CAS PubMed.
- G. Shen, A. Laaksonen, X. Lu and X. Ji, J. Phys. Chem. C, 2018, 122, 15464–15473 CrossRef CAS.
- L. B. Bhuiyan, S. Lamperski, J. Wu and D. Henderson, J. Phys. Chem. B, 2012, 116, 10364–10370 CrossRef CAS PubMed.
- H. Ers, M. Lembinen, M. Mišin, A. P. Seitsonen, M. V. Fedorov and V. B. Ivaništšev, J. Phys. Chem. C, 2020, 124, 19548–19555 CrossRef CAS.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- B. Wang, C. Moran, D. Lin, H. Tang, E. Gage and L. Li, ACS Appl. Nano Mater., 2019, 2, 5260–5265 CrossRef CAS.
- S. Di Lecce, A. A. Kornyshev, M. Urbakh and F. Bresme, ACS Nano, 2020, 14, 13256–13267 CrossRef CAS PubMed.
- F. Larsson, P. Andersson, P. Blomqvist and B.-E. Mellander, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- S. Che, R. Dao, W. Zhang, X. Lv, H. Li and C. Wang, Chem. Commun., 2017, 53, 3862–3865 RSC.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- G. A. Giffin, J. Mater. Chem. A, 2016, 4, 13378–13389 RSC.
- M. Antonietti, X. Chen, R. Yan and M. Oschatz, Energy Environ. Sci., 2018, 11, 3069–3074 RSC.
- M. V. Fedorov and A. A. Kornyshev, J. Phys. Chem. B, 2008, 112, 11868–11872 CrossRef CAS PubMed.
- Y. Su, T. Wang, F. Zhang, J. Huang, Z. Zhu, F. U. Shah, F. Xu and R. An, Langmuir, 2023, 39, 8463–8474 CrossRef CAS PubMed.
- J. Vatamanu, Z. Hu, D. Bedrov, C. Perez and Y. Gogotsi, J. Phys. Chem. Lett., 2013, 4, 2829–2837 CrossRef CAS.
- N. García Rey and D. D. Dlott, J. Phys. Chem. C, 2015, 119, 20892–20899 CrossRef.
- Y.-Z. Su, Y.-C. Fu, J.-W. Yan, Z.-B. Chen and B.-W. Mao, Angew. Chem., Int. Ed., 2009, 48, 5148–5151 CrossRef CAS PubMed.
- M. Urushihara, K. Chan, C. Shi and J. K. Nørskov, J. Phys. Chem. C, 2015, 119, 20023–20029 CrossRef CAS.
- Y.-J. Tu and J. G. McDaniel, J. Phys. Chem. C, 2021, 125, 20204–20218 CrossRef CAS.
- F. W. Richey, B. Dyatkin, Y. Gogotsi and Y. A. Elabd, J. Am. Chem. Soc., 2013, 135, 12818–12826 CrossRef CAS PubMed.
- I. A. Khan, Y.-L. Wang and F. U. Shah, J. Energy Chem., 2022, 69, 174–184 CrossRef CAS.
Footnote |
| † Rong An and Nanhua Wu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |