
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic metal coordinated to nitrogen-doped carbon electrocatalysts for proton exchange membrane fuel cells: a perspective on progress, pitfalls and prospectives
Angus
Pedersen
 *ab,
Alexander
Bagger
ac,
Jesús
Barrio
b,
Frédéric
Maillard
d,
Ifan E. L.
Stephens
a and
Maria-Magdalena
Titirici
*be
*ab,
Alexander
Bagger
ac,
Jesús
Barrio
b,
Frédéric
Maillard
d,
Ifan E. L.
Stephens
a and
Maria-Magdalena
Titirici
*be
aDepartment of Materials, Royal School of Mines, Imperial College London, London SW7 2AZ, England, UK. E-mail: a.pedersen19@imperial.ac.uk
bDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, England, UK. E-mail: m.titirici@imperial.ac.uk
cDepartment of Physics, Technical University of Denmark, Kongens Lyngby 2800, Denmark
dUniversity Grenoble Alpes, University Savoie-Mont-Blanc, CNRS, Grenoble-INP, LEPMI, 38000 Grenoble, France
eAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aobaku, Sendai, Miyagi 980-8577, Japan
First published on 5th October 2023
Abstract
Proton exchange membrane fuel cells require reduced construction costs to improve commercial viability, which can be fueled by elimination of platinum as the O2 reduction electrocatalyst. The past 10 years has seen significant developments in synthesis, characterisation, and electrocatalytic performance of the most promising alternative electrocatalyst; single metal atoms coordinated to nitrogen-doped carbon (M-N-C). In this Perspective we recap some of the important achievements of M-N-Cs in the last decade, as well as discussing current knowledge gaps and future research directions for the community. We provide a new outlook on M-N-C stability and atomistic understanding with a set of original density functional theory simulations.
Introduction
Low temperature proton exchange membrane fuel cells (PEMFCs) powered by green hydrogen offer high power density in stationary and transport application with zero CO2 emissions. One primary problem of PEMFCs is the kinetically sluggish cathodic O2 reduction reaction, which requires an efficient electrocatalyst to facilitate the reaction over 1000 s of hours. An ideal catalyst should have high turnover frequency (e− site−1 s−1), accessible volumetric and gravimetric active site density (site cm−3 and site g−1) and stability (electrochemical turnover number, e− site loss−1). Pt-based catalysts rank highest in all these categories for O2 reduction, hence their current commercial implementation. However, the US Department of Energy (DOE) identified that removing Pt from PEMFC cathodes will be key to achieving the ultimate PEMFC stack cost target of 30$ kW−1.1 Aside from cost, Pt is a critical and scarce material, with the vast majority mined in South Africa and Russia,2 where geopolitical tensions and national electricity shortages could lead to supply problems.To date, the most promising alternative to Pt-based cathodes are those constructed from transition metal atoms coordinated to N-doped C (M-N-C, where M = metal).3 These single atom catalysts have the possibility of 100% active site utilisation and a binding energy that can be tuned according to the metal and local ligand environment. Those based on Fe-N-C, with an active site resembling the Fe-N4 site in heme, exhibit the highest activity for O2 reduction, approaching that of Pt.4 Nonetheless, they currently possess insufficient PEMFC stability for application in light duty automotive vehicles, where an 8000 h operation target has been set by the US DOE.5 The performance of M-N-C catalysts has been exhaustively covered by recent reviews by Osmieri et al.6 and Specchia et al.7 Meanwhile, the main synthesis and characterisation methods and currently known degradation pathways have been summarised by Asset et al.,8 Bae et al.9 and Kumar et al.10 A wider perspective on different single atom catalysts for various electrochemical applications has been provided by Cherevko and coworkers.11 Here, as part of the Journal of Materials Chemistry A 10-year anniversary, we aim to provide our perspective on important advancements and milestones in the past 10 years of M-N-C for PEMFCs. We also identify current critical knowledge gaps, including atomistic modelling, as well as future research directions, with a discussion of stability and its descriptors.
10 years of achievements
Active site quantification
A suite of ex situ (CO cryo adsorption12 and acid leaching13) and in situ electrochemical methods from half cell (cyanide14 and nitrite stripping15) to PEMFC (Fourier-transform alternating current voltammetry (FTacV)16) have been established for active site quantification in M-N-Cs (Fig. 1a). This has enabled comparison of turnover frequencies and establishment of reactivity descriptors among reported catalysts.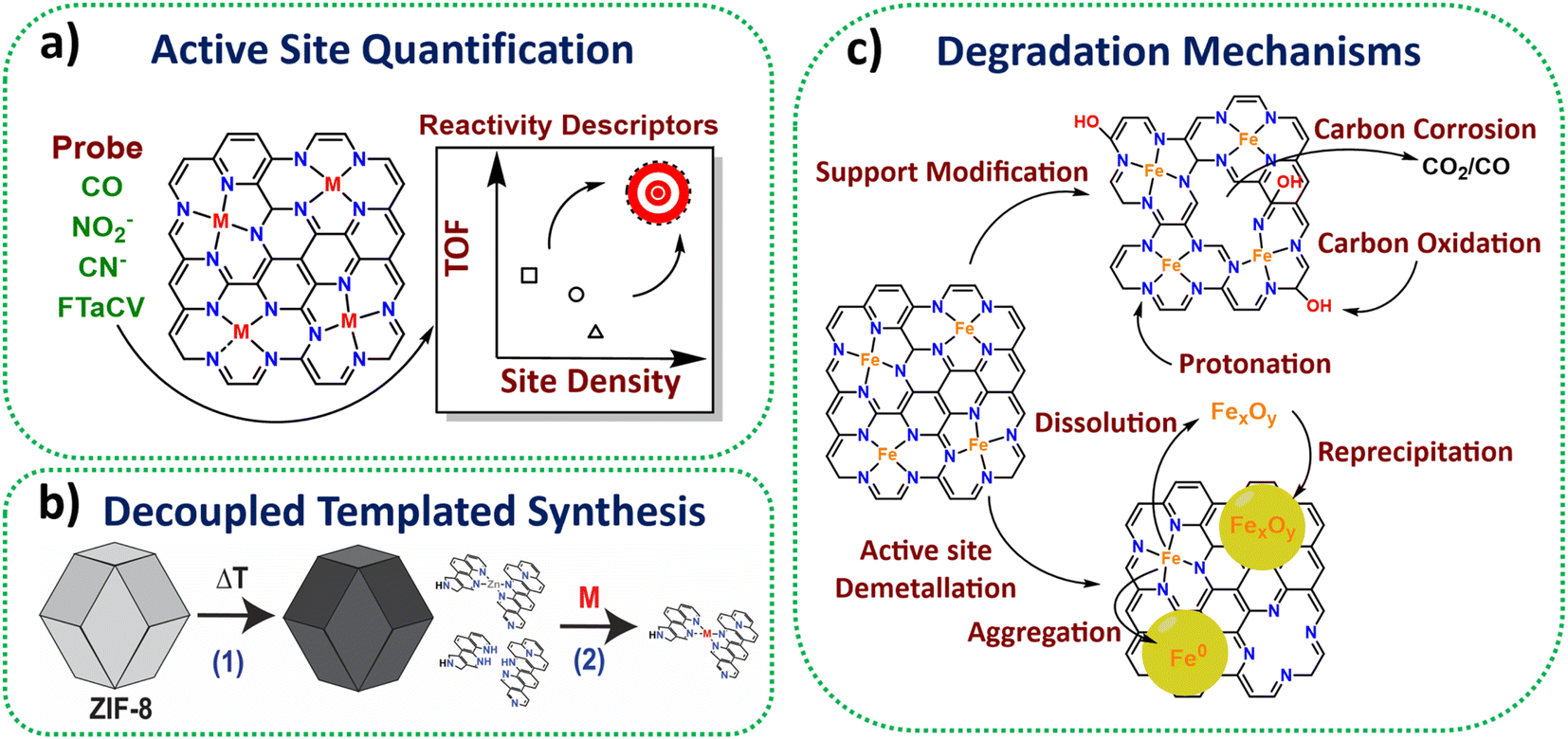 | ||
| Fig. 1 Schemes representing some highlighted achievements in method development and understanding in the past 10 years for M-N-Cs. (a) Active site quantification. (b) Decoupled synthesis consisting of pyrolysis of active site template materials, followed by active metal loading. (c) Identified degradation mechanisms of Fe-N-C. | ||
Synthesis towards high density active sites
Zeolitic framework-8 (ZIF-8) has served as an ideal active site template to load active single metal atoms, due to its Zn-N4 based structure, which is stable to high temperatures (∼900 °C), therefore serving as an ideal active template. Several studies have recently used ZIF-8 to create high active site density Fe-N-C catalysts with state-of-the-art activity based on decoupled pyrolysis and metal loading techniques (Fig. 1b),17,18 originally proposed by Fellinger and coworkers.19Active site utilisation
While high density active sites are desired, they also need to be reactant-accessible to benefit performance. Several works have achieved 100% active site utilisation based on ex situ CO cryo adsorption, although numbers of sites quantified from electrochemical methods are lower, typically <10%.20Wan et al. highlighted that electrochemical active utilisation (based on nitrite stripping) of ZIF-8 derived Fe-N-C could reach 40% at the lowest site density (1.3 × 1019 sites gM-N-C−1) using silica templating, although the utilisation fell with increasing site density.21 Attention has since turned to increasing the electrochemical active site utilisation and mass transport properties, while maintaining a high site density, by introducing more mesoporosity within the electrocatalyst, as shown in ours and others recent work.22,23 Alternative metal organic frameworks (MOFs) such as MIL-101 and MOF-5 have also been shown to possess improved mesoporosity compared to ZIF-8 upon pyrolysis, leading active M-N-Cs.24,25
Operando and in situ characterisation and mechanisms
In situ and operando X-ray absorption spectroscopy (XAS) has been used successfully to monitor Fe-N-C redox behaviour.26In situ Mössbauer has enabled identification of two types of Fe-N-C active sites, being either highly active but unstable, or less active but more stable.27 Meanwhile, operando Mössbauer has recently been shown to identify the type of Fe-N-C site and a new intermediate.28Operando mass spectrometry methods have proved insightful in determining carbon oxidation and metal dissolution pathways and key degradation mechanisms (Fig. 1c),29,30 with this understanding improving catalyst stability by informing methods for reducing active site demetallation.31Stability
Commercialised M-N-C (discussed further below) has been reported to operate >500 h,32 although the reported activity was not comparable to Pt-based catalysts. Recently, an improvement in PEMFC stability (>300 h) of a high activity Fe-N-C, comparable to Pt/C, has been achieved by coating an atomically thin layer of N-C on a highly active Fe-N-C.33 Well dispersed nanoparticles (CeOx, TaOx) and single atom radical scavengers have also proved effective in reducing Fe-N-C degradation from reactive oxygen species.34–36 Reactivation of Fe-N-C to extend catalyst lifetime has also been successfully demonstrated through in situ electrochemical reduction which reduces O species,37 resulting in a short-lived improved turnover frequency.38Benchmark materials, standardized PEMFC protocols, and cross laboratory studies
For any technological progress it is critical standardized protocols and benchmark commercial materials are developed to ensure equipment and researchers can reproduce results. The recent introduction of these in the Fe-N-C community39 will lead to accelerated research progress. Meanwhile, cross laboratory studies have helped to understand the varying properties and reactivity descriptors of Fe-N-C catalysts synthesised via different methods.20Commercialisation
Commercial development of M-N-Cs has been undertaken by Pajarito Powder, Nisshinbo, and recently Celcibus AB. In 2017, Ballard Power Systems, through collaboration with Nisshinbo, announced the first commercial implementation of M-N-C catalysts for low power application of emergency power/wifi backpack.32 The application was well suited since M-N-C are known to be poison resistant to impurities and contaminants,40 which are likely present in emergency situations, such as fires and volcano eruptions. This signalled a breakthrough maturation of research and the prospect of striving towards large but more demanding applications, such as transport. The following sections outline current missing knowledge and hurdles that need to be overcome for further practical applications.Current knowledge gaps
Life cycle assessment
It is naturally assumed M-N-Cs have a lower environmental impact than Pt-based catalysts, with claims of environmentally benign synthesis,42 although this has yet to be shown or quantified via life cycle assessments. The answer may not be so trivial since state-of-the-art M-N-C catalyst typically incorporate multiple process steps and the quantities of wasted and required PEMFC components, including environmentally impactful Nafion, will be higher than for Pt-based catalysts. Also, the lifetime of state-of-the-art M-N-Cs is still far below that of Pt-based catalysts. Factoring environmental impacts from the life cycle assessment in technoeconomic analyses to date (Fig. 2a), could make M-N-C based PEMFC more (or less) economically competitive. Value stream mapping could also be integrated with life cycle assessments to ensure sustainable manufacturing and scalability (see section: Scalable).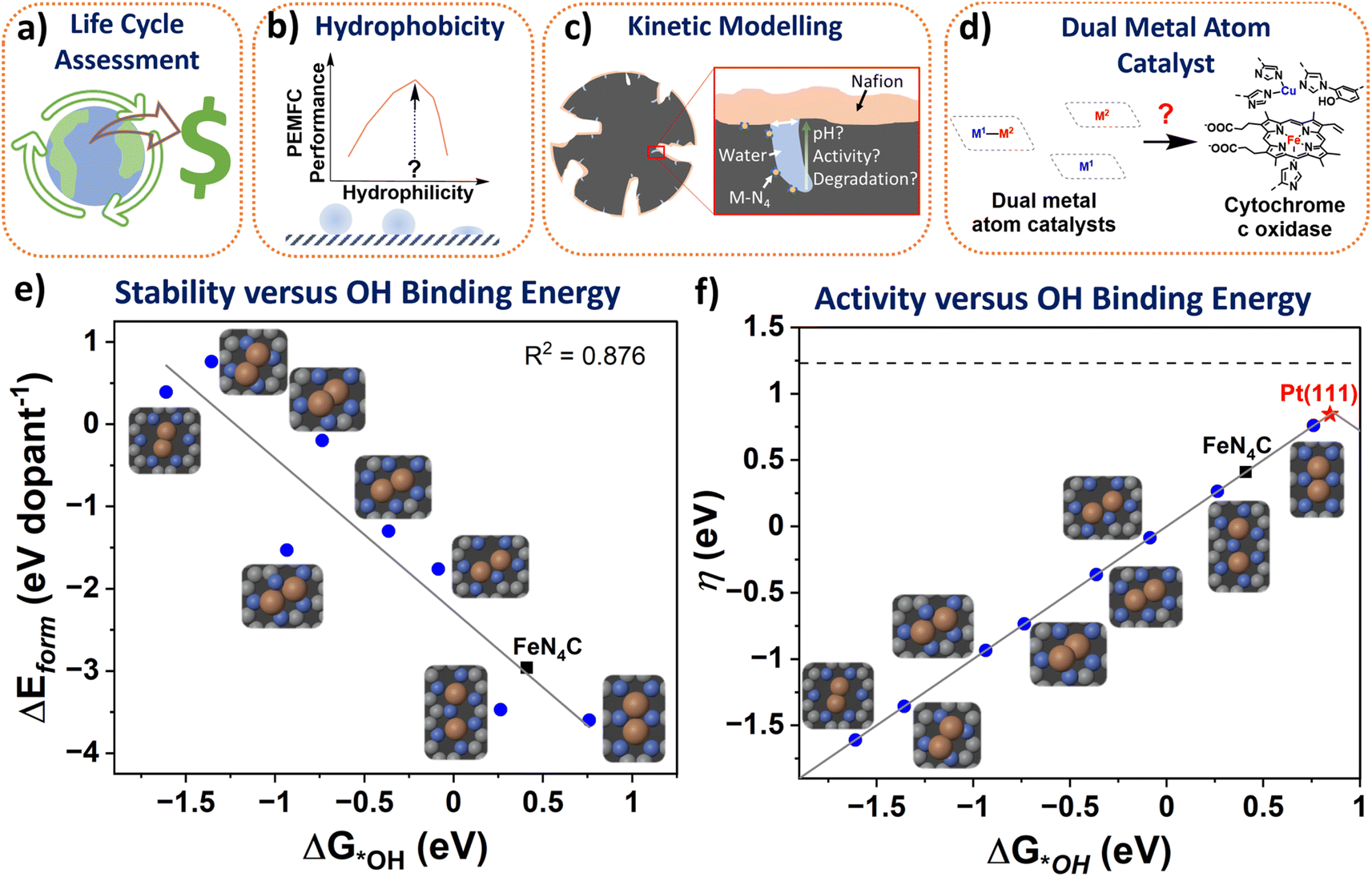 | ||
| Fig. 2 Schemes depicting knowledge gaps in the M-N-C literature. (a) Life Cycle Assessment and Monetized Impact. (b) Impact of hydrophobicity on PEMFC performance. (c) Representative kinetic modelling of active sites. (d) Dual metal atom catalysts mimicking enzymes for improved activity. (e) The formation energy of different dual site Fe2NxC (blue circles) and single atom FeN4C (black square) derived from Karmodak et al.41versus the calculated binding energy toward *OH calculated from DFT with revised Perdew–Burke–Ernzerhof functional. (f) The ORR volcano with Pt(111) at the top (red star), followed by the dual sites and reference FeN4C. ΔG*OH is the hydroxyl adsorption energy. QV2 is the most stable dual site and predicted to be the most active. Colours in inset figures represent: Orange – Fe, Blue – Nitrogen, Grey – Carbon. | ||
Translating from half-cell to PEMFC
Electrochemical half-cell systems do not accurately reflect degradation rates or mechanisms in real devices.43 This is unsurprising since degradation is a function of charge passed, where rotating disc electrodes operate at least an order of magnitude lower charge than a real device. For instance, a high-performance M-N-C at 0.6 VRHE in O2-saturated 0.1 M HClO4 in a 1600 rpm rotating disc electrode (RDE) would expect ca. 5 mA cm−2, which corresponds to 25 A gM-N-C−1 at 0.2 mgM-N-C cm−2. The same M-N-C at 0.6 VRHE in an optimised H2–O2 PEMFC could then reasonably show 1 A cm−2 at 4 mgM-N-C cm−2, equal to 250 A gM-N-C−1. Degradation rates and mechanisms also depend on temperature, where most half-cell tests are conducted at room temperature, rather than 80 °C as commonly used in PEMFCS.44 Critically, temperature affects the carbon oxidation reaction kinetics,29,45 which modifies electron delocalization and therefore turnover frequency of M-N-Cs.37,38,46 The quantitative effect of oxygen group chemistry on turnover frequency could be probed by high sensitivity temperature programmed desorption.Considering other catalysts, a Mn–Co spinel alkaline O2 reduction catalyst exhibited improved activity under fuel cell conditions compared to half-cell.47 Meanwhile, various Pt nanoparticle shapes have been proven highly active in RDE, but their activity does not translate to fuel cell performance.48 For Fe-N-C, some have reported higher activity in RDE compared to PEMFC, taking into account temperature effects.49,50 Meanwhile previous work by Jaouen et al. found a ca. factor five higher activity for one Fe-N-C in PEMFC compared to RDE, again accounting for temperature affects. Although a separate Fe-N-C synthesised via a different route did not express such discrepancy. This difference for the former was attributed to anion adsorption on protonated N from the H2SO4 electrolyte in RDE.51 They also found optimisation of catalyst ink formulation was also crucial to obtain more comparative O2 reduction activities between RDE and PEMFC.51 Methods for catalyst layer optimisation in PEMFC are available for M-N-Cs,52 although the optimisation process can still be lengthy and trial-and-error based. Also, it should be considered the electrode preparation step can affect the M-N-C properties and consequent structure–activity–stability correlations.53 The change from double to triple phase boundary from half-cell to PEMFC can also impact properties such as active site utilisation and mass transport limitations. Changes in the microenvironment also impact reaction kinetics54 (see: Microenvironment and local pH).
Fe-N-C redox and site density quantification
The reversible redox couple observed in some Fe-N-C catalysts at ca. 0.76 VRHE in acidic medium has been extensively confirmed as the Fe(II)/Fe(III) transition from Fe–Nx sites,26,27,55 with some exploration of electrolyte effects.56 This redox has been ascribed to arise from high spin Fe3+ sites.27 Still, it remains unclear why some Fe-N-C catalysts, containing Fe3+–Nx sites as measured by ex situ Mössbauer, exhibit Fe redox in PEMFC but not in RDE. Several factors could be the cause, such as: the redox being masked by pseudocapacitance, Fe3+ sites charge being neutralised by an electrolyte anion in RDE and/or electrochemical inaccessibility of Fe3+ sites from catalyst structure and morphology effects. A rigorous study exploring these various effects is required since in situ electrochemical site quantification from FTacV relies on a detectable redox.16 Meanwhile, in situ nitrite stripping has experienced conflicting claims in the number of electrons transferred during the electrochemical process (3–5 e−).15,57 It could be that different M–Nx sites produce different products (hydroxylamine or ammonium) with a corresponding different number of electrons. This requires further investigation. Additionally, nitrite stripping has been reported to probe non-single metal atom species such as the N/C framework58 and iron oxides,59 where the latter makes post-mortem active site quantification non-trivial.Hydrophobicity
Surface hydrophobicity of the carbon support of M–Nx sites plays an important role in the water management and therefore performance of PEMFCs. However, water vapor physisorption measurements are rarely conducted in the M-N-C community, with some exploration for electrochemical CO2 reduction.60 Correlating hydrophobicity with PEMFC results (Fig. 2b) would help to identify optimum hydrophobicity for M-N-C performance and avoid deleterious flooding. Also, understanding how oxidation of the carbon support changes hydrophobicity over time would prove insightful to tuning PEMFC operation.Electronic conductivity
While it is obvious electronic conductivity plays an important role in all electrocatalysis, surprisingly few studies measure the electronic conductivity of electrocatalysts as prepared or when incorporated in devices as electrodes. Boettcher and coworkers have thoroughly investigated the effect of electronic conductivity on earth-abundant oxygen evolution catalyst performance;61 however limited work has been carried out on M-N-Cs.62–64 Experimentally measured through-plane electronic conductivity has been reported to be three orders of magnitude lower than the in-plane.65 Additionally, since the thickness of M-N-Cs is typically ca. one order of magnitude higher than Pt/C, the impact of electronic conductivity becomes significant. For instance, as noted by Kulikovsky, for typical carbon support electronic conductivity of 20 S m−1 with 100 μm catalyst layer thickness would lead to 50 mV loss.62 For the same catalyst thickness, Jaouen et al. modelled different proton and electronic conductivity values, finding 80 mV loss for electronic conductivity of 20 S m−1.66 They concluded electronic conductivity of 100 S m−1 would be required to limit losses at high current density.66Jaouen and coworkers have also alluded to surface and local active site electronic conductivity effects.46 They suggested operando H2O2 generation which exfoliate graphene sheets or oxidise edges would decrease conductivity at graphene sheet edges, where active sites are preferentially located, potentially having a profound impact on turnover frequency.46 Expanding on previous work,67 understanding how concentrations and locations of O, N, active metal, surface area and pyrolysis temperature directly affect single carbon layers, particle, and bulk electronic conductivity would provide useful reference data. It would also be interesting to measure how conductivity differs ex situ and in situ and decreases over time with carbon corrosion and oxidation. This could then inform the subsequent effect on turnover frequency.46 Analysis of distribution of relaxation times from electrochemical impedance spectroscopy provides a complimentary pathway to deconvolute different effects of proton and electronic conductivity.68
Microenvironment and local pH
As our understanding of electrocatalysis improves, focus is shifting to understanding the microenvironment.69 Local pH effects have been considered thoroughly in alkaline CO2 reduction,70 and is beginning wider attention in electrocatalysis;71 however this idea has so far received little attention in acidic O2 reduction degradation mechanisms.72 While PEMFCs operate in a strongly bulk acidic environment, the pH has been modelled to increases down small mesopore channels, which are not in direct contact with Nafion and are instead filled with water.73 Active sites of M-N-Cs are thought to reside within micropores,20,74 which are inaccessible to direct Nafion contact and are instead filled with water during PEMFC operation (Fig. 2c). It is worth noting high surface area microporous M-N-C materials contain 10–20 wt% water under atmospheric conditions.19 Therefore the consumption of protons and O2 at high current densities may lead to local pH changes at active sites, as recently explored in O2 reduction down to pH 2 in RDE.75If local pH change does occur, this could have implications on the O2 reduction pathway, activity, and degradation route. For degradation pathway, considering the Pourbaix diagram of Fe, for certain potentials at increased pH, Fe3+ and Fe2+ form Fe2O3 species, which have been widely observed following stability tests.27,59 Meanwhile for activity, at pH 7, H+ is 10−7 M, which negatively affects kinetics.76
Atomistic understanding
The binding energy of reaction intermediates to Pt nanoparticle can be modelled by density functional theory (DFT) with certain accuracy due to their crystalline nature, which can be mimicked by single crystal studies. To date, the best model M-N-C systems are macrocycles (e.g., metal phthalocyanines or metal porphyrin),77 as these contain a well-defined structure in experiments and can be replicated atom by atom in quantum chemistry simulation. However, these macrocycles rapidly degrade or inactivate in acidic electrolyte. Additionally, the typical pyrolysis methods for making M-N-C catalysts create a variety of possible active sites with varying local carbon structures (defects, edges, oxygen groups), which modify the electron density. Another option is to accept the variety of sites in a pyrolysed catalyst and instead establish trends using one model, for example a M-N4-C site. Beyond establishing the atomic structure of the active site, the description of the electronic structures of this catalyst is not straight forward, as the M-N-C catalyst involve accessing multiple different magnetic spin-states. For example, prediction of the CO binding energy on these catalysts varies heavily with the exchange correlation functional.78 Additionally, modelling M-N-C for electrocatalysis encounter challenges of metal catalyst solvation effects and electrolyte effects.Dual metal atom active sites
We recently surveyed the literature on dual-metal atom catalysts.79 Enzymes and molecular catalysts based on dual metal atom sites have shown enhanced activity beyond state-of-the-art single atoms. Meanwhile, many reports suggest improved activity for catalysts putatively containing dual and single atom active sites compared to their equivalent materials containing just single atom sites. However, these catalysts still possess orders of magnitude lower turnover frequency than the enzymes they aim to mimic (Fig. 2d) and are not beyond state-of-the-art single atom catalysts. The reasoning for this remains unclear; perhaps dual metal atom sites produced to date are poisoned or unstable under reaction conditions,80 or are misidentified (see further discussion in section: Characterisation).The stability and activitity of dual metal atoms has been investigated by the formation energy found from DFT simulations in a series of different dual and single atomic metal in nitrogen-doped carbon pockets by Karmodak et al.41,81 Here, we investigated the same structures, focussing on FexNyC sites, adding calculations of the binding energy of these sites to OH (Fig. 2e). Interestingly, a clear trend is observed between active site stability and OH binding energy, with less stable sites binding OH stronger. Dual metal atoms in hexa-vacancy sites could have a directed experimental synthesis, as we and others have reported, based on a C2N-derived structure.82,83 Although, it appears Fe2 hexa-vacancy sites are far less stable (Fig. 2e) and would become instantly oxidised by strong OH binding under reaction conditions. The most stable dual metal atom site under the modelled conditions was quad-vacancy, closely mimicking single atom FeN4C sites, but with an improved weakened OH binding, approaching activity of Pt(111) (Fig. 2f). However, controllably creating the dual metal atom quad-vacancy site experimentally does not appear straightforward.
It should also be considered for M-N-C sites, there is an upper limit of ca. 0.9 VRHE in half cell (50 °C) before carbon oxidation begins.29 The upper operating limit on potential before debilitating carbon oxidation will occur is even lower in PEMFC conditions due to accelerated carbon corrosion kinetics at PEMFC operating temperatures of 80 °C.30,84 Consequently, the high activity at low overpotentials (<0.3 V) predicted by DFT for carbon-supported dual atom metal active sites85,86 would not be practical: at potentials positive of 1 V, the carbon support would corrode. Conceivably, the carbon could be stabilised if it were covered by an atomically thin and stable elemental layer, for instance, through atomic layer deposition.
Where next?
Stability
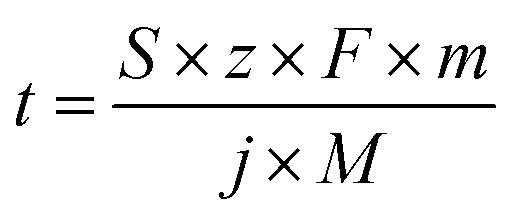 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), m is the mass of accessible active element (g cm−2), j is the applied current density (A cm−2), and M is the molar mass of the active element (g mol−1).
485 C mol−1), m is the mass of accessible active element (g cm−2), j is the applied current density (A cm−2), and M is the molar mass of the active element (g mol−1).
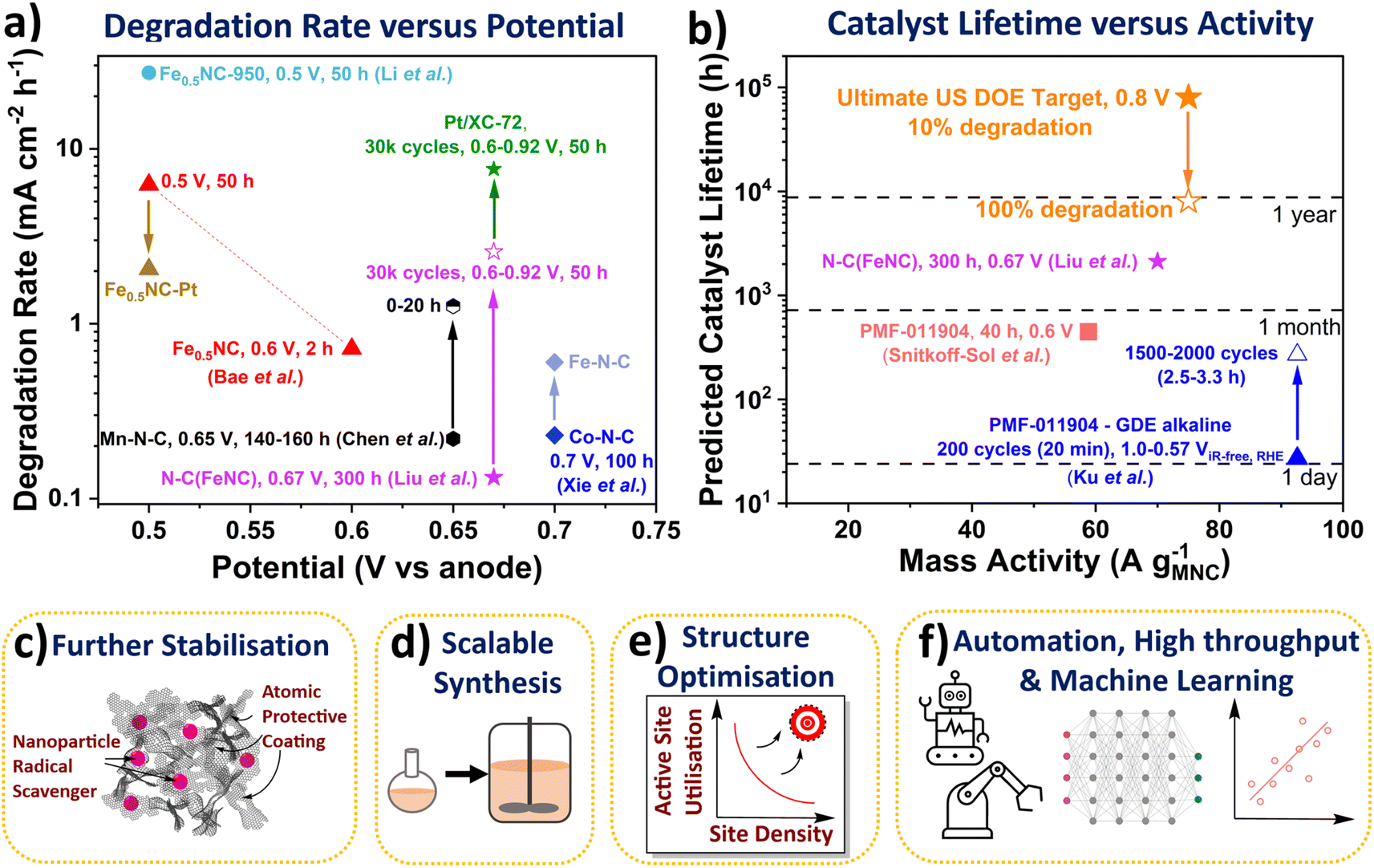 | ||
| Fig. 3 (a) Degradation rate of Fe-N-C and Pt/XC-72 catalysts in PEMFCs. Fuel cell potential represented versus hydrogen anode. All measurements were conducted at 80 °C. Liu et al.33 N-C(FeNC) 300 h at 0.67 V (H2/air 200 cm3 min−1) and 0.6 to ∼0.92 V with 3 s holds (30k cycles (50 h), H2/air 200 cm3 min−1, 1 bara). Bae et al.89 Fe0.5NC 2 h potential hold at 0.6 Vvs anode and 50 h at 0.5 Vvs anode. Li et al.27 Fe0.5NC-950 (H2/O2 60 cm3 min−1, 1 barg). Chen et al.42 Mn-N-C degradation rate between 140–160 h and 0–20 h holds at 0.65 Vvs anode (H2/air, 200 cm3 min−1, 1 barg). Xie et al.30 Co-N-C and Fe-N-C (H2/air, 1 barg). (b) Calculated catalyst lifetimes and measured mass activities. Ultimate US DOE light duty vehicle target of 8000 h (with 10% degradation) at 0.3 A cm−2 and 0.8 VRHE (equivalent to 75 A gM-N-C−1 based on 4 mgM-N-C cm−2 and 1 × 1020 sites gM-N-C−1). Snitkoff-Sol et al.16 Pajarito Powder FeNC (PMF 011904) 40 h at 0.6 V (H2/O2 300 cm3 min−1, 1 barg). Ku et al.90 PMF 011904 FeNC 1.0 to 0.57 ViR-free RHE (0 to −125 mA cm−2) with 3 s holds (GDE, 0.1 M NaOH). Schemes representing future research directions. (c) Methods for improved stability. (d) Scalable synthesis. (e) Structure optimisation for increased active site utilisation. (f) Automation, high throughput and machine and deep learning. | ||
The stability number can be calculated from post-mortem Mössbauer and inductively coupled plasma mass spectrometry of the catalyst and/or electrolyte, or via in situ PEMFC techniques,16 or from lifetime extrapolated from degradation rates. These stability numbers can then be used to calculate the catalyst lifetime (Fig. 3b). A stability number of 3.37 × 108 is required to reach the ultimate US DOE PEMFC target of 8000 h (with <10% degradation) at 0.3 A cm−2 and 0.8 V (equivalent to 75 A g−1 based on 4 mgM-N-C cm−2 and 1 × 1020 sites gM-N-C−1).5 The stability number and lifetime of M-N-C in acidic electrolyte or PEMFC has not been reported to date, however it can be calculated, as mentioned above. Extrapolating state-of-the-art nitrogen-carbon coated Fe-N-C (N-C(Fe-N-C)) of Liu et al. shows lifetime of 2121 h at 0.67 V is feasible, with initial ∼70 A g−1. The catalyst lifetime can also be extrapolated from changes in the site density, as shown by Elbaz and coworkers.16 For a commercial Fe-N-C (Pajarito Powder PMF-011904), the catalyst lifetime was predicted to be 447 h in PEMFC at 58.8 A g−1 (based on 40 h hold at 0.6 V), with an apparently linear degradation rate of sites.16 Meanwhile, in alkaline GDE, Ku et al. measured the stability number changing from ∼106 to ∼107 for the same commercial Fe-N-C (PMF-011904), when evaluating after 200 and 2000 cycles (3 s hold at 1 and 0.57 ViR-free, RHE).90 This equated to catalyst lifetime changing from 27 h to 270 h at 92.6 A g−1.90 Bae et al. also found a decrease in degradation rate over the initial 2 h measurement for their Fe-N-C in PEMFC, with negligible site density loss beyond 1.5 h.89 This highlights the catalyst lifetime from the stability number is an overestimation of PEMFC operation since not all catalyst degradation routes directly alter the number of active sites (e.g., attack from reactive oxygen species), and some activity recovery is also possible.37 It is therefore important to better understand differences between reversible and irreversible losses in PEMFCs. Additionally, decoupling contributions of different degradation mechanisms can identify critical parameters in M-N-C stability.84 Developing rates and descriptors for the different degradation mechanisms, while difficult, would be highly insightful for the community.
Theoretical tools for modelling material and active site stability with DFT have primarily been focused on thermodynamically deriving Pourbaix diagrams.91,92 Recent methods used for searching for acid stable oxygen electrocatalysts93–95 and metastable materials96 could be used for M-N-C catalyst to move beyond just the formation energies of the M-N-C sites. Noticeably, what the methods have in common is the starting point of simulating Pourbaix diagrams, which address the aqueous electrolyte potential and pH, but not the specific solvent species, such as anions and cations.
However, the degradation rate of this Co-N-C and a separate Mn-N-C (0.22 mA cm−2 h−1 after 160 h at 0.65 V)30,42 is greater than the recent state-of-the-art Fe-N-C by Liu et al. (0.13 mA cm−2 h−1 after 300 h at 0.67 V, labelled as (N-C)Fe-N-C in Fig. 3a).33 This Fe-N-C involved atomic N-C coating by chemical vapour deposition of an active Fe-N-C significantly enhanced stability, although with noticeable reduction in activity.33 The success of this method opens new avenues of research, including trying other potentially more stable coatings, and using more controlled deposition techniques for model studies, such as atomic layer deposition. Other alternatives to enhance stability could include modifying the catalyst support, for instance boron carbide has been previously used for Pt-based catalyst.98 Also, as mentioned earlier, nanoparticle radical scavengers have proved effective in minimising oxidative attack (Fig. 3c), although these nanoparticles could block active sites and may not be situated atomically close to active sites. More desirable scavengers would be single atoms scavengers doped within the catalyst support that are within proximity to the active site, due to the short lifetime of reactive oxygen species.35 Secondary metal atoms are often doped within M-N-C which improve activity and stability, although the exact reason for improved performance is only beginning to be elucidated.99 Pt atoms have been successfully incorporated into Fe-N-C with the performance improvement understand,89 although cheaper dopants are more desirable in future.
Aside from improving the degradation resistance of the catalyst, PEMFC operating conditions can also be tuned to minimize catalyst degradation. This can be achieved through model development, which identify key degradation parameters, as completed for a Pt-based catalyst.100
Synthesis
Still, these non-pyrolysed MOF-based materials do not represent the class of M-N-C. M-N-Cs would benefit from better understanding of the “black box” pyrolysis step. Comprehensive in situ pyrolysis studies have been limited in M-N-Cs, mainly led by Zenyuk and coworkers.107,108
000 PEMFC stacks year−1).109 Future directions of state-of-the-art M-N-C should therefore consider scalable routes (Fig. 3d), with minimal number of reaction steps and environmental impact, avoiding acid leaching steps where possible for commercialisation.
The lower mass activity of M-N-C compared to Pt-based catalysts necessitates ×10 thicker catalyst layers in PEMFCs to feasibly achieve high current demands. Further research is therefore required for designing thick electrodes with optimised pore structures and properties that minimise losses in proton and electronic conductivity, while maintaining sufficient O2 accessibility. Nature could provide inspiration for the catalyst layer design, as we have recently highlighted,110 and new architectures such as grooving,111 could be explored for M-N-Cs. Methods for thick electrode engineering, manufacturing and characterisation can also be adapted from battery literature.112 Meanwhile, better understanding of gas and water flow in thick M-N-C cathode layers is required to understand and optimise PEMFC performance. This could be realised through multi-phase modelling with deep learning, as recently shown for a Pt-based PEMFC system.113
While M-N-Cs are relatively restricted systems, the combined chemistry, material, and engineering space are so vast that traditional trial-and-error, high throughput and DFT methods are insufficient for exploration. Different approaches based on artificial intelligence could probe the space from a different perspective. Automation of characterisation, especially in microscopy, is also a growing direction.117
Recent studies on high entropy alloys have driven high throughput experiments coupled with machine learning can both refine our atomistic understanding of oxygen reduction activity and lead to the discovery of new catalyst materials;118 we envisage that such data driven hypothesis-based approaches could lead to even more active materials in future.119
Alternatively, increased active site densities will allow use of techniques which were previously insensitive due to their limit of detection. For instance, developments in laboratory-based X-ray absorption equipment, such as from EasyXAFS LLC, may be applicable for some high loading single atom M-N-Cs. Meanwhile, laboratory-based X-ray pair distribution function (XPDF) represents an under-utilised technique in the community, which could provide spatial resolution of the local atomic arrangement within M-N-Cs and long range order changes,121 and requires further exploration.
However, the community should be careful drawing conclusions from bulk characterisation techniques. As we discussed earlier in the section: Active site utilization, the vast majority (70–90%) of active sites in M-N-C are typically not electrochemically utilized. Therefore, operando characterisation techniques which probe the bulk M-N-C layers, such as Mössbauer spectroscopy, XAS or XPDF, will measure accessible and inaccessible active sites. This makes it difficult to deconvolute the contribution of activity and stability of different M–Nx sites. This challenge could be overcome by coupling in situ electrochemical probing techniques, such as FTacV, with bulk operando techniques.
For surface sensitive characterisation, time-of-flight secondary ion mass spectrometry (ToF-SIMS) is technique which has rarely been used. It can provide atomic surface chemical compositions of M-N-Cs, including light elements all the way up to hydrogen, with capabilities of depth profiling. ToF-SIMS can also distinguish single and dual metal atoms;122,123 however, quantification using ToF-SIMS is highly challenging.
Conclusions
In this Perspective we highlight significant steps taken by the community in improving knowledge of M-N-C electrocatalysts for PEMFCs over the last 10 years. Still, many stones remain unturned, and hurdles prevent further commercial M-N-C implementation. To this end, we discuss some of these unexplored research questions and future challenges. We present original DFT simulations which show correlation between stability (formation energy) and activity (OH binding energy) of dual atom Fe-N-C sites. We also highlight new perspectives on M-N-C stability via catalyst lifetime calculations, which illustrates the gap from state-of-the-art to reach ultimate US DOE targets.Methods
Density functional theory
The atomic M-N-C structures and formation energy were obtained from Karmodak et al., assuming a single sheet of graphene as the surrounding carbon structure.41 Here, an *OH intermediate was added to the structures and spin polarised for the clean and *OH structures. For relaxation computations GPAW code was used,127,128 with the following settings: (4, 4, 1) k-point, a grid spacing of 0.18, with the BEEF-vdW functional,129 and relax the structures to below 0.05 eV per atom. After relaxing the structures, the OH binding energy was calculated from the following equation:Here, 0.35 eV and −0.3 eV are thermodynamic and water stabilization corrections, respectively, obtained from Nørskov et al.130
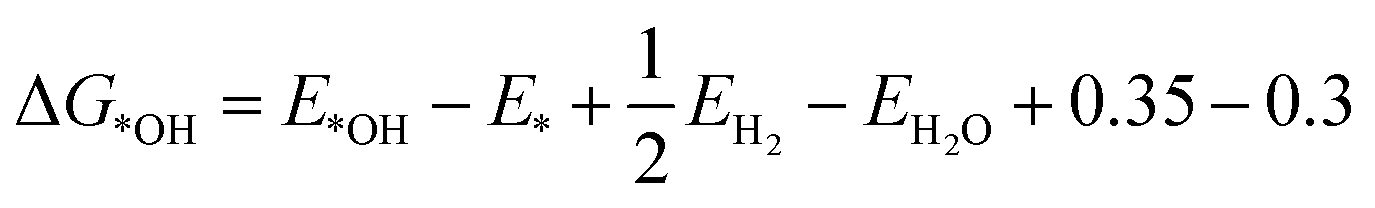
Author contributions
A. P. wrote the initial draft. J. B. and A. B. assisted writing the original draft. A. B. carried out DFT simulations. F. M., I. E. L. S. and M.-M. T provided supervision, and revised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Engineering and Physical Sciences Research Council (EPSRC) (EP/W031019/1 and EP/S023259/1), the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 866402). A. P. thanks the EPSRC Centre for Doctoral Training in the Advanced Characterisation of Materials (grant number EP/L015277/1). A. B. acknowledges support from the Carlsberg foundation (grant no. CF21-0114). J. B. acknowledges financial support from Imperial College London through the Imperial College Research Fellowship.References
- S. T. Thompson and D. Papageorgopoulos, Nat. Catal., 2019, 2, 558–561 CrossRef CAS.
- J. Matthey, PGM Market Report, May, 2022 Search PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office: Multi-Year Research, Development, and Demonstration Plan: 3.4, Fuel Cells, Washington, 2016 Search PubMed.
- L. Osmieri, J. Park, D. A. Cullen, P. Zelenay, D. J. Myers and K. C. Neyerlin, Curr. Opin. Electrochem., 2021, 25, 100627 CrossRef CAS.
- S. Specchia, P. Atanassov and J. H. Zagal, Curr. Opin. Electrochem., 2021, 27, 100687 CrossRef CAS.
- T. Asset, F. Maillard and F. Jaouen, in Supported Metal Single Atom Catalysis, ed. P. Serp and D. P. Minh, Wiley, 2022, pp. 531–582 Search PubMed.
- G. Bae, S. Han, H.-S. Oh and C. H. Choi, Angew. Chem., Int. Ed., 2023, 62, e202219227 CrossRef CAS PubMed.
- K. Kumar, L. Dubau, F. Jaouen and F. Maillard, Chem. Rev., 2023, 123(15), 9265–9326 CrossRef CAS PubMed.
- F. D. Speck, J. H. Kim, G. Bae, S. H. Joo, K. J. J. Mayrhofer, C. H. Choi and S. Cherevko, JACS Au, 2021, 1, 1086–1100 CrossRef CAS PubMed.
- F. Luo, C. H. Choi, M. J. M. Primbs, W. Ju, S. Li, N. D. Leonard, A. Thomas, F. Jaouen and P. Strasser, ACS Catal., 2019, 9, 4841–4852 CrossRef CAS.
- T.-P. Fellinger, D. Menga, A. G. Buzanich and F. Wagner, Angew. Chem., Int. Ed., 2022, 61, 1–5 Search PubMed.
- G. Bae, H. Kim, H. Choi, P. Jeong, D. H. Kim, H. C. Kwon, K.-S. Lee, M. Choi, H.-S. Oh, F. Jaouen and C. H. Choi, JACS Au, 2021, 1c00074 Search PubMed.
- D. Malko, A. Kucernak and T. Lopes, Nat. Commun., 2016, 7, 13285 CrossRef CAS PubMed.
- R. Z. Snitkoff-Sol, A. Friedman, H. C. Honig, Y. Yurko, A. Kozhushner, M. J. Zachman, P. Zelenay, A. M. Bond and L. Elbaz, Nat. Catal., 2022, 5, 163–170 CrossRef CAS.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- A. Mehmood, J. Pampel, G. Ali, H. Y. Ha, F. Ruiz-Zepeda and T.-P. Fellinger, Adv. Energy Mater., 2018, 8, 1701771 CrossRef.
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M.-T. Sougrati, P.-Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Energy Environ. Sci., 2020, 13, 2480–2500 RSC.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- J. Barrio, A. Pedersen, S. C. Sarma, A. Bagger, M. Gong, S. Favero, C. Zhao, R. Garcia-Serres, A. Y. Li, Q. Zhang, F. Jaouen, F. Maillard, A. Kucernak, I. E. L. Stephens and M. Titirici, Adv. Mater., 2023, 35, 2211022 CrossRef CAS PubMed.
- Y. Wang, M. J. Larsen, S. Rojas, M.-T. Sougrati, F. Jaouen, P. Ferrer, D. Gianolio and S. Berthon-Fabry, J. Power Sources, 2021, 514, 230561 CrossRef CAS.
- X. Xie, L. Peng, H. Yang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed.
- X. Xie, L. Shang, X. Xiong, R. Shi and T. Zhang, Adv. Energy Mater., 2022, 12, 2102688 CrossRef CAS.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- L. Ni, C. Gallenkamp, S. Wagner, E. Bill, V. Krewald and U. I. Kramm, J. Am. Chem. Soc., 2022, 144, 16827–16840 CrossRef CAS PubMed.
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. Ranjbar Sahraie, M.-T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, ACS Catal., 2016, 6, 3136–3146 CrossRef CAS.
- D. Banham, J. Y. Choi, T. Kishimoto and S. Ye, Adv. Mater., 2019, 31, 1–6 CrossRef PubMed.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- H. Xie, X. Xie, G. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. H. Phakatkar, M. Hong, M. Wu, R. Shahbazian-Yassar, V. Ramani, M. I. Al-Sheikhly, D. Jiang, Y. Shao and L. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS.
- Y. Chu, E. Luo, Y. Wei, S. Zhu, X. Wang, L. Yang, N. Gao, Y. Wang, Z. Jiang, C. Liu, J. Ge and W. Xing, Chem Catal., 2023, 3, 100532 CrossRef CAS.
- D. Shin, S. Bhandari, M. F. Tesch, S. A. Bonke, F. Jaouen, S. Chabbra, C. Pratsch, A. Schnegg and A. K. Mechler, J. Energy Chem., 2022, 65, 433–438 CrossRef CAS.
- P. Boldrin, D. Malko, A. Mehmood, U. I. Kramm, S. Wagner, S. Paul, N. Weidler and A. Kucernak, Appl. Catal., B, 2021, 292, 120169 CrossRef CAS.
- C. H. Choi, H. K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M. T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- H. Zhang, L. Osmieri, J. H. Park, H. T. Chung, D. A. Cullen, K. C. Neyerlin, D. J. Myers and P. Zelenay, Nat. Catal., 2022, 571–592 Search PubMed.
- D. Malko, T. Lopes, E. Symianakis and A. R. Kucernak, J. Mater. Chem. A, 2015, 4, 142–152 RSC.
- N. Karmodak, S. Vijay, G. Kastlunger and K. Chan, ACS Catal., 2022, 12, 4818–4824 CrossRef CAS PubMed.
- M. Chen, X. Li, F. Yang, B. Li, T. Stracensky, S. Karakalos, S. Mukerjee, Q. Jia, D. Su, G. Wang, G. Wu and H. Xu, ACS Catal., 2020, 10, 10523–10534 CrossRef CAS.
- K. Ehelebe, D. Escalera-López and S. Cherevko, Curr. Opin. Electrochem., 2021, 29, 100832 CrossRef CAS.
- K. Kumar, T. Asset, X. Li, Y. Liu, X. Yan, Y. Chen, M. Mermoux, X. Pan, P. Atanassov, F. Maillard and L. Dubau, ACS Catal., 2021, 11, 484–494 CrossRef CAS.
- V. Goellner, C. Baldizzone, A. Schuppert, M. T. Sougrati, K. Mayrhofer and F. Jaouen, Phys. Chem. Chem. Phys., 2014, 16, 18454–18462 RSC.
- V. Goellner, V. Armel, A. Zitolo, E. Fonda and F. Jaouen, J. Electrochem. Soc., 2015, 162, H403–H414 CrossRef CAS.
- Y. Wang, Y. Yang, S. Jia, X. Wang, K. Lyu, Y. Peng, H. Zheng, X. Wei, H. Ren, L. Xiao, J. Wang, D. A. Muller, H. D. Abruña, B. J. Hwang, J. Lu and L. Zhuang, Nat. Commun., 2019, 10, 1506 CrossRef PubMed.
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed.
- A. M. Damjanović, B. Koyutürk, Y.-S. Li, D. Menga, C. Eickes, H. A. El-Sayed, H. A. Gasteiger, T.-P. Fellinger and M. Piana, J. Electrochem. Soc., 2021, 168, 114518 CrossRef.
- S. Ünsal, M. Bozzetti, Y.-C. Chen, R. Girod, A. Berger, J. S. Diercks, S. Gialamoidou, J. Lyu, M. Medarde, H. A. Gasteiger, V. Tileli, T. J. Schmidt and J. Herranz, J. Electrochem. Soc., 2023, 170, 074502 CrossRef.
- F. Jaouen, V. Goellner, M. Lefèvre, J. Herranz, E. Proietti and J. P. Dodelet, Electrochim. Acta, 2013, 87, 619–628 CrossRef CAS.
- D. Malko, T. Lopes, E. A. Ticianelli and A. Kucernak, J. Power Sources, 2016, 323, 189–200 CrossRef CAS.
- V. A. Saveleva, K. Kumar, P. Theis, N. S. Salas, U. I. Kramm, F. Jaouen, F. Maillard and P. Glatzel, ACS Appl. Energy Mater., 2023, 6, 611–616 CrossRef CAS.
- J.-Q. Zhong, K.-J. Yan, J. Yang, W.-H. Yang and X.-D. Yang, ACS Catal., 2022, 12, 9003–9010 CrossRef CAS.
- L. Osmieri, R. K. Ahluwalia, X. Wang, H. T. Chung, X. Yin, A. J. Kropf, J. Park, D. A. Cullen, K. L. More, P. Zelenay, D. J. Myers and K. C. Neyerlin, Appl. Catal., B, 2019, 257, 117929 CrossRef CAS.
- X. Wang, M. Ferrandon, J. H. Park, J.-J. Shen, A. J. Kropf, H. Zhang, P. Zelenay and D. J. Myers, Electrochim. Acta, 2023, 443, 141934 CrossRef CAS.
- D. H. Kim, S. Ringe, H. Kim, S. Kim, B. Kim, G. Bae, H.-S. Oh, F. Jaouen, W. Kim, H. Kim and C. H. Choi, Nat. Commun., 2021, 12, 1856 CrossRef CAS PubMed.
- M. Gong, A. Mehmood, B. Ali, K.-W. Nam and A. Kucernak, ACS Catal., 2023, 13, 6661–6674 CrossRef CAS PubMed.
- K. Kumar, L. Dubau, M. Mermoux, J. Li, A. Zitolo, J. Nelayah, F. Jaouen and F. Maillard, Angew. Chem., Int. Ed., 2020, 59, 3235–3243 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- D. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. Li, H. Wang, Q. Zhang and S. W. Boettcher, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- T. Reshetenko, A. Serov, M. Odgaard, G. Randolf, L. Osmieri and A. Kulikovsky, Electrochem. Commun., 2020, 118, 106795 CrossRef CAS.
- A. Kulikovsky, Electrochem. Sci. Adv., 2021, 1, e2000023 CrossRef CAS.
- Y.-S. Li, D. Menga, H. Gasteiger and B. Suthar, J. Electrochem. Soc., 2023, 170(9), 094503 CrossRef.
- M. Ahadi, M. Tam, J. Stumper and M. Bahrami, Int. J. Hydrogen Energy, 2019, 44, 3603–3614 CrossRef CAS.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. Taek Chung, C. Marie Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- F. Charreteur, F. Jaouen, S. Ruggeri and J.-P. Dodelet, Electrochim. Acta, 2008, 53, 2925–2938 CrossRef CAS.
- S. Liu, Q. Meyer, C. Jia, S. Wang, C. Rong, Y. Nie and C. Zhao, Energy Environ. Sci., 2023, 16, 3792–3802 RSC.
- M. Schreier, P. Kenis, F. Che and A. S. Hall, ACS Energy Lett., 2023, 3935–3940 CrossRef CAS.
- A. S. Varela, Curr. Opin. Green Sustainable Chem., 2020, 26, 100371 CrossRef.
- A. Bonnefont, Curr. Opin. Electrochem., 2023, 39, 1–8 Search PubMed.
- G. Bae, M. W. Chung, S. G. Ji, F. Jaouen and C. H. Choi, ACS Catal., 2020, 10, 8485–8495 CrossRef CAS.
- I. V. Zenyuk and S. Litster, ECS Trans., 2013, 58, 27–35 CrossRef.
- F. Jaouen, M. Lefèvre, J.-P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed.
- B. N. Ruggiero, K. M. Sanroman Gutierrez, J. D. George, N. M. Mangan, J. M. Notestein and L. C. Seitz, J. Catal., 2022, 414, 33–43 CrossRef CAS.
- S. Rojas-Carbonell, C. Santoro, A. Serov and P. Atanassov, Electrochem. Commun., 2017, 75, 38–42 CrossRef CAS.
- J. H. Zagal and M. T. M. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed.
- S. Vijay, J. A. Gauthier, H. H. Heenen, V. J. Bukas, H. H. Kristoffersen and K. Chan, ACS Catal., 2020, 10, 7826–7835 CrossRef CAS.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2022, 12, 2102715 CrossRef CAS.
- W. Yang, Z. Jia, B. Zhou, L. Wei, Z. Gao and H. Li, Commun. Chem., 2023, 6, 1–6 CrossRef PubMed.
- N. Karmodak and J. K. Nørskov, Angew. Chem., Int. Ed., 2023 DOI:10.1002/anie.202311113.
- J. Barrio, A. Pedersen, J. Feng, S. Ch. Sarma, M. Wang, A. Y. Li, H. Yadegari, H. Luo, M. P. Ryan, M.-M. Titirici and I. E. L. Stephens, J. Mater. Chem. A, 2022, 10, 6023–6030 RSC.
- Z. Liang, D. Yang, P. Tang, C. Zhang, J. Jacas Biendicho, Y. Zhang, J. Llorca, X. Wang, J. Li, M. Heggen, J. David, R. E. Dunin-Borkowski, Y. Zhou, J. R. Morante, A. Cabot and J. Arbiol, Adv. Energy Mater., 2021, 11, 2003507 CrossRef CAS.
- S. Ünsal, R. Girod, C. Appel, D. Karpov, M. Mermoux, F. Maillard, V. A. Saveleva, V. Tileli, T. J. Schmidt and J. Herranz, J. Am. Chem. Soc., 2023, 145, 7845–7858 CrossRef PubMed.
- H. Wan, A. Jensen, M. Escudero-Escribano and J. Rossmeisl, ACS Catal., 2020, 10, 5979–5989 CrossRef CAS.
- T. Sours, A. Patel, J. Nørskov, S. Siahrostami and A. Kulkarni, J. Phys. Chem. Lett., 2020, 11, 10029–10036 CrossRef CAS PubMed.
- Q. Zhang, C. Harms, J. Mitzel, P. Gazdzicki and K. A. Friedrich, Curr. Opin. Electrochem., 2022, 31, 100863 CrossRef CAS.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- G. Bae, M. M. Kim, M. H. Han, J. Cho, D. H. Kim, M. Sougrati, J. Kim, K.-S. Lee, S. H. Joo, W. A. Goddard, H.-S. Oh, H. Kim, F. Jaouen and H. C. Choi, Nat. Catal., 2023, 1–11, DOI:10.1038/s41929-023-01039-7.
- Y.-P. Ku, K. Ehelebe, A. Hutzler, M. Bierling, T. Böhm, A. Zitolo, M. Vorokhta, N. Bibent, F. D. Speck, D. Seeberger, I. Khalakhan, K. J. J. Mayrhofer, S. Thiele, F. Jaouen and S. Cherevko, J. Am. Chem. Soc., 2022, 144, 9753–9763 CrossRef CAS PubMed.
- V. P. Glibin and J.-P. Dodelet, J. Electrochem. Soc., 2017, 164, F948 CrossRef CAS.
- E. F. Holby, G. Wang and P. Zelenay, ACS Catal., 2020, 10, 14527–14539 CrossRef CAS.
- Z. Wang, Y. R. Zheng, I. Chorkendorff and J. K. Nørskov, ACS Energy Lett., 2020, 5, 2905–2908 CrossRef CAS.
- G. T. K. K. Gunasooriya and J. K. Nørskov, ACS Energy Lett., 2020, 5, 3778–3787 CrossRef CAS.
- X. Guo, S. Zhang, L. Kou, C. Y. Yam, T. Frauenheim, Z. Chen and S. Huang, Energy Environ. Sci., 2023 10.1039/D3EE01723K , advance article.
- A. K. Singh, L. Zhou, A. Shinde, S. K. Suram, J. H. Montoya, D. Winston, J. M. Gregoire and K. A. Persson, Chem. Mater., 2017, 29, 10159–10167 CrossRef CAS.
- Y. Zhong, X. Liang, Z. He, W. Tan, J. Zhu, P. Yuan, R. Zhu and H. He, Appl. Catal., B, 2014, 150–151, 612–618 CrossRef CAS.
- C. Jackson, G. T. Smith, D. W. Inwood, A. S. Leach, P. S. Whalley, M. Callisti, T. Polcar, A. E. Russell, P. Levecque and D. Kramer, Nat. Commun., 2017, 8, 1–11 CrossRef PubMed.
- F. Luo, A. Roy, M. T. Sougrati, A. Khan, D. A. Cullen, X. Wang, M. Primbs, A. Zitolo, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2023, 145, 14737–14747 CrossRef CAS PubMed.
- R. K. Ahluwalia, X. Wang, J.-K. Peng, V. Konduru, S. Arisetty, N. Ramaswamy and S. Kumaraguru, J. Electrochem. Soc., 2021, 168, 044518 CrossRef CAS.
- G. Smith and E. J. F. Dickinson, Nat. Commun., 2022, 13, 1–6 Search PubMed.
- S. Minteer, J. Chen, S. Lin, C. Crudden, S. Dehnen, P. V. Kamat, M. Kozlowski, G. Masson and S. J. Miller, ACS Energy Lett., 2023, 8, 1793–1794 CrossRef CAS.
- K. Ehelebe, N. Schmitt, G. Sievers, A. W. Jensen, A. Hrnjić, P. Collantes Jiménez, P. Kaiser, M. Geuß, Y. P. Ku, P. Jovanovič, K. J. J. Mayrhofer, B. Etzold, N. Hodnik, M. Escudero-Escribano, M. Arenz and S. Cherevko, ACS Energy Lett., 2022, 7, 816–826 CrossRef CAS.
- V. Gridin, J. Du, S. Haller, P. Theis, K. Hofmann, G. K. H. Wiberg, U. I. Kramm and M. Arenz, Electrochim. Acta, 2023, 444, 142012 CrossRef CAS.
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, JACS Au, 2021, 1, 527–535 CrossRef CAS PubMed.
- R. G. Mariano, O. J. Wahab, J. A. Rabinowitz, J. Oppenheim, T. Chen, P. R. Unwin and M. Dinca, ACS Cent. Sci., 2022, 8, 975–982 CrossRef CAS PubMed.
- Y. Chen, Y. Huang, M. Xu, T. Asset, X. Yan, K. Artyushkova, M. Kodali, E. Murphy, A. Ly, X. Pan, I. V. Zenyuk and P. Atanassov, Mater. Today, 2022, 53, 58–70 CrossRef CAS.
- Y. Huang, Y. Chen, M. Xu, T. Asset, P. Tieu, A. Gili, D. Kulkarni, V. De Andrade, F. De Carlo, H. S. Barnard, A. Doran, D. Y. Parkinson, X. Pan, P. Atanassov and I. V. Zenyuk, Mater. Today, 2021, 47, 53–68 CrossRef CAS.
- B. D. James, DOE Hydrogen and Fuel Cells Program Review Fuel Cell Vehicle and Bus Cost Analysis, 2015 Search PubMed.
- J. Barrio, A. Pedersen, S. Favero, H. Luo, M. Wang, S. Ch. Sarma, J. Feng, L. T. T. Ngoc, S. Kellner, A. Y. Li, A. B. Jorge Sobrido and M.-M. Titirici, Chem. Rev., 2023, 123, 2311–2348 CrossRef CAS PubMed.
- C. H. Lee, W. J. M. Kort-Kamp, H. Yu, D. A. Cullen, B. M. Patterson, T. A. Arman, S. Komini Babu, R. Mukundan, R. L. Borup and J. S. Spendelow, Nat. Energy, 2023, 8, 685–694 CrossRef CAS.
- A. M. Boyce, D. J. Cumming, C. Huang, S. P. Zankowski, P. S. Grant, D. J. L. Brett and P. R. Shearing, ACS Nano, 2021, 15, 18624–18632 CrossRef CAS PubMed.
- Y. D. Wang, Q. Meyer, K. Tang, J. E. McClure, R. T. White, S. T. Kelly, M. M. Crawford, F. Iacoviello, D. J. L. Brett, P. R. Shearing, P. Mostaghimi, C. Zhao and R. T. Armstrong, Nat. Commun., 2023, 14, 745 CrossRef CAS PubMed.
- M. S. Ferrandon, J. H. Park, X. Wang, E. Coleman, A. Jeremy Kropf and D. J. Myers, Electrochim. Acta, 2023, 441, 141850 CrossRef CAS.
- M. R. Karim, M. Ferrandon, S. Medina, E. Sture, N. Kariuki, D. J. Myers, E. F. Holby, P. Zelenay and T. Ahmed, ACS Appl. Energy Mater., 2020, 3, 9083–9088 CrossRef CAS.
- S. Zhou, P. R. Shearing, D. J. L. Brett and R. Jervis, Curr. Opin. Electrochem., 2022, 31, 100867 CrossRef CAS.
- S. Mitchell, F. Parés, D. Faust Akl, S. M. Collins, D. M. Kepaptsoglou, Q. M. Ramasse, D. Garcia-Gasulla, J. Pérez-Ramírez and N. López, J. Am. Chem. Soc., 2022, 144, 8018–8029 CrossRef CAS PubMed.
- J. K. Pedersen, C. M. Clausen, O. A. Krysiak, B. Xiao, T. A. A. Batchelor, T. Löffler, V. A. Mints, L. Banko, M. Arenz, A. Savan, W. Schuhmann, A. Ludwig and J. Rossmeisl, Angew. Chem., 2021, 133, 24346–24354 CrossRef.
- M. B. Stevens, M. Anand, M. E. Kreider, E. K. Price, J. Z. Zeledón, L. Wang, J. Peng, H. Li, J. M. Gregoire, J. Hummelshøj, T. F. Jaramillo, H. Jia, J. K. Nørskov, Y. Roman-Leshkov, Y. Shao-Horn, B. D. Storey, S. K. Suram, S. B. Torrisi and J. H. Montoya, Energy Environ. Sci., 2022, 15, 3775–3794 RSC.
- S. Ch. Sarma, J. Barrio, M. Gong, A. Pedersen, A. Kucernak, M. Titirici and I. E. L. Stephens, Electrochim. Acta, 2023, 463, 142855 CrossRef CAS.
- C. Castillo-Blas, I. Romero-Muñiz, A. Mavrandonakis, L. Simonelli and A. E. Platero-Prats, Chem. Commun., 2020, 56, 15615–15618 RSC.
- D. M. Koshy, A. T. Landers, D. A. Cullen, A. V. Ievlev, H. M. Meyer, C. Hahn, Z. Bao and T. F. Jaramillo, Adv. Energy Mater., 2020, 10, 2001836 CrossRef CAS.
- S. C. Sarma, J. Barrio, A. Bagger, A. Pedersen, M. Gong, H. Luo, M. Wang, S. Favero, C. Zhao, Q. Zhang, A. Kucernak, M. Titirici and I. E. L. Stephens, Adv. Funct. Mater., 2023, 2302468 CrossRef CAS.
- J. S. Bates, M. R. Johnson, F. Khamespanah, T. W. Root and S. S. Stahl, Chem. Rev., 2023, 123, 6233–6256 CrossRef CAS PubMed.
- A. Y. Li, A. Pedersen, J. Feng, H. Luo, J. Barrio, J. Roman, K. K. Mimi Hii and M.-M. Titirici, Green Chem., 2022, 24, 7574–7583 RSC.
- X. Liu, J. Li, A. Zitolo, M. Gao, J. Jiang, X. Geng, Q. Xie, D. Wu, H. Zheng, X. Cai, J. Lu, F. Jaouen and R. Li, J. Am. Chem. Soc., 2023, 145, 3108–3120 CrossRef CAS PubMed.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035109 CrossRef.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens.Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |