DOI:
10.1039/D2TA09264F
(Review Article)
J. Mater. Chem. A, 2023,
11, 12659-12680
Molecular designs, synthetic strategies, and properties for porphyrins as sensitizers in dye-sensitized solar cells
Received
28th November 2022
, Accepted 13th March 2023
First published on 29th March 2023
Abstract
During the past decades, many efforts have been made to develop porphyrin sensitizers for dye-sensitized solar cells (DSSCs) due to their easy fabrication, low-cost production, and relatively high power conversion efficiency (PCE). Some of the molecular design principles and synthetic strategies have been proved to be able to construct high-performance porphyrin-based DSSCs. Herein, we focus on the promising and effective synthetic strategies for rationally designing porphyrins as sensitizers, and emphasize the crucial factors that have a profound impact on the photovoltaic performance to offer insightful views on the correlation between the molecular structure and photovoltaic performance.
 Yuzhe Zhang | Yuzhe Zhang is a PhD student under the supervision of Prof. Hiroshi Imahori at Kyoto University. He received his Bachelor's (2017) and Master's (2020) degrees in applied chemistry from Tianjin University. His current research is focused on porphyrin dye-sensitized solar cells. |
 Tomohiro Higashino | Tomohiro Higashino was born in 1986 in Osaka, Japan. He received his PhD in organic chemistry from Kyoto University in 2014. He was first an assistant professor (2013–2021) and then an associate professor (2021–present) in Prof. Imahori's group. His current interests include the development of novel π-conjugated materials and their applications. |
 Hiroshi Imahori | Hiroshi Imahori received his Doctor of Science from Kyoto University in 1990. After post-doctoral training at the Salk Institute for Biological Studies, he was first an assistant and then associate professor at Osaka University. Since 2002, he has been a full professor at Kyoto University. His current interests involve solar energy conversion and organic functional materials. |
10th anniversary statement
I would like to congratulate the Journal of Materials Chemistry on its 10th Anniversary. I previously served as a Deputy-Editor-in-Chief of the Journal Materials Chemistry A for four years and am still an Advisory Board member. At that time, the journal was steadily growing in terms of impact factor and submissions and I did not encounter any difficulties in the management. Throughout this activity, I have gotten to know many excellent researchers and RSC staff personally, and formed many relationships I still treasure. I hope the Journal of Materials Chemistry will continue to develop successfully long in to the future.
|
1. Introduction
Since the 19th century Industrial Revolution, fossil fuels have been the main resources in our daily life, which have driven the development of the world economy and the process of modernization. However, with the massive growth in the population of the world, the demand for fossil fuels keeps increasing, and will be inevitably be depleted in the future. Additionally, the excessive consumption of fossil fuels has led to a lot of serious environmental pollution and global warming in recent years. Thus, the impending energy crisis and environmental issues have become the key problems that need to be solved urgently in the world today. From these points of view, the development of sustainable and clean energy sources could be a reasonable solution. Solar energy, geothermal energy, wind energy, tidal energy, biomass energy, and nuclear energy possess their own advantages as alternative energy sources. Among these, the most abundant one is solar energy, which exceeds our global needs. Because solar energy is a substantially infinite energy source, photovoltaics, which can directly convert solar energy to electricity, have attracted considerable attention. In particular, since the present style of dye-sensitized solar cells (DSSCs)1 was reported by O'Regan and Grätzel2 in 1991, they have been developed as an alternative to conventional silicon-based solar cells in these past decades because of their ease of fabrication, low-cost production, high power conversion efficiency (PCE), and advantages in indoor lighting.
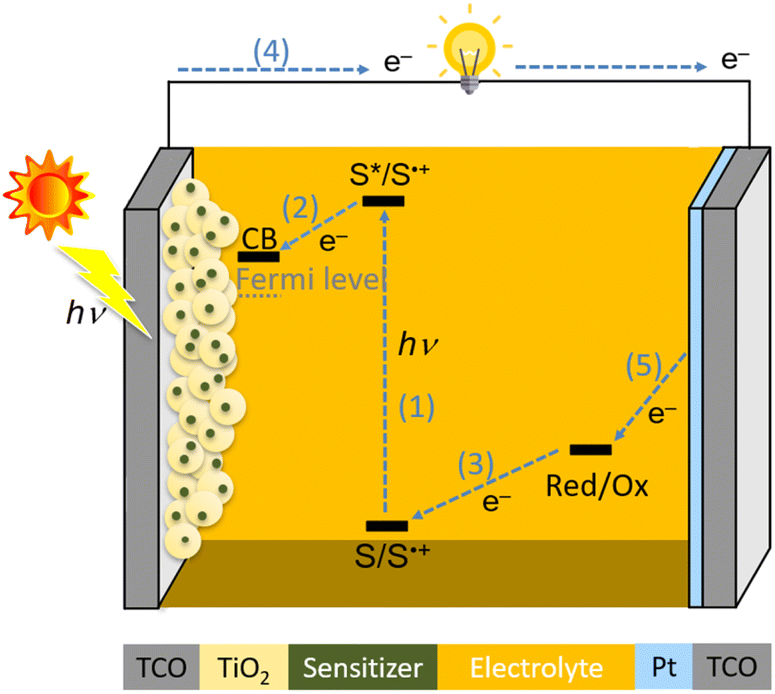
A schematic illustration of the components and operational principles of DSSCs is illustrated in Fig. 1. The typical device of DSSCs is based on a sandwich structure, which consists of the following components: a transparent conductive oxide (TCO) electrode, mesoporous TiO2, sensitizer (S), electrolyte solution, counter Pt electrode, and TCO. The photocurrent is generated by the following processes: (1) a sensitizer attached to TiO2 is excited from the ground state (S) to an excited state (S*) by harvesting sunlight; (2) the photoexcited sensitizer injects an electron into a conduction band (CB) of TiO2, whereby the oxidized sensitizer (S˙+) is generated; (3) the resultant oxidized sensitizer (S˙+) is reduced by redox shuttles in the electrolyte solution (Red: Co2+, Cu1+ complex, or I− species); (4) the electron in the CB is transferred to the counter electrode through an external circuit; (5) the oxidized redox shuttles (Ox: Co3+, Cu2+ complex, or I3− species) are reduced to regenerate the corresponding species at the surface of the counter electrode, and the circuit is thus completed.
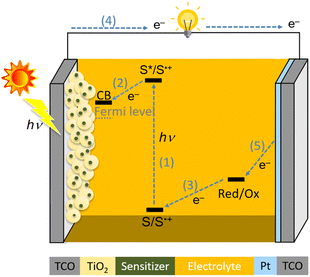 |
| | Fig. 1 Schematic illustration of the components and operational principles of a DSSC. | |
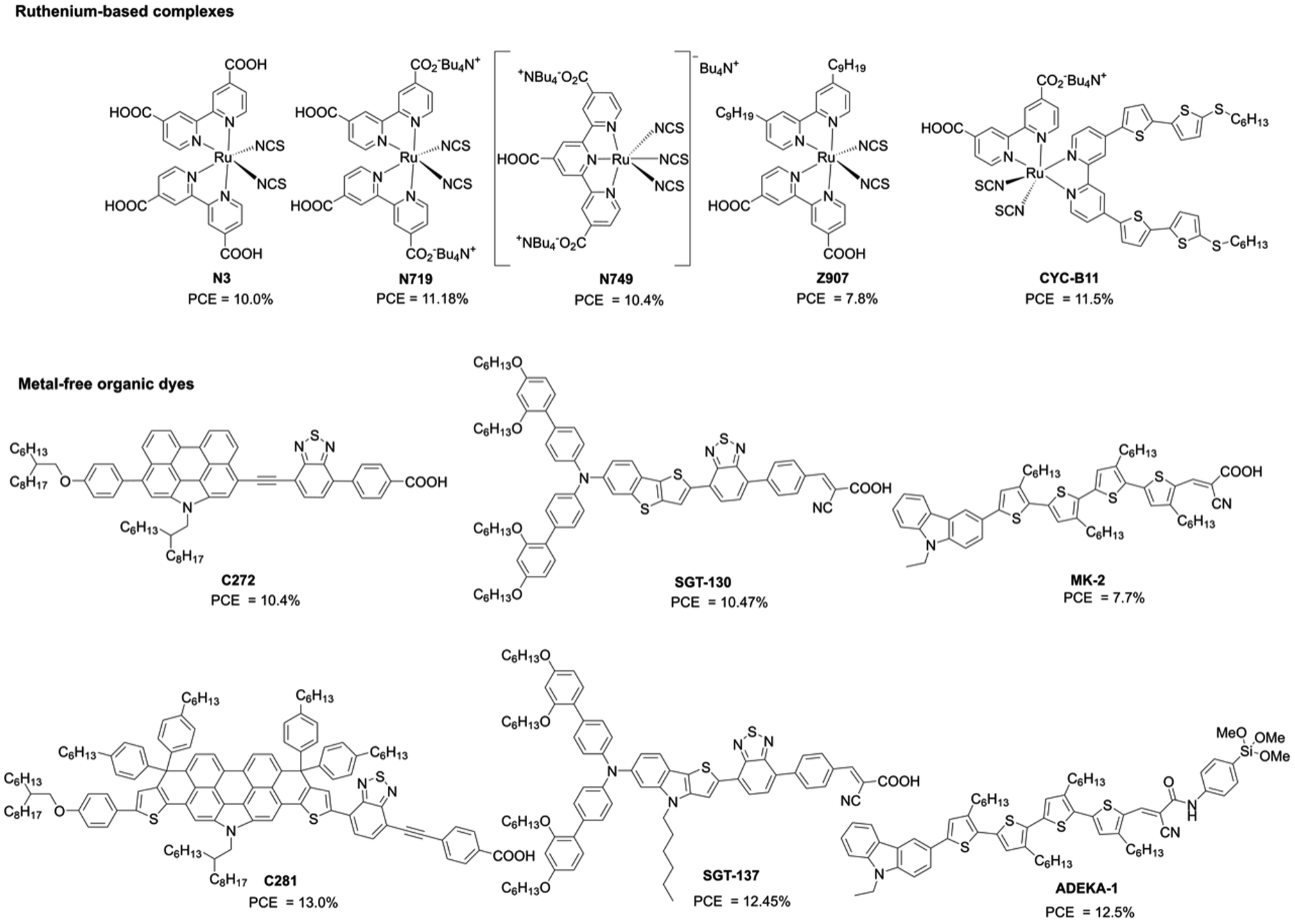
Sensitizers (or dyes) play an important role in the photocurrent generation and device performance of DSSCs. In the early stage of DSSCs, ruthenium-based complexes, such as N3 (ref. 3) and N719 (ref. 4) (Fig. 2), were applied to DSSCs and achieved excellent PCEs of up to 11% as a result of their excellent light-harvesting property, long excited-state lifetime, and high photostability. Furthermore, a lot of efforts have been directed towards the ligands for further optimization: for instance, work on the black dye N749 (ref. 5), whose broad absorption covers the entire visible spectrum, for extending it into the near-infrared (NIR) region (∼920 nm); the amphiphilic sensitizer Z907 (ref. 6), for exhibiting long-term thermostability with a polymer gel electrolyte; the heteroleptic ruthenium sensitizer CYC-B11 (ref. 7) with a hexylthio-bithiophene moiety in ancillary ligands, which possesses an excellent light-harvesting capacity (Fig. 2). However, the high price and limited supply of the ruthenium metal are problematic for its large-scale use in the industrial production of DSSCs. Therefore, ruthenium-free dyes, such as metal-free or cheap metal–organic dyes, have been explored towards high-performance DSSCs because their optical and electronic properties can be easily tuned by molecular modifications. To date, many metal-free organic dyes based on the donor–π-bridge–acceptor (D–π–A) structure have been developed and have attained excellent PCEs owing to their improved light-harvesting abilities by intramolecular charge transfer (ICT) from the donor to acceptor moieties (i.e. push–pull structures).8 The representative examples include C272 (ref. 9), SGT-130 (ref. 10), and MK-2 (ref. 11) (Fig. 2), which are comparable with traditional ruthenium-based complexes. In addition, D–π–A dyes provide wide possibilities for further modification on the donor, π-bridge, and acceptor moieties to achieve higher PCEs. For instance, C281 (ref. 12), SGT137 (ref. 13), and ADEKA-1 (ref. 14) (Fig. 2) have given PCEs of more than 12%.
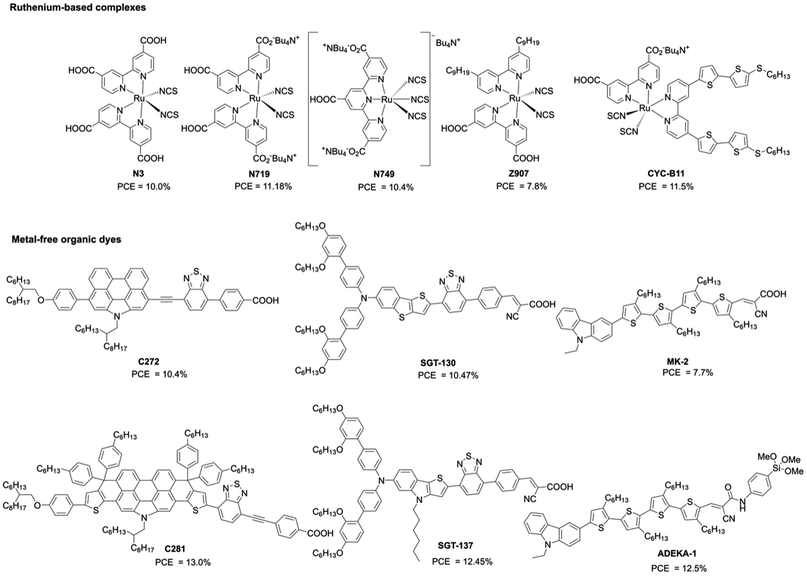 |
| | Fig. 2 . Chemical structures of some representative dyes for DSSCs. | |
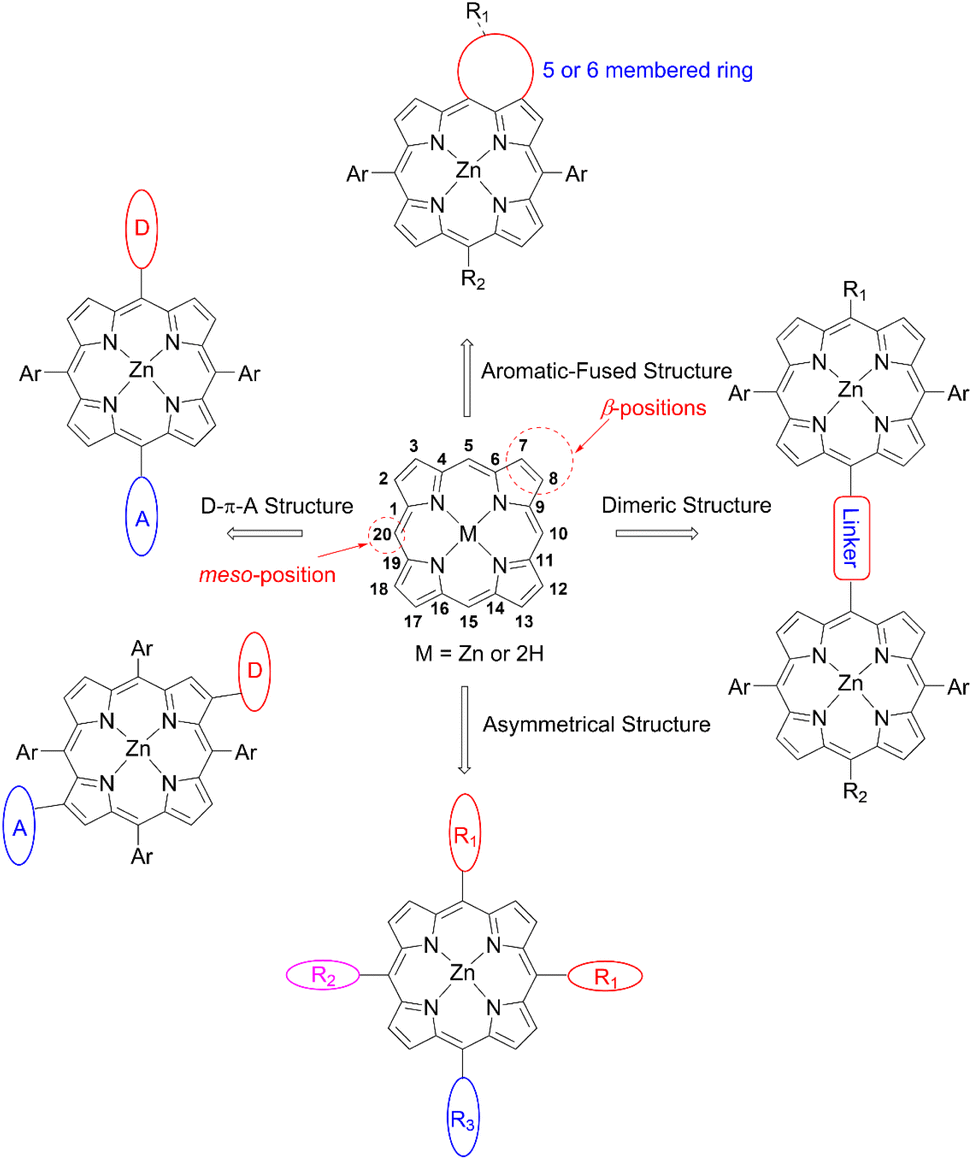
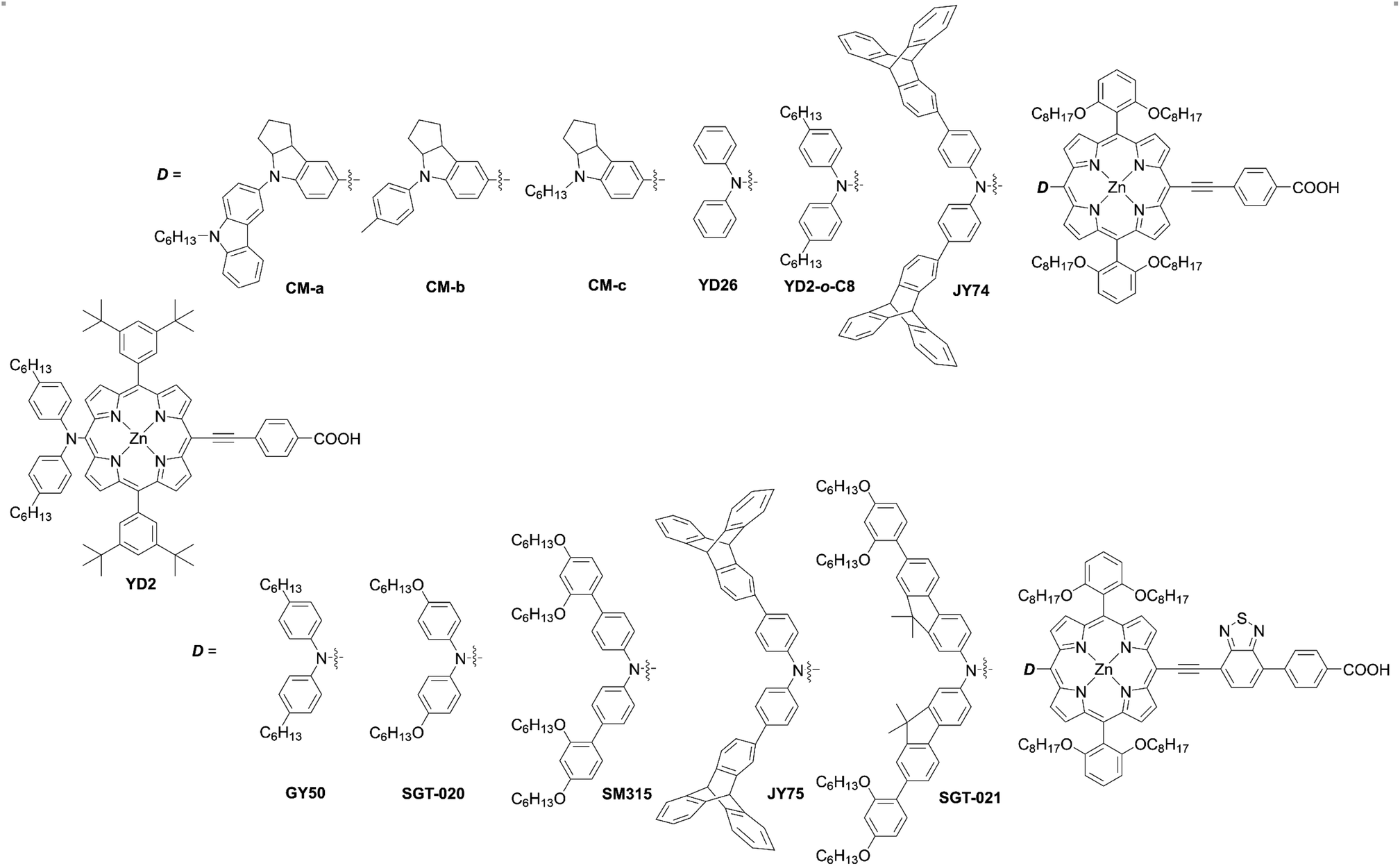
Porphyrin, an aromatic macrocycle with an 18π-electron system, has an intense Soret band (400–500 nm) and moderate Q-bands (500–600 nm) in the visible region, as well as extremely high molar absorption coefficients (ε ∼104–105 M−1 cm−1), surpassing other conventional dyes. Moreover, the introduction of substituents at the four meso- and eight β-positions of the porphyrin can easily tune the electronic, optical, electrochemical, and photophysical properties (Fig. 3). Additionally, porphyrins and related macrocycles are widely found in natural plants as important components for converting sunlight into chemical energy efficiently. Inspired by this elegant solar-energy-conversion process, researchers have attempted to utilize porphyrins as potential sensitizers for DSSCs. However, since the first application of porphyrins to DSSCs by Kay and Grätzel15 in 1993, work on porphyrin-based DSSCs stagnated (PCE < 7.1%) in the decades from 1990 to 2009 because of their weak light-harvesting abilities around 500 nm and over 600 nm. To overcome the bottleneck and kick start the work again in this field, porphyrin sensitizers have adopted a D–π–A structure, as seen in metal-free organic dyes. In 2010, Yeh et al. reported YD2 (ref. 16) (Fig. 4), bearing a bis(4-hexylphenyl)amine donor moiety and a 4-ethynylbenzoic acid acceptor moiety, obtaining a PCE of 11% with the I−/I3− redox couple. Although the I−/I3− redox couple is the most commonly used redox mediator, the open circuit voltage (VOC) value of DSSCs with the I−/I3− redox couple is limited to 0.7–0.8 V due to the large overpotential needed for dye regeneration.1 To surmount this VOC limitation, CoII/III trisbipyridyl ([Co(bpy)3]2+/3+) has been applied as a second-generation redox couple,1 since DSSCs with the [Co(bpy)3]2+/3+ redox shuttle were found to exhibit a high VOC value of up to 1.0 V owing to its more positive redox potential. It is noteworthy that the use of bulky substituents on dyes is essential to suppress the undesirable charge recombination (CR). Indeed, alkoxy-wrapped porphyrin dyes have been designed for DSSCs with the [Co(bpy)3]2+/3+ redox couple, and in this regard, the YD2 analogue YD2-o-C8 (ref. 17) (Fig. 4) showed a PCE of 11.9% with the [Co(bpy)3]2+/3+ redox couple (vide infra).
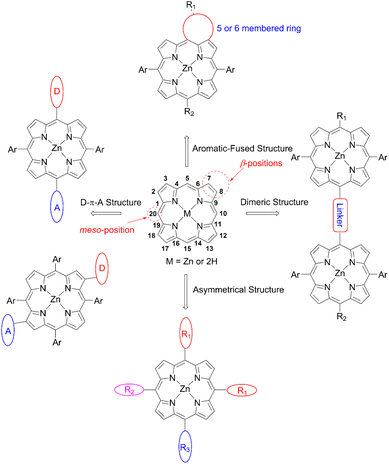 |
| | Fig. 3 Structure and numbering of the porphyrin molecule, and common design principles of porphyrins for DSSCs. | |
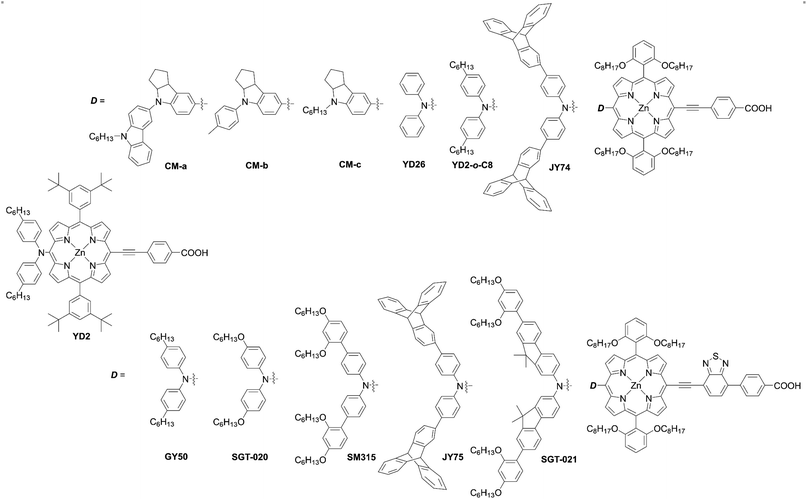 |
| | Fig. 4 Chemical structures of representative meso-functionalized D–π–A type porphyrins. | |
Further optimizations of the D–π–A structure led to the dyes SM315,18GY50 (ref. 19) (Fig. 4), and SGT-021 (ref. 20) (Fig. 4), which achieved impressive PCEs of up to ca. 13%. Although the PCEs of porphyrin-based DSSCs have increased gradually,21–27 there is still plenty of room for boosting the device performance by the rational molecular design of porphyrin sensitizers. Therefore, in this review article, we highlight the rational molecular designs, versatile synthetic strategies, and desired properties of high-performance porphyrin dyes in DSSCs. The representative photovoltaic parameters of porphyrin-based DSSCs are summarized in Tables 1 and 2.
Table 1 Photovoltaic parameters of porphyrin-based DSSCs with I−/I3− as the redox shuttle, under simulated AM 1.5 G full sun illumination (input power was 100 mW cm−2 without additional illumination)
| Dye |
J
SC (mA cm−2) |
V
OC (V) |
F. F.(fill factor) |
PCE (%) |
Ref. |
|
Coadsorbed with CDCA.
Obtained after ageing for several days.
Measured under 6000 lx intensity from a T5 fluorescent tube (input power was 1.70 mW cm−2).
Co-sensitized with Z1 (Fig. 19).
Co-sensitized with Z2 (Fig. 18).
Corrected on the basis of significant figures.
|
|
YD2
|
18.6 |
0.77 |
0.764 |
11 |
16
|
|
CM-a
|
19.56 |
0.668 |
0.651 |
8.5 |
29
|
|
CM-b
|
22.58 |
0.734 |
0.645 |
10.7 |
29
|
|
CM-c
|
20.88 |
0.726 |
0.641 |
9.7 |
29
|
|
YD2-o-C8
|
18.81 |
0.701 |
0.646 |
8.5 |
29
|
|
YD2-o-C8T
|
15.6 |
0.73 |
0.68 |
7.7 |
30
|
|
YD2-o-C8
|
17.3 |
0.78 |
0.65 |
8.8 |
30
|
|
ZnPSi2
|
9.60 |
0.698 |
0.702 |
4.7b |
32
|
|
YD2-o-C8
|
15.0 |
0.754 |
0.684 |
7.7b |
32
|
|
YD26
|
12.92 |
0.662 |
0.62 |
5.3f |
38
|
|
JY74
|
14.58 |
0.681 |
0.63 |
6.3f |
38
|
|
JY75
|
16.50 |
0.673 |
0.61 |
6.8f |
38
|
|
XW29
|
21.26 |
0.716 |
0.666 |
10.14 |
45
|
|
XW30
|
19.67 |
0.678 |
0.651 |
8.68 |
45
|
|
XW31
|
22.06 |
0.720 |
0.658 |
10.46 |
45
|
|
XW32
|
20.13 |
0.677 |
0.647 |
8.82 |
45
|
|
XW17
|
15.96 |
0.700 |
0.729 |
8.14 |
50
|
|
ZnPNTz
|
7.33 |
0.648 |
0.765 |
3.63 |
50
|
|
ZnPBA
|
16.26 |
0.713 |
0.719 |
8.3b |
51
|
|
ZnPBAT
|
19.33 |
0.719 |
0.724 |
10.1b |
51
|
|
YD2
|
17.05 |
0.742 |
0.718 |
9.1b |
51
|
|
ZnPBAT-o-C8
|
18.6 |
0.737 |
0.665 |
9.1b |
52
|
|
ZnPBAT-o-C8C4
|
15.9 |
0.673 |
0.684 |
7.3b |
52
|
|
ZnPBAT-o-C8C8
|
14.1 |
0.706 |
0.709 |
7.0b |
52
|
|
YD2-o-C8
|
16.4 |
0.777 |
0.676 |
8.6b |
52
|
|
21
|
11.6 |
0.563 |
0.62 |
4.1 |
53
|
|
22
|
14.0 |
0.586 |
0.58 |
4.7 |
53
|
|
23
|
8.9 |
0.528 |
0.65 |
3.1 |
53
|
|
24
|
12.9 |
0.598 |
0.60 |
4.6 |
53
|
|
25
|
10.5 |
0.586 |
0.65 |
4.0 |
53
|
|
26
|
11.1 |
0.600 |
0.59 |
3.9 |
53
|
|
27
|
13.3 |
0.519 |
0.61 |
4.2 |
53
|
|
28
|
11.7 |
0.482 |
0.53 |
3.0 |
53
|
|
SK7
|
13.25 |
0.663 |
0.745 |
6.54 |
54
|
|
YD2
|
15.42 |
0.694 |
0.727 |
7.78 |
54
|
|
SK7
|
0.739 |
0.584 |
0.778 |
19.7c |
54
|
|
YD2
|
0.721 |
0.602 |
0.783 |
20.0c |
54
|
|
ZnBD1
|
13.9 |
0.67 |
0.64 |
5.9f |
56
|
|
ZnBD2
|
18.4 |
0.71 |
0.57 |
7.5f |
56
|
|
fused-Zn-1
|
10.6 |
0.62 |
0.62 |
4.1 |
59
|
|
Zn-1
|
6.7 |
0.61 |
0.68 |
2.8 |
59
|
|
ZnQMA
|
11.2 |
0.72 |
0.68 |
5.2 |
61
|
|
ZnQDA
|
9.3 |
0.67 |
0.64 |
4.0 |
61
|
|
ZnPQI
|
13.9 |
0.68 |
0.71 |
6.8 |
62
|
|
ZnQMA
|
13.2 |
0.71 |
0.67 |
6.3 |
62
|
|
fused-ZnP
|
0.88 |
0.51 |
0.67 |
0.3 |
63
|
|
YH6
|
16.12 |
0.58 |
0.72 |
6.7f |
65
|
|
YH7
|
15.12 |
0.57 |
0.66 |
5.7f |
65
|
|
YH8
|
12.51 |
0.60 |
0.72 |
5.3 |
66
|
|
YH9
|
16.16 |
0.63 |
0.68 |
6.9 |
66
|
|
YH10
|
17.16 |
0.63 |
0.70 |
7.5 |
66
|
|
AfZnP
|
11.1 |
0.678 |
0.723 |
5.47 |
68
|
|
DfZnP
|
15.4 |
0.669 |
0.708 |
7.31 |
68
|
|
GY50
|
16.3 |
0.732 |
0.713 |
8.53 |
68
|
|
LD14
|
18.2 |
0.716 |
0.704 |
9.2 |
71
|
|
LDD1
|
18.8 |
0.692 |
0.677 |
8.8 |
71
|
|
LDD1+LD14
|
21.3 |
0.705 |
0.692 |
10.4 |
71
|
|
ZnPTD
|
14.7 |
0.595 |
0.712 |
6.22 |
72
|
|
LDD1
|
19.0 |
0.633 |
0.680 |
8.17 |
72
|
|
JY06
|
11.18 |
0.63 |
0.61 |
4.3f |
73
|
|
JY07
|
13.20 |
0.65 |
0.62 |
5.3f |
73
|
|
75
|
8.55 |
0.610 |
0.6996 |
3.65 |
80
|
|
76
|
7.21 |
0.600 |
0.6741 |
2.92 |
80
|
|
77
|
4.35 |
0.590 |
0.7206 |
1.85 |
80
|
|
78
|
8.37 |
0.600 |
0.7138 |
3.60 |
80
|
|
BC2H
|
5.95 |
0.60 |
0.75 |
2.7b |
81
|
|
YD2
|
17.6 |
0.72 |
0.70 |
8.9b |
81
|
|
LS-01
|
12.58 |
0.53 |
0.70 |
4.7f |
82
|
|
LS-11
|
16.13 |
0.52 |
0.64 |
5.4f |
82
|
|
H2PE1
|
5.26 |
0.54 |
0.73 |
2.1f |
82
|
|
LS-17
|
14.90 |
0.54 |
0.64 |
5.1f |
84
|
|
LS-43
|
14.67 |
0.52 |
0.61 |
4.6f |
84
|
|
LS-45
|
17.43 |
0.54 |
0.64 |
6.0f |
84
|
|
XW10
|
17.90 |
0.711 |
0.684 |
8.60 |
42
|
|
XW40
|
18.67 |
0.730 |
0.683 |
9.31 |
85
|
|
XW41
|
16.77 |
0.695 |
0.701 |
8.16 |
85
|
|
XW40
,
|
19.59 |
0.748 |
0.719 |
10.5f |
85
|
|
XW41
,
|
19.63 |
0.726 |
0.715 |
10.2f |
85
|
|
XW60
|
16.77 |
0.715 |
0.731 |
8.8 |
87
|
|
XW61
|
20.75 |
0.763 |
0.739 |
11.7 |
87
|
|
XW62
|
20.70 |
0.762 |
0.732 |
11.6 |
87
|
|
XW63
|
20.63 |
0.763 |
0.737 |
11.6 |
87
|
|
XW61
|
21.41 |
0.775 |
0.747 |
12.4 |
87
|
|
XW51
|
20.07 |
0.781 |
0.702 |
11.1 |
88
|
|
XW51
|
18.05 |
0.720 |
0.708 |
9.2 |
87
|
|
XW51
,
|
20.13 |
0.738 |
0.705 |
10.5 |
87
|
Table 2 Photovoltaic parameters of porphyrin-based DSSCs with [Co(bpy)3]2+/3+ as the redox shuttle, under simulated AM 1.5 G full sun illumination (input power was 100 mW cm−2 without additional illumination)
| Dye |
J
SC (mA cm−2) |
V
OC (V) |
F. F.(fill factor) |
PCE (%) |
Ref. |
|
99.5 mW cm−2.
Coadsorbed with HC-A4 (Fig. 19).
Coadsorbed with CDCA.
Co-sensitized with LEG4 (Fig. 12).
Corrected on the basis of significant figures.
|
|
YD2-o-C8
|
17.3 |
0.965 |
0.71 |
12e |
17
|
|
SM315
|
18.1 |
0.91 |
0.78 |
13e |
18
|
|
GY50
|
18.53 |
0.885 |
0.773 |
12.7e |
19
|
|
SGT-021b |
17.5 |
0.910 |
0.753 |
12.0 |
20
|
|
SGT-020b |
15.8 |
0.864 |
0.766 |
10.5 |
20
|
|
SM315
|
16.4 |
0.893 |
0.794 |
11.6 |
20
|
|
YD2-o-C8HA
|
10.0 |
0.83 |
0.77 |
6.4 |
31
|
|
YD2-o-C8
|
12.6 |
0.88 |
0.74 |
8.3 |
31
|
|
ZnPTC
|
8.46 |
0.757 |
0.760 |
4.87 |
33
|
|
YD2-o-C8
|
14.6 |
0.912 |
0.746 |
9.93 |
33
|
|
ZnEP1
|
11.1 |
0.77 |
0.69 |
5.9 |
57
|
|
ZnEP2
|
8.2 |
0.77 |
0.64 |
4.0 |
57
|
|
ZnBD1
|
13.1 |
0.78 |
0.68 |
7.1 |
58
|
|
ZnBD2
|
14.1 |
0.83 |
0.69 |
8.2 |
58
|
|
ZnPH1
|
10.8 |
0.77 |
0.69 |
5.7 |
58
|
|
ZnPH2
|
7.8 |
0.76 |
0.64 |
3.8 |
58
|
|
ZnPV1
|
12.2 |
0.77 |
0.70 |
6.6 |
58
|
|
ZnPV2
|
10.7 |
0.78 |
0.64 |
5.3 |
58
|
|
WW-3
|
9.81 |
0.744 |
0.767 |
5.6 |
67
|
|
WW-4
|
3.00 |
0.500 |
0.299 |
0.3 |
67
|
|
DfZnP
|
14.9 |
0.798 |
0.739 |
8.78 |
68
|
|
DfZnP-iPr
|
16.6 |
0.823 |
0.737 |
10.1 |
68
|
|
DfZnP-iPr
|
17.0 |
0.850 |
0.740 |
10.7 |
68
|
|
GY50
|
16.3 |
0.874 |
0.700 |
10.0 |
68
|
|
GY50
|
14.5 |
0.908 |
0.713 |
9.42 |
68
|
|
m-ZnPD
|
12.8 |
0.821 |
0.755 |
7.91 |
77
|
|
YD2-o-C8
|
15.1 |
0.908 |
0.738 |
10.1 |
77
|
|
mSJ1
|
10.551 |
0.833 |
0.762 |
6.69 |
86
|
|
mSJ2
|
5.468 |
0.845 |
0.752 |
3.48 |
86
|
|
mSJ3
|
3.729 |
0.814 |
0.768 |
2.33 |
86
|
|
bSJ1
|
12.525 |
0.823 |
0.779 |
8.03 |
86
|
|
bSJ2
|
16.856 |
0.849 |
0.759 |
10.7e |
86
|
|
bSJ3
|
16.483 |
0.836 |
0.755 |
10.4e |
86
|
|
YD2-o-C8
|
15.130 |
0.849 |
0.765 |
9.83 |
86
|
2. D–π–A-structured porphyrins
2.1
Meso-functionalized porphyrins
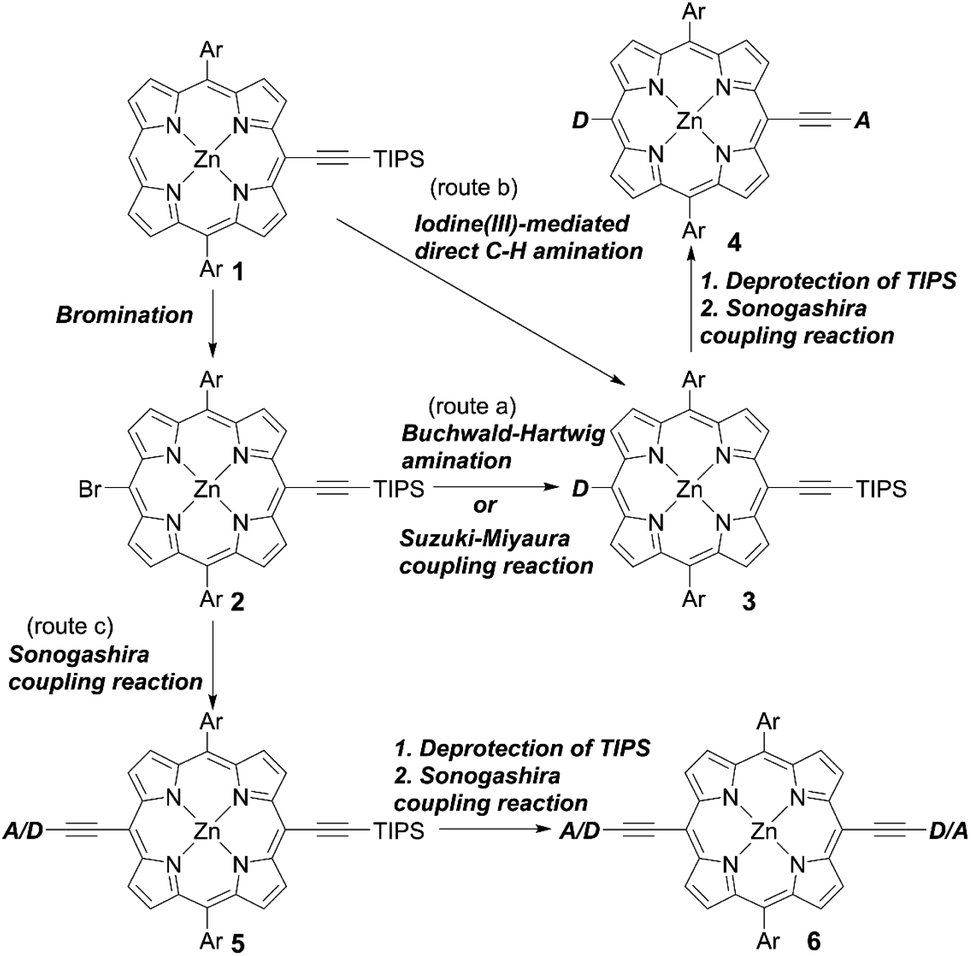
Adoption of a push–pull structure by donors and acceptors, respectively, is the most effective strategy in terms of the molecular design of high-performance porphyrin dyes because of their enhanced light-harvesting abilities and electron-injection efficiencies (ϕinj) based on ICT. Generally, push–pull porphyrins consist of a donor, π-porphyrin core, and acceptor. Because meso-substituted porphyrins can be relatively easily synthesized and meso-substituents can effectively affect their electronic structures, well-designed meso-substituted push–pull porphyrins have been extensively studied for application in DSSCs. Among them, trans-A2BC-type push–pull porphyrin dyes are representative examples. The common synthetic protocols are shown in Scheme 1. Route a, which was reported by Yeh et al. for the synthesis of YD2 (Fig. 4),28 is the most frequently adopted strategy. Here, the meso-(triisopropylsilyl)ethynyl porphyrin 1 undergoes bromination to give 2, which takes part in a subsequent Buchwald–Hartwig amination or Suzuki–Miyaura coupling reaction to introduce a donor moiety, such as diarylamino and indoline groups. Then, the triisopropylsilyl (TIPS) group of 3 is deprotected using tetrabutylammonium fluoride (TBAF), and then undergoes a Sonogashira coupling reaction to afford the trans-A2BC-type push–pull porphyrin dye 4. According to this strategy, benchmark push–pull porphyrin dyes, for example, YD2-o-C8, GY50, and SM315 (Fig. 4), have been synthesized. One of the advantages of this protocol is that we can easily change the donor and/or acceptor moieties to evaluate the effect of different molecular structures. Actually, the incorporation of additional electron-withdrawing benzothiadiazole (BTD) between the ethynyl and benzoic acid groups in GY50 and SM315 is an effective way to obtain high-performance sensitizers.
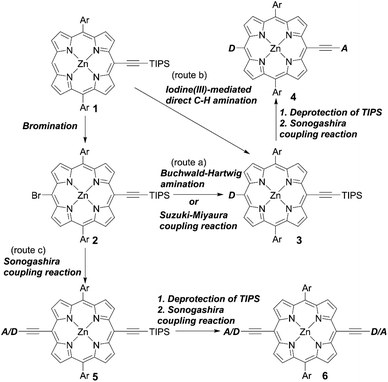 |
| | Scheme 1 Synthetic strategies for meso-functionalized D–π–A structured porphyrins. Ar = aryl substituents. | |
In 2017, Kim and co-workers20 designed and synthesized two porphyrin sensitizers, namely SGT-020 and SGT-021 (Fig. 4), according to this route a, and studied the effects of the electron-donating ability and bulkiness on the photovoltaic performance with SM315 as a reference. Among the three dyes, SGT-021 with the rigid and electron-rich fluorenyl moieties exhibited the highest PCE of 12.0% because of the improved short circuit current density (JSC) and VOC values. The electron-rich nature of fluorenyl moieties enhanced the light-harvesting ability due to the red-shifted absorption. In addition, the rigid fluorenyl moieties can ensure an efficient blocking effect and suppress undesirable CR. Wang and co-workers29 screened D–π–A-type porphyrin dyes with various donor moieties by using theoretical calculations and found that the indoline motifs may be suitable donor groups because of their nonplanar geometry and the large oscillator strength of the S0–S1 transition. Based on these screening results, they designed and synthesized three indoline-conjugated porphyrins: CM-a, CM-b, and CM-c (Fig. 4). As they expected, the molar extinction coefficients (ε) of the Soret and Q-bands for CM-a, CM-b, and CM-c were larger than those for YD2-o-C8, indicating the improvement of the light-harvesting abilities. Although the dye-loading amounts (Γ) for CM-a (2.1 × 10−7 mol cm−2), CM-b (1.4 × 10−7 mol cm−2), and CM-c (1.5 × 10−7 mol cm−2) were smaller than that for YD2-o-C8 (3.9 × 10−7 mol cm−2), which may result from the increasing nonplanarity and rigidity of the indoline unit compared to the diarylamino group of YD2-o-C8, their improved light-harvesting abilities compensated for their smaller dye-loading amounts. Consequently, the maximum PCEs for CM-a (8.5%), CM-b (10.7%), and CM-c (9.7%) were comparable or higher than that for YD2-o-C8 (8.5%). The highest PCE for CM-b could be attributed to its strongest absorption of both Soret and Q-bands. This value was a record for non-ruthenium iodine-based DSSCs without a co-sensitizer and co-adsorbate at that time.
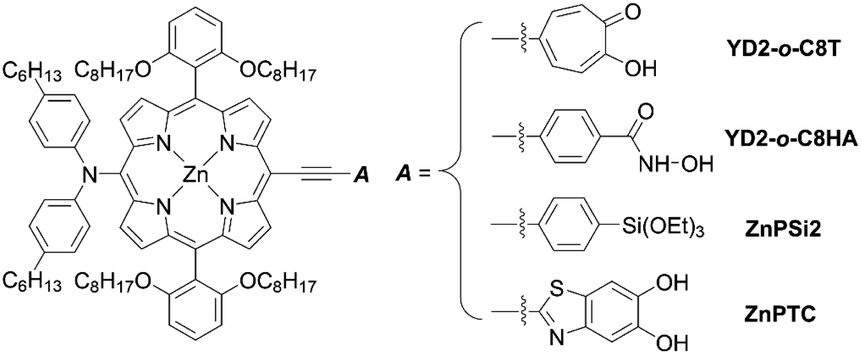
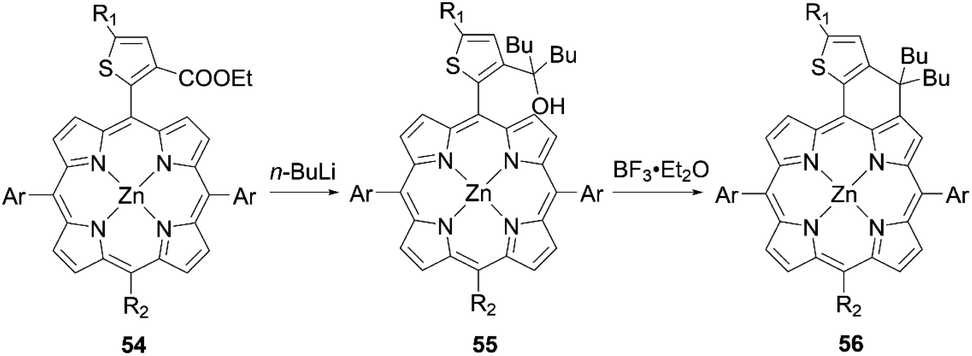
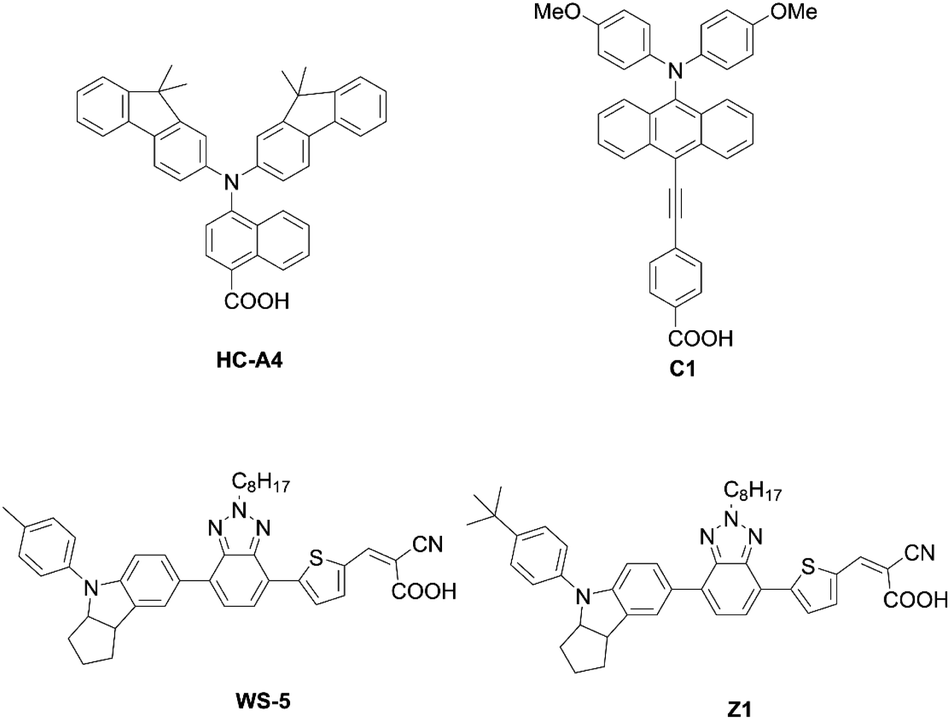
In addition to examining the effect of a donor moiety, the effect of an anchoring moiety in the electron-withdrawing group can be also easily discussed by this synthetic protocol. On the basis of the benchmark dye YD2-o-C8, Imahori and co-workers designed and synthesized a series of porphyrin dyes, namely YD2-o-C8T (ref. 30), YD2-o-C8HA (ref. 31), ZnPSi2 (ref. 32), and ZnPTC (ref. 33) (Fig. 5), by introducing tropolone, hydroxamic acid, triethoxysilyl, and thiazolocatechol moieties, respectively. The PCE values based on these dyes were in the order: YD2-o-C8T (7.7%) > YD2-o-C8HA (6.4%) > ZnPTC (4.87%) ≈ ZnPSi2 (4.7%). Although the PCEs for ZnPSi2 and ZnPTC were significantly lower than that for the carboxylic acid dye YD2-o-C8 (7.7–9.93%), the PCEs for YD2-o-C8T and YD2-o-C8HA were close to that for YD2-o-C8 (8.3–8.8%). The tropolone anchoring group showed superior long-term durability for iodine-based DSSCs, but it could not be applied to cobalt-based DSSCs due to the too-strong-coordination ability of the tropolone moiety to metal ions. On the other hand, the hydroxamic acid anchoring group was applicable to cobalt-based DSSCs and could attain high long-term durability. In addition, the PCE of 4.7% obtained by a ZnPTC-based DSSC is the record efficiency ever reported for DSSCs with catechol-based sensitizers, which had previously never exceeded a PCE of 2%. Generally, the stability of sensitizers, especially the excited state of sensitizers, play the critical role in the durability of DSSCs.34 Apart from the inherent nature of dyes, the binding ability of the anchoring groups also has a considerable effect on the long-term durability of DSSCs, which is regarded as a reason for the low durability of porphyrin-based DSSCs compared to ruthenium-based counterparts. This is because most porphyrin sensitizers possess only one carboxyl anchoring group, whereas ruthenium-based dyes typically have at least two carboxyl anchoring groups.35,36 For instance, the representative porphyrin-based dye SM315 exhibit good long-term durability and is not prone to degradation under continuous irradiation, whereas the desorption of a small amount of dyes results in a slightly decreased PCE. Therefore, stronger anchoring groups are expected to be developed for further improving device durability.
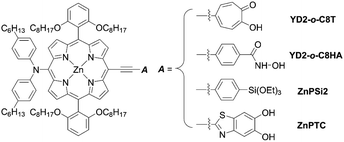 |
| | Fig. 5 Chemical structures of representative D–π–A type porphyrins with different anchoring groups. | |
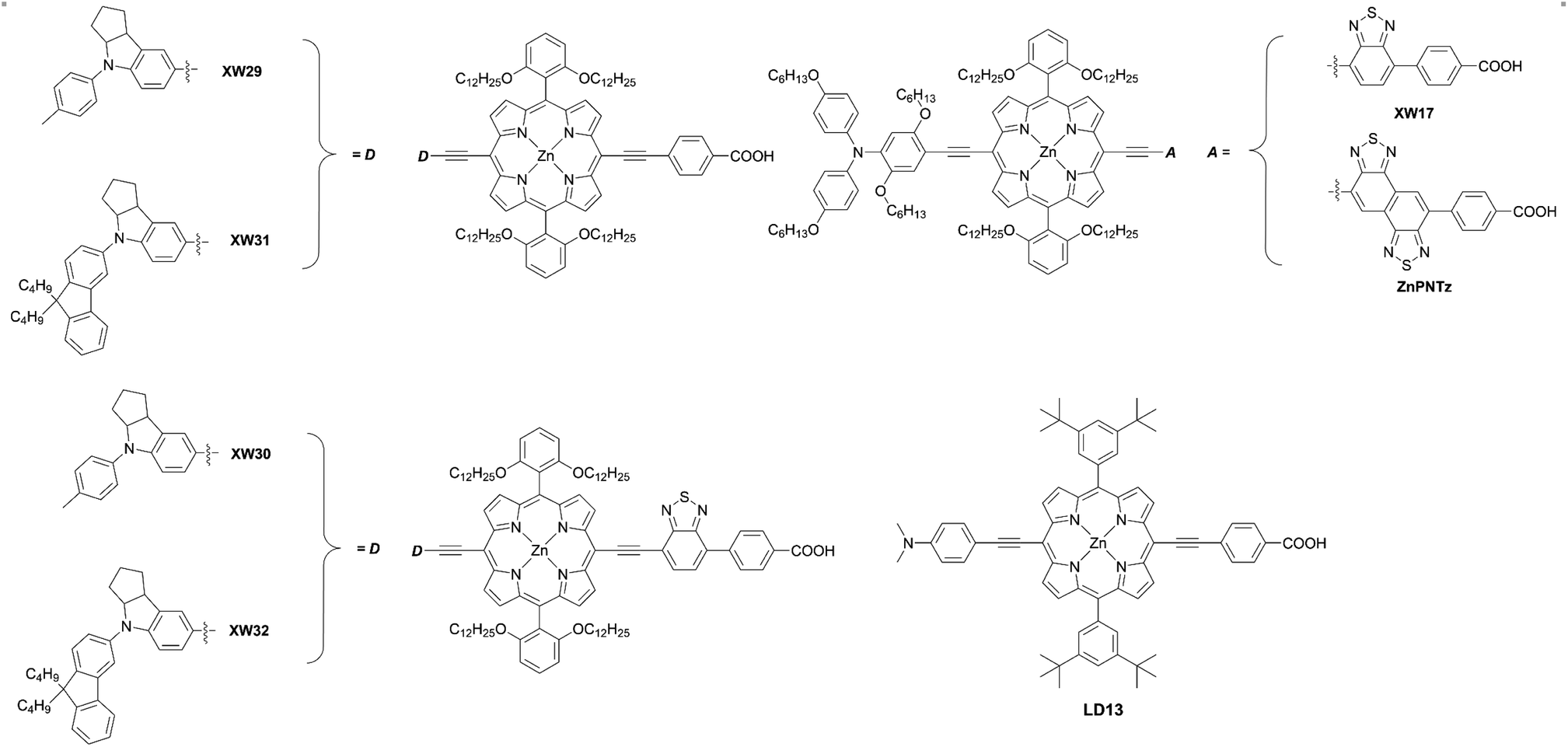
Besides, the iodine(III)-mediated direct C–H amination reaction37 of the meso-free porphyrin 1 and subsequent deprotection and Sonogashira coupling reactions of 3 can be an alternative method to synthesize trans-A2BC-type push–pull porphyrin dyes 4 (Scheme 1, route b). Compared to the Pd-catalyzed reactions of halogenated porphyrins (Scheme 1, route a), adopting the iodine(III)-mediated direct C–H amination reaction can reduce the number of synthetic steps and improve the overall reaction yield. Moreover, the conditions for this reaction are mild (i.e. there is no need for a dry solvent or nitrogen atmosphere) and the process is rapid, which can save time and economic costs. Recently, Zheng and co-workers38 first introduced triptycene groups at the diphenyl amine unit of YD26 to synthesize JY74, as well as JY75 (Fig. 4) with a BTD auxiliary electron-withdrawing acceptor. As mentioned above, the direct C–H amination simplified the synthetic steps and improved the yield compared with the process requiring the synthesis of a precursor for YD26 (70% vs. 54% for the optimized steps) (Fig. 4, 3 in Scheme 1, Ar = 2,6-di(octyloxyphenyl), D = diphenylamino).39JY75 exhibited a moderate PCE of 6.77%, which was increased by 26.8% and 7.6% compared to those of YD26 (5.34%) and JY74 (6.29%), respectively. Because of the well-known rigid and shape-persistent structure of triptycene, its inherent steric effect prevented penetration of the redox shuttle to the surface of TiO2 efficiently. Furthermore, the introduction of the electron-withdrawing BTD unit further enhanced the JSC value, resulting in the obvious improvement in device efficiency.
Incorporating an additional ethynyl group into the donor side is a useful strategy for enlarging the π-conjugation to overcome the insufficient absorption of porphyrins around 500 nm and over 600 nm. Instead of the Buchwald–Hartwig amination or Suzuki–Miyaura coupling reaction, the Sonogashira coupling reaction of 2 and subsequent deprotection and Sonogashira coupling reaction of 5 could furnish the push–pull porphyrin dyes 6 with two ethynyl moieties (Scheme 1, route c). This strategy was established by Lin et al. for the synthesis of LD13 (Fig. 6).40 According to this protocol, Xie and co-workers synthesized various D–π–A-type porphyrin sensitizers with red-shifted Q-bands.41–44 They evaluated the effects of the donor and/or acceptor moieties and found that the introduction of suitable donors and/or acceptors could broaden the absorption band due to the enlarged π-conjugation and strengthened ICT process, resulting in improved device efficiency. Importantly, the introduction of bulky alkyl chains was able to supress dye aggregation and CR caused by the large π-conjugation framework, and further optimize the cell performance. In 2018, Xie and co-workers45 reported a series of porphyrin sensitizers XW29–XW32 (Fig. 6) with indoline derivatives as the donor moieties. For XW29 and XW31 without the additional BTD acceptor, high PCEs exceeding 10% were attainable. Furthermore, a high PCE of 10.46% was observed by XW31, in comparison with that of 10.14% for XW29. Both the JSC and VOC values for XW31 could be improved by introducing the dialkylfluorene unit, as a result of its red-shifted absorption, increased dye-loading amounts, and suppressed CR processes. Specifically, dialkylfluorene-substituted ones (XW31: 1.14 × 10−7 mol cm−2, XW32: 1.67 × 10−7 mol cm−2) exhibited slightly higher dye-loading amounts than the methylphenyl-substituted analogues (XW29: 9.7 × 10−8 mol cm−2, XW30: 1.49 × 10−7 mol cm−2), respectively, which may be attributed to the increased dye solubility and promoted dye adsorption process by the additional alkyl chains.46 However, XW30 and XW32 with the additional BTD group afforded lower PCEs of 8.68% and 8.82%, respectively, although they showed higher dye-loading amounts compared to XW29 and XW31. This trend could be explained through the increase in their dye aggregation, consistent with the higher Γ values, and charge-recombination effects owing to the introduction of the strong electron-drawing BTD moiety. As we demonstrated before,47–49 adsorbed Zn-porphyrins on TiO2 are heavily tilted to the surface, even if they are well-packed on TiO2 (40°∼70° from the surface normal). The tilt angle of Zn-porphyrins on TiO2 depends on the dye-loading amount, molecular length, bulkiness, interaction between specifically incorporated atoms (N, F, S atoms) and TiO2, and the adsorption conditions (time, immersion solvent, etc.). Thus, electron transfer (ET) between the porphyrin and TiO2 occurs through a through-space mechanism rather than a through-bond mechanism. Namely, a greater tilted geometry of the porphyrin on TiO2 leads to faster electron injection from the excited porphyrin to TiO2 and faster CR from TiO2 to the oxidized porphyrin, thus lowering the PCE. Overall, this trend mentioned above can be rationalized by plausible fast CR in XW30 and XW32 due to their longer molecular lengths and resultant more tilted adsorption orientation on TiO2. Meanwhile, Imahori and co-workers50 first introduced the naphtho-[1,2-c:5,6-c′]bis[1,2,5]-thiadiazole (NTz) unit into porphyrins-based DSSCs (ZnPNTz) (Fig. 6). The stronger electron-withdrawing ability of the NTz moiety than the widely used BTD was expected to enhance the light-harvesting ability. In comparison with XW17 (Fig. 6),43ZnPNTz showed a red-shifted (14 nm) and broadened Q-band, and the absorption edge exceeded 800 nm. However, a lower PCE of 3.63% was observed for ZnPNTz than that for XW17 (8.14%). The lower PCE of ZnPNTz may have resulted from the inefficient electron injection derived from its inferior ET parameters as well as much faster CR, as a result of the longer molecular length of ZnPNTz than that of XW17.47–49
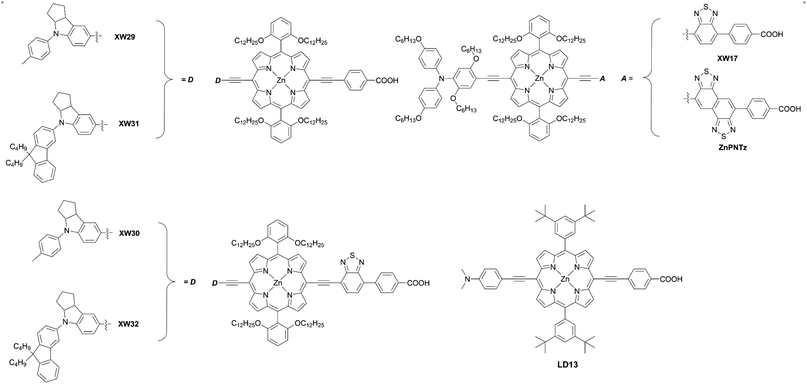 |
| | Fig. 6 Chemical structures of representative meso-functionalized D–π–A type porphyrins with an acene bridge. | |
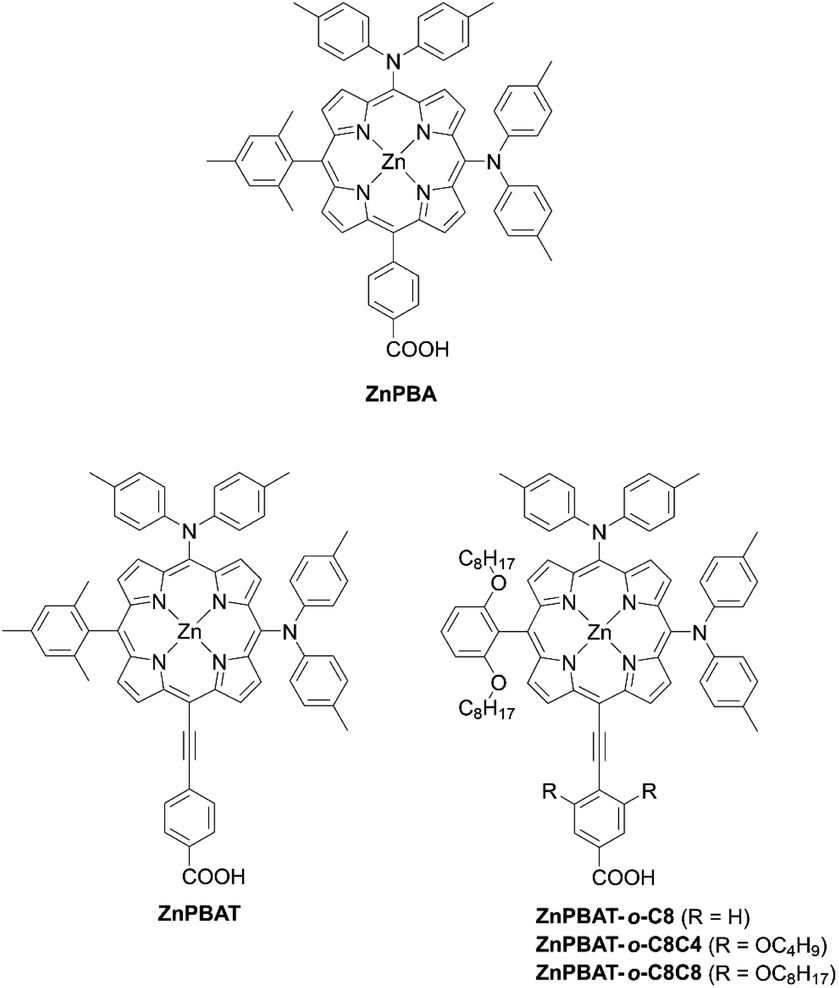
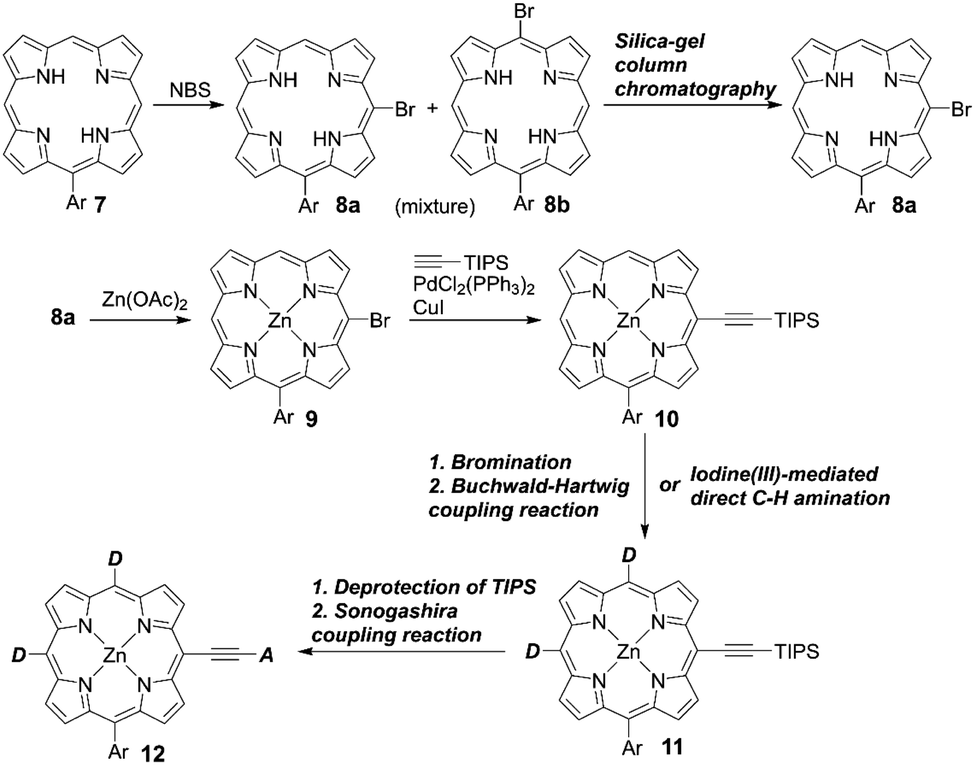
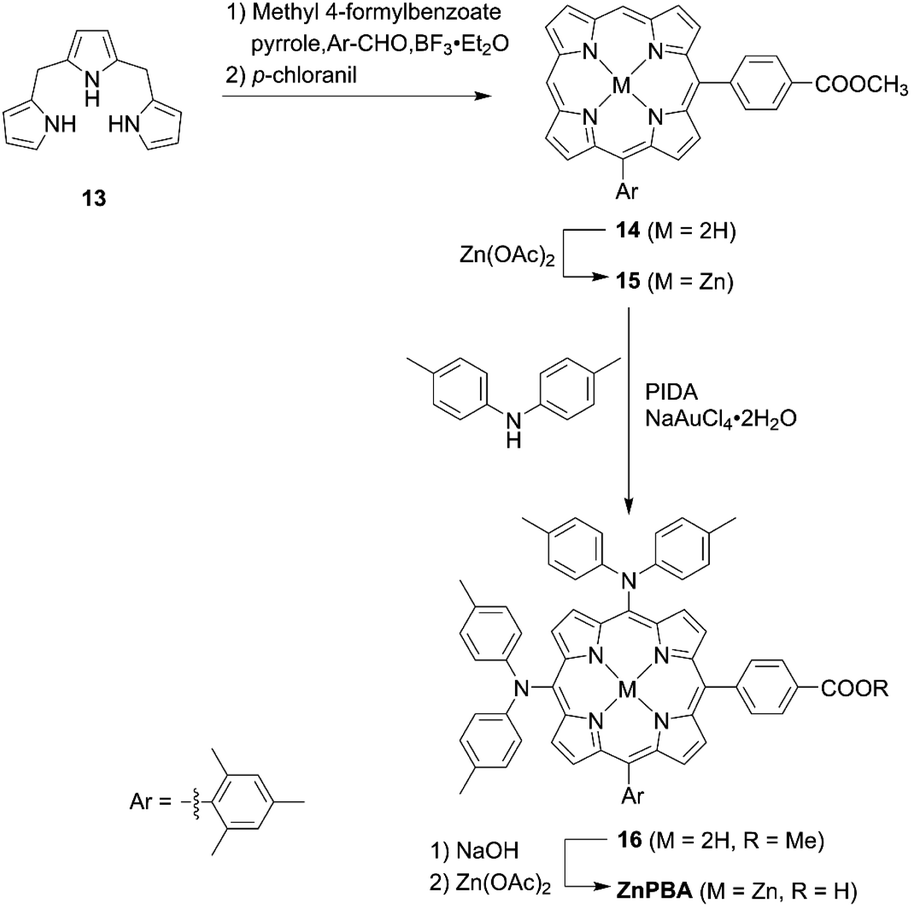
In contrast to trans-A2BC-type push–pull porphyrin dyes, cis-A2BC-type push–pull porphyrin dyes with two donor moieties are more appealing because of the more favourable orbital distribution in the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for ET processes as well as improved light-harvesting ability by their asymmetrical molecular structure (vide infra). Despite the prospect of cis-A2BC-type push–pull porphyrin dyes, it is a synthetic challenge to introduce three different substitutions at desired meso-positions. In 2013, Imahori and co-workers first reported the cis-A2BC-type push–pull porphyrin dyes ZnPBA and ZnPBAT (Fig. 7).51 First, 5-substituted porphyrin 7 was employed as the starting material. The bromination of the free base porphyrin 7 afforded two isomers of the mono-brominated porphyrins, 8a and 8b, which could be separated by conventional silica-gel column chromatography. After the metalation of 8a, the Sonogashira coupling reaction of 9 provides 10. Then, bromination and Buchwald–Hartwig coupling reactions, or a iodine(III)-mediated direct C–H amination reaction, gave the corresponding cis-A2BC-type porphyrins 11. Finally, deprotection of the TIPS moiety, followed by the Sonogashira coupling reaction and hydrolysis furnished the cis-A2BC-type push–pull porphyrin dyes 12 with the two diarylamino moieties (Scheme 2). They synthesized ZnPBAT according to this protocol. The reference ZnPBA can be prepared by a direct C–H amination of the cis-AB-type porphyrin 15 and subsequent hydrolysis and metalation of 16, which could be obtained by the acid-catalyzed condensation of meso-free tripyrrane 13 and metalation of 14 (Scheme 3). Both ZnPBA and ZnPBAT revealed broadened and red-shifted absorption spectra relative to YD2 as a result of the introduction of the additional diarylamino moieties. In addition, the Q-bands of ZnPBAT were red-shifted compared to ZnPBA due to an effective π-extension by the triple bond. Because of the improved light-harvesting ability of ZnPBAT, the DSSC with ZnPBAT achieved the highest incident photon-to-current efficiency (IPCE) and resulted in the best PCE among these three dyes (ZnPBA (8.3%) < YD2 (9.1%) < ZnPBAT (10.1%)). Moreover, a ZnPBAT-based cell exhibited long-term durability and only a 10% decay of the overall device efficiency under exposure to full sunlight for 300 h at 25 °C, which may be attributed to the loss of the volatile solvent in the electrolyte solution and desorption of the sensitizers. Later, Imahori and co-workers further attempted to optimize its molecular structure by introducing alkoxyl groups into the electron-withdrawing anchoring group and meso-phenyl group, giving ZnPBAT-o-C8, ZnPBAT-o-C8C4, and ZnPBAT-o-C8C8 (Fig. 7).52 Although the introduction of the alkoxy groups reduced dye aggregation and enhanced the blocking effect effectively, they caused intermolecular alkoxy–alkoxy interactions, resulting in an unexpected aggregation tendency on TiO2. Additionally, the number of alkoxy chains at the meso-substituted benzene rings had a significant effect on the Γ values [YD2-o-C8 (7.9 × 10−11 mol cm−2) < ZnPBAT-o-C8C4 = ZnPBAT-o-C8C8 (8.7 × 10−11 mol cm−2) ≈ ZnPBAT-o-C8 (8.8 × 10−11 mol cm−2)]. Note that the Γ values reported by Imahori and co-workers were estimated taking into account the actual surface area of TiO2 particles in a porous TiO2 film instead of the projected area of the TiO2 film. Overall, the PCEs of these four dyes were in the order: ZnPBAT-o-C8C8 (7.0%) < ZnPBAT-o-C8C4 (7.3%) < YD2-o-C8 (8.6%) < ZnPBAT-o-C8 (9.1%).
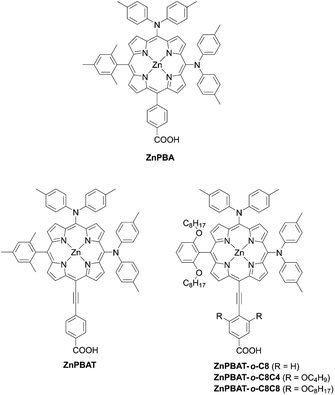 |
| | Fig. 7 Chemical structures of representative cis-A2BC-type D–π–A type porphyrins. | |
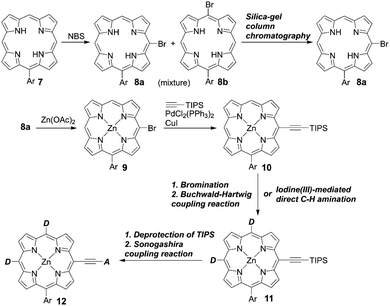 |
| | Scheme 2 Synthetic strategy for cis-A2BC-type porphyrins. Ar = aryl substituents; NBS = N-bromosuccinimide. | |
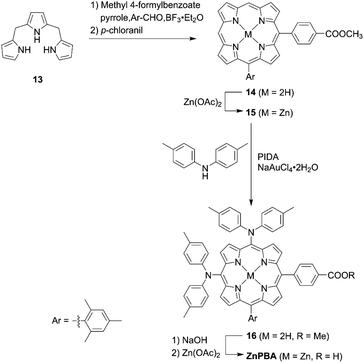 |
| | Scheme 3 Synthesis of ZnPBA. PIDA = phenyliodine(III) diacetate. | |
2.2 β-Functionalized porphyrins
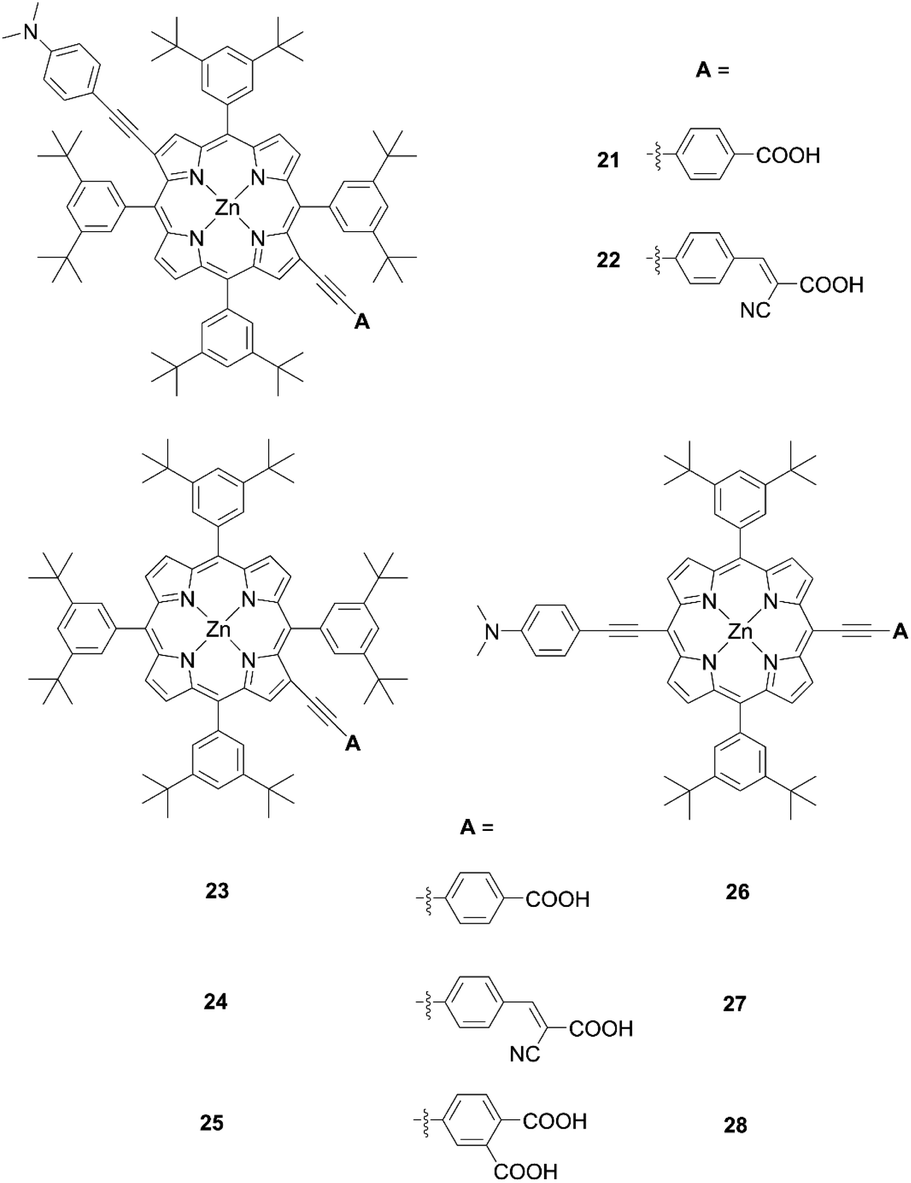
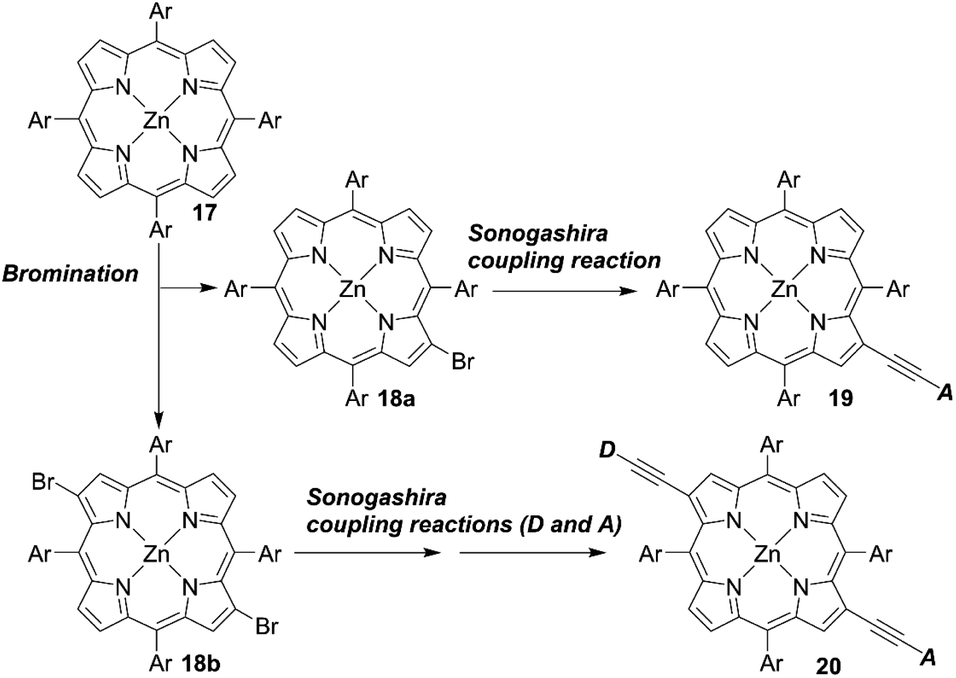
Opposite from meso-substituted porphyrins, β-substituted porphyrin dyes are still limited due to their synthetic difficulties in lower symmetry porphyrins. Nevertheless, many efforts have been directed towards developing β-functionalized push–pull porphyrins. Generally, β-brominated or β-borylated porphyrins are key precursors because a variety of substituents can be introduced by transition metal-catalyzed coupling reactions. In 2013, Pizzotti and co-workers53 reported a series of β-monosubstituted (23–25, Fig. 8) or disubstituted push–pull meso-tetraarylporphyrins (21 and 22, Fig. 8). As illustrated in Scheme 4, the β-brominated intermediates 18a and 18b were synthesized from 17 by β-bromination with NBS. Then, the Sonogashira coupling reactions of 18a and 18b provided the β-functionalized porphyrins 19 and 20, respectively. Although meso-substituted analogues 26–28 (Fig. 8) exhibited lower HOMO–LUMO energy gaps, the β-monosubstituted or disubstituted ones showed comparable or better device performance, which were in accordance with their enhanced IPCE spectra. Among 21–28, dye 22 with a cyanoacrylic acid as the anchoring group and N,N-dimethylaminophenylethynyl moiety as the donor group gave a moderate PCE of 4.7%. The higher PCE may originate from the localization of the LUMO in the electron-withdrawing anchoring group and that of the HOMO in the electron-donating group, which would accelerate electron injection from the dye to TiO2 and slow down CR between the dye radical cation and TiO2.
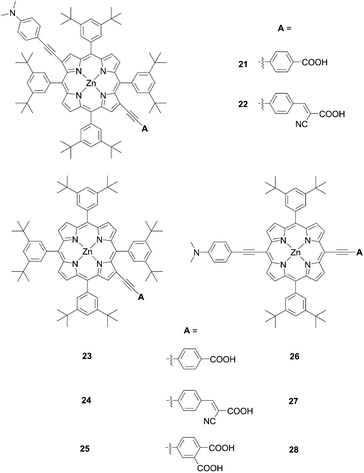 |
| | Fig. 8 Chemical structures of 21–28. | |
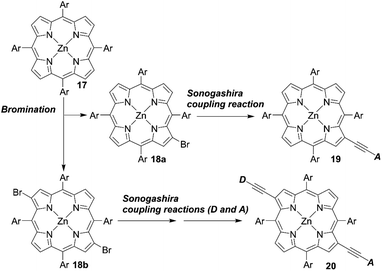 |
| | Scheme 4 Synthetic strategies for β-functionalized D–π–A-type porphyrins. Ar = aryl substituents. | |
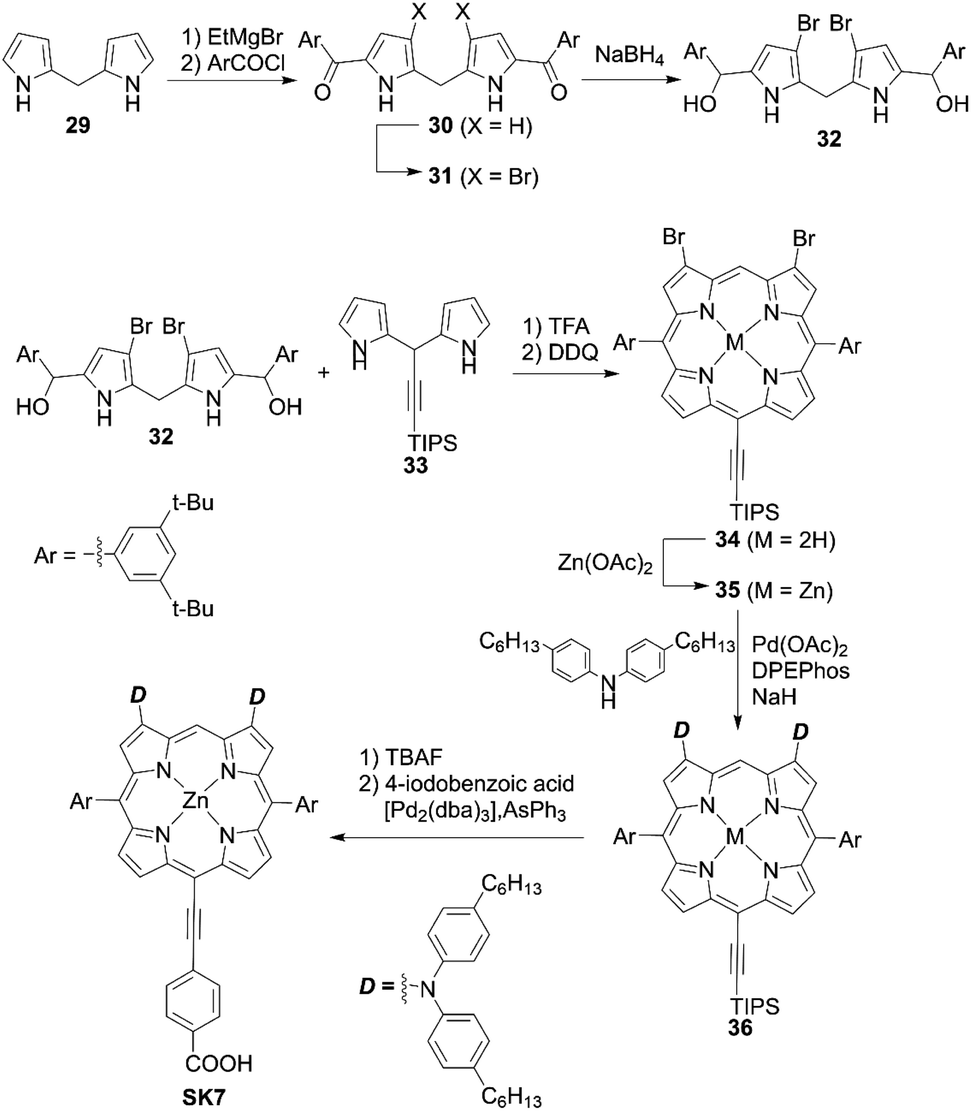
Yeh and co-workers54 prepared the β-dibrominated porphyrin precursor 34 by an acid-catalyzed condensation of the corresponding dipyrromethanes 32 and 33 and subsequent oxidization by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (Scheme 5). The Grignard reagent-mediated acylation of dipyrromethane 29 with 3,5-di-tert-butylbenzoyl chloride afforded the diacyldipyrromethane 30. After the bromination of 30, the reduction of 31 with NaBH4 yielded the key bis(hydroxymethyl)dippyromethane 32. After the metalation of 34, the Buchwald–Hartwig amination of 35 furnished the β-aminoporphyrin 36. Deprotection of the TIPS moiety and a subsequent Sonogashira coupling reaction gave the porphyrin dye SK7 (Scheme 5). These reactions are notable as they show that the β-dibromoporphyrins 34 and 35 may become key precursors for various β-difunctionalized porphyrin sensitizers. The introduction of two amino groups at the β-positions can have a considerable impact on the a1u orbital but not the a2u orbital, which leads to a decrease in the degeneracy of HOMO/HOMO−1. Consequently, the Soret band of SK7 appeared as two split peaks (λmax = 424, 491 nm), which made up for the weak absorption of typical porphyrins around 450–550 nm and thereby enhance the light-harvesting ability. However, the dye-loading amount for SK7 (6.08 × 10−8 mol cm−2, where the thickness of TiO2 was 15 μm, which was the same as for YD2) was two-thirds of that for YD2 (9.48 × 10−8 mol cm−2) (Fig. 4). The lower dye-loading amount was responsible for the lower JSC value (13.25 vs. 15.42 mA cm−2) and resultant lower PCE (6.54% vs. 7.78%) under 1 sun illumination. Although SK7 possessed two bulky diarylamino groups, a loose packing of SK7 on TiO2 was obtained under the T5 light source, but the PCE of the SK7-based device was nearly 97% that of YD2 (84% under 1 sun). This could be rationalized by the much more matched emission wavelengths of the incident light sources.
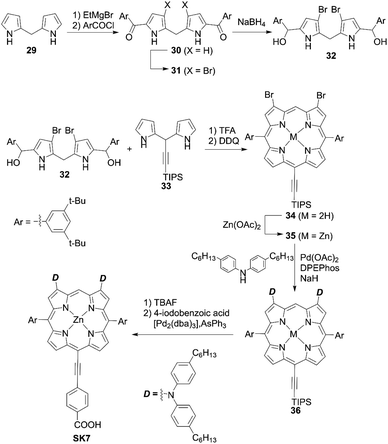 |
| | Scheme 5 Synthesis of SK7 with two donor moieties at β-positions. TFA = trifluoroacetic acid; DPEPhos = bis[(2-diphenylphosphino)phenyl]ether; [Pd2(dba)3] = tris(dibenzylideneacetone)dipalladium(0). | |
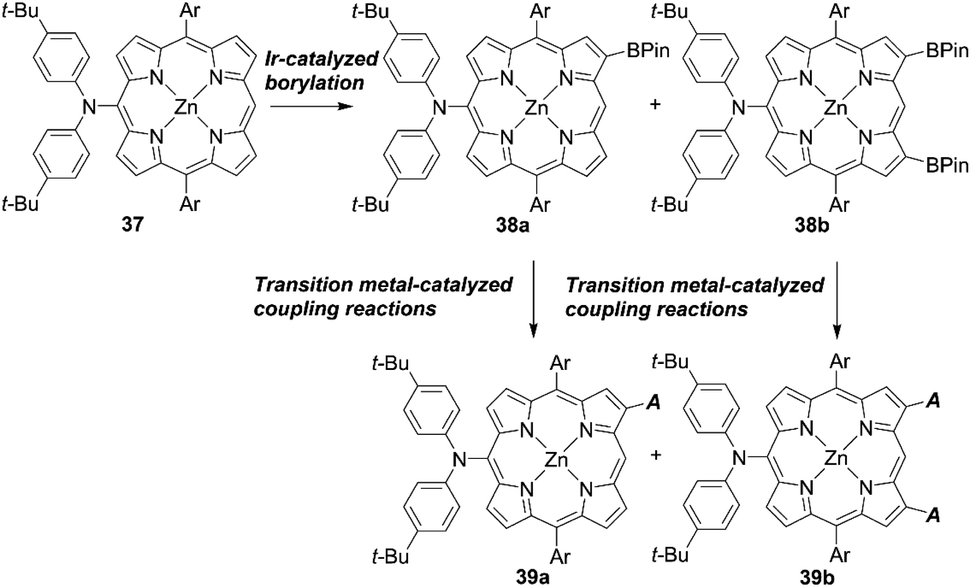
Osuka and co-workers55 presented a highly regioselective C–H borylation reaction of meso-arylporphyrins using an iridium catalyst. Inspired by this work, Sessler, Kim, and co-workers designed several β-functionalized porphyrins with the meso-diarylamino group.56,57 As illustrated in Scheme 6, the borylation reaction of meso-aminoporphyrin 37 yielded a mixture of the mono- or bis-borylated porphyrins 38a and 38b. Then, acceptor moieties were introduced by transition metal-catalyzed coupling reactions to give the D–π–A-type porphyrins 39a and 39b. Especially, they developed a process for the direct palladium-catalyzed oxidative alkynylation of β-borylated porphyrins, instead of going via a common bromo-precursor, for the preparation of push–pull-structured ethynylbenzoic acid porphyrin derivatives. Although the nature of the terminal alkynes had a considerable effect on the reaction yield (i.e. electron-deficient alkynes were less reactive than electron-rich ones), the simplified synthetic routes, i.e. omitting the bromination of β-borylated porphyrins, were beneficial for improving the overall yield and synthetic costs. Initially, ZnBD1 and ZnBD2 (Fig. 9) achieved moderate efficiencies of 5.9% and 7.5% with the I−/I3− redox couple, respectively.56 After optimization with a cobalt-based electrolyte, ZnBD2 attained a record PCE of 8.2% among doubly β-butadiene-linked porphyrin sensitizers for DSSCs.58 The introduction of additional β-linkages gave rise to a red-shifted and broadened absorption spectrum for ZnEP2 compared to ZnEP1 (Fig. 9). Despite its enhanced light-harvesting ability, the ZnEP2-based DSSC exhibited a lower PCE (4.0%) than that of the ZnEP1-based DSSC (5.9%). The lower PCE for ZnEP2 was attributed to the lower Jsc value, which may be rationalized by the low dye-loading amount (ca. one-third) relative to ZnEP1. Moreover, the dihedral angle between the porphyrin core and the ethynylphenyl rings was larger in ZnEP2 (17.4°) than that in ZnEP1 (0.7°) with the optimized structures. The larger dihedral angle could disrupt the effective electronic communication through the ethynylphenyl π-bridge and retard efficient electron injection. Considering the importance of the rigidity of the two anchoring groups in the photovoltaic performance, they synthesized a series of new β-functionalized push–pull-type porphyrins (ZnPHn and ZnPVn: n = 1 and 2, Fig. 9).58 The DSSCs with ZnPHn bearing the rigid π spacer provided lower PCEs than those with ZnPVn. The low light-harvesting abilities of ZnPHn were mainly responsible for the decreased JSC values.
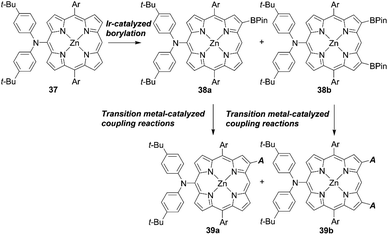 |
| | Scheme 6 Synthetic strategies for β-functionalized porphyrins via Ir-catalyzed borylation. Ar = aryl substituents. | |
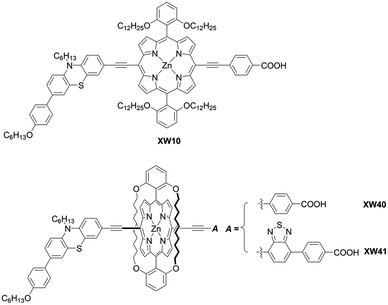 |
| | Fig. 9 Chemical structures of ZnBD1-2, ZnPH1-2, and ZnPV1-2. | |
Furthermore, the dihedral angles between the β-aryl rings and porphyrin core of ZnPHn were ca. 43°, which led to a weak electronic coupling between the LUMO of ZnPHn and the CB of TiO2 as well as between the acceptor moiety and porphyrin core. Consequently, the ϕinj values may become lower for ZnPHn than those for ZnPVn. In contrast, the dihedral angles between the carboxyphenyl rings and porphyrin core were significantly small for ZnPVn, which enabled strong electronic coupling between the acceptor moieties and porphyrin core. This efficient electronic communication resulted in large orbital distributions on the acceptor moiety of LUMO, leading to the high ϕinj values. Furthermore, the CR from the CB of TiO2 was expected to be slower over a longer distance as a result of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond π-spacer. Therefore, the DSSCs with ZnPVn exhibited higher PCEs than those with ZnPHn. These studies indicated that the flexible and small alkenyl π-conjugated spacers play an important role in the molecular structures and electron coupling, which offers a valuable guide for rational molecular design.
C double bond π-spacer. Therefore, the DSSCs with ZnPVn exhibited higher PCEs than those with ZnPHn. These studies indicated that the flexible and small alkenyl π-conjugated spacers play an important role in the molecular structures and electron coupling, which offers a valuable guide for rational molecular design.
3. Aromatic-fused porphyrins
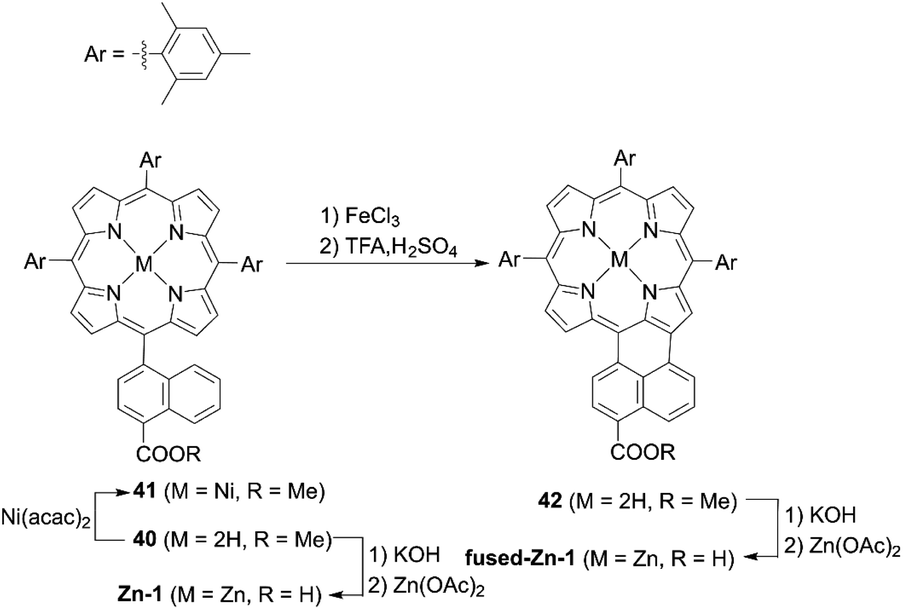
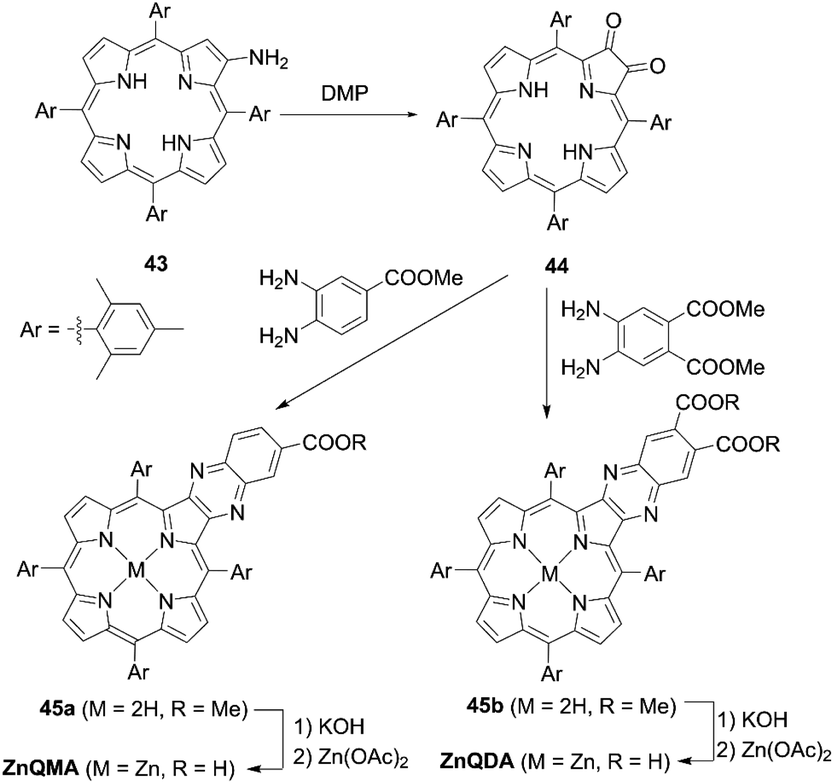
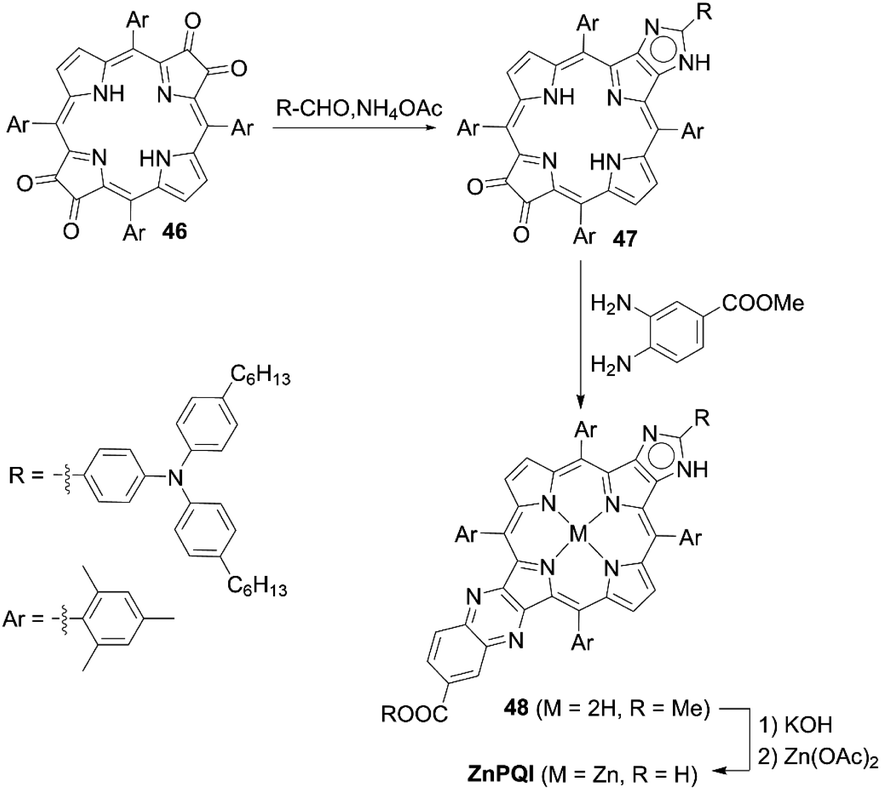
The incorporation of an aromatic-fused structure into a porphyrin core is a promising strategy for obtaining highly efficient DSSCs because of the extended π-system and resultant red-shifted absorption. In 2007, Imahori and co-workers59,60 synthesized a meso-naphthyl-fused porphyrin, fused-Zn-1, which was the first example of a fused porphyrin-based DSSC. The intermediate 42 could be prepared by the metalation of 40 and successive treatment of the precursor 41 with FeCl3 and TFA. Then, a subsequent hydrolysis and Zn(II)-metalation afforded the target molecule (Scheme 7). Compared to the non-fused counterpart Zn-1 (Scheme 7), the Soret and Q-bands of fused-Zn-1 were broadened and red-shifted by ∼60 and 130 nm. A higher PCE of 4.1% for the fused-Zn-1 than that of 2.8% for Zn-1 was obtained. Then, Imahori and co-workers61 continued to present the first example of two β,β′-edge quinoxaline-fused zinc porphyrins: ZnQMA with a PCE of 5.2% and ZnQDA with a PCE of 4.0%. The inferior PCE of ZnQDA was mainly due to the lower electron injection and charge-collection efficiency related to the weaker coplanarity of the two carboxylic acid groups with the porphyrin plane and the higher degree of proton transfer from ZnQDA to TiO2 (Scheme 8). The key intermediate 44 could be synthesized by oxidation of the precursor 43 with Dess–Martin periodinane (DMP). ZnQMA and ZnQDA were derived from the condensation of intermediate 44 with the substrates with different numbers of ester groups, followed by the subsequent hydrolysis and metalation reactions of 45a and 45b. Later, they reported the fused porphyrin ZnPQI (ref. 62) with a push–pull structure. ZnPQI could be synthesized through the condensation of 47 with methyl 3,4-diaminobenzoate, followed by the hydrolysis and metalation reactions of 48, in which 47 was afforded by the acid-catalyzed coupling reaction of intermediate 46 with the corresponding triarylamine (Scheme 9). As expected, the absorption spectrum of ZnPQI was further red-shifted. A moderate PCE of 6.8% was obtained by the DSSC with ZnPQI. This was slightly higher than that with ZnQMA, implying the importance of the push–pull structure even in aromatic-fused porphyrins for DSSCs.
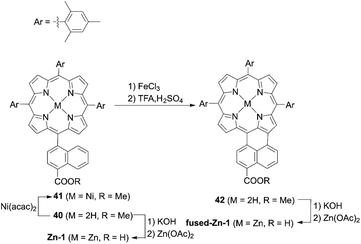 |
| | Scheme 7 Synthesis of fused-Zn-1. Ni(acac)2 = nickel(II) bis(acetylacetonate). | |
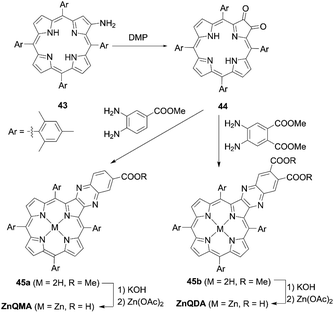 |
| | Scheme 8 Syntheses of ZnQMA and ZnQDA. | |
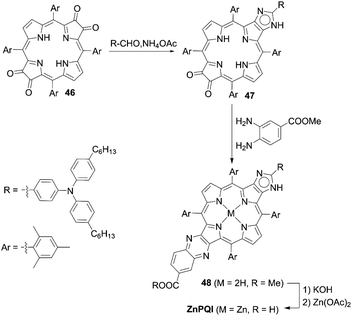 |
| | Scheme 9 Synthesis of ZnPQI. | |
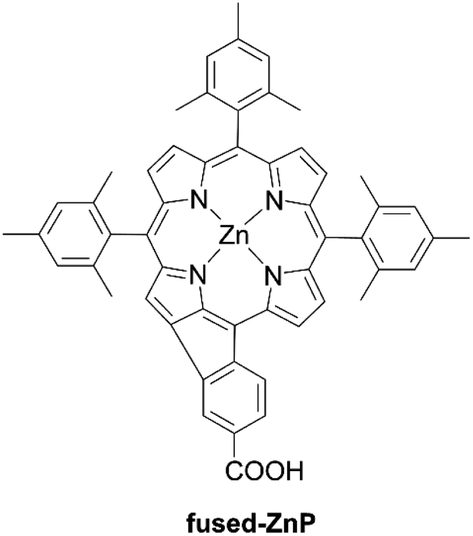
Besides, Imahori et al. reported that the simple fusion reaction between the β-position and neighbouring benzene ring with an anchoring carboxylic acid made a fused five-membered porphyrin, fused-ZnP, with an absorption edge of 800 nm (Fig. 10).63 Unfortunately, the PCE (0.30%) was very low, probably due to very fast nonradiative decay of the excited singlet state to the ground state by the directly fused structure.
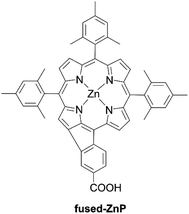 |
| | Fig. 10 Chemical structure of fused-ZnP. | |
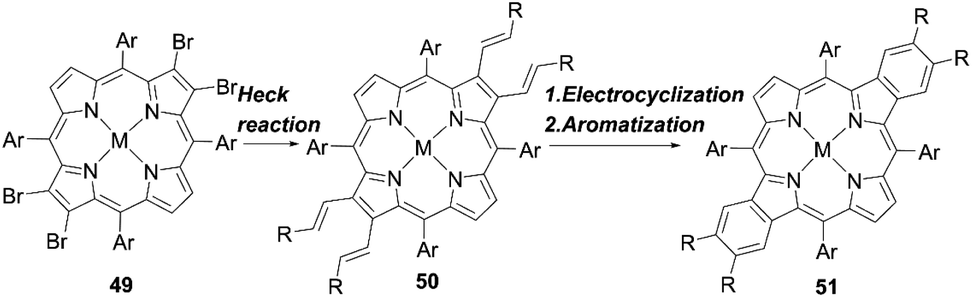
Other fused porphyrins with larger π-systems were also reported to further enhance the light-harvesting abilities. Wang and co-workers64 provided an efficient method for the synthesis of functionalized opp-dibenzoporphyrin by introducing alkenyl groups to the porphyrin periphery. Their synthetic strategy consisted of the Heck reaction of 49 to generate the intermediate 50, and subsequent electrocyclization and aromatization reactions to yield 51 in one pot (Scheme 10). This could overcome the drawbacks of most synthetic methods for benzoporphyrins, i.e. the lack of functional groups introduced at the fused aromatic rings and the synthetic difficulties because of the fewer purification processes during the one-pot approach.
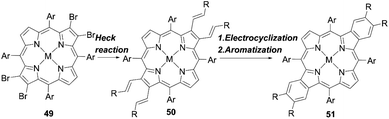 |
| | Scheme 10 Synthetic strategy for functionalized opp-dibenzoporphyrins. Ar = aryl substituents; M = Ni or Zn; R = phenyl or carbomethoxy. | |
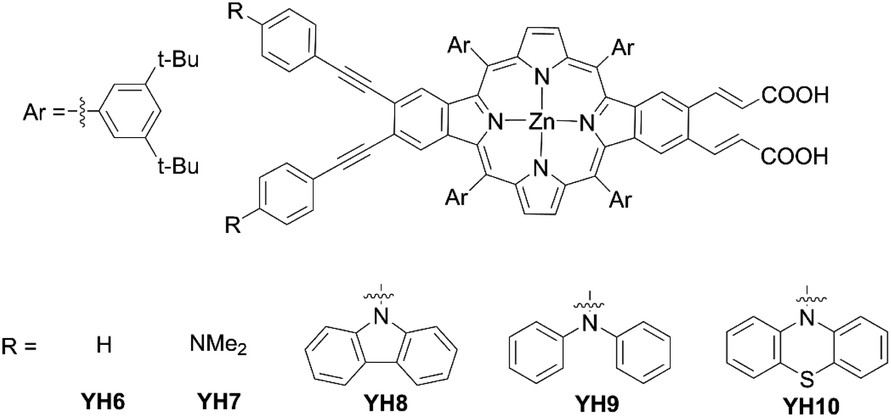
Then, a series of β-functionalized push–pull zinc opp-dibenzoporphyrins were synthesized and applied in DSSCs. In 2019, they designed and synthesized two dibenzoporphyrins: YH6 and YH7 (Fig. 11).65YH7 displayed clearly segregated HOMO/LUMO and fast electron injection owing to the enhanced push–pull character caused by the incorporation of both phenylethynyl bridges and amino groups. However, the non-push–pull porphyrin YH6 had a higher PCE of 6.70% than that of YH7 (5.72%), which could be attributed to the illumination-induced photodegradation of YH7 in the presence of the redox shuttle I−/I3−. They supposed that tertiary amino groups were responsible for this severe photodegradation. They further designed and synthesized dibenzoporphyrin dyes YH8 with the carbazole moiety, YH9 with the diphenylamine moiety, and YH10 with the phenothiazine moiety, in which aliphatic amine was replaced with arylamine (Fig. 11).66 In contrast to a YH7-based device, the YH8-10-based device revealed good stability similar to YH6 during the photovoltaic measurement, because the bulky tertiary arylamine protected the amine group from interacting with the redox shuttle on the TiO2 surface. Moreover, it also benefited from the exceptional light-harvesting ability allowing YH9 and YH10 to yield moderate PCEs of 6.9% and 7.5%, respectively, which were comparable to that (7.7%) of the N719-based DSSC. Especially, the reduced CR for the YH10-based device contributed to its higher PCE, because the orbital distribution in its HOMO was further away from the TiO2 surface.
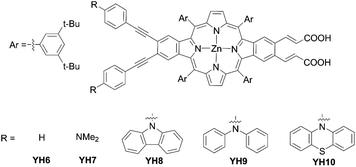 |
| | Fig. 11 Chemical structures of YHn (n = 6–10). | |
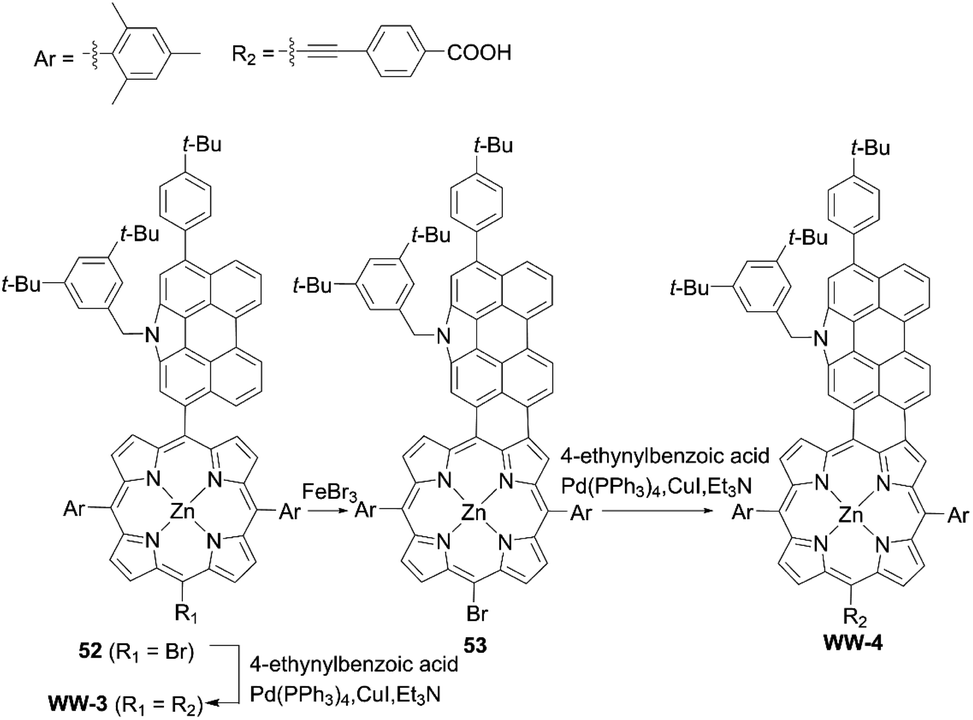
Although fused porphyrins possess extremely enhanced light-harvesting abilities, and some of the absorption edge reach more than 1000 nm, their corresponding DSSCs suffer from the low PCEs stemming from serious dye aggregation, the mismatch of the HOMO–LOMO energy levels, and short lifetimes of the singlet excited-states. For instance, Wu and co-workers67 presented N-annulated perylene (NP)-fused porphyrin WW-4 (Scheme 11), which was prepared by an FeBr3-promoted intramolecular oxidative cyclodehydrogenation of the precursor 52, followed by the subsequent Sonogashira coupling reaction of 53. WW-4 displayed a maximum absorption at 792 nm (ε = 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 M−1 cm−1) and two intense absorption bands at 444 nm (ε = 55200 M−1 cm−1) and 540 nm (ε = 69900 M−1 cm−1). However, despite its excellent light-harvesting ability, the WW-4-based DSSC showed a much lower PCE of 0.3% than the WW-3-based reference (Scheme 11, 5.6%). The low PCE for WW-4 could be explained by the insufficient driving force for electron injection, caused by its low-lying LUMO energy level (−3.60 eV) in comparison with that of WW-3 (−3.09 eV).
400 M−1 cm−1) and two intense absorption bands at 444 nm (ε = 55200 M−1 cm−1) and 540 nm (ε = 69900 M−1 cm−1). However, despite its excellent light-harvesting ability, the WW-4-based DSSC showed a much lower PCE of 0.3% than the WW-3-based reference (Scheme 11, 5.6%). The low PCE for WW-4 could be explained by the insufficient driving force for electron injection, caused by its low-lying LUMO energy level (−3.60 eV) in comparison with that of WW-3 (−3.09 eV).
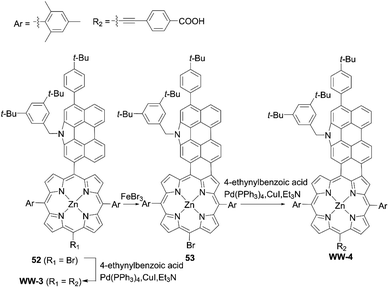 |
| | Scheme 11 Synthesis of WW-4. | |
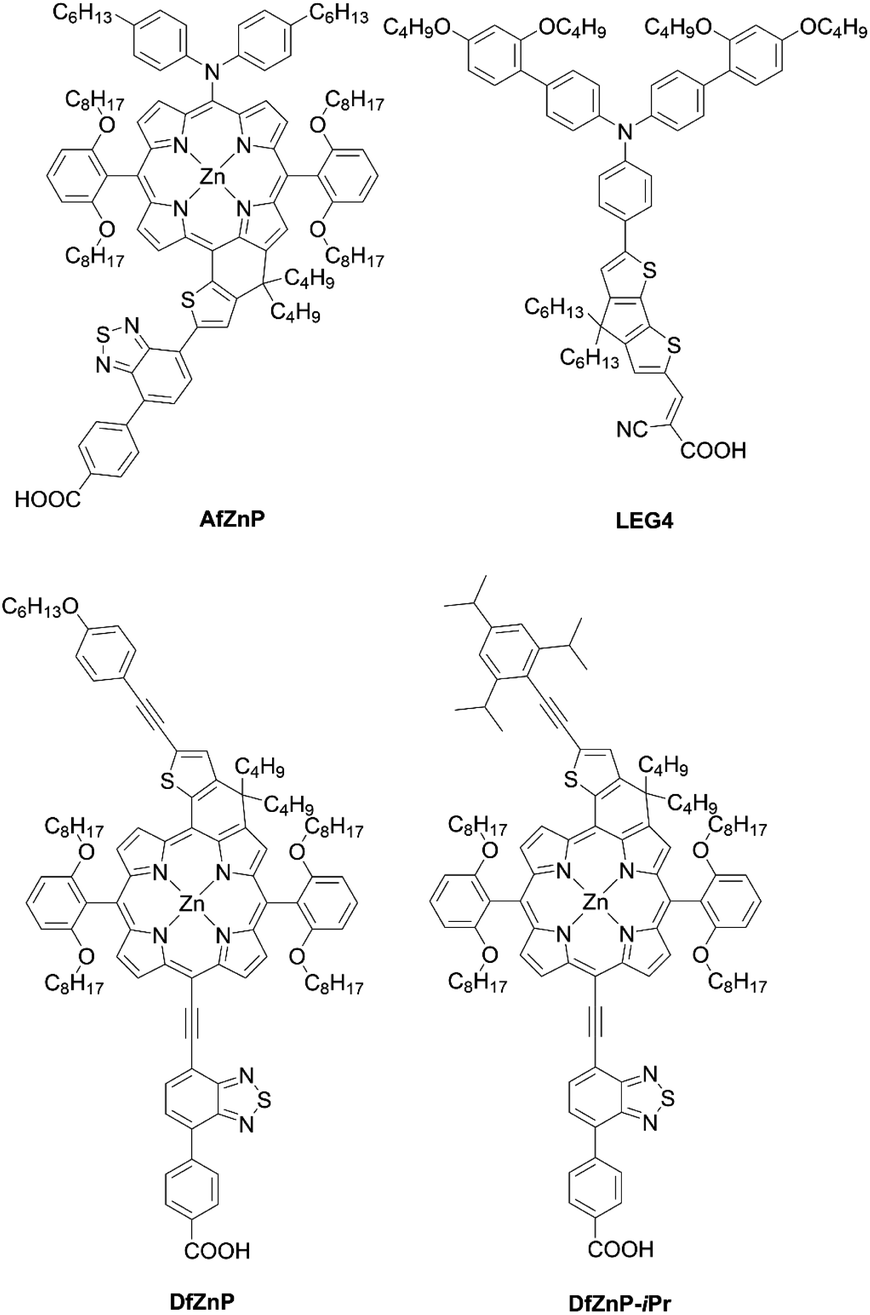
To surmount the obstacles for aromatic-fused porphyrin dyes, Imahori and co-workers68 created a series of substituted methylene-bridged thiophene-fused porphyrin sensitizers AfZnP, DfZnP, and DfZnP-iPr (Fig. 12). The substituted methylene-bridged thiophene-fused structure was expected to modulate the electronic communication between the porphyrin core and thiophene moiety, while suppressing the dye aggregation on TiO2 by the bulkiness around the porphyrin. In the synthetic scheme, meso-thienyl porphyrin 54 with an ester moiety on the meso-thienyl moiety was a key intermediate. The treatment of 54 with n-BuLi afforded the alcohol 55, which underwent an intramolecular Friedel–Crafts reaction to yield methylene-bridged thiophene-fused porphyrin 56 (Scheme 12). It was found that the fusion of thiophene to the porphyrin core on the donor side instead of the acceptor side was more efficient in order to enhance not only its molar extinction coefficients, but also light-harvesting ability. Meanwhile, an increase in the length of the linkers between a porphyrin core and TiO2 and closer HOMO orbital distribution to TiO2 by the introduction of the thiophene to the acceptor side would lead to fast CR because of the tilted geometry and close orbital distribution to TiO2.47–49 The more tilted geometry also resulted in a lower Γ value of AfZnP (AfZnP (4.8 × 10−11 mol cm−2) < DfZnP-iPr (7.5 × 10−11 mol cm−2) ≈ DfZnP (7.6 × 10−11 mol cm−2)). Therefore, AfZnP bearing the thiophene ring fused on the acceptor side exhibited a lower PCE than those for DfZnP and DfZnP-iPr containing the fused structure on the donor side. In addition, the DSSC with DfZnP-iPr achieved a promising PCE of 10.1% with the [Co(bpy)3]2+/3+ redox shuttle as a result of the larger steric hindrance and resultant blocking effect on CR between TiO2 and the redox shuttle by 2,4,6-triisopropylphenyl group. The PCE was comparable with the one for GY50 (PCE = 10.0%) under the optimized conditions. Remarkably, co-sensitization with the complementary LEG4 (Fig. 12) further boosted the PCE to 10.7%, a record for fused-porphyrin sensitizers in DSSCs. Such a combination of a push–pull structure, fused structure, and co-sensitization would be a very promising strategy for further enhancing the PCE in DSSCs.
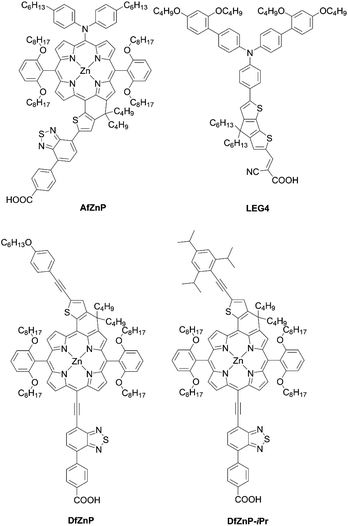 |
| | Fig. 12 Chemical structures of AfZnP, DfZnP, DfZnP-iPr, and LEG4. | |
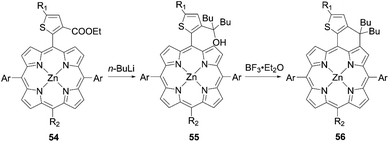 |
| | Scheme 12 Synthetic strategy for methylene-bridged thiophene-fused porphyrins. Ar = 2,6-di(octyloxy)phenyl; R1 = H or alkynyl substituents; R2 = diarylamino or TIPS group. | |
4. Porphyrin dimers
Covalently linked porphyrin dimers have attracted much attention because of their extended π-conjugated systems and excellent light-harvesting abilities arising from the double number and exciton coupling of the porphyrins. Since the first report on porphyrin dimers for DSSCs by Segawa69 and Officer,70 various porphyrin dimers for DSSCs have been designed and synthesized. Considering the success of push–pull-type porphyrin dyes, push–pull-type porphyrin dimers are attractive targets for high-performance DSSCs.
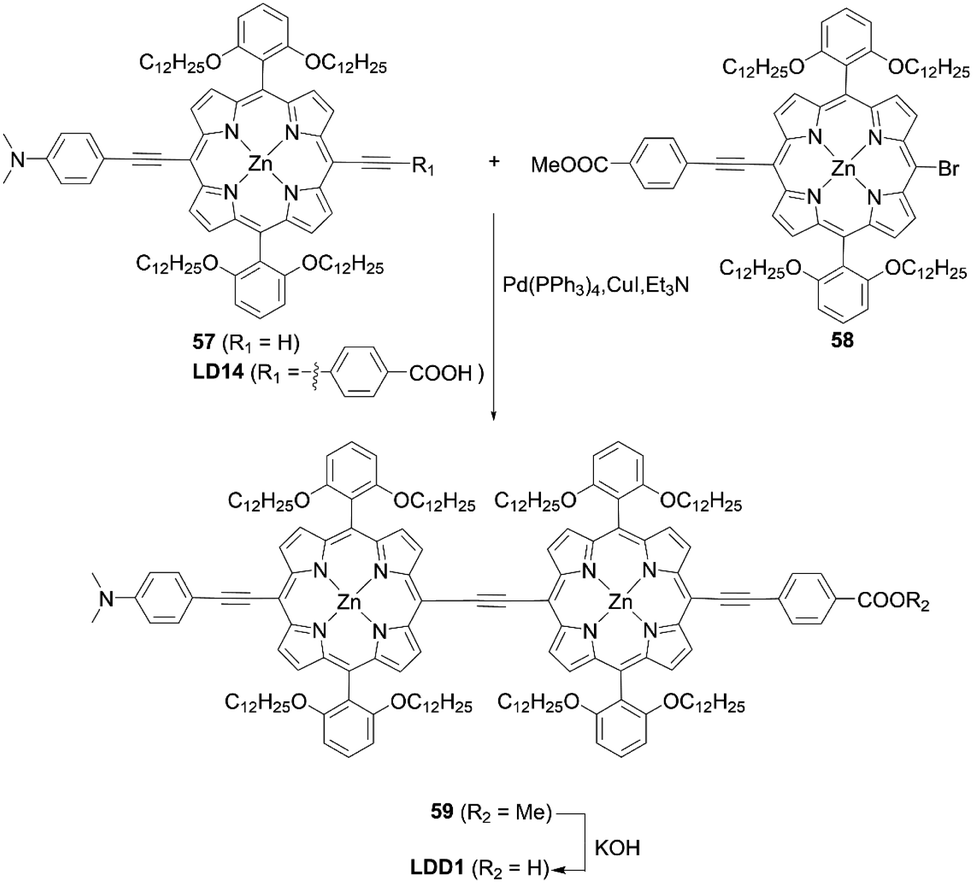
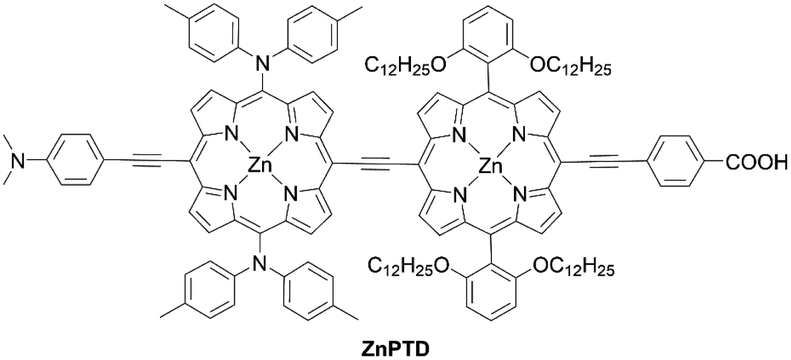
Lin and co-workers71 designed and synthesized an ethynyl-bridged dimeric porphyrin dye LDD1 based on the D–π–A-type porphyrin LD14 (Scheme 13), through a Sonogashira coupling between the donor-side porphyrin 57 and the acceptor-side porphyrin 58 and the subsequent hydrolysis reaction of 59. As expected, LDD1 revealed a wide absorption spectrum extending beyond 800 nm, but its PCE of 8.8% was lower than that for the LD14-based device (9.2%) due to serious dye aggregation. In addition, after the co-sensitization of LDD1 with LD14 under the optimized molar ratio, the PCE rose to 10.4% owing to the further improved light-harvesting ability reaching 900 nm. Furthermore, Imahori and co-workers72 reported the porphyrin dimer ZnPTD to replace the two meso-phenyl groups with two diarylamino groups on the basis of LDD1, expecting there would be an improvement in the light-harvesting ability (Fig. 13). Similarly, the absorption edge of ZnPTD was extended up to 900 nm, but the PCE of the DSSC with ZnPTD (6.22%) was lower than that with LDD1 (8.17%). The decrease in the PCE may be ascribed to the more tilted geometry of ZnPTD on TiO2 and thereby the faster CR between the injected electrons in the CB of TiO2 and the porphyrin radical cation.47–49 Given that the ethynyl-linked porphyrin dimers exhibit excellent light-harvesting abilities, the suppression of dye aggregation and/or control of dye geometry on TiO2 by a co-adsorbent/co-sensitizer and/or by the pretreatment of TiO2 may boost their PCEs.
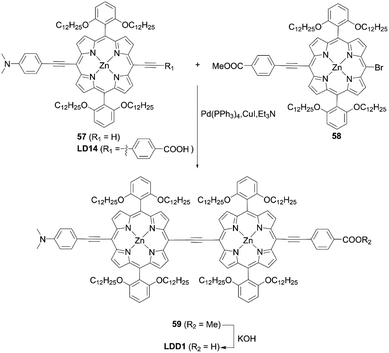 |
| | Scheme 13 Synthesis of LDD1. | |
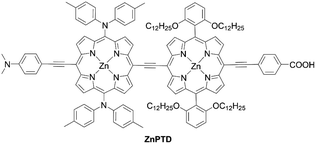 |
| | Fig. 13 Chemical structure of ZnPTD. | |
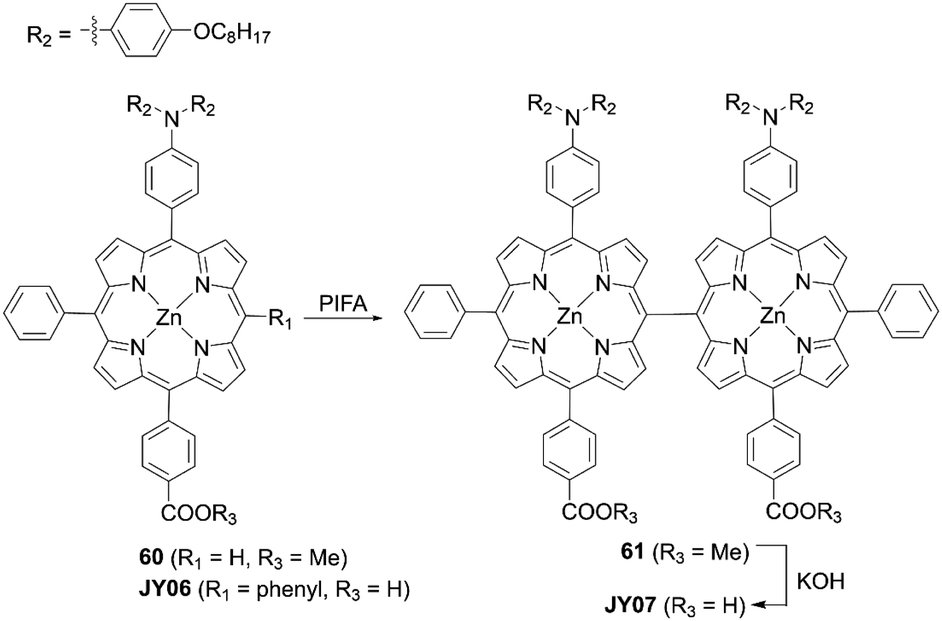
Zheng and co-workers73 presented the novel double D–π–A meso–meso directly linked porphyrin dimer JY07 (Scheme 14), which could be synthesized by the treatment of the meso-free porphyrin precursor 60 with phenyliodine(III) bis(trifluoroacetate) (PIFA) and the subsequent hydrolysis of 61. In comparison with the monomer JY06, the dimer JY07 exhibited broader and split Soret bands, as well as red-shifted and enhanced Q-bands due to the strong exciton coupling between the adjacent porphyrins, indicating the improved light-harvesting capability. Besides, the orthogonal conformation of JY07 suppressed the dye aggregation and CR. Therefore, the dimer JY07 showed much higher VOC (0.65 V vs. 0.63 V) and JSC (13.20 mA cm2vs. 11.18 mA cm2) values. As a result, the PCE of 5.33% for JY07 was larger than the 4.30% for JY06. Moreover, JY07 showed improved photostability due to the presence of two anchoring groups relative to JY06.
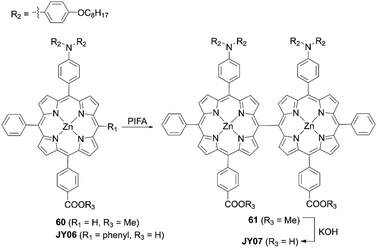 |
| | Scheme 14 Synthesis of JY07. | |
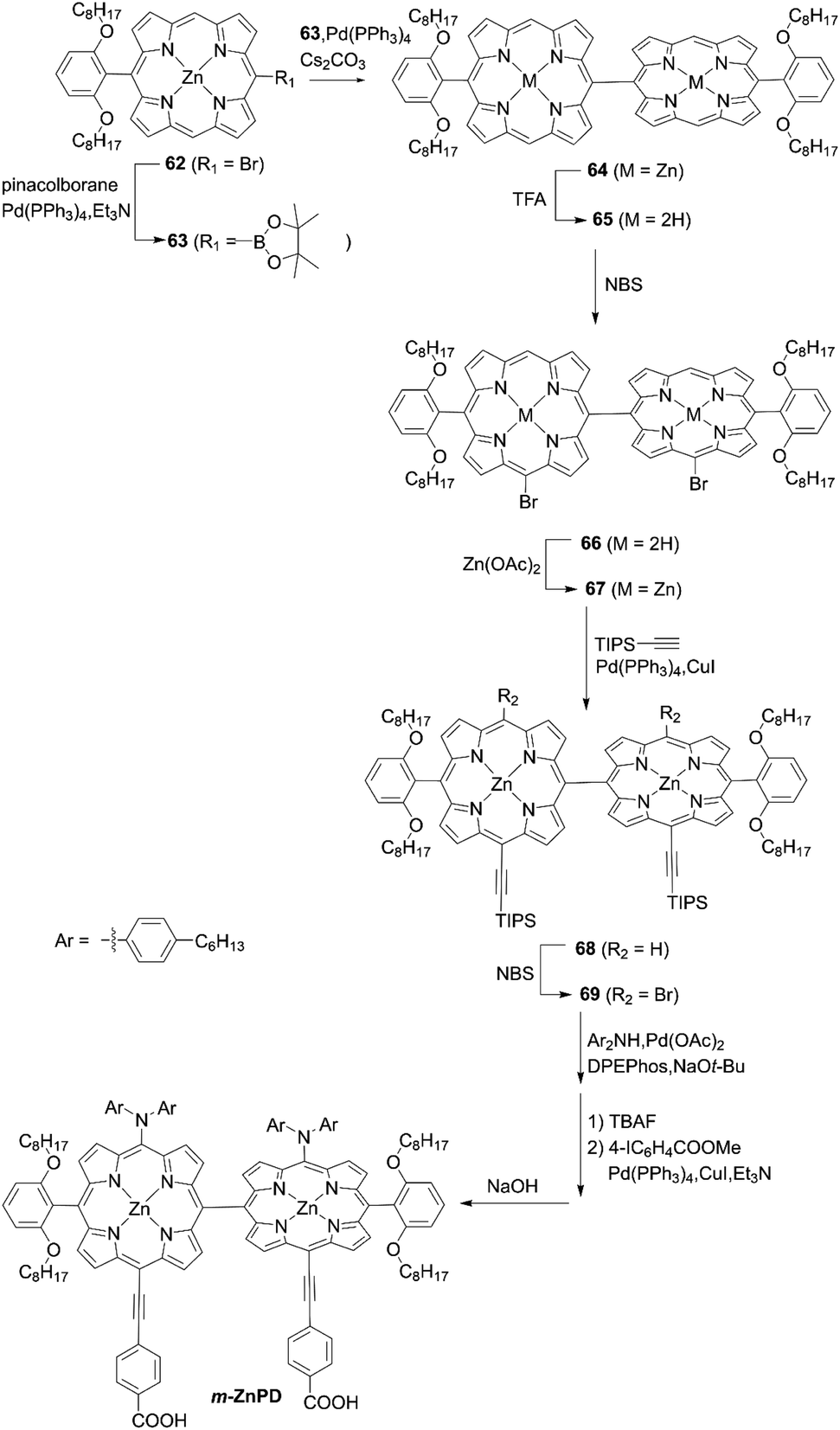
As observed in the synthesis of JY07, the oxidative homocoupling of meso-free porphyrins with an oxidant silver hexafluorophosphate (AgPF6)74,75 or PIFA76) is a powerful tool for the synthesis of meso–meso directly linked porphyrin oligomers. However, such oxidative homocoupling is mainly limited to the reaction of partially meso-aryl porphyrins. Namely, the widely used meso-functional substituents (alkynyl and/or amino groups) may not be tolerated. In this context, it is challenging to create a rational synthetic strategy for meso–meso directly linked porphyrin dimers suitable for DSSCs. Imahori and co-workers77 established a general synthetic strategy for ABC–ABC-type meso–meso directly linked porphyrin dimers with different substituents at the meso-positions. They had previously reported that the bromination of 5-substituted porphyrin afforded two bromoporphyrin isomers, which could be separated by conventional silica-gel column chromatography (see 8a and 8bScheme 2). Accordingly, the borylation of bromo group of 62 and subsequent Suzuki–Miyaura coupling reaction of 63 and demetallation reaction of 64 yielded the meso-free porphyrin dimer 65 as a key precursor (Scheme 15). Stepwise regioselective functionalization provided the partially meso-free meso-AB–AB type porphyrin dimer 67via66 and 67; then finally the meso-ABC–ABC-type porphyrin dimer m-ZnPD could be obtained from 68 and 69. The DSSC with m-ZnPD reached a record PCE of 7.91% for DSSCs based on orthogonal dimeric D–π–A-type organic sensitizers. This relatively high PCE was mainly attributable to the split Soret bands, red-shifted Q-bands, and resultantly improved light-harvesting ability in the visible and near-infrared regions. Additionally, the strong binding ability from the double anchoring of m-ZnPD on TiO2 was beneficial for improving the durability of DSSCs in comparison with the reference YD2-o-C8 with a single anchoring group. However, the rigid orthogonal dimeric structure of m-ZnPD may form a large void space on TiO2 that allows the redox shuttle to approach TiO2, accelerating CR and lowering the PCE relative to YD2-o-C8 (10.1%). It is noteworthy that this general synthetic strategy is applicable to ABC–ABC-type meso–meso directly linked porphyrin dimers for various applications.
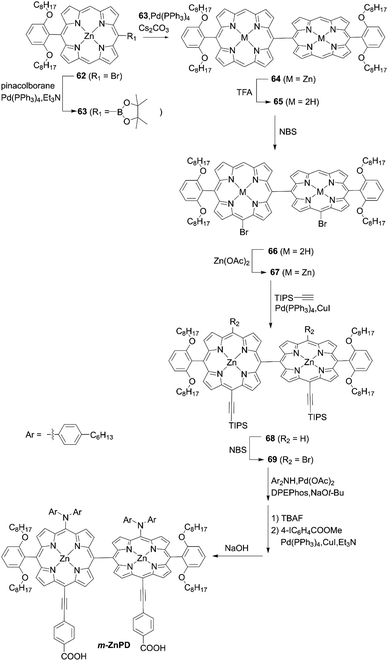 |
| | Scheme 15 Synthetic strategy for m-ZnPD. | |
5. Chlorins and bacteriochlorins
Porphyrins have an allowed S0 → S2 transition (Soret band) and a forbidden S0 → S1 one (Q-bands). Lowering the symmetry of porphyrins leads to less allowed S0 → S2 transition and allowed S0 → S1 one, matching better with the solar energy distribution on earth. In this regard, partially hydrogenated chlorin (17,18-dihydroporphyrin) and bacteriochlorin (7,8,17,18-tetrahydroporphyrin) derivatives have attracted considerable interests and are regarded as potential dye sensitizers owing to their excellent light-harvesting abilities at wavelengths >600 nm. Tamiaki and co-workers78,79 demonstrated the utility of modified chlorophylls or bacteriochlorophylls as dyes in DSSCs. However, it is still a challenge to synthesize chlorins and bacteriochlorins for DSSCs due to their molecular structures with lower symmetry.
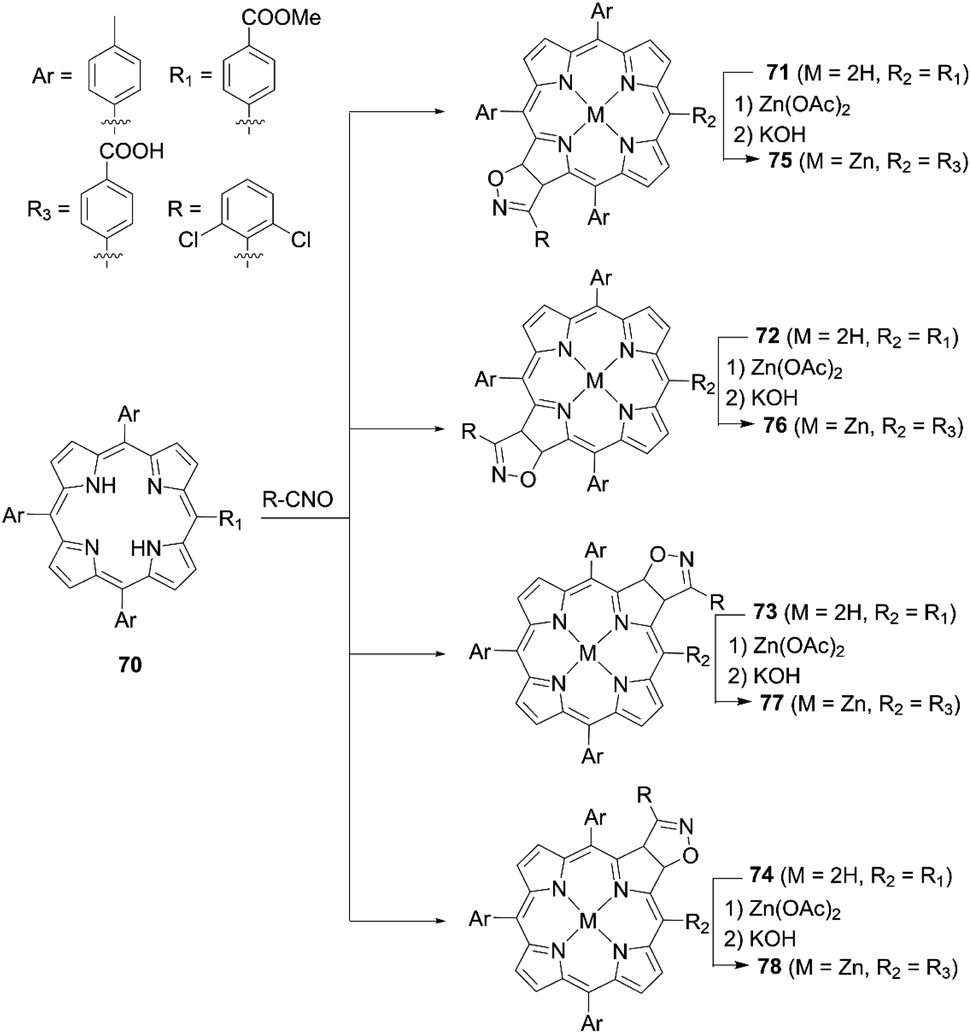
Recently, Feng and co-workers80 synthesized a series of isomeric chlorin-type sensitizers according to the 1,3-dipolar cycloaddition reaction between the free base porphyrin 70 and nitrile oxide, as well as the subsequent metalation and hydrolysis reactions of 71, 72, 73, and 74, respectively (Scheme 16). They found that the PCEs for 76 (2.92%) and 77 (1.85%) were lower than those for 75 (3.65%) and 78 (3.60%) because of the dye aggregation on TiO2 and inhibition of dye adsorption onto TiO2, associated with the steric hindrance by the 2,5-dichlorophenyl group.
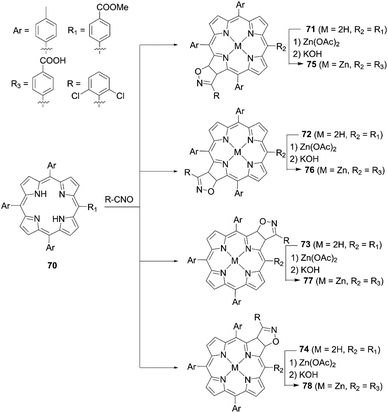 |
| | Scheme 16 Syntheses of 75–78. | |
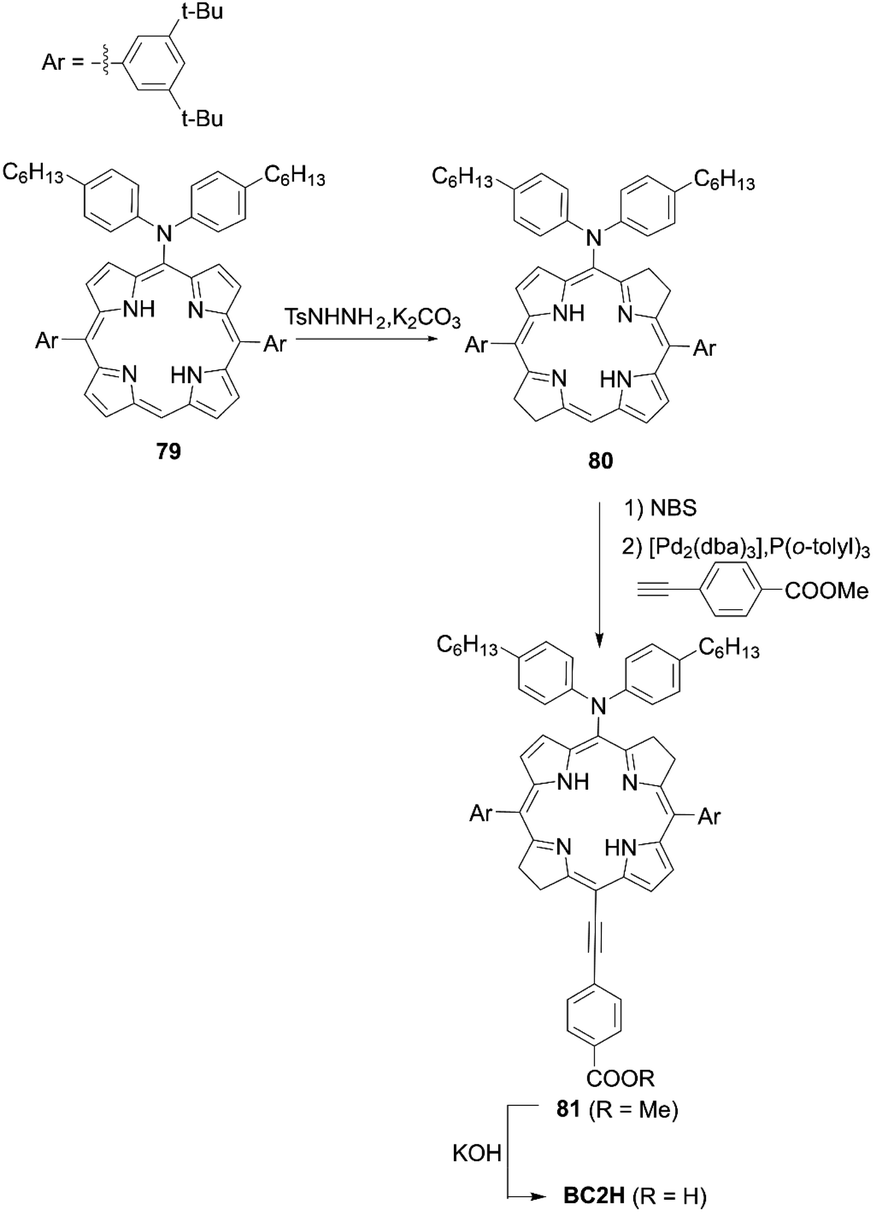
Imahori and co-workers81 introduced the push–pull structure into bacteriochlorin for the first time for DSSCs. The push–pull-type bacteriochlorin BC2H is depicted in Scheme 17. The reduction of meso-aminoporphyrin 79 with an excess amount of p-toluenesulfonyl hydrazide (TsNHNH2) provided the meso-amino bacteriochlorin 80. Subsequent bromination and the Sonogashira coupling reaction then afforded the push–pull structure of 81. Finally, the hydrolysis reaction gave BC2H, showing broad absorption with a wavelength edge of up to 800 nm. Regrettably, the PCE (2.7%) was lower compared to its porphyrin analogue YD2 (8.9%) under the optimized conditions. The lower PCE was mainly resulted from the serious dye aggregation of BC2H on TiO2, which was consistent with the higher Γ value of BC2H (1.0 × 10−10 mol cm−2) than YD2 (0.9 × 10−10 mol cm−2), and the resultant low ϕinj value. Nevertheless, it should be noted that BC2H revealed excellent stability under an ambient atmosphere, even for several months.
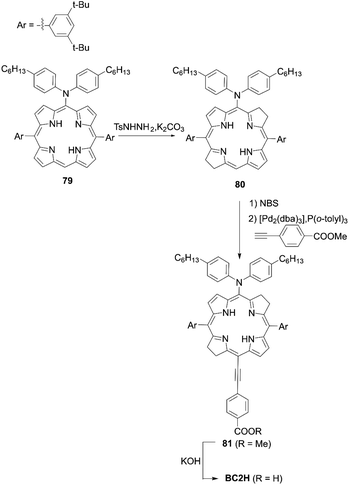 |
| | Scheme 17 Synthesis of BC2H. P(o-tolyl)3 = tris(o-tolyl)phosphine. | |
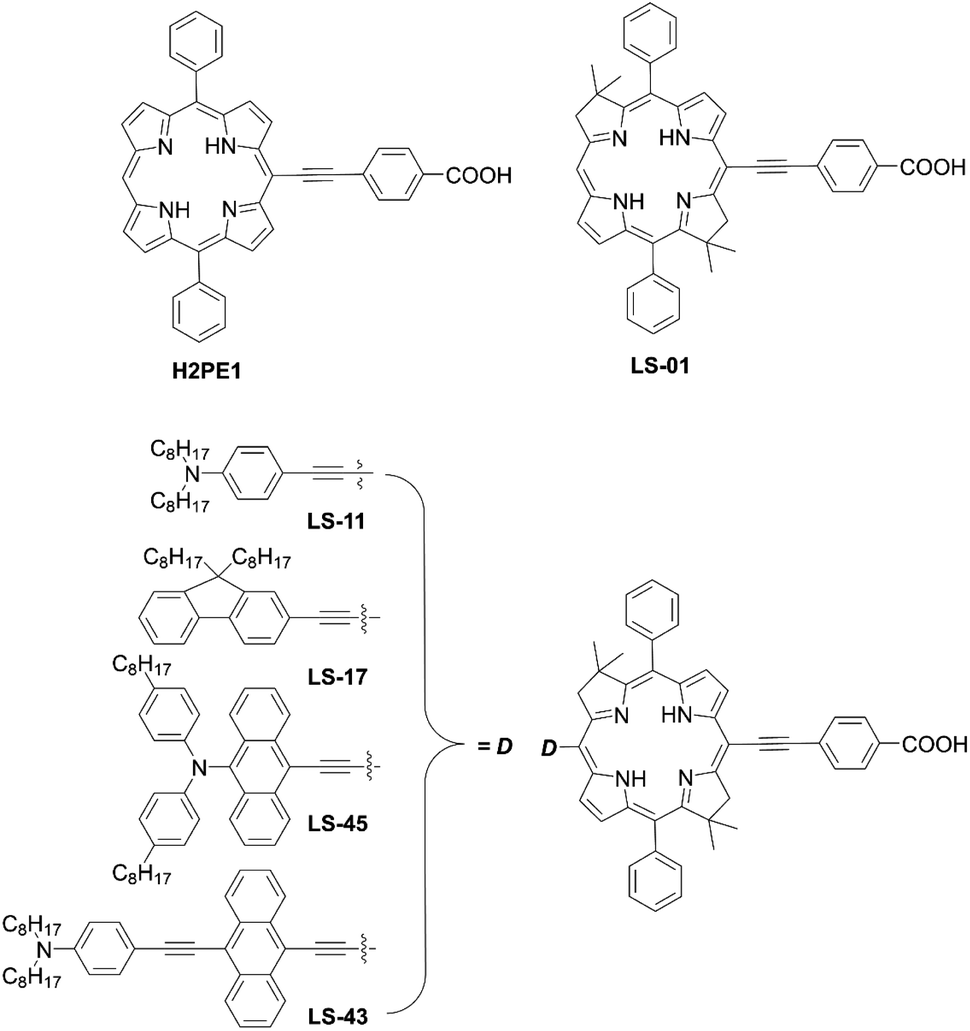
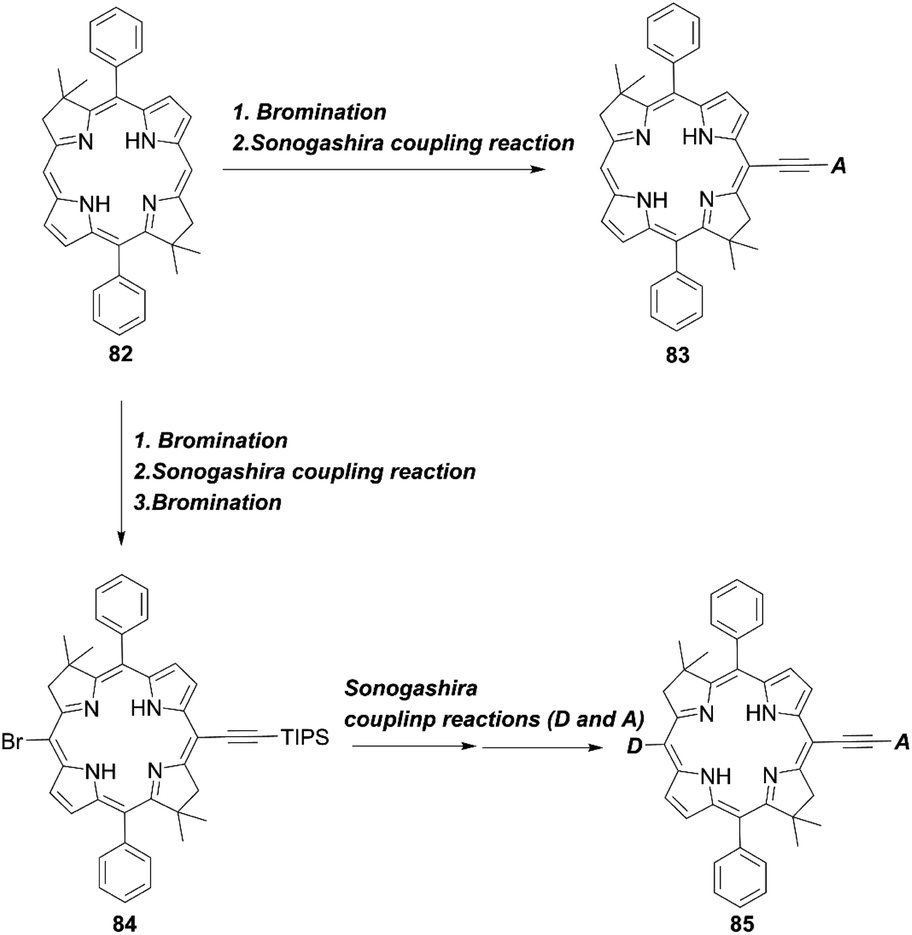
In 2017, Lin and co-workers82 reported bacteriochlorin-based sensitizers with two geminal dimethyl groups: LS-01 and LS-11 (Fig. 14). The key intermediate 82 was prepared by modifying the protocol established by Lindsey et al.83 Then, an anchoring group/push–pull structure was introduced by bromination and the Sonogashira coupling reaction of 82, or a similar synthetic strategy for 82 and 84, as shown in Scheme 1, route c, to yield 83 and 85 (Scheme 18). As expected, LS-01 and LS-11 revealed red-shifted and enhanced Qx- and Qy-bands relative to the corresponding porphyrin H2PE1, which were useful for the enhanced IPCE and resulted in larger current densities and PCEs (LS-01 (4.67%), LS-11 (5.36%), and H2PE1 (2.06%)). However, in terms of the durability, the LS cells exhibited a large decay in device efficiencies compared to the H2PE1 cell, which may be ascribed to the desorption of dyes on TiO2 and the resulting decreased JSC. Later, they continued to report three new meso-substituted, push–pull bacteriochlorins,84 coded as LS-17, LS-43, and LS-45, respectively (Fig. 14), where the PCE increased in the order of LS-43 (4.63%) < LS-17 (5.16%) < LS-45 (6.04%). The superior performance of LS-45, especially larger than that for LS-11, resulted from the larger JSC and VOC values, caused by the broadened and red-shifted absorption spectrum and larger CR resistance, respectively. Importantly, this general strategy can be a guideline for the further optimization of meso-substituted, air-stable bacteriochlorins.
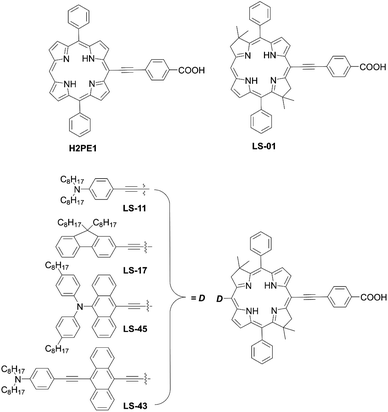 |
| | Fig. 14 Chemical structures of H2PE1, LS-01, LS-11, LS-17, LS-43, and LS-45. | |
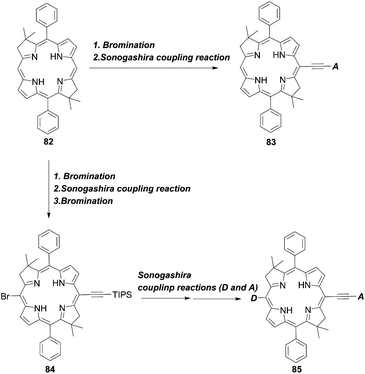 |
| | Scheme 18 Synthetic strategy for meso-functionalized bacteriochlorins. | |
6. Porphyrins created by unique synthetic strategies
6.1 Multi-fence porphyrins
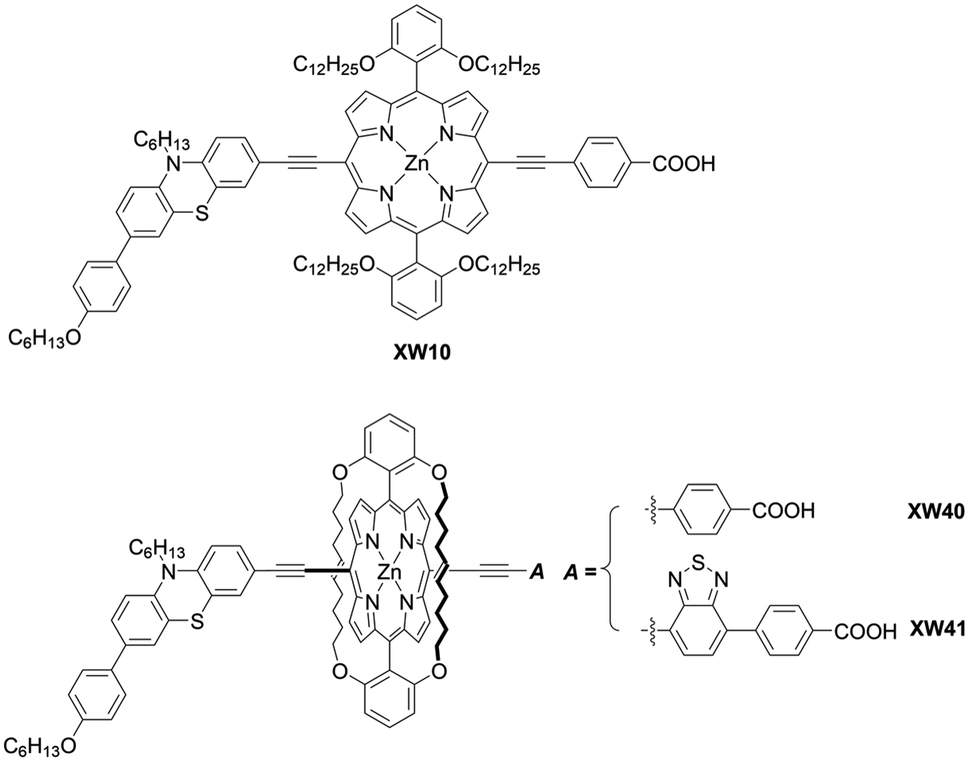
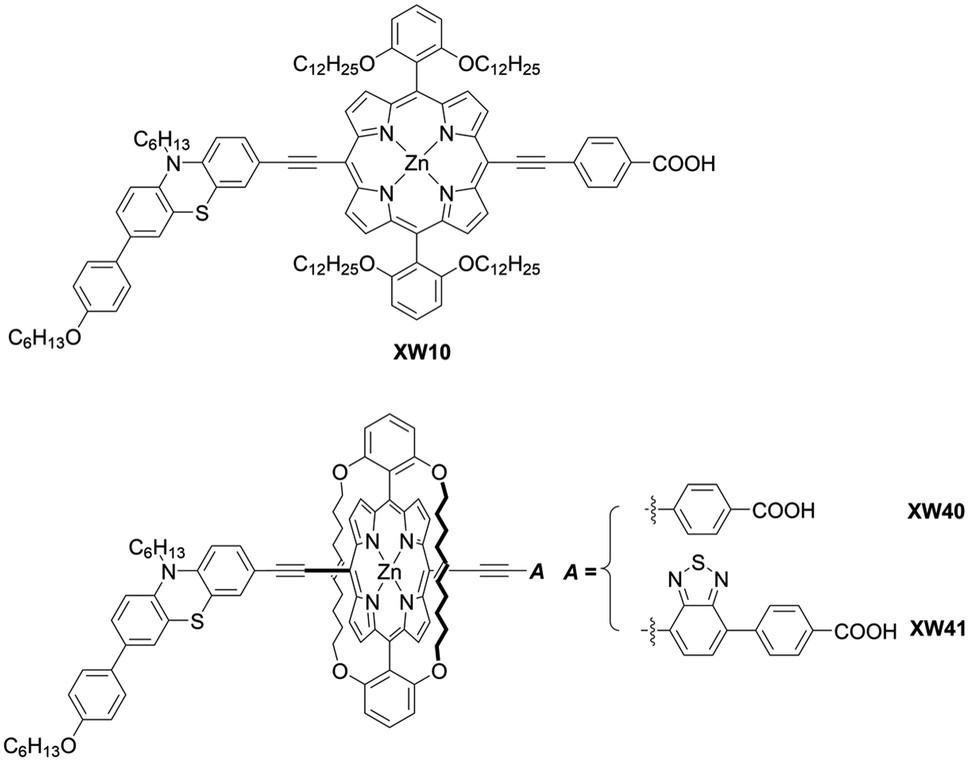
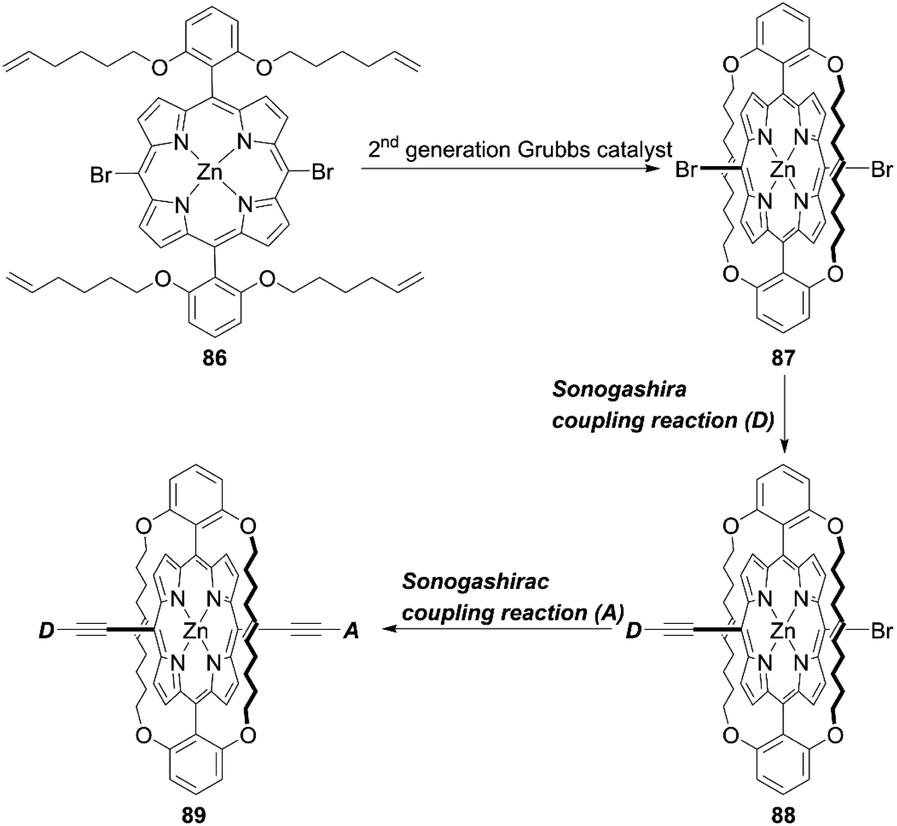
As corroborated in bulky YD2-o-C8, the introduction of long alkoxy chains at meso-aryl substituents is an effective strategy to ensure a high blocking effect of CR between the redox couple and TiO2 as well as to suppress the aggregation of porphyrins on TiO2 because of the wrapping of the porphyrin core by multiple long alkoxy chains. Xie and co-workers85 proposed a new class of doubly strapped porphyrin sensitizers, as represented by XW40 and XW41 (Fig. 15). In their synthetic scheme, 2,6-bis(octyloxy)phenyl groups at the meso-positions were replaced with 2,6-bis(hex-5-en-1-yloxy)phenyl groups (Scheme 19). The ring-closing metathesis reaction of precursor 86 with the 2nd generation Grubbs catalyst provided the doubly strapped porphyrin 87, and then stepwise Sonogashira coupling reactions starting from 87 afforded 88 and the target molecule 89 with the push–pull structure. In contrast with the porphyrin XW10 wrapped with four long alkoxyl chains (Fig. 15), XW40 obtained a higher VOC value (0.711 V vs. 0.730 V), because the introduction of the circle chain further inhibited the dye aggregation and CR. Additionally, a higher JSC value (18.67 mA cm−2vs. 17.90 mA cm−2) was also attained because of the more compact structure of XW40 and resulting increased dye-loading amount. Overall, XW10 and XW40 attained PCEs of 8.60% and 9.31%, respectively. Meanwhile, XW41 with BTD as the auxiliary electron-withdrawing unit possessed a broader response spectral range than XW40, but the dye aggregation caused by the expansion of the π-conjugation led to a having a lower PCE of 8.16%. Enhanced efficiencies of 10.55% and 10.19% were obtained for XW40- and XW41-based DSSCs, respectively, through a combined approach of co-adsorption (CDCA) and co-sensitization (Z1, Fig. 19).
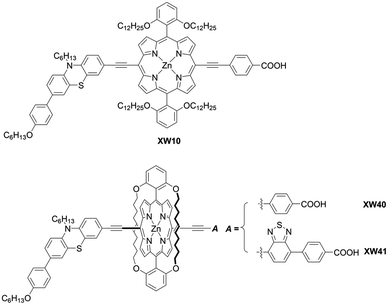 |
| | Fig. 15 Chemical structures of XW40, XW41, and XW10. | |
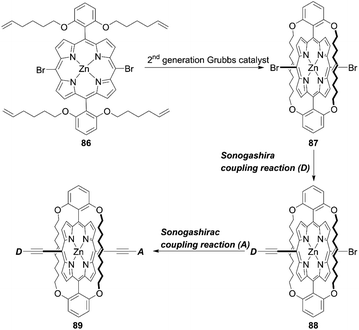 |
| | Scheme 19 Synthetic strategy for doubly strapped porphyrins. | |
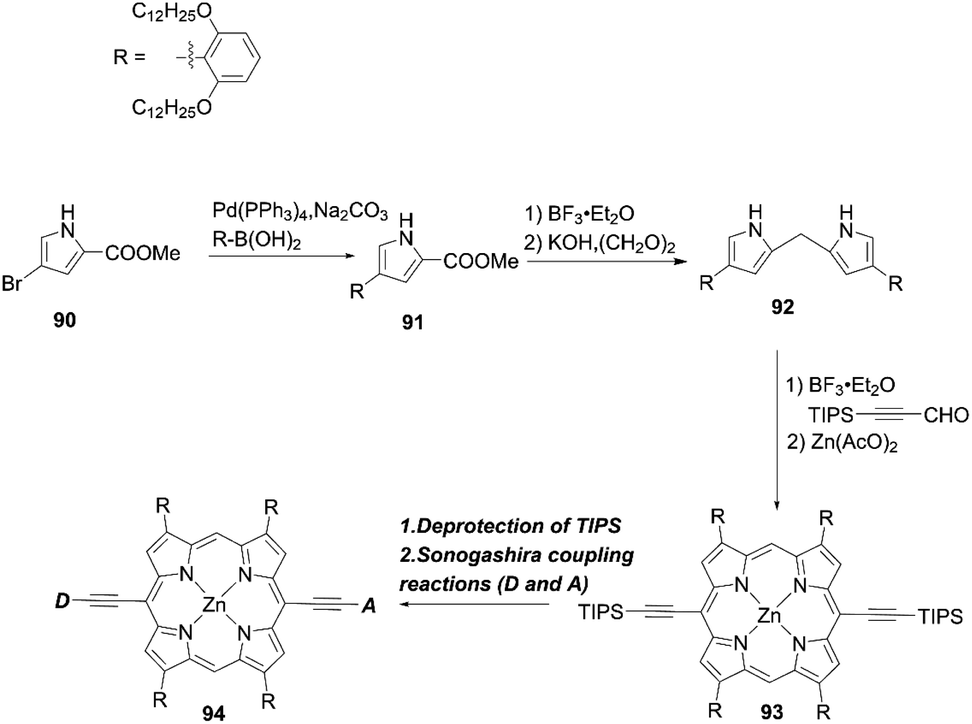
Yeh and co-workers86 put forward a new elegant approach to reduce dye aggregation and the CR process in which four β-phenyl rings were substituted with long alkoxyl groups at the β-positions of the porphyrin core. The key intermediate pyrrole 91 was prepared according to a Suzuki coupling reaction using the corresponding bromopyrrole 90 and boronic acid (Scheme 20). Then, subsequent condensation and decarboxylated reactions afforded the dipyrromethane 92. Finally, the target molecule 94 was obtained by the acid-catalyzed condensation reaction of 92 with 3-(triisopropylsilyl)-1-propynal and the ensuing metalation, by the deprotection of 93 and a final Sonogashira coupling reaction.
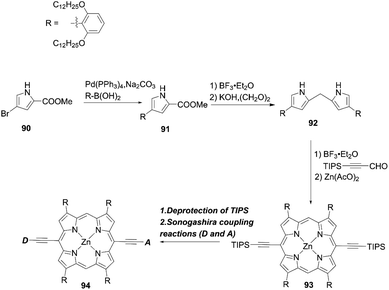 |
| | Scheme 20 Synthetic strategy for double-fence porphyrins. | |
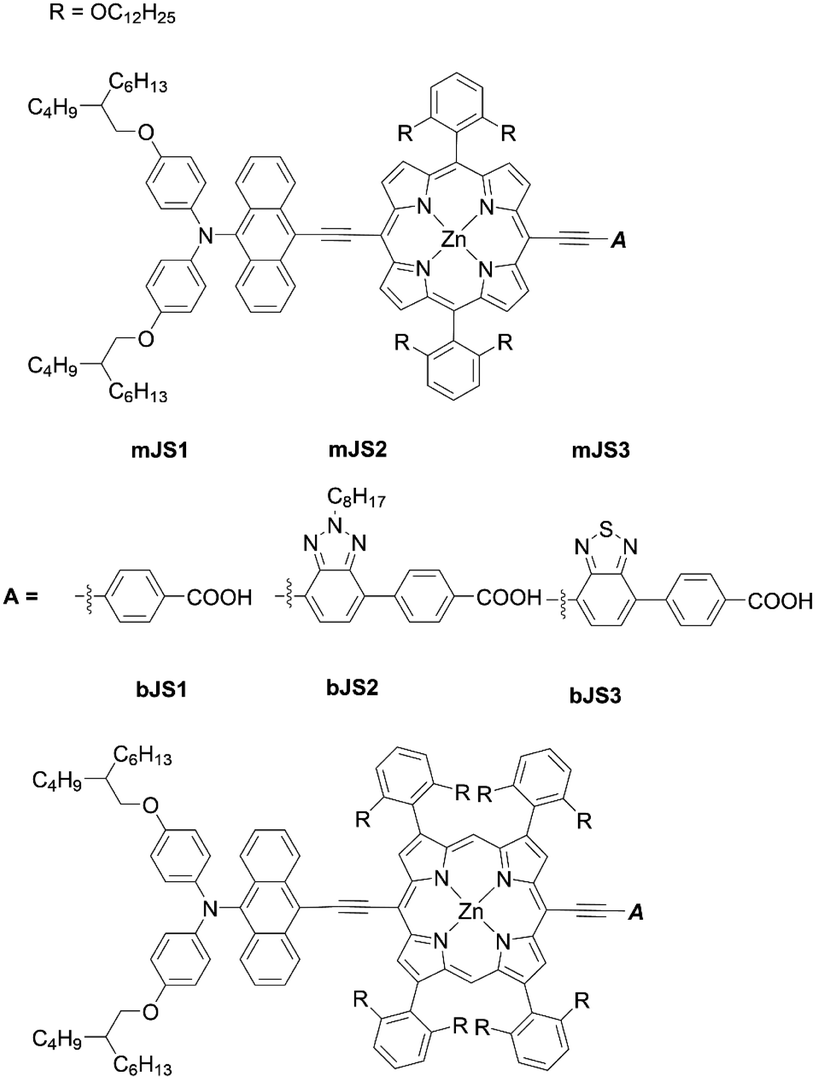
The double-fence porphyrin sensitizers bJS1–bJS3 (Fig. 16) achieved much higher PCEs (8.03–10.69%) than their single-fence analogues mJS1–mJS3 (2.33–6.69%). Among the three double-fence porphyrins bJS1–bJS3 in Fig. 16, the incorporation of benzotriazole and BTD units in the anchoring acceptor moiety for bJS2 and bJS3 caused further red-shifted absorption spectra, leading to high PCEs of 10.69% and 10.42%, respectively. These results exemplify that the introduction of multiple bulky substituents into the β-positions is an alternative fascinating strategy for the development of high-performance porphyrin-based DSSCs.
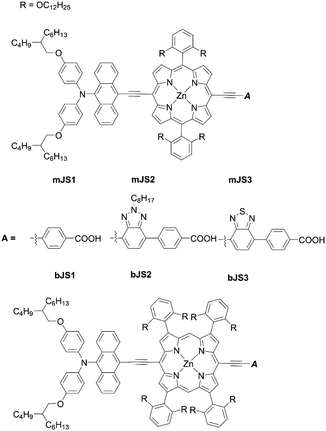 |
| | Fig. 16 Chemical structures of mJS1-3 and bJS1-3. | |
6.2 Porphyrins covalently linked to a co-sensitizer
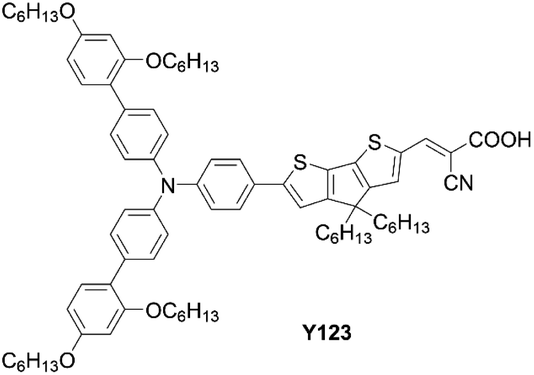
Co-sensitization is widely used in DSSCs as an effective approach to improve the PCE. In general, two or more different dyes with complementary optical absorption characteristics are used to fully cover the solar spectrum, but generally it is necessary that the dyes have little intermolecular interactions on the TiO2 surface. For example, the co-sensitization of DfZnP-iPr+LEG4 and YD2-o-C8+Y123 (Fig. 17), achieved higher PCEs than their single porphyrin systems without the addition of co-sensitizers as a consequence of their better light-harvesting abilities in the visible and near-infrared regions. However, traditional co-sensitization methods need many complicated optimization steps for attaining suitable adsorption conditions, while serious competitive adsorption frequently occurs between the main dye and the co-sensitizer.
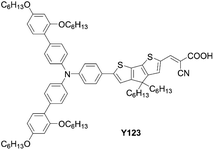 |
| | Fig. 17 Chemical structure of Y123. | |
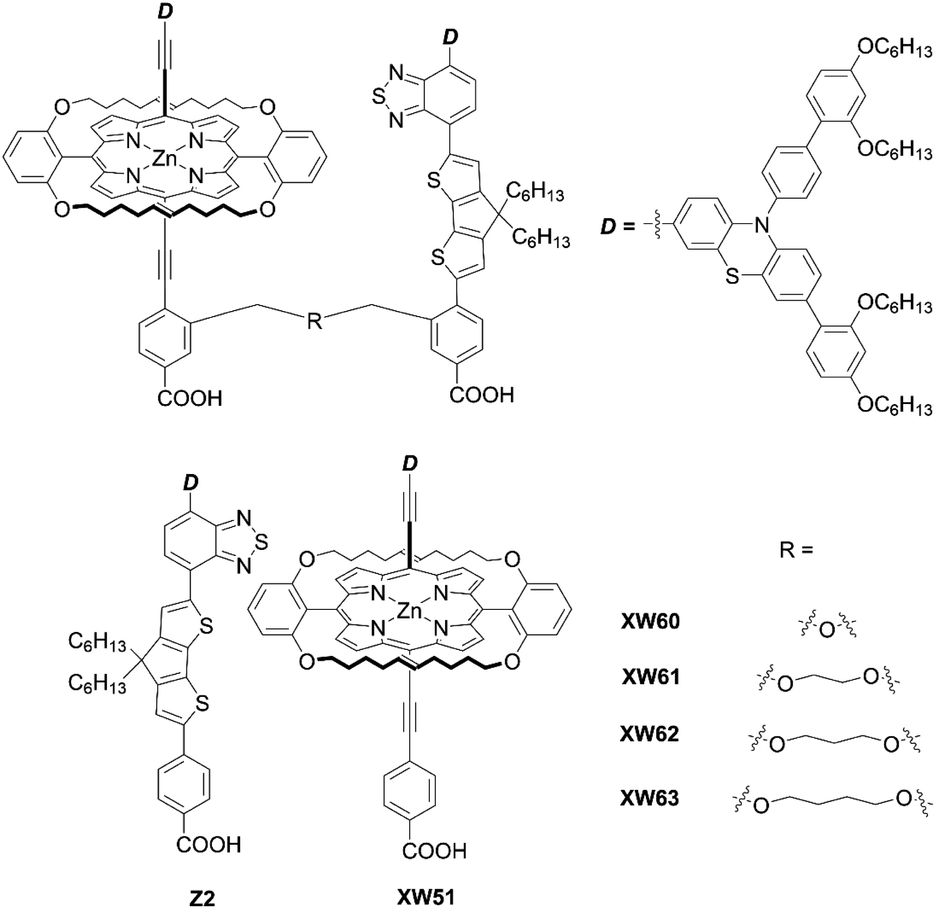
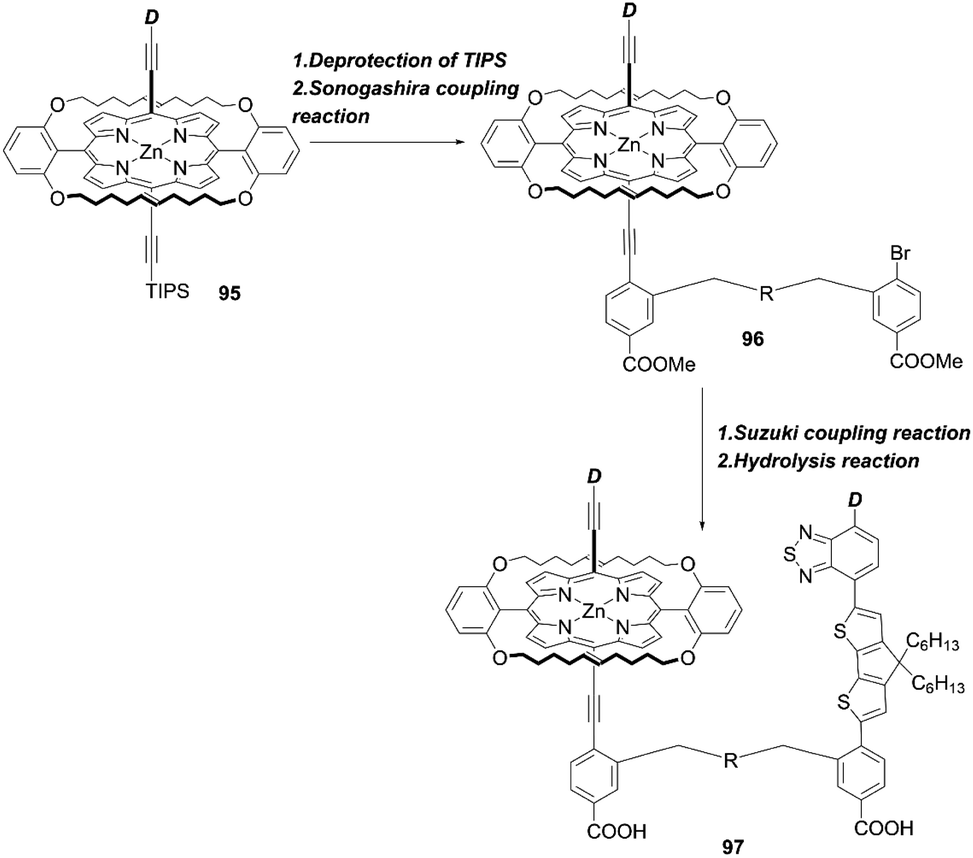
Recently, Xie and co-workers87 suggested an exotic approach for co-sensitization by designing “concerted companion dyes” (CC dyes), reporting XW60-XW63 (Fig. 18). These dyes could be synthesized as shown in Scheme 21. Structurally, their difference was the length of the alkoxyl linker between the acceptor sides of Z2 and XW51 (Fig. 18). The key intermediate 96 was obtained by deprotection of the precursor 95 and a subsequent Sonogashira coupling reaction. Then, the further Suzuki coupling and hydrolysis reactions of 96 afforded the target molecule 97. According to the DSSC obtained with the conventional co-sensitization method, where the TiO2 film was immersed in the solution of XW51 and Z2 in sequence, the DSSC with XW51+Z2 displayed a PCE of 9.2%. After optimizing the co-sensitization time and co-adsorption condition with CDCA, the PCE was slightly improved to 10.5%, but was still lower than the PCE of 11.1% obtained for the DSSC with XW51. This result may be explained by the drastic desorption of XW51 dye from TiO2 in the adsorption process of Z2 onto TiO2. Furthermore, during the co-sensitization process, some vacant sites generated by the desorption of XW51 from TiO2 would not be filled with Z2 due to the mismatched spatial structure, leading to the occurrence of undesirable CR through the vacant hole on TiO2. Interestingly, all the CC dyes except XW60 exhibited enhanced PCEs of 11.6–11.7%. The reason for the decreased PCE obtained by the XW60-based DSSC may be due to the unfavourably tilted adsorption of the Z2 component caused by the short bridging group.47–49 On the other hand, the fact that nearly the same PCEs were achieved by the device with XW61–63 indicated that the longer bridging group was unnecessary for highly efficient CC dyes. Furthermore, the co-adsorption of XW61 with CDCA afforded a record PCE of 12.4% for iodine electrolyte-based DSSCs.
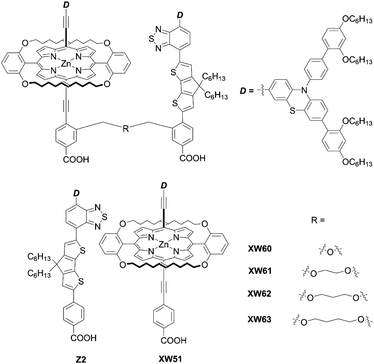 |
| | Fig. 18 Chemical structures of XW60-63, XW51, and Z2. | |
 |
| | Fig. 19 Chemical structures of HC-A4, C1, WS-5, and Z1. | |
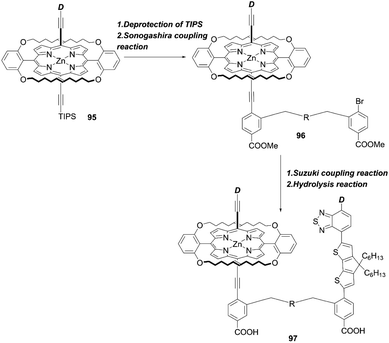 |
| | Scheme 21 Synthetic strategy for CC dyes.R = alkoxy linker. | |
7. Summary and outlook
Many DSSCs have been developed hopefully as an alternative to silicon-based solar cells due to their ease of fabrication, cost-effectiveness, and environmental friendliness. Very recently, the highest PCE reported exceeded 15%.89 Dyes play a crucial role in cell performance, and a lot of synthetic efforts have been paid to the development of new dyes showing high photovoltaic performance. In this context, porphyrins have drawn much attention because of their extraordinary high molar absorption coefficients of Soret and Q-bands around 400–600 nm. If the absorption is flattened at the wavelength of 400–800 nm, such porphyrins could be extremely promising in terms of their light-harvesting abilities. In this review article, we highlighted the synthetic strategies for preparing well-tailored porphyrins for DSSCs, aiming at a better light-harvesting property as well as reducing the aggregation on TiO2 and increasing the blocking effect of the CR between TiO2 and the electrolyte solution. It should be pointed out that it is still difficult to control the dye geometry on TiO2, which affects the ET kinetics on TiO2 and thereby the PCE, essentially because of the involvement of complicated multiple factors (vide supra). The following approaches have been taken for achieving a better light-harvesting property: (1) a push–pull D–π–A structure with suitable electron-donating and electron-withdrawing groups to strengthen ICT; (2) enlarging the π-conjugation, such as by fusion with peripheral aromatic rings; (3) lowering the symmetry of the porphyrins core by chemical modification. However, it should be noted here that elongation of the π-conjugation is always accompanied with serious dye aggregation, and sometimes leads to a more tilted geometry on the TiO2 surface and a fast undesirable CR process. The introduction of bulky groups or alkyl/alkoxyl chains at the donor, porphyrin core, or acceptor is the general synthetic strategy to override these obstacles, but the try-and-error nature of experimentation is still a challenging process to optimize the properties. More importantly, the co-sensitization of dyes with complementary absorption is indispensable because even well-designed porphyrins have still a relatively weak light-harvesting property at 500–600 nm. Recently, copper(I/II) complexes have emerged as the third-generation redox couples27,89,90 thanks to mitigation of the VOC loss by the use of copper(I/II) complexes and by suppression of the nonradiative CR.91 Generally, the first oxidation potential of most zinc porphyrin dyes usually lies at +0.8–1.0 V (versus NHE), which is similar to the redox potential of conventional copper redox shuttles. As a result of the mismatched energy levels, there is insufficient driving force for dye regeneration, which always leads to poor device performance. Accordingly, it is important to develop new porphyrins and copper redox shuttles that possess sufficient driving force for ET from the redox shuttle to the porphyrin radical cation (≥0.2 eV). Along this line, we achieved a PCE of 5.07% by selecting a suitable combination of an existing porphyrin and copper redox shuttle.90 Shifting the redox potential of the copper redox shuttles to a negative direction by adopting electron-donating ligands could be an option for a breakthrough. Besides, efficient synthetic strategies are needed to reduce the economic costs of dye synthesis towards large-scale commercial production. Although several novel reactions have been adopted to shorten the synthetic pathways (i.e. route b in Scheme 1), the number of required synthetic steps for obtaining high-performance porphyrins is still large (i.e. more than 10 steps for SM315 from the commercial starting materials), which would lower the overall yield. Additionally, the use of noble metal catalysts (i.e. Pd-based complexes) should be optimized for further cost reduction.
It is noteworthy that, in recent years, machine learning (ML) has been developed rapidly. In this regard, there has been promising data-driven approaches reported for predicting the performance of new porphyrins92 or organic dyes93,94 to accelerate the discovery of potential sensitizers for DSSCs. Researchers have constructed a rational quantitative structure–property relationship (QSPR) model by ML based on reported dyes rather than first-principles equations, which can be used to predict the PCE of candidate dyes. ML-based approaches can greatly decrease the time, costs, and labour requirements compared to the common “trial and error” approach. Although the utility is still limited to the available reported data of sensitizers (102–103, which is much less than the widely preferred “big data”), we believe that it will become an effective tool for the design of dye sensitizers with high PCEs after further improvement.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the JSPS (KAKENHI Grant Numbers JP20H05841 (T. H.) JP22K05066 (T. H.), JP20H05831 (H. I.), and JP20H05832 (H. I.)). Y. Zhang (CSC No. 202006250042) is grateful for the supports from the China Scholarship Council (CSC).
Notes and references
- A. Hagfeldt, G. Boschloo, L.-C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. K. Nazeeruddin, F. D. Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, T. Bessho and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS PubMed.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336–14341 CrossRef CAS.
- C.-Y. Chen, M.-K. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- Y.-Z. Wu, W.-H. Zhu, S. M. Zakeeruddin and M. Grätzel, ACS Appl. Mater. Interfaces, 2015, 7, 9307–9318 CrossRef CAS PubMed.
- Z.-Y. Yao, H. Wu, Y.-M. Ren, Y.-C. Guo and P. Wang, Energy Environ. Sci., 2015, 8, 1438–1442 RSC.
- Y. K. Eom, I. T. Choi, S. H. Kang, J. Lee, J. Kim, M. J. Ju and H. K. Kim, Adv. Energy Mater., 2015, 5, 1500300 CrossRef.
- N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS PubMed.
- Z.-Y. Yao, H. Wu, Y. Li, J.-T. Wang, J. Zhang, M. Zhang, Y.-C. Guo and P. Wang, Energy Environ. Sci., 2015, 8, 3192–3197 RSC.
- Y. K. Eom, S. H. Kang, I. T. Choi, Y. Yoo, J. Kim and H. K. Kim, J. Mater. Chem. A, 2017, 5, 2297–2308 RSC.
- K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379–6381 RSC.
- A. Kay and M. Grätzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C.-Y. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973–2977 CrossRef CAS.
- S. H. Kang, M. J. Jeong, Y. K. Eom, I. T. Choi, S. M. Kwon, Y. J. Yoo, J. H. Kim, J. Kwon, J. H. Park and H. K. Kim, Adv. Energy Mater., 2017, 7, 1602117 CrossRef.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS PubMed.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- H. Imahori, T. Umeyama, K. Kurotobi and Y. Takano, Chem. Commun., 2012, 48, 4032–4045 RSC.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- J.-M. Ji, H.-R. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518–14545 RSC.
- K.-W. Zeng, Z.-F. Tong, L. Ma, W.-H. Zhu, W.-J. Wu and Y.-S. Xie, Energy Environ. Sci., 2020, 13, 1617–1657 RSC.
- T. Higashino and H. Imahori, ACS Energy Lett., 2022, 7, 1926–1938 CrossRef CAS.
- C.-W. Lee, H.-P. Lu, C.-M. Lan, Y.-L. Huang, Y.-R. Liang, W.-N. Yen, Y.-C. Liu, Y.-S. Lin, E. W.-G. Diau and C.-Y. Yeh, Chem. Eur. J., 2009, 15, 1403–1412 CrossRef CAS PubMed.
- C.-M. Li, L. Luo, D. Wu, R.-Y. Jiang, J.-B. Lan, R.-L. Wang, L.-Y. Huang, S.-Y. Yang and J.-S. You, J. Mater. Chem. A, 2016, 4, 11829–11834 RSC.
- T. Higashino, Y. Fujimori, K. Sugiura, Y. Tsuji, S. Ito and H. Imahori, Angew. Chem., Int. Ed., 2015, 54, 9052–9056 CrossRef CAS PubMed.
- T. Higashino, Y. Kurumisawa, N. Cai, Y. Fujimori, Y. Tsuji, S. Nimura, D. M. Packwood, J. Park and H. Imahori, ChemSusChem, 2017, 10, 3347–3351 CrossRef CAS PubMed.
- T. Higashino, S. Nimura, K. Sugiura, Y. Kurumisawa, Y. Tsuji and H. Imahori, ACS Omega, 2017, 2, 6958–6967 CrossRef CAS PubMed.
- T. Higashino, H. Iiyama, Y. Kurumisawa and H. Imahori, ChemPhysChem, 2019, 20, 2689–2695 CrossRef CAS PubMed.
- A. Hinsch, J. M. Kroon, R. Kern, I. Uhlendorf, J. Holzbock, A. Meyer and J. Ferber, Prog. Photovolt: Res. Appl., 2001, 9, 425–438 CrossRef CAS.
- R. B. Ambre, G.-F. Chang and C.-H. Hung, Chem.
Commun., 2014, 50, 725–727 RSC.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- D.-M. Shen, C. Liu, X.-G. Chen and Q.-Y. Chen, J. Org. Chem., 2009, 74, 206–211 CrossRef CAS PubMed.
- M. Yan, Q.-H. Wang, Y.-Z. Zhu, M.-L. Han, Y.-Q. Yan and J.-Y. Zheng, J. Photochem. Photobiol. A, 2021, 416, 113325 CrossRef CAS.
- H.-H. Chou, K. S. K. Reddy, H.-P. Wu, B.-C. Guo, H.-W. Lee, E. W.-G. Diau, C.-P. Hsu and C.-Y. Yeh, ACS Appl. Mater. Interfaces, 2016, 8, 3418–3427 CrossRef CAS PubMed.
- C.-F. Lo, S.-J. Hsu, C.-L. Wang, Y.-H. Cheng, H.-P. Lu, E. W.-G. Diau and C.-Y. Lin, J. Phys. Chem. C, 2010, 114, 12018–12023 CrossRef CAS.
- Y.-Q. Wang, B. Chen, W.-J. Wu, X. Li, W.-H. Zhu, H. Tian and Y.-S. Xie, Angew. Chem., Int. Ed., 2014, 53, 10779–10783 CrossRef CAS PubMed.
- Y.-S. Xie, Y.-Y. Tang, W.-J. Wu, Y.-Q. Wang, J.-C. Liu, X. Li, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055–14058 CrossRef CAS PubMed.
- Y.-Y. Tang, Y.-Q. Wang, X. Li, H. Ågren, W.-H. Zhu and Y.-S. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 27976–27985 CrossRef CAS PubMed.
- G.-S. Yang, Y.-Y. Tang, X. Li, H. Ågren and Y.-S. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 36875–36885 CrossRef CAS PubMed.
- H.-L. Song, J. Zhang, J.-M. Jin, H.-F. Wang and Y.-S. Xie, J. Mater. Chem. C, 2018, 6, 3927–3936 RSC.
- C.-L. Wang, C.-M. Lan, S.-H. Hong, Y.-F. Wang, T.-Y. Pan, C.-W. Chang, H.-H. Kuo, M.-Y. Kuo, E. W.-G. Diau and C.-Y. Lin, Energy Environ. Sci., 2012, 5, 6933–6940 RSC.
- H. Imahori, S. Kang, H. Hayashi, M. Haruta, H. Kurata, S. Isoda, S. E. Canton, Y. Infahsaeng, A. Kathiravan, T. Pascher, P. Chábera, A. P. Yartsev and V. Sundström, J. Phys. Chem. A, 2011, 115, 3679–3690 CrossRef CAS PubMed.
- S. Ye, A. Kathiravan, H. Hayashi, Y. Tong, Y. Infahsaeng, P. Chabera, T. Pascher, A. P. Yartsev, S. Isoda, H. Imahori and V. Sundström, J. Phys. Chem. C, 2013, 117, 6066–6080 CrossRef CAS.
- H. Imahori, Bull. Chem. Soc. Jpn., 2023 DOI:10.1246/bcsj.20230031.
- T. Higashino, Y. Kurumisawa, S. Nimura, H. Iiyama and H. Imahori, Eur. J. Org. Chem., 2018, 2537–2547 CrossRef CAS.
- K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ito, P. Chabera, V. Sundström and H. Imahori, Chem. Eur. J., 2013, 19, 17075–17081 CrossRef CAS PubMed.
- T. Higashino, K. Kawamoto, K. Sugiura, Y. Fujimori, Y. Tsuji, K. Kurotobi, S. Ito and H. Imahori, ACS Appl. Mater. Interfaces, 2016, 8, 15379–15390 CrossRef CAS PubMed.
- G. D. Carlo, A. O. Biroli, M. Pizzotti, F. Tessore, V. Trifiletti, R. Ruffo, A. Abbotto, A. Amat, F. D. Angelis and P. R. Mussini, Chem. Eur. J., 2013, 19, 10723–10740 CrossRef PubMed.
- K. Sudhir, K. S. K. Reddy, Y.-C. Liu, H.-H. Chou, K. Kala, T.-C. Wei and C.-Y. Yeh, ACS Appl. Mater. Interfaces, 2018, 10, 39970–39982 CrossRef PubMed.
- H. Hata, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8264–8265 CrossRef CAS PubMed.
- The names of the dyes were previously given as tda-1b-d-Zn and tda-2b-bd-Zn, respectively. See: M. Ishida, S. W. Park, D. Hwang, Y. B. Koo, J. L. Sessler, D. Y. Kim and D. Kim, J. Phys. Chem. C, 2011, 115, 19343–19354 CrossRef CAS.
- M. Ishida, D. Hwang, Y. B. Koo, J. Sung, D. Y. Kim, J. L. Sessler and D. Kim, Chem. Commun., 2013, 49, 9164–9166 RSC.
- M. Ishida, D. Hwang, Z. Zhang, Y. J. Choi, J. Oh, V. M. Lynch, D. Y. Kim, J. L. Sessler and D. Kim, ChemSusChem, 2015, 8, 2967–2977 CrossRef CAS PubMed.
- M. Tanaka, S. Hayashi, S. Eu, T. Umeyama, Y. Matano and H. Imahori, Chem. Commun., 2007, 2069–2071 RSC.
- S. Hayashi, M. Tanaka, H. Hayashi, S. Eu, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 15576–15585 CrossRef CAS.
- S. Eu, S. Hayashi, T. Urneyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 4396–4405 CrossRef CAS.
- H. Hayashi, A. S. Touchy, Y. Kinjo, K. Kurotobi, Y. Toude, S. Ito, H. Saarenpää, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, ChemSusChem, 2013, 6, 508–517 CrossRef CAS PubMed.
- S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846–847 CrossRef CAS.
- R. Deshpande, L. Jiang, G. Schmidt, J. Rakovan, X.-P. Wang, K. Wheeler and H. Wang, Org. Lett., 2009, 11, 4251–4253 CrossRef CAS PubMed.
- Y. Hu, W. A. Webre, M. B. Thomas, A. Moss, S. N. Hancock, J. Schaffner, F. D'Souza and H. Wang, J. Mater. Chem. A, 2019, 7, 10712–10722 RSC.
- Y. Hu, A. Alsaleh, O. Trinh, F. D'Souza and H. Wang, J. Mater. Chem. A, 2021, 9, 27692–27700 RSC.
- J. Luo, M.-F. Xu, R.-Z. Li, K.-W. Huang, C.-Y. Jiang, Q.-B. Qi, W.-D. Zeng, J. Zhang, C.-Y. Chi, P. Wang and J.-S. Wu, J. Am. Chem. Soc., 2014, 136, 265–272 CrossRef CAS PubMed.
- Y. Kurumisawa, T. Higashino, S. Nimura, Y. Tsuji, H. Iiyama and H. Imahori, J. Am. Chem. Soc., 2019, 141, 9910–9919 CrossRef CAS PubMed.
- J. T. Dy, K. Tamaki, Y. Sanehira, J. Nakazaki, S. Uchida, T. Kubo and H. Segawa, Electrochemistry, 2009, 77, 206–209 CrossRef CAS.
- A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles, K. C. Gordon, L. Du, R. Katoh, A. Furube and D. L. Officer, J. Am. Chem. Soc., 2009, 131, 15621–15623 CrossRef CAS PubMed.
- J.-W. Shiu, Y.-C. Chang, C.-Y. Chan, H.-P. Wu, H.-Y. Hsu, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, J. Mater. Chem. A, 2015, 3, 1417–1420 RSC.
- T. Higashino, K. Sugiura, Y. Tsuji, S. Nimura, S. Ito and H. Imahori, Chem. Lett., 2016, 45, 1126–1128 CrossRef CAS.
- T. Zhang, X. Qian, P.-F. Zhang, Y.-Z. Zhu and J.-Y. Zheng, Chem. Commun., 2015, 51, 3782–3785 RSC.
- A. Osuka and H. Shimidzu, Angew. Chem., Int. Ed. Engl., 1997, 36, 135–137 (
Angew. Chem.
, 1997
, 109
, 93–95
) CrossRef CAS.
- N. Yoshida, H. Shimidzu and A. Osuka, Chem. Lett., 1998, 27, 55–56 CrossRef.
- L.-M. Jin, L. Chen, J.-J. Yin, C.-C. Guo and Q.-Y. Chen, Eur. J. Org. Chem., 2005, 3994–4001 CrossRef CAS.
- T. Higashino, Y. Kurumisawa, H. Iiyama and H. Imahori, Chem. Eur. J., 2019, 25, 538–547 CrossRef CAS PubMed.
- X.-F. Wang, H. Tamiaki, L. Wang, N. Tamai, O. Kitao, H. Zhou and S. Sasaki, Langmuir, 2010, 26, 6320–6327 CrossRef CAS PubMed.
- X.-F. Wang, O. Kitao, H. Zhou, H. Tamiaki and S. Sasaki, J. Phys. Chem. C, 2009, 113, 7954–7961 CrossRef CAS.
- X.-J. Liu, C.-J. Li, X. Peng, Y.-Z. Zhou, Z. Zeng, Y.-C. Li, T.-Y. Zhang, B. Zhang, Y. Dong, D.-M. Sun, P. Cheng and Y.-Q. Feng, Dyes Pigm., 2013, 98, 181–189 CrossRef CAS.
- T. Higashino, Y. Tsuji, Y. Fujimori, K. Sugiura, S. Ito and H. Imahori, Chem. Lett., 2015, 44, 1395–1397 CrossRef CAS.
- S. Chakraborty, H.-C. You, C.-K. Huang, B.-Z. Lin, C.-L. Wang, M.-C. Tsai, C.-L. Liu and C.-Y. Lin, J. Phys. Chem. C, 2017, 121, 7081–7087 CrossRef CAS.
- H.-J. Kim and J. S. Lindsey, J. Org. Chem., 2005, 70, 5475–5486 CrossRef CAS PubMed.
- S. Chakraborty, M.-C. Tsai, X.-D. Su, X.-C. Chen, T.-T. Su, C.-K. Tsao and C.-Y. Lin, RSC Adv., 2020, 10, 6172–6178 RSC.
- K.-W. Zeng, Y.-Y. Lu, W.-Q. Tang, S.-L. Zhao, Q.-Y. Liu, W.-H. Zhu, H. Tian and Y.-S. Xie, Chem. Sci., 2019, 10, 2186–2192 RSC.
- C.-C. Chen, J.-S. Chen, V. S. Nguyen, T.-C. Wei and C.-Y. Yeh, Angew. Chem., Int. Ed., 2021, 60, 4886–4893 CrossRef CAS PubMed.
- K.-W. Zeng, Y. Chen, W.-H. Zhu, H. Tian and Y.-S. Xie, J. Am. Chem. Soc., 2020, 142, 5154–5161 CrossRef CAS PubMed.
- K.-W. Zeng, W.-Q. Tang, C.-J. Li, Y.-Y. Chen, S.-L. Zhao, Q.-Y. Liu and Y.-S. Xie, J. Mater. Chem. A, 2019, 7, 20854–20860 RSC.
- Y. Ren, D. Zhang, J. Suo, Y. Cao, F. T. Eickemeyer, N. Vlachopoulos, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Nature, 2023, 613, 60–65 CrossRef CAS PubMed.
- T. Higashino, H. Iiyama, I. Nishimura and H. Imahori, Chem. Lett., 2020, 49, 936–939 CrossRef CAS.
- H. Imahori, Y. Kobori and H. Kaji, Acc. Mater. Res., 2021, 2, 501–514 CrossRef CAS.
- C.-C. Fan, M. Springborg and Y.-Q. Feng, Phys. Chem. Chem. Phys., 2019, 21, 5834–5844 RSC.
- H. Li, Z. Zhong, L. Li, R. Gao, J. Cui, T. Gao, L. H. Hu, Y. Lu, Z.-M. Su and H. Li, J. Comput. Chem., 2015, 36, 1036–1046 CrossRef CAS PubMed.
- Y.-P. Wen, L.-L. Fu, G.-Q. Li, J. Ma and H.-B. Ma, Sol. RRL, 2020, 4, 2000110 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Hiroshi
Imahori
*a and
Hiroshi
Imahori