
Understanding the role of Tris(2-aminoethyl)amine in stabilizing mixed-cation perovskites under humid and thermal stress conditions†
Priyanka
Chhillar
,
Amit
Kumar
,
Bhanu Pratap
Dhamaniya‡
,
Kartiki
Chandratre
,
Saurabh
Pareek
,
Sandeep Kumar
Pathak
and
Supravat
Karak
 *
*
Department of Energy Science and Engineering, Indian Institute of Technology Delhi, Hauz Khas, New Delhi-110016, India. E-mail: supravat@dese.iitd.ac.in
First published on 20th November 2023
Abstract
Mixed organic–inorganic lead halide perovskites have emerged as promising optoelectronic materials for photovoltaic applications in the span of a few years. Mixed cation FA1−xCsx perovskites have been widely explored as a resolution to the phase instability issue in FAPbI3 and CsPbI3 perovskites. In this study, the impact of Tris(2-aminoethyl)amine (TAEA) on FA0.83Cs0.17Pb(I0.90Br0.10)3 absorber layer stability under humid and thermal stress conditions has been investigated. Interestingly, the incorporation of TAEA in the perovskite precursor has resulted in enhanced structural and optoelectronic properties. Moreover, TAEA addition has led to greatly enhanced thermal and ambient stability. TAEA containing FA0.83Cs0.17Pb(I0.90Br0.10)3 films retained their black phase even after 180 days of exposure to a highly humid environment with RH ∼ 60–70% and more than 7 days on exposure to continuous thermal stress at 85 °C along with high humidity and illumination. Therefore, the perovskite films fabricated with the optimized concentration of TAEA in FA0.83Cs0.17Pb(I0.90Br0.10)3 perovskite have great potential for application in high performance devices with excellent thermal and ambient stability.
Introduction
In the past decade, organic–inorganic lead halide perovskites have turned out to be a cost-efficient and promising alternative photovoltaic technology due to their excellent semiconductor properties such as tunable bandgap across a wide range, large charge carrier diffusion lengths (∼1 μm), low exciton binding energies and large absorption coefficients (104 to 105 cm−1) in the solar spectrum region.1–3 Within a few years of development, a record power conversion efficiency of 25.8% has been attained in perovskite solar cells.4 Methylammonium lead iodide (MAPbI3) was the initial perovskite composition that was studied widely and several other perovskite material compositions have also been studied to date owing to the poor stability of MAPbI3 perovskite material. In MAPbI3 the organic MA part is highly hygroscopic and also thermally unstable, preventing the widespread commercialization of this material.5–8 MAPbI3 easily dissociates into CH3NH2, HI, and PbI2 on exposure to moisture.9,10Therefore, formamidinium lead iodide [FAPbI3, HC(NH2)2PbI3] has been investigated as an alternative light harvesting perovskite material in most of the high-performance photovoltaic devices due to its superior thermal and photo-stability and favorable band gap of 1.47 eV.11,12 However, FAPbI3 suffers from phase transition from the black photoactive α-FAPbI3 phase which is stable only above 150 °C to the yellow non-photoactive hexagonal phase δ-FAPbI3 at room temperature and in a humid atmosphere.13,14 Various approaches have been formulated for stabilization of the α-FAPbI3 phase by mixing an amount of MAI into FAPbI3.15,16 However, this mixed-organic cation perovskite composition (MAxFA1−xPbI3) might not attain thermal and ambient stability due to the presence of a volatile organic part.17 So, as an alternative researchers have followed various approaches to stabilize the α-FAPbI3 phase by using inorganic monovalent cations such as Cs+, K+, or Rb+, ensuring improved stability and device performance.18–24 Although there are various studies employing mixed multiple cations such as FAMACs,20 FAMACsRb,20,25 or FAMACsK20 to stabilize the photoactive phase, however, the presence of volatile MA components makes them susceptible to instability at high temperatures.26 Therefore, MA-free FA-Cs mixed-cation perovskite films need to be developed and investigated.
It is reported that the optimum concentration of Cs in FA1−xCsx has to be in the range 0.10 < x < 0.20, for which the Goldschmidt tolerance factor is also within the range to form a stable perovskite structure and thus preferred to form a black α-FAPbI3 phase. However, when the content of Cs ions is increased to higher values of up to 30%, the appearance of a diffraction peak corresponding to α-CsPbI3 is noticed.27 α-CsPbI3 which is a photoactive phase is stable only at temperatures above 320 °C and transforms to the photoactive γ-CsPbI3 phase when quenched to room temperature in an inert atmosphere.28–34 However, α-CsPbI3 transforms abruptly to the non-photoactive δ-CsPbI3 on exposure to atmospheric moisture.22,35,36 Therefore, α-CsPbI3 is more sensitive to atmospheric moisture, which renders it unsuitable for photovoltaic applications.37,38 FA0.83Cs0.17PbX3 perovskite composition is widely studied and employed as an absorber layer in solar cells.39–42
In this study, we have explored the effect of Tris(2-aminoethyl)amine (TAEA) on the thermal and moisture stability of MA-free mixed-cation perovskite compositions FA0.83Cs0.17Pb(I0.90Br0.10)3. The effect of different concentrations of TAEA added in FA0.83Cs0.17Pb(I0.90Br0.10)3 precursor was studied extensively to find the optimum composition. Addition of TAEA in FA0.83Cs0.17Pb(I0.90Br0.10)3 leads to an enhancement in the absorbance and optoelectronic properties of the host perovskite material. XRD measurements were conducted to explore the degradation of perovskite film under thermal stress and a highly humid ambient environment. TAEA incorporation has resulted in greatly enhanced ambient and thermal stability. XPS and FTIR measurements were carried out to examine the interaction of TAEA with FA0.83Cs0.17Pb(I0.90Br0.10)3 perovskite crystal lattice. This work provides insights into designing a long-term stable double cation perovskite composition for photovoltaic applications.
Results and discussion
XRD patterns were recorded to study the impact of introducing different amounts of TAEA in FA0.83Cs0.17Pb(I0.90Br0.10)3 (denoted as FACs from hereon) perovskite as shown in Fig. 1a. Characteristic peaks at 2θ values 14.02°, 19.94°, 24.44°, 28.34°, 31.76°, 34.82°, 40.46°, and 43.06° which correspond to the (001), (011), (111), (002), (012), (112), (022), and (033) planes respectively are detected in all the perovskite films corresponding to the photoactive α-FAPbI3 phase.43 XRD analysis confirmed that all the films crystallize into the α-phase irrespective of the concentration of added TAEA. Also, no additional peak was observed in any of the TAEA-containing films towards the low 2θ values signifying the absence of any 2D phase formation.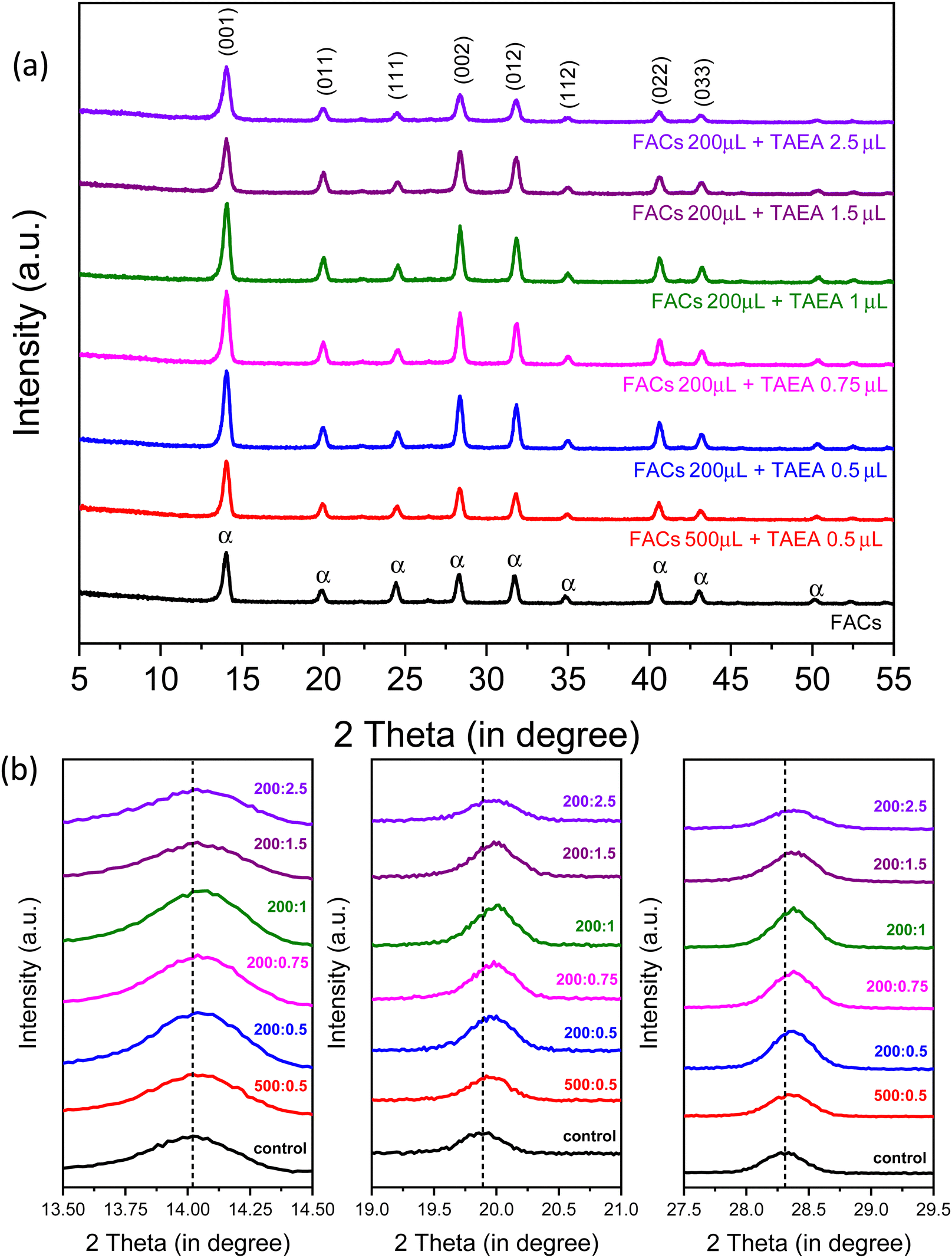 | ||
| Fig. 1 Crystallography analysis of FA0.83Cs0.17Pb(I0.90Br0.10)3 thin film. (a) XRD for FA0.83Cs0.17Pb(I0.90Br0.10)3 film containing different concentrations of TAEA added inside perovskite precursor. (b) XRD peak shift for 2θ around 14°, 20°, and 28° for TAEA containing films with different concentrations of TAEA added in FA0.83Cs0.17Pb(I0.90Br0.10)3 perovskite precursor. | ||
With the increase in the concentration of TAEA, an enhancement in the XRD peak intensity is noticed until the concentration where 1 μL of TAEA is added into 200 μL of the FACs precursor, and beyond this the peak intensity is almost similar to the control FACs film. This confirms that the addition of TAEA leads to an improvement in the crystallinity of the FACs films. Fig. 1b shows the individual XRD peak shift in peaks at around 2θ values 14°, 20°, and 28°. With the incorporation of TAEA in FACs perovskite the XRD peaks shift towards higher 2θ values which confirms the presence of TAEA.
Absorption spectra were recorded to assess the impact of the incorporation of TAEA in FACs perovskite on the absorbance properties. An absorption profile with an absorption edge at around 780 nm corresponding to a bandgap of around 1.6 eV is detected for all the films. The UV-Vis absorption spectra shown in Fig. 2a show an increase in the absorbance with the addition of TAEA in FACs perovskite. The maximum absorbance is observed for the FACs perovskite film containing 1 μL of TAEA in 200 μL of perovskite precursor. Fig. 2b shows the corresponding Tauc plots of the UV-Vis spectra shown in Fig. 2a. It is observed that with the increase in the TAEA concentration, there is a very slight increase in the bandgap of the perovskite from 1.590 to 1.616 eV.
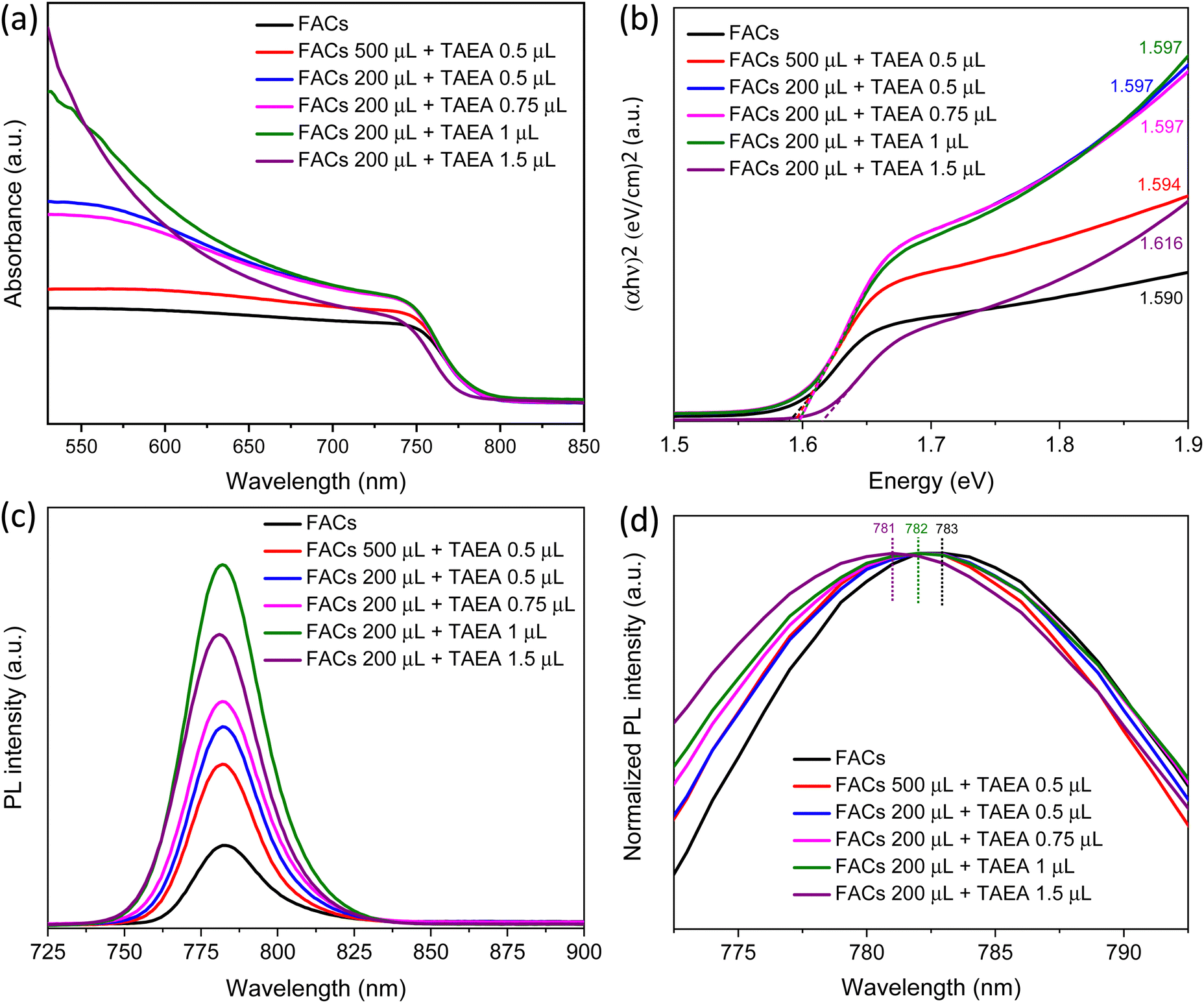 | ||
| Fig. 2 UV-Vis absorption and PL spectra. (a) UV-Vis absorption spectra and (b) Tauc plot for FACs films with different concentrations of TAEA added in perovskite precursor. (c) PL spectra and (d) a normalized PL plot for FACs films with different concentrations of TAEA added in perovskite precursor. | ||
Steady-state photoluminescence (PL) was recorded to observe the effect of TAEA addition on the optoelectronic properties of the FACs perovskite. An increase in the PL intensity is observed with the incorporation of TAEA when compared with the control film as shown in Fig. 2c. All the TAEA containing films show better photoluminescence than the pristine FACs film, indicating a great reduction in the defect-mediated non-radiative recombination. In accordance with the increase in the crystallinity of the film as noticed from the XRD spectra, the PL intensity also increases with the increase in the concentration of TAEA in perovskite precursor until reaching a concentration where 1 μL of TAEA is added into 200 μL of precursor. However, beyond this concentration, the PL intensity starts to decrease. So, as evident from the XRD analysis, UV-Vis absorption spectra and PL spectra the optimum concentration of TAEA in the FACs precursor is considered to be 1 μL in 200 μL, respectively. Normalized PL is plotted as shown in Fig. 2d to examine the peak shift with the incorporation of TAEA. There is a negligible blue shift in the peak position of the order of ∼1–2 nm which is in accordance with the aforementioned very slight increase in the bandgap detected from the UV-Vis absorption spectra.
The size of TAEA is large as compared to FA and Cs, therefore it is less likely that it will substitute the A-site cations in the FACs perovskite lattice. Additionally, there is no sign of 2D phase formation from the XRD, UV-Vis, and PL studies. There are no low angle peaks observed in the XRD patterns with 2θ less than 10°, which carries the signature of 2D phase formation.44,45 Additionally, the single PL peak and lack of any excitonic peak in the UV-Vis spectra clearly hint towards the absence of a 2D phase.45–47 So, most likely the TAEA is present on the surface of the perovskite. Therefore, the shift in the XRD peaks towards higher 2θ values can possibly be caused by the compressive strain present in the TAEA containing films. The strain could be induced due to the interaction of TAEA with the perovskite lattice during or after crystallization, introduced distortions in the octahedral environment surrounding the metal cations (Pb). The presence of TAEA on the surface of the perovskite crystals may affect the local atomic arrangement and crystal symmetry, causing shifts in the XRD peaks. Therefore, we have carried out Williamson–Hall analysis to calculate the strain from XRD patterns and the calculated strain is plotted below in Fig. S1 (ESI†). The plot shows an increase in the microstrain with an increase in the concentration of TAEA in the FACs precursor.
The impact of TAEA addition on the FACs film morphology was observed using field emission scanning electron microscopy (FESEM) and is presented in Fig. 3. Fig. 3a and b show the morphology of the fresh FACs film with high and low magnifications, respectively. The FACs film shows a rough morphology with a lot of large pinholes in the order of a few μm as shown in the low magnification image, which could be detrimental to the device performance. This could be the reason for the aforementioned lowest PL intensity observed in the FACs film. As TAEA is added inside the precursor of FACs the pinholes start to decrease as shown in Fig. 3d, f and h until the concentration where 0.75 μL of TAEA is added into 200 μL of perovskite precursor. When the concentration of TAEA is further increased to 1 μL per 200 μL of FACs precursor, the pinholes are completely removed and we obtain a uniform and compact film. However, with a further increase in the concentration of TAEA the pinholes reappear as shown in Fig. 3l. This could possibly be the reason for the aforementioned highest PL observed in the 1 μL TAEA concentration which starts reducing with a further increase in TAEA concentration.
 | ||
Fig. 3 FESEM morphology of fresh FACs film (a) high magnification (50k×), (b) low magnification (5k×); FACs film with TAEA with a volumetric concentration 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 (c) high magnification, (d) low magnification; FACs film with TAEA with a volumetric concentration 200:0.5 (e) high magnification, (f) low magnification; FACs film with TAEA with a volumetric concentration 200:0.75 (g) high magnification, (h) low magnification; FACs film with TAEA with a volumetric concentration 200:1 (i) high magnification, (j) low magnification; FACs film with TAEA with a volumetric concentration 200:1.5 (k) high magnification, and (l) low magnification. :0.5 (c) high magnification, (d) low magnification; FACs film with TAEA with a volumetric concentration 200:0.5 (e) high magnification, (f) low magnification; FACs film with TAEA with a volumetric concentration 200:0.75 (g) high magnification, (h) low magnification; FACs film with TAEA with a volumetric concentration 200:1 (i) high magnification, (j) low magnification; FACs film with TAEA with a volumetric concentration 200:1.5 (k) high magnification, and (l) low magnification. | ||
It is also observed from the high magnification images presented in Fig. 3a, c, e, g, i and k that the grain size is reduced with the incorporation of TAEA. Micro-strain induced by introducing TAEA could have resulted in the reduced crystallite size. Additionally, the films become more compact and smoother with the increase in the concentration of TAEA. This could be ascertained from the atomic force microscopy (AFM) images shown in Fig. 4. The 2D and 3D AFM images confirm that the incorporation of TAEA leads to smoothening of the films as the root mean square roughness (Rrms) is reduced with the increase in the concentration of TAEA. The lowest Rrms = 58.47 nm is detected in the film containing 1 μL of TAEA per 200 μL of FACs precursor, which is increased to a value of 61.72 nm on further increase in TAEA concentration. So, from FESEM and AFM it is witnessed that the film containing 1 μL of TAEA per 200 μL of FACs precursor renders a smooth, uniform, compact, and pinhole free film, which is critical for efficient planar heterojunction-devices.
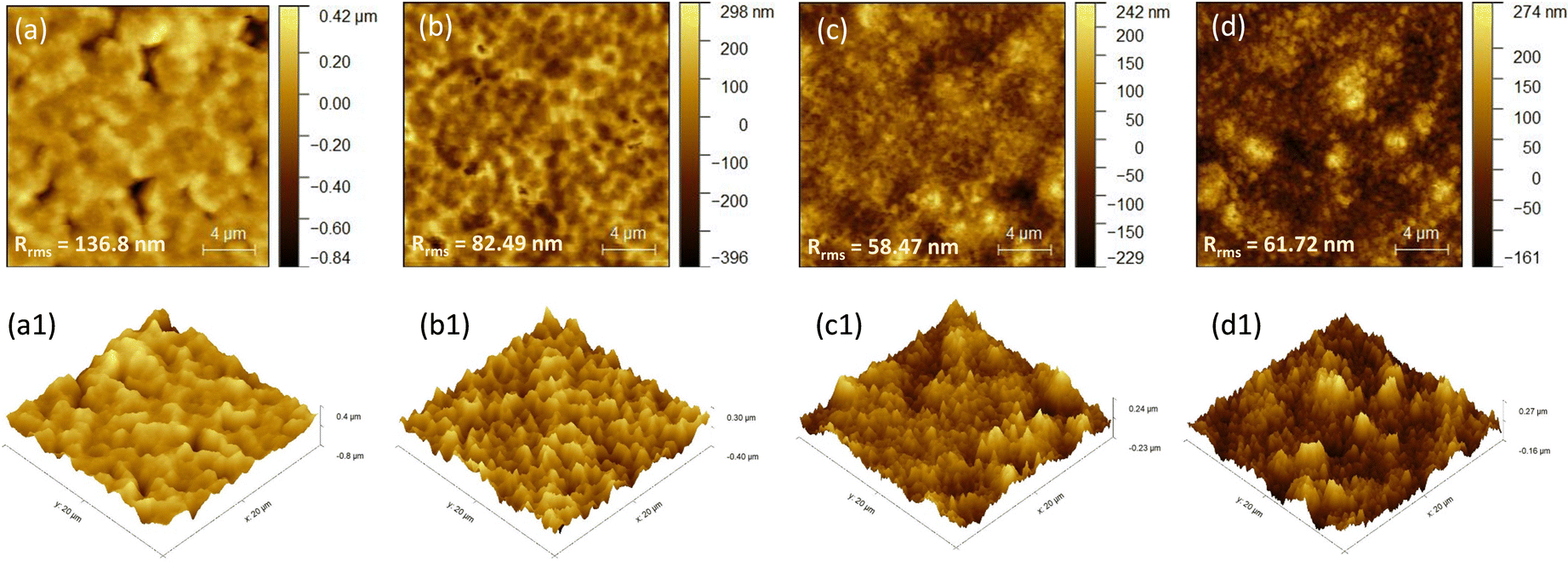 | ||
| Fig. 4 AFM images. (a) 2D and (a1) 3D AFM topography images of FACs film, (b) 2D and (b1) 3D AFM topography images of FACs film with TAEA with a volumetric concentration of 200:0.5, (c) 2D and (c1) 3D AFM topography images of FACs film with TAEA with a volumetric concentration of 200:1, and (d) 2D and (d1) 3D AFM topography images of FACs film with TAEA with a volumetric concentration of 200:1.5. | ||
Scanning laser confocal microscopy (SLCM) was also performed to examine the spatial distribution of luminescence intensity. Images in Fig. 5a–f represent the luminescence maps of the FACs films with different concentrations of TAEA. The pristine FACs film is shown to have a number of dark spots which depict the non-radiative emission sites. These dark spots mainly represent the defect sites at grain boundaries and the pinholes in the film as observed from the FESEM image in Fig. 3b. Additionally, the fluorescence density of 1.71 × 106 which is calculated over an area of 16384 μm2 in pristine FACs film is also the lowest among all the films as presented in Fig. 5g. With the addition of a small amount of TAEA in FACs (0.5 μL in 200 μL) the black spots are significantly reduced as can be seen from Fig. 5b and consequently there is an increase in the fluorescence density (= 1.95 × 106) as well. Moreover, a further increase in the concentration of TAEA in FACs precursor leads to greatly reduced black spots, and the fluorescence density is increased to a value of 2.09 × 106. This is in agreement with the aforementioned increase in steady-state PL intensity and reduction in the pinholes with the increase in the concentration of TAEA as depicted in Fig. 2c and 3.
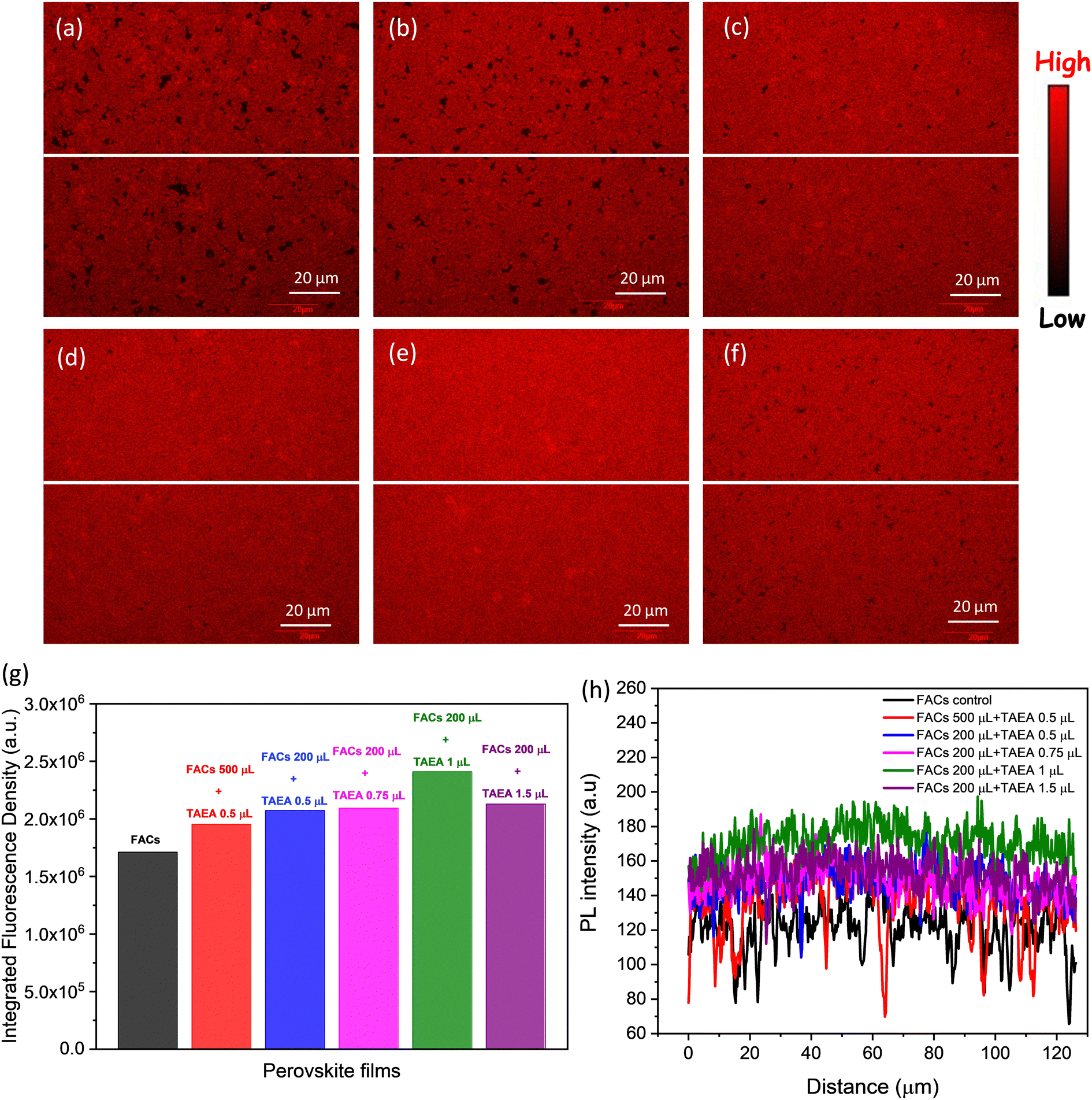 | ||
| Fig. 5 SLCM PL intensity maps of (a) FACs film, FACs film containing (b) 0.5 μL of TAEA in 500 μL of FACs precursor, (c) 0.5 μL of TAEA in 200 μL of FACs precursor, (d) 0.75 μL of TAEA in 200 μL of FACs precursor, (e) 1 μL of TAEA in 200 μL of FACs precursor, and (f) 1.5 μL of TAEA in 200 μL of FACs precursor. (g) Integrated fluorescence density in the films calculated in the 128 μm × 128 μm area of the sample. (h) PL line profile plotted along the breadth of the samples as denoted by the white horizontal lines in (a)–(f). | ||
The film with 1 μL in 200 μL of FACs shows the most uniform and brightest emission distribution among the other compositions without any visible pinholes, consistent with the PL data shown in Fig. 2c. This film shows the highest fluorescence density of 2.41 × 106 among all the films. The observation from the spatial PL is consistent with previous observations and this is the optimum composition showcasing the best structural, morphological, and optoelectronic properties. Further increase in the TAEA concentration leads to the reappearance of black spots and reduced fluorescence density (= 2.13 × 106), as observed from Fig. 5f and g.
To assess the heterogeneity of PL emission across the sample, a line profile was plotted across the breadth of the film as depicted by the white line in Fig. 5a–f. The FACs film shows the most inhomogeneous distribution of PL intensity across the sample with a lot of dips and showcases the lowest PL intensity. With the increase in the concentration of TAEA in the precursor, the PL intensity becomes more homogeneous and the overall PL intensity across the film is increased. Inspiringly, the highest PL intensity across the film is detected for the film with 1 μL of TAEA in 200 μL of FACs precursor, which is consistent with the aforementioned observations.
To understand the degradation of perovskite solar cells and find failures, stability testing under enhanced environmental stressing conditions such as high humidity, elevated temperature, and illumination becomes crucial. Therefore, all the films were kept outside under ambient temperature with RH ∼ 60–70% to assess the impact of TAEA on the stability of the FACs films. Digital photographs of the films with time are presented in Fig. 6a. All the fresh films are initially found to be blackish in color, indicating the crystallization of films in the photoactive α-phase. The pristine FACs film started turning yellow towards the edges and corners after 110 days of exposure to the ambient environment and after 150 days a major portion of the film had turned yellow. However, all the FACs films containing different TAEA concentrations show a stable photoactive phase even after 180 days of exposure to light and highly humid conditions.
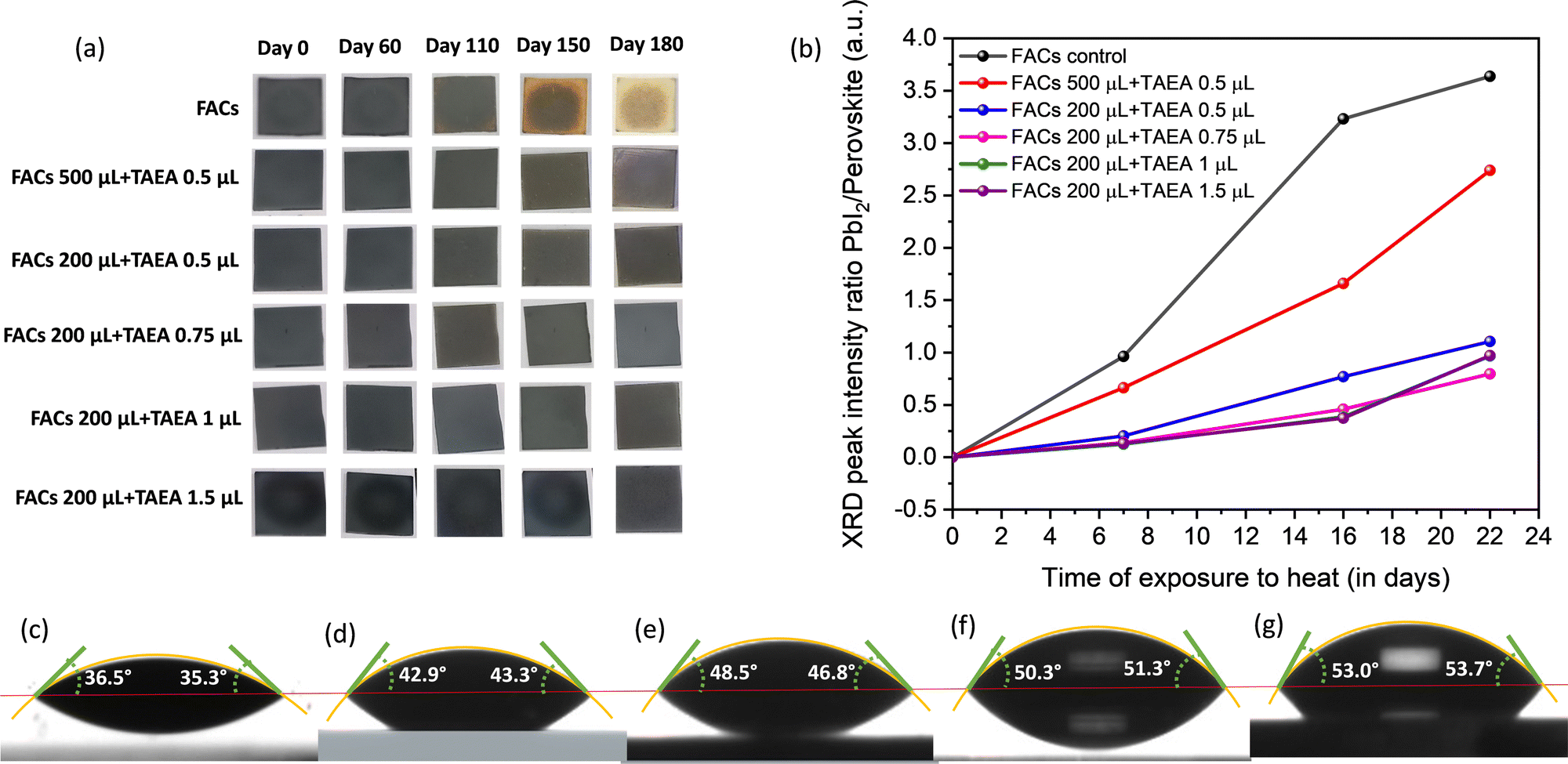 | ||
| Fig. 6 (a) Digital photographs of FACs films with different concentrations of TAEA upon exposure to an ambient atmosphere with RH ∼ 60–70%. (b) XRD peak intensity ratio of PbI2/perovskite versus time of exposure plot for FACs films with different concentrations of TAEA exposed to continuous heat at 85 °C in ambient atmosphere. Contact angle measurement for (c) the FACs film, and the FACs film containing (d) 0.5 μL of TAEA in 500 μL of FACs precursor, (e) 0.5 μL of TAEA in 200 μL of FACs precursor, (f) 1 μL of TAEA in 200 μL of FACs precursor, and (g) 1.5 μL of TAEA in 200 μL of FACs precursor. | ||
The XRD patterns of the FACs film and the optimized FACs film containing 1 μL of TAEA are presented in Fig. S2 (ESI†). It is clearly evident from the XRD pattern that both the films retain the initial α-phase until 60 days of exposure to ambient conditions. However, the pristine FACs film started degrading after 110 days as can be ascertained from the appearance of peaks corresponding to PbI2, δ-FAPbI3, and δ-CsPbI3 at 2θ values ≈ 12.9°, 11.2°, and 9.86°, respectively. Whereas, the TAEA-containing film retains the α-FAPbI3 phase even after 110 days of exposure to light and highly humid conditions, which is in accordance with the physical appearance of the films in terms of no color change noticed as shown in Fig. 6a. It is observed that the UV-Vis absorption spectra shown in Fig. S3 (ESI†) show a negligible decrease in the absorbance of the TAEA-containing films even after 110 days of exposure to RH ∼ 60–70%. This ascertains the fact that the film retains the initial perovskite phase even after exposure to highly humid conditions.
Water contact angle measurement was carried out to explore the surface wetting properties of the FACs films with different concentrations of TAEA as shown in Fig. 6c–g. With an increase in the concentration of TAEA the contact angle is increased signifying the decrease in the affinity to water with increasing amounts of TAEA. So, it is evident from the observations that the greater the amount of TAEA in the precursor the more the film surface is resistant to water ingress due to the presence of alkyl chains in TAEA. This explicates the reason for the higher ambient stability achieved in the TAEA containing films when compared to the pristine FACs film.
Apart from ambient stability, thermal stability is also crucial and one of the most challenging parameters for perovskite materials to survive. Hence, all the films were kept at continuous heat at 85 °C in an ambient environment with high humidity RH ∼ 60–70% to explore the thermal stability. XRD patterns were recorded at different intervals of time to assess the amount of degradation and the impact on the structural properties upon exposure to heat, as presented in Fig. S4 (ESI†). It is observed that after 7 days of continuous exposure to the ambience and heat the pristine FACs film shows the appearance of a diffraction peak corresponding to PbI2 which is of almost equal intensity as that of the main (001) perovskite peak as shown in Fig. S4a (ESI†). However, as we keep increasing the amount of TAEA in the FACs films the intensity of the PbI2 peak compared to the (001) perovskite peak keeps reducing and it is almost negligible for higher concentrations of TAEA starting from 0.75 μL of TAEA in 200 μL of FACs precursor. This can be clearly visualized from the ratio of PbI2/perovskite XRD peak intensity versus the time plot shown in Fig. 6b. After 16 days of exposure to heat and humidity, two low intensity peaks pertaining to δ-FAPbI3 and δ-CsPbI3 start to appear along with the PbI2 peak in the pristine FACs film, whose intensity has increased over further heating past this time. However in the TAEA containing films these peaks are absent even after 22 days of continuous heating. These observations clearly signify that the presence of TAEA in the FACs films has a remarkable effect on slowing down the degradation process in the presence of both heat and high humidity. Additionally, the incorporation of TAEA has also resulted in resisting the α → δ phase transition in the perovskite for a much longer duration, which is the biggest limitation in FA and Cs based perovskite materials.
X-ray photoelectron spectroscopy (XPS) studies were carried out as presented in Fig. 7, to gain insight into the chemical composition of FACs perovskite films with and without TAEA. The two Pb2+ 4f peaks are detected at binding energies (BEs) of 142.9 and 138 eV for the pristine FACs film which can be assigned to 4f5/2 and 4f7/2 core levels as shown in Fig. 7a. Along with these, two weak shoulder peaks at lower BEs of 141.3 and 136.3 eV are also detected that correspond to the metallic Pb0 4f. Metallic lead (Pb0) is detrimental to device performance as its presence indicates the presence of defect sites, resulting in non-radiative recombination.48,49 Pb0 reflects the presence of iodide and cation vacancies in the perovskite. With the addition of TAEA, there is a shift of the Pb2+ peaks towards higher BEs and the Pb0 peak intensity has reduced to almost negligible magnitude. This shows that the TAEA incorporation is affecting the chemical environment of the Pb and also inhibiting the defects, hence contributing to the greatly decreased Pb0 peak.
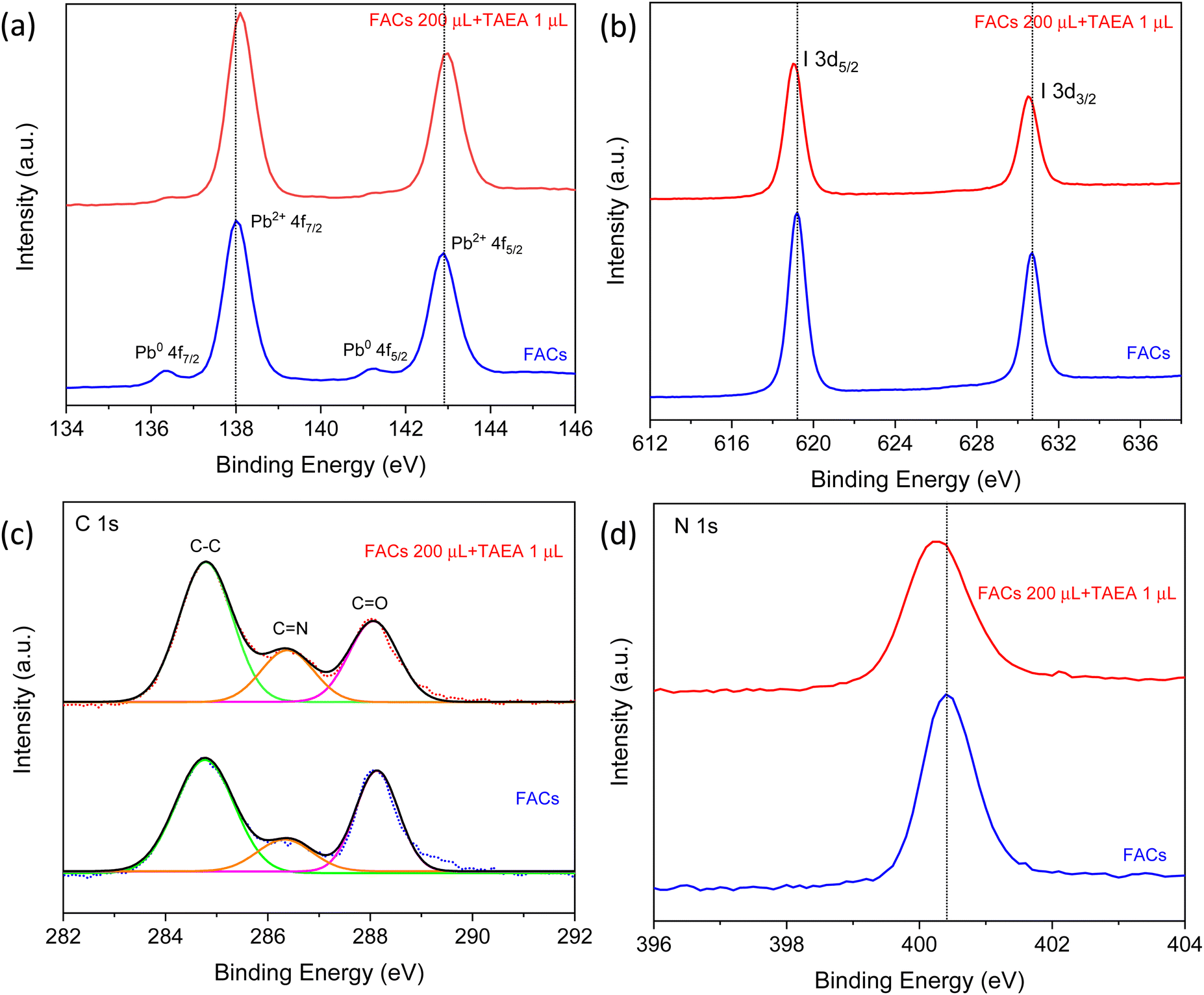 | ||
| Fig. 7 XPS spectra of (a) Pb 4f, (b) I 3d, (c) C 1s, and (d) N 1s for FACs perovskite films with and without TAEA. | ||
The peaks at 630.7 and 619.2 eV in the pristine FACs films are associated with I 3d3/2 and I 3d5/2, respectively, as shown in Fig. 7b. The addition of TAEA results in the shifting of these peaks towards lower BEs indicating the interaction of I− with TAEA. It is previously reported that an organic additive containing –NH2 can form a N–H⋯X H-bond with the halide ions and hence helps in restricting the migration of halide ions pertaining to the ionic nature of perovskites.50 This suppression of the migration of I− results in reducing the iodide defects and enhancing the hydrophobicity of perovskite.
The BEs in C 1s XPS spectra at 284.8, and 286.4 eV as presented in Fig. 7c are assigned to the C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH2+, respectively from FA+. The peak at 288 eV corresponds to CO, which reflects the degradation of perovskite film on exposure to the ambient environment.39 The peak area of CO visibly decreases with the addition of TAEA which implies that the target film is more resistant to H2O or O2 and hence exhibits improved film stability. The N 1s peak at BE 400.4 eV shifts slightly towards the lower BE value of 400.3 eV, indicating that the BE of N in FA is affected by the interaction of TAEA with the lattice as shown in Fig. 7d.
NH2+, respectively from FA+. The peak at 288 eV corresponds to CO, which reflects the degradation of perovskite film on exposure to the ambient environment.39 The peak area of CO visibly decreases with the addition of TAEA which implies that the target film is more resistant to H2O or O2 and hence exhibits improved film stability. The N 1s peak at BE 400.4 eV shifts slightly towards the lower BE value of 400.3 eV, indicating that the BE of N in FA is affected by the interaction of TAEA with the lattice as shown in Fig. 7d.
Amine additives are shown to be capable of modulating the crystallization process. TAEA can coordinate with the metal cations present in the perovskite structure such as Pb2+ through its amino groups, which can act as ligands to form coordination bonds with the metal cations. This facilitates a better nucleation and crystallization process and leads to the formation of well-ordered and more crystalline films. The amino groups in TAEA can also bind to the undercoordinated surface Pb ions in the perovskite, forming coordination bonds.51–53 TAEA can passivate the crystallographic defect and trap states present on the surface of perovskite films, reducing the non-radiative recombination and improving the overall optical and electronic properties of the perovskite material. This enhanced surface passivation prevents the formation of unwanted surface defects, leading to enhanced crystallinity. This is also ascertained from the Pb2+ 4f peak shift and reduced metallic Pb0 peak observed in XPS spectra of TAEA containing the FACs film shown in Fig. 7a.
TAEA is a large sized molecule and hence cannot fit into the voids of the [PbI6]4− polyhedral frame. Therefore, it is highly likely that TAEA is not substituting the A-site cations in the FA0.83Cs0.17Pb(I0.90Br0.10)3 lattice and lies on the surface. Water contact angle measurements shown in Fig. 6c–g show an increase in the contact angle with an increase in the concentration of TAEA in the perovskite precursor, which signifies the increased hydrophobicity of the surface with increased TAEA concentrations. So, the possibility of the presence of TAEA on the surface is very high, which makes the surface more resistant to water ingress and thus provides a rational explanation for the enhanced ambient stability.
The amino groups in TAEA (–NH2) can coordinate with metal cations such as Pb2+ in the perovskite structure. This can involve the formation of coordination bonds between the nitrogen atoms of the amino groups and the metal cations as shown in the schematic shown in Fig. 8a. It is previously reported that –NH2 containing additives tend to form N–H⋯X hydrogen bonds (H-bonds) with the halides in the perovskite structure (I and Br).54 This strong interaction between –NH2 and the halide, suppresses I− ion migration as shown by the schematic in Fig. 8a. Ion migration can lead to the creation of defects and degradation of the crystal structure. Thermal decomposition of the perovskite can occur, especially in the presence of moisture and oxygen. High-quality, defect-free films with well-defined crystal structures are more stable under thermal stress. Due to the longer bond length of TAEA it can cross-link the two adjacent perovskite units on the surface making H-bonds with the halides. Therefore, two branches of TAEA may bond with the surface of FACs perovskite and the third is protruding out of the surface as shown in Fig. 8a. Therefore, TAEA can act as a protective layer, shielding the perovskite from moisture and oxygen, which are known to cause degradation in perovskite materials. This protection is essential for retaining the α-phase at room temperature and highly humid conditions for the long term as observed from the degradation studies shown in Fig. 6a, b and Fig. S2, S4 (ESI†).
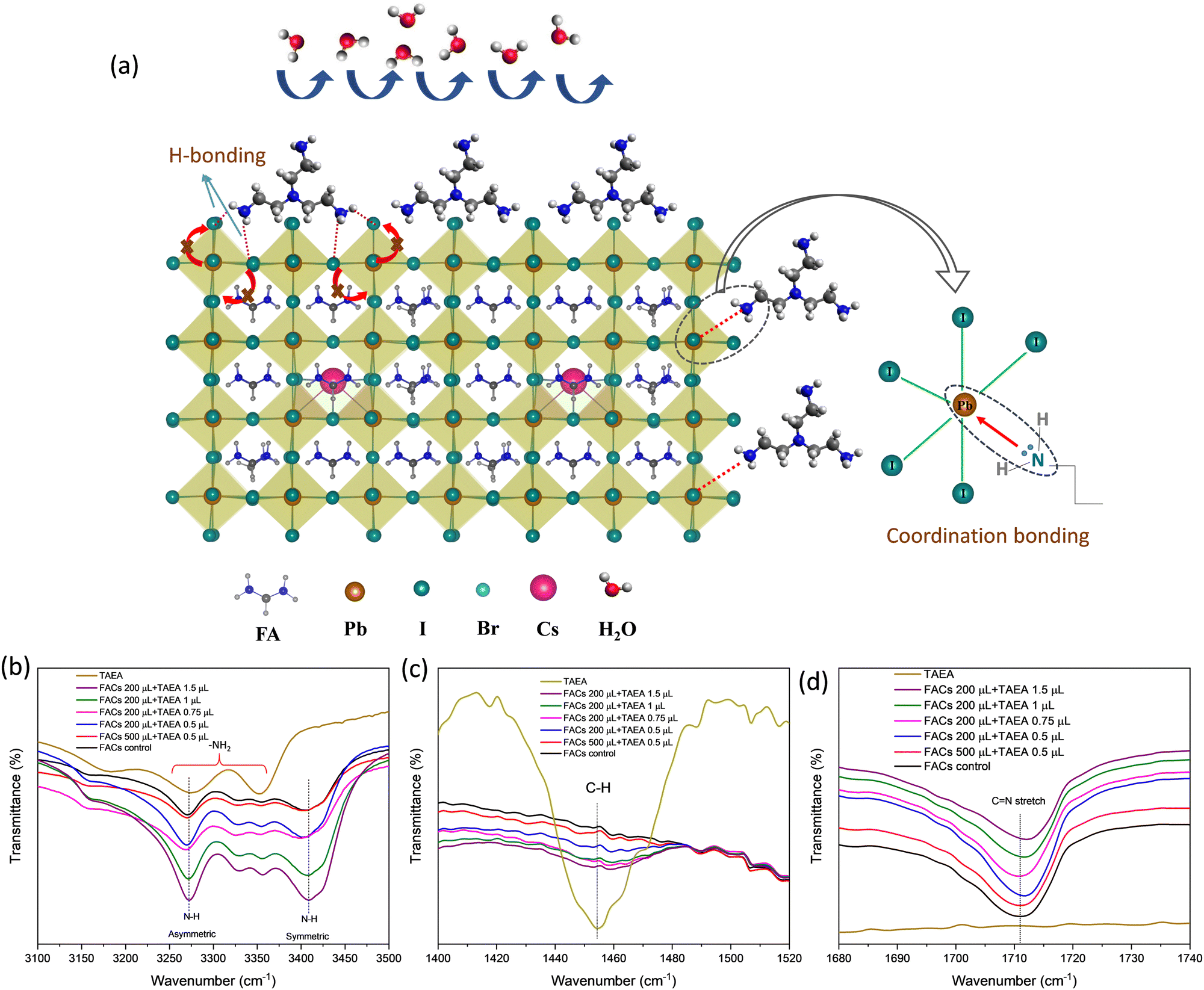 | ||
| Fig. 8 (a) Schematic showing the interaction of TAEA with the FACs lattice. FTIR spectra showing the TAEA and FACs perovskite film containing different concentrations of TAEA showing (b) N–H stretching, (c) C–H stretching, and (d) CN stretching. | ||
To trace the interaction between TAEA and FACs perovskite lattice, Fourier Transform Infrared spectroscopy (FTIR) was performed as shown in Fig. 8b–d. FTIR spectra of TAEA presented in Fig. 8b show broad peaks in the range 3200–3400 cm−1 corresponding to the –NH2 vibrational modes. FTIR peaks corresponding to N–H stretching are detected in all the FACs films. FTIR peaks in FACs films at 3270 cm−1 and 3408 cm−1 are associated with the asymmetric and symmetric stretching vibrations of the N–H bond of FA+, respectively.55 An increase in the TAEA concentration leads to the broadening of the peak and an increase in the peak intensity, which could account for the H-bond formation between the –NH2 in TAEA with the perovskite lattice.56 The FTIR peak at around 1458 cm−1 shown in Fig. 8c corresponds to the stretching due to the C–H bond in TAEA. This peak is not present in the control FACs film, however, as we go on increasing the concentration of TAEA in FACs this peak starts to appear. It again validates the fact that TAEA has bonded with the perovskite and is present in the film. The peak intensity is greatly reduced with the incorporation of TAEA in the FACs film, owing to the interaction between TAEA and the FACs lattice. The characteristic FTIR peak near 1711 cm−1 corresponds to the CN stretching present due to the presence of FA+. This stretching is not present in TAEA as shown in Fig. 8d. However, when TAEA is added into the FACs perovskite this peak is shifted which hints towards interaction between the FA+ and TAEA. The hydrogen atom from the amino group in TAEA can form a hydrogen bond with the nitrogen atom of the amide group in FA. The hydrogen atom is attracted to the lone pair of electrons on the nitrogen atom, creating a hydrogen bond. This bonding can suppress the volatilization of FA from perovskite and further improve the stability.
Conclusions
In summary, we have presented a systematic study of the effect of different concentrations of TAEA on the FACs-based perovskite. The incorporation of TAEA in the FACs precursor has led to enhanced structural, absorbance, and optoelectronic properties. The optimum concentration of TAEA in FACs is found to be 1 μL in 200 μL. TAEA incorporation results in reduced pinholes and hence yields a compact, smother, and uniform morphology. TAEA containing FACs films are stable for more than 180 days in a highly humid and illuminated ambient environment, whereas the pristine FACs begins to degrade within 110 days of exposure. The incorporation of TAEA in FACs films has also led to a drastic enhancement in thermal stability. TAEA containing FACs films are stable for more than 7 days under continuous exposure to extreme conditions of high humidity, illumination, and thermal stress at 85 °C. So, this study provides a scientific approach for long-term stabilization of the most commonly studied and promising mixed cation FACs perovskite composition, paving the way for its mass commercialization as an efficient and long run stable perovskite technology.Experimental section
Materials and methods
:1. The precursor solution was stirred for 15 min at 70 °C prior to deposition. TAEA was added into FA0.83Cs0.17Pb(I0.90Br0.10)3 precursor in different volume-by-volume ratios of FA0.83Cs0.17Pb(I0.90Br0.10)3/TAEA (500 μL:0.5 μL, 200 μL:0.5 μL, 200 μL:0.75 μL, 200 μL:1 μL, and 200 μL:1.5 μL) and shaken vigorously on a shaker for a couple of hours to obtain a clear solution.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to acknowledge the Department of Science and Technology, India for providing funding. P. C. acknowledges IIT Delhi for providing Senior Research Fellowship. We would also like to acknowledge Nanoscale Research Facility (NRF) and Central Research Facility (CRF) at the Indian Institute of Technology, Delhi for characterization measurements.Notes and references
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 1979, 342, 341–344 CrossRef PubMed.
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T. W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582–587 Search PubMed.
- National Renewable Energy Laboratory NREL. Best-Research Cell-Efficiencies, 2023.
- T. A. Berhe, W. N. Su, C. H. Chen, C. J. Pan, J. H. Cheng, H. M. Chen, M. C. Tsai, L. Y. Chen, A. A. Dubale and B. J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. De Angelis and H. G. Boyen, Adv. Energy Mater., 2015, 15, 1500477 CrossRef.
- A. Rana, P. Sharma, A. Kumar, S. Pareek, S. Waheed, R. K. Singh and S. Karak, IEEE Trans. Electron Devices, 2021, 68, 3907–3913 CAS.
- M. Bag, L. A. Renna, R. Y. Adhikari, S. Karak, F. Liu, P. M. Lahti, T. P. Russell, M. T. Tuominen and D. Venkataraman, J. Am. Chem. Soc., 2015, 137, 13130–13137 CrossRef CAS PubMed.
- B. P. Dhamaniya, P. Chhillar, B. Roose, V. Dutta and S. K. Pathak, ACS Appl. Mater. Interfaces, 2019, 25, 22228–22239 CrossRef PubMed.
- P. Chhillar, B. P. Dhamaniya, V. Dutta and S. K. Pathak, ACS Omega, 2019, 4, 11880–11887 CrossRef CAS PubMed.
- J. W. Lee, D. J. Seol, A. N. Cho and N. G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS PubMed.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- A. Binek, F. C. Hanusch, P. Docampo and T. Bein, J. Phys. Chem. Lett., 2015, 6, 1249–1253 CrossRef CAS PubMed.
- N. Pellet, P. Gao, G. Gregori, T. Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- F. Yao, P. Gui, C. Chen, B. Li, R. Li, C. Tao, Q. Lin and G. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 39875–39881 CrossRef CAS PubMed.
- R. Chen, Y. Wu, Y. Wang, R. Xu, R. He, Y. Fan, X. Huang, J. Yin, B. Wu, J. Li and N. Zheng, Adv. Funct. Mater., 2021, 11, 2008760 CrossRef.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, A. J. Pearson, G. Divitini, S. Cacovich, B. Philippe, H. Rensmo, C. Ducati, R. H. Friend and S. D. Stranks, ACS Energy Lett., 2018, 3, 2671–2678 CrossRef CAS PubMed.
- J. W. Lee, D. H. Kim, H. S. Kim, S. W. Seo, S. M. Cho and N. G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- S. Masi, A. F. Gualdrón-Reyes and I. Mora-Seró, ACS Energy Lett., 2020, 5, 1974–1985 CrossRef CAS.
- S. Maniyarasu, J. C.-R. Ke, B. F. Spencer, A. S. Walton, A. G. Thomas and W. R. Flavell, ACS Appl. Mater. Interfaces, 2021, 13, 43573–43586 CrossRef CAS PubMed.
- D. P. Mcmeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- Y. Hu, E. M. Hutter, P. Rieder, I. Grill, J. Hanisch, M. F. Aygüler, A. G. Hufnagel, M. Handloser, T. Bein, A. Hartschuh, K. Tvingstedt, V. Dyakonov, A. Baumann, T. J. Savenije, M. L. Petrus and P. Docampo, Adv. Energy Mater., 2018, 8, 1703057 CrossRef.
- J. Yang, X. Liu, Y. Zhang, X. Zheng, X. He, H. Wang, F. Yue, S. Braun, J. Chen, J. Xu, Y. Li, Y. Jin, J. Tang, C. Duan, M. Fahlman and Q. Bao, Nano Energy, 2018, 54, 218–226 CrossRef CAS.
- F. X. Liang, J. Z. Wang, Z. X. Zhang, Y. Y. Wang, Y. Gao and L. B. Luo, Adv. Opt. Mater., 2017, 22, 1700654 CrossRef.
- P. Chhillar, B. P. Dhamaniya, S. K. Pathak and S. Karak, ACS Appl Electron. Mater., 2022, 4, 5368–5378 CrossRef CAS.
- K. Wang, Z. Jin, L. Liang, H. Bian, H. Wang, J. Feng, Q. Wang and S. (Frank) Liu, Nano Energy, 2019, 58, 175–182 CrossRef CAS.
- H. Zhao, J. Xu, S. Zhou, Z. Li, B. Zhang, X. Xia, X. Liu, S. Dai and J. Yao, Adv. Funct. Mater., 2019, 27, 1808986 CrossRef.
- B. Zhao, S. F. Jin, S. Huang, N. Liu, J. Y. Ma, D. J. Xue, Q. Han, J. Ding, Q. Q. Ge, Y. Feng and J. S. Hu, J. Am. Chem. Soc., 2018, 140, 11716–11725 CrossRef CAS PubMed.
- R. J. Sutton, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, F. Giustino and H. J. Snaith, ACS Energy Lett., 2018, 3, 1787–1794 CrossRef CAS.
- N. K. Cho, H. J. Na, J. Yoo and Y. S. Kim, Commun. Mater., 2021, 1, 30 CrossRef.
- W. Zhu, A. A. Khan, M. M. Rana, R. Gautheron-Bernard, N. R. Tanguy, N. Yan, P. Turban, S. Ababou-Girard and D. Ban, ACS Omega, 2022, 7, 10559–10567 CrossRef CAS PubMed.
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J. M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni, Y. Sanehira, M. Ikegami and T. Miyasaka, Chem. Mater., 2018, 30, 6668–6674 CrossRef CAS.
- G. E. Eperon, G. M. Paternò, R. J. Sutton, A. Zampetti, A. A. Haghighirad, F. Cacialli and H. J. Snaith, J. Mater. Chem. A Mater., 2015, 3, 19688–19695 RSC.
- J. Tian, Q. Xue, Q. Yao, N. Li, C. J. Brabec and H. L. Yip, Adv. Energy Mater., 2020, 10 Search PubMed.
- Y. Wang, J. Song, J. Ye, Y. Jin, X. Yin, Z. Su, L. Hu, Y. Wu, C. Qiu, H. Wang, W. Yan and Z. Li, Chem. Commun., 2023, 43, 6580–6583 RSC.
- Z. Wang, Q. Lin, F. P. Chmiel, N. Sakai, L. M. Herz and H. J. Snaith, Nat. Energy, 2017, 9, 1–10 Search PubMed.
- D. Vaitukaityte, Z. Wang, T. Malinauskas, A. Magomedov, G. Bubniene, V. Jankauskas, V. Getautis and H. J. Snaith, Adv. Mater., 2018, 45, 1803735 CrossRef PubMed.
- W. Rehman, D. P. McMeekin, J. B. Patel, R. L. Milot, M. B. Johnston, H. J. Snaith and L. M. Herz, Energy Environ. Sci., 2017, 10, 361–369 RSC.
- G. Murugadoss, P. Arunachalam, S. K. Panda, M. Rajesh Kumar, J. R. Rajabathar, H. Al-Lohedan and M. D. Wasmiah, J. Mater. Res. Technol., 2021, 12, 1924–1930 CrossRef CAS.
- X. Ding, H. Chen, Y. Wu, S. Ma, S. Dai, S. Yang and J. Zhu, J. Mater. Chem. A Mater., 2018, 6, 18258–18266 RSC.
- Y. Jiang, J. Yuan, Y. Ni, J. Yang, Y. Wang, T. Jiu, M. Yuan and J. Chen, Joule, 2018, 2, 1356–1368 CrossRef.
- F. Li, Y. Pei, F. Xiao, T. Zeng, Z. Yang, J. Xu, J. Sun, B. Peng and M. Liu, Nanoscale, 2018, 10, 6318–6322 RSC.
- K. Wang, Z. Li, F. Zhou, H. Wang, H. Bian, H. Zhang, Q. Wang, Z. Jin, L. Ding and S. Liu, Adv. Energy Mater., 2019, 42, 1902529 CrossRef.
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S. H. Jang, J. Zhang, X. Lu, D. Lei, C. S. Lee, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed.
- Y. Zhang and N. G. Park, ACS Energy Lett., 2022, 7, 757–765 CrossRef CAS.
- F. Zhang, J. Song, B. Cai, X. Chen, C. Wei, T. Fang and H. Zeng, Sci. Bull., 2021, 66, 2189–2198 CrossRef CAS PubMed.
- M. Li, R. Sun, J. Chang, J. Dong, Q. Tian, H. Wang, Z. Li, P. Yang, H. Shi, C. Yang, Z. Wu, R. Li, Y. Yang, A. Wang, S. Zhang, F. Wang, W. Huang and T. Qin, Nat. Commun., 2023, 14, 573 CrossRef CAS PubMed.
- W.-Q. Wu, Z. Yang, P. N. Rudd, Y. Shao, X. Dai, H. Wei, J. Zhao, Y. Fang, Q. Wang, Y. Liu, Y. Deng, X. Xiao, Y. Feng and J. Huang, Science, 2019, 5, eaav8925 CAS.
- X. Hou, S. Huang, W. Ou-Yang, L. Pan, Z. Sun and X. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 35200–35208 CrossRef CAS PubMed.
- W. Feng, Y. Tan, M. Yang, Y. Jiang, B. X. Lei, L. Wang and W. Q. Wu, Chem, 2022, 8, 351–383 CAS.
- X. He, T. Wu, X. Liu, Y. Wang, X. Meng, J. Wu, T. Noda, X. Yang, Y. Moritomo, H. Segawa and L. Han, J. Mater. Chem. A Mater., 2020, 8, 2760–2768 RSC.
- I. Zojaji, A. Esfandiarian and J. Taheri-Shakib, Adv. Colloid Interface Sci., 2021, 289, 102314 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02644b |
| ‡ Present address: U.R.Rao Satellite Centre, Indian Space Research Organisation, Bengaluru 560017, Karnataka, India. |
| This journal is © The Royal Society of Chemistry 2023 |