
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal incorporation: electrochemical, structure, and chemical composition effects on nickel oxyhydroxide oxygen-evolution electrocatalysts†
Raul A.
Marquez
 a,
Emma
Kalokowski
a,
Michael
Espinosa
a,
Jay T.
Bender
b,
Yoon Jun
Son
b,
Kenta
Kawashima
a,
Chikaodili E.
Chukwuneke
a,
Lettie A.
Smith
a,
Hugo
Celio
d,
Andrei
Dolocan
d,
Xun
Zhan
d,
Nathaniel
Miller
c,
Delia J.
Milliron
ab,
Joaquin
Resasco
b and
C. Buddie
Mullins
*abdef
a,
Emma
Kalokowski
a,
Michael
Espinosa
a,
Jay T.
Bender
b,
Yoon Jun
Son
b,
Kenta
Kawashima
a,
Chikaodili E.
Chukwuneke
a,
Lettie A.
Smith
a,
Hugo
Celio
d,
Andrei
Dolocan
d,
Xun
Zhan
d,
Nathaniel
Miller
c,
Delia J.
Milliron
ab,
Joaquin
Resasco
b and
C. Buddie
Mullins
*abdef
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: mullins@che.utexas.edu
bMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA
cDepartment of Geosciences, The University of Texas at Austin, Austin, Texas 78712, USA
dTexas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA
eCenter for Electrochemistry, The University of Texas at Austin, Austin, Texas 78712, USA
fH2@UT, The University of Texas at Austin, Austin, Texas 78712, USA
First published on 6th February 2024
Abstract
Understanding how electrode materials evolve in energy conversion and storage devices is critical to optimizing their performance. We report a comprehensive investigation into the impact of in situ metal incorporation on nickel oxyhydroxide oxygen evolution reaction (OER) electrocatalysts, encompassing four multivalent cations: Fe, Co, Mn, and Cu. We found that adding trace amounts of these cations to alkaline electrolytes alters the electrocatalytic and energy storage properties of NiOxHy films after electrochemical conditioning. As opposed to the well-known increase in OER activity induced by Fe, in situ incorporation of trace Co and Mn cations increases the total capacitance, while Cu incorporation does not proceed. We show that increasing Fe and Co concentrations leads to a maximum electrochemical performance attributed to a saturation threshold in the metal uptake. Depth profiling measurements reveal that metal incorporation is confined to the surface of the film, resulting in an interstratified structure that partially retains the more active, disordered phase at the surface. Building upon solid-state chemistry principles, we provide an in-depth discussion of four critical factors determining the occurrence of in situ metal incorporation, underscoring its nature as a cation exchange process. To further support this concept, we manipulate cation exchange by shifting the solubility equilibrium and via ion complexation. By providing a better understanding of in situ metal incorporation, our results underscore its potential as a strategy for manipulating the surface chemical composition, thus advancing the development of electrochemical energy materials.
Broader contextWith an increasing global population, the demand for high-value chemicals and energy services will continue to grow. Meeting this demand while simultaneously decreasing our impact on the environment is a huge challenge. Electrification offers a promising route for storing and converting renewable electricity into chemicals and energy carriers. Electrocatalysts are central to this transition as they increase chemical reaction rates and selectivities. One notable example is electrochemical water splitting for hydrogen production, a process whose performance is limited by the slow kinetics of the oxygen evolution reaction (OER). Earth-abundant transition metals have shown promise in achieving higher OER rates. Understanding how the chemical structure and composition influence these materials is critical to optimizing their performance. A discovery that revolutionized this field was the dramatic improvement in OER activity due to the intentional, or incidental, incorporation of iron into nickel and cobalt oxyhydroxide electrocatalysts. This finding raised questions about the true active phase in OER materials. While iron incorporation is now widely acknowledged, there is still a need for a comprehensive understanding of the factors governing the in situ incorporation of trace metal cations and their impacts on the electrochemical, structural, and electronic properties of OER electrocatalysts. This study aims to shed light on these aspects and establish a better depiction of in situ metal incorporation as a cation exchange process. |
Introduction
Energy conversion technologies offer a promising avenue for harnessing intermittent renewable energy to produce high-value chemicals and energy carriers.1,2 However, these technologies face significant challenges in enhancing the performance of electrocatalytic reactions. A critical bottleneck for devices relying on water-splitting reactions is the oxygen evolution reaction (OER). The OER performance is hampered by its inherently slow kinetics and the scarcity of suitable metal constituents for use in acidic media.3,4 Conducting the OER in alkaline media allows for using first-row transition metal oxides, which are almost as efficient and stable as precious metals and far more abundant and cost-effective.5–7First-row transition metals also have interesting properties that make them exceptional materials for clean-energy and environmental applications, such as adequate electrical conductivity and high chemical and mechanical stability.8,9 As a result, they have become critical components in various electrochemical energy devices, like alkaline electrolyzers, capacitors, and batteries. Moreover, the wide range of compounds that can be made from these metals offers many physical and electrochemical properties to explore.10 However, while extensive research has gone into enhancing their OER performance, there is still much more to discover, especially regarding how these materials behave under different conditions in systems containing multiple metals.
Transition metal cations are often intentionally added to nickel oxides and hydroxides to tune their electrochemical and structural properties.11,12 The impact of metal incorporation on OER performance has been examined for numerous metals. Ni(OH)2 films co-precipitated with Ce, Fe, and La have proven to be highly active OER electrocatalysts, while co-precipitation with isovalent metals like Cd, Pb, and Zn has resulted in poor performance.13 Furthermore, Co and Mn are typically used to increase the energy storage properties of Ni-based materials. Replacing Ni2+ with other metal cations can affect the discharge potential, cycle stability, and overall charge capacity.14,15 Moreover, incorporating metal cations stabilizes the turbostratic α-phase of nickel hydroxide, which is desirable for energy storage due to its higher charge storage capability.15
The effects of these additives depend on the specific transition metal used. Whereas adding Fe increases the oxidation potential of Ni2+ close to the OER onset,16 Co lowers the average oxidation potential of nickel hydroxide.15–18 Cobalt also improves the electronic conductivity and charge acceptance.15,19 These improvements in electrocatalytic activity and energy storage are believed to be due to structural and electronic effects, which suggests the importance of the local environment. For example, cations with higher oxidation states like Co3+ and Fe3+ have an electron-withdrawing influence on Ni, thereby impacting its redox properties.14,20 These insights highlight how sensitive these Ni-based electrocatalysts are to their local environment, leading to critical changes in their chemistry and functional properties.
Furthermore, it is crucial to investigate how these materials adapt to dynamic environments. During the OER in alkaline media, the initial bulk crystalline transition metal oxide phase undergoes a surface transformation to form its respective oxyhydroxide. These materials are composed of layered two-dimensional nanosheets, which serve as the active phase for OER electrocatalysis.9,21 Because these nanosheets harbor the catalytic sites, studying their structural and chemical transformations under catalytically-relevant environments is crucial. However, investigating these surface changes is experimentally challenging. In contrast to the traditional idea that catalytic environments are static, it is critical to recognize that the electrocatalytic interface is, in fact, highly dynamic and shaped by the immediate environment.22
The electrolyte composition is vital in shaping the intrinsic electrocatalytic activity and selectivity.23,24 A significant breakthrough occurred when Corrigan showed that incorporating trace Fe impurities from the electrolyte into nickel oxide films drastically enhances the OER activity.25 Subsequent studies revealed critical insights to guide the design of transition metal-based OER electrocatalysts. These included elucidation of the significant structural and electronic effects of Fe on NiOOH,26–28 a similar incorporation impact on CoOOH,9 the low OER activity of β-NiOOH,27,29 and evidence of strong catalyst-support interactions that lead to Co incorporation.19 Whether incidental or deliberate, in situ metal incorporation is critical for studying energy materials based on transition metals. Therefore, understanding how metal incorporation impacts nickel oxyhydroxides is invaluable for advancing energy storage and electrocatalysis research.
Here, we investigate in situ transition metal incorporation and its influence on the electrochemical, structure, and chemical properties of NiOxHy electrocatalysts. To provide an in-depth understanding of in situ metal incorporation at the trace level, we investigate four multivalent cations (Fe, Co, Mn, and Cu) relevant to Ni electrochemistry. Our findings reveal the unique effects of each cation on the electrocatalytic and energy storage properties of NiOxHy films and highlight the comparatively limited incorporation of Mn and Cu in contrast to Fe and Co. We demonstrate that in situ metal incorporation is confined to the surface, leading to the formation of an interstratified structure with distinct electronic and chemical properties. We discuss the factors that influence metal incorporation as a cation exchange process. Finally, we show that in situ metal incorporation can be manipulated by adjusting the solubility equilibria and using ion complexation, demonstrating its flexibility.
Experimental
Electrocatalytic film preparation and characterization
Typical NiOxHy films were electrodeposited on Ni foil (NF) substrates to avoid interference from substrate effects.19,30–32 Prior to deposition, substrates were cleaned in ethanol and 3 M HCl under sonication, rinsed with 18.2 MΩ cm deionized water, and dried. Films were cathodically deposited using a two-electrode setup and a 20 mM nickel(II) nitrate aqueous solution (pH ∼ 3).9,13,27,31 A typical film had a thickness of ∼550 nm, resulting from deposition at −1 mA cm−2 for 300 s (charge: 300 mC cm−2). As suggested by the Gewirth group, electrodeposition with consistent uniformity was carried out in three cycles of 100 s each, separated by rest intervals of 10 s to minimize concentration gradients.33 When co-depositing Fe and Co into the films, the total metal concentration in the solution was maintained at 20 mM. For Fe-containing films, FeCl2·4H2O and NaNO3 were used as the source materials, whereas cobalt(II) nitrate was utilized for the co-deposition of Co.27,31Thin films (charge: 50 mC cm−2, ∼80 nm thick) were deposited specifically for surface-enhanced Raman spectroscopy (SERS) and coulovoltammetry (QV) measurements. Films for SERS were also deposited on roughened Au substrates. To identify these types of electrodes, the electrode naming scheme consists of the substrate (NF or Au) followed by the metal composition and phase. Metal hydroxides denote as-deposited or aged films, while oxyhydroxides (e.g., NiOOH) specifically denote films conditioned electrochemically. Thin films are denoted by a “t” preceding their chemical composition.
Film thickness and roughness measurements of the as-deposited films were conducted via non-contact profilometry (NCP) with a Keyence VK-X1100 optical profilometer. The electrochemically active surface area (ECSA) of the NF substrate was estimated by measuring the double-layer capacitance (Cdl) using cyclic voltammetry (CV) scans. Measurements were conducted following previous guidelines.34,35 Further details on substrate pretreatment, deposition, electrode nomenclature, and film characterization can be found in the ESI.†
Electrochemical measurements
Measurements were conducted using a Gamry Reference 620 potentiostat/galvanostat and a three-electrode cell made of polytetrafluoroethylene (PTFE). A graphite rod (Gamry) served as the counter electrode to avoid incidental Pt incorporation.36 All potentials were measured against the Hg/HgO reference electrode. The electrode was calibrated and converted to the reversible hydrogen electrode (RHE).37 The KOH electrolyte was prepared, standardized, and purified in line with previous guidelines.27,38 The electrolyte was purged with high-purity O2 for 30 min before testing, and a constant flow was maintained in the headspace throughout electrochemical testing.Electrodes were conditioned electrochemically via chronopotentiometry (CP) at 2.5 mA cm−2 for at least 12 h to form a thin and compact hydrous oxide layer on the surface.39 Linear sweep voltammetry (LSV) and CV scans were recorded at a scan rate of 5 mV s−1 and corrected for 85% uncompensated resistance (Ru) using the positive feedback mode.40,41Ru was determined using electrochemical impedance spectroscopy (EIS) by identifying the minimum point of total impedance at high frequencies (Fig. S1, ESI†).27,28,31 The OER overpotential (η) was calculated using the equation η = Ei − Erev where Ei is the measured potential (corrected for 85% Ru) and Erev is the OER reversible potential (0.306 V vs. Hg/HgO).26 Tafel slopes were estimated via multistep chronoamperometry (MUSCA) and chronopotentiometry (MUSCP),42 with corrections made for 100% iR drop using Ru values from EIS (typically 2.2 ± 0.2 Ω).
To examine the energy storage properties, electrodes were conditioned electrochemically via galvanostatic charge–discharge (GCD) at 2.5 mA cm−2 for 1600 cycles. For each GCD cycle, a constant current was applied to charge/discharge the electrode between the starting and full-charge potentials, the latter corresponding to the OER onset potential determined via QV (more details in the ESI†).43 The total capacitance (Ctot) was estimated from GCD using the Gamry Echem Analyst software. CV scans at varying sweep rates were conducted to determine the energy storage behavior.44 All currents were normalized to the projected geometric area, determined using NCP.
Potentiostatic EIS measurements were performed to identify the charge transfer resistance (Rct) linked to the OER and the Cdl. EIS was measured in the frequency range from 0.1 to 106 Hz at an amplitude of 5 mV from 0.50 to 0.75 V vs. Hg/HgO. A modified Randles equivalent circuit was fitted to estimate the Rct and Cdl values (Fig. S2, ESI†).45 The Cdl was subtracted from Ctot to determine the redox capacitance (Credox). Detailed testing conditions are outlined in the ESI.†
Material characterization
Grazing incidence X-ray diffraction (GIXRD) patterns were recorded on a Rigaku Ultima IV diffractometer equipped with a Cu Kα radiation source and set to a thin-film configuration with an incident angle of 0.3°. Scanning electron microscopy (SEM) images and energy-dispersive X-ray spectroscopy (EDX) elemental maps were captured using a Thermo Scientific Apreo 2 microscope at a 10 kV accelerating voltage. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging and electron energy loss spectroscopy (EELS) measurements were conducted in a JEOL NEOARM probe-corrected microscope with aberration correction at an operating voltage of 80 kV.Time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profiles and high-resolution elemental maps were acquired with an ionTOF GmbH TOF.SIMS 5 instrument. For depth profiling in negative polarity, a Cs+ beam (500 V, 40 nA) was used for sputtering 300 × 300 μm2 areas, followed by a Bi+ analysis beam to scan 100 × 100 μm2 areas at the crater's center. An O2+ sputtering ion beam (1 kV, 40 nA) was used instead of Cs+ for depth profiling in positive polarity.
X-ray photoelectron spectroscopy (XPS) analyses were conducted on a PHI VersaProbe 4 instrument using a Mg Kα source to avoid peak overlap issues between Ni LMM Auger and Fe 2p peaks.27 The charge neutralizer was intentionally not used for post-conditioned samples to prevent partial reduction of the NiOOH phase. Binding energy was calibrated using the C 1s peak of adventitious hydrocarbons at 284.8 eV, and spectral fitting followed established methods.46–48
Raman spectroscopy measurements were performed in a Horiba LabRAM ARAMIS confocal Raman microscope operating with 532 and 633 nm lasers. SERS experiments were conducted on Au screen-printed electrodes (Metrohm DropSens, 220BT). Solution-mode inductively coupled plasma mass spectrometry (ICP-MS) analyses were conducted on an Agilent 7500ce spectrometer equipped with a quadrupole mass analyzer and a collision/reaction cell. We employed a special KOH electrolyte dilution routine, as outlined previously.38 For more details on characterization conditions and sample preparation protocols, please refer to the ESI.†
Results and discussion
OER activity varies depending on the incorporated trace metal
Ni(OH)2 films deposited on Ni foil substrate consist of randomly distributed grains approximately 5 μm in diameter and 550 nm in thickness (Fig. S3a–d, ESI†). SEM images of the surface reveal a nanosheet morphology (Fig. S3e, ESI†), mainly composed of Ni and O from EDX elemental mapping (Fig. S3f, ESI†). The GIXRD pattern of as-deposited NF–Ni(OH)2 corresponds with that of α-Ni(OH)2, with clear additional reflections from the Ni foil substrate (Fig. S3g, ESI†). These characteristics align with those seen in layered double hydroxides (LDHs), which feature stacked layers of edge-sharing NiO6 octahedra separated by water and charge-balancing anions.27 The film's morphology differs significantly from the Ni foil substrate, which exhibits a roughness factor of 1.04, relative to its projected geometric area according to ECSA measurements (Fig. S4a and b, ESI†).NF–NiOOH electrodes were tested in purified 1 M KOH electrolyte. ICP-MS analysis of the purified electrolyte confirms the removal of Fe and Co impurities (Fig. S5, ESI†). Anodic LSV and CV scans were obtained before and after electrochemical conditioning (Fig. S6a and b, ESI†). The OER current decreases after conditioning in purified KOH. Simultaneously, electrochemical signatures characteristic of a mixture of γ- and β-NiOOH were observed.26,38 The Ni2+/3+ redox peak current is notably higher than that of the NF substrate (Fig. S4c and d, ESI†), indicating that the NiOxHy film primarily contributes to the observed response. The average OER Tafel slope measured from MUSCA and MUSCP was approximately 115 mV dec−1 (Fig. S6c-f, ESI†), in line with values reported for Fe-free KOH.26,45 The electrochemical performance metrics for each testing condition discussed in this section are summarized in Table S1 of the ESI.†
As shown in Fig. 1a, the OER currents vary significantly after conditioning NF–NiOOH electrodes in purified 1 M KOH electrolyte spiked with 100 ppb of Mn+ (Mn+: Fe3+, Co2+, Mn2+, and Cu2+). Adding Fe to the KOH electrolyte yields the lowest OER overpotential (i.e., ∼330 mV) to reach a current density of 10 mA cm−2 (Fig. 1b). In contrast, the overpotentials for electrodes exposed to Co, Mn, and Cu cations are statistically indistinguishable. From LSV scans before and after conditioning (Fig. S7, ESI†), only Fe incorporation results in a substantial enhancement of the OER activity, and the current increase obscures the peak at ∼1.54 V vs. RHE, which results from overcharging the β phase into γ-NiOOH.26,38
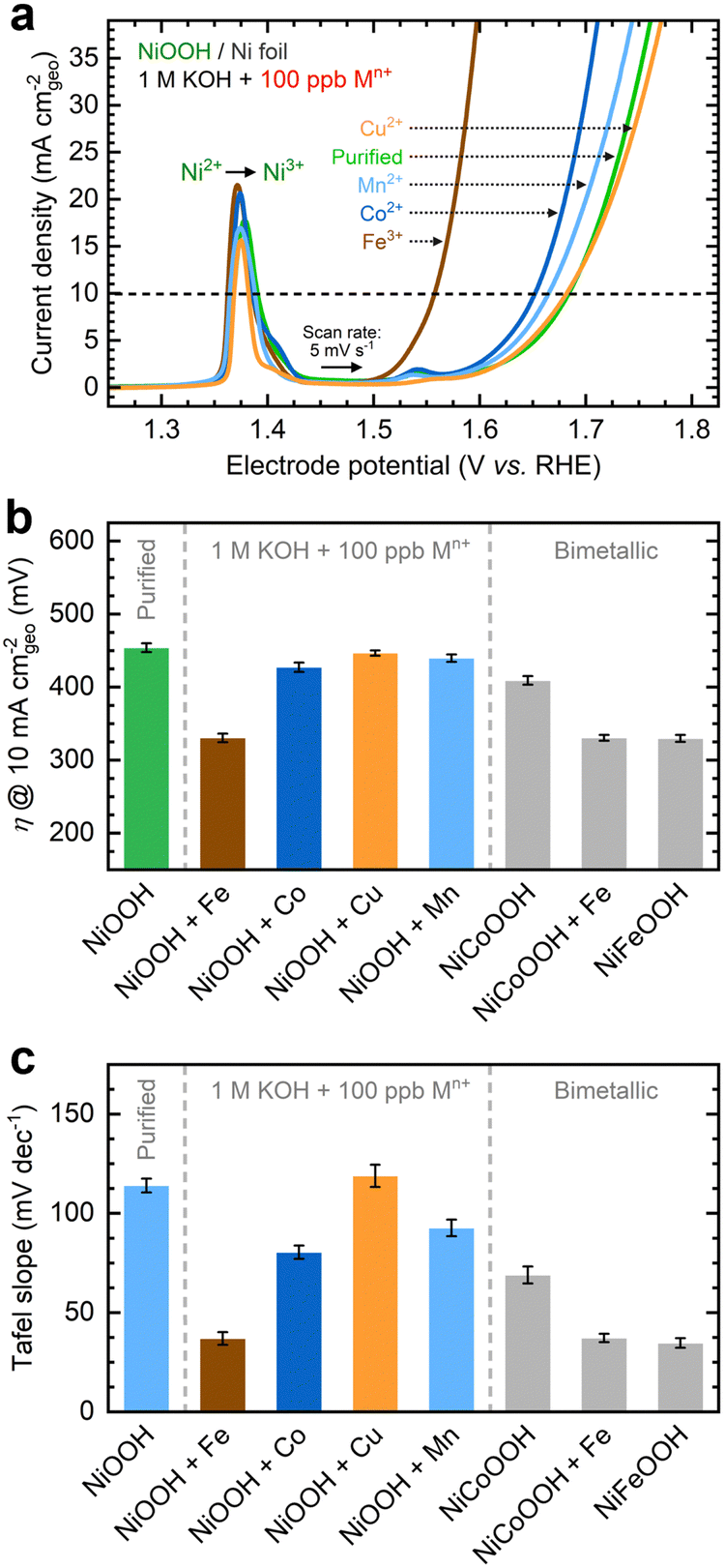 | ||
Fig. 1 OER performance of NF–NiOOH electrodes after CP conditioning in purified 1 M KOH electrolyte spiked with transition metal cations (100 ppb Mn+): (a) linear sweep voltammograms showing variations of the OER current with transition metal cations; (b) corresponding OER overpotentials at 10 mA cmgeo−2; (c) average Tafel slopes determined from MUSCA and MUSCP. Bimetallic hydroxide films (Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M ratio = 1:1) are included as control samples in (b) and (c). :M ratio = 1:1) are included as control samples in (b) and (c). | ||
Using the QV method,43 the OER onset potential for NF-t-NiOOH electrodes conditioned in purified KOH was found to be approximately 1.57 V vs. RHE (Fig. S8, ESI†), and it overlaps with the Ni overcharging peaks (see supporting note of Fig. S6, ESI†). Films conditioned in KOH electrolyte containing Co, Mn, and Cu cations exhibit similar OER onsets, while Fe incorporation shifts the onset potential cathodically to 1.45 V vs. RHE (Fig. S9, ESI†). Without QV measurements, it is challenging to estimate the OER onset obscured by the Ni2+/3+ redox peak, which shifted anodically with Fe incorporation.
Contrary to prior work in which films were deposited by metal co-precipitation or incorporation via CV cycling, we do not observe a significant Ni2+/3+ redox peak shift after CP conditioning (Fig. 1a). We attribute this result to a less pronounced bulk incorporation into our relatively thick films, suggesting that metal incorporation is restricted to a thin oxyhydroxide layer on the surface, leaving the composition of the inner bulk material unchanged.13,18,39 Note that CV scans of thin films (∼80 nm) exclusively used for QV measurements exhibit a redox peak shift (Fig. S9, ESI†), implying that metal incorporation has a more pronounced effect on thin films, allowing cations to infiltrate more fully into the bulk material.39
As displayed in Fig. 1c, the OER Tafel slopes vary with the choice of transition metal cation. Incorporating Fe, Co, and Mn cations results in Tafel slopes of approximately 37, 80, and 93 mV dec−1, respectively. In contrast, Cu incorporation yields a Tafel slope of ∼110 mV dec−1, statistically indistinguishable from that of the film conditioned in purified KOH. NF–NiFeOOH and NF–NiCoOOH electrodes tested in purified and Fe-containing KOH electrolytes show OER overpotentials (Fig. 1b) and Tafel slopes (Fig. 1c) consistent with those of NF–NiOOH electrodes conditioned electrochemically in KOH containing Fe or Co cations. Contrary to previous results,12 conditioning NiCoOOH in Fe-containing KOH lowers the OER overpotential and Tafel slope to be close to those of Fe-containing films, indicating that Fe incorporation enhances the OER regardless of the film.
Incorporating Co and Mn enhances the energy storage properties
Next, we investigated the energy storage properties of NF–NiOOH electrodes conditioned in purified KOH electrolyte spiked with transition metal cations. The energy storage behavior was examined by obtaining CV scans at multiple sweep rates. As shown in Fig. S10 and Table S1 of the ESI,† all films exhibit a b value close to 0.55. This finding indicates that charge storage in NF–NiOOH follows a battery-type behavior and is not pseudocapacitive, according to the peak current's linear correlation with the square root of the scan rate (i ∼ ν0.5).44,49Fresh NF–NiOOH electrodes were conditioned through GCD cycling. As shown in Fig. 2a, the charge–discharge profiles after 1600 cycles differ for films conditioned with different transition metal cations. Cycling in Fe-containing KOH shortens the GCD profile compared to purified KOH electrolyte, while Mn- and Co-containing KOH extend it. Fig. 2b highlights the full charge and half-discharge potentials derived from the GCD profiles in Fig. 2a. Using the OER onset potential as a starting value, we adjusted the full charge potential to achieve close to 100% coulombic efficiency, minimizing OER interference. As displayed in Fig. 2a, cycling in Fe-containing KOH, which leads to Fe incorporation into the NiOxHy film, requires a lower full charge potential, limiting the charge extracted from the redox reaction. In contrast, the full charge potential for electrodes cycled in purified KOH containing Co, Mn, and Cu cations remains consistent, aligning with the OER onset potential estimated from QV. The half-discharge potential was determined from the discharge curve. Fe incorporation increases the half-discharge potential, while Co lowers it, in line with previous research.14–17 Moreover, while Co is followed closely by Mn, Cu does not lead to significant differences compared to films conditioned in purified KOH. Thus, Co- and Mn-containing films allow greater charge storage before reaching the OER onset.
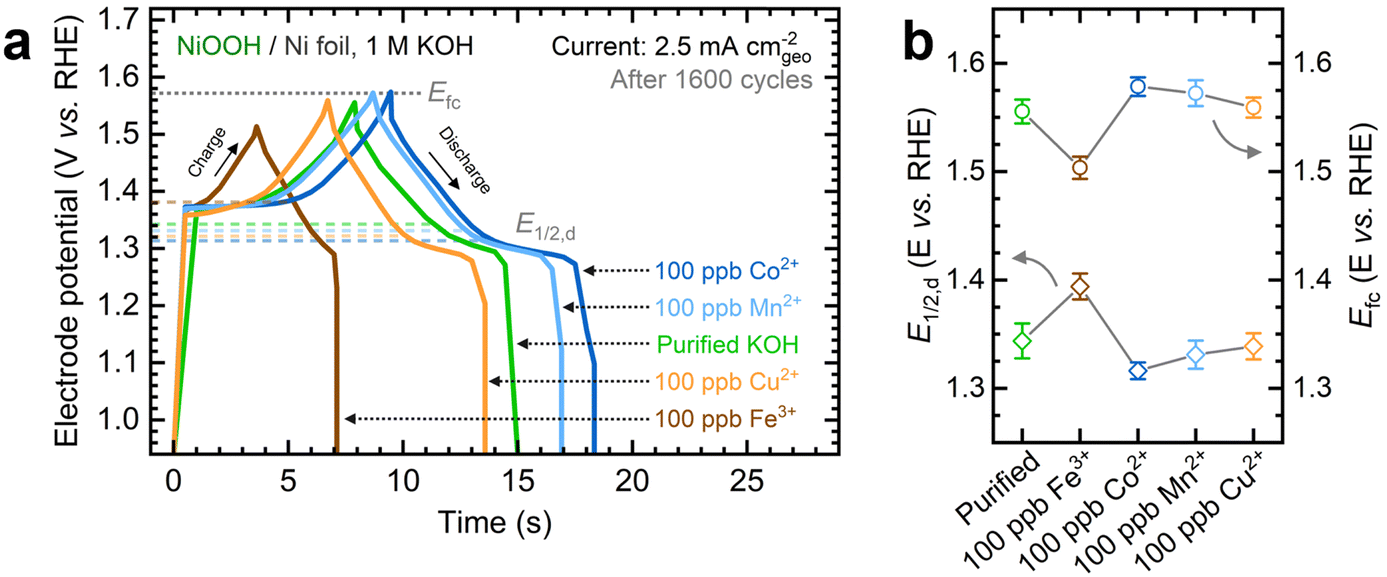 | ||
| Fig. 2 Influence of in situ trace metal incorporation on the energy storage properties of NF–NiOOH electrodes: (a) galvanostatic charge–discharge profiles at 2.5 mA cmgeo−2 after 1600 charge–discharge cycles; (b) comparison of the half-discharge (E1/2,d) and full-charge (Efc) potentials after GCD conditioning in purified 1 M KOH electrolyte spiked with transition metal cations (100 ppb Mn+). Uncertainty bars indicate the standard deviation derived from three replicate measurements. | ||
We used GCD conditioning to estimate additional energy storage metrics, including total capacitance, coulombic efficiency, capacity retention, and energy efficiency. Conditioning NF–NiOOH electrodes in purified KOH leads to a marked drop in the total capacitance and capacity retention within the first 200 cycles (Fig. S11 and Table S1, ESI†). This behavior is characteristic of the α/γ to β/β transition in Ni(OH)2/NiOOH, as less charge can be extracted from the more compact β phases formed after aging in alkaline media.16,38 Although electrodes conditioned in transition metal-containing KOH also exhibit an initial decrease in the total capacitance (Fig. S12, ESI†), the final value varies based on the incorporated cation. Conditioning in Co- and Mn-containing KOH results in a capacity retention 25% higher than in purified KOH, while Fe-containing KOH yields about 30% lower retention. These findings indicate that the in situ incorporation of trace metals affects the energy storage properties after GCD conditioning.
We conducted EIS analysis to estimate the Rct and Cdl after conditioning NF–NiOOH electrodes. Nyquist and Bode plots for electrodes conditioned in metal-containing KOH show clear differences (Fig. S13, ESI†). The Rct, which reflects the kinetics of the OER and is associated with the diameter of the semicircle in the lower-frequency range, decreases upon incorporating Fe, Co, and Mn. In contrast, the Rct values for Cu-containing and purified KOH electrolytes are comparable (Table S1, ESI†). These findings are consistent with trends observed for the OER overpotential and demonstrate that only Fe incorporation enhances the OER kinetics.
We subtracted the Cdl obtained via EIS analysis from the Ctot derived from GCD conditioning to estimate the redox contribution, Credox. Fig. 3a depicts the separate contributions of Credox and Cdl to the total capacitance for NF–NiOOH electrodes conditioned in KOH electrolyte containing different transition metal cations, whereas Fig. 3b highlights their relative contributions. Cdl values for NF–NiOOH electrodes conditioned in both purified and metal-spiked KOH are statistically similar, indicating no significant alteration in the surface area. This result implies that trends in geometric OER activity are not influenced significantly by differences in the ECSA (Fig. 1). Among the metal cations tested, incorporating Co yields the most significant increase of Credox, followed closely by Mn. As shown in Fig. 3b, the Credox contribution to the total capacitance in NF–NiOOH electrodes represents approximately 85% and 80% after in situ incorporation of trace Co and Mn, respectively. In contrast, films conditioned with Fe, Cu, and purified KOH show no appreciable change in Credox.
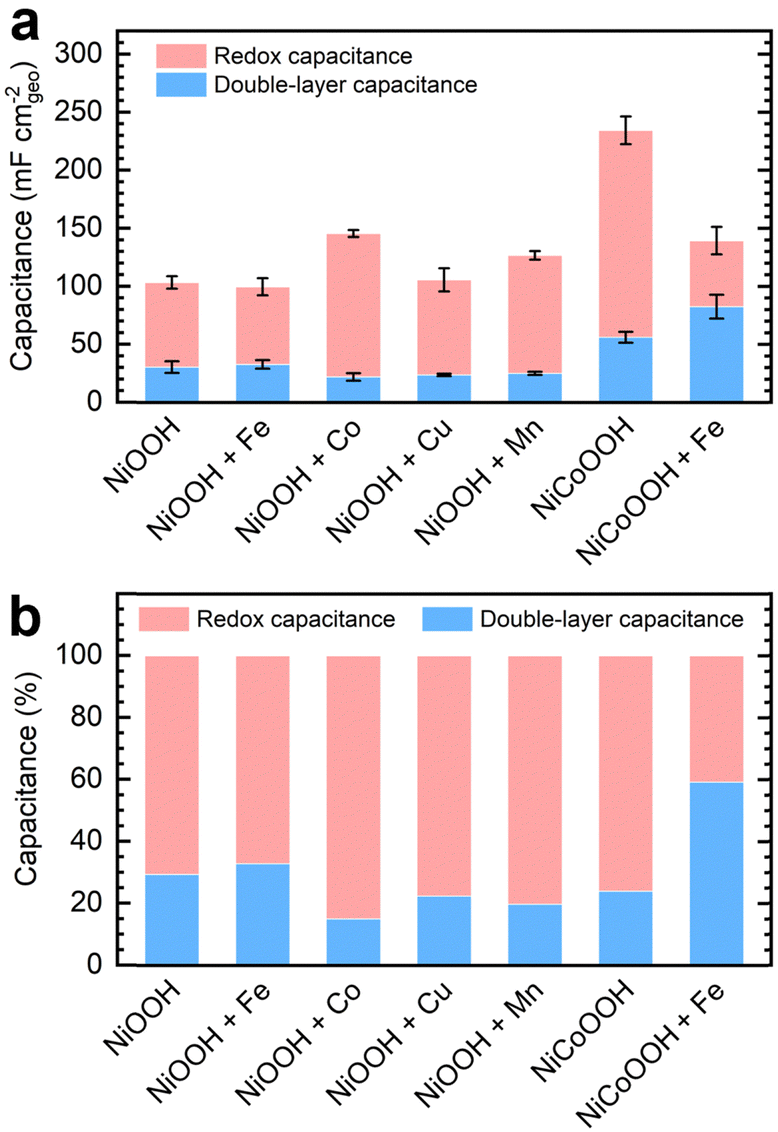 | ||
| Fig. 3 (a) Capacitance contributions of NF–NiOOH electrodes after GCD conditioning in purified 1 M KOH electrolyte spiked with transition metal cations (100 ppb Mn+); (b) relative contribution of each type of capacitance (Credox and Cdl) to the total capacitance (Ctot). | ||
Additionally, we evaluated the energy storage properties of control NF–NiCoOOH electrodes. The capacitance contributions for the control NF–NiFeOOH electrode are not reported due to the overlapping of the Ni2+/3+ redox peak with the OER current (Fig. S14, ESI†). The NF–NiCoOOH electrode exhibits the most significant improvement (Fig. 3 and Fig. S15, ESI†), which is consistent with the benefits of using Co as an additive for energy storage materials.14,15 However, despite increasing the OER activity (Fig. S16, ESI†), Fe incorporation drastically decreases the Credox for the NF–NiCoOOH electrode. This finding confirms that in situ Fe incorporation, even at the trace level, is counterproductive for energy storage applications.
In situ Fe and Co incorporation ceases to enhance electrochemical performance beyond a saturation threshold
Fe and Co exhibited the most significant electrochemical performance effects on NiOxHy films among the transition metals examined in this study. To expand upon these observations, we evaluated the effects of increasing the Fe and Co concentrations on measured catalytic and energy storage properties. Focusing first on the impact of Fe incorporation, NF–NiOOH electrodes were conditioned in purified 1 M KOH spiked with Fe concentrations ranging from 100 to 1000 ppb (Fig. S17a, ESI†). As shown in Fig. 4a, increasing the Fe concentration enhances the OER activity, nearly stabilizing at 1000 ppb. Similar to Fig. 1a, there is no shift of the Ni redox peak with increasing Fe concentration, implying that Fe incorporation remains restricted to the surface.39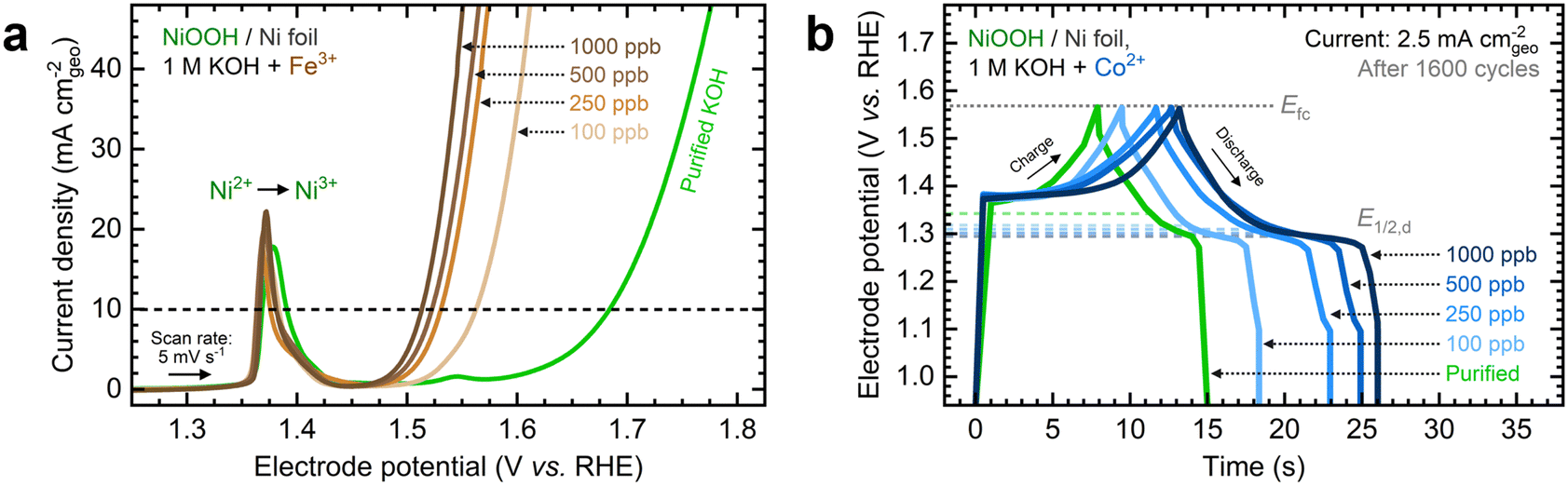 | ||
| Fig. 4 Influence of the transition metal cation concentration on the electrochemical performance of NF–NiOOH electrodes: (a) linear sweep voltammograms showing the systematic increase of the OER current with Fe3+ concentration; (b) GCD curves showing the increase of the total capacitance with Co2+ concentration. | ||
The steady-state overpotential from CP conditioning shows a plateau above 500 ppb Fe (Fig. S17b, ESI†). EIS measurements align with these trends (Fig. S17c and d, ESI†), with 1000 ppb Fe resulting in the lowest Rct. OER overpotentials at 10 mA cm−2 and Tafel slopes are also statistically similar above 500 ppb (Fig. S17e and f, ESI†). These findings highlight the critical effect of in situ incorporation of trace metals; even 100 ppb of Fe results in a dramatic increase in OER activity, while further addition of Fe does not improve the OER significantly.
Next, we studied the impact of increasing the Co concentration on the energy storage properties of NF–NiOOH electrodes. GCD profiles depicted in Fig. 4b reveal that increasing the Co concentration leads to lower half-discharge potentials, thereby enabling more charge extraction at the same full-charge threshold. Adding more Co does not enhance the OER; in fact, the OER overpotential increases when Co levels exceed 100 ppb (Fig. S18a, ESI†). Furthermore, the total capacitance obtained after GCD conditioning increases with the Co concentration (Fig. S18b, ESI†). EIS analyses align with OER trends (Fig. S18c and d, ESI†). The OER overpotentials at 10 mA cm−2 and Tafel slopes remain relatively consistent across different Co concentrations (Fig. S19a and b, ESI†), with the data for 1000 ppb Co closely resembling purified KOH. Regarding capacitance contributions calculated from GCD (Fig. S19c and d, ESI†), adding more Co to the electrolyte increases Credox without modifying Cdl until Credox stabilizes above 500 ppb Co. A summary of these figures of merit at increasing cation concentrations is provided in Table S2 in the ESI.†
These findings reveal that adding Fe and Co to the KOH electrolyte improves the electrochemical properties of NiOxHy films only up to a certain point. Beyond this threshold, the metal cation uptake is likely hindered by the saturation of surface sites and the segregation of less active phases. This concept has been noted previously for in situ Fe incorporation and co-deposited NiFe catalysts.26,50 Our results confirm that, in addition to Fe incorporation, this saturation limit also applies to in situ incorporation of trace Co, leading to significant effects on the energy storage properties of NiOxHy films.
Previous research has underscored the role of metal compositions in maximizing the electrochemical performance of NiOxHy. Louie and Bell showed that OER activity varies with Fe content in co-deposited NiFe films, with the lowest overpotential observed at 40% Fe.28 Klaus and coworkers noted a decrease in turnover frequency after exceeding 11% Fe content, leading to the formation of a less active, Fe-rich phase.26 Friebel et al. reported a maximum OER activity at 25% Fe, beyond which further Fe co-deposition resulted in the segregation of an inactive γ-FeOOH phase.50 Görlin et al. found peak OER activity at 50% Fe in bimetallic NiFe nanoparticles.51 Furthermore, exceeding 45% co-precipitated Co in NiOOH leads to significant capacity decay.12
Prior studies using bulk NiFe catalysts report higher Fe contents and volcano plot-like OER activity trends with Ni:Fe composition.28,50,51 In contrast, our findings reveal a performance plateau at trace Fe and Co concentrations (<1 ppm). Similarly, Klaus et al. reported a performance decay and critical saturation at lower Fe contents (11% Fe) after aging NiOxHy films with Fe present at <1 ppm in the KOH electrolyte.26 We attribute these contrasting results to the metal incorporation mechanism, whether through bulk synthesis or in situ incorporation of dissolved cations. It has been claimed that OER catalysis should be considered as a volume, not surface, process based on experiments involving bulk NiFe catalysts.51 However, we argue that the electrochemical performance, whether surface-restricted or volume-restricted, depends on the location of the incorporated metals. The solubility of Fe and Co in the Ni host appears to differ between bulk and surface incorporation, which could explain the lower saturation limit for the latter. This claim is supported by understanding in situ incorporation as a cation exchange process that creates an interstratified structure confined to the surface (vide infra).
In situ metal incorporation induces the formation of an interstratified structure at the surface of the film
We investigated the influence of in situ metal incorporation on the structure of NiOxHy films. First, we utilized Raman spectroscopy to study the structural disorder in Ni(OH)2 polymorphs induced by metal incorporation. The O–H stretching modes can serve as fingerprints to estimate the proportions of α and β phases influenced by impurities and defects.52,53 We probed this spectral region using NF–Ni(OH)2 electrodes as the hydroxyl band is absent in conditioned samples (i.e., NF–NiOOH).54 We placed the as-deposited NF–Ni(OH)2 electrodes in purified KOH electrolyte spiked with transition metal cations for one week to allow incorporation into Ni(OH)2; this process is referred to as aging.26,27 As shown in Fig. S20 (ESI†), the α and β phases are easily discernible from their characteristic internal stretching modes at 3642 and 3580 cm−1, respectively.The Raman spectra of the electrodes upon aging are shown in Fig. 5a and b. The pristine film mainly exhibits a broad peak corresponding to the α-phase at 3642 cm−1, while clear signals for the β-phase are absent. The aged films display distinct β-phase peaks alongside reduced α-phase peaks. The intensity of these peaks varies depending on the specific transition metal cation used for aging, a variation most clearly observed when considering the ratio between the 3580 and 3642 cm−1 bands for each sample (Fig. 5c). Co-incorporated films exhibit the lowest β/α peak ratio, followed closely by films aged with Fe and Mn. Thus, a greater proportion of the disordered α-phase is retained after aging in KOH spiked with Co, Fe, and Mn. In contrast, the phase proportions in films aged with Cu remain unchanged compared to those aged in purified KOH, reaffirming the earlier finding that Cu does not readily incorporate into NiOxHy films.
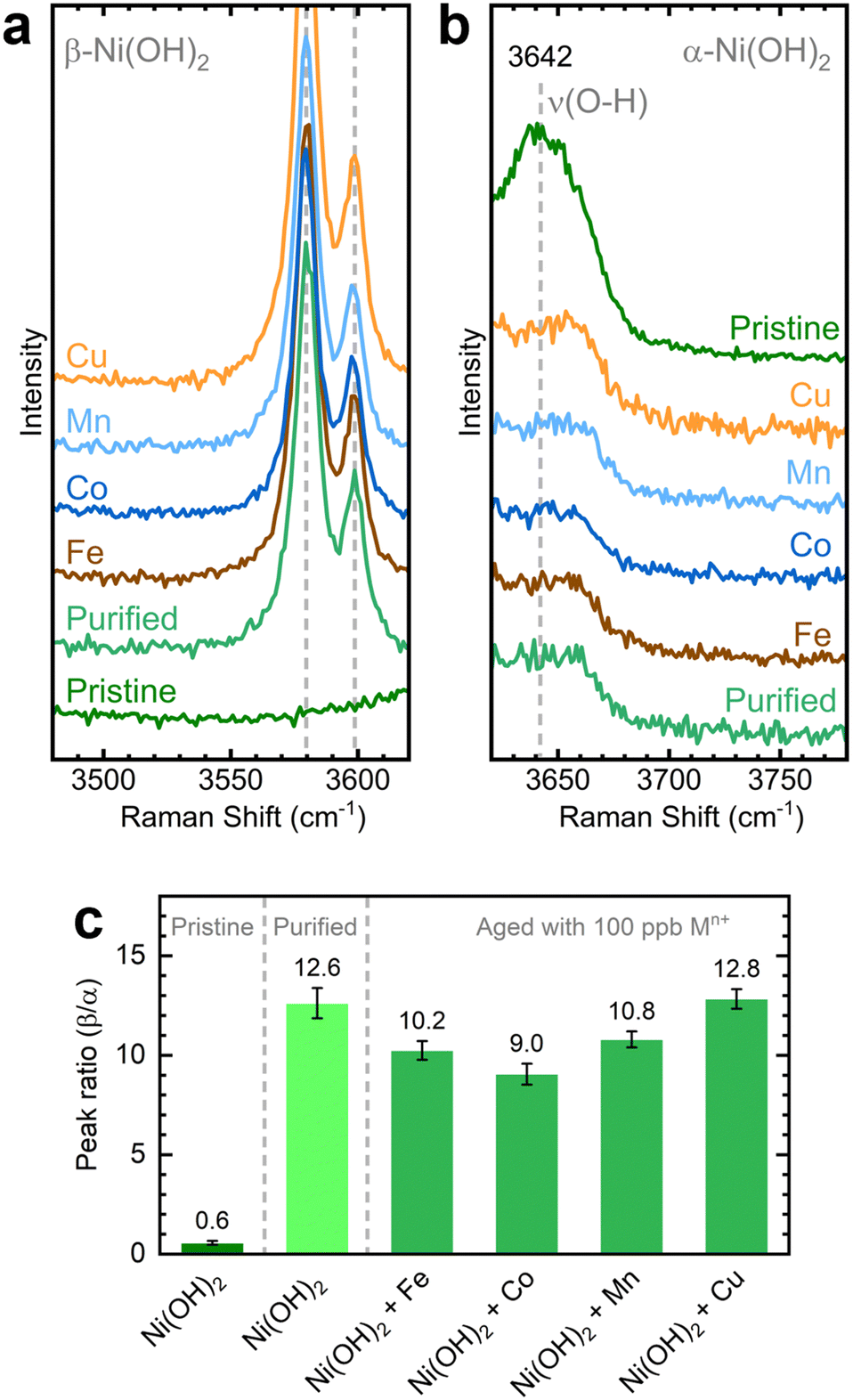 | ||
| Fig. 5 Raman spectra of the (a) β-Ni(OH)2 and (b) α-Ni(OH)2 regions collected over NF–Ni(OH)2 electrodes after aging for 7 days in purified 1 M KOH spiked with different transition metal cations (100 ppb Mn+); (c) corresponding ratios of the β-Ni(OH)2 and α-Ni(OH)2 peaks across different transition metal cations. Spectra are offset on the y-axis for clarity. Uncertainty bars indicate the standard deviation derived from five replicate measurements. Laser: 532 nm. | ||
Next, we conducted TOF-SIMS analysis in negative polarity on NF–NiOOH electrodes after CP conditioning in Fe-, Co-, and Mn-containing KOH to gain deeper insights into the spatial distribution of transition metals confirmed to incorporate from alkaline electrolytes. TOF-SIMS was used to create depth profiles of secondary ion fragments that were ejected following the sputtering of the NiOxHy film. NCP was used to estimate the sputtering rate and approximate depths (Fig. S21, ESI†).
As displayed in Fig. 6a, 2D maps and 3D depth profile renderings of secondary Ni− and Fe− ion fragments show that Fe predominantly resides in the NiOxHy grains (i.e., red “islands” in Fig. 6a) and is primarily confined to the surface. The depth profile in Fig. 6b shows a gradual decrease in the concentration of the Fe− fragment beyond ∼50 nm, while the levels of Ni-based ion fragments remain consistent. Similarly, Co incorporation is restricted to the surface, as shown in Fig. 6c, with the concentration of the Co− fragments decreasing drastically within the initial 30 nm of the film (Fig. 6d). Moreover, mappings and depth profiles of Mn-incorporated films exhibit a similar distribution for Mn− ion fragments (Fig. S22, ESI†). Data acquired in positive polarity corroborate the surface-confined incorporation of Fe, Co, and Mn cations (Fig. S23, ESI†).
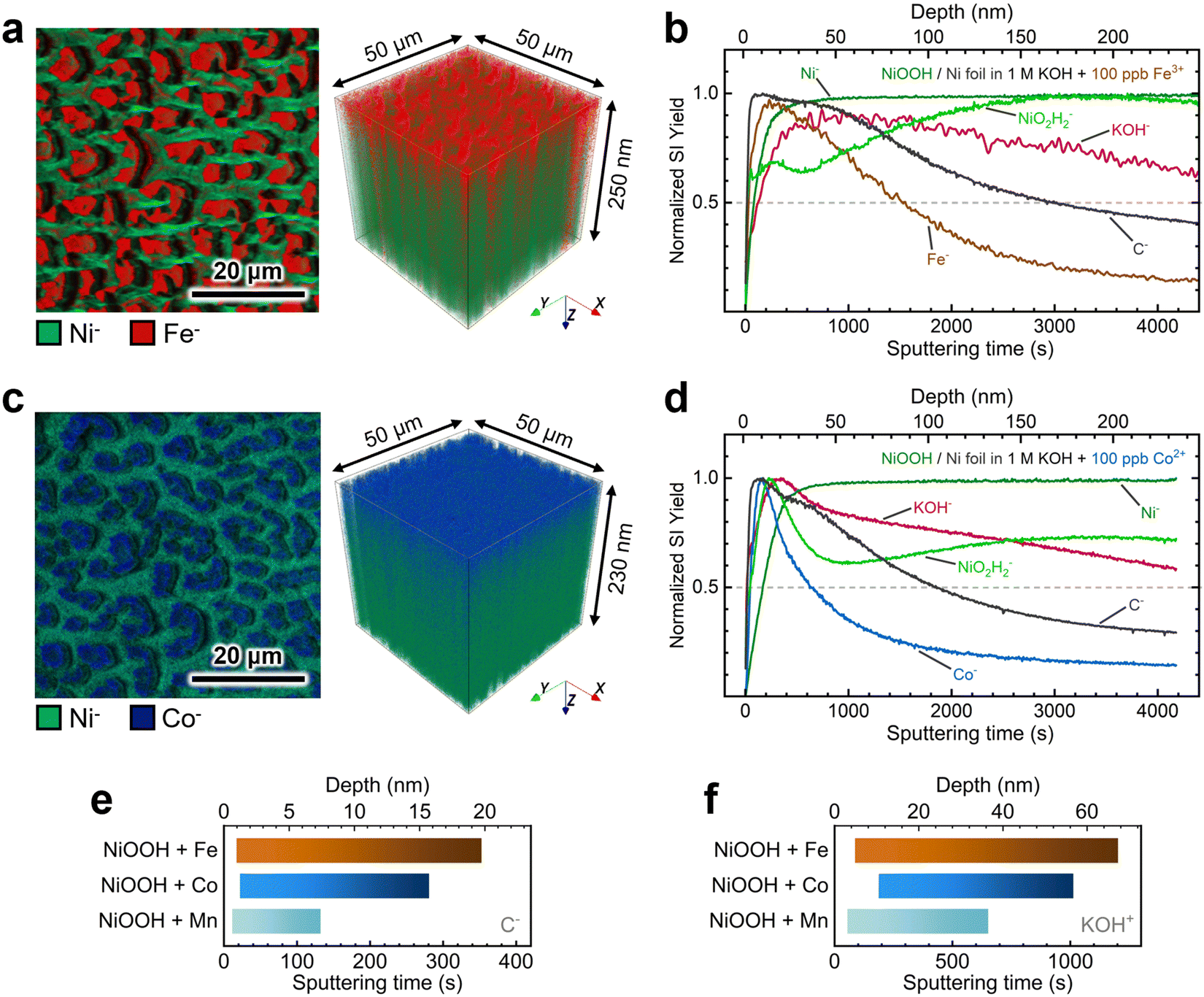 | ||
| Fig. 6 TOF-SIMS characterization of NF–NiOOH electrodes after CP conditioning in purified 1 M KOH electrolyte spiked with Fe and Co: (a) high-resolution 2D maps and 3D depth profile renderings showing the spatial distribution of Ni− and Fe− markers and (b) depth profiles (normalized to maximum) of secondary-ion fragments acquired from electrodes exposed to 100 ppb of Fe3+; (c) high-resolution 2D maps and 3D depth profile renderings showing the spatial distribution of Ni− and Co− markers and (d) depth profiles (normalized to maximum) of secondary-ion fragments acquired from electrodes exposed to 100 ppb of Co2+; calculated average range of intercalation of (e) C− (negative ion mode) and (f) KOH+ (positive ion mode) markers in electrodes exposed to 100 ppb of Fe3+, Co2+, and Mn2+. | ||
The depth profiles in Fig. 6 and Fig. S23 in the ESI† reveal consistent distributions of C− and KOH+ fragments throughout the films. These fragments can serve as proxies for intercalated CO32− and K+ ions, respectively.16 We examined the spatial distribution of these ion fragments to elucidate differences in ion intercalation among films containing different metal cations. Using the absolute secondary-ion yield for C− (Fig. S24, ESI†) and KOH+ (Fig. S25, ESI†), we determined statistically significant intercalation ranges for each metal incorporation condition (Fig. S26, ESI†). As revealed in Fig. 6e, C− fragments exhibit a broader and deeper distribution range in Co- and Fe-incorporated films. In contrast, the Mn-incorporated film shows a narrow range confined to the surface (∼7 nm). The KOH+ fragment distribution displays a similar trend, with KOH+ ions penetrating further into the film, as shown in Fig. 6f.
These findings suggest that Fe and Co incorporation promotes greater intercalation of anions and cations, thus promoting the retention of the α-phase.15,55 Despite results being collected from samples with different environments (i.e., aging vs. CP conditioning), Raman spectroscopy and TOF-SIMS results reveal that incorporating metal cations into NiOxHy films leads to a more disordered structure. This structural effect explains the improvement in the electrocatalytic and energy storage properties. According to previous research, utilizing Co as an additive for Ni batteries stabilizes the turbostratic α-phase,15 allowing the extraction of more electrons according to the Bode scheme.38,56 Dette and coworkers have shown that Co oxyhydroxide nanosheets exhibit greater structural stability than their nickel oxyhydroxide counterparts due to reduced mechanical stress accumulation during conditioning.21 Fe incorporation has been shown to reduce the amount of β-NiOOH formed after aging in Fe-unpurified KOH.26 Intercalated ions may further boost the electrocatalytic performance by stabilizing high-valent metal sites and enhancing ionic conductivity.57,58
The degree of intercalation is reduced in films containing Mn, implying a lesser extent of Mn incorporation into the NiOxHy structure. This observation is consistent with the lower electrochemical performance of Mn-incorporated NF–NiOOH electrodes compared to Fe and Co. Another possible explanation could be the slower oxidation rate of Mn3+ to Mn4+, thereby requiring fewer anions to balance the charge.15 Co-precipitated Mn tends to be unstable and eventually forms the less electrochemically active β-phase.16 Thus, Mn incorporation could be significantly limited compared to Fe and Co cations.
Building upon the observed structural effects, a comprehensive description is illustrated in Fig. 7. The Ni(OH)2/NiOOH structure consists of neutral NiO2 layers formed by edge-sharing NiO6 octahedra. The interlayer space between these slabs serves as a host for the reversible intercalation of protons, alkali ions, water molecules, and anions during electrochemical cycling.59 Transition metal cations such as Co3+ or Fe3+ are incorporated via cation exchange, replacing the Ni2+ centers in the NiO6 octahedra and rendering the slabs cationic in the reduced state. To maintain charge neutrality, negatively-charged species like OH− and CO32− fill the interlayer space.15,60 This arrangement resembles a hydrotalcite-like structure, where the intercalated species form ionic bonds with the slab, acting as structural “pillars” that stabilize the overall framework. The density of these pillars is directly influenced by the negative charge of the intercalating anions and the number of cations that have been incorporated into the NiO2 layer. Consequently, when only small numbers of cations are incorporated, the interlayer space may not achieve full occupancy due to insufficient intercalated anions.59
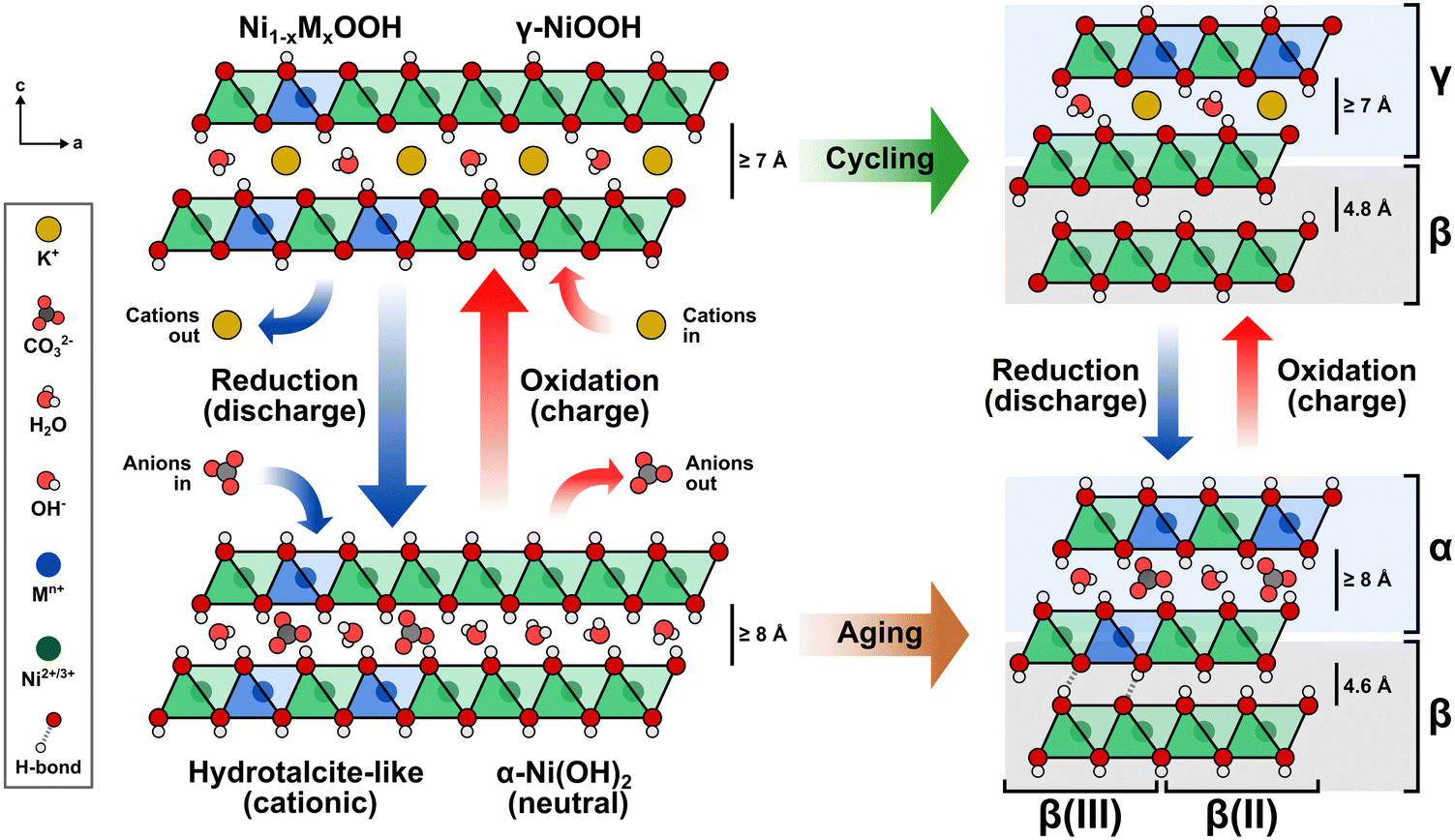 | ||
| Fig. 7 Transition metal incorporation induces structural transformations of layered Ni(OH)2/NiOOH to an interstratified structure. The hydrotalcite-like structure evolves into a mixed α/β phase composition upon aging and a mixed γ/β phase composition upon electrochemical cycling/conditioning. | ||
Protons are removed upon oxidation, increasing the electrostatic repulsion between oxygen layers in adjacent NiO2 slabs and resulting in the intercalation of K+ ions.15,55,59,61,62 Thus, repeated electrochemical cycling between Ni(OH)2 and NiOOH alters the type of intercalated ion. The α-phase is less stable in alkaline media; therefore, extended aging or electrochemical cycling eventually transforms the disordered phase into the more compact and less active β-phase, which hinders the intercalation of anions and cations.11
Intercalated anions balancing the additional positive charge occupy specific interlayer regions, thus creating an interstratified structure. The arrangement of these intercalated species correlates directly with the spatial distribution of the incorporated transition metal cations, leading to a blend of compact (β/β) and disordered (α/γ) phases.11,14,53,63
A de-mixing phenomenon can explain the saturation at increasing cation concentrations. As the number of dissolved metal cations capable of replacing Ni increases in the electrode–electrolyte interface, the Ni sites available at the NiOxHy surface decrease. When the cation exchange process cannot continue, the metal cations start to nucleate and segregate into a separate phase on the surface, which may be inactive, as in the case of γ-FeOOH.26,50 This phenomenon is more likely to occur at higher temperatures, elevated KOH concentrations, and smaller particle sizes.59 Additionally, proton deficiencies emerge where anions cannot intercalate into the hydrotalcite structure to neutralize the positive charge given by the incorporated metal cations, resulting in a β(III)-phase.11
The formation of an interstratified structure at the surface of the NiOxHy film explains the increase in structural disorder upon in situ metal incorporation, the confinement of incorporated foreign metal cations near the surface, and the correlation between incorporated metals and proxies for intercalated CO32− and K+ ions. Although the β-phase predominantly forms in Ni(OH)2/NiOOH after extended aging or electrochemical conditioning, our findings suggest that in situ metal incorporation preserves the interstratified structure to a depth of a few tens of nanometers from the surface, leaving the inner bulk of the film accounting for most of the β-phase formed after aging/conditioning. Although the bulk Fe or Co content may seem low, these reside primarily within the interstratified structure near the surface, which could already be saturated with incorporated metals. This concept reinforces that electrochemical performance is not strictly surface- or volume-restricted but depends on active site localization.
In situ metal incorporation influences the electronic properties and chemical composition of NiOxHy films
Next, we investigated the influence of Fe and Co incorporation on the electronic properties and chemical composition of NiOxHy films. XPS analysis reveals marked differences between the as-deposited NF–Ni(OH)2 and NF–NiOOH electrodes, including a subtle shift in the primary Ni 2p peak toward higher binding energies (Fig. S27a, ESI†). The valence band (VB) spectra also show notable differences between pre- and post-conditioned samples (Fig. S27b, ESI†). The NF–Ni(OH)2 electrode sample exhibits a sharp feature at lower binding energies (∼3.1 eV), indicative of localized Ni 3d states, and a less prominent feature around 4.1 eV, characteristic of O 2p states. In contrast, the post-conditioned sample lacks the sharp feature at around 3.1 eV and displays a broader feature at 4.11 eV, typical of NiOx(OH)y.64,65 Further examination after sputtering the post-conditioned film with Ar+ to expose inner layers yields Ni 2p and VB spectra similar to those of the surface and a subtle shift in the valence band maximum (VBM).Spectral fitting of hydroxide and oxyhydroxide components was conducted on the Ni 2p and O 1s peaks,46,48 as displayed in Fig. S28 in the ESI.† The Ni 2p peak displays a Ni(OH)2 component for the as-deposited sample. In contrast, the post-conditioned sample comprises a mixture of Ni(OH)2 and NiOOH. As for the O 1s spectra, both samples feature a component at approximately 533 eV, which can be attributed to adsorbed water. Both display a high binding energy feature at around 531.4 eV, which is characteristic of OH− species. The post-conditioned sample also shows a low binding energy feature near 529.5 eV, ascribed to metal-bonded oxygen within an oxide crystal lattice (O2−), typical of NiOOH.64,66 These findings suggest that the post-conditioned film is primarily composed of NiOOH at the surface.
XPS analysis was also conducted on NF–NiOOH electrodes conditioned in KOH spiked with transition metal cations. Only signals for Fe and Co were revealed from XPS survey scans (Fig. S29, ESI†), suggesting that any Mn and Cu incorporation, if present, was below the limits of detection by XPS. Therefore, our XPS analysis focused solely on films containing Fe and Co.
Fig. S30 in the ESI† displays the XPS results after CP conditioning, including a suitable fitting of components based on prior literature.46,47 From Ni 2p spectra, Fe- and Co-containing films exhibit a blend of Ni(OH)2 and NiOOH. The O 1s spectra show the presence of lattice O2−, suggesting that the film surface is rich in oxides/oxyhydroxides. Fe 2p spectral fitting for the Fe-containing film indicates oxidation states consistent with FeOOH and Fe2O3.47 The Co 2p spectrum for the Co-containing film shows distinct oxidation states matching CoOOH and Co3O4. These results are consistent with prior research, confirming the presence of Co3+ and Fe3+ after OER conditioning.50,67,68
We examined the effects of Fe and Co incorporation in the XPS VB region. As illustrated in Fig. 8a, post-conditioned samples exhibit broader peaks than as-deposited NF–Ni(OH)2, suggesting the presence of oxide, hydroxide, and oxyhydroxide species.64,65 In Co-containing NF–NiOOH, the feature near 1.3 eV correlates with 3d Co3+ situated in octahedral sites (t62g), whereas the band from 2 to 5 eV includes Co2+ states.67,69,70 Moreover, the VBM varies across the films studied. In pristine NF–Ni(OH)2 and Fe-containing NF–NiOOH electrodes, the VBM shifts towards higher binding energies, moving away from the Fermi level (EF). In contrast, for pure and Co-containing NF–NiOOH, the VBM shifts towards the EF, a characteristic feature of p-type semiconductors.65,68,71,72 Proximity of the VBM to the EF suggests enhanced electrical conductivity. In the case of Co-containing films, this is due to a higher hole concentration in the valence band. These observations align with prior studies showing that the VBM of Fe oxides shifts further from the EF while the VBM of Co oxides lies closer.71,73,74
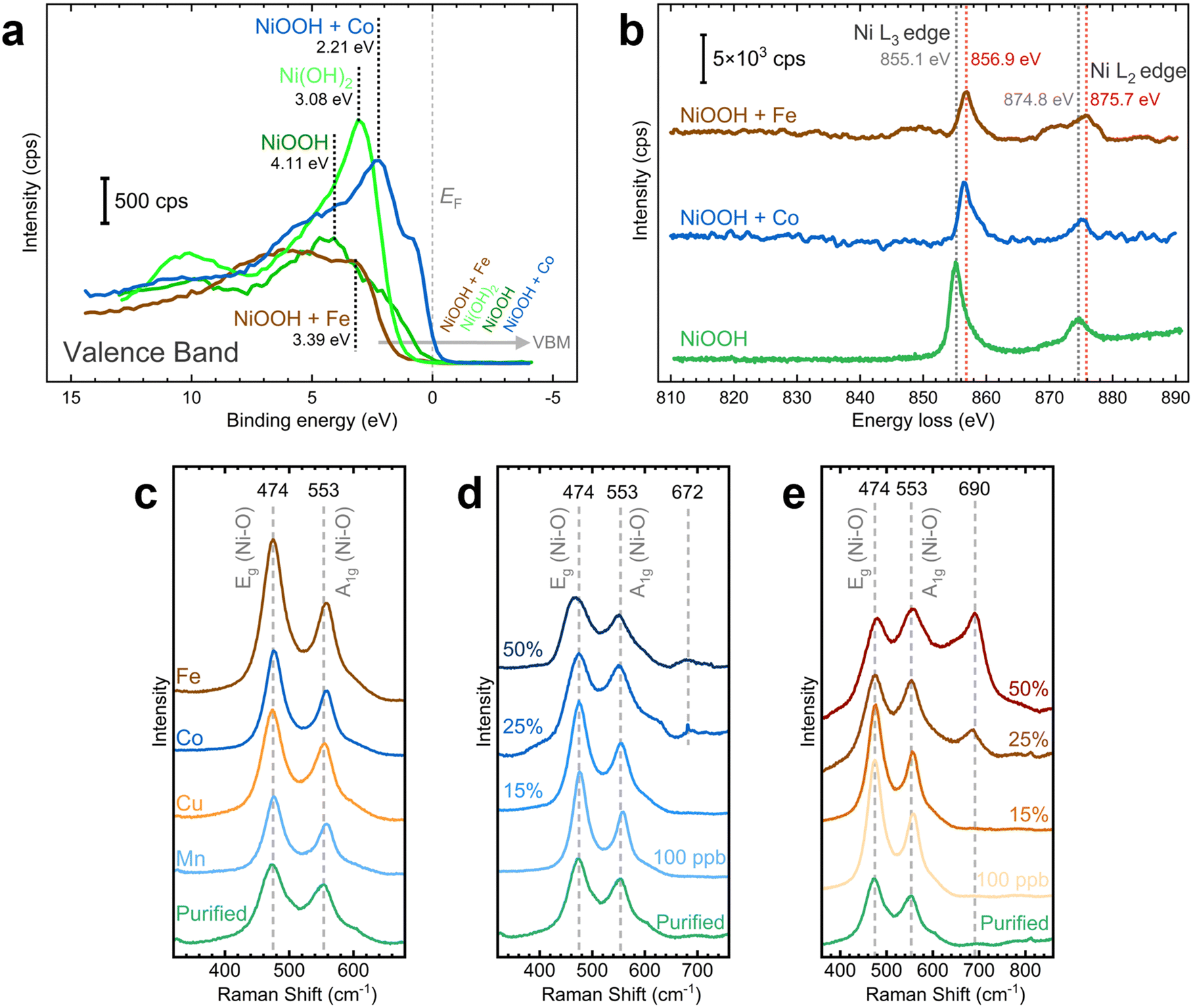 | ||
| Fig. 8 Chemical and electronic transformations of NiOOH electrodes after CP conditioning: (a) XPS valence band (VB) spectra for NF–NiOOH electrodes; as-deposited NF–Ni(OH)2 is shown as comparison. (b) EELS spectra showing energy loss shift of the Ni L3 and L2 edges with Co and Fe incorporation on NF–NiOOH electrodes; (c) Raman spectra collected over Au-t-NiOOH electrodes after CP conditioning; Raman spectra at increasing (d) Co and (e) Fe contents. Metal content percentages are presented in terms of average atomic percent (at%) from EDX measurements. Laser: 633 nm. SERS samples were conditioned in purified 1 M KOH electrolyte spiked with 100 ppb Mn+. A metal cation concentration of 500 ppb Mn+ was used for conditioning XPS and EELS samples. | ||
To gain further insights into the electronic behavior of these films, the electronic states of the VB spectra near the EF were separated into their O 2p and metal 3d contributions.10,68,75 Post-conditioned electrodes display a reduced contribution from peak C, representing the antibonding transition metal 3d state, ultimately determining the VBM (Fig. S31, ESI†). Peak C shifts closer to the EF with Co incorporation but moves away from the EF upon Fe incorporation, reaching a position nearly identical to that in the NF–Ni(OH)2 sample. Co incorporation also broadens the bonding O 2p states (peak A) and the nonbonding O 2p states (peak B). Co incorporation positions the EF closer to the O 2p states, decreasing the energy gap between O 2p and metal 3d states. These effects lead to greater overlap of the O 2p and metal 3d bands, suggesting a higher degree of covalency in the metal–oxygen bond.68
For a more accurate description of this effect, we determined the peak position, FWHM, and area of these components (Table S3, ESI†). Co incorporation results in peaks closer to the EF, reduced energy separation between O 2p and metal 3d bands, and broader O 2p states, all contributing to more significant O 2p–metal 3d band overlap.68 In contrast, Fe incorporation appears to decrease the electronic conductivity, in line with previous research.13 Still, the significant broadening of O 2p states compared to pure NiOOH after Fe incorporation suggests that the Fe–O bond is also more covalent.14,76 These results support claims that conductivity is not responsible for the OER activity enhancement with in situ Fe incorporation.27
We conducted TEM-EELS measurements to examine the electronic effects of Fe and Co incorporation. HAADF STEM images of particles sampled from NF–NiOOH electrodes reveal a low crystallinity and a phase structure similar to NiOOH (Fig. S32, ESI†). STEM-EDX elemental mapping confirms the uniform distribution of Fe and Co within the particles after conditioning (Fig. S33, ESI†). EELS spectra show clear Co L2,3 and Fe L2,3 edges, verifying the incorporation of Co and Fe (Fig. S34, ESI†). Fig. 8b shows Ni L2,3-edge spectra for pure and metal-containing NF–NiOOH electrodes. The Ni L2- and L3-edge peaks shift towards higher energy losses upon Fe and Co incorporation, in line with prior research.20 These results suggest that Fe and Co alter the electronic environment of Ni via electron-withdrawing effects.
Building on prior studies employing Raman spectroscopy to explore the influence of metal incorporation on the electronic properties of NiOxHy electrocatalysts,26,77,78 we carried out ex situ measurements on NF–NiOOH electrodes. Samples exhibited weak signals, with distinct bands only evident for Fe-containing samples (Fig. S35, ESI†). Thus, we deposited NiOOH films onto roughened gold substrates to amplify the Raman scattering.79 As displayed in Fig. 8c, Raman spectra collected over Au-t-NiOOH electrodes conditioned in KOH containing transition metal cations reveal two pronounced bands at approximately 474 and 553 cm−1. These bands can be attributed to the δ(O–Ni–O) bending (Eg) and ν(O–Ni–O) stretching (A1g) vibrational modes, respectively.54,80
Close inspection of Fig. 8c reveals subtle differences in the relative intensities of the bands. This observation becomes more discernible when the ratios between these bands are plotted for each transition metal (Fig. S36, ESI†). Peak ratios for Fe-, Co-, and Mn-containing films are significantly higher than pure NiOOH. These relative peak intensities serve as indicators for various factors such as structural disorder, hydrogen and vacancy content, and the nickel oxidation state.26,81
Before discerning the effects of Fe incorporation, previous research had already noted that the ratio of these two bands changed after aging.77,81 Subsequent investigations by Klaus and coworkers confirmed that these alterations can be attributed explicitly to Fe incorporation.26 However, unlike previous findings by Klaus, we report an increase in the peak ratio from 1.08 in purified KOH to 1.19 in Fe-containing KOH. We primarily attribute this divergence to the different electrochemical setups employed (in situ vs. ex situ). Nevertheless, our results suggest that in addition to Fe incorporation, in situ incorporation of trace Co and Mn also influences the Ni–O bond.
SERS measurements were conducted to examine changes in the chemical composition with increasing Fe and Co contents. Fe and Co were co-deposited with Ni onto gold substrates for these tests. SERS measurements were then performed after CP conditioning. As displayed in Fig. 8d, the intensities and widths of the Ni–O bands change with increasing Co content. A graph showing the peak ratio at increasing Co contents reveals a peak ratio maximum at 100 ppb of Co (Fig. S37a, ESI†). This ratio starts to decline when the Co content exceeds 15%. Additionally, a new band emerges at around 672 cm−1 beyond 25% Co content, which is characteristic of the Co–O A1g vibrational mode found in Co3O4 and CoOOH.82,83 As shown in Fig. 8e, band broadening also occurs for Fe incorporation. The peak ratio reaches a maximum at 15% Fe content and drops sharply beyond that point (Fig. S37b, ESI†). Likewise, a new band becomes evident at roughly 690 cm−1, which could be attributed to the A1g vibrational mode of the Fe–O bond, commonly found in γ-FeOOH, γ-Fe2O3, Fe3O4, and NiFe2O4.28,84,85
These findings indicate that remarkable alterations in chemical composition occur upon reaching higher Fe and Co contents, consistent with our previous discussion on the metal uptake saturation threshold. However, due to the challenging nature of our amorphous films, we cannot definitively determine the chemical composition of the metal oxide/oxyhydroxide phase that forms at higher Fe and Co contents.
Previous research has provided insights into the composition of these segregated phases with increasing Fe and Co contents through X-ray absorption spectroscopy (XAS). According to Kim et al., co-depositing Fe and Co with Ni up to approximately 10 mol% results in single-phase materials, with Co3+ and Fe3+ replacing Ni sites in the NiOxHy lattice.18 However, exceeding 25% Fe content leads to increased Fe–O distances and the formation of a segregated γ-FeOOH phase.50 Fe-containing electrocatalysts experience a marked decline in OER performance once phase segregation occurs, given that Fe oxides and oxyhydroxides exhibit poor OER catalytic activity.9,50
Given our current limitations to provide a precise composition, we can only speculate that an oxide/oxyhydroxide phase segregates from the interstratified structure at high metal contents. A more comprehensive investigation employing advanced characterization is required to gain deeper insights into the surface segregation phenomenon and accurately characterize the composition of the resulting phase. Nonetheless, our proposition of an interstratified surface structure driven by cation exchange aligns with the concept of surface segregation of less active phases, explaining the decrease in performance observed at higher metal uptakes.
Factors influencing in situ metal incorporation
It is essential to grasp in situ metal incorporation as a cation exchange process influenced by the characteristics of the host lattice, the metal cation, and its presence in the electrolyte. Based on prior research and principles of solid-state chemistry, we identify four factors that explain in situ metal incorporation into layered NiOxHy materials via cation exchange.The enhanced stability of certain metal cations can be better understood through crystal field theory and the preference of metal ions for occupying octahedral sites.87 This affinity can be estimated using the crystal field stabilization energy (CFSE) for the octahedral geometry and the octahedral site stabilization energy (OSSE). Data for CFSE and OSSE values for Ni, Co, Fe, Mn, and Cu in various oxidation states are shown in Table S4 in the ESI.† Given the high oxidation states of first-row transition metals, these cations are expected to favor a low-spin state.55,88 From Table S4 (ESI†), Ni4+, Co3+, Fe2+, Fe3+, and Mn2+ exhibit the highest stability and are most likely to occupy octahedral sites. Conversely, cations like Mn4+ and Cu2+ show more positive OSSE values, suggesting a stronger inclination to reside in tetrahedral (interlamellar) sites.
Jahn–Teller distortions, stemming from the degeneration of electronic ground states, could also play a role in determining site stability. According to Table S4 (ESI†), low-spin Co3+ exhibits no distortion, whereas Fe3+ and Mn3+ show weak distortion. In contrast, Ni3+ and Cu2+ display strong distortion, which could lead to significant alterations in the crystal structure. The ionic radius and coordination number are also linked with these observations. From Table S4 (ESI†), the ionic radius decreases as the oxidation state increases. High-valent cations tend to be surrounded by more negatively charged ligands, leading them to prefer octahedral sites.
Even when metal cations exhibit lower oxidation states, dissolved O2 can readily oxidize these cations, thus facilitating cation exchange.11,14,16,86 This effect could account for the formation of Co3+ in our study despite initially spiking Co2+ into the KOH electrolyte. Cation oxidation could further accelerate during the OER as the evolved O2 saturates the electrolyte and oxidizes dissolved, lower-valent metal cations. Therefore, in the presence of O2, dissolved Fe3+, Co3+, and Mn3+ cations are more likely to exist than their M2+ counterparts.
Based on these observations, Co3+ and Fe3+ are ideal candidates for cation exchange. Low spin Co3+ offers high structural stability due to the absence of Jahn–Teller distortions and exhibits a strong affinity for octahedral sites. This accounts for its high incorporation rate and strong covalency with O2−. While Fe3+ does display some weak distortion, it also strongly favors octahedral sites, and its covalency can stabilize neighboring NiO6 octahedra.18,50 In contrast, Cu2+ has a low affinity for octahedral sites, is prone to strong distortion, and possesses an ionic radius larger than that of Ni2+/3+ cations. Thus, it is unlikely that Cu2+ would engage in effective cation exchange, which explains the lack of electrochemical, structural, and electronic effects after conditioning NiOxHy in Cu-containing KOH electrolyte.
Mn incorporation appears to be more complex. Mn shows no tendency to adsorb on the NiOOH surface,86 and is considered unstable when co-precipitated deliberately.14,89 Mn3+ is more likely to undergo cation exchange than Mn2+, as Mn3+ shows only weak distortion, and its ionic size is suitable for occupying octahedral sites. However, contrary to the electron-withdrawing effects of Fe3+ and Co3+, incorporating Mn3+ would be expected to decrease the oxidation state of Ni3+, as Mn3+ has a notable reducing character.11,16,63 The Mn3+/4+ eg band is at a higher energy level than the Ni3+/4+ eg band.87 Thus, electrons from Mn3+ would move to the Ni3+/4+ band, leading to the oxidation of Mn3+ to Mn4+ and the reduction of Ni3+ to Ni2+.
The presence of Mn4+ in the lattice may have mixed implications. Although Mn4+ helps to relax the structure due to the absence of Jahn–Teller distortions (Table S4, ESI†),11 it is smaller in size and has less affinity for octahedral sites, resulting in higher mobility towards tetrahedral sites. While Mn4+ remains part of the lattice, its effects could differ from what one might initially expect. Previous research using iodometric techniques has confirmed the presence of Mn4+ in co-precipitated nickel hydroxides.11 Other cations like Zn2+ also show a preference for tetrahedral coordination in brucite-type nickel hydroxide, leading to a hydrotalcite-like structure and enhanced structural stability.90
Based on our results and existing research, however, Mn does not enhance charge acceptance as Co does, nor does it enhance OER catalysis like Fe, likely owing to its structural instability.13 Still, incorporating Mn into tetrahedral sites could offer some structural stability. Recent studies indicate that tetrahedral sites also play a role in shaping the electronic properties of layered hydroxides.91 Further research is needed to clarify if tetrahedral sites occupied by transition metal cations affect the electrocatalytic and energy storage performance of MOxHy materials.
Our results indicate that Co3+ and Fe3+ ions maintain stability even during extended periods of aging or conditioning. The absence of Co redox peaks in our LSV and CV scans suggests that these cations maintain a stable oxidation state, thereby contributing to structural stability.12,13,16,18,88 Prior XAS studies have reported that Co and Fe oxidation states remain unchanged during OER.18 Isovalent cations such as Al3+ also stabilize the α-phase.76 Thus, keeping oxidation states constant could be essential in achieving high stability. While it seems that Fe3+ remains constant in our study, prior research indicates that it oxidizes to Fe4+.14,55,76 Further investigations are needed to confirm these changes.
Finally, having metal centers with higher oxidation states is advantageous for enhancing performance. Cations with high oxidation states and strong electron-withdrawing effects could account for changes observed in the intensities of the Ni–O bands (Fig. 8c).11 This effect is typically used to explain the high activity of Fe-containing OER catalysts.50 Additionally, the improved electronic conductivity in Co-containing NiOOH can be attributed to the introduction of holes into the partially-filled, low-spin Co3+/4+: t2g6−x band, as well as direct metal-to-metal interactions across the shared octahedral edges within the slab.87,92 In contrast, the evenly-filled bands seen in Mn4+ result in semiconducting behavior.
Removal of the metal cations due to dissolution can also occur during electrochemical conditioning.16 Thus, local dissolution and redeposition processes could lead to partial segregation and reprecipitation of unstable phases.59 Note that Fe and Mn are particularly susceptible to such effects (Fig. S38, ESI†). It is worth noting that these phenomena could also apply to materials co-precipitated intentionally with transition metals that might be unstable. Mn3+ faces significant dissolution issues as it forms Mn2+ and Mn4+ when protons are present. Since protons participate in the proton-coupled Ni(OH)2/NiOOH transition, even local pH variations could drive Mn disproportionation. This reaction typically results in Mn4+ remaining in the solid phase while Mn2+ is leached into the solution.87,89 The redissolution of soluble species into the electrolyte could subsequently lead to the precipitation of insoluble oxides and hydroxides. Thus, caution is advised when selecting transition metals for catalyst development.
Changes in the oxidation state and migration to tetrahedral sites may become evident only after extended conditioning. For instance, the Mn3+ to Mn4+ transition described earlier could unfold differently during aging or conditioning. Mn disproportionation might also manifest after sustained exposure to fluctuating pH. This consideration becomes significant when evaluating materials used in intermittent energy conversion systems like water electrolyzers. Thus, prolonged conditioning is essential to track down incorporation and dissolution processes.39,89 This observation also applies to co-deposited films, which may undergo significant changes until the resulting active phases are formed. Further research is needed to gain a more comprehensive understanding of how various transition metals experience chemical and structural changes during testing, which is a critical step for advancing energy conversion and storage technologies.
Manipulating the electrolyte composition reverses the in situ incorporation of trace metals
To shed light on the dynamic nature of metal incorporation, we conducted experiments to examine the reversibility of cation exchange by altering the solubility equilibria. First, we conditioned NF–NiOOH electrodes via CP using Fe-containing KOH electrolyte. Then, we replaced this electrolyte with purified KOH and conducted a second CP conditioning step of identical duration. Conditioning in Fe-containing KOH decreases the potential by nearly 70 mV in 12 h, and subsequent conditioning in purified KOH increases the potential by 20 mV (Fig. S39a, ESI†). Fig. 9a shows that the OER activity increases after conditioning with Fe-containing KOH and declines after conditioning in purified KOH. The same trend is observed from CV scans (Fig. S39b, ESI†) and EIS analysis (Fig. S39c, ESI†).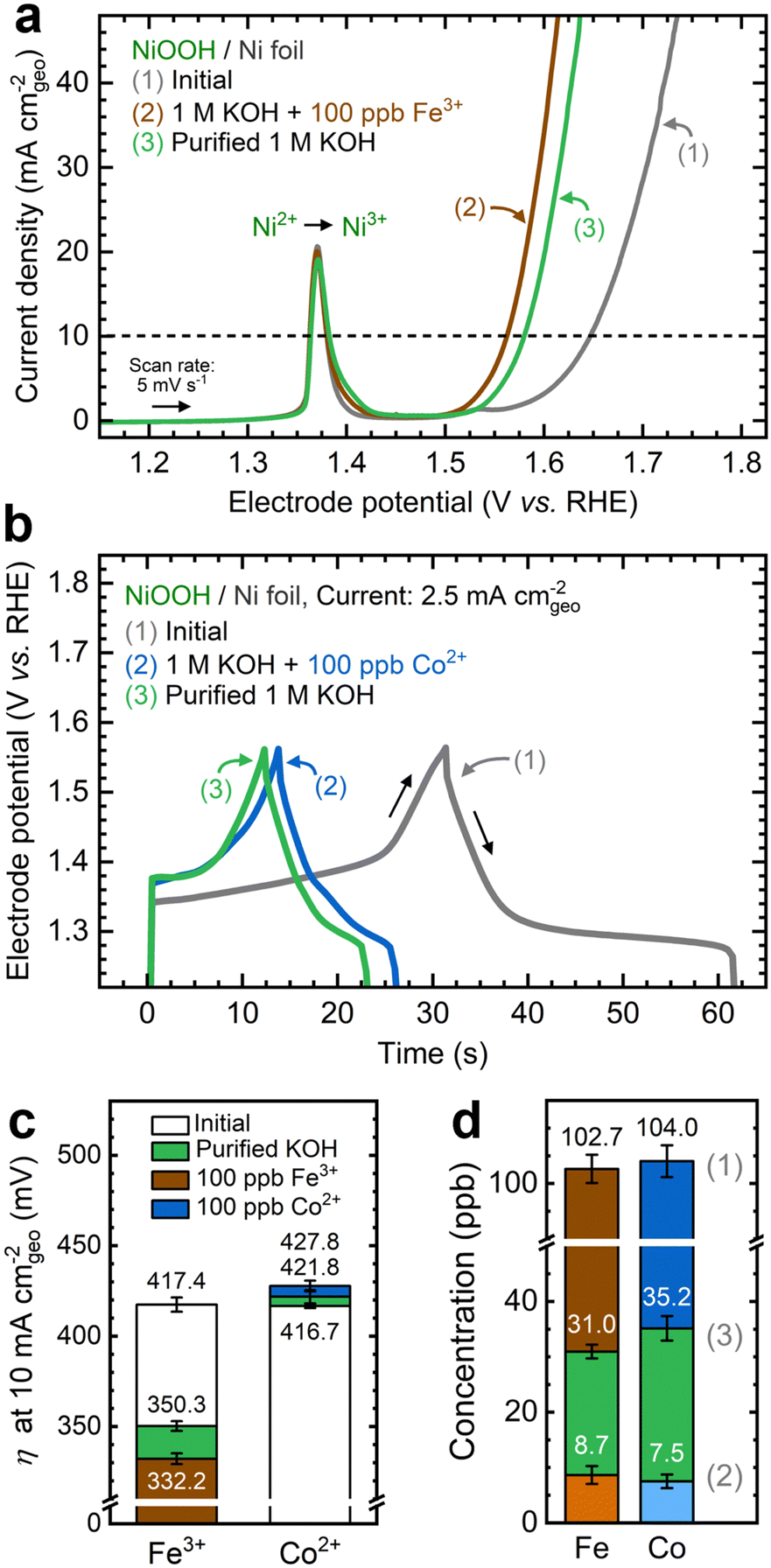 | ||
| Fig. 9 Reversing in situ metal incorporation on NF–NiOOH electrodes by substituting the electrolyte: (a) linear sweep voltammograms showing the change of the OER current (1) before CP conditioning, (2) after the first conditioning step in Fe-containing KOH, and (3) after a second conditioning step in purified KOH; (b) charge–discharge profiles (1) before GCD conditioning, (2) after the first conditioning step in Co-containing KOH, and (3) after a second conditioning step in purified KOH; (c) OER overpotentials at 10 mA cmgeo−2 after each step; (d) Fe and Co concentrations in the KOH electrolyte measured after each step using solution-mode ICP-MS. | ||
Following the same strategy, we conditioned NF–NiOOH electrodes via GCD in Co-containing and purified KOH. As shown in Fig. 9b, the stored charge decreases until stabilization when cycling in Co-containing KOH, which is expected due to the α to hydrotalcite-like phase transition. However, GCD cycling in purified KOH further narrows the GCD profile. Cycling in Co-containing KOH stabilizes the total capacitance to around 164 mF cm−2, whereas subsequent cycling in purified KOH leads to an additional 18% decrease (Fig. S40a, ESI†). The OER current remains unchanged (Fig. S40b, ESI†), and minor redox peak changes occur during CV scans (Fig. S40c, ESI†). Nyquist plots also remain virtually identical, indicating comparable OER activity (Fig. S40d, ESI†), and the total capacitance estimated from GCD declines by approximately 15% (Fig. S40e, ESI†).
We monitored OER performance metrics across the two-step conditioning process. As shown in Fig. 9c, the OER overpotential experiences marked fluctuations when Fe is added to or removed from the electrolyte. In contrast, removing Co from the electrolyte does not significantly alter the OER. Comparable trends can be seen for the Tafel slope and Rct (Fig. S41, ESI†). These findings suggest that removing Fe in the second step reverses the OER enhancement initially brought on by Fe incorporation. While removing Co from the electrolyte does not impact OER performance, it decreases the charge capacity.
These findings indicate that the second conditioning step in purified KOH triggers the redissolution of Fe and Co back into the electrolyte. ICP-MS analysis reveals that Fe and Co concentrations in the electrolyte decrease below 10 ppb after the first step, then rise above 30 ppb upon conditioning in purified KOH (Fig. 9d). This result suggests that conditioning in a metal-free electrolyte alters the solubility equilibrium, leading to transition metal dissolution from the film and affecting the electrochemical performance.
Chelation inhibits the in situ incorporation of trace metals
Another strategy to manipulate the solubility equilibria is ion complexation. Previous studies have utilized ethylenediaminetetraacetic acid (EDTA) to prevent metal impurities from depositing onto CO2 reduction catalysts.93 Following a similar approach, we added varying concentrations of EDTA to KOH electrolytes containing Fe and Co ions before conditioning. Given its strong affinity for high-valent transition metals (Table S6, ESI†), we aimed to investigate whether EDTA could inhibit cation exchange.As shown in Fig. 10a, adding EDTA to Fe-containing KOH decreases the OER current. Moreover, CP profiles stabilize at higher potentials in the presence of EDTA (Fig. S42a, ESI†). Nyquist plots also show larger semicircles as EDTA concentration increases (Fig. S42b, ESI†). According to ICP-MS analysis, adding EDTA to the KOH electrolyte keeps the Fe concentration relatively stable (Fig. S42c, ESI†). Increasing the EDTA concentration correlates with increased OER overpotentials and Tafel slopes (Fig. S42d and e, ESI†). Adding EDTA to Co-containing KOH reduces the total capacitance during GCD conditioning (Fig. 10b). Higher EDTA concentrations narrow the GCD profiles and marginally diminish OER performance (Fig. S43, ESI†). ICP-MS data further verify that EDTA stabilizes Co concentrations in the electrolyte (Fig. S43d, ESI†). While OER metrics remain primarily unchanged, a slight decrease in redox capacitance is observed as EDTA concentration increases (Fig. S44, ESI†).
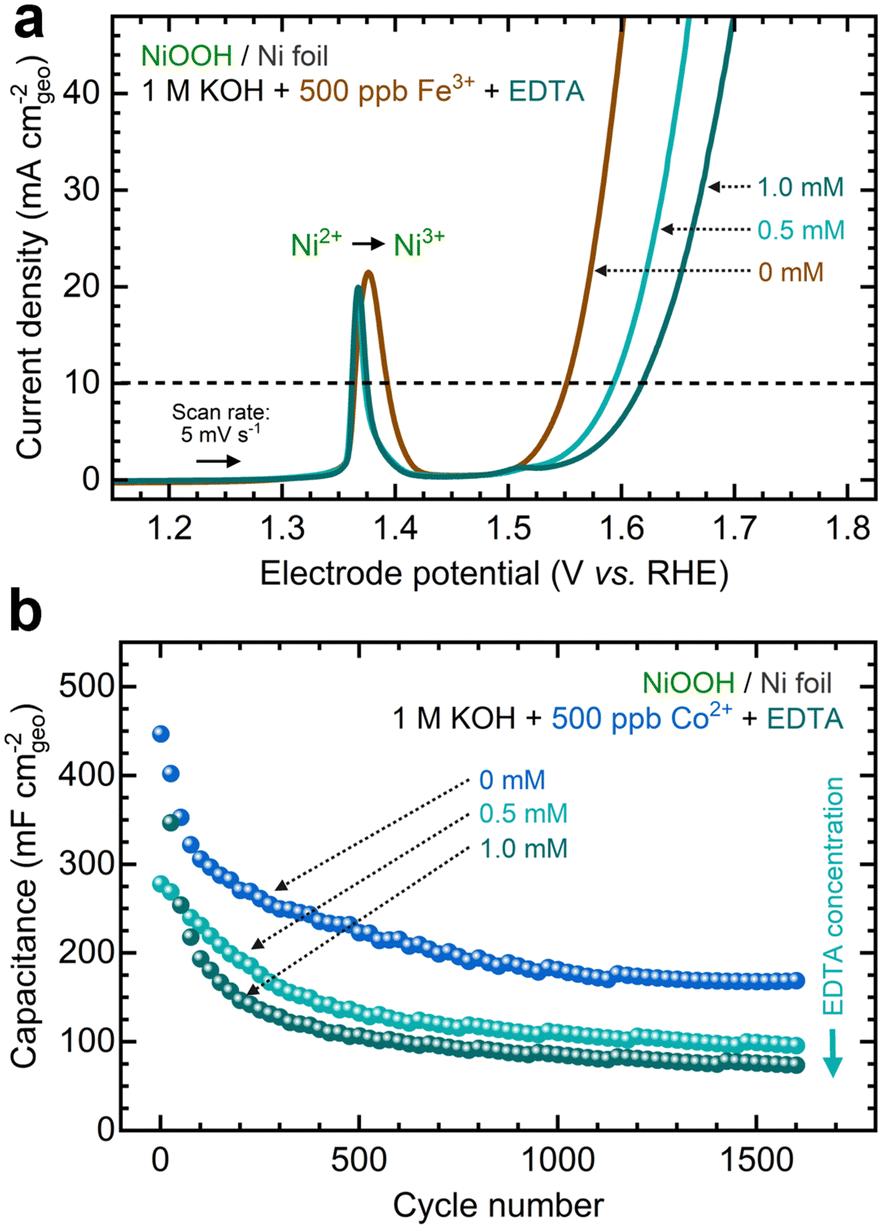 | ||
| Fig. 10 Inhibiting in situ metal incorporation on NF–NiOOH electrodes via metal complexation: (a) linear sweep voltammograms showing the decrease of the OER current with EDTA concentration in 1 M KOH electrolyte spiked with 500 ppb Fe3+; (b) GCD curves showing the decrease of the total capacitance with EDTA concentration in 1 M KOH electrolyte spiked with 500 ppb Co2+. | ||
These results suggest that ion complexation inhibits in situ metal incorporation, resulting in electrochemical performance similar to NF–NiOOH electrodes conditioned in purified KOH electrolyte. It is worth noting that, given its strong affinity for Ni cations, EDTA may not be ideal for this purpose (Table S6, ESI†). We observed Ni dissolution when EDTA was introduced (Figs. S42c and S43d, ESI†). More selective chelating agents should be explored to inhibit metal incorporation via ion complexation.
Conclusions
This work outlines the significant impact of the in situ incorporation of trace metals on the electrochemical, structural, and chemical properties of Ni(OH)2/NiOOH materials. In addition to previous findings on OER activity enhancement induced by Fe incorporation, we revealed that in situ incorporation of trace Co boosts the energy storage capabilities of NiOxHy films. Mn also showed partial incorporation, marginally improving electrochemical performance. Conditioning in Cu-containing KOH electrolyte exhibited no discernible electrochemical, structural, or compositional effects, indicating that it does not readily incorporate into NiOxHy films. Moreover, experiments conducted at increasing Fe and Co concentrations indicate that metal cation uptake reaches a saturation threshold and ceases to enhance electrochemical performance.Depth profiling results revealed that metal incorporation is confined to the surface and correlates with the intercalated ion distribution in the interstratified structure. This finding confirms the concept of an interstratified structure induced by in situ metal incorporation: while the original α-phase primarily transforms into a denser, less active β-phase, incorporated Fe, Co, and Mn cations preserve the disordered, more active α/γ phase near the surface. According to XPS VB analysis, electrodes conditioned with Co cations exhibited improved electronic conductivity and greater covalency. SERS measurements also suggest the formation of separated oxide/oxyhydroxide phases above 15 at% Fe and Co contents.
Building on the concept of cation exchange, we discussed four determinants of metal incorporation: the metal oxidation state and preference for the octahedral site, chemical solubility, morphology, and the electrochemical conditioning method. By manipulating the cation exchange process, we showed that metal incorporation can be reversed by shifting the solubility equilibria and inhibited through metal complexation.
This study emphasizes the significance of cation exchange processes in shaping electrocatalytic material properties. Our findings confirm that even trace-level metal incorporation can significantly affect the electrochemical, structural, and chemical properties. We urge the community to recognize this effect and be cautious to avoid incidental incorporation of trace impurities or cross-contamination, which could lead to erroneous descriptions of electrocatalytic and energy storage performance. Additionally, we underscore the potential of cation exchange reactions as a strategy for designing better materials. Given that it is confined to the surface, this process can be harnessed to target surface sites and craft specialized catalysts, while reducing metal loadings.12 Future research should focus on understanding the stability of incorporated metals and their influence on the local environment.
Author contributions
R. A. Marquez: conceptualization, investigation, methodology, formal analysis, writing – original draft, visualization. E. Kalokowski: investigation, writing – review & editing. M. Espinosa: investigation, writing – review & editing. J. T. Bender: investigation, writing – review & editing. Y. J. Son: conceptualization, writing – review & editing. K. Kawashima: conceptualization, writing – review & editing. C. E. Chukwuneke: writing – review & editing. L. A. Smith: writing – review & editing. H. Celio: investigation. A. Dolocan: investigation, formal analysis. X. Zhan: investigation, formal analysis. N. Miller: investigation. D. J. Milliron: writing – review & editing, resources, supervision. J. Resasco: writing – review & editing, supervision. C. B. Mullins: writing – review & editing, resources, funding acquisition, project administration. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the National Science Foundation via CHE Grant 2102307, MRI Grant 2117623 for the acquisition of the VersaProbe 4 scanning XPS instrument, and the Welch Foundation through Grants F-1436, F-2076, and F-1848 for their generous support. R. A. M. (CVU 919871) acknowledges CONAHCYT for his Doctoral scholarship award.References
- Z. Yan, J. L. Hitt, J. A. Turner and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12558–12563 CrossRef CAS PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, J. Mater. Chem. A, 2019, 7, 14971–15005 RSC.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- L. Guerlou-Demourgues and C. Delmas, J. Power Sources, 1994, 52, 275–281 CrossRef CAS.
- R. D. Armstrong, G. W. D. Briggs and E. A. Charles, J. Appl. Electrochem., 1988, 18, 215–219 CrossRef CAS.
- D. A. Corrigan and R. M. Bendert, J. Electrochem. Soc., 1989, 136, 723 CrossRef CAS.
- P. Axmann and O. Glemser, J. Alloys Compd., 1997, 246, 232–241 CrossRef CAS.
- R. Oesten, M. Wohlfahrt-Mehrens, S. Ströbele, M. Kasper and R. A. Huggins, Ionics, 1996, 2, 293–301 CrossRef CAS.
- L. Guerlou-Demourgues, C. Denage and C. Delmas, J. Power Sources, 1994, 52, 269–274 CrossRef CAS.
- M. Oshitani, Y. Sasaki and K. Takashima, J. Power Sources, 1984, 12, 219–231 CrossRef CAS.
- S. Kim, D. A. Tryk, M. R. Antonio, R. Carr and D. Scherson, J. Phys. Chem., 1994, 98, 10269–10276 CrossRef CAS.
- H. Gu, G. Shi, H.-C. Chen, S. Xie, Y. Li, H. Tong, C. Yang, C. Zhu, J. T. Mefford, H. Xia, W. C. Chueh, H. M. Chen and L. Zhang, ACS Energy Lett., 2020, 5, 3185–3194 CrossRef CAS.
- Y. Dou, C.-T. He, L. Zhang, M. Al-Mamun, H. Guo, W. Zhang, Q. Xia, J. Xu, L. Jiang, Y. Wang, P. Liu, X.-M. Chen, H. Yin and H. Zhao, Cell Rep. Phys. Sci., 2020, 1, 100077 CrossRef.
- C. Dette, M. R. Hurst, J. Deng, M. R. Nellist and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2019, 11, 5590–5594 CrossRef CAS PubMed.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- J. T. Bender, A. S. Petersen, F. C. Østergaard, M. A. Wood, S. M. J. Heffernan, D. J. Milliron, J. Rossmeisl and J. Resasco, ACS Energy Lett., 2023, 8, 657–665 CrossRef CAS.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377 CrossRef CAS.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- A. M. Smith, L. Trotochaud, M. S. Burke and S. W. Boettcher, Chem. Commun., 2015, 51, 5261–5263 RSC.
- C. Gort, P. W. Buchheister, M. Klingenhof, S. D. Paul, F. Dionigi, R. van de Krol, U. I. Kramm, W. Jaegermann, J. P. Hofmann, P. Strasser and B. Kaiser, ChemCatChem, 2023, 15, e202201670 CrossRef CAS.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- P. Chakthranont, J. Kibsgaard, A. Gallo, J. Park, M. Mitani, D. Sokaras, T. Kroll, R. Sinclair, M. B. Mogensen and T. F. Jaramillo, ACS Catal., 2017, 7, 5399–5409 CrossRef CAS.
- T. T. H. Hoang and A. A. Gewirth, ACS Catal., 2016, 6, 1159–1164 CrossRef CAS.
- Y. Yoon, B. Yan and Y. Surendranath, J. Am. Chem. Soc., 2018, 140, 2397–2400 CrossRef CAS PubMed.
- D. M. Morales and M. Risch, J. Phys. Energy, 2021, 3, 034013 CrossRef CAS.
- R. Chen, C. Yang, W. Cai, H.-Y. Wang, J. Miao, L. Zhang, S. Chen and B. Liu, ACS Energy Lett., 2017, 2, 1070–1075 CrossRef CAS.
- K. Kawashima, R. A. Márquez, Y. J. Son, C. Guo, R. R. Vaidyula, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Catal., 2023, 13, 1893–1898 CrossRef CAS.
- R. A. Márquez, K. Kawashima, Y. J. Son, G. Castelino, N. Miller, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Energy Lett., 2023, 8, 1141–1146 CrossRef.
- Y. J. Son, S. Kim, V. Leung, K. Kawashima, J. Noh, K. Kim, R. A. Marquez, O. A. Carrasco-Jaim, L. A. Smith, H. Celio, D. J. Milliron, B. A. Korgel and C. B. Mullins, ACS Catal., 2022, 12, 10384–10399 CrossRef CAS.
- Y. J. Son, R. A. Marquez, K. Kawashima, L. A. Smith, C. E. Chukwuneke, J. Babauta and C. B. Mullins, ACS Energy Lett., 2023, 8, 4323–4329 CrossRef CAS.
- W. Zheng, ACS Energy Lett., 2023, 1952–1958 CrossRef CAS.
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS.
- R. Sanchis-Gual, A. Seijas-Da Silva, M. Coronado-Puchau, T. F. Otero, G. Abellán and E. Coronado, Electrochim. Acta, 2021, 388, 138613 CrossRef CAS.
- Y. Gogotsi and R. M. Penner, ACS Nano, 2018, 12, 2081–2083 CrossRef CAS PubMed.
- Y. J. Son, K. Kawashima, B. R. Wygant, C. H. Lam, J. N. Burrow, H. Celio, A. Dolocan, J. G. Ekerdt and C. B. Mullins, ACS Nano, 2021, 15, 3468–3480 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- J. F. Jackovitz, Proceedings of the Symposium on the Nickel Electrode, The Electrochem. Soc., New Jersey, 1982 Search PubMed.
- L. Demourgues-Guerlou, L. Fournès and C. Delmas, J. Solid State Chem., 1995, 114, 6–14 CrossRef CAS.
- H. Bode, K. Dehmelt and J. Witte, Electrochim. Acta, 1966, 11, 1079–1087 CrossRef CAS.
- X. Xu, C. Li, J. G. Lim, Y. Wang, A. Ong, X. Li, E. Peng and J. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 30273–30282 CrossRef CAS PubMed.
- R. A. Marquez-Montes, K. Kawashima, Y. J. Son, J. A. Weeks, H. H. Sun, H. Celio, V. H. Ramos-Sánchez and C. B. Mullins, J. Mater. Chem. A, 2021, 9, 7736–7749 RSC.
- C. Delmas, C. Faure, L. Gautier, L. Guerlou-Demourgues, A. Rougier, R. M. Dell, R. A. Huggins, R. Parsons, B. C. H. Steele and A. C. Vincent, Philos. Trans. R. Soc., A, 1997, 354, 1545–1554 Search PubMed.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Müller, Energy Environ. Sci., 2016, 9, 1734–1743 RSC.
- A. V. der Ven, D. Morgan, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2005, 153, A210 CrossRef.
- P. Oliva, J. Leonardi, J. F. Laurent, C. Delmas, J. J. Braconnier, M. Figlarz, F. Fievet and A. de Guibert, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
- L. Guerlou-Demourgues and C. Delmas, J. Electrochem. Soc., 1996, 143, 561 CrossRef CAS.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- S. Diplas, T. Knutsen, S. Jørgensen, A. E. Gunnæs, T. Våland, T. Norby, A. Olsen and J. Taftø, Surf. Interface Anal., 2008, 40, 826–829 CrossRef CAS.
- B. Payne, M. Biesinger and N. Mcintyre, J. Electron Spectrosc. Relat. Phenom., 2009, 175, 55–65 CrossRef CAS.
- N. Weidler, S. Paulus, J. Schuch, J. Klett, S. Hoch, P. Stenner, A. Maljusch, J. Brötz, C. Wittich, B. Kaiser and W. Jaegermann, Phys. Chem. Chem. Phys., 2016, 18, 10708–10718 RSC.
- L. Heymann, M. L. Weber, M. Wohlgemuth, M. Risch, R. Dittmann, C. Baeumer and F. Gunkel, ACS Appl. Mater. Interfaces, 2022, 14, 14129–14136 CrossRef CAS PubMed.
- L. Qiao, H. Y. Xiao, H. M. Meyer, J. N. Sun, C. M. Rouleau, A. A. Puretzky, D. B. Geohegan, I. N. Ivanov, M. Yoon, W. J. Weber and M. D. Biegalski, J. Mater. Chem. C, 2013, 1, 4628–4633 RSC.
- T. J. Chuang, C. R. Brundle and D. W. Rice, Surf. Sci., 1976, 59, 413–429 CrossRef CAS.
- G. Velegraki, I. Vamvasakis, I. T. Papadas, S. Tsatsos, A. Pournara, M. J. Manos, S. A. Choulis, S. Kennou, G. Kopidakis and G. S. Armatas, Inorg. Chem. Front., 2019, 6, 765–774 RSC.
- M. Cui, X. Ding, X. Huang, Z. Shen, T.-L. Lee, F. E. Oropeza, J. P. Hofmann, E. J. M. Hensen and K. H. L. Zhang, Chem. Mater., 2019, 31, 7618–7625 CrossRef CAS.
- H. Yang, Y. Mao, M. Li, P. Liu and Y. Tong, New J. Chem., 2013, 37, 2965–2968 RSC.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Hävecker, A. Knop-Gericke, G. Rupprechter, B. Klötzer and J. Fleig, J. Phys. Chem. C, 2016, 120, 1461–1471 CrossRef CAS PubMed.
- W. T. Hong, K. A. Stoerzinger, B. Moritz, T. P. Devereaux, W. Yang and Y. Shao-Horn, J. Phys. Chem. C, 2015, 119, 2063–2072 CrossRef CAS.
- M. Balasubramanian, C. A. Melendres and S. Mini, J. Phys. Chem. B, 2000, 104, 4300–4306 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- A. Audemer, A. Delahaye, R. Farhi, N. Sac-Epée and J.-M. Tarascon, J. Electrochem. Soc., 1997, 144, 2614 CrossRef CAS.
- H.-C. Chen, C.-H. Chen, C.-S. Hsu, T.-L. Chen, M.-Y. Liao, C.-C. Wang, C.-F. Tsai and H. M. Chen, ACS Omega, 2018, 3, 16576–16584 CrossRef CAS PubMed.
- C. A. Melendres, W. Paden, B. Tani and W. Walczak, J. Electrochem. Soc., 1987, 134, 762 CrossRef CAS.
- R. Kostecki and F. McLarnon, J. Electrochem. Soc., 1997, 144, 485–493 CrossRef CAS.
- C.-W. Tang, C.-B. Wang and S.-H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS.
- Y. Li, W. Qiu, F. Qin, H. Fang, V. G. Hadjiev, D. Litvinov and J. Bao, J. Phys. Chem. C, 2016, 120, 4511–4516 CrossRef CAS.
- L. Stagi, J. A. De Toro, A. Ardu, C. Cannas, A. Casu, S. S. Lee and P. C. Ricci, J. Phys. Chem. C, 2014, 118, 2857–2866 CrossRef CAS.
- K. Fominykh, D. Böhm, S. Zhang, A. Folger, M. Döblinger, T. Bein, C. Scheu and D. Fattakhova-Rohlfing, Chem. Mater., 2017, 29, 7223–7233 CrossRef CAS.
- A. J. Tkalych, J. M. P. Martirez and E. A. Carter, J. Electrochem. Soc., 2018, 165, F907 CrossRef CAS.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- Y. Li, H. Jiang, Z. Cui, S. Zhu, Z. Li, S. Wu, L. Ma, X. Han and Y. Liang, J. Phys. Chem. C, 2021, 125, 9190–9199 CrossRef CAS.
- F. D. Speck, P. G. Santori, F. Jaouen and S. Cherevko, J. Phys. Chem. C, 2019, 123, 25267–25277 CrossRef CAS.
- J.-H. Choy, Y.-M. Kwon, K.-S. Han, S.-W. Song and S. H. Chang, Mater. Lett., 1998, 34, 356–363 CrossRef CAS.
- R. Sanchis-Gual, D. Hunt, C. Jaramillo-Hernández, A. Seijas-Da Silva, M. Mizrahi, C. Marini, V. Oestreicher and G. Abellán, ACS Catal., 2023, 13, 10351–10363 CrossRef CAS PubMed.
- R. T. M. van Limpt, M. Lavorenti, M. A. Verheijen, M. N. Tsampas and M. Creatore, J. Vac. Sci. Technol., A, 2023, 41, 032407 CrossRef CAS.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, supporting figures and tables, additional characterization data. See DOI: https://doi.org/10.1039/d3ee03617k |
| This journal is © The Royal Society of Chemistry 2024 |