
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical upcycling and recycling of plastics: achievements and future opportunities
Olga G.
Mountanea†
,
Elpida
Skolia†
and
Christoforos G.
Kokotos
 *
*
Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis 15771, Athens, Greece. E-mail: ckokotos@chem.uoa.gr
First published on 2nd July 2024
Abstract
The pervasive effects of plastic waste pollution affect both humanity and the environment, thus innovative and environmentally-friendly methods for recycling of plastics are crucially needed. The application of light to degrade or transform plastics into valuable products has gained significant attention. Numerous researchers have explored irradiation to achieve the photocatalytic breakdown of highly resilient plastic waste components into valuable monomers, which can be utilized for the synthesis of novel materials of synthetic or pharmacological interest. Many of these techniques have resulted in H2 evolution, while efforts were also made to reduce carbon emissions. In some cases, light was combined with additional energy sources, leading to development of photothermal or photoelectrochemical processes. With this tutorial review, our aim is to offer an overview of these novel photochemical upcycling protocols for the degradation of polymers, aspiring toward the introduction of novel processes in the near future.
Introduction
The indispensability of plastics in modern society spans in multiple aspects.1 Their remarkable versatility is overshadowed by their resistance to degradation, coupled with reckless disposal practices, leading to one of the world's most discussed challenges: plastic waste accumulation. The ever-increasing production and mismanagement of plastic waste has led to a global crisis, with massive volumes of accumulating plastics.2,3 Currently, 9% of plastic waste is recycled, while the majority is discarded in landfills or the environment. If present trends persist, by 2050, the world will struggle with 12 billion metric tons of plastic waste.2 The pervasive presence of plastics has markedly heightened environmental pollution and climate change, resulting in significant socio-economic challenges. Reducing plastic emissions and employing effective waste management are crucial to combat plastic pollution (Fig. 1A).3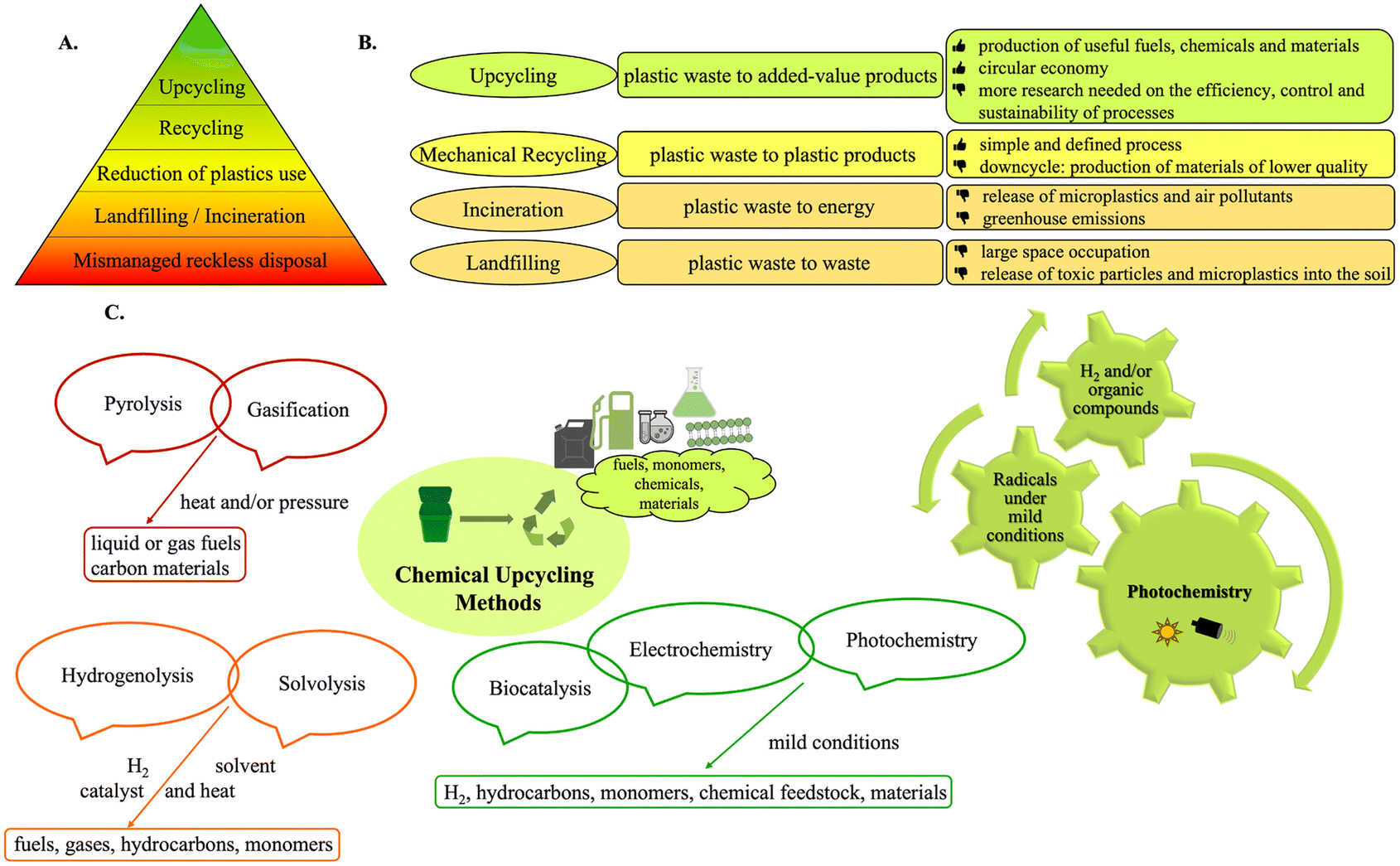 | ||
| Fig. 1 A. Pyramidal diagram of a zero-waste hierarchy for plastics management; B. common methods for managing plastics; C. widely used upcycling methods. | ||
The primary methods for managing plastics present challenges (Fig. 1B).4 Landfills require vast space and perpetuate pollution. Incineration leads to generation of microplastics, as well as increased carbon and toxic pollutant emissions.4,5 Mechanical recycling often results in a downcycling effect, diminishing quality and imposing limits on the number of cycles and the presence of contaminants.6 In contrast, chemical upcycling aims at converting plastics into high-added-value compounds (Fig. 1C). Representing a “plastic-based refinery” strategy, chemical upcycling holds promise in addressing environmental pollution, reducing carbon emissions and promoting circular economy.5 Pyrolysis and gasification have also been developed. More recent methods, like hydrogenolysis, solvolysis or biocatalysis, cleave plastics into smaller molecules, while electrocatalysis and photocatalysis leverage electricity or light to promote chemical reactions.1,4–8
The use of photocatalysis is promising, since visible light or sunlight is an ideal energy source to facilitate chemical transformations, previously considered challenging or unattainable.9,10 Photocatalysis is environmentally-friendly and cost-effective, enabling highly efficient decomposition of plastics without high energy input demand or harsh reaction conditions.4
The processes involved in initiating reactions through light can be categorised into three main types: (a) reactions initiated by electron transfer, also known as photoredox reactions, (b) reactions initiated by hydrogen atom transfer (HAT), and (c) reactions initiated by energy transfer (ET).9–11 Hydrogen atom transfer (HAT) stands as a fundamental and widely observed phenomenon in both chemical reactions and biological systems, presenting an effective strategy for cleaving C(sp3)–H bonds. This process entails the coordinated movement of both an electron and a proton (H˙ ≡ H+ + e−) from the substrate, referred to as the hydrogen donor, to an acceptor referred to as the abstractor.9 References in the literature pertaining to the HAT process can be divided into two distinct approaches: indirect HAT (i-HAT) and direct HAT (d-HAT). In the former, the photocatalyst (PCSET) absorbs light and, upon excitation, generates the excited chemical species (X˙, a radical or radical ion) responsible for hydrogen abstraction via a single electron transfer (SET) step. Conversely, in the d-HAT process, the photocatalyst (PCHAT) directly triggers HAT upon becoming excited. In essence, the PC*HAT acts as the X˙.9
When exposed to irradiation, macromolecules’ degradation is triggered, causing chain scission, branching, cross-linking or oxidation reactions.4 This approach allows for the selective manipulation of traditionally inert aliphatic C–H bonds, but also facilitates acceleration of the stable, non-polar C–C bond activation, essential for plastics depolymerisation, which typically spans hundreds of years under natural conditions.4,8,9 The conversion of plastic waste into high-value fuels, organic molecules or functionalized materials via light-driven plastic upcycling represents an effective approach that significantly mitigates the environmental impact of plastic production, contributing to a sustainable circular economy.1,4–8,11,12
In the past few years, especially during 2023–2024,4,11–13 there has been a surge in literature discussing novel approaches utilising light for the photochemical upcycling of plastics, accompanied by numerous review articles detailing the reactions and conditions of these methods. This review delves into the utilisation of light to transform polymers into valuable small molecules, rather than simply breaking them down. It offers a comprehensive examination of the most commonly employed polymers, with a particular focus on polystyrene, presenting a more in-depth analysis of this widely-used polymer, including new photochemical protocols—some of which are being reported for the first time in a review. Additionally, this review elucidates degradation mechanisms and evaluates the effectiveness of these methodologies. To facilitate a thorough understanding of the unique characteristics of each depolymerisation process, we conduct a comparative analysis of various methodologies specifically applied to polystyrene, leveraging the wealth of research available in this domain. The objective of this tutorial review is to motivate and assist researchers keen on investigating light-driven upcycling approaches for diverse polymer plastics.
Polystyrene
The photochemical upcycling of polystyrene (PS) presents several advantages, particularly due to its potential to yield PhCO2H, a versatile compound with numerous industrial applications, making it an attractive target for chemical upcycling. Since 1984,14 research on photo-induced degradation of PS has markedly increased, since it is an efficient technique for converting plastics into higher-value compounds (Scheme 1A–D). Hu's introduction of a pioneering metal-based photochemical protocol in 2021, employing FeCl2 in solution under 400 nm LED irradiation and oxygen to yield PhCO2H, underscored innovative approaches toward sustainable solutions for the plastic pollution crisis.15 In the same vein, Zeng used FeCl3 for the photochemical oxidation of alkyl aromatics to carboxylic acids under blue LED irradiation and O2.16 The incorporation of a photocatalytic system (solid FeCl3, tetrabutylammonium chloride and 2,2,2-trichloroethanol) demonstrated the versatility of the protocol, showcasing its application in the oxidative degradation of polystyrene.16 Similarly, Oh and Stache exploited the use of Fe(III), leading to the formation of chlorine radicals.17 This innovative approach using 10 wt% FeCl3 dissolved in acetone proved effective as they successfully converted polystyrene into a 23 mol% benzoyl product mixture. Their protocol converted commercial PS products, like packaging materials. Additionally, a gram-scale photoflow-based process was developed (Scheme 1A).17 Later, they delved into polystyrene's photochemical degradation via halogen radicals.18 They investigated the different outcome regarding the use of Cl˙ or Br˙ to the production of PhCO2H or acetophenone, respectively, revealing that Cl˙ yielded exclusively PhCO2H, while Br˙ led to the formation of acetophenone and PhCO2H in nearly equal amounts. These observations are due to 2° C–H bond activation primarily yielding PhCO2H, while 3° C–H bond cleavage results in either PhCO2H or acetophenone. Br˙ mainly triggers 3° C–H bonds, due to a hydrogen atom transfer (HAT) on HBr [as a result of its low bond dissociation energy (BDE)], attributing an H atom to a methyl radical on the chain end, leading to acetophenone, whilst addition of LiBr led to acetophenone over PhCO2H.18 Chlorine and bromine radicals exhibit high reactivity, facilitating the efficient decomposition of PS into smaller molecular fragments. Cl-based compounds, being easily accessible and cost-effective, can successfully serve as radical initiators in photochemical upcycling processes. Even though Br˙ shows superior selectivity in contrast to Cl˙, which arises from its lower radical reactivity, bromine-based compounds tend to be pricier and less accessible than their chlorine counterparts, potentially constraining their feasibility in large-scale applications. While both chlorine and bromine radicals demonstrate promising outcome in the photochemical PS degradation, they also elicit concerns regarding environmental repercussions and product selectivity.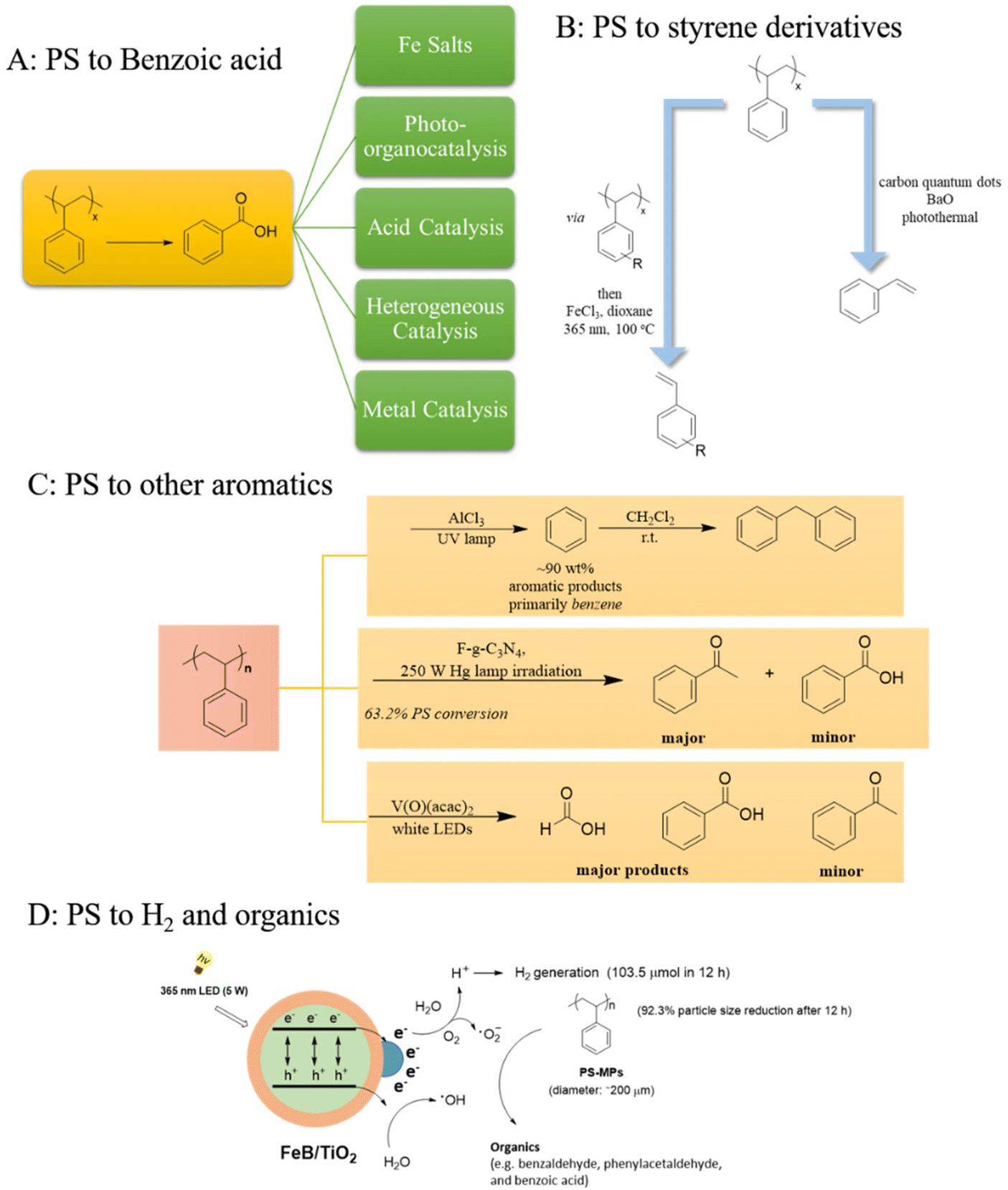 | ||
| Scheme 1 A. Photochemical PS upcycling to benzoic acid; B. photochemical PS upcycling to styrene derivatives; C. photochemical conversion of polystyrene to aromatic compounds; D. photochemical protocol towards the transformation of PSMPs to H2. | ||
Even though iron is considered among the most sustainable metals, the cost of its removal from fine chemicals requires alternative approaches.
In all these examples, chlorine or bromine salts function as photocatalysts, consistently facilitating the reactions and subsequent product formation (Scheme 2). The use of irradiation is essential for generating reactive radicals. Following a catalytic cycle, the catalyst is also regenerated, ensuring continuous activity. In the given mechanisms, FeCl3 plays a crucial role in facilitating a photoinduced ligand-to-metal charge transfer (LMCT) process, as well as participating in single electron transfer (SET) reactions. More specifically, when FeCl3 is exposed to light, it undergoes a photoinduced LMCT, a process which involves the transfer of an electron from the chloride ion (Cl−) to the iron centre (Fe3+). This electron transfer results in the generation of chlorine radicals (Cl˙) and the reduction of Fe3+ to Fe2+. The chlorine radical (Cl˙) is a highly reactive species that can initiate succeeding hydrogen atom transfer (HAT) processes. Subsequently, the generated chlorine radical (Cl˙) can abstract a hydrogen atom from polystyrene, leading to the formation of organic radicals. These organic radicals can undergo further transformations to yield the desired products. At the same time, regeneration of Fe(III) occurs, enabling the formation of superoxide anion radicals O2˙−. After the initial generation of Cl˙, the Fe2+ species needs to be regenerated to Fe3+ to sustain the catalytic cycle. This regeneration occurs via a single electron transfer (SET) process between molecular oxygen (O2) and the generated Fe2+, meaning that Fe2+ donates an electron to O2, forming Fe3+ and superoxide anion radical (O2˙−), which is also highly reactive and can participate in deep oxidation processes, leading the intermediate organic radicals or other species in the reaction mixture to further oxidation and the formation of final products (Scheme 2). Thus, in most cases, the metal salt is mostly used as a source of halogen radical, in particular chlorine radical, which are known for their abilities as HAT catalysts, but also facilitates O2˙− generation through oxidation of the metal. This is primarily due to the fact that when chlorine radicals abstract hydrogen atoms from hydrocarbons, the resulting HCl formation is more stable, since most C–H bond dissociation energies are generally lower than the H–Cl bond formation energy. Consequently, chlorine radicals can easily break the weaker C–H bonds in hydrocarbons and form new bonds with hydrogen atoms. The higher bond dissociation energy (BDE) of H–Cl (BDE, H–Cl = 103 kcal mol−1), compared to most C–H bonds, make the hydrogen abstraction process thermodynamically favorable.17 In the meantime, the generation of singlet oxygen from the reduced metal is a side process, which also facilitates the generation of reactive species enabling the promotion of the degradation mechanism. Even though the above mentioned observations can be applied to most of the photochemical protocols, in Hu's proposed mechanism,15 while the possibility of an i-HAT mechanism via a Cl radical was not dismissed, the process primarily involves Fe(II) being oxidised to Fe(III), which acts as the active photocatalyst under an oxygen atmosphere. Upon excitation, Fe(III)* facilitates the desired initial oxidative C–H bond cleavage through a SET process.15
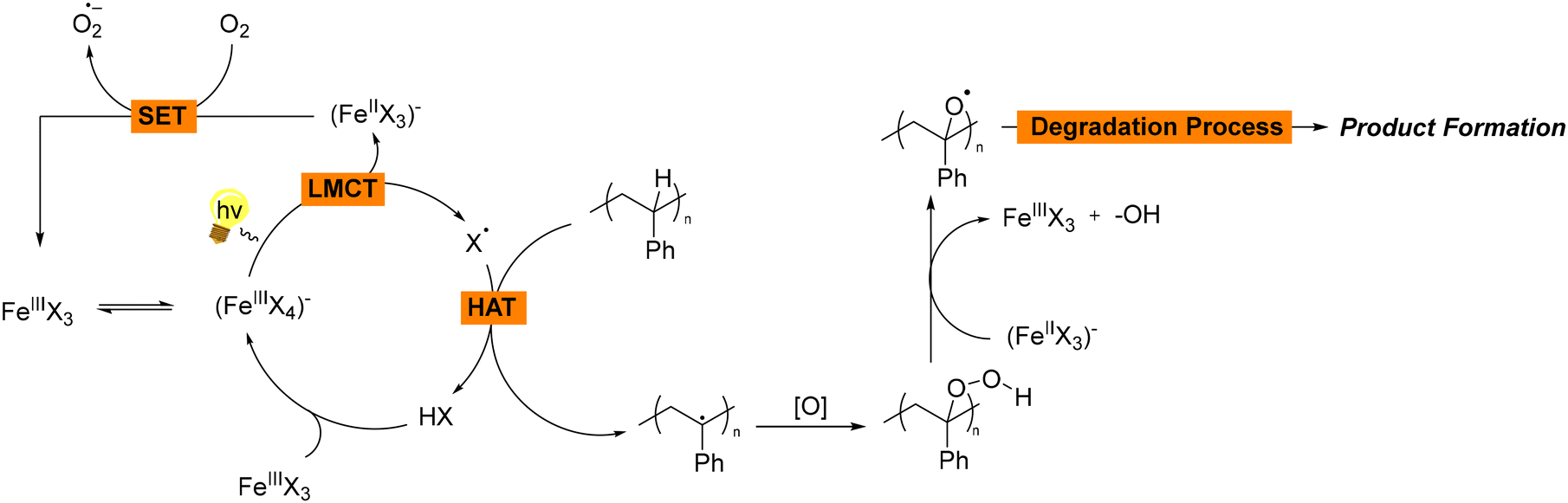 | ||
| Scheme 2 General reaction mechanism for the photochemical upcycling of polystyrene using Fe salts as the photocatalysts. | ||
In 2022, Reisner studied fluorenone for the light-induced degradation of polystyrene to PhCO2H. Utilising blue LED irradiation at 50 °C led to the formation of ∼40% PhCO2H and ∼20% other aromatic products, although the use of diluted H2SO4 was a significant drawback.19 In the same spirit, Huang et al. reported the light-driven, acid-catalysed methodology for the photochemical upcycling of polystyrene to HCO2H, PhCO2H and acetophenone, using pTsOH·H2O under 405 nm irradiation and O2.20 Although strong acids usually serve as effective oxidizing agents in industrial processes, their corrosive nature can pose both environmental and safety challenges requiring careful handling.
Additionally, Das exploited HAT chemistry generated from N-bromosuccinimide and CF3SO2Na for the upcycling of polystyrene under 390 nm irradiation.21 The protocol was applied to real-life plastics, leading to PhCO2H in high yields, while a tandem reforming of PhCO2H into different aromatics, not only widened the study's scope, but also accentuated the practicality and adaptability embedded in the devised approach.21 In 2022, Liu introduced a photochemical degradation of polystyrene to value-added aromatics (Scheme 1C).22 The authors proposed the photodegradation of PS waste under UV irradiation with subsequent transformation to diphenylmethane. Moreover, an industrial-scale techno-economic analysis took place, suggesting high economic viability for the elimination of end-of-life plastics.22 These photochemical processes are highly desirable, due to their ability to convert polymer plastics into valuable monomers, without the use of expensive or hazardous catalysts, while also requiring mild reaction conditions. Most significantly, they effectively upcycled polystyrene into PhCO2H and benzene. Thus, polystyrene can be efficiently transformed into PhCO2H or other valuable monomers, achieving optimal yields under appropriate reaction conditions, not only improving the economic feasibility of the upcycling process, but also maximising valuable compound production.
Metal-free catalysis emerged as a promising substitute for traditional metal-based catalytic systems, providing opportunities for selectivity, and versatility across a spectrum of reaction conditions and substrates. Additionally, it facilitates successful reactions without relying on typically toxic metals, while the relatively lower cost of organocatalysts, compared to the high expenses associated with metal derivatives, further support the efficacy of this method. In this spirit, Kokotos introduced another attractive metal-free aerobic upcycling process of polystyrene to PhCO2H, using anthraquinone under 390 nm irradiation and air as the sole oxidant.23 A noteworthy application was the synthesis of two pharmaceuticals, salicylic acid and acetylsalicylic acid, from the upcycled PhCO2H. This underscores the prospect of implementing sustainable approaches in the pharmaceutical industry, while expanding the application of green chemistry to the production of valuable compounds.23
An alternative method for the PS upcycling involves employing heterogeneous catalysis.24,25 In contrast to homogeneous catalysts that are mainly soluble in the reaction mixture, heterogeneous catalysts remain separate from the reaction products, facilitating simpler separation and facile catalyst recovery. Furthermore, heterogeneous catalysis promotes cleaner and more sustainable processes by enabling the utilisation of mild reaction conditions, thereby reducing energy consumption and minimising by-products’ generation. Titanium dioxide, being insoluble in both aqueous and organic solvents, exhibits enhanced stability which makes it suitable for a multitude of applications. Additionally, it displays photocatalytic characteristics, allowing it to expedite chemical reactions upon exposure to light. In 2023, Yang introduced a FeB/TiO2 photocatalyst using 365 nm LED, where oxygenated aromatics, including benzaldehyde, phenylacetaldehyde and PhCO2H were detected from microplastics (Scheme 1D).26 Additionally, Li introduced a potassium stearate—N,N-diethyl-3-(trimethoxysilyl)-propan-1-amine system for the enhancement of the catalytic activity of TiO2 under 370 nm irradiation and 1 bar O2.27
Also in 2023, Chen proposed a two-step photocatalytic method for the degradation of styrofoam. Initially, styrofoam waste was functionalised by addition of acyl or bromine groups on the polymeric chain (Scheme 1B).28 Subsequent depolymerisation of reformed PS, using solid FeCl3 under 365 nm UV irradiation and heating at 100 °C enabled the formation of acylated and brominated styrene at >50% yield.28 Although the method effectively assesses the functionalisation of polymers into acylated and brominated polystyrene, its sustainability is questioned primarily in the case of the generation of halogenated compounds, which raise significant environmental concerns. Specifically, the production of brominated polymers poses risks to the environment, due to the potential release of bromine-containing compounds. Moreover, the presence of brominated polymers complicates recycling processes, diminishing the overall recyclability of the materials. Furthermore, although the method distinguishes itself from other processes typically involving the degradation of polystyrene, it lacks specificity regarding the yields of the newly formed compounds. While functionalisation and subsequent depolymerisation are primarily observed via FT-IR and 1H-NMR, no additional information regarding the precise yield of the newly formed monomers is provided, thereby complicating the estimation of the process efficacy.
The field of quantum dots has gained tremendous interest, highlighted by the Nobel prize in Chemistry in 2023. In the same year, Stache proposed a novel photothermal protocol, based on the activation of carbon quantum dots, which upon photoexcitation, produce thermal gradients, capable of depolymerisation of commercial plastics.29 This elegant method constitutes the first carbon dots-based photothermal process for the chemical recycling of polymers. Photothermal processes involve the use of light absorbers, which convert light energy directly into heat, thereby driving chemical reactions through localized heating. This method contrasts with the combined use of heat and irradiation, where an external heat source is employed alongside light irradiation. In photothermal processes, materials such as nanoparticles, absorb light and convert it into thermal energy via non-radiative relaxation, subsequently inducing thermal reactions like polymer degradation. Conversely, the combination of heat and irradiation utilises light to excite photocatalysts, generating reactive species (e.g., electron–hole pairs or radicals), while heat is provided by an external source. The primary distinction lies in the heat source: photothermal processes rely solely on light-induced heat generation, whereas combined approaches rely on an external heat source, supplementing the photochemical effects of light. It's imperative to note that these terms should not be used interchangeably, as they entail distinct mechanisms and operational principles. For the purpose of our review, to underscore the differentiation between these methodologies, we employ the term “synergistic thermal-photochemical approach” when referring to the integrated utilisation of heat and irradiation to maintain consistency and clarity in terminology and reserve the term “photothermal” for processes exclusively reliant on light-induced heat generation. The photothermal protocol was well-applied to several polymers, including PLA and polystyrene, as well as PMMA [poly(methylmethacrylate)], leading to L-lactide, styrene and methyl methacrylate monomers, respectively under white LED irradiation in good yields and in short time,29 while Li proposed a metal-free photochemical benzylic C(sp3)–H oxidation, using 6-bromobenzo[d]thiazole as the photosensitizer, under 390–400 nm LED irradiation and air for the aerobic oxidation of methylarenes and benzyl derivatives. Slight modifications to the protocol enabled photo-oxidation of PS to PhCO2H in 58% yield.30
Exploring relatively uncommon photocatalysts, Jiang and Soo both suggested the synergistic photocatalytic degradation of different plastics, including polystyrene by employing uranyl- or vanadium-based complexes.31,32 The methods were applied to commercial polymers, while the incorporation of continuous flow chemistry further highlighted the adaptability and scalability of these approaches.31,32 Lastly, within the range of innovative methods, acridinium salts, though common photocatalysts for organic transformations, had not been used for plastic upcycling, until Ong et al. exploited their known ability to reach highly oxidising excited states through visible light photo-activation for the low-energy visible light oxidative degradation of polystyrene.33 They anticipated that photoexcitation of acridinium salts could offer better control, resulting in improved product selectivity. The catalyst efficiently converted polystyrene into PhCO2H (33% under 390 nm irradiation, O2 atmosphere, 10 mol% catalyst and aq. HCl). Interestingly, when the HCl concentration was increased (200 mol%), the yield rose (46%), even with a reduced catalyst loading (5 mol%). Subsequent in silico screening aimed at identifying superior photocatalysts. The study pinpointed a fluorinated acridinium catalyst, whose synergistic action with HCl allowed for low catalyst loadings (<5 mol%), yielding 55% PhCO2H. Real-world PS waste could be employed, while the low toxicity of acridinium salts is advantageous, regarding trace metal contamination in upcycled PhCO2H.33
In 2024, Kokotos and colleagues introduced two new photochemical protocols utilising bromine radicals for converting polystyrene polymer into benzoic acid through synergistic i-HAT catalysis.34,35 The initial method involved a mild, easy-to-perform photochemical aerobic process, exploiting the combined effects of HAT catalysis and aerobic oxidation, utilising ambient air as the oxidizing agent. Employing a catalytic system comprising of NBS/2,4-diethylthioxanthone under UVA 390 nm LED irradiation, this method achieved a benzoic acid yield of up to 54% from the upcycling of everyday polystyrene items.34 The resulting benzoic acid was easily recovered through acid/base wash and extraction, following a purification process pattern similar to their previous protocol. Furthermore, investigations into a large-scale reaction of 2 g and catalyst recycling experiments were conducted, alongside a comprehensive analysis using high-resolution mass spectrometry (HRMS), which identified numerous intermediates, confirming the proposed reaction pathways.34 Similarly, the second protocol introduced a practical photochemical approach for the aerobic upcycling of polystyrene through synergistic indirect HAT catalysis, utilising a BrCH2CN/thioxanthone system and 390 nm irradiation in the presence of air.35 Both polystyrene and everyday polystyrene-based products were transformed into benzoic acid, yielding up to 49% following a simple base-acid workup. An analytical HRMS study was conducted to identify potential intermediates (Scheme 3C), and recycling experiments were carried out. Notably, the environmental sustainability of this methodology was assessed by calculating environmental metrics, such as the environmental factor (E), reaction mass efficiency (RME), and process mass intensity (PMI). These metrics were then compared with those of other photochemical protocols, demonstrating the sustainability of the protocol, mainly attributed to the elimination of column chromatography, high yield of benzoic acid, and utilisation of air as the oxidant, which significantly enhanced atom economy, compared to processes requiring an oxygen balloon. Furthermore, while a 2 g scale reaction was successfully conducted, the photochemical protocol was upscaled to a 20 g reaction, yielding 45% of benzoic acid. Although only 10 mol% of the catalyst was required, the amount of thioxanthone was halved to 5 mol% for economic reasons, which still yielded satisfactory results, though necessitating an extension of the reaction period to 14 days.35
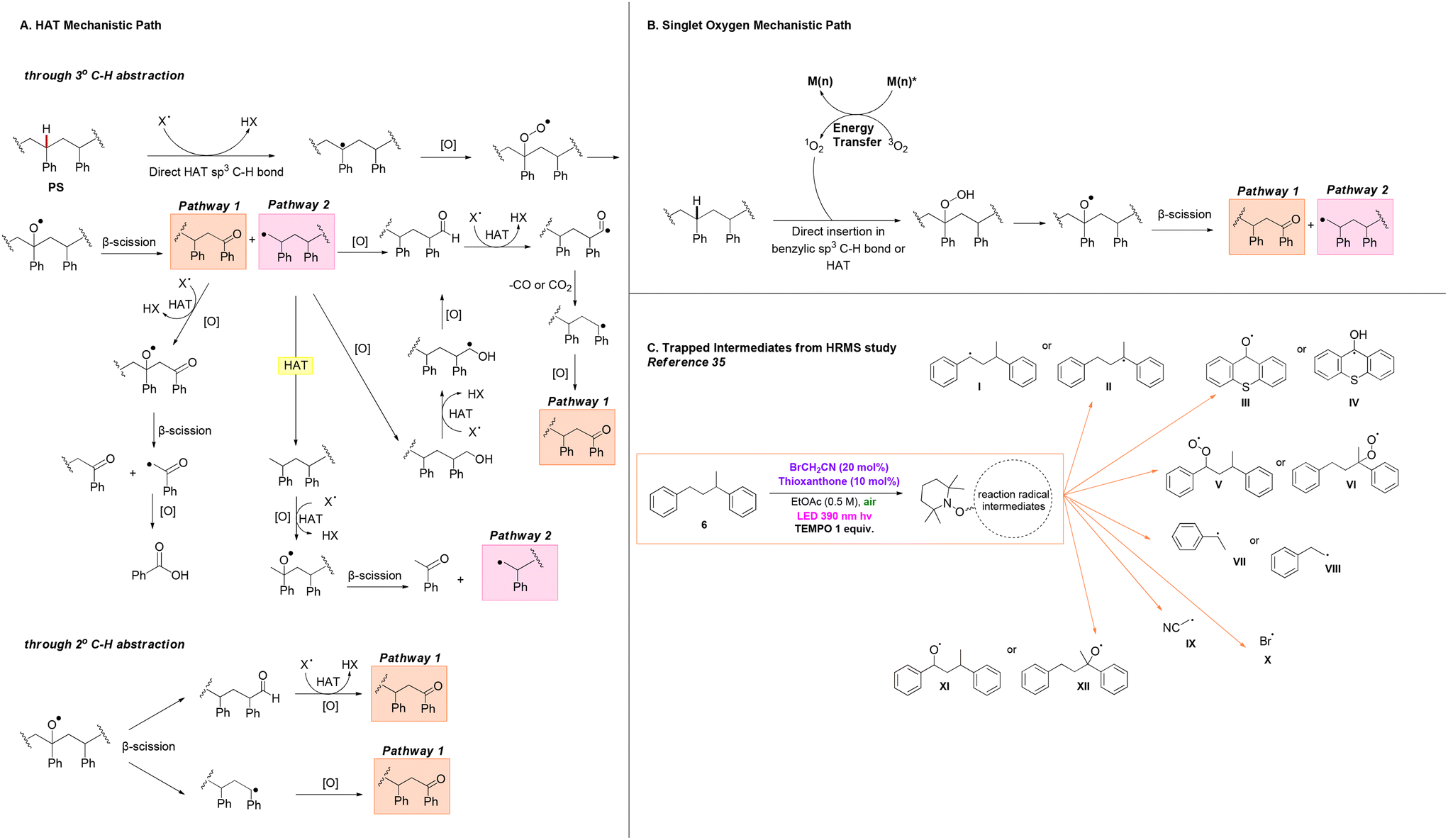 | ||
| Scheme 3 A. Plausible HAT-mediated mechanistic pathways for the photochemical upcycling of polystyrene through 3° and 2° C–H abstraction; B. plausible singlet oxygen-mediated mechanistic pathway for the photochemical upcycling of polystyrene; C. intermediates observed via DI-HRMS of the photochemical oxidation of model substrate 1,3-diphenylbutane using BrCH2CN-thioxanthone as the catalytic system. | ||
After examining all previous protocols on the photochemical upcycling of polystyrene, we concluded that in most cases where polystyrene is used as the starting material for the production of benzoic acid, numerous β-scission and oxidation events occur along the polymer backbone. Based on established studies, we found that Oh and Stache, in their mechanistic investigations, proposed a detailed and accurate mechanism that elegantly explains the processes occurring during the irradiation of the polymer in the presence of a catalyst (in this case halogen radicals were used as HAT catalysts, Scheme 3A and B).18 Photochemical upcycling of polystyrene in the presence of a photocatalyst can proceed via two mechanistic pathways: HAT (hydrogen atom transfer) or singlet oxygen pathways. In the first case, degradation occurs via 3° C–H or 2° C–H abstraction along the polymer chain (Scheme 3A).18
Following 3° HAT and oxidation with oxygen, β-chain scission in the polystyrene backbone produces an aromatic ketone at one chain end (pathway 1) and a 1° alkyl radical at the other chain end (pathway 2). In pathway 1, X˙ (typically derived from HAT events within the catalytic system) abstracts a β-C–H to produce a benzylic radical, which subsequently oxidises to an oxygen-centred radical in the presence of oxygen. Following another β-chain scission event, a polymer fragment with an aromatic ketone and an α-acyl radical is formed. The latter, is then oxidised, leading to decarbonylation or decarboxylation, ultimately resulting in the formation of benzoic acid.18
Pathway 2 involves three different routes for the conversion of PS fragments into small molecules. First, the alkyl radical can be oxidised to form an aldehyde or, alternatively, a primary alcohol which, subsequently oxidises to an aldehyde. Another HAT event leads to the formation of an acyl radical, followed by decarbonylation or decarboxylation, yielding a 2° benzylic radical on the PS fragment. In the presence of O2, the benzylic radical can form an aromatic ketone, which could eventually lead to production of benzoic acid following pathway 1. Additionally, the 1° alkyl radical has the potential to abstract a proton either through intermolecular HAT on another PS backbone, intramolecular 1,5-HAT on the same PS backbone, or HAT on the catalyst, leading to the formation of a methyl-group chain end on the PS fragment. Sequential HAT, oxidation, and β-chain scission events, could lead to formation of acetophenone, along with another 1° alkyl radical, which could then re-enter pathway 2.18 When 2° C–H abstraction occurs, β-chain scission of the oxygen-centered PS radical results in the generation of aldehyde and benzylic radical chain ends. By following the procedures described earlier, subsequent aromatic ketone groups could be formed. These ketone groups, upon proceeding along pathway 1, could lead to the formation of benzoic acid.18
Upon irradiation of polystyrene, another mechanistic pathway is possible, where initiation of the process happens through generation of singlet oxygen by the excited state of the catalyst. Singlet oxygen exhibits the capability to either directly insert at the activated benzylic position or react similarly through HAT to the benzylic C–H bond of PS,20 leading to formation of intermediates bearing the peroxy moiety. Breakdown of these high-energy peroxy intermediates has the potential to generate reactive oxygen species (ROS), such as oxygen-centred PS radicals, hydroxyl radicals, and superoxide radical anions, which possess high oxidative activity and act as potent hydrogen atom abstractors. Subsequent β-chain scission events, once again, conclude in formation of intermediates following pathways 1 and 2 (Scheme 3B).23 These mechanisms facilitate β-scission and oxidation events on the polymer backbone in a similar manner across various catalysts.
Through a comprehensive analysis of adducts corresponding to the intermediates of the previous mechanisms, certain scientific studies have contributed to the accumulation of experimental evidence supporting these pathways, thus validating the theoretical hypotheses (Scheme 3C).23,34,35
To ensure a comprehensive understanding of the distinctive features of each depolymerisation process, we have chosen to undertake a comparative analysis of the diverse methodologies applied specifically to polystyrene, since there is plenty of research available in this field, providing us with ample material for a thorough investigation and meaningful comparison (Table 1). As already mentioned, photochemical upcycling of polystyrene (PS) into valuable products represents a promising strategy for effective plastic waste management. Various catalytic methods, including use of iron or other metal salts, photoorganocatalysts, acid catalysis or heterogeneous catalysis, have been explored for this purpose, each presenting its own set of advantages and disadvantages.
| Entry | Catalytic system/conditions | Yield% of BA in reaction | Main products/comments on practical aspects | Ref. |
|---|---|---|---|---|
| Iron salts protocols | ||||
| 1 | FeCl2 – O2 balloon – LED 400 nm | Up to 68% | Benzoic acid – thorough mechanistic studies – no scale-up demonstration | 15 |
| 2 | FeCl3/TBACl/Cl3CCH2OH – O2 balloon – blue LED (390 nm) | Up to 67% | Benzoic acid – wide spectrum application to the oxidation of alkyl aromatics (including 1°, 2°, and 3° alkyl aromatics) to carboxylic acids – no real-life applications | 16 |
| 3 | FeCl3 – O2 balloon – white LED | Up to 16% | Benzoic acid (in mixture with other benzoyl products) – low yields – flow application – promising but room for improvement | 17 |
| 4 | FeCl3 – 100 °C – UV 365 nm | n/a | p-Br or Ac styrene – modification of polymer first, brominated derivatives and microplastics release risk, elevated T – no efficacy data | 28 |
| Organic molecules protocols | ||||
| 5 | p-TsOH·H2O – O2 balloon – blue LED (405 nm) | 51% | Benzoic acid – toxic benzene as solvent – flow application | 20 |
| 6 | NBS/CF3SO2Na – O2 balloon – LED 390 nm | Up to 73% | Benzoic acid – high yields, gram scale application and subsequent synthesis of bioactive molecules and compounds with high market value | 21 |
| 7 | Fluorenone/H2SO4 – O2 balloon – blue LED (450 nm) | ∼40% | Benzoic acid – use of corrosive strong acid – gram-scale application to substrate as well as real-world plastics | 19 |
| 8 | Anthraquinone – open-air reaction – LED 390 nm | Up to 59% | Benzoic acid – real-life applications, synthesis of pharmaceuticals and 2-gram scale-up, ambient air as O2 source | 23 |
| 9 | 6-Bromobenzo[d]thiazole/HCl – open air reaction – 390–400 nm LEDs | 58% | Benzoic acid – no real-life and gram applications, ambient air as O2 source | 30 |
| 10 | Ph-Acr-Ph·BF4 or FPh-Acr-Np·BF−4/HCl-O2 balloon – LED 390 nm | Up to 55% | Benzoic acid – DCE as solvent, limited applications and no scale-up data | 33 |
| 11 | NBS/2,4-diethylthioxanthone – open air reaction – 390 nm LED | Up to 54% | Benzoic acid – several real-life applications and scale-up, ambient air as O2 source, simple isolation, extensive HRMS mechanistic studies | 34 |
| 12 | BrCH2CN/thioxanthone –open air reaction – 390 nm LED | Up to 49% | Benzoic acid – several real-life applications and scale-up of 2 g and 20 g, ambient air as O2 source, simple isolation, sustainability evaluation, extensive HRMS mechanistic studies | 35 |
| Metal-based protocols | ||||
| 13 | AlCl3 – UV reactor equipped with 12 light bulbs (253.7 nm) | 91.9 wt% benzene (GC) | Benzene (then modified to diphenylmethane) – scale-up demonstration – proof of concept with socioeconomic analysis | 22 |
| 14 | UO2(NO3)2·6H2O/HCl – O2 balloon – bue LED (460 nm) | 46% | Benzoic acid – wide application to different polymers – successful to copolymers – uranium-based catalyst raises concerns on sustainability | 31 |
| 15 | V(O)(acac)2 – O2 balloon – white LED(s) | 33.3%/33.0% | Formic/benzoic acid – broad plastic categories application, long reaction times (>4 days), flow application for scale-up | 32 |
| Heterogeneous protocols | ||||
| 16 | g-C3N4 – 150 °C – 10 bar O2–Xe lamp | Up to 74% | Benzoic acid – elevated T and P, harsh and expensive Xe lamp | 24 |
| 17 | F-g-C3N4 – 2 bar O2–Hg lamp or sunlight | 72.8% PS conv. 81.6% AP/18.4% BA | Acetophenone and benzoic acid – mildly elevated P, Hg lamp, use of sunlight is possible | 25 |
| 18 | FeB/TiO2 – reactor equipped with LED 365 nm | H2 (103.5 μmol) | H 2 and organic compounds – complex mixture without selectivity and yield data | 26 |
| 19 | PSA-1-TiO2 or DTSPA-0.6-TiO2 – 1 bar O2 LED 370 nm | Up to 44% | Benzoic acid and monomers – real plastic application, short reaction time, no scale-up demonstrated | 27 |
| 20 | CQDs, BaO or ZnO – photothermal – white LED chip | Up to 50% | Styrene – novel approach, short reaction times, real-life applications – suitable for different polymers | 29 |
Commencing with iron salts, they present numerous advantages as catalysts in diverse chemical reactions, primarily attributed to their abundance and affordability, rendering them highly appealing choices and offering cost-effective catalytic solutions. Furthermore, their environmental compatibility is noteworthy, given iron's relatively low toxicity compared to other metals, thus aligning with endeavours towards environmental sustainability. Notably, the versatility of iron salts in participating across a wide spectrum of chemical transformations highlights their desirability as catalysts in various photochemical processes. Specifically, ferric chlorides are recognised for their role in generating chlorine radicals under light exposure, a pivotal process in initiating polymer radical formation, as chlorine radicals act effectively as hydrogen abstraction catalysts. Nonetheless, there are drawbacks to consider. Notably, the catalytic process may lead to the formation of by-products, complicating purification processes, and iron salts may lack high selectivity among photochemical reactions. Moreover, while iron halides typically serve as effective catalysts for diverse photochemical transformations, they may at times necessitate the presence of synergistic compounds for enhanced reaction yields, and limited solubility in certain reaction solvents poses additional challenges. Despite these limitations, the beneficial characteristics of iron salts position them as valuable contenders for catalysing various chemical transformations, with their application in polystyrene upcycling frequently resulting in the isolation of benzoic acid in high yields.
Other metal salts, including those derived from titanium, vanadium or uranium, offer distinct advantages as catalysts in various chemical reactions due to their high efficiency, robust light absorption, and catalytic properties. These salts can facilitate a wide range of transformations, enhancing their versatility and applicability across different reaction systems. However, there are certain drawbacks to consider. Cost and availability are significant factors, as some metal salts can be expensive and less accessible. Additionally, the toxicity of many metal-derived compounds presents environmental and health risks, requiring careful handling and disposal protocols. For example, even though the less toxic isotope 238U is used in Jiang's uranyl-based protocol31 (where naturally occurring uranium consists of 238U, 235U, and 234U, with the latter two being highly radioactive), the industrial application of such processes still raises concerns about environmental impact.
Photoorganocatalysts offer unique benefits as catalysts in diverse chemical reactions. One significant advantage lies in their lower toxicity, compared to metal-based catalysts. Additionally, the structure of photoorganocatalysts can be modified by introducing different functional groups, enhancing their properties. These modifications often result in high selectivity for specific chemical reactions. Furthermore, photoorganocatalysis typically yields high conversion rates in organic transformations, thanks to the homogeneous nature of the reaction mixtures. This is primarily attributed to the excellent solubility of the reactants, which greatly facilitates the photochemical processes. These processes usually do not require extreme reaction conditions, such as high pressures or temperatures, and are easy to handle. However, challenges exist in their utilisation. These include limited applicability at an industrial scale, posing challenges in terms of both cost efficiency and scalability. Additionally, thorough optimisation of reaction conditions, such as the choice of solvent, irradiation wavelength, catalytic amount, and reaction time, can be time-consuming.
Over the years, acid catalysis has emerged as an appealing option for various chemical transformations. It offers numerous advantages in facilitating chemical reactions, particularly in polymer degradation processes. Acids are effective in activating substrates, especially in polymer degradation methods, contributing to enhanced reaction efficiency. Additionally, many acids possess strong oxidising properties, allowing them to actively participate in intermediate reactions during the generation of monomers or oligomers. This strong activation boosts overall reaction efficiency, resulting in improved yields and reaction rates. Furthermore, the widespread availability and low cost of acidic reaction counterparts make them accessible for a broad range of applications. However, the corrosive nature of acids complicates their handling, especially on an industrial scale. Careful management of reactions and thorough purification of the reactions’ mixtures are necessary precautions. Moreover, although acids are highly effective in the photochemical degradation of polymers, their application may be limited in processes, where specific compound formation is desired. Their reactive nature can pose risks of unwanted side reactions or by-product formation.
Heterogeneous catalysis presents numerous benefits for facilitating chemical reactions, especially in industrial processes. One major advantage of heterogeneous catalysts is their ease of separation from the reaction mixture, allowing for efficient reuse without loss of catalytic activity. Additionally, their stability enhances their utility. However, there are several challenges associated with their use. For instance, heterogeneous catalysts often exhibit low solubility in many reaction solvents, which may hinder their effectiveness in processes, such as polymer degradation. This low solubility can also lead to difficulties in trapping reactants or products onto the catalyst surface, potentially impeding reaction completion. Furthermore, the characterisation of heterogeneous catalysts can be time-consuming, and issues with yield reproducibility may arise. Additionally, these catalysts often require the application of heat or high pressures to facilitate reaction completion, while certain carbon nanomaterials, in particular, exhibit reduced absorption of visible light, compared to semiconductor materials like TiO2, this characteristic can constrain their photocatalytic efficacy under specific circumstances.
It's evident that each catalyst type comes with its own distinct set of advantages and drawbacks, when it comes to photochemical plastic upcycling. Consequently, selecting the most suitable catalyst requires careful consideration of various factors, such as reaction conditions, availability of reactants, cost, and environmental impact. It's crucial to weigh both the benefits and drawbacks of each method thoroughly to make the right choice for the upcycling process.
Polyethylene/polypropylene
The upcycling of linear polymers found in plastics, such as polyethylene (PE) and polypropylene (PP), presents significant challenges, due to their resistance to degradation and the limited availability of valuable compounds upon breakdown. PE and PP are solely composed by long and resistant hydrocarbon chains, while the absence of functional groups, such as ester or hydroxyl groups, which usually participate in degradation processes, enhance the polymers’ resistance to degradation even further. Furthermore, the chemicals generated through their upcycling processes are predominantly mixtures of gaseous compounds, presenting challenges in handling and raising doubts about their practical utility. In 2021, Reisner investigated the photodegrative activity of platinum-doped nanocatalysts to convert pure succinic and glutaric acid to ethane and propane as main products, and ethylene and propylene, as secondary decarboxylation products, respectively (Scheme 4A).36 Despite the involvement of low energy input, less complicated product mixtures and easy separation of gases from liquid phase, the process yielded a poor 1.0% of the aforementioned hydrocarbons, while Xing designed a hybrid nanocatalyst for the upcycling of polyolefins and real-life plastics, namely PE, PE bags, PP, PP surgical masks and PVC plastic wrap into HCO2H under visible-light irradiation and oxygen atmosphere.37 At this point, it is worth mentioning that most of the existing processes require pretreatment steps to facilitate the breakdown of polymeric bonds, resulting in upcycling primarily through the transformation of these breakdown compounds. Liu compared polarised and non-polarised KNbO3 nanosheet to degrade PE, PP and real-world plastics. The polarised material exhibited higher degradation efficiency, thanks to its enhanced photoelectrical properties, while real-life PE and PP plastics displayed even higher CO2 production than pure polymers, with the authors speculating that particle size might play a role.38 Zhang's group fabricated an heterogeneous catalyst for the PEG microplastics’ upcycling,39 while the degradation of polyolefins has been reported using metal-based catalysts.40 These processes share a similar mechanistic background, initiated by the generation of exited e− and holes (h+) on the heterogeneous catalyst by excitation. These lead to the generation of reactive oxygen species (ROS), namely, O2˙− and ˙OH, which are responsible for the polymer chain cleavage by the formation of carbon-centred radicals on the polymer chain. The oligomer radical intermediates formed continue to undergo cleavage until they are combined forming small organic molecules, that are over-oxidised to CO2 and H2O. This is also the main drawback of such processes, since lack of control over the oxidation process leads to a non-valuable gaseous product.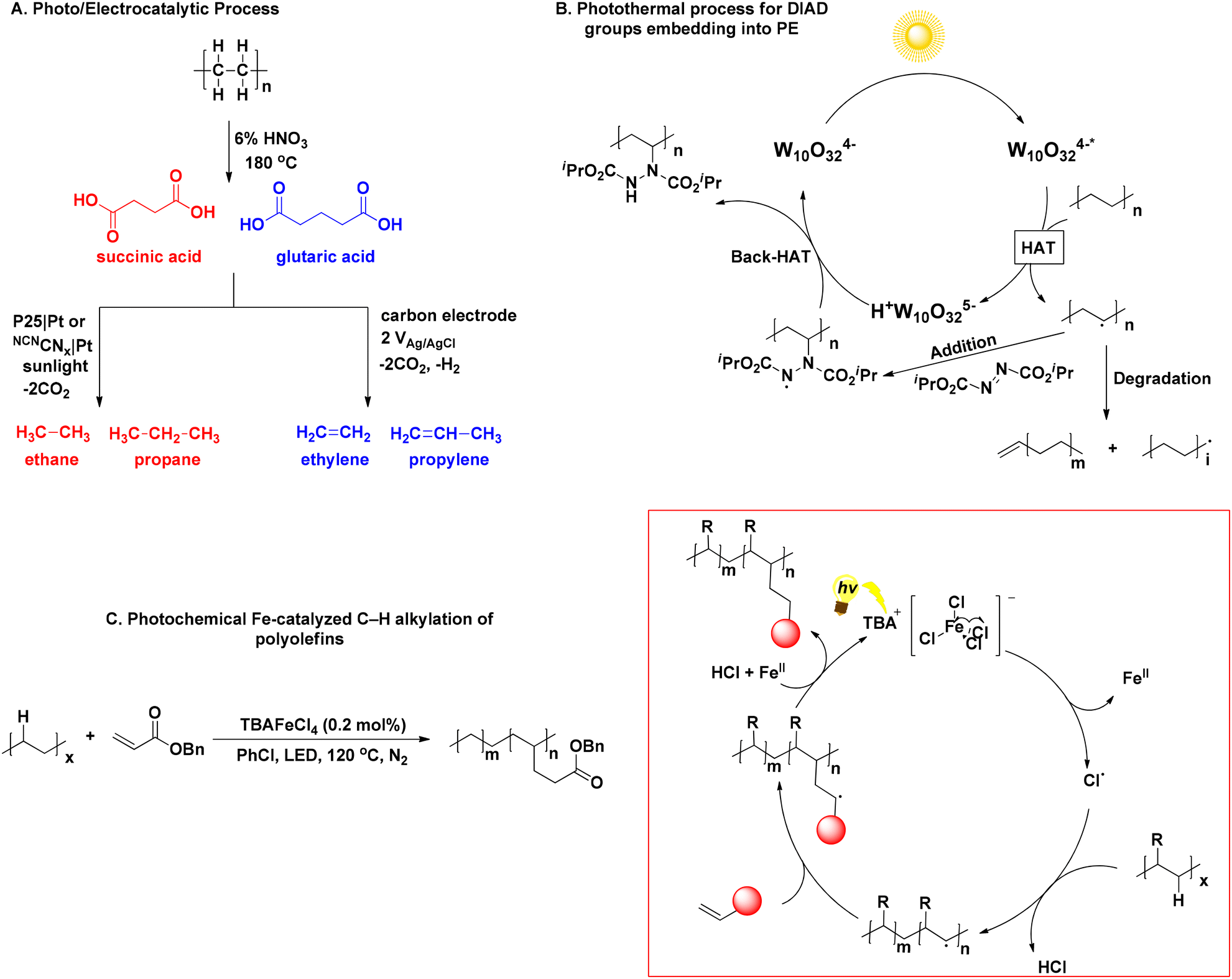 | ||
| Scheme 4 A. Photoelectrocatalytic PE degradation; B. photothermal process for embedding DIAD into PE; C. light-induced polyolefin upcycling to modified polymers. | ||
Opting for an alternative route, Kong designed a synergistic thermal-photochemical process to embed diisopropyl azodicarboxylate (DIAD) groups into PE and commodity plastics, using TBADT as the photocatalyst and sunlight irradiation (Scheme 4B).41 Resultantly, modified DIAD-grafted PE was obtained, while the molecular weight of the newly-formed material was decreased, due to photochemical β-scission. The authors asserted that the modified polymers have the potential utility as compatibilizers in blending polyolefin-starch. This blending process generates degradable polyolefin plastics, representing a significant step towards addressing the pervasive issue of plastic pollution.41 Another approach for the light-induced polyolefin upcycling to modified (multi)functionalized polymers was recently presented, exploiting the photochemical properties of TBAFeCl4, which once again acted as a chlorine radical generator, abstracting a hydrogen atom from the polymeric chain, resulting in the formation of an intermediate hydrocarbon radical, under 390 nm LED at 120 °C to introduce an electron-withdrawing group onto the polymer backbone (Scheme 4C).42 The process was successful for PE, PP, PS and real-life plastics. Furthermore, even employment of poor levels of functionalization into the polymers led to altered materials’ physical properties, such as increase of toughness and hydrophilicity.42 These particularly intriguing approaches present solutions to address the challenge posed by the inert, hydrophobic hydrocarbon chains found in PE and PP (Table 2). By altering these chains, the properties of the polymers can be modified, resulting in the creation of novel polymers with improved characteristics.
| Polymer/entry | Catalytic system/conditions | Yield% of reaction | Main products/comments on practical aspects | Ref. | |
|---|---|---|---|---|---|
| PE/PP | 1 | (i) HNO3, 80 °C | Succinic acid: P25|Pt: main hydrocarbon → ethane (56.3 μmol g−1 h−1) | Ethane and propane (major products) from succinic and glutaric acid – not isolable gaseous hydrocarbons – significant H2 and CO2 formation – acid pretreatment leads to molecules that are then transformed photochemically – flow application | 36 |
| (ii) P25|Pt or NCNCNx|Pt – solar simulator (AM 1.5G, 100 mW cm−2) with water filter (removing infrared light) | NCNCNx|Pt: main hydrocarbon → ethane (7.2 μmol g−1 h−1) | ||||
| Glutaric acid: P25|Pt: main hydrocarbon → propane (17.1 μmol g−1 h−1) | |||||
| NCNCNx|Pt: main hydrocarbon → propane (4.9 μmol g−1 h−1) | |||||
| 2 | V-Substituted phosphomolybdic acid clusters/g-C3N4 nanosheets (VPOM/CNNS) – O2 balloon – 300 W Xe lamp | Formic acid evolution for PE → (24.66 mmol g−1 h−1) for PP → (26.68 mmol g−1 h−1) | Formic acid – no up-scale reaction – only one real-life example for each polymer | 37 | |
| 3 | Polarized KNbO3 (P-KNbO3) nanosheet and several heterogeneous and metal-based catalysts – 150 W Xe lamp with AM 1.5G filter | CO2 production rates for P-KNbO3 for photocatalytic degradation PE and PP are 17.4 mg g−1 h−1 and 28.3 mg g−1 h−1, respectively, vs. 8.2 mg g−1 h−1 and 19.6 mg g−1 h−1 of KNbO3 | CO2 – enhanced photocatalytic efficiency of P-KNbO3 due to external electric field induced KNbO3 polarisation | 38 | |
| 4 | Ag2O nanoparticle encapsulated Fe based MOF (Ag2O/Fe-MOF) – 300 W Xe lamp with AM 1.5G filter | 1.7 mmol g−1 h−1 H2 | Microplastics to H2 – real-life-plastics derived MPs – no data on organic molecules produced – no scale-up | 39 | |
| 5 | Pretreatment: 110 °C (due to insolubility) – V(O)(acac)2 – O2 balloon – white LED(s) | 14.8% formic acid for HDPE | Formic acid for PE and formic/acetic acid mixture for PP – long reaction times – CO2 by-product – limited real-life plastic application | 32 | |
| 9.9% formic acid for LDPE | |||||
| 29.6% formic acid for PP | |||||
| 22.9% acetic acid for PP | |||||
| 6 | Pretreatment in HNO3, 180 °C – MoS2-tipped CdS nanorods – 300 W Xe lamp illumination equipped with an AM 1.5 solar simulator | 1.13 ± 0.06 mmol g−1 h−1 H2, 196.2 ± 1.76 μmol g−1 h−1 CH4, 1.86 ± 0.04, 0.78 ± 0.20, and 7.60 ± 0.60 μmol g−1 h−1 (C2H6, C3H8, C5H12) | Pretreatment-derived mixture of glutaric and succinic acid afforded formic acid, H2 from H2O splitting and 1- to 5-carbon hydrocarbons – increased contents of formic acid and acetic acid after photoreforming of pretreated PE are possibly originated from the photo-oxidation reaction of dicarboxylic acids (i.e., glutaric acid and adipic acid) – no real-life application for PE | 46 | |
| Upcycling to novel materials | 7 | TBADT, DIAD, sunlight and 110 °C | 80–144% weight of grafted units relative to the PE/11–20 grafted DIAD groups per 100 ethylene | PE modification to DIAD-functionalised polymer – 1,1,2,2-tetrachloroethane dangerous solvent – although sunlight is an abundant and renewable energy source, daylight is not always available – high T – real-life plastic application – no scale-up | 41 |
| 8 | TBAFeCl4, polar olefins, UVA LED (390–395 nm) and 120 °C | 4–11% level of functionalisation | Alkylated polyolefins – diverse polar olefin scope – multi-polar polyolefins synthesis through modularly polar group assembly via C–H bond modification – real-plastic applications – scale-up application | 42 | |
Polyethylene terephthalate and polylactic acid
Photoreforming is the use of light to generate H2 from a substrate (electron donor) and water (reduction to H2) (Scheme 5A).43 Photochemical PET reforming has received increased interest, where usually an alkaline pretreatment to release monomers is followed by photoreforming of the aqueous mixture. Usually, PET reforming is studied along with PLA, thus the oxidation refers to ethylene glycol (terephthalic is inert to oxidation) and lactate, respectively (Scheme 5B). The issue of product selectivity presents a significant challenge in PET and PLA photoreforming, since it usually leads to the generation of a complex mixture of outputs incidentally, rather than relying on the careful design to afford high-value products. Despite the potential yield of hydrogen, a valuable fuel source, the current technology for its utilisation and safe handling is still in its infancy. Moreover, the necessity for an additional pretreatment step, which most of the times entails harsh reaction conditions, blurs the distinction between genuine recycling efforts and mere workaround solutions, as well as diminishes the sustainability of the process. However, photoreforming is a popular research topic at this moment, thanks to its potential.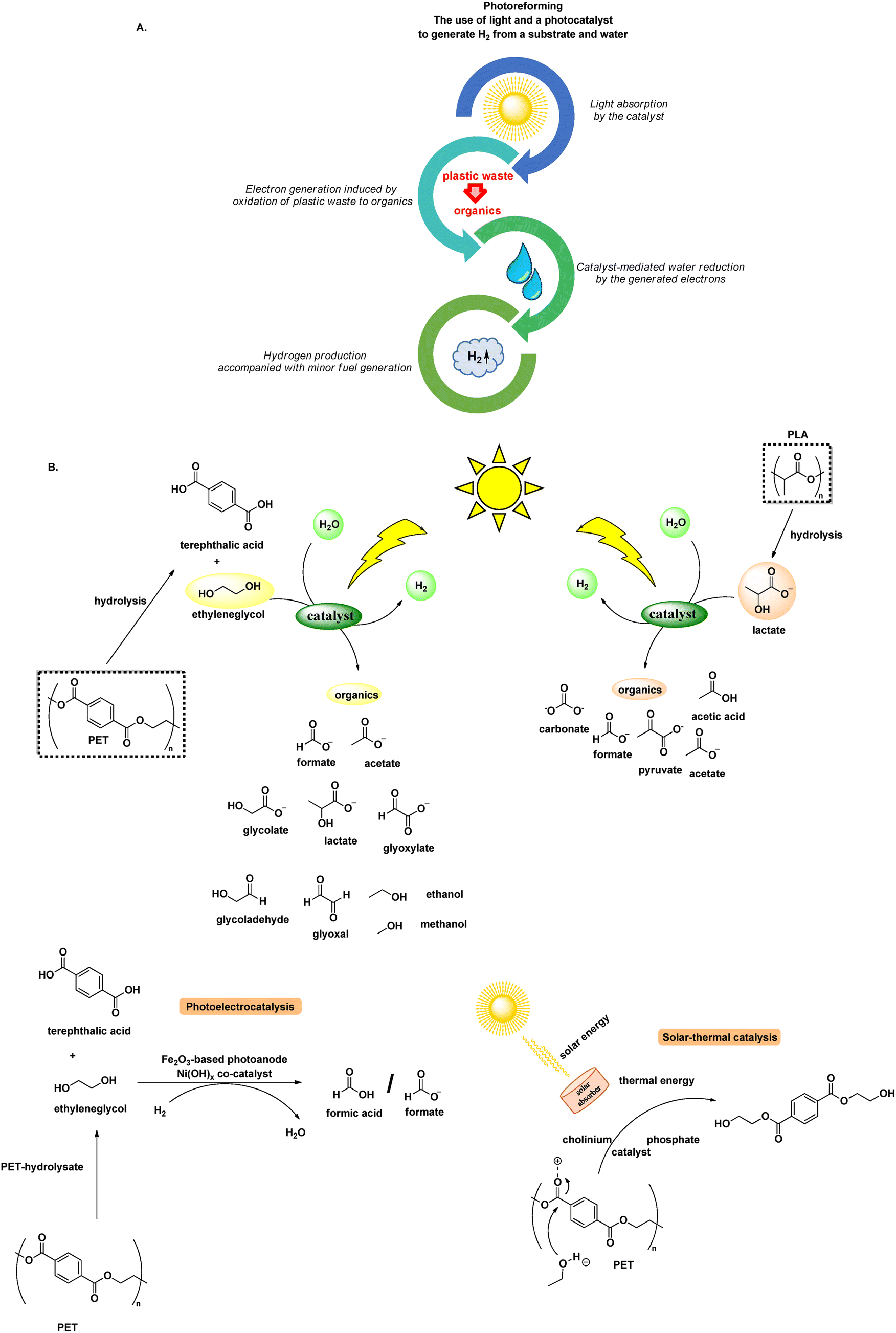 | ||
| Scheme 5 A. Representation of photoreforming; B. common light-driven protocols for the photochemical conversion of PET and PLA to H2 and organic compounds. | ||
Laying the foundations for polyester photoreforming, Reisner designed the mild photoreforming of PET and PLA, utilising a CdS/CdOx quantum dot photocatalyst. Along with the release of H2, organic molecules were produced. It was observed that alkaline pre-treatment and centrifugation significantly boosted photoreforming efficiency, since hydrolysis enables formation of monomers, while PLA, being soluble in aq. NaOH, does not need pre-treatment. Regarding the organic products of photoreforming, lactate monomer was oxidized to pyruvate or its derivatives, while oxidation of ethylene glycol led to a mixture of formate, glycolate, ethanol and acetate.44 However, the use of highly toxic cadmium was necessary.44 Later, the same group, designed a similar process avoiding Cd, using a carbon nitride/nickel phosphide photocatalytic system (CNx/Ni2P). This process offers a more benign and easily-scalable alternative, albeit with a comparably lower efficiency. In both cases, no greenhouse gases are released.43
Carbon nitride is a useful platform for photoreforming catalyst development. Yang synthesised carbonised polymer dots-graphitic carbon nitride (CPDs-CN) composites for the upcycling of plastics, coupled with H2 production,45 while Cd has also reappeared in the literature regarding PET photoreforming by Qiu's group, who designed MoS2-tipped CdS nanorods to simultaneously upcycle plastics via water reduction to H2.46 Many studies focus on PET-to-H2 photoreforming, not significantly taking into consideration the production of high-added-value chemicals, using heterogeneous fabricated catalytic systems, like in situ formation of carbon nanotubes/carbon nitride/NiMo (CN-CNTs-NiMo) hybrids enabling H2 production, as well as glyoxal/glyoxalate and formate, from PET and PLA,47 MXene/ZxC1−xS nanocomposites for the photochemical H2, glycolate, acetate, ethanol and methanol production, followed by a PET bottle degradation48 and defect-rich chalcogenide nanosheet-coupled photocatalysts, producing H2, along with acetates and pyruvate-based products from PLA and formate, acetate and glycolate from PET.49
Photoelectrochemistry constitutes a modern tool in plastics’ reforming, combining photoreforming's selectivity and electroreforming's efficiency. A recent example uses a Fe2O3/Ni(OH)x visible-light active photoanode. The system's oxidation ability is tuned to selectively produce formic acid/formate and avoid overoxidation to CO2. Employment of a PET hydrolysate in the reaction mixture enhanced production of PET's monomers, while H2 was produced, due to water splitting.50
Another photothermal approach with low energy investment and carbon emission, similarly to Stache's approach described above for the upcycling of PS,29,51 for the depolymerisation of PET to bis-2-hydroxyethyl terephthalate (BHET) in ethylene glycol was designed by Chen, utilizing a solar absorber of carbon nanotubes pre-modified by polydopamine (CNT-PDA), to convert simulated sunlight to heat, creating a localised heating effect and allowing a low depolymerisation temperature (150 °C). The addition of cholinium phosphate introduced a nucleophilic addition–elimination mechanism to yield BHET.51 Natural sunlight was successfully utilised, while the authors successfully repolymerised the yielded BHET to PET.51 This process holds considerable promise, as it demonstrates how the output of reforming can be effectively utilised to re-synthesize the polymer, aligning harmoniously with the principles of the circular economy and exemplifying genuine recycling in action (Table 3).
| Polymer/entry | Catalytic system/conditions | Yield% of reaction | Main oxidation products/comments on practical aspects | Ref. | |
|---|---|---|---|---|---|
| PET | 1 | CdS/CdOx quantum dots in alkaline solution after pretreatment – simulated solar light (AM 1.5G, 100 mW cm−2) | Overall conversion <40% for all polymers 12.4 ± 2 mmolH2 gCdS−1 h−1 | H2 and mixture of organic compounds from pretreatment-derived ethylene glycol, namely formate, glycolate, ethanol, acetate, lactate – controlled to not produce CO2 – real-plastic application, proof-of-concept – use of highly toxic Cd | 44 |
| 2 | Alkaline pretreatment then CNx/Ni2P – simulated solar light (AM 1.5G, 100 mW cm−2) | (62% pretreatment yield to ethylene glycol) 82.5 ± 7.3 μmolH2 gsub−1 | H2 and acetate, formate, glycolate, glyoxal – upscale and real-life applications – extended reaction time | 43 | |
| 3 | KOH, 120 °C pretreatment – carbonised polymer dots-graphitic carbon nitride (CPDs-CN) composites – 300 W Xe lamp | H2 production rate of 1034 ± 134 μmol g−1 h−1 | H2 from H2O splitting and complex mixture consisting of glycolaldehyde, glycolic acid, formic acid, ethanol, acetaldehyde and acetic acid | 45 | |
| 4 | KOH pretreatment – MoS2-tipped CdS nanorods – 300 W Xe lamp illumination equipped with an AM 1.5 solar simulator | 3.90 ± 0.07 mmol g−1 h−1 H2, 5.96 ± 0.02 and 0.95 ± 0.01 mmol L−1 after 5 h formate and acetate | H2 from H2O splitting and formate, acetate, glycolate from pretreatment-derived ethylene glycol – real-life applications | 46 | |
| 5 | CN-CNTs-NiMo hybrids – 500 W Xe lamp (simulated solar light, 95 mW cm−2) | H2 (90 mmol g−1 h−1) in 10 M KOH aqueous solution | H2 from H2O splitting, glyoxal and glycolate – real plastic application, no scale-up | 47 | |
| 6 | MXene/ZxC1−xS nanocomposites – 300 W Xe lamp | Up to 14.17 mmol g−1 h−1 H2 | H2 and glycolate, formate, acetate, methanol etc. | 48 | |
| 7 | Alkaline pretreatment – defect-rich chalcogenide nanosheets – 300 W xenon lamp | 31.38 mmol gcat−1 h−1 H2 in 2 M KOH 62.4 μmol yield of organic acids | H2 and formate, acetate, glycolate – real plastic application, no scale-up | 49 | |
| 8 | Fe2O3/Ni(OH)x photoanode – PET hydrolysate in reaction mixture for depolymerisation | Up to ∼100% faradaic efficiency of formic acid/formate H2 from water splitting, formic acid/formate without CO2 formation – photoelectrochemical method, improved selectivity with lower energy consumption | 50 | ||
| 9 | Solar absorber of carbon nanotubes pre-modified by polydopamine (CNT-PDA) and cholinium phosphate in ethylene glycol – simulated sunlight (600 mW cm−2) | Up to 82% BHET (100% conversion of PET) | Bis-2-hydroxyethyl terephthalate (BHET) – light to thermal energy approach, selective process, depolymerisation, repolymerization application, lower energy consumption data, large-scale application in natural sunlight | 51 | |
| 10 | Ag2O nanoparticle encapsulated Fe based MOF (Ag2O/Fe-MOF) – 300 W Xe lamp with AM 1.5G filter | 1.9 mmol g−1 h−1 H2 | Microplastics to H2 – real-life-plastics derived MPs – no data on organic molecules produced – no scale-up | 39 | |
| 11 | UO2(NO3)2·6H2O/HCl/HFIP – O2 balloon – blue LED (460 nm) | Up to 88% | Terephthalic acid without pre-treatment and strong conditions – flow application up to kg-scale – uranium-based catalyst raises concerns on sustainability | 31 | |
Although the photodegradation of PLA is usually enclosed within the scope of other polymers studies, many groups have studied its upcycling alone. Huang reported a NixCo1−xP/reduced graphene oxide/g-C3N4 (NixCo1−xP/rGO/CN)-based photocatalytic protocol for the photoreforming of PLA into value-added compounds.52 Formate and acetate were identified as the main products, while the production of H2 was detected.52In 2023, Zhang launched the use of metal tungstates (FeCoNiCuZn)WO4, which were incorporated into polyacrylonitrile (PAN) nanofibers, enabling enhanced photocatalytic activity for the conversion of PLA microparticles, obtained by basic pretreatment, into AcOH under 300 W Xe illumination and simulated natural environment conditions (Table 4).53 The same group employed a zinc oxide/UiO66-NH2 heterojunction to the photo-conversion of PLA and PVC, as well as commercially-available polymer-based products, into AcOH under irradiation, whereas H2 generation was observed.54 The photocatalytic process was compatible with LDPE and PET, suggesting a promising avenue for addressing a wider range of plastic waste.54
| Polymer/entry | Catalytic system/conditions | Yield% of reaction | Main products/comments on practical aspects | Ref. | |
|---|---|---|---|---|---|
| PLA | 1 | NixCo1−xP/reduced graphene oxide/g-C3N4 (NixCo1−xP/rGO/CN) – alkaline solution – 300 W Xe lamp | Up to 576.7 μmol h−1 g−1 | H2 from H2O splitting, formate and acetate – focus is given on H2 production, gram-scale reactions | 52 |
| 2 | Alkaline pretreatment – (FeCoNiCuZn)WO4 in polyacrylonitrile nanofibers (PAN-NFs) | Up to 770.24 mg gcat−1 | Acetic acid (propionic acid and propanol were also detected) – employment of PLA microparticles after pretreatment, all reactions are gram-scale in a filter reactor with a pump and a feed tank to establish a continuous flow condition | 53 | |
| PAN-NFs for the in situ growth of highly arrayed, triangular XWO4, which served as a photocatalytic membrane for filtration of plastic. – 300 W Xe lamp | |||||
| 3 | ZnO/UiO66-NH2 heterojunction – 300 W Xe lamp | 14.4% acetic acid [the turnover number (TON) and turnover frequency (TOF) of acetic acid generation of ZnO/UiO66-NH2 were 17.92 and 0.51 h−1, respectively. The conversion rate of PLA was 57.10 mg gcat−1 h−1] | Acetic acid – all reactions are gram-scale, real-plastic application | 54 | |
| 4 | CQDs, ZnO – photothermal – white LED chip | Up to 95% | Lactide – novel approach, short reaction time, real-life applications, suitable for different polymers | 29 | |
| 5 | CdS/CdOx quantum dots in alkaline solution – simulated solar light (AM 1.5G, 100 mW cm−2) | Overall conversion <40% for all polymers 64.3 ± 14.7 mmolH2 gCdS−1 h−1 | H2 and pyruvate or derivatives from hydrolysis-derived sodium lactate | 44 | |
| 6 | Alkaline pretreatment then CNx/Ni2P – simulated solar light (AM 1.5G, 100 mW cm−2) | (72% of PLA solubilized to lactate) 178 ± 12 μmolH2 gsub−1 | H2 and acetate, formate – extended reaction times for better efficacy | 43 | |
| 7 | KOH pretreatment – MoS2-tipped CdS nanorods – 300 W Xe lamp illumination equipped with an AM 1.5 solar simulator | 6.68 ± 0.10 mmol g−1 h−1 H2, (alkalinity degree of 10 M) after 5 h 90% of PLA to lactate after pretreatment | H2 from H2O splitting and formate as well as CO32− (accumulated concentrations of 5.37 ± 0.67 and 23.50 ± 2.43 mmol L−1) – no real-life application for PLA | 46 | |
| 8 | Alkaline pretreatment – defect-rich chalcogenide nanosheets – 300 W xenon lamp | 39.76 mmol gcat−1 h−1 H2 78.1 μmol yield of organic acids | H2 and 13.6 μmol of acetates and 64.5 μmol of pyruvate-based products plus carbonates, including pyruvate and derivatives – real plastic application, no scale-up | 49 | |
| 9 | Pt/Bi12O17Cl2 – 300 W xenon lamp 100 mW cm−2 | 474.3 mg gcat−1 & 287.6 mg gcat−1 (for formic acid and acetic acid, respectively, in 10 h) | Formic and acetic acid main products, 6–7 products detected in total after 10 h reaction (for 60SP-BOC) – all gram-scale reactions | 57 |
Hydroxylated polymers
Research has demonstrated significant advancements in the field of photochemical polymer upcycling, yet is mainly focused on non-polar polyolefins. Meanwhile, less is known about the depolymerisation of synthetic polymers, like hydroxylated macromolecules. In 2019, Soo presented an appealing photochemical protocol, using a vanadium (V)-based photocatalyst that enabled C–C bond β-scission of unactivated aliphatic alcohols, exploiting oxygen as the oxidant under visible light irradiation (Scheme 6A).55 This method paved the way for the synthesis of oxygenated hydrocarbons, using aromatic, aliphatic or unsaturated alcohols and was also applied to several hydroxyl-terminated polymers, PE and PE-copolymers, enabling the formation of alkyl formates and formic acid as products.55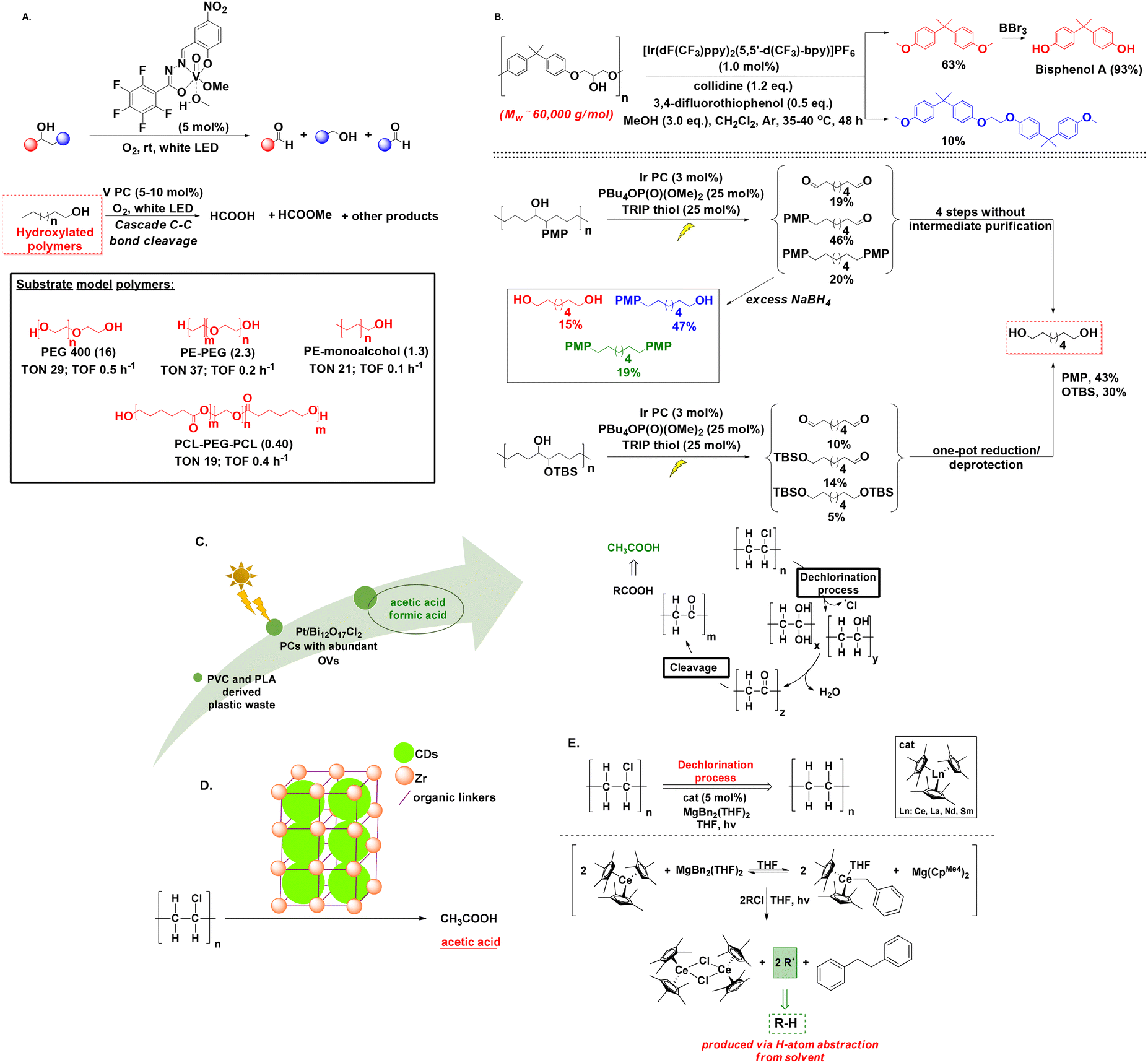 | ||
| Scheme 6 A. Vanadium (V)-based photocatalytic degradation of aliphatic alcohols; B. iridium-based photo-degradation of hydroxylated polymers; C. design of a Pt/Bi12O17Cl2 photocatalytic system for upcycling PVC and PLA; D. Zr-MOF framework catalyst for the photochemical upcycling of PVC; E. photochemical PVC dechlorination. | ||
In 2021, Knowles introduced the photo-degradation of commercially available hydroxylated polymers via proton-coupled electron transfer (PCET) for the generation of α,ω-bifunctional compounds (Scheme 6B).56 The authors studied the phenoxy-resin of bisphenol A, utilizing a combination of [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 as the photocatalyst, collidine as the base and 3,4-difluorothiophenol, whereas addition of MeOH to the reaction mixture accelerated the photocatalytic transformation through the formation of hemiacetal intermediates, which enabled O–H PCET and subsequent β-scission, promoting the generation of dimethyl bisphenol A as the main product. Treatment of the latter with BBr3 led to bisphenol A, while irradiation of phenoxy resin in the presence of other polymers, like PVC or PS, led to only a slight decrease of the degradation yield.56
Polyvinylchloride
In 2023, Zhang introduced a Pt/Bi12O17Cl2 photocatalytic system with abundant oxygen vacancies (OVs) for the upcycling of PVC and PLA into AcOH and HCO2H (Scheme 6C).57 The catalyst displayed great PVC dechlorination efficiency reaching 78.1% within 10 h, which can be attributed to the considerably lower C–Cl bond energy, compared to C–C and C–H bonds.57 The same group additionally investigated MOF chemistry for the conversion of PVC into value-added carboxylates (Scheme 6D).58A slightly different approach was proposed by Arnold studying the dechlorination of PVC using lanthanide complexes (Scheme 6E).59 A Ce(III) cyclopentadiene-based photocatalyst was employed for the removal of chlorine from PVC alongside MgBn2(THF)2 under 440 nm irradiation, leading to C–Cl bond cleavage and subsequent polyethylene formation at a 79% rate, while no polymer chain breakage was detected.59 Although not polymer degradation-related, we chose to include this elegantly-applied photochemical protocol, since removal of chlorine from PVC can offer a range of environmental and health benefits preventing release of chlorine-containing compounds.
Polyacrylate-based polymers
Recent exploration of photocatalysis for polymethacrylates and polyacrylates degradation aims at producing oligomers, rather than small organic molecules, potentially reducing plastic waste by offering avenues for faster degradation or transformation into other valuable products, like copolymers or oils. These methods usually follow a pathway involving carbon-centred radical formation via co-monomers and subsequent backbone C–C bond β-scission. Although, not entirely within the concept of upcycling, the following few selected examples demonstrate the broad potential of different photochemical approaches in the polymer upcycling endeavour.60In 2022, Sumerlin and Seidel introduced a pioneering method for polymer modification and degradation through the photocatalytic direct decarboxylation of carboxylic acids, using a violet LED light source, operating at a wavelength of 395 nm and an acridine photocatalyst (Scheme 7A and B). This technique generates carbon-centred radicals in polymer chains, facilitating the synthesis of unique copolymers, such as acrylate-olefin copolymers and those with pendent alkenes, while also enabling polymer degradation. By utilising decarboxylation in the presence of a hydrogen atom donor (i.e. PhSH), statistical acrylate-ethylene and acrylate-propylene copolymers, which are challenging to obtain through direct polymerisation, can be formed. Additionally, decarboxylation of methacrylic acid units within polymethacrylates can trigger the degradation of polymer backbones. Furthermore, a dual catalytic approach, combining an alternative acridine photocatalyst with a cobaloxime catalyst, is employed to produce unique copolymers with pendent alkenes. The process is advantageous as it operates under mild conditions without requiring preactivation of acid groups.61
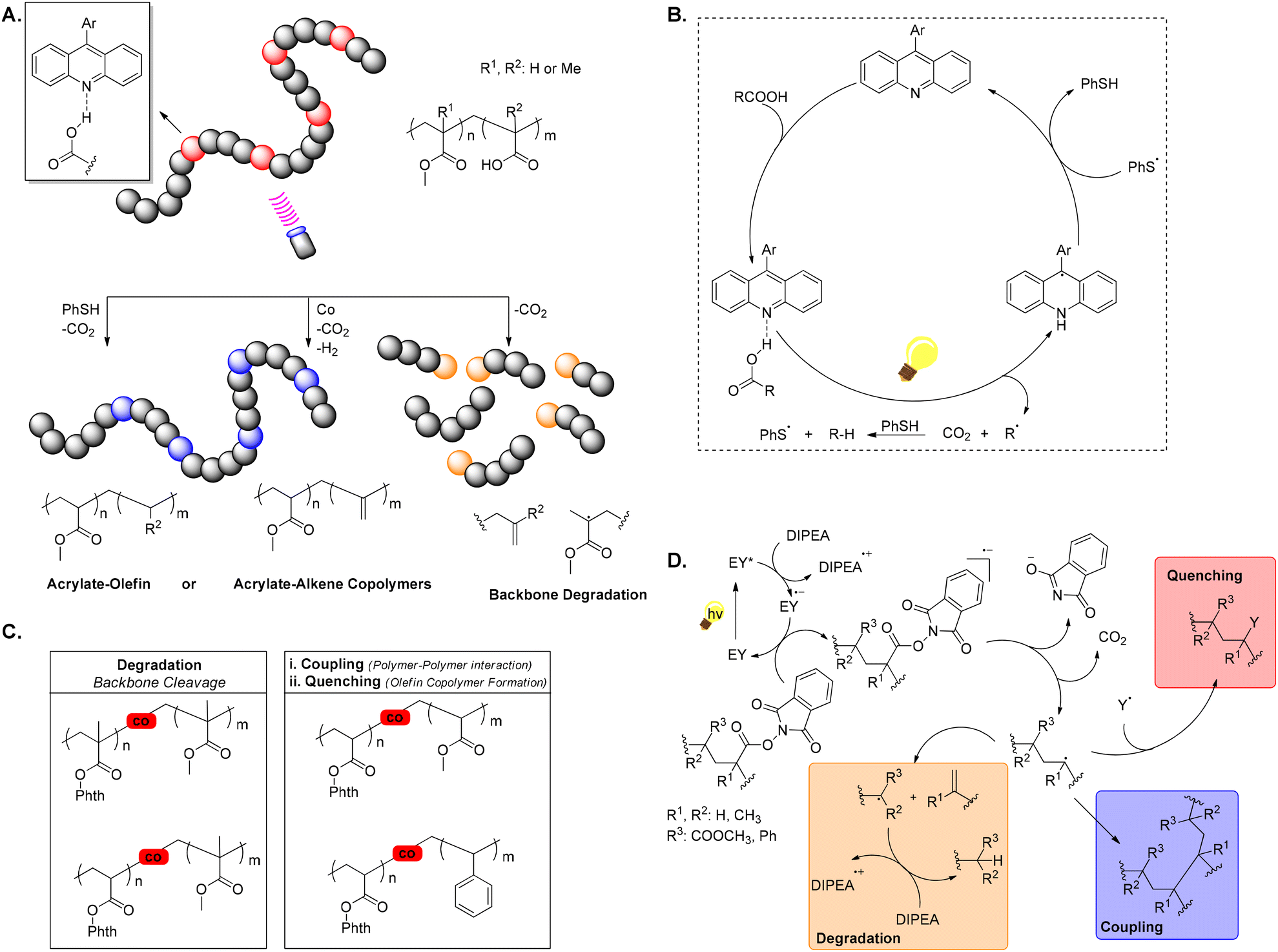 | ||
| Scheme 7 A. Direct decarboxylation of copolymers with AA or MAA units, resulting in the creation of acrylate-olefin or acrylate–alkene copolymers, or the breakdown of polymethacrylate backbones; B. catalytic cycle demonstrating acridine-catalysed photodecarboxylation and the subsequent capping of radicals by PhSH; C. synopsis of observed outcomes for copolymers containing phthalimide-ester groups following SET-induced decarboxylative backbone radical generation; D. proposed mechanism outlining single-electron transfer (SET)-mediated decarboxylative backbone generation in polymers, emphasizing the three possible outcomes of the backbone radical: quenching, degradation, and coupling. | ||
At the same time, another approach was studied by the Summerlin group. A photocatalytic strategy using a low mole percent of a redox-active comonomer, specifically N-(acyloxy)phthalimide, to induce backbone cleavage, upon light exposure, was developed. When the N-(acyloxy)phthalimide comonomer receives an electron from a single-electron transfer (SET) donor [in this case Eosin Y (EY)], it undergoes decarboxylation, producing a backbone-centred radical. Depending on this radical and the neighbouring monomer substitution, a thermodynamically favoured β-scission pathway leads to backbone cleavage. This method significantly reduces molecular weight in methacrylate, whereas acrylate- and styrene-based redox-active copolymers primarily underwent polymer coupling reactions as indicated by increased molecular weight observed by GPC. Introducing a hydrogen donor suppressed these couplings, resulting in statistical styrene-ethylene copolymers (Scheme 7C and D). However, this method did not achieve degradation of polystyrene or polyacrylates.62a The same group also attempted a post-polymerisation modification strategy to create statistical olefin-acrylate copolymers by applying an activated monomer ideal for radical copolymerisation with polar vinyl monomers (e.g., acrylates) and capable of post-polymerisation modification to reveal an olefin repeat unit. Under green light, SET from an organic photocatalyst (EY) to a polymer containing redox-active phthalimide ester units generates reactive carbon-centred radicals on the polymer backbone. Statistical olefin-acrylate copolymers were produced by decarboxylation in the presence of a hydrogen atom donor, which caps the backbone radical with a hydrogen atom, resulting in an ethylene or propylene repeat unit.62b One year later, Sumerlin, Seidel, and colleagues developed another photochemical convenient method for degrading polyacrylate homopolymers through a one-pot sequential dehydrodecarboxylation and ozonolysis process to generate mid-chain radicals, leading to β-scission, consistent with previously proposed mechanisms. In this sequential process, by partially hydrolysing ester side chains, carboxylic acids are introduced along the polymer backbone, which are further converted into alkenes and, then, oxidatively cleaved. This approach is compatible with a wide range of polymers made from vinyl monomers, including copolymers of acrylic acid with acrylates, acrylamides, and styrene-derived molecules.62c
In the same spirit, Ouchi explored the degradation of polymers, demonstrating two different photochemical protocols. In the first scenario, the carbon–boron (C–B) bond of a vinyl boronate unit was used as a trigger for main-chain degradation of the copolymer. This was achieved by generating radicals from the C–B bond through the addition of a base, such as sodium methoxide, which interacts with Lewis acidic boron, followed by photocatalysis with an organic photocatalyst. Although the C–B bond in the side chain and the carbon–carbon (C–C) bond in the main chain are thermally stable, the copolymer underwent main-chain degradation via β-scission, when the C–B bond was unlocked by dual stimuli. The degradation was controllable by switching the light on and off, while control experiments confirmed the crucial role of the C–B bond in triggering this degradation process.63a Ouchi and coworkers also explored vinyl ether as a comonomer in radical copolymerisation in radical copolymerization of (meth)acrylate via a hydrogen atom transfer (HAT) approach, using UV irradiation combined with heat and benzophenone as the catalyst.63b
While the aforementioned methodologies prioritise polymer modification and degradation, the upcycling of polyacrylate-based polymers into high-value molecules remains unaddressed. To address this gap, protocols facilitating the conversion of these polymers back to their monomeric constituents have been developed. The Summerlin group explored photoassisted radical depolymerisation as a method to recycle polymers back into monomers. Depolymerisation of reversible addition–fragmentation chain transfer (RAFT)-synthesized poly(methyl methacrylate) (PMMA) with various end groups (trithiocarbonate, dithiocarbamate, p-substituted dithiobenzoate) was examined, using UV, blue or green light irradiation. UV light, in particular, significantly accelerated the depolymerisation of their synthesized polymers, achieving up to 87% efficiency within 1 hour at 100 °C. Notably, as the wavelength shifted from the visible range to the UV range, the depolymerisation rate significantly increased. This approach demonstrates promising results not only for designing recyclable materials, but also significantly enhances the potential of polymethacrylate depolymerisation systems to achieve plastic life-cycle circularity.64
In a similar vein, Anastasaki's group demonstrated the light-accelerated depolymerisation of polymethacrylates using Eosin Y as the photocatalyst. Experimental findings revealed that light exposure and ppm concentrations of EY can enhance depolymerisation rates and conversions compared to conventional thermal methods, typically conducted at high temperatures (such as 120 °C). Screening of different solvents and light wavelengths revealed that the combination of green light and Eosin Y resulted in the most substantial enhancement in depolymerisation conversions, while a variety of solvents, which are not typically utilised in such processes, proved compatible with the photocatalytic procedure, with DMSO achieving notably higher depolymerisation conversions (up to 82%) in an 8 h period of time.65 The same group also presented a photocatalytic RAFT-controlled radical depolymerisation method with precise temporal control under visible light for PMMA. This technique achieves a stepwise decrease in molecular weight by enhancing deactivation of polymer chain-ends, allowing controlled depolymerisation at lower temperatures. The approach offers promising opportunities for chemical recycling of polymers, providing valuable mechanistic insights and on-demand access to polymer properties.66 Continuing in a similar fashion, the group, in collaboration with Matyjaszewski, presented a novel photocatalytic ATRP depolymerisation method for poly(benzylmethacrylate) using iron-based catalysts, FeCl2 and FeCl3, at low ppm concentrations. This approach allows temporal control of the depolymerisation rate by switching light on and off, preventing depolymerisation during dark periods, providing high-yield monomer recovery and end-group fidelity preservation at reduced temperatures, compared to thermal depolymerisations.67
Miscellaneous
In this review, we focused on the most fundamental categories of plastics and included less commonly upcycled plastics, such as hydroxylated polymers, PVC, and polyacrylates. Despite significant advances in photochemical upcycling for some polymers, several remain underexplored in this context. For instance, while there are a few documented examples of photochemical upcycling of polyurethanes,43,44 polyamides have virtually no such studies. This is particularly surprising given the widespread use of polyamides in various industries. Limited existing literature primarily addresses the degradation of polyamides via photochemistry rather than their upcycling. Remarkably, FeCl3 has been studied for the degradation of polyamides, highlighting the potential of iron salts to facilitate the upcycling of a wider scope of polymers.68 This promising approach suggests that future research could successfully expand photochemical upcycling methods to include polyamides, addressing a significant gap in sustainable plastic waste management. The potential use of such catalysts could significantly expand the scope of photochemical upcycling, offering new pathways for converting previously underexplored waste into valuable materials. As the field of upcycling is relatively new, exploring and developing these techniques further could lead to more sustainable methods for managing plastic waste, ultimately reducing environmental pollution and contributing to the advancement of a circular economy.Photochemical upcycling processes: commonalities and differences
Categorizing catalysts or methodologies by polymer-type proves challenging, due to the extensive variety utilised for various objectives, resulting in diverse outcomes. While there are no rigid categories, there are identifiable patterns in the selection of photocatalysts based on the chemical structure of the polymer and the desired outcome of the upcycling process. Heterogeneous catalysts dominate the landscape of photochemical upcycling, due to their stability, reusability, and capacity to facilitate complex reactions under light irradiation. Three wide categories of catalysts are recurring in the literature regarding photochemical plastic upcycling, while their preference for such processes stems from their known photochemical properties. These are metal oxides, g-C3N4 and MOFs. Metal oxides, like titanium dioxide (TiO2) and zinc oxide (ZnO), are frequently used, since they efficiently generate electron–hole pairs when exposed to light. These pairs promote photochemical processes, where the electrons typically participate in reduction reactions and the holes facilitate the oxidation of plastic polymers. The versatility of these catalysts lies in their ability to utilise light energy to activate and transform plastic waste, but they typically operate under UV light, limiting their applicability, since UV light irradiation is harsher and requires higher energy than visible light. This limitation has led to the modification of these oxides, providing a variety of protocols that study modified metal–oxide nanoparticles and their application in plastic upcycling. Graphitic carbon nitride (g-C3N4) has emerged as another significant heterogeneous catalyst in photochemical upcycling, since it can absorb visible light, making it a promising candidate for solar-driven processes. g-C3N4 generates electron–hole pairs similarly to metal oxides, where the photogenerated holes oxidize the plastic polymers, contributing to the upcycling process. However, the efficiency of g-C3N4 can be limited by rapid recombination of photogenerated electron–hole pairs, which necessitates further modifications, such as doping with metals or coupling with other semiconductors, to improve its performance. Metal–organic frameworks (MOFs) represent another novel class of heterogeneous catalysts with high surface areas and tunable pore structures, which can be tailored for specific catalytic reactions. MOFs, composed of metal ions coordinated to organic ligands, are utilised in photochemical upcycling, due to their ability to incorporate various metal centers and organic functionalities. This tunability allows for the optimisation of electron–hole generation and separation, enhancing their efficiency. However, the stability of MOFs under reaction conditions can be a concern, and their relatively high cost compared to traditional metal oxides may limit their widespread application.Polyethylene terephthalate (PET) emerges as a significant polymer in the domain of photochemical upcycling, due to its extensive utilisation. Nevertheless, PET poses distinct challenges that require pretreatment before engaging in upcycling procedures. Across most of the majority of the reported procedures, it is clear that PET must undergo hydrolysis to break down its polymer chains into monomers, such as terephthalic acid (TPA) and ethylene glycol (EG), which are more amenable to further transformation. After hydrolysis, terephthalic acid (TPA) remains largely inert to any transformation procedure and is insoluble in most solvents, rendering it a passive component in the process. Introducing an additional pretreatment step in the upcycling process can inadvertently undermine its efficiency and viability, since not only it escalates the energy and resource requirements, but also prolongs the processing duration, potentially compromising the economic feasibility of PET upcycling endeavours. Furthermore, the reliance on hydrolysis as a pretreatment method presents additional hurdles, particularly concerning waste management and the disposal of reaction byproducts. The byproducts generated from hydrolysis, such as residual acids and oligomers, pose environmental concerns and require careful handling and treatment to mitigate their impact. Consequently, while PET remains an attractive feedstock for upcycling initiatives, due to its ubiquity and potential value, the need for extensive pretreatment procedures highlights the urgency of developing alternative approaches. Streamlining the upcycling process by minimising or eliminating pretreatment steps, not only enhances the sustainability and scalability of plastic waste management efforts, but also ensures the competitiveness of PET upcycling as a viable solution to the global plastic pollution crisis. Another prevalent characteristic of processes involving polyethylene terephthalate (PET) is prioritising hydrogen production over selectively generating value-added molecules. Often, for these processes, the polymer is used as a sacrificial agent for hydrogen generation from water splitting, rather than an organic-molecule platform, resulting in complex mixtures of organic acids, without consideration given to their separability. While hydrogen production, especially from PET, has received significant attention in photochemical upcycling, it is crucial to acknowledge the potential for diverse product outcomes from different plastic polymers. The same characteristics are sometimes applicable to polyolefins (PE/PP) and PLA as well, however, it is noteworthy that for the majority of processes, small organic acids, like acetic acid emerge as the primary product.
Conversely, polystyrene (PS) presents unique opportunities for the production of more intricate and valuable products, owing to its distinctive chemical structure. This characteristic has garnered wider attention and focus on PS in the realm of photochemical upcycling. Unlike polyethylene terephthalate (PET), which typically undergoes non-selective transformation of ethylene glycol, PS upcycling offers the potential for a careful conversion to valuable products, such as the oxidation product benzoic acid or styrene monomers, each possessing diverse industrial applications. The distinct chemical composition of PS enables the careful selection and design of processes that involve hydrogen atom transfer (HAT) catalysis, followed by oxidation. This sequential process allows for selective conversion of PS into one or two specific products. Through HAT catalysis, PS can be selectively broken down into radical intermediates, which can then undergo controlled oxidation to yield desired end products. Such precision in product formation is facilitated by the inherent reactivity of PS molecules, bearing a benzylic position. This inherent reactivity allows for more efficient and controlled reactions, making homogeneous catalysis more suitable for PS upcycling, compared to other polymers with less reactive chemical structures. While, this provides a plausible explanation for the preference for homogeneous catalysis in PS upcycling processes, heterogeneous catalysts being engineered with active sites on their surface, can accommodate the diverse chemical structures found in PET, PE, and PP. This adaptability enables heterogeneous catalysts to effectively catalyse reactions in these complex and varied environments. Furthermore, the presence of active sites on the catalyst surface could enhance the accessibility of reactants to these catalytic sites, thereby promoting efficient reaction rates. In the context of upcycling processes involving polymers, such as PET, PE/PP etc., where mixtures and gases are generated through heterogeneous catalysis, diverse approaches are employed for expressing yields. These approaches often involve rates and multiple parameters, rendering the evaluation of upcycling efficacy, particularly concerning organic compounds, and the comparison between methodologies challenging. The expansion on the various conditions of these processes, which may vary across different studies and are occasionally presented as incomplete proof-of-concepts, contributes to a sense of disarray within the literature.
Future opportunities
The rapid advancements in the photochemical upcycling of plastics over the past few years highlight a promising future for this field. One of the most critical areas for future exploration is the expansion of the range of polymers that can be effectively depolymerised under photocatalytic conditions. Currently, the applicability of several approaches is limited to certain polymers. Developing new photocatalysts and optimising reaction conditions to target more polymer categories will be essential. Enhancing the scalability and efficiency of photochemical processes through innovations in flow chemistry will also be pivotal in transitioning from laboratory-scale experiments to industrial applications. Addressing the environmental and practical challenges associated with current methodologies is equally crucial. Many present strategies rely on toxic solvents and catalysts, necessitating the development of more environmentally benign alternatives. There is a pressing need for versatile strategies that enable the direct degradation of commodity polymers, eliminating high-temperature approaches for depolymerisation, which could improve efficiency and reduce energy consumption. Accurately evaluating the effectiveness of processes remains challenging, due to difficulties in analysing their yields. Overcoming these limitations requires innovative solutions that simplify the procedure and improve output yields of products that can readily be utilised for industrial use. Carefully optimising the choice of wavelengths for photochemical reactions and conducting thorough studies on their effects is also essential to further enhance process effectiveness. Many existing methods produce product mixtures that are difficult to separate, and some require acidic or thermal degradation of the polymer, adding extra steps to the process. Integrating photochemical upcycling technologies into existing waste management systems and scaling up processes from laboratory-scale demonstrations to industrial applications are ongoing endeavours. These efforts are essential for promoting resource efficiency, diminishing waste generation and mitigating environmental impacts, ultimately contributing to a more sustainable approach to plastic waste management. Furthermore, the integration of computational approaches for predicting and designing efficient photocatalytic protocols presents an exciting opportunity to accelerate advancements in this field. However, a significant gap remains: most laboratory scientists lack knowledge of industrial-scale machinery and the intricacies of industrial processes. Bridging this gap is crucial for leveraging laboratory innovations into viable industrial applications. Laboratory-scale protocols, while often innovative, frequently lack the applicability required for industrial implementation. This is because many parameters critical to industrial success—such as cost, yield, generation of unwanted byproducts, and toxicity—must be thoroughly considered. Thus, to design and develop effective photocatalytic procedures, it is essential to incorporate an understanding of industrial requirements from the outset. By doing so, we can ensure that new protocols are not only scientifically advanced, but also practical and scalable, paving the way for their adoption in real-world applications and significantly contributing to environmental sustainability.Conclusion
Photochemical upcycling of plastics is a promising area of research aimed at addressing the environmental challenges posed by plastic waste. Until now, significant progress has been achieved in the realm of photochemical conversion of polymers into high-added-value compounds. Initially, researchers have combined innovative photocatalytic materials capable of effectively breaking down plastic polymers when exposed to light. The utilisation of light in these photochemical processes proves highly efficient, as it is demonstrated to be cost-effective, facilitating the decomposition of plastics without necessitating high energy inputs or harsh reaction conditions. Furthermore, these processes can selectively depolymerise plastics into their constituent monomers, instead of entirely converting them into smaller fragments. It is noteworthy that photochemical upcycling of plastics promotes resource efficiency, diminishes waste generation and mitigates environmental impact. Concurrently, endeavours are ongoing to integrate photochemical upcycling technologies into existing waste management systems and industrial processes with researchers endeavouring to scale up photochemical upcycling processes from laboratory-scale demonstrations to industrial applications. Addressing these critical questions and challenges is not only essential for advancing the field of photochemical upcycling of plastics, but also for realising its full potential as a sustainable solution for plastic waste management. Considering its possibilities prompts the question: “Is photochemical upcycling the innovative solution we have been seeking to address the issue of plastic overaccumulation?”Author contributions
C. G. K. planned the manuscript, O. G. M. and E. S. prepared the original draft, C. G. K. assisted in preparing the original draft and made the final editing of the manuscript. The manuscript was written through contributions of all authors.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a grant, which is financed by “Greece 2.0 Basic Research Financing Action (Horizontal support of all Sciences) Sub-action 2 Funding Projects in Leading-Edge Sectors” (grant number 15949).References
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Adv. Mater., 2022, 34, 2100843 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- W. Li, W. Zhao, H. Zhu, Z.-J. Li and W. Wang, J. Mater. Chem. A, 2023, 11, 2503–2527 RSC.
- X. Chen, Y. Wang and L. Zhang, ChemSusChem, 2021, 14, 4137–4151 CrossRef CAS PubMed.
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- K. Faust, P. Denifl and M. Hapke, ChemCatChem, 2023, 15, e202300310 CrossRef CAS.
- M. R. Karimi Estahbanati, X. Y. Kong, A. Eslami and H. S. Soo, ChemSusChem, 2021, 14, 4152–4166 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- Visible Light Photocatalysis in Organic Chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH Verlag GmbH & Co. KGaA, 2018 Search PubMed.
- E. Skolia, O. G. Mountanea and C. G. Kokotos, Trends Chem., 2023, 5, 116–120 CrossRef CAS.
- F. Eisenreich, Angew. Chem., Int. Ed., 2023, 62, e202301303 CrossRef CAS PubMed.
- For selected recent reviews published in 2023–2024, see: (a) Y. Qin and S. Das, Chimia, 2023, 77, 830–835 CrossRef CAS PubMed; (b) J. Ran, A. Talebian-Kiakalaieh, S. Zhang, E. M. Hashem, M. Guo and S.-Z. Qiao, Chem. Sci., 2024, 15, 1611–1637 RSC; (c) L. Wimberger, G. Ng and C. Boyer, Nat. Commun., 2024, 15, 2510 CrossRef CAS PubMed; (d) D. Sajwan, A. Sharma, M. Sharma and V. Krishnan, ACS Catal., 2024, 14, 4865–4926 CrossRef CAS.
- I. Mita, T. Takagi, K. Horie and Y. Shindo, Macromolecules, 1984, 17, 2256–2260 CrossRef CAS.
- M. Wang, J. Wen, Y. Huang and P. Hu, ChemSusChem, 2021, 14, 5049–5056 CrossRef CAS PubMed.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, ACS Catal., 2023, 13, 10968–10975 CrossRef CAS.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Chem, 2022, 8, 2472–2484 CAS.
- Z. Xu, F. Pan, M. Sun, J. Xu, N. E. Munyaneza, Z. L. Croft, G. (G.) Cai and G. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203346119 CrossRef CAS PubMed.
- N. F. Nikitas, E. Skolia, P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Green Chem., 2023, 25, 4750–4759 RSC.
- R. Cao, M.-Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- R. Ghalta, R. Bal and R. Srivastava, Green Chem., 2023, 25, 7318–7334 RSC.
- J. He, L. Han, W. Ma, L. Chen, C. Ma, C. Xu and Z. Yang, iScience, 2023, 26, 10683 Search PubMed.
- Z. Peng, R. Chen and H. Li, ACS Sustainable Chem. Eng., 2023, 11, 10688–10697 CrossRef CAS.
- B. Gautam, M.-R. Huang, C.-C. Lin, C.-C. Chang and J.-T. Chen, Polym. Degrad. Stab., 2023, 217, 110528 CrossRef CAS.
- L. H. Kugelmass, C. Tagnon and E. E. Stache, J. Am. Chem. Soc., 2023, 145, 16090–16097 CrossRef CAS PubMed.
- N. Xu, X. Peng, C. Luo, L. Huang, C. Wang, Z. Chen and J. Li, Adv. Synth. Catal., 2023, 365, 142–147 CrossRef CAS.
- J. Meng, Y. Zhou, D. Li and X. Jiang, Sci. Bull., 2023, 68, 1522–1530 CrossRef CAS PubMed.
- C. Li, X. Y. Kong, M. Lyu, X. T. Tay, M. Đokić, K. F. Chin, C. T. Yang, E. K. X. Lee, J. Zhang, C. Y. Tham, W. X. Chan, W. J. Lee, T. T. Lim, A. Goto, M. B. Sullivan and H. S. Soo, Chem, 2023, 9, 2683–2700 CAS.
- A. Ong, Z. C. Wong, K. L. O. Chin, W. W. Loh, M. H. Chua, S. J. Ang and J. Y. C. Lim, Chem. Sci., 2024, 15, 1061–1067 RSC.
- E. Skolia, O. G. Mountanea and C. G. Kokotos, ChemSusChem, 2024, e202400174 CrossRef PubMed.
- O. G. Mountanea, E. Skolia and C. G. Kokotos, Chem. – Eur. J., 2024, e202401588 CrossRef PubMed.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and T. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- C. Xing, G. Yu, J. Zhou, Q. Liu, T. Chen, H. Liu and X. Li, Appl. Catal., B, 2022, 315, 121496 CrossRef CAS.
- F. Liu, X. Zhuang, Z. Du, Y. Dan, Y. Huang and L. Jiang, Appl. Catal., B, 2022, 318, 121897 CrossRef CAS.
- J. Qin, Y. Dou, F. Wu, Y. Yao, H. R. Andersen, C. Hélix-Nielsen, S. Y. Lim and W. Zhang, Appl. Catal., B, 2022, 319, 121940 CrossRef CAS.
- Y. Zhang, M.-Y. Qi, Z.-R. Tang and Y. J. Xu, ACS Catal., 2023, 13, 3575–3590 CrossRef CAS.
- S. Kong, C. He, J. Dong, N. Li, C. Xu and X. Pan, Macromol. Chem. Phys., 2022, 223, 2100322 CrossRef CAS.
- Z. Zhang, Y. Zhang and R. Zeng, Chem. Sci., 2023, 14, 9374–9379 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- M. Han, S. Zhu, C. Xiang and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- M. Du, Y. Zhang, S. Kang, X. Guo, Y. Ma, M. Xing, Y. Zhu, Y. Chai and B. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- X. Gong, F. Tong, F. Ma, Y. Zhang, P. Zhou, Z. Wang, Y. Liu, P. Wang, H. Xheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121143 CrossRef CAS.
- B. Cao, S. Wan, Y. Wang, H. Guo, M. Ou and Q. Zhong, J. Colloid Interface Sci., 2022, 605, 311–319 CrossRef CAS PubMed.
- S. Zhang, H. Li, L. Wang, J. Liu, G. Liang, K. Davey, J. Ran and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 6410–6419 CrossRef CAS PubMed.
- X. Li, J. Wang, T. Zhang, T. Wang and Y. Zhao, ACS Sustainable Chem. Eng., 2022, 10, 9546–9552 CrossRef CAS.
- Y. Liu, Q. Zhong, P. Xu, H. Huang, F. Yang, M. Cao, L. He, Q. Zhang and J. Chen, Matter, 2022, 5, 1305–1317 CrossRef CAS.
- J.-Q. Yan, D.-W. Sun and J.-H. Huang, Chemosphere, 2022, 286, 131905 CrossRef CAS PubMed.
- F. Wu, Y. Dou, J. Zhou, J. Qin, T. Jiang, Y. Yao, C. Hélix-Nielsen and W. Zhang, Chem. Eng. J., 2023, 470, 144134 CrossRef CAS.
- J. Qin, Y. Dou, J. Zhou, V. M. Candelario, H. R. Andersen, C. Hélix-Nielsen and W. Zhang, Adv. Funct. Mater., 2023, 33, 2214839 CrossRef CAS.
- S. Gazi, M. Đokić, K. F. Chin, P. R. Ng and H. S. Soo, Adv. Sci., 2019, 6, 1902020 CrossRef CAS PubMed.
- S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef CAS PubMed.
- F. Wu, C. Li, Y. Dou, J. Zhou, T. Jiang, Y. Yao, N. Y. Lee, S. Y. Lim, C. Hélix-Nielsen and W. Zhang, Sci. Total Environ., 2023, 902, 165899 CrossRef CAS PubMed.
- J. Qin, Y. Dou, J. Zhou, D. Zhao, T. Orlander, H. R. Andersen, C. Hélix-Nielsen and W. Zhang, Appl. Catal., B, 2024, 341, 123355 CrossRef CAS.
- A. E. Kynman, S. Christodoulou, E. T. Ouellette, A. Peterson, S. N. Kelly, L. Maron and P. Arnold, Chem. Commun., 2023, 59, 10924–10927 RSC.
- K. Parkatzidis, H. S. Wang and A. Anastasaki, Angew. Chem., Int. Ed., 2024, 63, e202402436 CrossRef CAS PubMed.
- A. Adili, A. B. Korpusik, D. Seidel and B. S. Sumerlin, Angew. Chem., Int. Ed., 2022, 61, e202209085 CrossRef CAS PubMed.
- (a) J. B. Garrison, R. W. Hughes and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 441–446 CrossRef CAS PubMed; (b) J. B. Garrison, R. W. Hughes, J. B. Young and B. S. Sumerlin, Polym. Chem., 2022, 13, 982–988 RSC; (c) A. B. Korpusik, A. Adili, K. Bhatt, J. E. Anatot, D. Seidel and B. S. Sumerlin, J. Am. Chem. Soc., 2023, 145, 10480–10485 CrossRef CAS PubMed.
- (a) H. Makino, T. Nishikawa and M. Ouchi, Macromolecules, 2023, 56, 8776–8783 CrossRef CAS; (b) T. Kimura and M. Ouchi, Angew. Chem., Int. Ed., 2023, 62, e202305252 CrossRef CAS PubMed.
- J. B. Young, J. I. Bowman, C. B. Eades, A. J. Wong and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 1390–1395 CrossRef CAS PubMed.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. D. A. Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC.
- V. Bellotti, H. S. Wang, N. P. Truong, R. Simonutti and A. Anastasaki, Angew. Chem., Int. Ed., 2023, 62, e202313232 CrossRef CAS PubMed.
- K. Parkatzidis, N. P. Truong, K. Matyjaszewski and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 21146–21151 CrossRef CAS PubMed.
- Y. Zhong, L. Zhuo and W. Lu, J. Environ. Sci., 2024, 139, 473–482 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |