
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew advances in chiral nanographene chemistry
Hannah V.
Anderson
 ,
Nicolai D.
Gois
and
Wesley A.
Chalifoux
*
,
Nicolai D.
Gois
and
Wesley A.
Chalifoux
*
Department of Chemistry, University of Nevada, Reno, 1664 N. Virginia St., Reno, NV 89557, USA. E-mail: wchalifoux@unr.edu
First published on 6th June 2023
Abstract
Chiral nanographenes are an intriguing class of polycyclic aromatic hydrocarbon structures, where combining the unique properties of inherently chiral molecules with the optical, electronic, and even magnetic properties of nanographenes can allow for new potential applications in the field of nanoelectronics. Being able to fine-tune these properties through rational synthetic design is a key part of the field of nanoelectronic development, and there have been many new advances made, both in the past and very recently, to prepare novel chiral nanographenes. Here, we will highlight several of the more recent, noteworthy, and lesser-reviewed contributions made in chiral nanographene preparation.
 Hannah V. Anderson | Hannah V. Anderson graduated from Northwest Nazarene University in 2018 with her B.S. in Chemistry. She is currently pursuing her Ph.D. under the supervision of Professor Wesley Chalifoux at the University of Nevada, Reno. Hannah is the leading author for this review, and her specific research interests are directed towards preparing open-shell polycyclic aromatic hydrocarbon systems. |
 Nicolai D. Gois | Nicolai D. Gois received his B.A. in Chemistry from California State University, Stanislaus in 2020. He is currently pursuing a Ph.D. under the supervision of Prof. Wesley Chalifoux at the University of Nevada, Reno. Nicolai's research is focused on the synthesis of pyrene-based polycyclic aromatic hydrocarbons with interesting optical and electronic properties. |
 Wesley A. Chalifoux | Dr Wesley A. Chalifoux received is B.Sc. from the University of Alberta (Alberta, Canada) in 2004 where he conducted undergraduate research under the direction of Professor Rik. R. Tykwinski. During his undergraduate training, he did a one-year industrial internship at Raylo Chemicals (Edmonton) on an NSERC IURA award. He was an NSERC graduate scholar and Alberta Ingenuity scholar at the University of Alberta (Alberta, Canada) where he obtained his PhD in 2010 under the supervision of Professor Rik R. Tykwinski. The focus of his dissertation was the synthesis and study of polyynes to gain insight into the properties of the elusive sp-allotrope of carbon known as carbyne. Dr Chalifoux worked as an NSERC postdoctoral fellow at Columbia University under the supervision of James L. Leighton from 2010–2012. He joined the Department of Chemistry at the University of Nevada, Reno in 2012 where he worked as an Associate Professor until 2023. In 2023, he moved to the University of Alberta, Canada as a Professor of Chemistry. His research focus is method development and organic synthesis of novel polycyclic aromatics, nanographenes, and graphene nanoribbons. |
Introduction
Nanographenes are a broad class of polycyclic aromatic hydrocarbon structures, loosely defined as finite graphitic units primarily composed of fused six-membered sp2-hybridized carbons with sizes between 1–100 nm.1,2 A multitude of work has been done in the field of bottom-up nanographene synthesis since the pioneering contributions of Clar,3,4 and Scholl,5,6 and the interesting physical properties of these small graphene units have sparked a widespread interest in developing structures that could have potential applications in nanoelectronics, optoelectronics, and spintronics.7–10One particular area of interest is directed toward the development of non-planar, contorted, or twisted structures that exhibit inherent chirality. Many synthetic techniques have been employed for the development of these chiral, nonplanar nanographenes, including, but not limited to, alkyne-based methods11,12 and Scholl oxidation. Scholl chemistry in particular has been utilized extensively in the preparation of chiral nanographenes, and has recently been thoroughly reviewed by Miao et al. in the context of preparing contorted structures.13 Due to the prevalence of this method in recent literature, this review will focus primarily on structures prepared through non-Scholl techniques, in order to highlight more unique or less common methods. The contortion of these structures from planar to non-planar, and the introduction of chirality, produces a new set of optoelectronic, photophysical, and physical properties to be explored, opening up a new area of development and potential application in nanoelectronics.14–17 Here, we will highlight and discuss some of the notable and recent advances that have been made in the field of chiral nanographene design and synthesis.
Twistacenes
The members of the acene family are most commonly defined as polycyclic aromatic hydrocarbons that have fused six-membered rings organized in a linear fashion, and these systems are almost always planar. However, inducing a twist along the backbone of these acene systems produces a unique class of nonplanar nanographenes, the twistacenes. A 2006 review was published outlining some of the more notable twistacenes,18 and more recently, a more comprehensive overview on chiral acenes has been published.19One noteworthy example of a chiral twistacene was produced by Pascal et al. in 2006. The group reported the synthesis of twisted 9,10,11,20,21,22-hexaphenyltetrabenzo[a,c,l,n]pentacene 1, by reacting 1,3-diphenylphenanthro[9,10-c]furan precursors with biaryne equivalents in the presence of n-butyllithium, followed by low-valent titanium-catalyzed deoxygenation (Fig. 1a).20 These structures had a dramatic end-to-end twist of 144°, and the enantiomers were able to be resolved cleanly via chiral HPLC. The rate of racemization for these compounds was slow at 27 °C, with a half-life for the loss of enantiomeric excess at 6.2 hours (ΔG‡rac = 23.8 kcal mol−1). Pascal's work was later expanded by Kilway and coworkers who synthesized twisted hexacene analog 2 with a record-breaking end-to-end twist of 184° (Fig. 1b).21
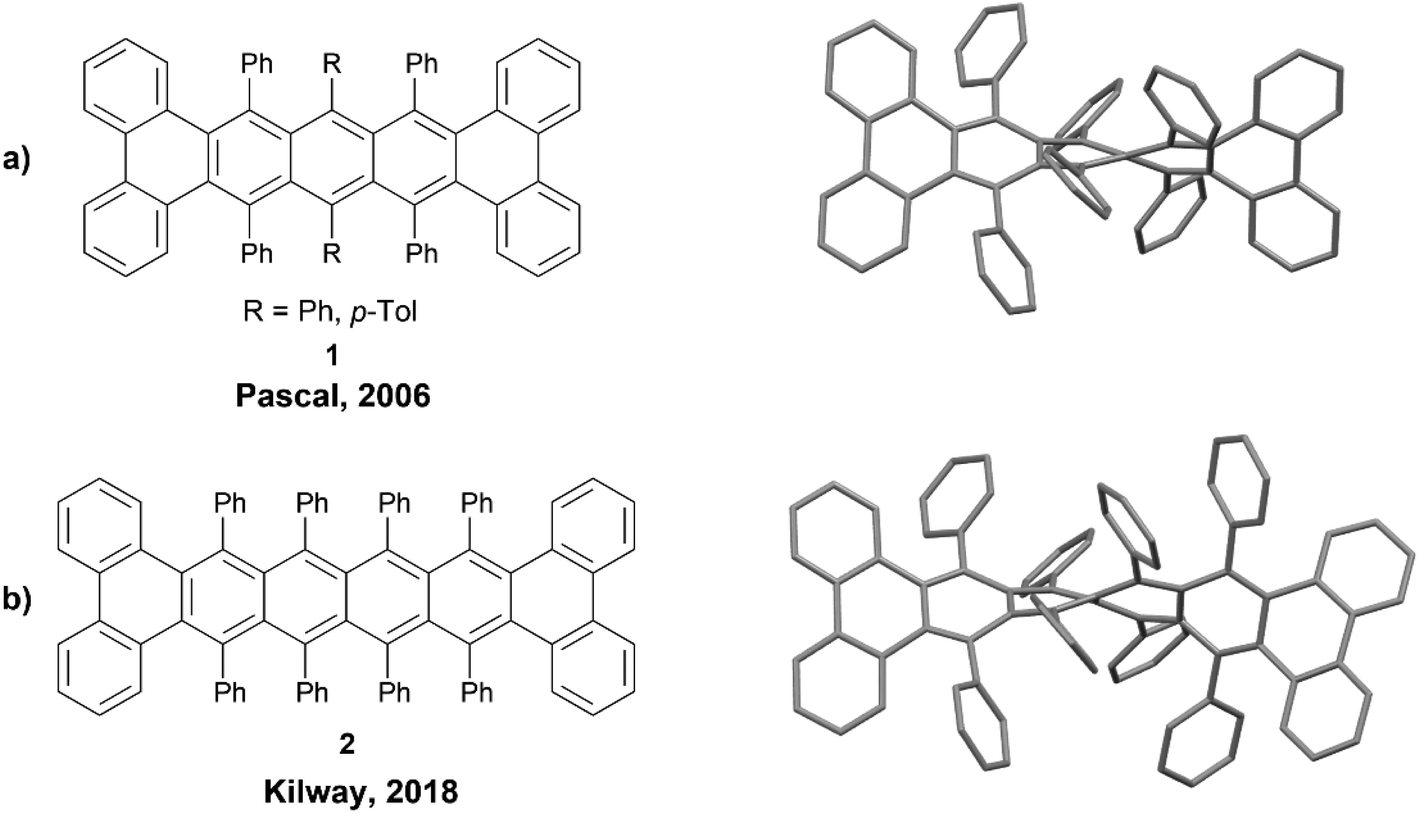 | ||
| Fig. 1 (a) Twistacene 1 synthesized by Pascal and coworkers with published crystal structure; (b) Twistacene 2 synthesized by Kilway and coworkers with published crystal structure. Hydrogens omitted for clarity. | ||
A couple of years later, Pascal et al. reported the synthesis of configurationally stable twisted PAH structures 5 and 8 using a similar protocol as previously reported (vide supra) (Scheme 1).22 Enantiomers of 5 and 8 were separable by supercritical fluid chromatography (SFC) on chiral supports. The authors reported specific rotations of +320° and −330° for (+)-5 and (−)-5, respectively, and ±23° for (+)-8 and (−)-8. Strong circular dichroism (CD) signals were also observed, with values of Δε as high as 5% of ε (Δε ≈ 700 M−1 cm−1vs. ε = 14 000 M−1 cm−1) for compound 8. Moderately strong circularly polarized luminescence (CPL) was also observed, with dissymmetry values of 1.4 × 10−3 and 0.8 × 10−3 for 5 and 8, respectively.
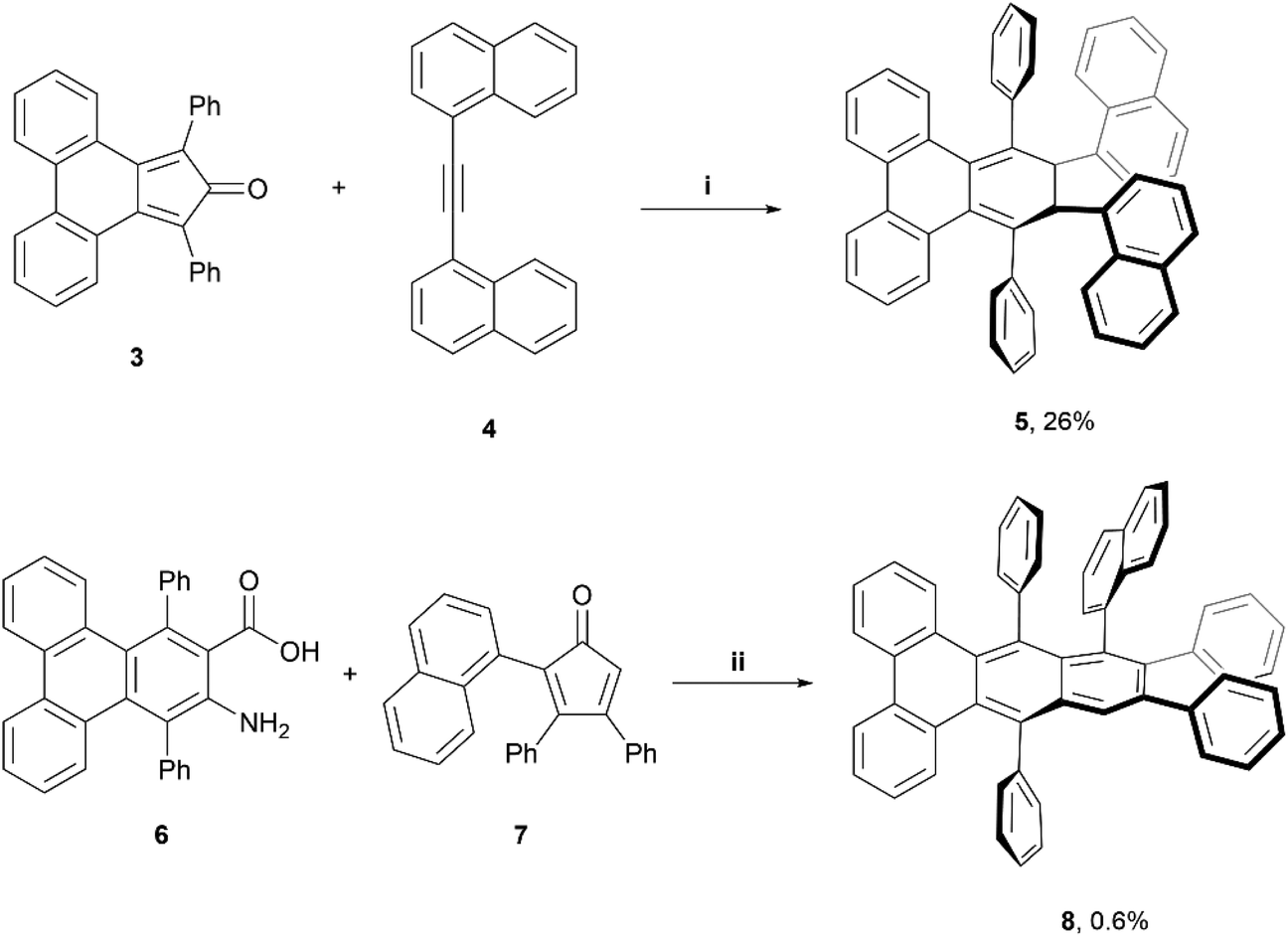 | ||
| Scheme 1 Synthesis of twistacene compound 5 and 8; (i) 3 (1.0 equiv.), 4 (1.0 equiv.), Ph2O (1 mL), 300 °C, 1 h.; (ii) 6 (1.0 equiv.), 7 (1.1 equiv.), isoamyl nitrite (2.0 equiv.), DCE, reflux, 1 h. | ||
In 2015, Pascal and coworkers produced some of the first “hairpin furans”, a class of helically-twisted nanographenes centered around a furan ring (Fig. 2a).23 This hairpin structure 9 combines the twist of a sterically-hindered acene with the twist of an expanded helicene. Compound 9 was prepared through multiple steps including Diels–Alder cycloaddition, decarbonylation, and dehydrogenation, followed by an oxidative cyclodehydrogenation step to produce 9 in low yield. Single crystals of a racemic mixture of 9 were obtained and it was observed that the twisted terminal phenanthryl moieties of each structure are roughly parallel to each other with an intramolecular spacing of about 4 Å (Fig. 2b). Interestingly, it was found that the enantiomeric pairs adopt a centrosymmetric arrangement, with each enantiomer nestling a phenanthrene group within the cleft of its enantiomeric counterpart.
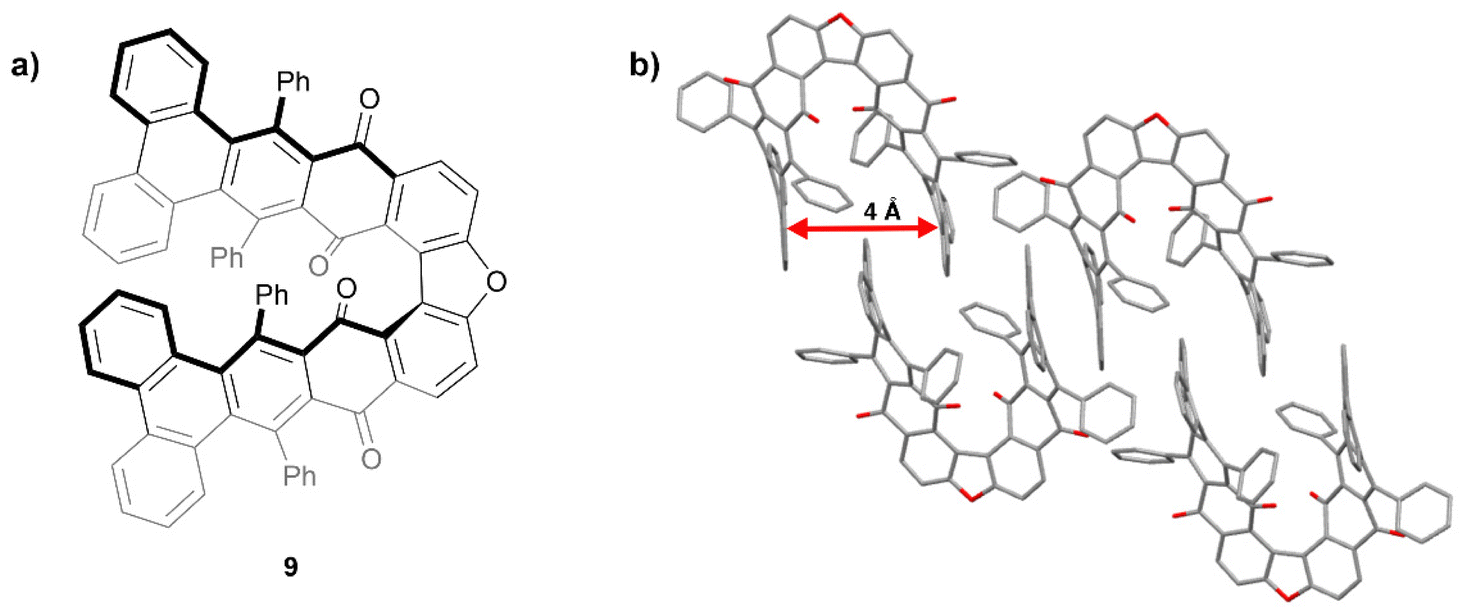 | ||
| Fig. 2 (a) Hairpin furan 9 synthesized by Pascal and coworkers; (b) published crystal structure of 9 showing twisting and packing. Hydrogens omitted for clarity. | ||
The Chalifoux research group has previously utilized a Brønsted acid catalyzed four-fold alkyne benzannulation reaction for the rapid expansion of π-systems. This technique has been used to make chiral peropyrenes 11a–d from p-terphenyl-2,2′′,6,6′′-tetrayne precursors 10a–d (Scheme 2). The sterically congested bay regions of these structures result in a contorted backbone with an end-to-end twist angle of 28°, which was unambiguously determined through X-ray crystallographic analysis.24 The enantiomers of derivative 11b were separated via chiral HPLC, and were analyzed with many methods including Raman, fluorescence, and circularly polarized spectroscopy. Most notably, 11b produced a large specific rotation value with [α]25589 = +1438 (c = 8.69 × 10−3 g L−1 in CH2Cl2), similar to that observed in helicene systems, and the CD spectrum displayed strong Cotton effects with Δε = ±100 M−1 cm−1 at 300 nm. The racemization inversion barrier was experimentally determined to be 29 kcal mol−1, which agreed well with quantum mechanical calculations.
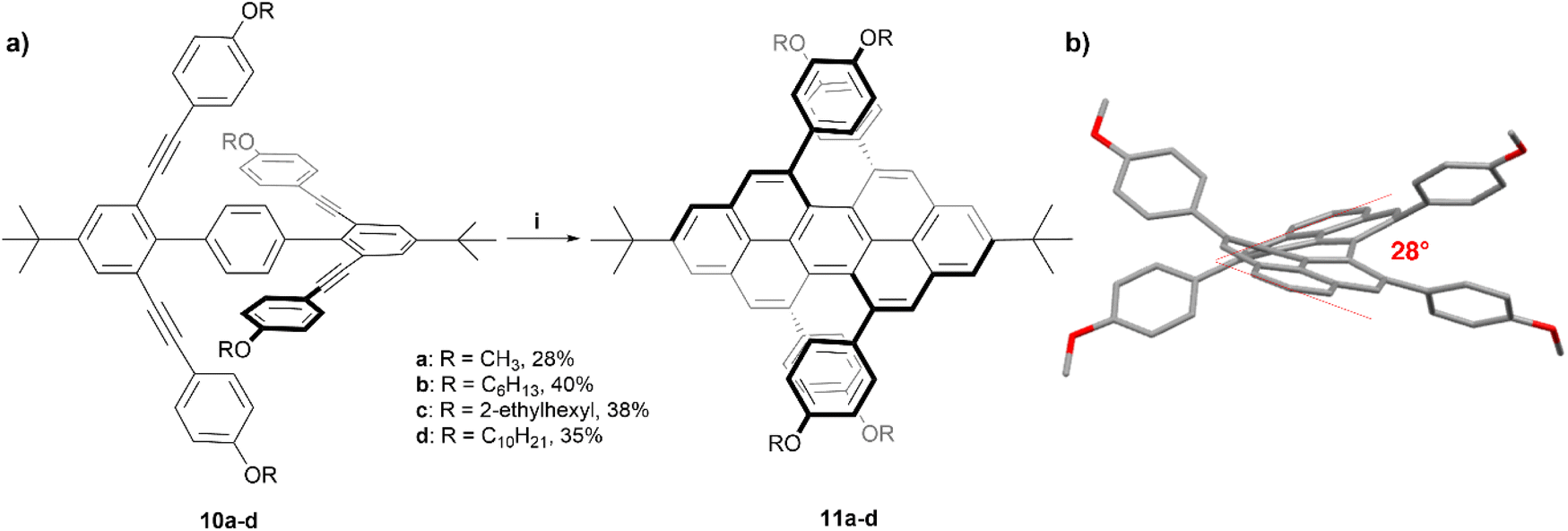 | ||
| Scheme 2 (a) Synthesis of chiral peropyrene 11; (i) 10 (1.0 equiv.), TfOH (0.2 equiv.), DCM, −40 °C, 30 min; (b) published crystal structure of 11b showing twisting. Hydrogens and alkyl groups omitted for clarity. | ||
Helicenes
Helicenes are a specialized and well-known class of helical nanographenes, identified by consecutively ortho-fused rings in a circular, helical arrangement. Structures that are composed of just a helicene backbone are referred to as [n]helicenes, where n denotes the number of rings contributing to the spiraling backbone. Introducing specific variations to the general structure of helicene can give rise to many distinct classes: (a) increasing the inner diameter of the helicene shape beyond that of [n]helicenes leads to expanded helicene derivatives, (b) laterally extending the helicene units by fusing additional aromatic rings to the backbone produces π-extended helicene derivatives, (c) placing multiple helicene positions within the same structure gives rise to multiple helicene derivatives, and (d) adding heteroatoms somewhere within a helicene-like structure results in heterohelicene derivatives. [n]Helicenes are by far the most extensively studied class of chiral nanographenes, with a multitude of new contributions being made since the early works of Lednicer and Newman,25 Katz,26 Laarhoven and Prinsen,27 and Martin,28 amongst others. Many more recent reviews have been written on the subject of helicenes and their derivatives, covering synthesis, properties and applications29–34 and several focus on particular classes, including multiple helicenes,35,36 and heterohelicenes.12,37,38 For the purpose of this review, discussion of [n]helicenes and simple heterohelicenes will be omitted due to their extensive presence in previous literature. This section will instead focus mainly on the more recently synthesized and lesser reviewed helicene classes, and other chiral nanographenes with helicene-like topology.Expanded helicenes
In 2017, the Tilley group was among the first to define the term and contribute to the preparation of expanded helicenes.39 Two expanded [11]helicene derivatives (not pictured) and one [13]helicene derivative (13) were prepared from hexa-alkyne starting material 12, which was synthesized through a straightforward four-step method. The alkynes were cyclized fully to the final helical product 13 in impressive yields (78–91%) through iridium-catalyzed [2 + 2 + 2] cyclization, based on a previously-known method (Scheme 3).40 Interestingly, 1H NMR analysis, along with X-ray crystallography data indicated that the [13]helicene 13 exists not only as the monomeric structure, but that it also readily forms a homochiral, π-stacked dimer of two interlocked enantiomers.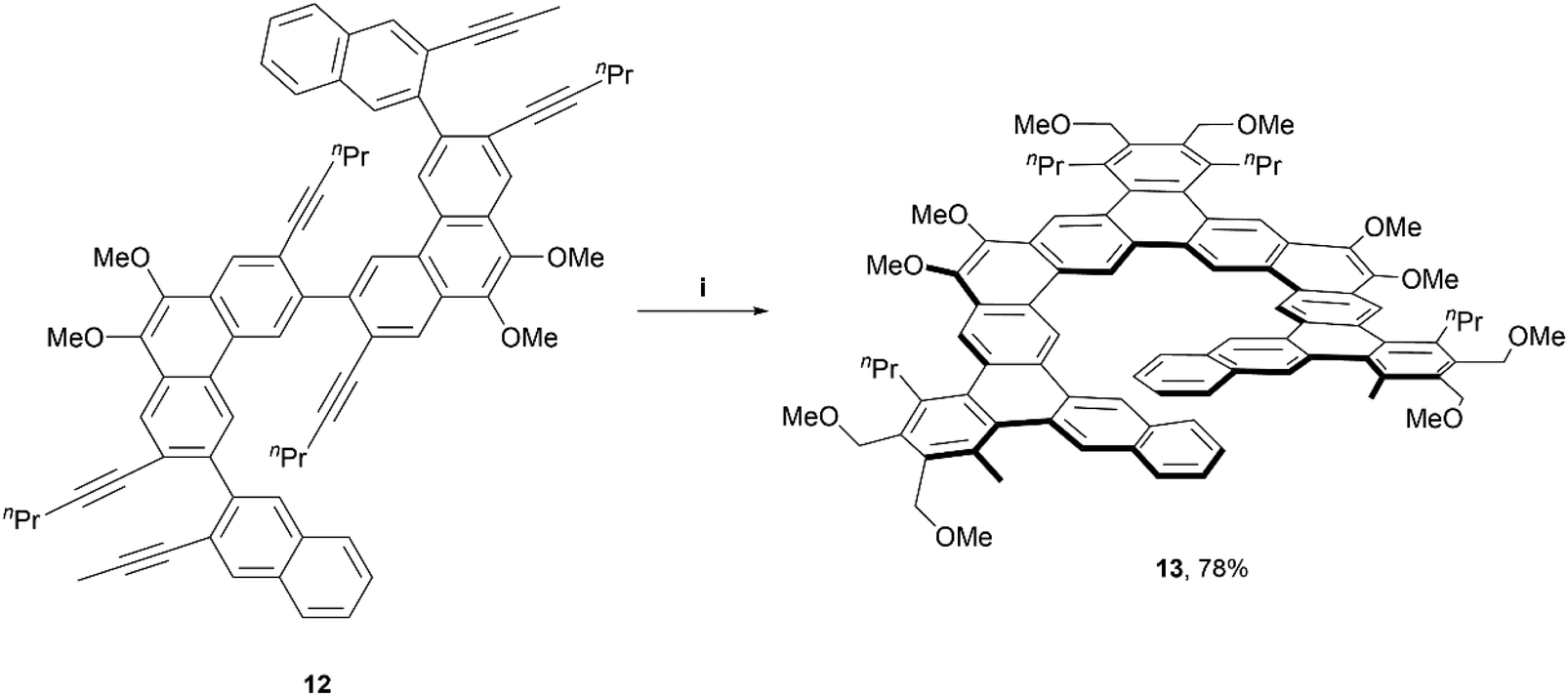 | ||
| Scheme 3 Synthesis of expanded, chiral helicenes; (i) 12 (1.0 equiv.), 1,4-dimethoxy-2-butyne (6.0 equiv.), Ir[(cod)Cl]2 (0.03 equiv.), DPPE (0.06 equiv.), benzene, 80 °C, 2 h. | ||
In 2020, the same group utilized similar chemistry to produce even larger and more complex structures. The [11]helicene precursor 14, was prepared using similar methods to their previous work, and underwent two divergent transformations to produce unique, chiral macrocycles 15a–b and 16 (Scheme 4).41 The spatial orientation of the two alkynes of 14 suggested the possibility of a [2 + 2 + n] cycloaddition to form a bridge between the two terminal ends of the system, by reacting with a zirconocene complex, followed by in situ treatment of either SeCl2(2,2′-bipyridine) to produce structure 15a, or dimethyl acetylenedicarboxylate (DMAD) to give structure 15b. The figure-eight shaped macrocycle 16 was produced by treatment of 14 with molybdenum catalyst along with the loss of 4-octyne. Its impressive structure was unambiguously confirmed with NMR, MALDI-TOF mass spectrometry, and X-ray crystallography. The enantiomerization barriers for the products were studied by variable temperature 1H NMR, and the upper bounds of enantiomerization energy for 14 and 16 were found to be low (11.9 and 12.5 kcal mol−1, respectively). However, the barrier for 15a showed a moderate energy increase (approximately 16.6 kcal mol−1 at 65 °C), and 15b displayed a significantly higher value, with the lower bound estimated around 20.2 kcal mol−1 (later specified by CD analysis as 22.1 kcal mol−1). Due to the higher estimated energy of inversion, enantiomers of 15b were able to be resolved via chiral HPLC at 25 °C, but interconversion still occurred at room temperature, preventing analysis of enantiopure samples. Although the low racemization barrier prevented the separation of enantiomers of 16, single crystals of the racemic product were obtained so chirality could be confirmed. The crystal packing behavior of these large, figure-eight structures was unusual – the crystals were organized into two separate parallel, spiraling, interconnected helices, each of which is homochiral and therefore displays opposite handedness.
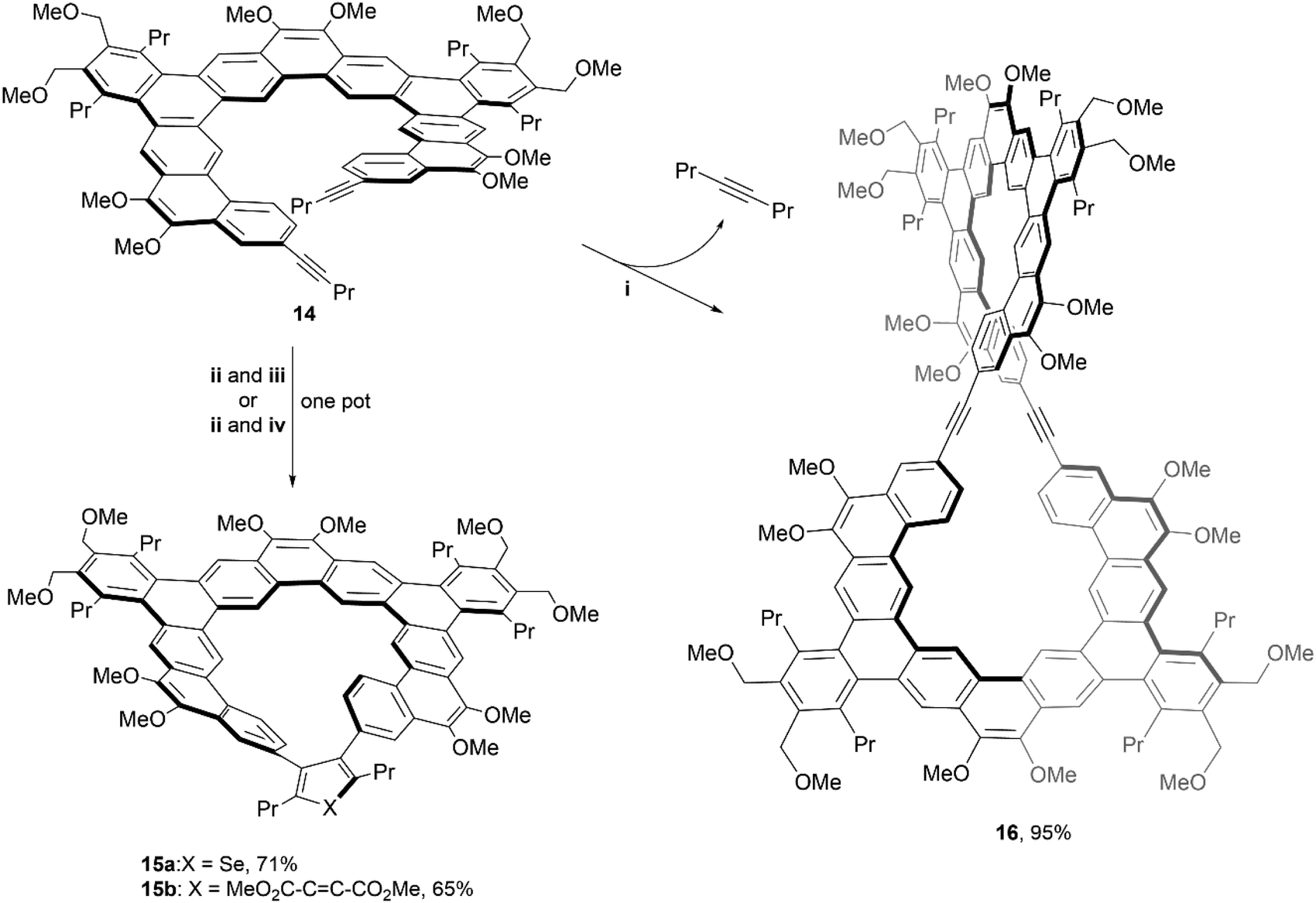 | ||
Scheme 4 Synthesis of [11]helicene 15 and of a figure-eight shaped macrocycle 16; (i) 14 (1.0 equiv.), EtC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mo(OC(CH3)(CF3)2)3(DME) (0.1 equiv.), 5 Å MS, toluene, 80 °C; (ii) 14 (1.0 equiv.), Cp2Zr(pyr)(Me3SiCCSiMe3) (1.2 equiv.), toluene, 22 °C; (iii) SeCl2(bipy) (2.0 equiv.), 22 °C; (iv) DMAD (3.0 equiv.), CuCl (3.0 equiv.), DMPU (6 equiv.), 22 °C. Mo(OC(CH3)(CF3)2)3(DME) (0.1 equiv.), 5 Å MS, toluene, 80 °C; (ii) 14 (1.0 equiv.), Cp2Zr(pyr)(Me3SiCCSiMe3) (1.2 equiv.), toluene, 22 °C; (iii) SeCl2(bipy) (2.0 equiv.), 22 °C; (iv) DMAD (3.0 equiv.), CuCl (3.0 equiv.), DMPU (6 equiv.), 22 °C. | ||
In recent years, Toyota and coworkers produced a helical structure, composed of three fused anthracene units, through a multistep synthesis ending with a platinum-catalyzed alkyne cyclization (Scheme 5).42 Enantiomers of the diphenyl derivative 18b were resolved by chiral HPLC, and both enantiomers exhibited strong Cotton effects at 352 nm in the CD spectra, and kinetic analysis predicted an enantiomerization barrier of ΔG‡363 of 28.9 kcal mol−1, significantly larger than the 8.1 kcal mol−1 that was estimated for the unsubstituted analog 18a. The group later followed up with further exploration of this structure by oxidizing derivatives of 18 to introduce ketones into the structure. This oxidation allowed for further dynamic studies of the helical inversion of the structure, which was explored via variable-temperature 1H NMR and DFT calculations.43 The inversion barrier for these more hindered structures was experimentally determined by the coalescence method to be 18.4 kcal mol−1, a slightly higher value than the calculated 16.5 kcal mol−1, with both values being notably higher than the estimated barrier (8.12 kcal mol−1) for the unsubstituted, non-oxidized precursor 18a, but still smaller than the disubstituted variant 18b.
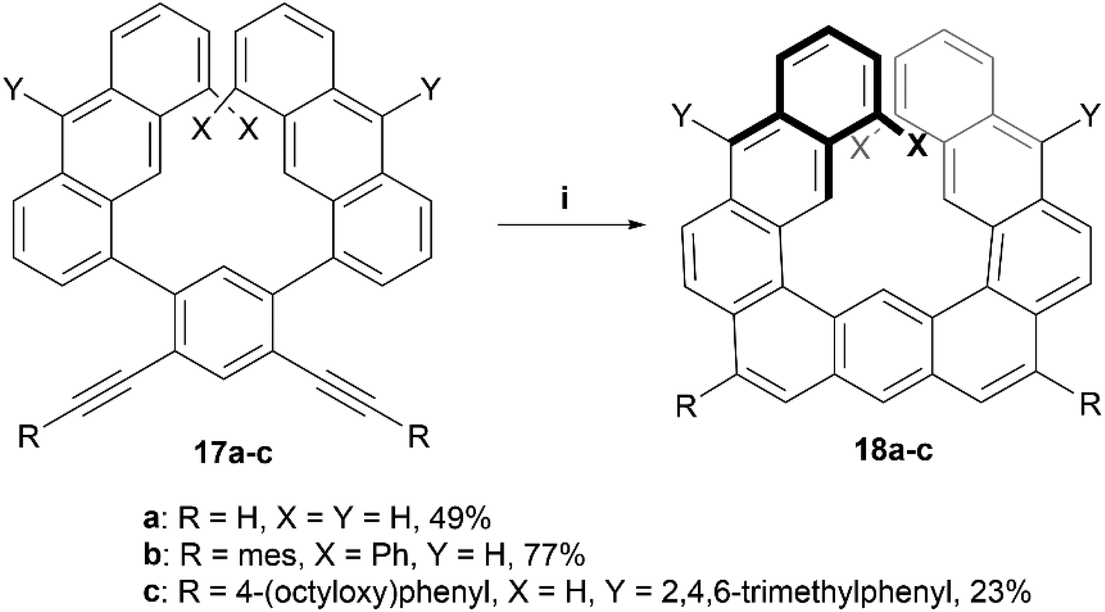 | ||
| Scheme 5 Synthesis of helicene structure 18; (i) 17 (1.0 equiv.), PtCl2 (0.25 equiv.), P(C6F5)3 (0.2 equiv.), toluene, 80 °C, 24–95 h. | ||
In 2021, Wu and coworkers produced the synthesis of a large ribbon-shaped macrocycle with a structure analogous to an expanded helicene, through a process including Suzuki coupling followed by Bi(OTf)3-mediated benzannulation, concluded with a final oxidative dehydrogenation to achieve the large figure-eight shaped structure 19 (Fig. 3a). The racemization barrier for a partially fused analogue of this structure was estimated to be very high, about 82.5 kcal mol−1, which was strong evidence for a final rigid structure of 19. Enantiomers were successfully separated by chiral HPLC, and the products displayed persistent chirality, with moderate absorption dissymmetry factor values (|gabs|) ranging from 5 × 10−3 to 7 × 10−3.44 A similar synthetic method was later used in 2023 by the same group to prepare a series of large, spiraling expanded helicene derivatives containing 11, 19, 27 and 35 cata-fused benzene rings. These structures displayed significant chiroptical responses, with |gabs| values as high as 2.4 × 10−2.45
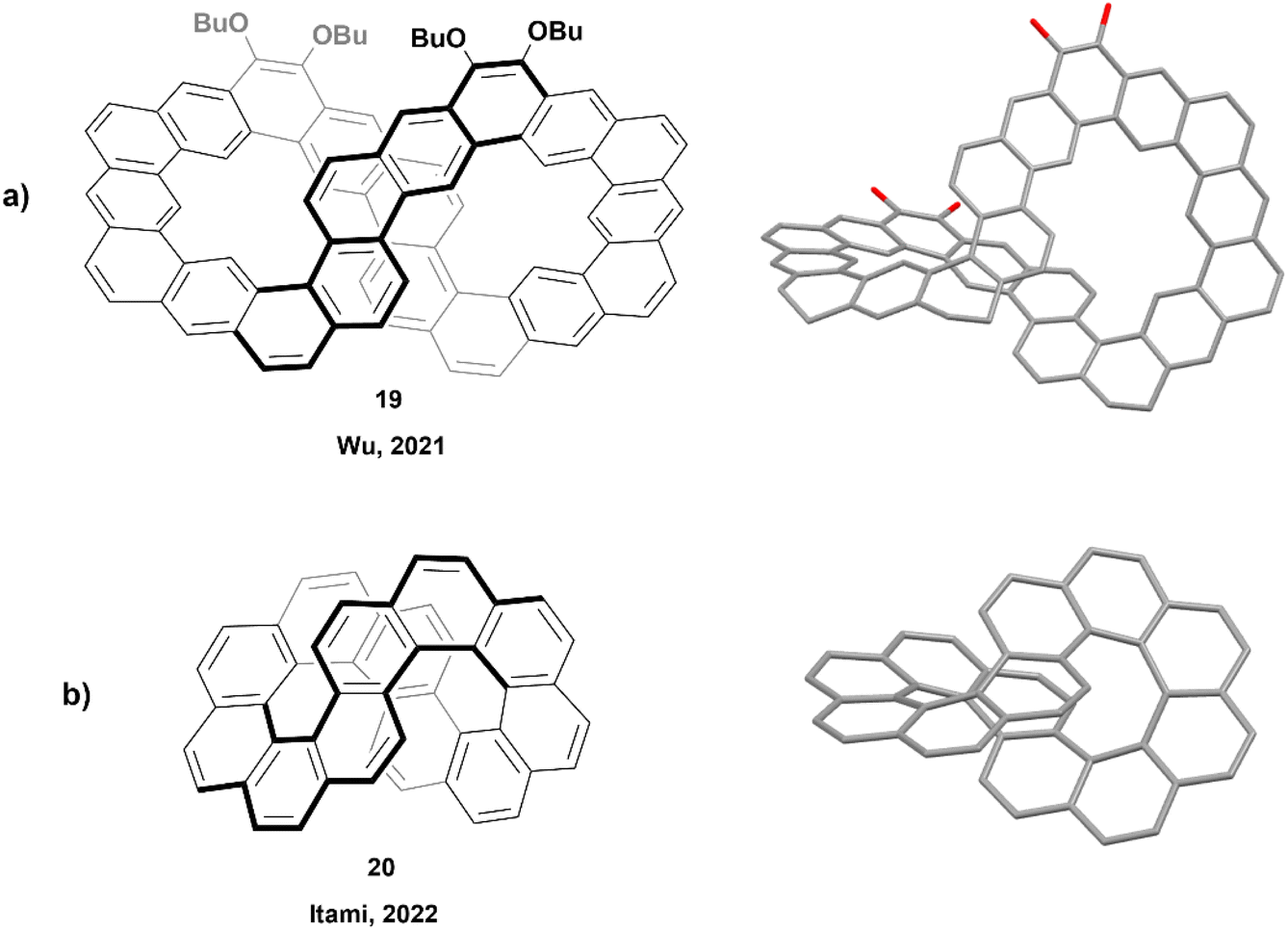 | ||
| Fig. 3 (a) Large, twisted carbon nanobelt 19 synthesized by the Wu group with published crystal structure; (b) infinitene 20 synthesized by the Itami group with published crystal structure. Hydrogens and alkyl chains omitted for clarity. | ||
The next year, Itami and coworkers reported the synthesis of a smaller nanobelt-like analogue, similar to Wu's, with an unambiguous figure-eight shape, aptly referred to as infinitene (Fig. 3b).46 The synthetic method involved multiple steps, finishing with a photocyclization to receive the final structure 20 in excellent yield (89%). The twisted structure of the final product was unambiguously confirmed by X-ray crystallography, and it was observed that the distance between the central positions of the middle twist was remarkably small (3.152–3.192 Å), as compared to the distance between the terminal rings of the analogous unsubstituted [7]helicene (3.804 Å).47 The enantiomers were separated via chiral HPLC, and chiroptical studies were performed. The circular dichroism and anisotropy factors (Δε and gCD) were measured, with Δε values ranging between 3.0 M−1 cm−1 at 484 nm and 69.0 M−1 cm−1 at 375 nm, and gCD values between 3.5 × 10−4 and 4.8 × 10−3. These reported values were notably smaller than that of other similar [n]helicenes, such as (P)-[6]helicene, which displays Δε values of −267 M−1 cm−1 at 246 nm and +259 M−1 cm−1 at 324 nm, and |gCD| values in the range of 5.0 × 10−3–9.3 × 10−3.
In 2018, Hirose, Matsuda, and coworkers reported the synthesis of expanded helicene 21 with a structure analogous to kekulene (Fig. 4).48 The synthetic procedure was largely based around a 6-fold ring-closing olefin metathesis reaction, and the final chiral structure was observed to have an impressively large helical diameter of 10.2 Å, which was unambiguously determined via X-ray crystallography. The product was recovered as a racemic mixture of the (P) and (M) enantiomers, and they were observed to pack in an alternating pattern in the crystal structure. The racemization barrier for this structure was predicted to be low (ΔG‡ = 13.0 kcal mol−1 at 25 °C), preventing the separation of enantiomers, but there was no configurational disorder present in the crystalline structures of the (P)-21 and (M)-21 structures, suggesting that the helical inversion can be controlled well within the solid or crystalline state.
 | ||
| Fig. 4 Analog of kekulene 21 synthesized by Hirose, Matsuda and coworkers with published crystal structure. Hydrogens omitted for clarity. | ||
π-Extended helicenes
In 2018, the Chalifoux group published a method of expanding the scope of multiple-alkyne benzannulation, by using a Lewis acid catalyst, indium(III) chloride, rather than the previously relied-on Brønsted acid catalysts (trifluoroacetic acid/triflic acid). This method allowed for the cyclization of alkynes attached to electron-rich, electron-neutral, and electron-poor aryl groups, and ultimately allowed for the preparation of new chiral peropyrene derivatives that had previously been inaccessible.49 This method was used in 2019 to prepare chiral pyreno[a]pyrene-based helicene hybrids 22 (Fig. 5), by utilizing Brønsted acid catalyzed four-fold benzannulation.50 Enantiomers of 22b and 22c were easily separated using chiral HPLC, and various chiroptical properties were analyzed. Both structures had a similar CD response, with an absorption dissymmetry ratio (gabs) of ∼3.5 × 10−3. Interestingly, 22 displayed glum values of approximately ∼0.9 × 10−3 and ∼1.5 × 10−3 (for 22b and 22c) respectively, which were notably higher values than observed in simple helicenes, and slightly higher even than chiral peropyrenes, which were previously studied within the Chalifoux group.24 Interestingly, displayed glum values of approximately ∼0.9 × 10−3 and ∼1.5 × 10−3 for 22b and 22c respectively, which were notably higher values than observed in simple helicenes, and slightly higher even than chiral peropyrenes, which were previously studied within the Chalifoux group.24 Not long afterwards, the same group used the InCl3-mediated benzannulation method to prepare substituted naphtho[1,2-a]pyrene derivatives 23 with persistent chirality at low temperatures, allowing for variable temperature dynamic HPLC separation of enantiomers (Fig. 5).51 It was observed that the single enantiomers of these structures were prone to racemization at room temperature, and the racemization barriers for all of the synthesized derivatives were confirmed to be fairly low, all lying within a narrow range of 19.39 to 21.36 kcal mol−1.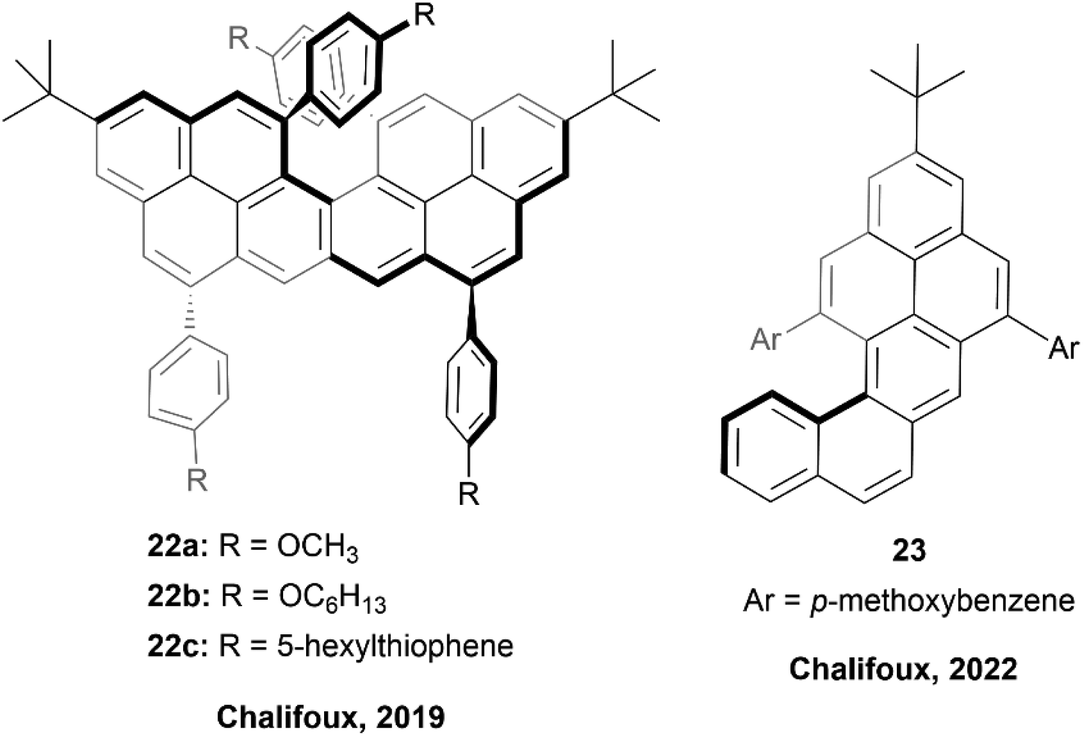 | ||
| Fig. 5 Pyreno[a]pyrene based helicene hybrid 22 and naphtho[1,2-a]pyrene 23 both synthesized by the Chalifoux group. | ||
In 2015, Juríček and coworkers presented a synthetic route to prepare cethrene, an interesting C-shaped helical analogue of the diradicaloid zethrene family (Scheme 6a). Cethrene is notably the first structure to combine the unique magnetic properties of a diradicaloid species with the optical properties of helically chiral structures.52 Obtaining the target cethrene 26 was challenging however, because upon attempting the final step by oxidizing with p-chloranil, the reactive diradicaloid cethrene underwent relatively fast rearrangement to planar structure 27. The formation and rearrangement of cethrene 26 was monitored by 1H NMR to confirm the transformation was occurring as proposed. Although 26 could not be isolated, the enantiomers of the precursor 24 were separated and analyzed by chiral HPLC. Mirror-image CD responses were recorded, and the racemization barrier was found to be approximately 25 kcal mol−1 at 25 °C, slightly higher than that of [5]helicene (24.1 kcal mol−1) at the same temperature.
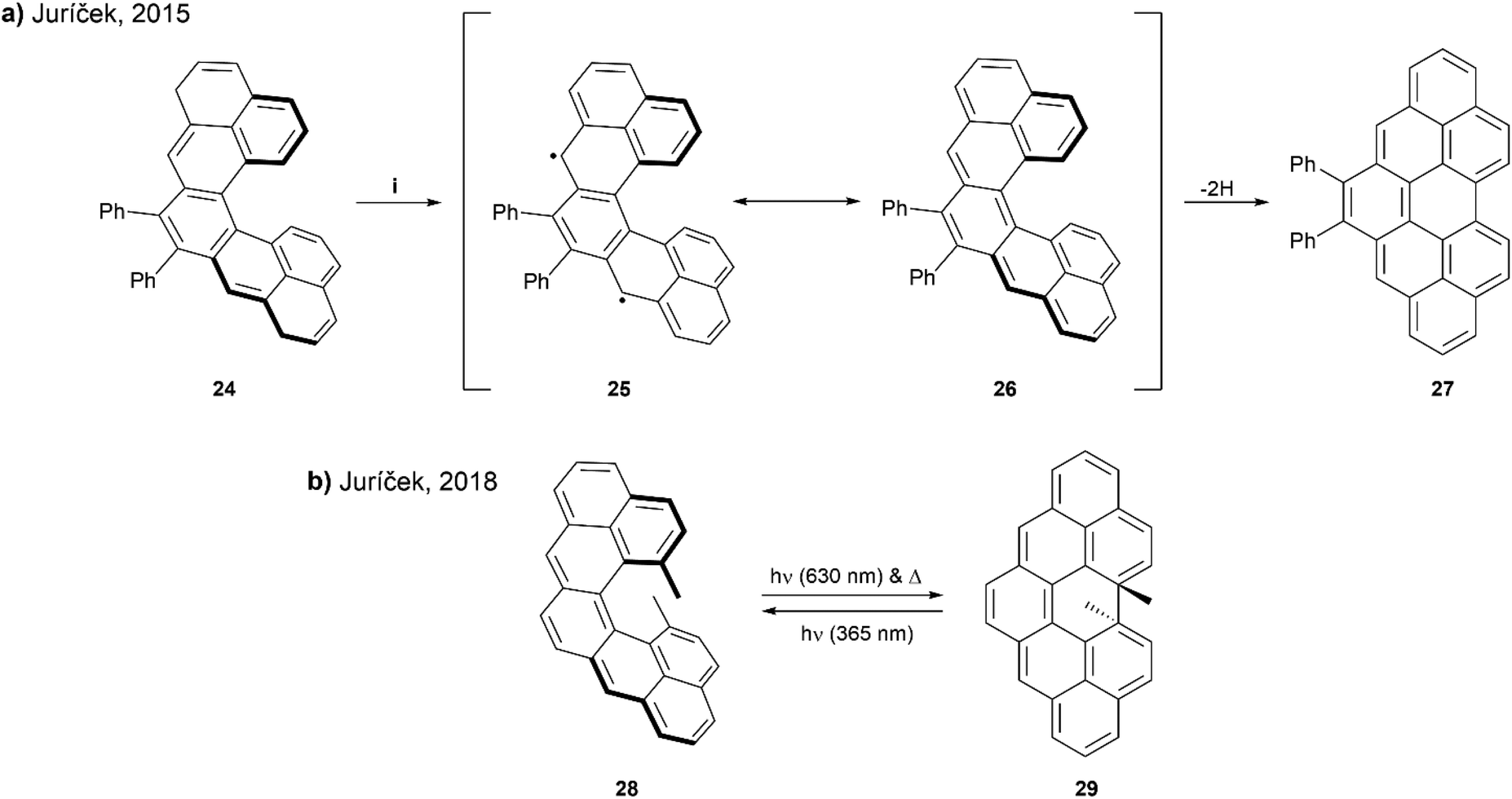 | ||
| Scheme 6 (a) Ring closure of cethrene 24; (i) p-chloroanil, DCM-d2, rt, 10 min; (b) ring closure of cethrene 28. | ||
A few years later, the Juríček group continued this research by preparing a more sterically-congested derivative, 13,14-dimethylcethrene 28 to explore the reversibility of the ring-closing transition. The sought to improve the stability of the cyclized structure 29, and increase configurational stability of the [5]helicene core (Scheme 6b).53 It was observed that the transition from open to closed (28 to 29) was reversible – the ring-closing to 29 could proceed with either light (630 nm) or heat, and the reverse ring-opening to 28 could also be induced photochemically (365 nm). This reversible transition could be monitored by several spectroscopic methods, such as NMR, UV-vis, and CD, and distinct optical and chiroptical changes could be observed due to the altered HOMO/LUMO and singlet–triplet (ST) gaps, as well as the changing degree of helicity. Enantiomers of 29 were separable by chiral HPLC, and absolute configurations were assigned with the assistance of time-dependent DFT calculations. Notably, when analyzed by electron paramagnetic resonance (EPR) spectroscopy as a way to determine the ST gap, compound 28 was found to be EPR silent at room temperature, indicating a larger ST gap than the unsubstituted analog, which was EPR active (ΔST = 6 kcal mol−1). This could largely be explained by the greater degree of helical twist in structure 28, caused by steric pressure from the methyl groups, which increases the distance (3.37 Å) between the fjord carbons as compared with the unsubstituted cethrene derivative (3.03 Å). Due to this increased distance, the level of through-space orbital overlap of frontier molecular orbitals (FMOs) is reduced, leading to an increased singlet–triplet gap.
Recently, Juríček, Ravat, and coworkers produced the synthesis of a variation on their previous work with cethrene derivatives – nonacethrene. The goal of preparing this nonacethrene structure was largely to reduce or prevent the ring-closure effect seen previously with cethrene, as well as extend the π-system in an attempt to lower the singlet/triplet gap and produce an EPR signal.54 Nonacethrene precursor 30 was prepared through a multistep synthetic process, and separated into enantiomerically-enriched samples through stationary-phase chiral HPLC, where the chiral structure of (P)-30 was unambiguously confirmed by X-ray crystal analysis (Scheme 7). When 30 was treated with p-chloroanil in an attempt to prepare the target nonacethrene structure 31, unexpected chiral side products 32, 33, and 34 formed instead, in ratios that were dependent on reaction times and the amount of oxidant added. It was proposed that these unexpected products were formed either from a radical cascade mechanism from 30, or a pericyclic reaction of 31, which ultimately react further to form 33 and 34. A number of 2D NMR studies were employed to elucidate and confirm the structures of the side products, and synthetic experiments with the enantioenriched starting materials were carried out in order to identify the configurational isomers for each species. Notably, the CPL response for the enantiomerically enriched expanded structure (P,P)-34 produced a brightness value of 8.3 M−1 cm−1, and had a moderate absorption dissymmetry factor (gabs) of 1.13 × 10−3 at 502 nm. Interestingly, 34 produced the same value for the luminescence dissymmetry factor (glum) of 1.13 × 10−3 at 520 nm. A glum/gabs ratio of 1.0 is unique amongst helicenes, and a value of ∼0.6 is more common, which is largely attributed to the flexibility of the systems. A glum/gabs value of 1.0 is likely due to the very rigid structure of 34, induced by the two sp3-locked centers.
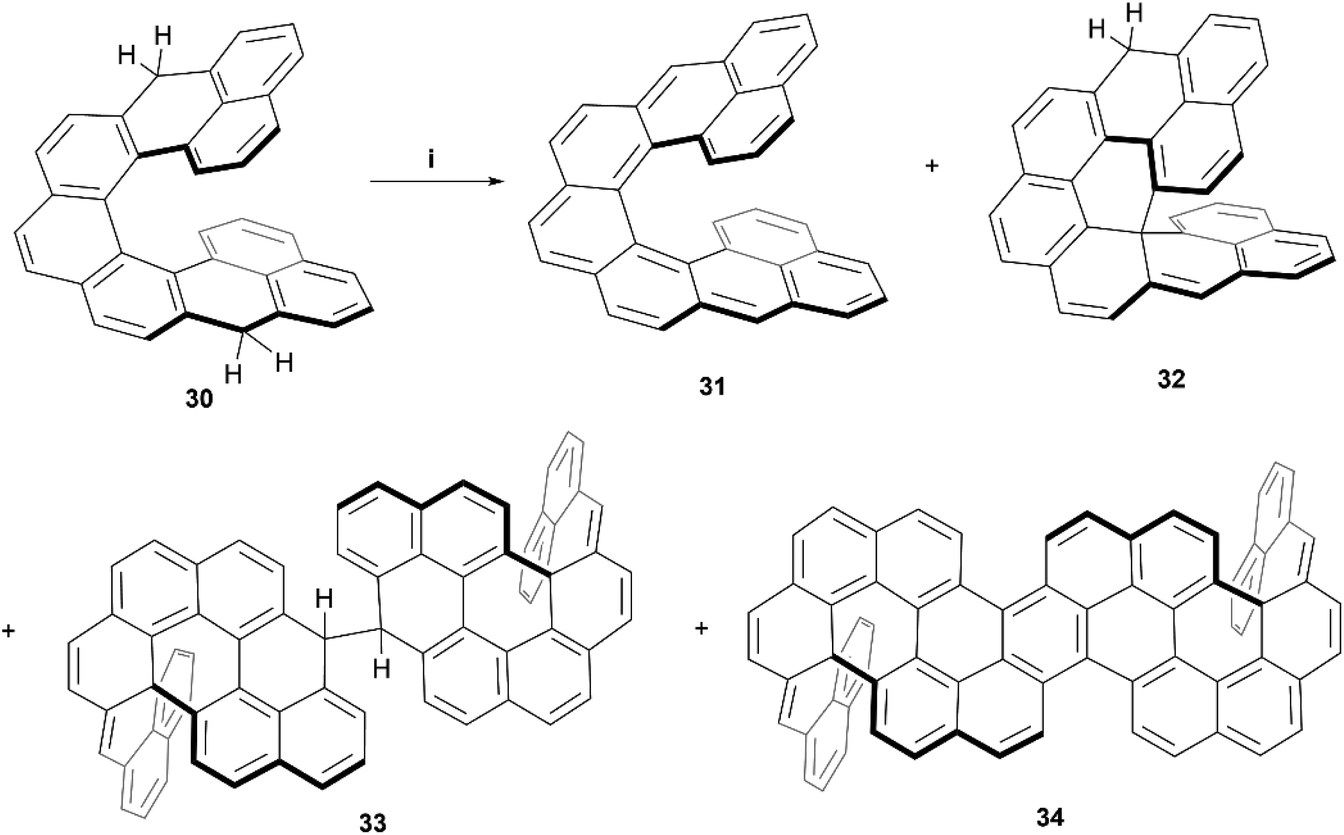 | ||
| Scheme 7 Synthesis of nonacethrene and unintended side products; (i) compound 30 (1.0 equiv.), p-chloroanil (3.5 equiv.), benzene-d6 (3.6 equiv.), rt, 16 h. | ||
In 2020, Stará and Starý produced an excellent account summarizing the use of [2 + 2 + 2] alkyne cyclizations in the preparation of extended helicenes.55 Being a highly exergonic process, this method is ideal in the synthesis of strained systems. One particular work of interest from this group was the synthesis of a pyrene-based dibenzo[7]helicene 36 through Ni0/PCy3-catalysed [2 + 2 + 2] cycloisomerisation of a carefully designed triyne precursor 35 (Scheme 8).56 Enantiomeric resolution of the racemic 36 was possible via liquid chromatography on a chiral column, and these products were found to exhibit fluorescence quantum yields of 10%, and displayed a large Stokes shift (296 nm). Exploring this process even further, a diastereoselective synthetic method was employed by incorporating a chiral moiety into the precursor 38, which resulted in the formation of diastereomer 39, of M helicity, with good regioselectivity. Interestingly, the crystal structure of this product 39 showed a significantly more flattened structure than compound 36, with the pyrene units bowing towards each other more significantly than DFT calculations would suggest, which was largely attributed to significant intramolecular forces within the system. Like 36, structure 38 also exhibited notable fluorescence (ΦF = 0.17), and a large Stokes shift (203 nm), and both structures seem to form remarkably stabilized intramolecular excimer states upon excitation.
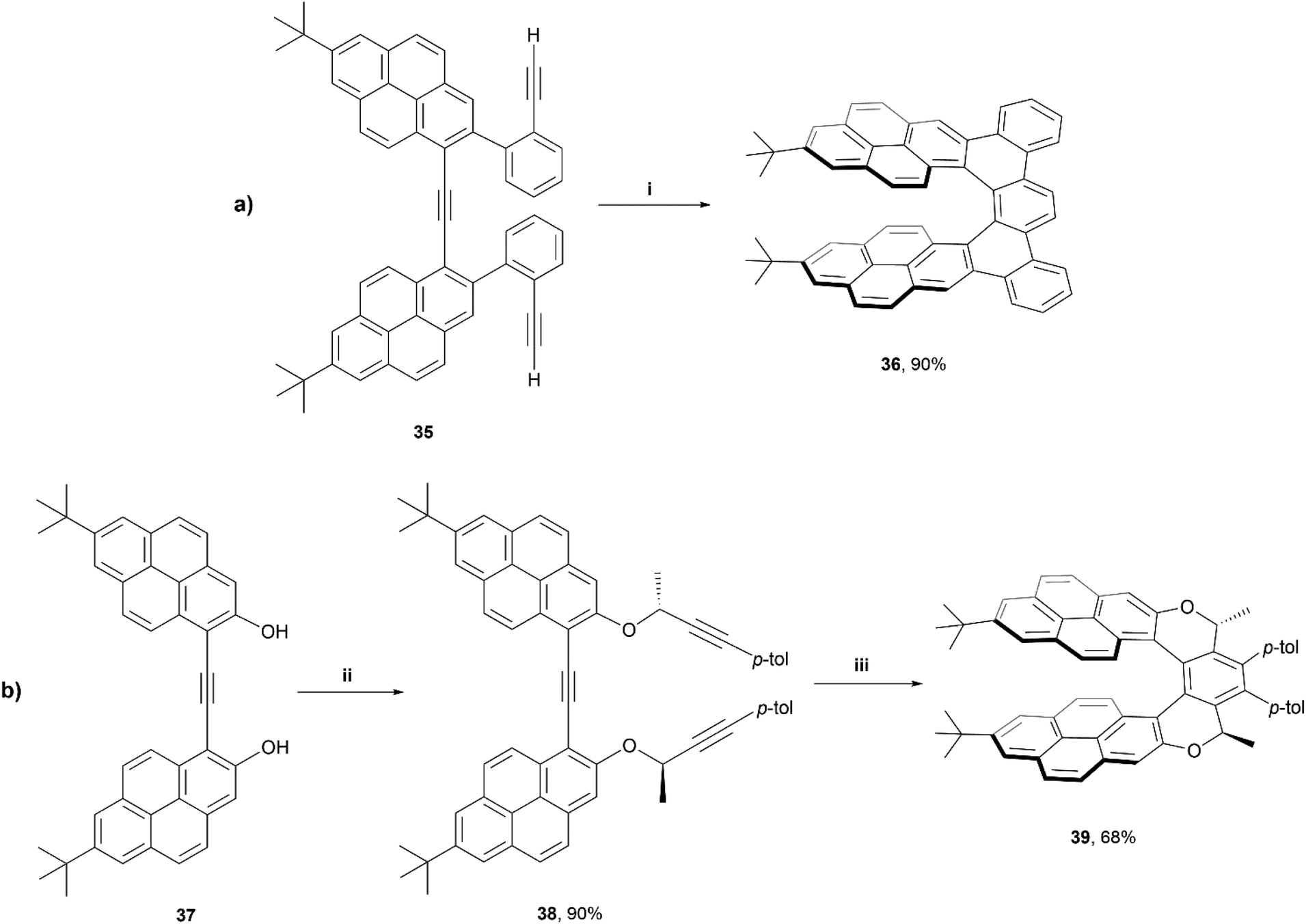 | ||
| Scheme 8 (a) Synthesis of expanded helicene 36; (i) 35 (1.0 equiv.), Ni(cod)2 (0.2 equiv.), PCy3 (0.5 equiv.), THF, rt, 24 h; (b) synthesis of expanded helicene 39; (ii) 37 (1.0 equiv.), (S)-4-(4-methylphenyl)-3-butyn-2-ol (2.4 equiv.), diisopropyl azodicarboxylate (2.4 equiv.), PPh3 (2.2 equiv.), benzene, rt, 4.5 h; (iii) 38 (1.0 equiv.), [CpCo(CO)(fum)] (1.0 equiv.), 1-butyl-2,3-dimethylimidazolium tetrafluoroborate, THF, microwave, 150 °C, 10 min. | ||
Very recently, Tanaka and coworkers reported a regiodivergent synthesis of several hexabenzocoronene (HBC)-based π-extended helicene derivatives through rhodium-catalyzed intramolecular trimerization, followed by Scholl oxidation.57 A wide range of compounds were targeted – [5]helicene, [6]helicene, [7] helicene, and double [6]helicene derivatives were all produced in varying yields throughout the synthetic process, with only a few pictured below (Scheme 9). Further Scholl oxidation of these products resulted in another wide array of helical derivatives. The enantioselective rhodium-catalyzed [2 + 2 + 2] addition, utilizing a chiral ligand ((S)-segphos), produced (−)-41 in moderate yields, with moderate-to-good enantiomeric ratios (highest was 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 for 41) along with some small yields of the racemic [2 + 1 + 2 + 1] cycloaddition products (pentabenzoanthracenes). Derivatives 41 and 42 displayed very similar fluorescence quantum yield values (8% at 330 and 340 nm, respectively), and 41 had a moderate absorption dissymmetry ratio (gabs) value of 1.2 × 10−3 at 381 nm, and a specific optical rotation value of −354°. After Scholl oxidation of 41, compound 43 displayed an increase in quantum yield (12% at 450 nm) and specific optical rotation (−1144°), while the gabs values remained almost the same (1.1 × 10−3 at 431 nm). Notably, the final HBC-based [6]helicene derivatives 43 and 44 displayed π-stacking-induced chiral self-recognition behaviour in solution, which allowed for measurement of enantiomeric ratios by 1H NMR spectroscopy without the need for any chiral additives.
:9 for 41) along with some small yields of the racemic [2 + 1 + 2 + 1] cycloaddition products (pentabenzoanthracenes). Derivatives 41 and 42 displayed very similar fluorescence quantum yield values (8% at 330 and 340 nm, respectively), and 41 had a moderate absorption dissymmetry ratio (gabs) value of 1.2 × 10−3 at 381 nm, and a specific optical rotation value of −354°. After Scholl oxidation of 41, compound 43 displayed an increase in quantum yield (12% at 450 nm) and specific optical rotation (−1144°), while the gabs values remained almost the same (1.1 × 10−3 at 431 nm). Notably, the final HBC-based [6]helicene derivatives 43 and 44 displayed π-stacking-induced chiral self-recognition behaviour in solution, which allowed for measurement of enantiomeric ratios by 1H NMR spectroscopy without the need for any chiral additives.
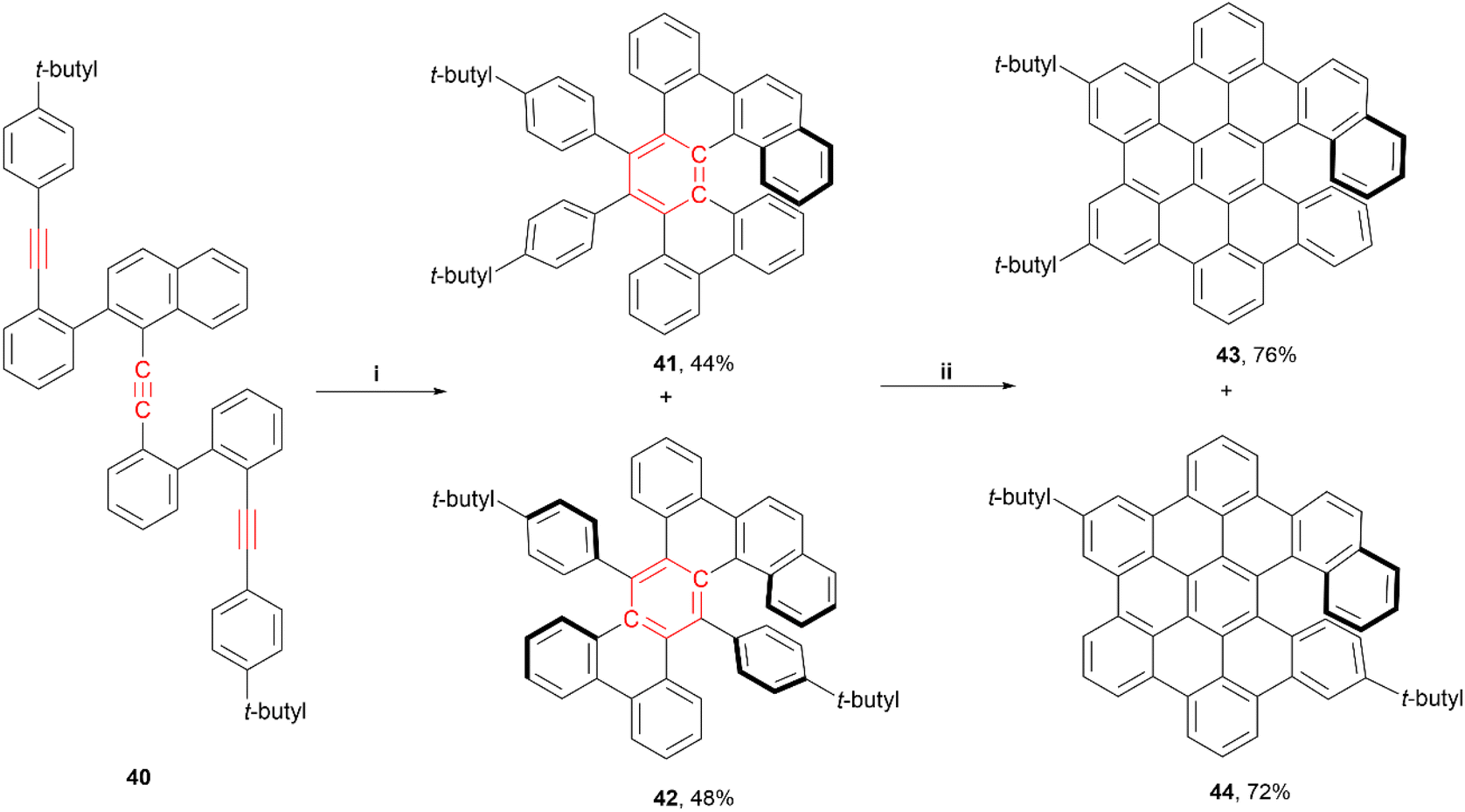 | ||
| Scheme 9 (i) (S)-Segphos (0.4 equiv.) in DCM, [Rh(cod)2]BF4 (0.4 equiv.) in DCM, rt, 1 h, then 40 (1.0 equiv.) in DCE, 80 °C, 40 h; (ii) 41 or 42 (1.0 equiv.), DDQ (4.5 equiv.) in DCM, TfOH, 0 °C, 1 h. | ||
π-Extended heterohelicenes
In recent years, Cauteruccio et al. synthesized and studied the properties of a series of functionalized thiahelicenes through two approaches, starting from the same precursors. Structures 45a–c were produced by a two-step synthesis, involving Suzuki coupling followed by a Mallory-like reaction to produce benzo-fused thiahelicene derivatives. A different procedure, one-step palladium-catalyzed alkyne annulation, was used to produce a wide variety of simple thiahelicenes, as well as the more complex pyrene-containing structure 46 (Fig. 6).58 Enantiomers of the [6]helicene structures 45a–c were able to be resolved easily via chiral HPLC, with some separated into peaks with enantiomeric purities as high as 96%. Structure 46, with a smaller [5]helicene moiety, was too configurationally unstable to be separated in the same way, but the helical chiral structure, and the presence of two enantiomers, was unambiguously confirmed through X-ray crystallography. The racemization energy barriers were calculated and ranged from low/moderate values for structures 45a–c, where 45c was the highest (34 kcal mol−1), 45b was the lowest (26 kcal mol−1), and 45a was in the middle (29 kcal mol−1). As expected, the barrier for 46 was low, at around 18 kcal mol−1.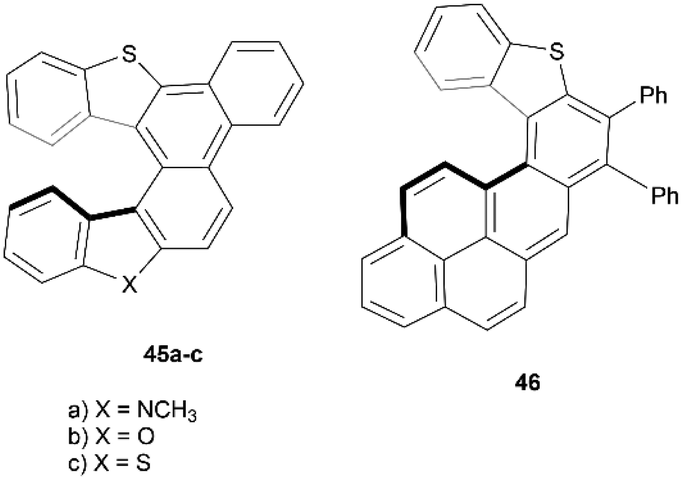 | ||
| Fig. 6 Functionalized, expanded thiahelicenes 45 and 46 synthesized by Cauteruccio and coworkers. | ||
A synthesis of dithia-quasi[8]circulene was reported recently by Avarvari, Pop, and coworkers, by first preparing dithia[7]helicene 49 through a three-step synthesis, followed by oxidative dehydrocyclization to cyclize into final product 48 (Scheme 10).59 The warped and nonplanar configuration of the structures was determined through single crystal analysis of dithia[7]helicene 49 and of quasi[8]circulene 48. The sum of the internal torsion angles of 49 add up to approximately 76°, indicating a higher degree of twisting than for the all-carbon analogues of [5]helicene, which have torsion angle sums ranging from 56.9 to 61.9°. Quasi[8]circulene 48 displays an even more dramatic total degree of twisting of around 101.95°. Structure 49 displays one of the highest degrees of stereochemical stability for its kind (simple dihetero- and trihetero-[7]helicenes), with an experimentally-determined enantiomerization barrier of 36.9 kcal mol−1. Structure 48 also claims a high degree of stereochemical stability, with an experimental value of 30.4 kcal mol−1 – one of the largest values amongst other unsubstituted quasi-circulenes. Separation of enantiomers was successful for both products by chiral HPLC on chiral stationary phases, and CD analysis indicated that the two species 48 and 49 had similar CD intensities despite their structural differences.
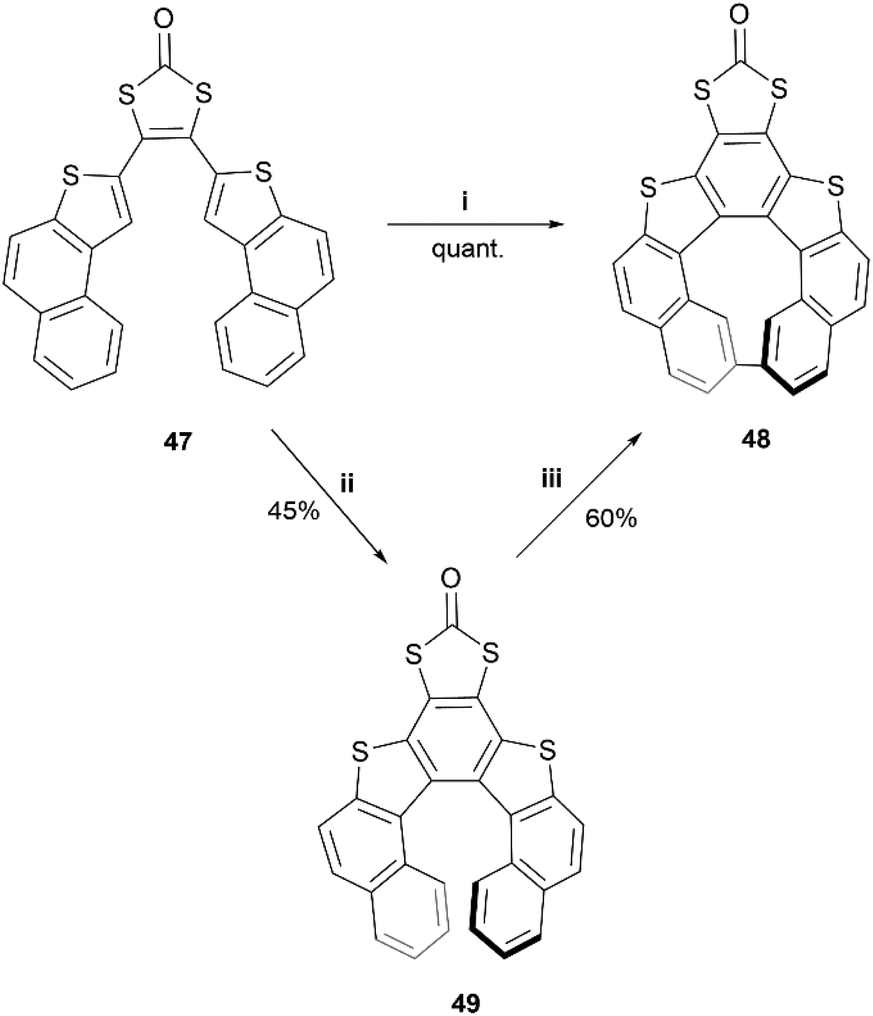 | ||
| Scheme 10 Synthesis of dithia-quasi[8]circulene compounds; (i) 47 (1.0 equiv.) in CHCl3, FeCl3 (10.0 equiv.) in MeNO2, rt, 1 h; (ii) 47 (1.0 equiv.) in DCM, PIFA (1.2 equiv.), BF3·Et2O (2.4 equiv.), DCM, −78 °C, 1 h; (iii) 48 (1.0 equiv.) in toluene, DDQ (5.0 equiv.), scandium triflate (5.0 equiv.), rt to 100 °C, 24–44 h. | ||
Recently, the Chalifoux group produced the synthesis of a chiral dipyrenoheterole derivative through Suzuki coupling followed by an InCl3/AgNTf2 mediated fourfold alkyne benzannulation, resulting in compounds 51a–c (Scheme 11).60 The enantiomers were separated by HPLC on a semipreparative chiral column, and after analysis, displayed CD and CPL values comparable to other chiral, highly contorted organic structures, with moderate gabs values ranging from 8 × 10−4 (479 nm) at the lowest to 5.1 × 10−3 (351 nm) at the highest, and glum values of 1.1 × 10−3 (from 480 to 620 nm) for compound 51a and 8 × 10−4 (from 480 to 620 nm) for compound 51b.
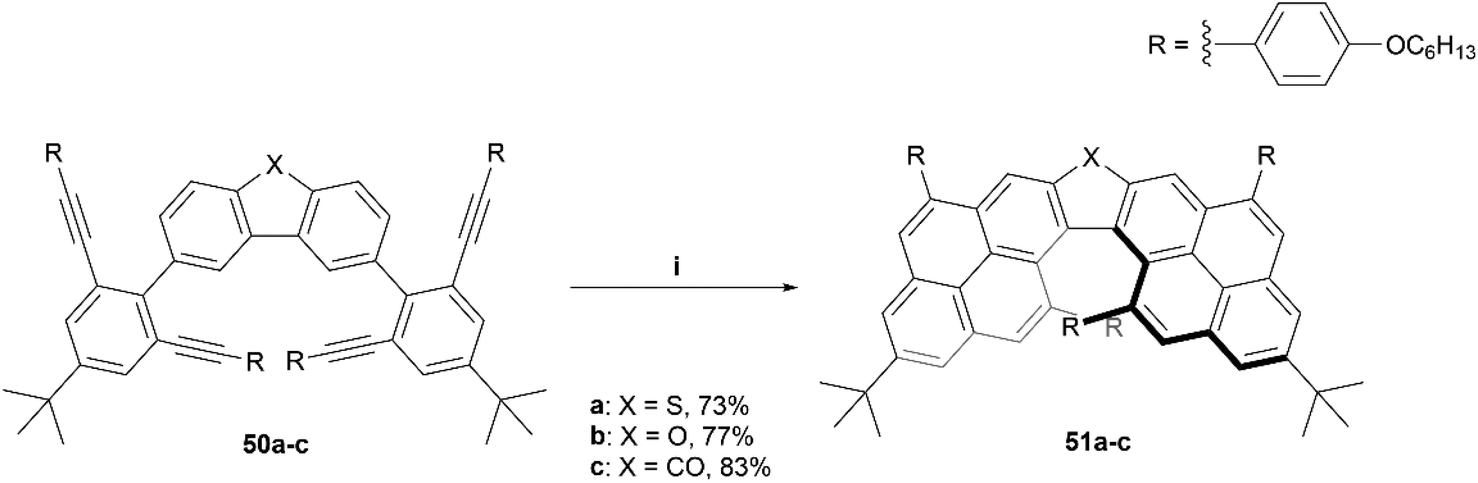 | ||
| Scheme 11 Synthesis of chiral dipyrenoheteroles. 51; (i) 50 (1.0 equiv.), InCl3 (0.4 equiv.), AgNTf2 (0.5 equiv.), toluene, 100 °C, 1–25 h. | ||
In 2012, Hatakeyama, Nakamura, and coworkers produced a new semiconductor material possessing helical chirality, synthesized through borylation of a diarylamine, followed by a bora-Friedel–Crafts type cyclization, resulting in helical structure 52 (Fig. 7).61 The helical structure was confirmed through X-ray crystallography, and a unique packing structure was revealed in the racemic crystal, in which single enantiomers pack in a head-to-tail fashion with fairly small intermolecular distances (CH–π distances of 2.9–3.3 Å), with the opposite-handed stacks lining up with each other in an alternating fashion. Resolution of (P)- and (M)-52 enantiomers was successful via chiral HPLC, and the enantiopurity of these samples remained unchanged even after heating to 275 °C, displaying excellent thermal configurational stability. Interestingly, the crystal packing structure of the single enantiomers was observed to be significantly different than in the racemic crystal, with the enantiomers arranging in a more face-to-face parallel columnar arrangement instead of head-to-tail. Both the homochiral and heterochiral (racemic) 52 possessed interesting carrier-transport properties, with rac-52 displaying high hole mobility (μh = 4.6 × 10−4 cm2 V−1 s−1), but no notable electron mobility, whereas (P)-52 displays higher electron mobility (μe = 4.5 × 10−3 cm2 V−1 s−1) than hole mobility (μh = 7.9 × 10−4 cm2 V−1 s−1). These results suggest that rac-52 and homochiral 52 are good p- and n-type semiconductors, respectively, and the differences in charge-transport properties could largely be explained by the unique crystal packing differences between the single enantiomer and the racemate.
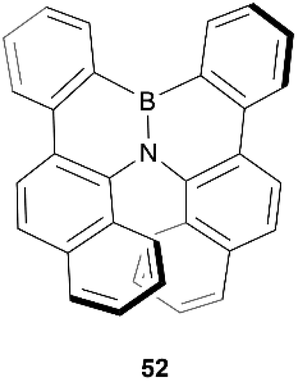 | ||
| Fig. 7 Azaboradibenzo[6]helicene 52 synthesized by Hatakeyama, Nakamura and coworkers. | ||
In the last few years, Alcarazo and coworkers have explored the use of gold-catalysts for enantioselective synthesis of carbohelicenes and other chiral structures.62–65 Recently, this method was expanded by Cauteruccio and Alcarazo and applied to thiophene-based structures, and a series of dithia[5]helicene derivatives 54 were produced through highly enantioselective gold-catalyzed alkyne hydroarylation of alkyne precursors 53 (Scheme 12).66 The enantioselectivity of the transformation from 53 to 54 arises from the use of a specific chiral gold complex catalyst, which has previously been shown to induce excellent selectivity in the hydroarylation of alkynes.63 HPLC analysis showed that the enantioselectivity of the first trial was excellent (92% ee), and a very wide scope of 20 alkyne derivatives of 53, possessing different substituents, were screened, resulting in varying moderate-to-excellent yields (53–98%) and varying enantioselective success (33–98% ee). The general trends seemed to show that electronics of the substituents seemed to have little effect on enantioselectivity, but that placing substituents in the meta- or ortho- positions of the terminal phenyl rings significantly reduced the enantioselectivity of the reaction. The synthesis was then further expanded by testing the directing-group effect of the thienyl groups of 54 on substitution steps. When 54 (R = 4-methylphenyl) was exposed to N-bromosuccinimide (NBS), bromine substitutions occurred in the positions ortho to the R-groups, allowing for Suzuki coupling of phenyl groups, followed by Scholl cyclization, resulting in the final dithia[9]helicene 55 (Scheme 12). 55 was recovered in fairly low yields (28–29%) when produced from both rac-54 and enantiopure 54, and no erosion of enantiopurity was observed throughout the sequence when starting with enantiopure 54. The yield of 55 was found to be improved by utilizing an excess of iron(III) chloride as the oxidant in the Scholl step but resulted in monochlorination of the helicene backbone.
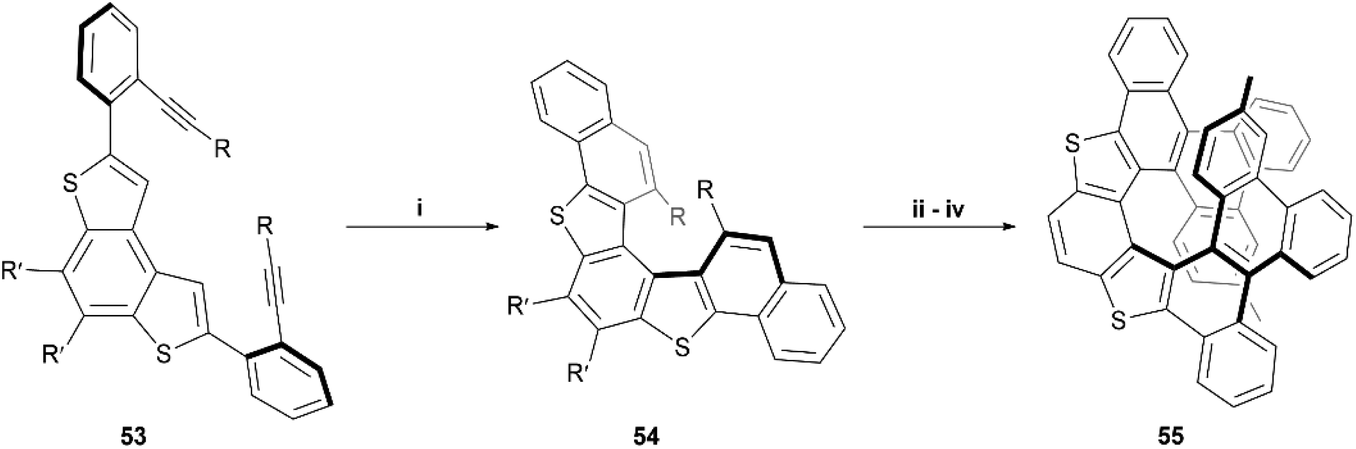 | ||
| Scheme 12 Synthesis of dithia[5]helicene derivatives; (i) 53 (1.0 equiv.), Au2+ catalyst (0.05 equiv.), AgSbF6 (0.05 equiv.), DCM, −20 °C, 48 h; (ii) 54 (1.0 equiv.), NBS (3.0 equiv.), rt, CHCl3; (iii) PhB(OH)2 (4.0 equiv.), Pd(dppf)Cl2 (0.1 equiv.), KF (4.0 equiv.), toluene/MeOH (1:1), 70 °C, 9 h; (iv) DDQ (5.0 equiv.), MeSO3H, DCM, 0 °C. 20 derivatives of 54, with different R- and R′-substituents, were prepared – see the cited publication for specifics. | ||
In 2020, Mateo-Alonso and coworkers reported a straightforward three-step synthesis of three sterically congested, nitrogenated benzodipentaphenes containing expanded helicene moieties (Fig. 8).67 One structure was planar, but two contorted products 56 and 57 were recovered in low yields (11% and 3%, respectively). The particularly low yield of 57 can largely be explained by the remarkably sterically crowded overlap of two pairs of triisopropylsilyl (TIPS) acetylene groups. Single crystals of compound 57 were obtained, and a highly warped double expanded helicene structure was confirmed in the regions of TIPS-acetylene overlap, but 57 appeared to be present only as the achiral meso (P/M) isomer. Crystals of 56 were not obtained, but optimized calculations indicated that the structure has the appearance of a planar structure along the Z-shaped section, and twists out of planarity along the U-shaped helicene unit. The confirmational stability of the meso structure of 57 appeared to be high, with no observable change in the 1H NMR spectrum after heating to 120 °C. Interestingly, 56 and 57 both had remarkably high fluorescence quantum yields (70% and 73%, respectively), which is in contrast with previous observations of twisted structures, where a greater degree of warping of the aromatic system tends to decrease the fluorescence quantum yield.
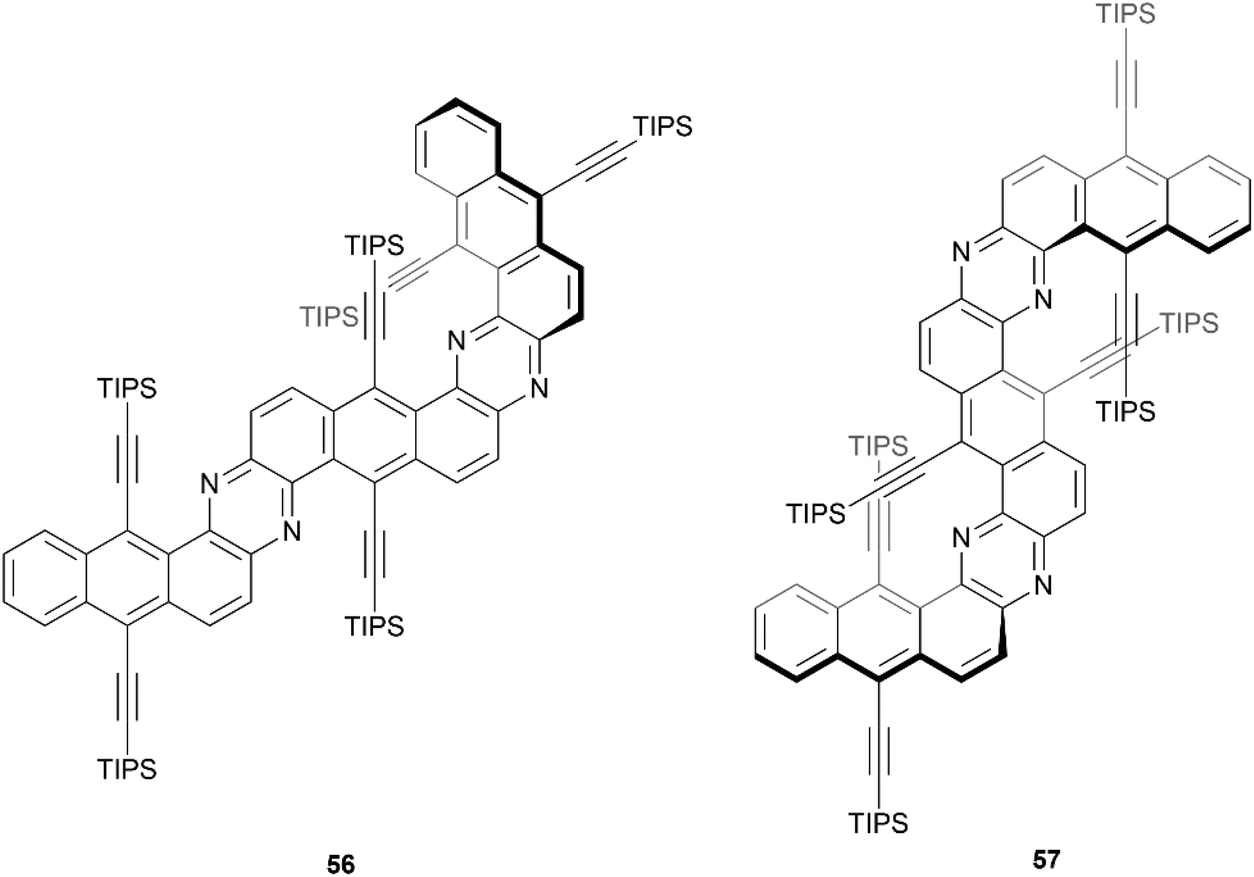 | ||
| Fig. 8 π-expanded helicenoid 56 and double π-expanded helicenoid 57 synthesized by Mateo-Alonso and coworkers. | ||
Recently, Gulevskaya and coworkers reported a five-step synthesis of novel carbazole-based helicenes, fused with an azine ring (Fig. 9).68 The key synthetic steps include Suzuki–Miyaura and Sonogashira couplings, ICl-mediated alkyne cyclization, followed by an additional Sonogashira coupling, and an acid-induced alkyne cyclization, resulting in derivatives 58a–c. All derivatives were configurationally stable enough for HPLC separation of (P) and (M) enantiomers, and the chiral, nonplanar structures were also confirmed by X-ray analysis. It was noted that the crystal packing structure of racemic 58c was unique, where alternating enantiomers stacked in a helical, corkscrew fashion. X-ray analysis also indicated that the interplanar angle between the two terminal rings for compound 58c (62°) was slightly higher than that of the parent carbazole-[6]helicene (without the azine ring moiety) (56.7°), which was largely attributed to the slight steric strain induced by the C–H group that would be absent from the parent helicene. Interestingly, the other derivatives 58a and 58b showed an opposite trend, where the interplanar angles were smaller (50.5° and 52.8°, respectively) than the parent structure, which was largely attributed to attractive interactions of the nitrogen groups of the azine ring with the neighboring hydrogens of the helicene.
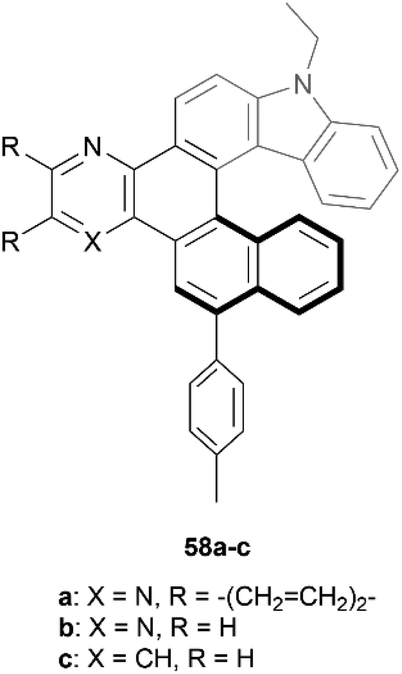 | ||
| Fig. 9 Carbazole-based [6]helicene fused with an azine ring 58 synthesized by Gulevskaya and coworkers. | ||
Imide-based chiral nanographenes
One particular class of heteronanographenes are structures containing imide moieties. Imide-functionalized nanographenes, particularly perylene diimide (PDI) based structures, have been extensively explored for their impressive optical and electronic properties. Combining the properties of imides with the chiroptical properties of chiral molecules provides exciting opportunities for unique behavior and applications. Early strides in this field were made by the Würthner69,70 and Müllen71 groups, and recently this class of structure has been thoroughly reviewed by Ravat and Saal,72 and Yin, Guan, and coworkers.73 Here we will highlight a few notable works.Recently, the Lin research group produced the synthesis of a series of S- and C-shaped substituted PDI-based helicenes, 59 and 60, respectively (Fig. 10).74 Single crystals of both structures were obtained as racemic mixtures, confirming the chiral nature of the structures. The enantiomers for the C-shaped structure 60 were separable by chiral HPLC, and appeared to be remarkably conformationally stable, with no observable changes in the CD spectra after heating an enantiopure sample to 250 °C. In contrast, enantiomers of the S-shaped analogue 59 were not separable by HPLC, indicating that the racemization barrier for these is too low for separation. Interestingly, the crystal structure of one of the enantiomerically pure 60 derivatives was observed to adopt a unique spiral-staircase-like π-stacking motif rather than the usual face-to-face stacking that is typical in most helical structures.
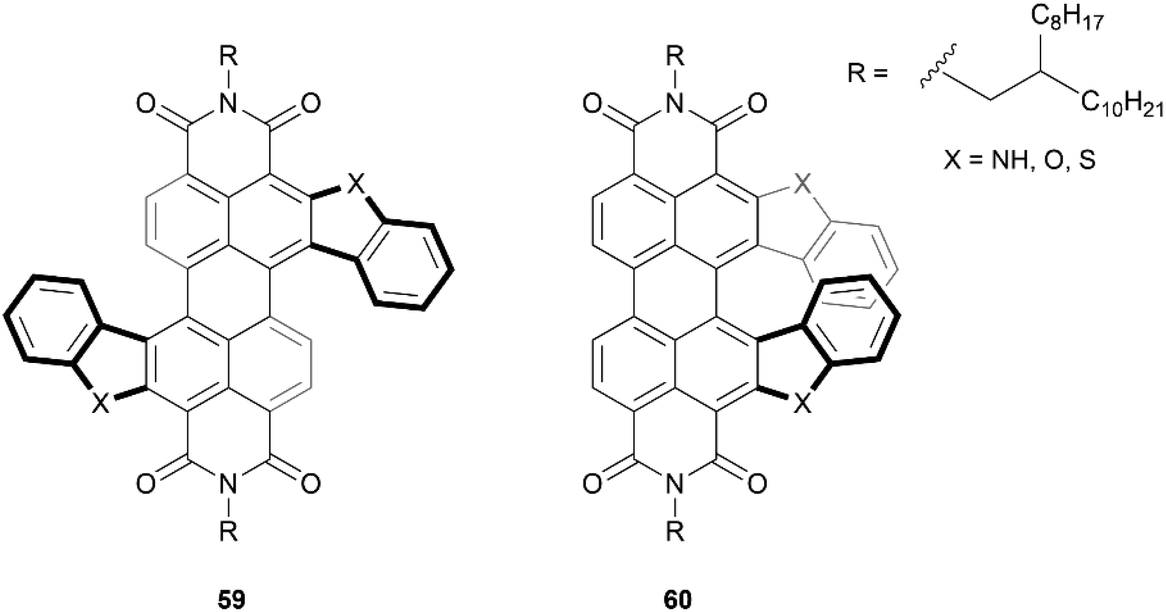 | ||
| Fig. 10 S-shaped PDI double-heterohelicene 59 and C-shaped PDI heterohelicene 60 synthesized by Lin and coworkers. | ||
In 2019, Wang et al. produced a novel nonplanar nanographene 61, which they aptly named corannurylene pentapetalae, inspired by the five-fold flower-like appearance of the final structure (Fig. 11). This unique structure was synthesized through a two-step process – a five-fold Suzuki–Miyaura coupling of brominated PDI units to a corannulene core, followed by photocyclic cycloaromatization to form the final flower-like structure.75 The complexity of the structure could lead to eight possible stereoisomers, or four pairs of enantiomers. Two pairs of these possible enantiomers were successfully separated through HPLC, the (P,P,P,P,P)- and (M,M,M,M,M)-61a enantiomers, in which all PDI units are arranged in a symmetric up-and-down pattern, and (P,P,P,P,M)- and (M,M,M,M,P)-61b, where three PDI units are arranged in the same up-and-down fashion, but one unit is completely up and a neighboring unit is completely down. Single crystals of the racemic mixtures of these compounds were grown, and their structures unambiguously confirmed by X-ray crystallography. The energy difference between the two stereoisomers is notable, where 61b is higher in energy than 61a by 5.53 kcal mol−1, but after attempts to convert 61b to the lower-energy 61a (heating to 250 °C for 6 hours), there was no observable change in the 1H NMR spectra, indicating excellent configurational stability.
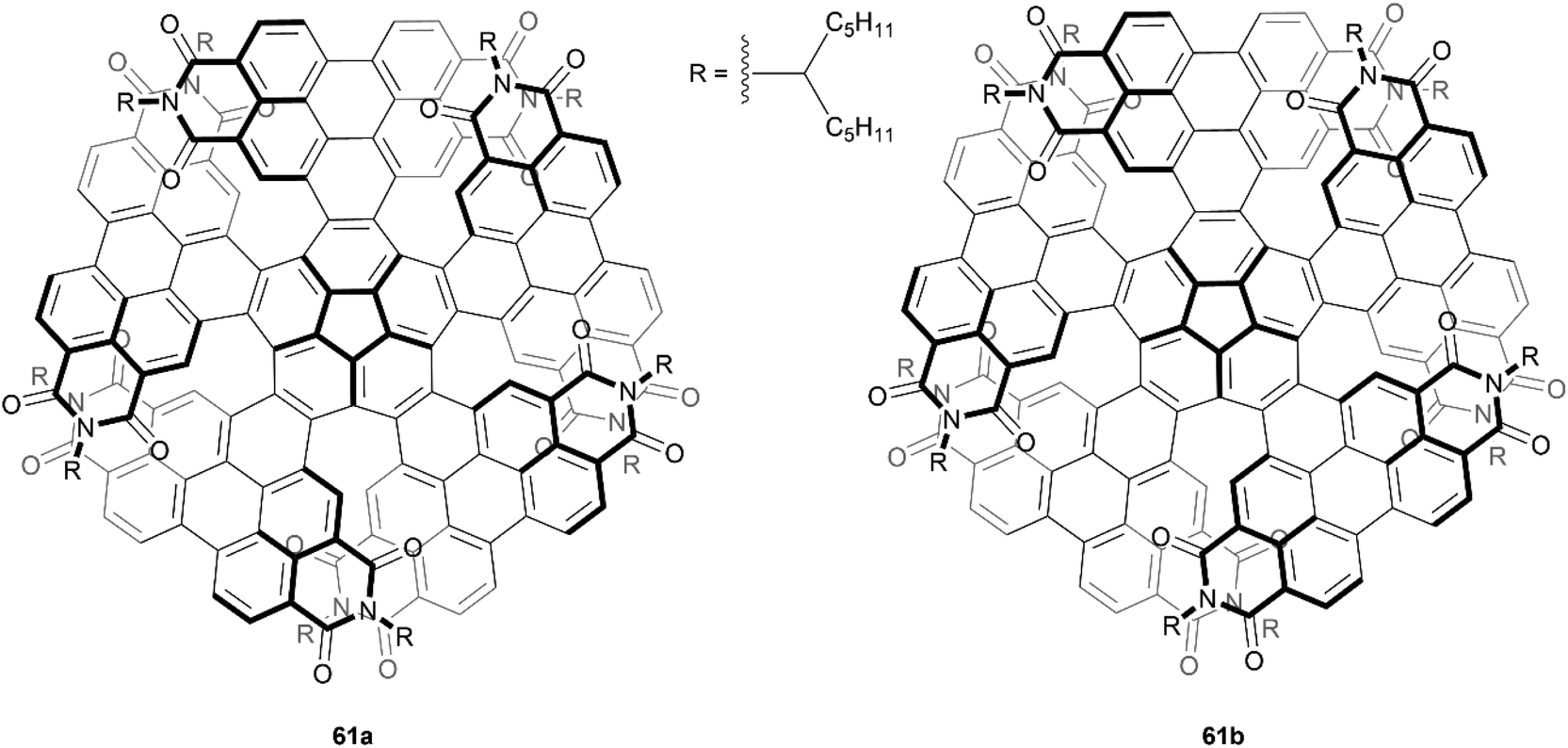 | ||
| Fig. 11 Corannurylene pentapetalae 61 stereoisomers, synthesized by Wang and coworkers. | ||
An even larger and similarly complex chiral PDI-based structure was recently synthesized by Wang, Li, and Liu. This structure was built with an impressive scaffold of 204 carbons and 9 PDI subunits, and also contains twelve [5]-helicene units, centered around four core positions (Fig. 12).76 The synthetic strategy for preparation involved a series of Suzuki–Miyaura couplings and ring-closing photocyclic aromatization steps, and the final structure 62 displayed excellent solubility and air stability. Although there were 256 possible stereoisomers (or 128 possible pairs of enantiomers) that could have formed from the many overlapping PDI units, theoretical calculations and extensive NMR experiments seemed to suggest that only one pair was actually produced, specifically the (3M,3M,3M,3M) and (3P,3P,3P,3P) enantiomers (where 3M = MMM and 3P = PPP). These enantiomers were separable by chiral HPLC, and the structures were found to have a large degree of enantiomeric stability, with no racemization or configurational inversion observed even when heated to 200 °C. Notably, when the CD properties were studied, it was observed that the intensity of the peak at 256 nm produced by 62 was approximately 6 times as intense as the peak produced by the smaller analogue 63, which had previously been synthesized by the same group in 2016, and has since been widely reviewed.77
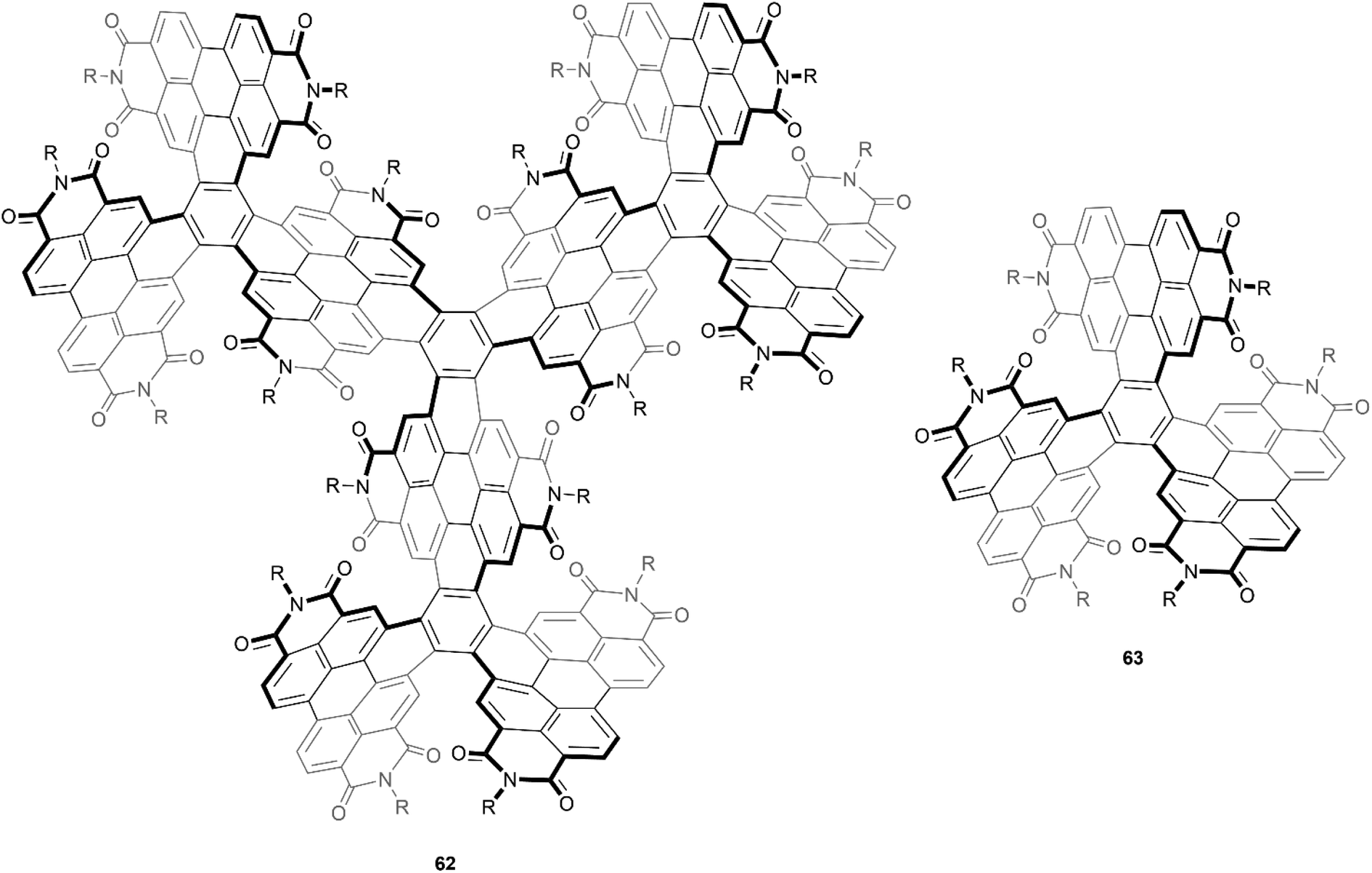 | ||
| Fig. 12 Large, complex chiral PDI-based compound 62, and smaller analogue 63, both synthesized by Wang and coworkers. | ||
Stępień and coworkers previously reported a successful synthesis of a hexapyrrolohexaazacoronene (HPHAC) derivative through FeCl3 oxidative cyclization of a hexapyrrolylbenzene precursor, producing a nonplanar but achiral final structure.78 However, the same group later discovered a more stereo-controlled method of preparing a similar structure by utilizing more sterically hindered bay positions. It was noted that this new steric strain could be synthetically overcome by utilizing bromine electrophiles rather than more high-potential oxidants.79 Interestingly, it was observed that by using different bromine electrophiles, the orientation of the “fan blades” was different, leading to a wide scope of different diastereomers. When N-bromosuccinimide (NBS) was the oxidant, three neatly symmetric derivatives of 64a were recovered in good yields (85–88%). But the use of bromine (Br2) in place of NBS produced an interesting change in stereoselectivity, and one derivative of the less symmetric stereoisomer 64b was the major product (86%) (Fig. 13). The enantiomers for all derivatives of 64a were separable by chiral HPLC, and CD analysis of the purified enantiomers displayed high Δε values (up to ca. 800 m−1 cm−1) and significant anisotropy factors g (up to 7 × 10−3). All recovered enantiomers displayed good thermal stability, with no sign of configurational changes when heated to 100 °C in toluene for 1 h. Separation of the enantiomers of 64b was less successful, but g-normalized CD spectra of scalemic samples suggested that the maximum Δε values are reduced by a factor of 2/3 as compared to 64a, which was suggested to indicate that the contributions of individual helicene moieties may be somewhat additive to the CD spectrum for the whole molecule. The less symmetric 64b showed no evidence of converting to the more symmetric 64a when heated to 120 °C, despite DFT calculations indicating that it is the higher-energy species by 7.2 kcal mol−1, indicating an impressive degree of configurational stability.
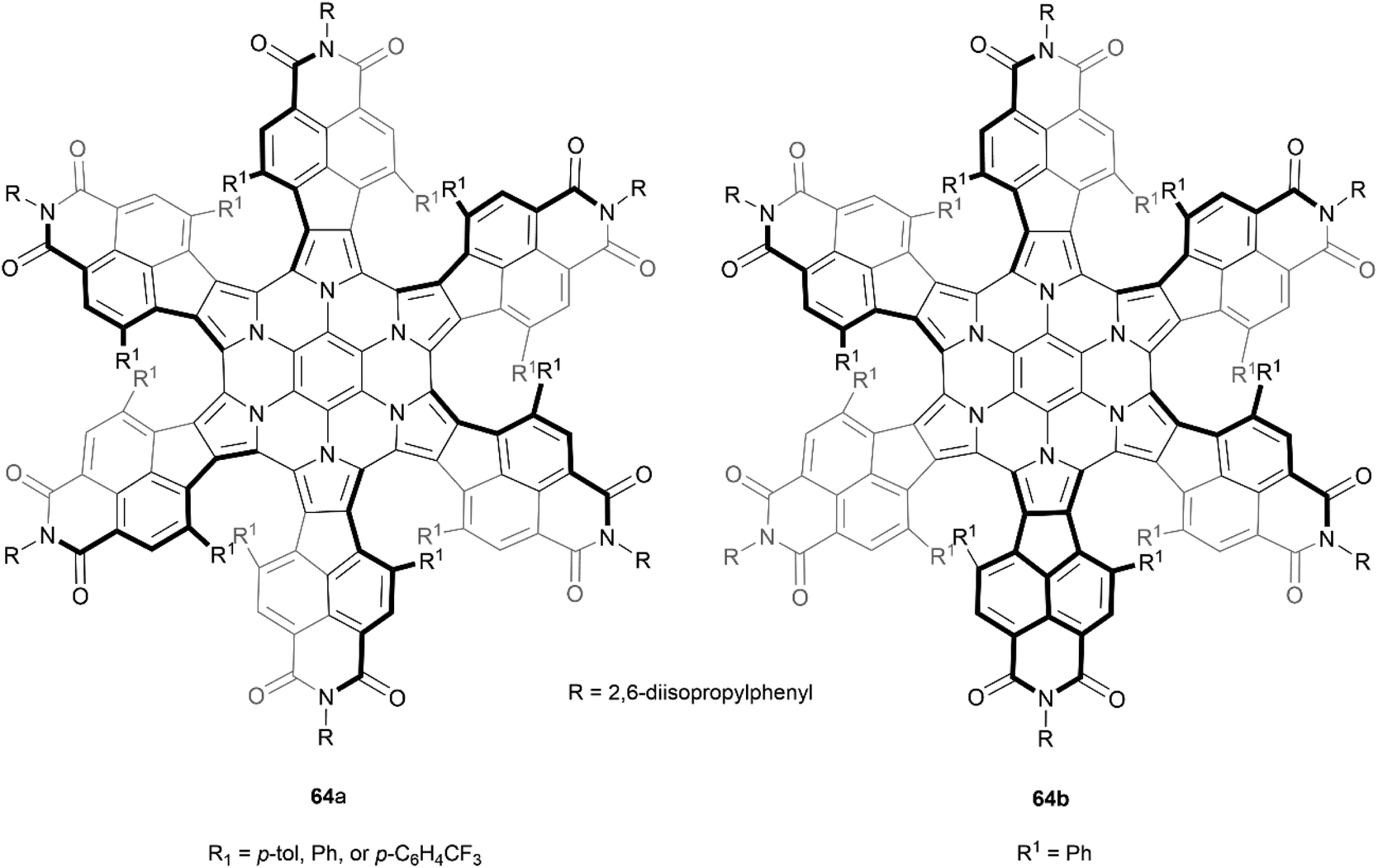 | ||
| Fig. 13 Recovered configurational isomers of azacoronene propeller 64, synthesized by Stępień and coworkers. | ||
In 2020, Wang, Jiang, and coworkers reported a synthetic approach towards PDI-based double-helicene hybrid structures with six possible stereoisomers – two pairs of enantiomers, and two mesomers.80 The synthetic method involves Suzuki–Miyaura coupling of borylated [6]helicene 65 with varying brominated PDI cores (66–68), followed by iodine-mediated photocyclization, to produce the final products: single helicene 69, and double helicenes 70 and 71 (Scheme 13). Notably, three total stereoisomers of both 70 and 71 were produced through this method – two racemic mixtures of enantiomers (rac-70 and rac-71), and two achiral meso-forms (meso-70 and meso-71). The meso and racemic mixtures were separable as intermediates, before the final cyclization step (step ii – Scheme 13), allowing for cyclization of separate species and accounting for the high final yields of rac-71 and meso-71. The crystal structures for each conformation of 70 and 71 (both the racemic and meso structures) showed unique packing arrangements due to the subtle configurational differences of the helicene “wings”. The racemic mixtures of 70 and 71 were separated successfully into their respective enantiomeric pairs. Notably, the enantiomers of 70 displayed more significant chiroptical responses than the those of 71, with relatively large dissymmetry factors of |gabs| of 1.2 × 10−2 and a |glum| of 2 × 10−3, which was largely attributed to the superhelicene-like shape of 70. Additionally, due to the PDI moieties, all the final structures exhibited outstanding quantum fluorescence yields between 29–34%.
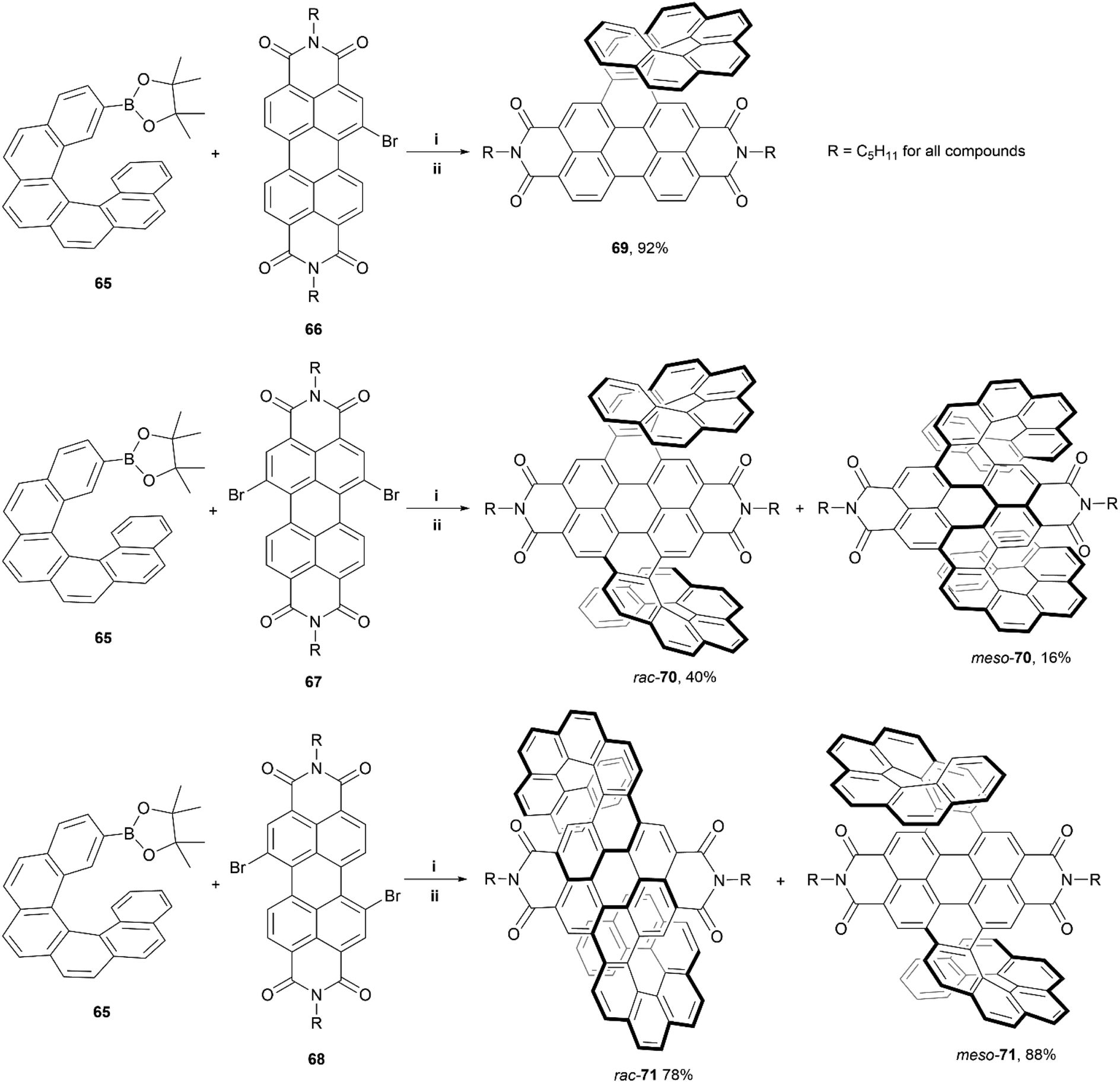 | ||
| Scheme 13 Synthesis of PDI–helicene hybrid compounds; (i) 66, 67, or 68 (1.0 equiv.), 65 (1.2 equiv.), Pd(dppf)Cl2 (0.1 equiv.), K3PO4, THF, 90 °C; (ii) I2, hν, toluene, 100 °C. Note: Precyclized intermediates were separated via column chromatography prior to step ii. | ||
The Nuckolls group has previously pioneered a large number of reliable methods for selectively functionalizing PDI units, which paved the way for the group to design and prepare a significant number of contorted, twisted, and chiral functionalized PDI-based nanographenes. Many aspects of their work have been thoroughly summarized and discussed in their recent publication.81 One of their most notable works included a synthetic design wherein PDI moieties were combined with [6]helicene units to produce a large, discrete helical nanoribbon 72 (Fig. 14).82 It was observed that 72 exhibits an outstanding molar electronic circular dichroism (ECD), with an |Δε|max value of 1920 M−1 cm−1 at 420 nm – the highest recorded value for any compound. This structure can also claim the record for the longest discrete helicenoid structure, with a backbone composed of four axially fused [6]carbohelicenes (24 benzene rings).
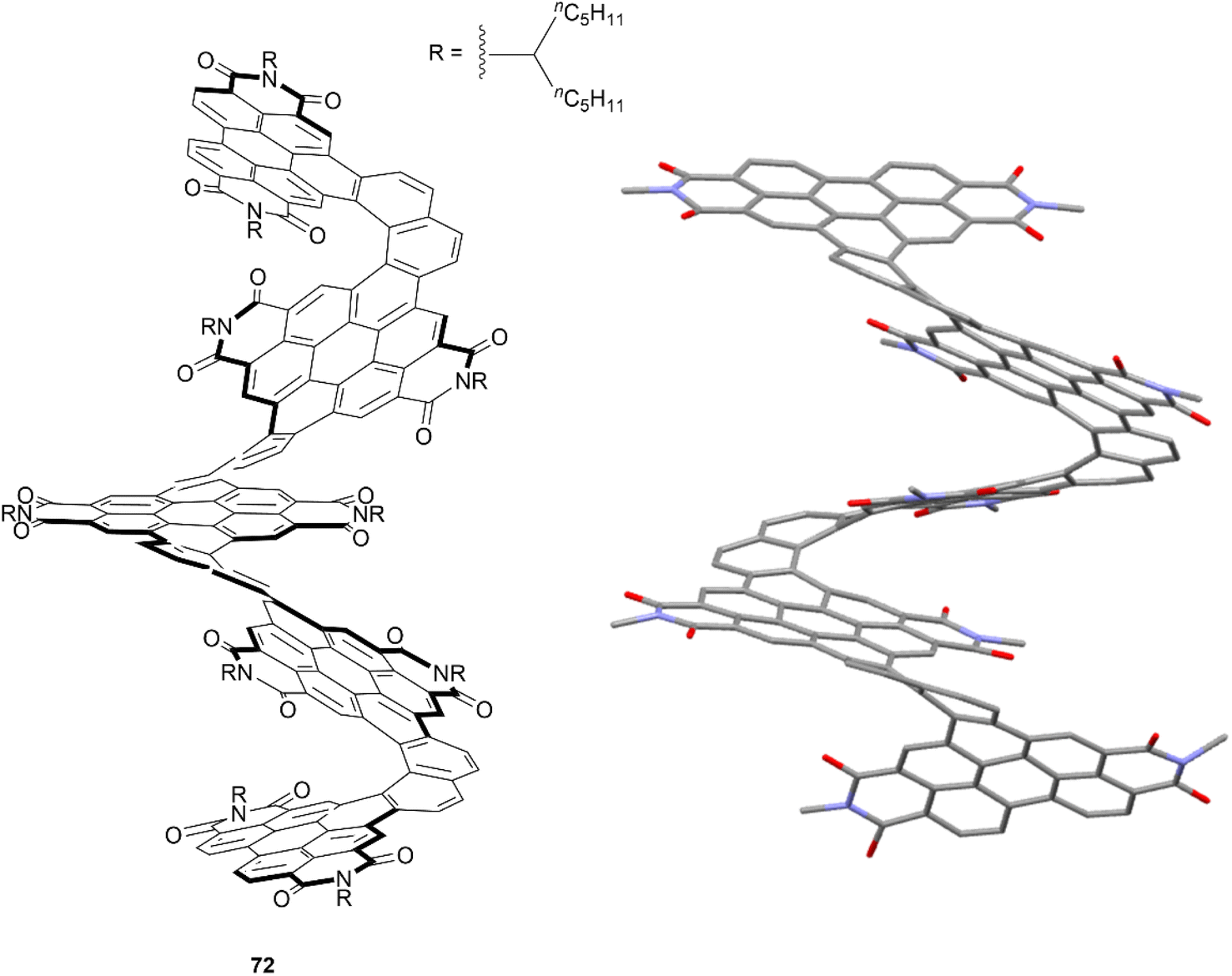 | ||
| Fig. 14 Helical nanoribbon 72 synthesized by Nuckolls and coworkers with published crystal structure. Hydrogens and alkyl groups omitted for clarity. | ||
The Würthner group has also made significant contributions to the design and synthesis of imide-based structures, being the first to introduce stable axial chirality to perylene bisimides (PBIs) through the introduction of sterically demanding substituents on the bay regions of the PBI core.83 These structures have since found applications in organic solar cells and circularly polarized light (CPL) detection devices.24,84–86 The group recently published a procedure for the straightforward preparation of a scope of sterically-congested PBIs through Suzuki coupling of various aryl groups to a 1,6,7,12-tetrachloro PBI core 73 to prepare aryl-substituted derivatives 74a–c (Scheme 14).86 Three derivatives are displayed here, but a small scope of nine total products, with different aryl (Ar) and alkyl (R) groups, were synthesized in varying yields (22–75%). Single crystals of products 74a–c were obtained, and X-ray analysis unambiguously confirmed the twisting of the core structure due to the bulky substituents. The solid state configurations for all the structures of 74 contained both the (P) and (M) enantiomers, and the degrees of twisting were observed to be 34.3°, 36.6°, and 29.5° for 74a, 74b, and 74c, respectively. Chiral separation via HPLC was successful for compounds 74a and 74b, but was unsuccessful for 74c, likely because the rotation of the large pyreno substituents allows for a complex number of isomers to form, complicating the separation process.
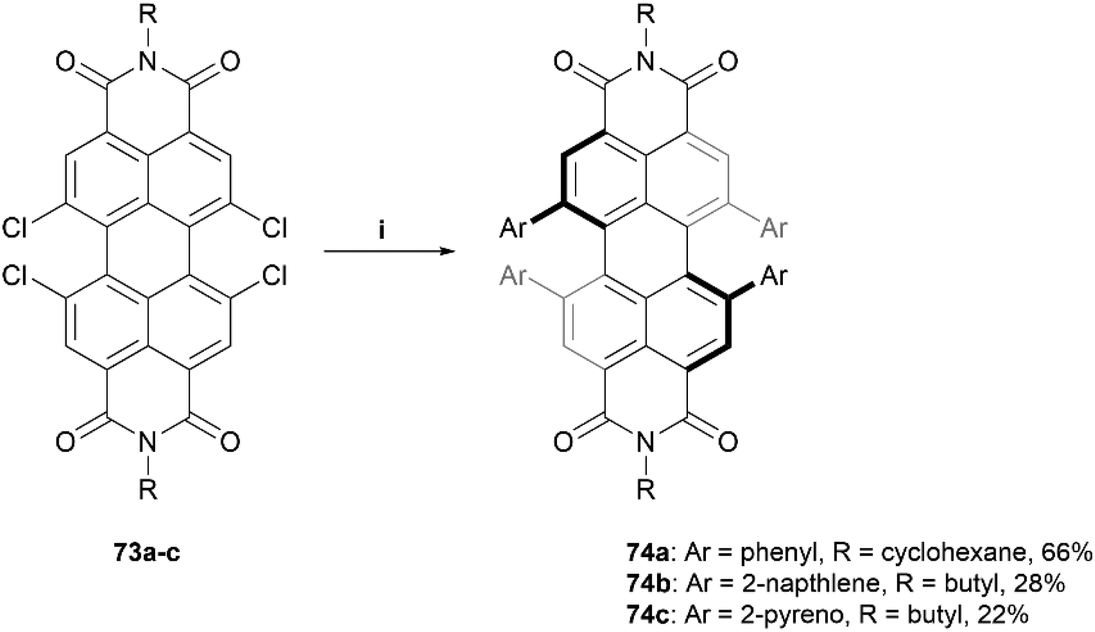 | ||
| Scheme 14 Synthesis of sterically-congested PBIs 74a–c; (i) 73 (1.0 equiv.), Ar-B(OH)2 (20.0 equiv.), Pd(PPh3)4 (0.3 equiv.), K2CO3 (10.5 equiv.), toluene, EtOH, H2O, 80 °C, overnight. | ||
This work was later expanded by the same group, from PBI's to quaterrylene bisimides (QBIs), and introduced the first higher order rylene structure with configurationally stable axial chirality, which is directly imprinted into the synthetic building blocks via enantiopure synthesis.87 Two QBI structures of different lengths were produced – racemic final product 78, through a two-step synthetic process, and enantiomerically pure final product 81 through a three-step process (Scheme 15). Crystal packing analysis showed that (P,P)-81 had an impressive end-to-end twist of 76°, and both structures 78 and 81 displayed sharp emission with a small Stokes shift in the far NIR region at 862 nm and 903 nm, respectively. Additionally, 78 and 80 displayed fluorescence quantum yields of 1.5% and 0.9%, respectively (impressive for such small band gap chromophores), and large chiroptical responses with a unisignated CD signal, with Δε of up to 67 M−1 cm−1 at 849 nm.
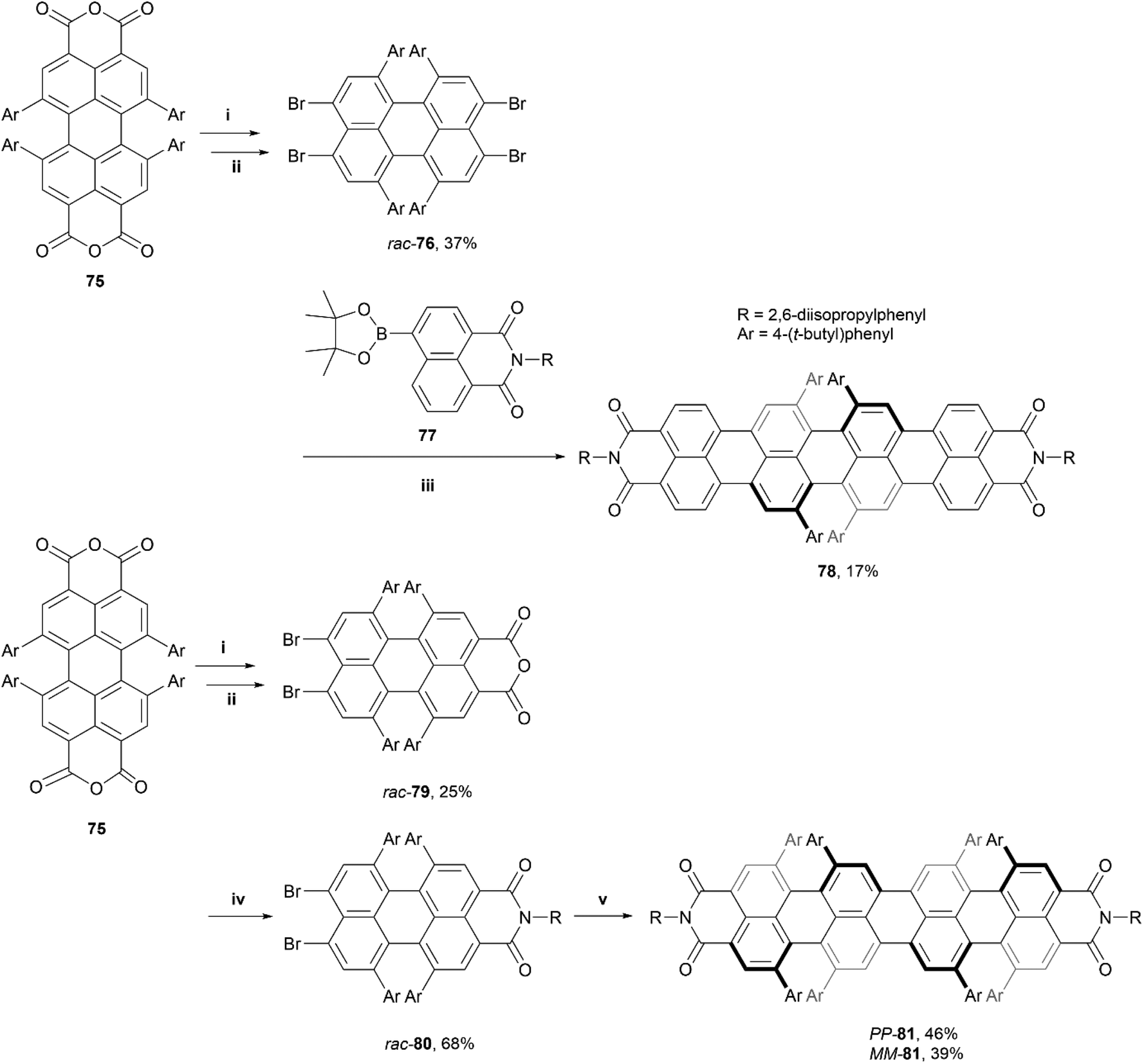 | ||
| Scheme 15 Synthesis of quaterrylene bisimides; (i) 75 (1.0 equiv.), NaOH (6.0 equiv.), H2O, THF, 85 °C; (ii) Br2 (40.0 equiv.), 85 °C, 16 h; (iii) rac-76 (1.0 equiv.), 77 (2.2 equiv.), [Pd2(dba)3]·CHCl3 (0.2 equiv.), PCy3·HBF4 (0.8 equiv.), Cs2CO3 (6.0 equiv.), 1-chloronaphthalene, 160 °C, 20 h; (iv) rac-79 (1.0 equiv.), R-NH2 (15.0 equiv.) in acetic acid, NMP, 110 °C, 72 h; (v) rac-80 (2.0 equiv.), Zinc powder (13.2 equiv.), [Pd2(dba)3]·CHCl3 (0.12 equiv.), DMF, 60 °C, 18 h. | ||
Multiple helicenes
The Alabugin group has previously utilized an alkyne-based “folding” technique – a radical-initiated cascading cyclization of a carefully designed series of alkynes – to make a wide variety of nanographenes.88 This technique was later used to prepare contorted structures containing multiple helicenes, ultimately producing two diastereomeric derivatives of 83 in a roughly 1:1 mixture, which were able to be separated through crystallization in CD3CN (Scheme 16).89 It was determined, both experimentally and computationally, that the chirality of the helicene moieties in 83 were strongly coupled, and the two diastereomers had “matching” chirality (P,P or M,M), where the two possible mismatched diastereomers (M,P or P,M) were notably higher in energy (∼5 kcal and ∼8 kcal higher), and therefore not observed in the final products.
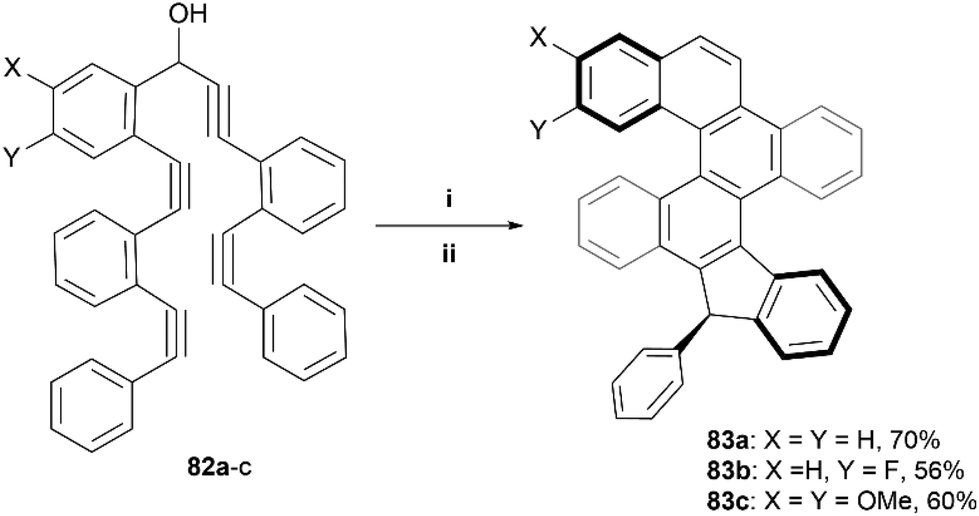 | ||
| Scheme 16 Synthesis of nanographenes via alkyne-based “folding” technique; (i) 82 (1.0 equiv.), Bu3SnH (1.4 equiv.), AIBN (0.3 equiv.), toluene 110 °C, 14 h; (ii) Aq. HCl (2 M), rt, 3 h. | ||
In 2021, Wang and coworkers produced a synthesis for a B,N-embedded double [7]helicene 83, through a synthetic route involving a four-fold electrophilic aromatic substitution and a nucleophilic substitution, followed by a final intramolecular C–H borylation to receive a small scope of derivatives in yields ranging from 23–53% (Fig. 15).90 These structures displayed significant chiroptical responses from 300–700 nm, with a maximum gabs of 3.3 × 10−2 at 502 nm (a record high for helicenes in the visible region), tunable CPL responses in the red to near-infrared regions, and high photoluminescence quantum yields (up to 100%). The enantiomers of all derivatives 84a–c were obtained through preparative chiral HPLC, and the configurational stability of 84a was tested by heating to 300 °C for 3 h – no change was observed in the NMR spectra after heating, suggesting excellent stability and allowing for further chiroptical studies. CD analysis of the three derivatives produced strong chiroptical responses, with maximum |gabs|values reaching 3.3 × 10−2 (502 nm), 3.1 × 10−2 (518 nm), and 2.6 × 10−2 (526 nm) for 84a, 84b, and 84c, respectively, representing some of the highest |gabs| values for helicenes in the visible range.
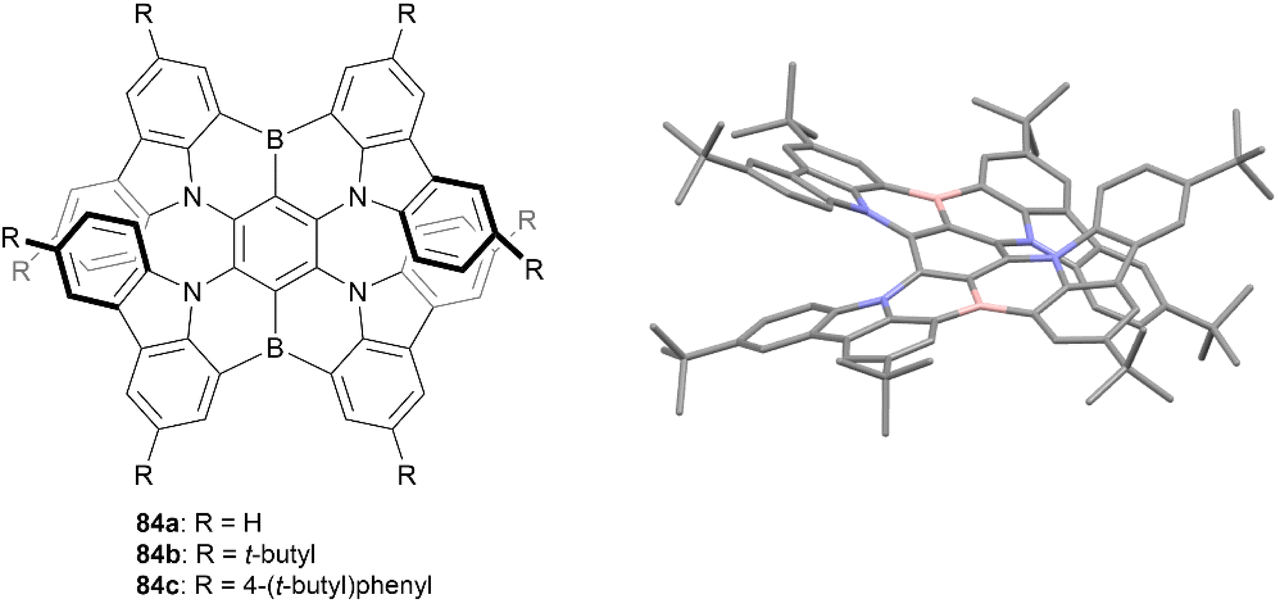 | ||
| Fig. 15 B,N-Embedded double [7]helicene 84 synthesized by Wang and coworkers with published crystal structure of 84b. Hydrogens omitted for clarity. | ||
Recently, Ikai, Yashima, and coworkers produced a quantitative, chemoselective, and diastereospecific synthesis of double- and triple-expanded helicenes.91 The synthesis was carried out through a TFA-promoted cascading alkyne cyclization technique, beginning with a carefully designed anthracene-based precursor 85, and ultimately producing a twisted, expanded helicene-like product 86 in high yield (88%). Double-expanded helicene 87 and triple-expanded helicene 88 were prepared with analogous methods (also in high yields), by utilizing the corresponding alkyne starting materials (Scheme 17). The enantiomers were all separable through chiral HPLC, and moderate CD values were measured, with the maximum luminescence dissymmetry factor |glum| for 88 (9.4 × 10−3) being more than three times that of the double helicene 87 (2.7 × 10−3). Notably, the triple-expanded derivative 88 was found to exhibit the highest CPL brightness among other known CPL-active carbo- and hetero-helicenes and helicenoids, at 255 M−1 cm−1. This value is also approximately ten times higher than other high-level CPL-active multiple helicenes (∼28.2 M−1 cm−1). It was observed that the CD signals of the enantiomerically pure 86 and 87 gradually decreased in chloroform at 0 °C with no change in absorption spectra, indicating that racemization was occurring. This makes sense as the racemization energies for 86 and 87 in toluene were fairly low, estimated as 17.6 kcal mol−1 and 19.1 kcal mol−1, respectively. Structure 88 however, displayed no altered optical behavior even after heating in toluene at 80 °C for 24 hours, indicating a remarkable configurational stability considering the expected flexibility of the expanded structure. The specific energy of racemization was unfortunately not able to be quantitatively determined due to the poor thermal instability of 88 at temperatures over 100 °C.
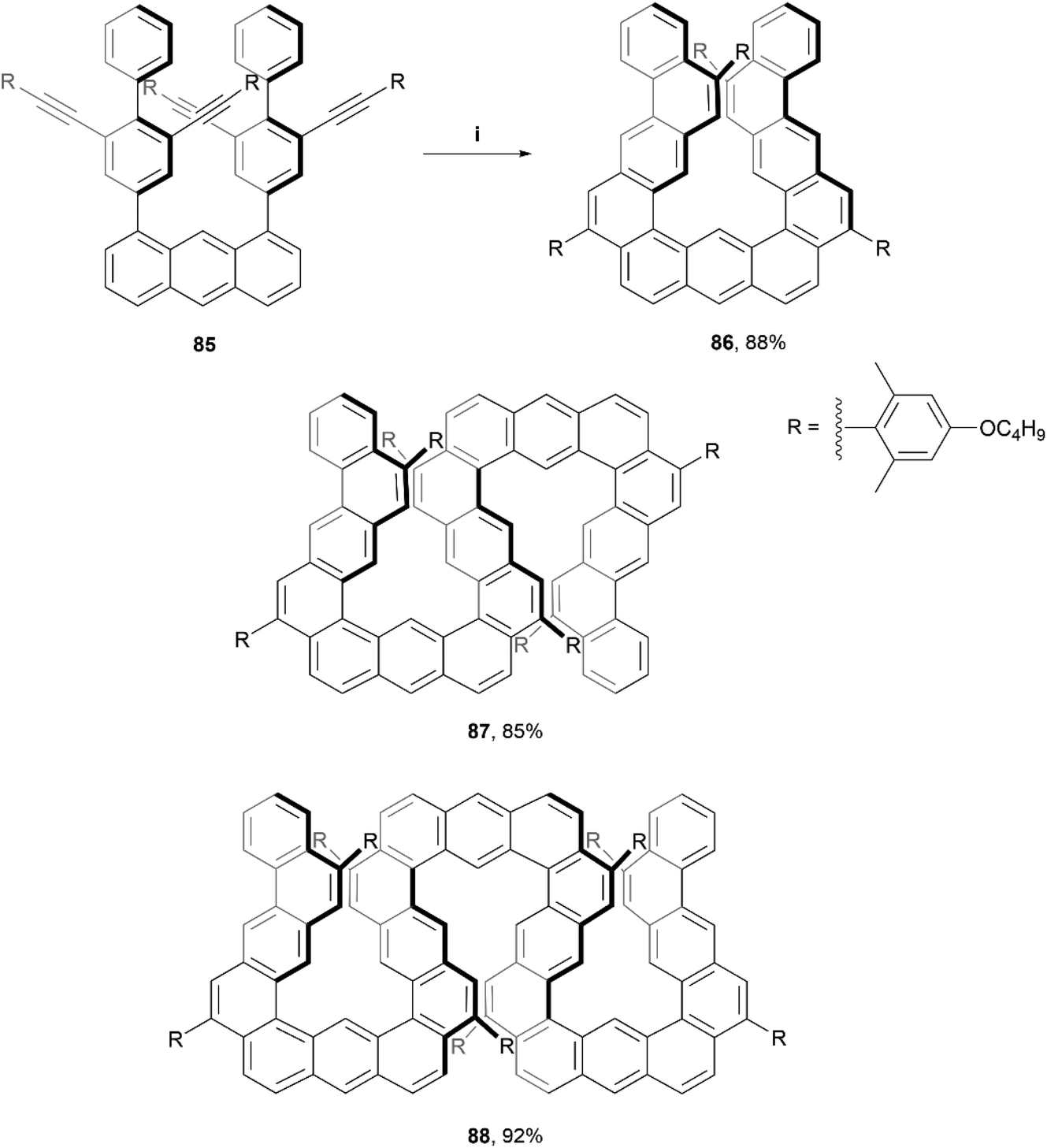 | ||
| Scheme 17 Synthesis of expanded helicenes; (i) 85 (1.0 equiv.), DCM:TFA (40:1, v:v), rt, 1 h. 87 and 88 prepared through analogous methods. | ||
In 2021, Miao et al. reported the preparation of a novel quadruplehelicene 89, containing two [6]helicene and two [7] helicene units, through a multiple-step synthesis involving a final key Scholl oxidation for an eight-fold cyclization (Fig. 16).92 The final structure 89 has nine possible stereoisomers – four pairs of enantiomers and one meso compound, and through single-crystal analysis and chiral resolution, it was determined that the synthesized product was a racemic mix of (P[6], P[7], P[6], P[7])-89 and (M[6], M[7], M[6], M[7])-89, which showed remarkable resistance to thermal isomerization. The products also exhibited unusual near-infrared (NIR) absorption and emission, with maxima at 848 nm and 977 nm, respectively, and the isolated enantiomers exhibited electronic circular dichroism in both the NIR and visible-light regions.
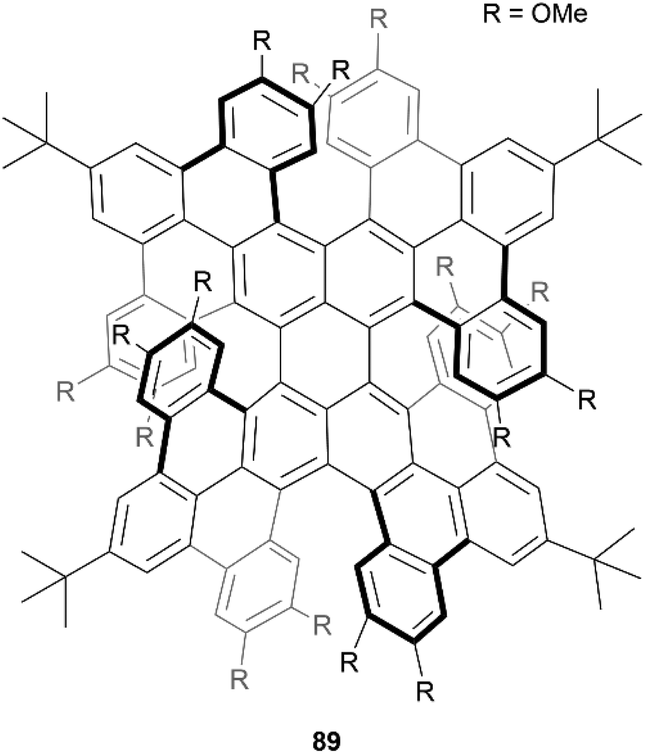 | ||
| Fig. 16 Quadruple helicene 89 synthesized by Miao and coworkers. | ||
In 2021, Yashima, Yamakawa, and coworkers reported a one-step synthesis of a series of double, triple, and quadruple helicenes, through an acid-mediated intramolecular alkyne cyclization (Scheme 18).93 The quadruple [4]helicene 91 was the intended product, as the alkyne cyclization was predicted to occur primarily on the 5 and 11 positions of the chrysene starting material, but it was found that the expected product was only formed in a yield of 14–26%, and side products triple helicene 92 (one [5]helicene and two [4]helicenes) and double [5]helicene 93 were also formed in low-to-moderate yields (60–61% and 13–26%, respectively). Enantiomers for quadruple helicene 91 were unable to be separated, likely due to the low inversion barriers of the four [4]helicene units, but double helicene 93 was successfully separated via HPLC at low temperature (0 °C), exclusively into the (P[5],P[5]) and (M[5],M[5]) configurations with no sign of the meso (P[5],M[5])-93 being present. When heated to higher temperatures (40–80 °C) in toluene, the CD signals of (M[5],M[5])-93 decreased over time, and the activation energy (Ea) for the structure was estimated to be 26.1 kcal mol−1, slightly higher than that of [5]helicene (Ea = 23.5 kcal mol−1), suggesting that the dynamic helicities of two [5]helicene moieties are coordinated with each other. Triple helicene 92 was also able to be resolved by HPLC at an even lower temperature (−20 °C), and two pairs of peaks, one minor and one major were observed to elute. DFT calculations indicated that the major peaks were diastereomers (P[5],P[4],M[4] and M[5],M[4],P[4])-92, which was later confirmed by X-ray analysis, and the minor peaks were assigned to the next most stable pair (P[5],M[4],M[4] and M[5],P[4],P[4])-92. CPL analysis of the two species that were able to be separated, 92 and 93, produced maximum luminescence dissymmetry factors of 5.0 × 10−3 and 2.5 × 10−3, respectively, which are of comparable value to other previously reported helicene-based structures.
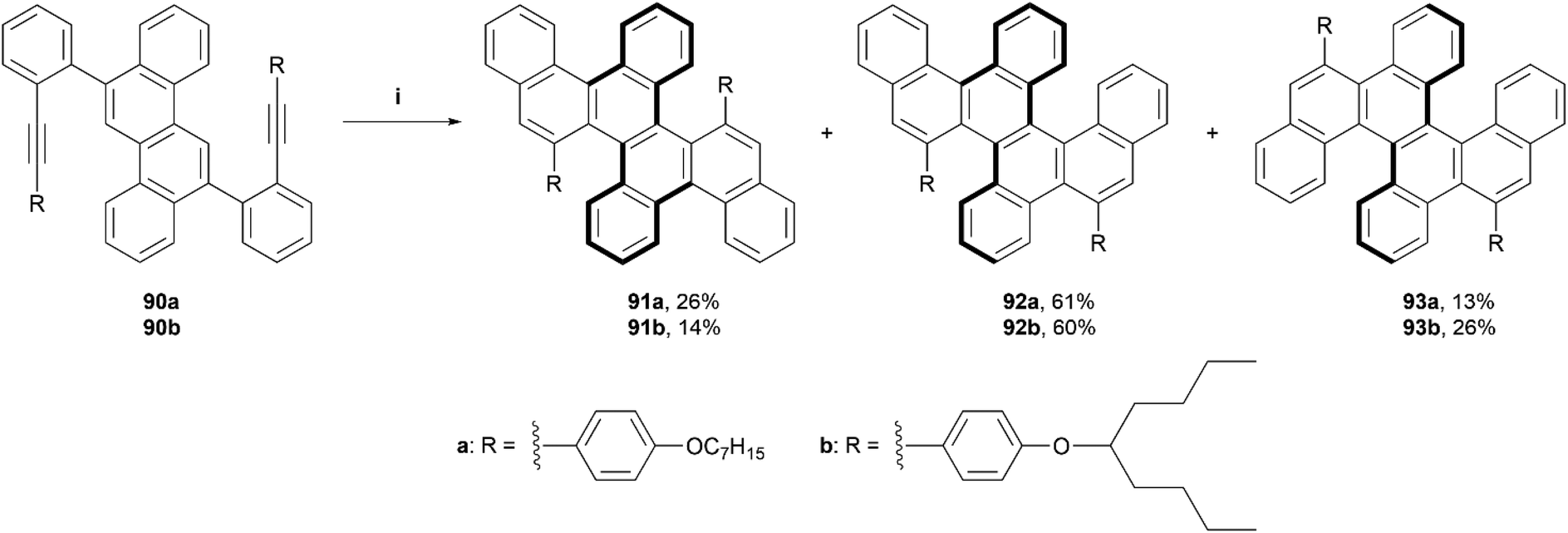 | ||
| Scheme 18 Synthesis of double, triple and quadruple helicenes; (i) 90 (1.0 equiv.), DCM:TFA (98:2, v:v), 25 °C, 12 h. | ||
In 2021, Sun, Chen, and coworkers reported a synthesis of a twisted and stable derivative of the zethrene family – a class of nanographenes being studied for their intriguing magnetic properties (Fig. 17).94 This structure was synthesized through a straightforward two-step synthesis involving a Suzuki–Miyaura coupling, followed by a nucleophilic addition/ring cyclization/oxidation sequence to produce the final product 94. Single crystal X-ray diffraction unambiguously confirmed that the structure was chiral, due to the inclusion of two helical benzo[4]helicene units which cause a torsional strain angle of about 20° within the helicene unit. Two enantiomers could be observed in the crystal subunit, of (P,P) and (M,M) configuration, and DFT calculations indicated that the chiral-to-meso (P,M) interconversion barrier was very low (8.20 kcal mol−1), which indicated that enantiomeric separation would be difficult at room temperature.
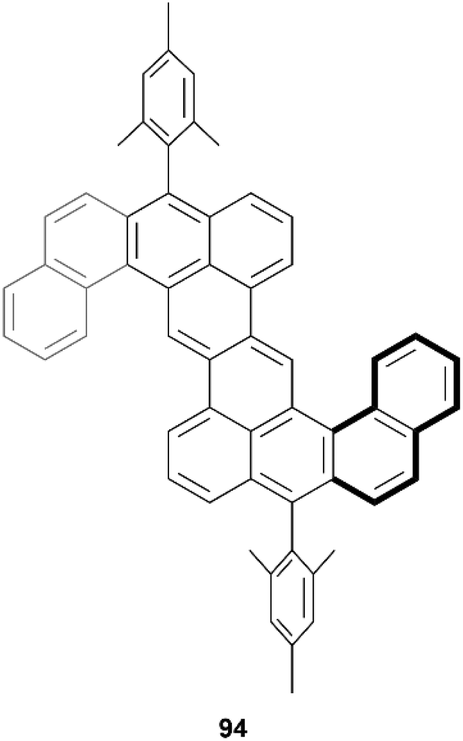 | ||
| Fig. 17 5,6:12,13-Dinaphthozethrene 94 synthesized by Sun, Chen and coworkers. | ||
In 2019, Ravat and coworkers published a synthesis based around the well-reviewed works of Kamikawa95 and Gingras,96 who utilized palladium and nickel-catalyzed [2 + 2 + 2] cycloadditions, respectively, to synthesize structures containing six [5]helicene subunits. Ravat and coworkers used a similar nickel-catalyzed [2 + 2 + 2] cycloaddition method, but using [7]helicene rather than [5]helicene coupling groups in an attempt to improve the configurational stability of the structure (Scheme 19).97 This method proved to be successful at producing the final propeller-shaped structure 100, containing three [5]helicene moieties and three [7]helicene moieties in 57% yield. It was found that when starting with (M)-9,10-dibromo[7]helicene 99, the reaction produced only one diastereomer, (M,M,M,P,P,P)-100. The stereospecific structure was confirmed by a variety of 1D and 2D NMR spectroscopy studies and single-crystal X-ray analysis, and the diastereomeric configuration was also supported by calculations, as the (M,M,M,P,P,P)-100 was determined to be 15 kcal mol−1 lower in energy than the next highest energetically favorable configuration, (M,M,M,P,P,M)-100. The other diastereomer, (P,P,P,M,M,M)-100, was also able to be formed cleanly through the same synthetic process by using the (P)-9,10-dibromo[7]helicene 99 rather than the (M). (M,M,M,P,P,P)-100 produced a small, negative specific optical rotation (OR) value of −222°, due to the presence of both the (P) and (M) moieties. The overall negative value can be explained by the huge negative contribution of the (M)-[7]helicene units, which have previously been shown to have a specific OR value of −6200°.98
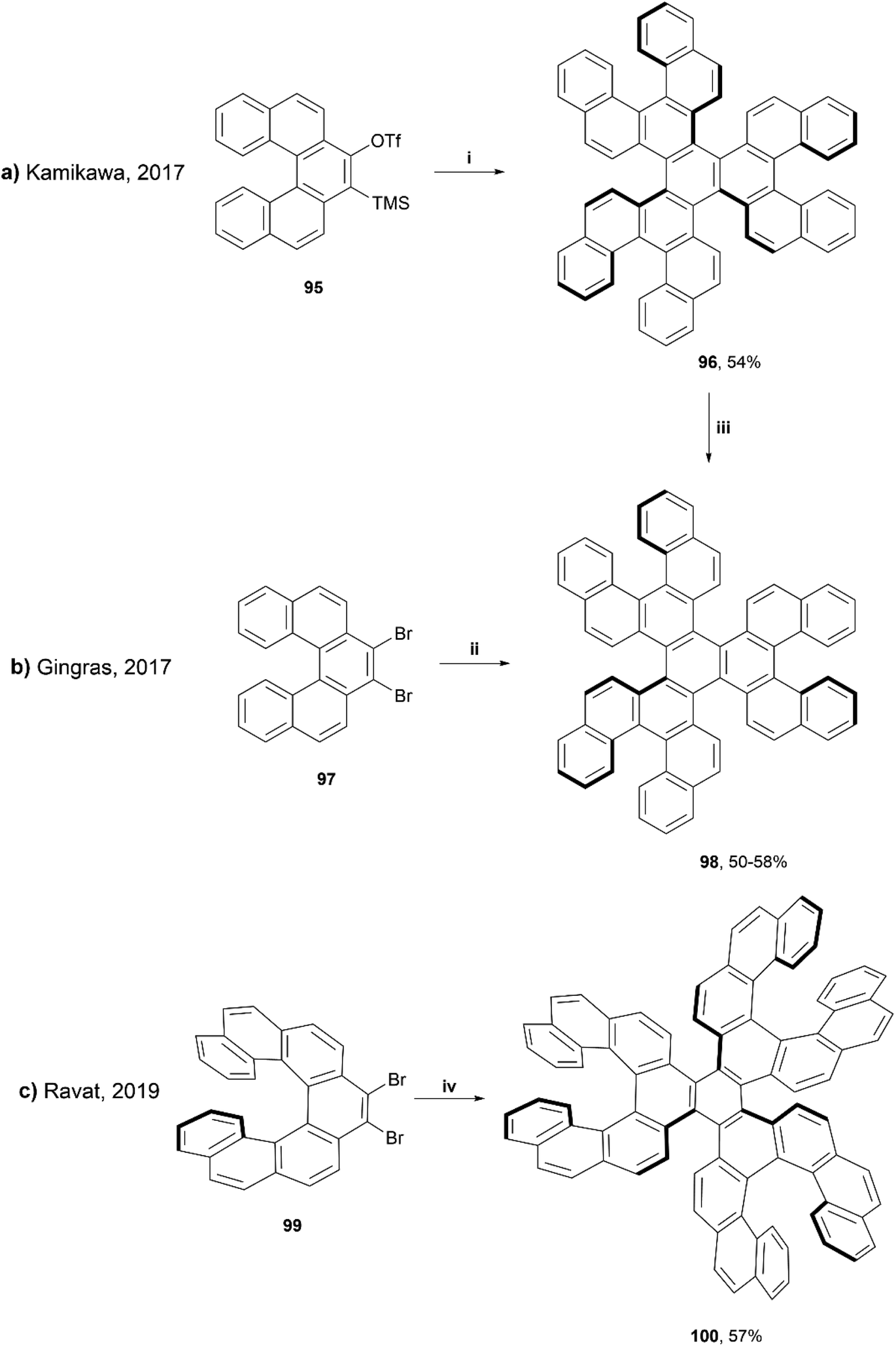 | ||
| Scheme 19 Synthesis of nanographenes containing six [5]helicene units; (i) 95 (1.0 equiv.), CsF (6.0 equiv.), Pd2(dba)3·CHCl3 (0.1 equiv.), MeCN, rt, 24 h; (ii) 97 (1.0 eq.), Ni(cod)2 (1.45 equiv.), 2,2′-bipyridine (1.93 equiv.), cis-1,5-cyclooctadiene (5.7 equiv.), THF, microwave 120 °C, 12 min; (iii) 96 (1.0 equiv.), toluene, 100 °C, 3 h; (iv) 99 (1.0 equiv.), Ni(cod)2 (1.5 equiv.), 2,2′-bipyridine (2.1 equiv.), cis-1,5-cyclooctadiene (5.7 equiv.), THF, 110 °C, 30 min. | ||
In 2018, Tanaka et al. reported a series of dibenzo[7]helicenes, that were synthesized through a rhodium-catalyzed enantioselective intramolecular [2 + 2 + 2] cycloaddition reaction (Scheme 20a).99 It was observed that when using a rhodium(I)/(R)-BINAP complex, dibenzo[7]helicene (+)-102 was synthesized in good yield (64%), and a small amount of the [2 + 1 + 2 + 1] cycloaddition product 103 (6%) was also recovered. Interestingly, when the same conditions were used, but with (S)-difluorphos instead of (R)-BINAP as the ligand, the yield of the side product 103 increased to 21% and the yield of the expected (−)-102 dropped to 38%. This phenomenon was hypothesized to be due to the electron-deficient (S)-difluorphos ligand accelerating the reductive elimination step in the proposed mechanism, which encourages the formation of 103. Enantiopure (+)-102 and (−)-102 were able to be separated in ee of 99% and 85%, respectively, through silica-gel preparative thin-layer chromatography. Twisted anthracene product 103 was recovered in an enantiomeric ratio of 50:50, and were configurationally stable enough to be separated by HPLC at room temperature. Compound 103 was then subjected to Scholl cyclization conditions, which resulted in twisted double dibenzo[6]helicene 104 in moderate yield (Scheme 20b), and enantiomers were cleanly resolved by chiral HPLC. This compound had excellent thermal configurational stability, as extended heating of (−)-104 at 130 °C for 16 hours didn't result in any racemization or epimerization.
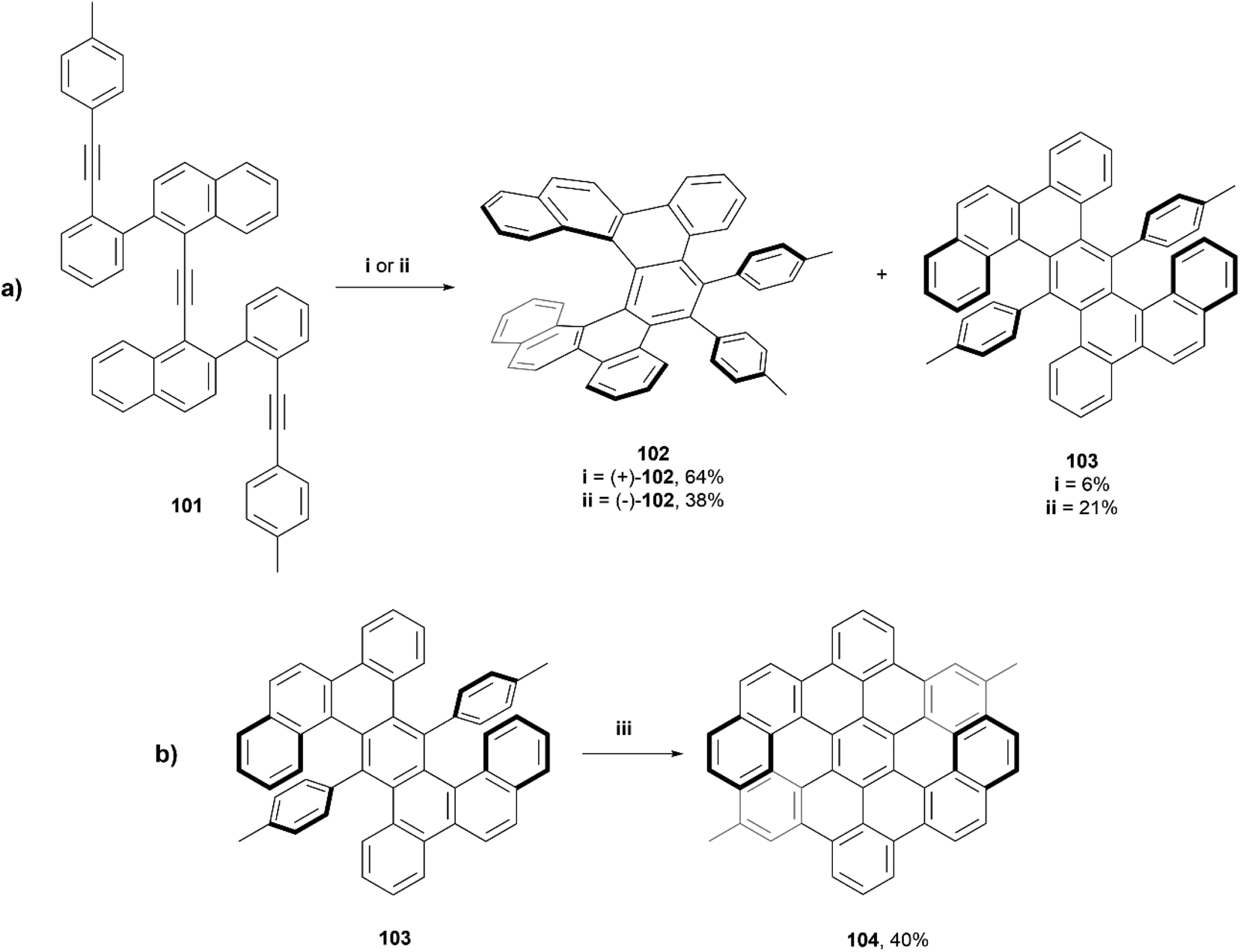 | ||
| Scheme 20 (a) Synthesis of dibenzo[7]helicenes; (i) 101, [Rh(cod)2]OTf, (R)-BINAP, DCE, 80 °C, 16–90 h; (ii) 101, [Rh(cod)2]BF4, (S)-difluorphos, DCE, 80 °C, 16–90 h; (b) Scholl reaction of minor product of (a) (iii) 103 (1 equiv.) in DCM, FeCl3 (10 equiv.) in MeNO2, rt, 1 h. | ||
X-ray structures of (+)-102, (±)-102, and (±)-103 were obtained and the twisted structures were unambiguously confirmed. The chiroptical properties of the single and double helicenes 102 and 104 were analyzed, and it was observed by CPL spectroscopy that the luminescence dissymmetry ratio (glum) value for single helicene 102 was significantly larger than that of the double helicene 104, −2.2 × 10−3vs. 7.4 × 10−4 respectively. Notably, the fluorescence quantum yields for 102 and 103 (6% and 4%, respectively) were slightly higher than that of unsubstituted [7]helicene (2.1%), but 104 showed a remarkably higher value (75%) than the recorded value for double dibenzo[7]helicene (34%). This is largely explained by the luminophore of 104 being a peropyrene moiety, which are known to be strongly luminescent molecules.
By utilizing Scholl oxidation methods, Narita and Müllen have prepared other twisted double [7]helicene structures somewhat analogous to 104.100,101 And by using a similar synthetic method, Narita and Hu reported the preparation of a twisted, laterally π-extended double [9]helicene analogue in 2023.102
Recently, Zhang and coworkers reported the preparation of a series of double-heterohelicenes containing an N–B–N zigzag edge. The final three chiral compounds (105, 106 and 107) were produced through a multiple-step synthesis in good yields, and pure enantiomers were successfully separated for all three structures (Fig. 18).103 Single-crystals were obtained for each, which unambiguously confirmed the nonplanar nature of the structures, where the two bulky sides twist away from each other around the axis of the central benzene ring, with a central torsion angle of up to 30°. These products were found to exhibit significant hydrogen-bonding capability, the effects of which are transferrable to the helical moieties in the structure, as was determined through CPL and CD experiments, which displayed a red-shift in the presence of fluoride anions.
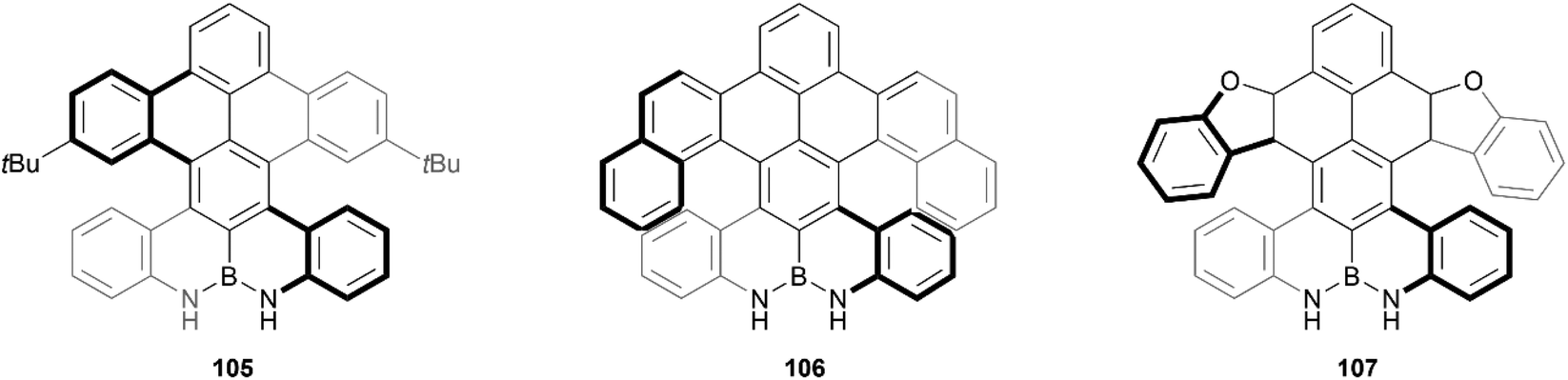 | ||
| Fig. 18 Three derivatives of double-heterohelicenes containing N–B–N zigzag edge (105, 106, and 107) synthesized by Zhang and coworkers. | ||
Irregular/nonalternant nanographenes
In recent years, several reviews104,105 and communications106 have been written based around irregular nanographenes, in which five-, seven-, or eight-membered rings are embedded within a graphitic structure. These irregularities can cause unique geometric effects, and often are able to induce saddle- or bowl-shaped chirality in otherwise flat structures.In 2018, Siegel and coworkers synthesized a small series of indeno-fused corannulene structures, intended to show how different fusion arrangements would affect the properties and overall structure (Scheme 21).107 The derivatives of precursor 108 (where X = Cl or F) were prepared through a Suzuki coupling of 1,6-dibromo-2,5-dimethylcorannulene with the appropriately halogenated phenyl boronic acid. Treatment of F-108 with silyl cation produced a mixture of 109, 110, and 111 with small or trace yields, but similar treatment of Cl-108 resulted in formation of only 109 and 110, in higher yields, but no 111 appeared to form. Structure 111, the only chiral structure of the three, was able to be resolved into enantiomers by chiral HPLC, and its bowl-to-bowl inversion barrier was estimated to be approximately 26 kcal mol−1 at 62 °C, with a half-life of approximately 2 hours at a similar temperature.
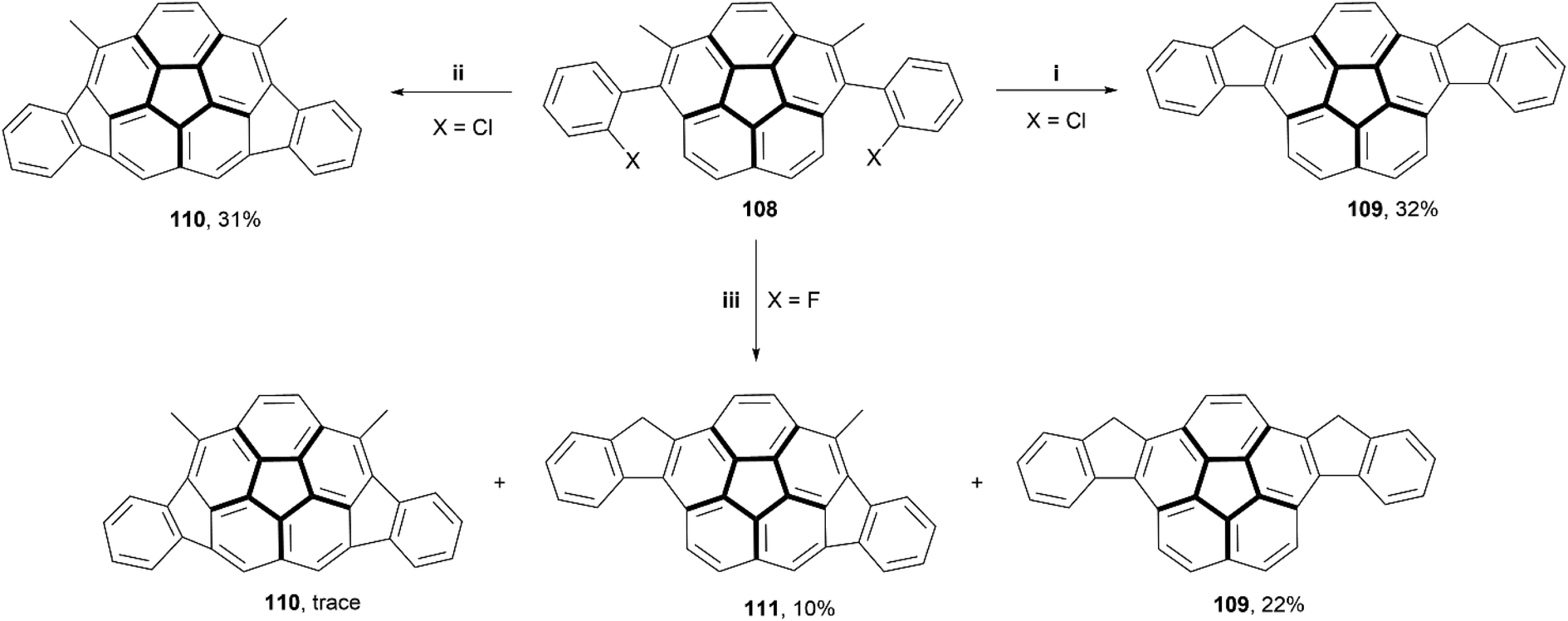 | ||
| Scheme 21 Synthesis of indeno-fused corannulenes; (i) 108 (1.0 equiv.), Pd(OAc)2 (0.4 equiv.), [1,3-bis(2,6-diisopropylphenyl)-imidazolium chloride] (0.16 equiv.), K2CO3 (4.3 equiv.), NMP, 125 °C, 20 min; (ii) 108 (1.0 equiv.), Pd(PCy3)2Cl2 (0.4 equiv.), DBU (16.7 equiv.), DMA, microwave 160 °C, 30 min; (iii) 108 (1.0 equiv.), [iPr3Si][CHB11H5Br6] (0.12 equiv.), DMDMS (1.6 equiv.), PhCl, microwave 120 °C, 10 h. | ||
Recently, Liu and coworkers reported a method for the formation of azulene units in PAH systems through dehydration condensation of aryl-substituted cyclopentadienes and ortho-haloaryl aldehydes, followed by palladium-catalyzed C–H cross-coupling.108 This process facilitated the creation of four nonalternant pentacene and hexacene isomers, which exhibited unique optical properties (Fig. 19). Single-crystal X-ray analysis of three of the four structures (112, 113, and 114), and optimized calculations for 115, confirmed that the interaction between the 6-7-6 membered rings creates a helically twisted bay region, shifting two sides of the seven-membered ring out of the plane in opposite directions to reduce steric strain.
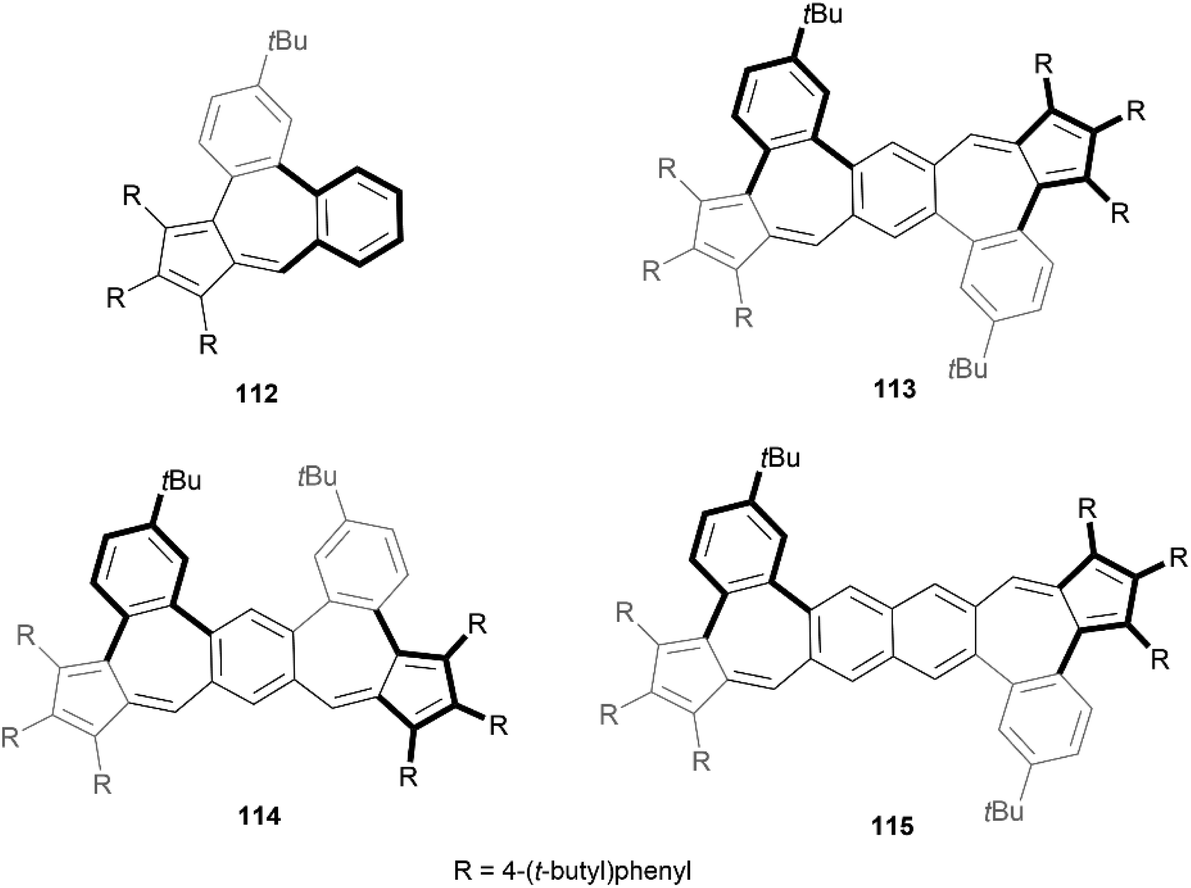 | ||
| Fig. 19 Helically twisted structures 112, 113, 114, and 115 synthesized by Liu and coworkers. | ||
In 2021, Fei et al. published the synthesis of three 7-5-7 “defective” nanographenes, two of which were found to be chiral (Fig. 20).109 These two structures adopted different geometries (saddle-shaped, dual saddle-shaped), and were found to exhibit closed-shell antiaromatic behavior, leading to high air-stability as compared to similar more open-shell, unstable 5–7 ring structures. Single crystals of 116 and 117 were grown and their structures and geometries were unambiguously determined by X-ray single crystal analysis. Compound 116 adopts a nearly flat geometry, with one of the seven-membered rings shifting the adjacent ring out of the plane with a dihedral angle of 21.9°, while the other seven-membered ring remains planar, and the M and P enantiomers were observed to exist together in the crystal structure in a 1:1 ratio. Compound 117 adopts a more saddle-shaped geometry, where steric interactions from the neighboring benzene rings shift the heptagonal rings out of the plane with dihedral angles of 21.6° and 20.4°, and the enantiomeric pairs arrange into a columnar pattern with interplanar spacing of as low as 3.28 Å. DFT calculations of 116 indicate that the structure can go through an M- to -P conversion through a flat transition state, with a very low free activation barrier of 3.08 kcal mol−1, indicating that the product exists as a racemic mixture and separation is unlikely at room temperature. A similar conclusion was drawn for 117, where the isomerization barrier was calculated as 2.69 kcal mol−1, also very low and indicative of an inability to separate the enantiomers.
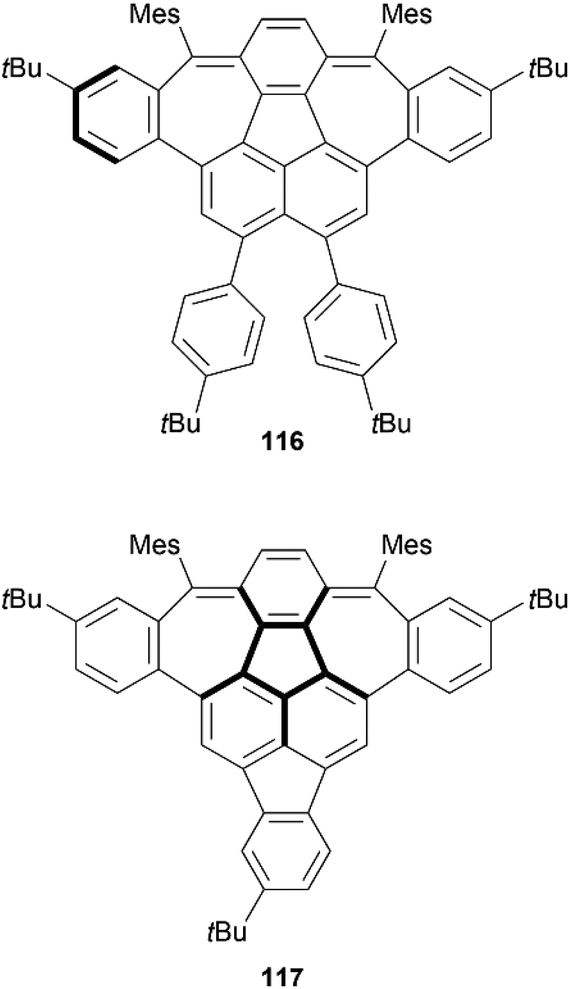 | ||
| Fig. 20 Chiral nanographenes 116 and 117 synthesized by Fei and coworkers. | ||
The Würthner group published a general synthetic method for the preparation of irregular structures containing both bowl and saddle-like curvatures, through the use of hydroboration, electrophilic borylation, dehydrogenation and [5 + 2] annulation methodologies.110 These structures generally contain both a five-membered ring and a seven-membered ring, causing unique geometric warping of the system. A scope of four structures were successfully prepared by this method in low-to-moderate yields (19–33%), and all were thoroughly characterized (Scheme 22). Crystals of 119c were obtained by sublimation, and the twisted structure could be unambiguously confirmed, with a positive curvature on the corannulene and a negatively curved seven-membered ring. The bowl of 119c was observed to be 0.76 Å deep, shallower than that of corannulene with a bowl depth of 0.87 Å.
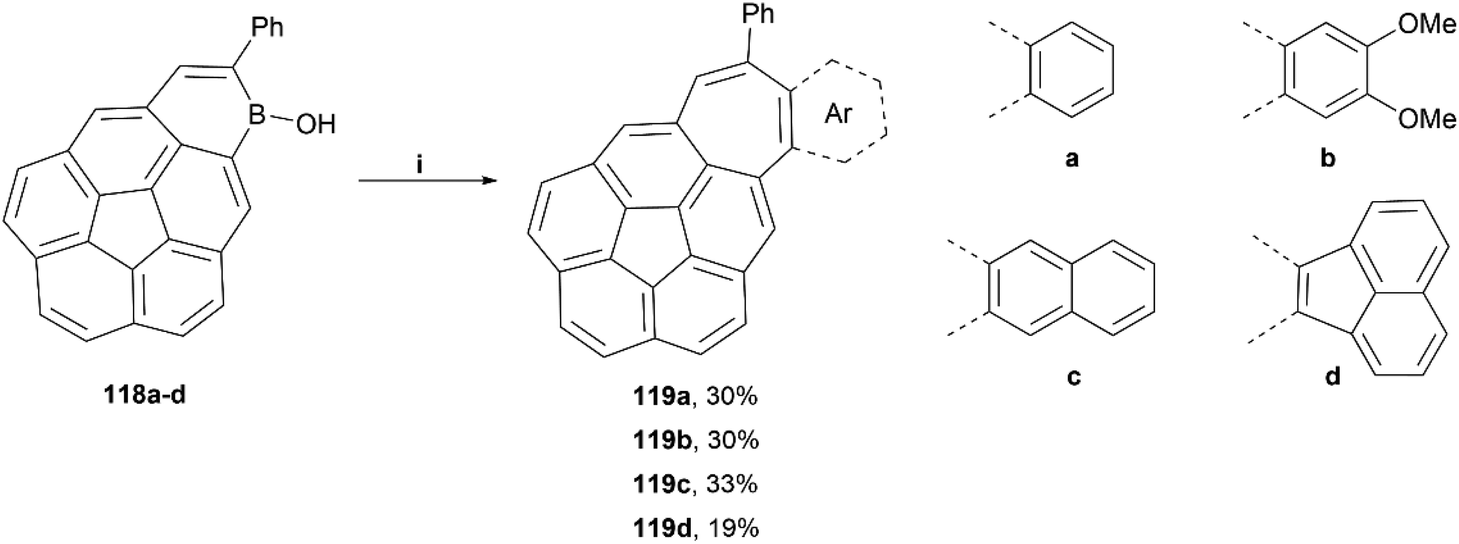 | ||
| Scheme 22 Synthesis of expanded corannulene-type compounds 119a-d; (i) 118 (1.0 equiv.), dibromoarene (2.0 equiv.), [Pd2(dba)3]·CHCl3 (0.05 equiv.), P(tBu)3·HBF4 (0.11 equiv.), Cs2CO3 (3.3 equiv.), H2O (40 equiv.), tAmOH, 100 °C, 42 h. | ||
In 2019, the Mastalerz group reported a three-step synthesis of a unique monkey-saddle-shaped nanographene, based around an irregular structure containing three five-membered rings and three eight-membered rings (Fig. 21).111 The curvature of 120 was unambiguously confirmed by X-ray analysis, and enantiomers were resolved via chiral HPLC separation. The enantiomers were calculated to have high degrees of stereochemical stability, with a notably high inversion barrier of approximately 24 kcal mol−1, and a half-life of one day at room temperature (t1/2 = 23 ± 1h). This group has made a variety of other contorted and chiral compounds based around irregular nanographene structures.112–114
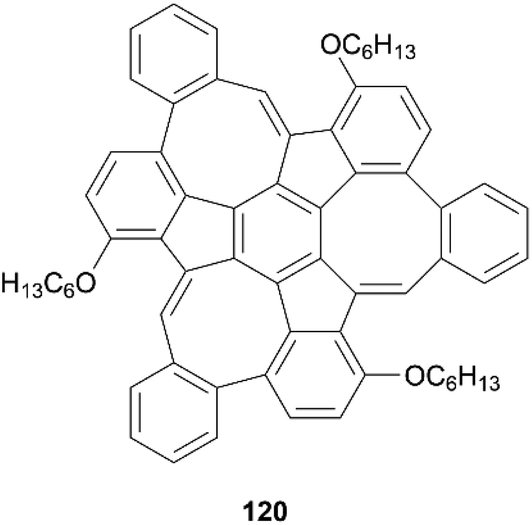 | ||
| Fig. 21 Monkey-saddle-shaped nanographene 120 synthesized by Mastalerz and coworkers. | ||
Conclusions
The field of chiral nanographene preparation has certainly been widely explored, but new advances are still frequently being made to introduce new structures with unique applications and properties. One particular area that still needs further work is in the development of enantioselective synthetic methods to prepare nanographene materials. Altering commonly used synthetic methods, or even developing entirely new ones, in order to prepare enantiopure or enantioenriched chiral nanographenes poses an intriguing challenge to be explored within the community. Several broad classes of chiral nanographenes, such as twistacenes, expanded helicenes, π-extended helicenes, multiple helicenes, imide-based nanographenes, heterohelicenes, and irregular nanographenes, are areas of study with their own unique challenges and properties. Their inherent nonplanar structures introduce a variety of altered properties, such as crystal packing, light polarizing, and emission/absorbance abilities, and the diversity of these structures allows for great opportunities to fine-tune their properties and allow for application in many areas of organic- and nanoelectronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0022178.References
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, From Nanographene and Graphene Nanoribbons to Graphene Sheets: Chemical Synthesis, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, New advances in nanographene chemistry, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- E. Clar and J. F. Stephen, The synthesis of 1:2, 3:4, 5:6, 7:8, 9:10, 11:12-hexabenzocoronene, Tetrahedron, 1965, 21, 467–470 CrossRef CAS.
- E. Clar and W. Schmidt, Localised vs delocalised molecular orbitals in aromatic hydrocarbons, Tetrahedron, 1979, 35, 2673–2680 CrossRef CAS.
- R. Scholl, C. Seer and R. Weitzenböck, Perylen, ein hoch kondensierter aromatischer Kohlenwasserstoff C20H12, Ber. Dtsch. Chem. Ges., 1910, 43, 2202–2209 CrossRef CAS.
- R. Scholl and C. Seer, Abspaltung aromatisch gebundenen Wasserstoffes unter Verknüpfung aromatischer Kerne durch Aluminiumchlorid, 6. Mitteilung: Versuche mit Phenol-äthern und mit Diphenyl-methan, Ber. Dtsch. Chem. Ges. (A and B), 1922, 55, 330–341 CrossRef.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Synthetic organic spin chemistry for structurally well-defined open-shell graphene fragments, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- W. Pisula, X. Feng and K. Müllen, Charge-Carrier Transporting Graphene-Type Molecules, Chem. Mater., 2011, 23, 554–567 CrossRef CAS.
- K. K. Baldridge and J. S. Siegel, Of Graphs and Graphenes: Molecular Design and Chemical Studies of Aromatic Compounds, Angew. Chem., Int. Ed., 2013, 52, 5436–5438 CrossRef CAS PubMed.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Contorted Polycyclic Aromatics, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed.
- K. M. Magiera, V. Aryal and W. A. Chalifoux, Alkyne benzannulations in the preparation of contorted nanographenes, Org. Biomol. Chem., 2020, 18, 2372–2386 RSC.
- A. V. Gulevskaya and D. I. Tonkoglazova, Alkyne-Based Syntheses of Carbo- and Heterohelicenes, Adv. Synth. Catal., 2022, 364, 2502–2539 CrossRef CAS.
- Y. Zhang, S. H. Pun and Q. Miao, The Scholl Reaction as a Powerful Tool for Synthesis of Curved Polycyclic Aromatics, Chem. Rev., 2022, 122, 14554–14593 CrossRef CAS PubMed.
- M. Rickhaus, M. Mayor and M. Juríček, Chirality in curved polyaromatic systems, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC.
- J. R. Brandt, F. Salerno and M. J. Fuchter, The added value of small-molecule chirality in technological applications, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS.
- J. M. Fernández-Garciá, P. J. Evans, S. Filippone, M. Á. Herranz and N. Martín, Chiral Molecular Carbon Nanostructures, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef PubMed.
- M. Rickhaus, M. Mayor and M. Juríček, Strain-induced helical chirality in polyaromatic systems, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC.
- R. A. Pascal, Twisted Acenes, Chem. Rev., 2006, 106, 4809–4819 CrossRef CAS PubMed.
- A. Nitti, G. Preda and D. Pasini, in Chiral Building Blocks in Asymmetric Synthesis, 2022, pp. 551–582 Search PubMed.
- J. Lu, D. M. Ho, N. J. Vogelaar, C. M. Kraml, S. Bernhard, N. Byrne, L. R. Kim and R. A. Pascal, Synthesis, Structure, and Resolution of Exceptionally Twisted Pentacenes, J. Am. Chem. Soc., 2006, 128, 17043–17050 CrossRef CAS PubMed.
- R. G. Clevenger, B. Kumar, E. M. Menuey and K. V. Kilway, Synthesis and Structure of a Longitudinally Twisted Hexacene, Chem. – Eur. J., 2018, 24, 3113–3116 CrossRef CAS PubMed.
- R. S. Walters, C. M. Kraml, N. Byrne, D. M. Ho, Q. Qin, F. J. Coughlin, S. Bernhard and R. A. Pascal, Configurationally Stable Longitudinally Twisted Polycyclic Aromatic Compounds, J. Am. Chem. Soc., 2008, 130, 16435–16441 CrossRef CAS PubMed.
- X. Geng, J. T. Mague, J. P. Donahue and R. A. Pascal, Hairpin Furans and Giant Biaryls, J. Org. Chem., 2016, 81, 3838–3847 CrossRef CAS PubMed.
- W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Varganov and W. A. Chalifoux, Chiral Peropyrene: Synthesis, Structure, and Properties, J. Am. Chem. Soc., 2017, 139, 13102–13109 CrossRef CAS PubMed.
- M. S. Newman and D. Lednicer, The Synthesis and Resolution of Hexahelicene1, J. Am. Chem. Soc., 1956, 78, 4765–4770 CrossRef CAS.
- L. Liu and T. J. Katz, Simple preparation of a helical quinone, Tetrahedron Lett., 1990, 31, 3983–3986 CrossRef CAS.
- W. H. Laarhoven and W. J. C. Prinsen, Carbohelicenes and heterohelicenes, Top. Curr. Chem., 1984, 125, 63–130 CrossRef CAS.
- R. H. Martin, The Helicenes, Angew. Chem., Int. Ed. Engl., 1974, 13, 649–660 CrossRef.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Advances in helicene derivatives with circularly polarized luminescence, Chem. Commun., 2019, 55, 13793–13803 RSC.
- M. Jakubec and J. Storch, Recent Advances in Functionalizations of Helicene Backbone, J. Org. Chem., 2020, 85, 13415–13428 CrossRef CAS PubMed.
- Y. Shen and C.-F. Chen, Helicenes: Synthesis and Applications, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- T. Mori, Chiroptical properties of symmetric double, triple, and multiple helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- J. Storch, J. Žádný, V. Církva, M. Jakubec, J. Hrbáč and J. Vacek, in Helicenes, 2022, pp. 1–52 Search PubMed.
- A. Urbano, Recent Developments in the Synthesis of Helicene-Like Molecules, Angew. Chem., Int. Ed., 2003, 42, 3986–3989 CrossRef CAS PubMed.
- C. Li, Y. Yang and Q. Miao, Recent Progress in Chemistry of Multiple Helicenes, Chem. – Asian J., 2018, 13, 884–894 CrossRef CAS PubMed.
- K. Kato, Y. Segawa and K. Itami, Symmetric Multiple Carbohelicenes, Synlett, 2019, 30, 370–377 CrossRef CAS.
- F. Pop, N. Zigon and N. Avarvari, Main-Group-Based Electro- and Photoactive Chiral Materials, Chem. Rev., 2019, 119, 8435–8478 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P), Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- G. R. Kiel, S. C. Patel, P. W. Smith, D. S. Levine and T. D. Tilley, Expanded Helicenes: A General Synthetic Strategy and Remarkable Supramolecular and Solid-State Behavior, J. Am. Chem. Soc., 2017, 139, 18456–18459 CrossRef CAS PubMed.
- S. Kezuka, S. Tanaka, T. Ohe, Y. Nakaya and R. Takeuchi, Iridium Complex-Catalyzed [2 + 2 + 2] Cycloaddition of α,ω-Diynes with Monoynes and Monoenes, J. Org. Chem., 2006, 71, 543–552 CrossRef CAS PubMed.
- G. R. Kiel, K. L. Bay, A. E. Samkian, N. J. Schuster, J. B. Lin, R. C. Handford, C. Nuckolls, K. N. Houk and T. D. Tilley, Expanded Helicenes as Synthons for Chiral Macrocyclic Nanocarbons, J. Am. Chem. Soc., 2020, 142, 11084–11091 CrossRef CAS PubMed.
- K. Fujise, E. Tsurumaki, G. Fukuhara, N. Hara, Y. Imai and S. Toyota, Multiple Fused Anthracenes as Helical Polycyclic Aromatic Hydrocarbon Motif for Chiroptical Performance Enhancement, Chem. – Asian J., 2020, 15, 2456–2461 CrossRef CAS PubMed.
- K. Morioka, K. Wakamatsu, E. Tsurumaki and S. Toyota, Synthesis, Structures, and Properties of Helically Fused Anthraquinones with Unusually Close Carbonyl-Carbonyl Contacts, Chem. – Eur. J., 2022, 28, e202103694 CrossRef CAS PubMed.
- W. Fan, T. Matsuno, Y. Han, X. Wang, Q. Zhou, H. Isobe and J. Wu, Synthesis and Chiral Resolution of Twisted Carbon Nanobelts, J. Am. Chem. Soc., 2021, 143, 15924–15929 CrossRef CAS PubMed.
- G.-F. Huo, T. M. Fukunaga, X. Hou, Y. Han, W. Fan, S. Wu, H. Isobe and J. Wu, Facile Synthesis and Chiral Resolution of Expanded Helicenes with up to 35 cata-Fused Benzene Rings, Angew. Chem., Int. Ed., 2023, 135, e202218090 CrossRef.
- M. Krzeszewski, H. Ito and K. Itami, Infinitene: A Helically Twisted Figure-Eight [12]Circulene Topoisomer, J. Am. Chem. Soc., 2022, 144, 862–871 CrossRef CAS PubMed.
- M. J. Fuchter, M. Weimar, X. Yang, D. K. Judge and A. J. P. White, An unusual oxidative rearrangement of [7]-helicene, Tetrahedron Lett., 2012, 53, 1108–1111 CrossRef CAS.
- Y. Nakakuki, T. Hirose and K. Matsuda, Synthesis of a Helical Analogue of Kekulene: A Flexible π-Expanded Helicene with Large Helical Diameter Acting as a Soft Molecular Spring, J. Am. Chem. Soc., 2018, 140, 15461–15469 CrossRef CAS PubMed.
- W. Yang, R. R. Kazemi, N. Karunathilake, V. J. Catalano, M. A. Alpuche-Aviles and W. A. Chalifoux, Expanding the scope of peropyrenes and teropyrenes through a facile InCl3-catalyzed multifold alkyne benzannulation, Org. Chem. Front., 2018, 5, 2288–2295 RSC.
- R. Bam, W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Four-Fold Alkyne Benzannulation: Synthesis, Properties, and Structure of Pyreno[a]pyrene-Based Helicene Hybrids, Org. Lett., 2019, 21, 8652–8656 CrossRef CAS PubMed.
- P. Sitaula, R. J. Malone, G. Longhi, S. Abbate, E. Gualtieri, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano and W. A. Chalifoux, π-Extended Helical Nanographenes: Synthesis and Photophysical Properties of Naphtho[1,2-a]pyrenes, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12931703.v1.
- P. Ravat, T. Šolomek, P. Ribar and M. Juríček, Biradicaloid with a Twist: Lowering the Singlet-Triplet Gap, Synlett, 2016, 27, 1613–1617 CrossRef CAS.
- P. Ravat, T. Šolomek, D. Häussinger, O. Blacque and M. Juríček, Dimethylcethrene: A Chiroptical Diradicaloid Photoswitch, J. Am. Chem. Soc., 2018, 140, 10839–10847 CrossRef CAS PubMed.
- D. Čavlović, D. Häussinger, O. Blacque, P. Ravat and M. Juríček, Nonacethrene Unchained: A Cascade to Chiral Contorted Conjugated Hydrocarbon with Two sp3-Defects, JACS Au, 2022, 2, 1616–1626 CrossRef PubMed.
- I. G. Stará and I. Starý, Helically Chiral Aromatics: The Synthesis of Helicenes by [2 + 2 + 2] Cycloisomerization of π-Electron Systems, Acc. Chem. Res., 2020, 53, 144–158 CrossRef PubMed.
- M. Buchta, J. Rybáček, A. Jančařík, A. A. Kudale, M. Buděšínský, J. V. Chocholoušová, J. Vacek, L. Bednárová, I. Císařová, G. J. Bodwell, I. Starý and I. G. Stará, Chimerical Pyrene-Based [7]Helicenes as Twisted Polycondensed Aromatics, Chem. – Eur. J., 2015, 21, 8910–8917 CrossRef CAS PubMed.
- F. Morita, J. Nogami, A. J. Araujo Dias, S. Kinoshita, Y. Nagashima and K. Tanaka, Regiodivergent Synthesis and π-Stacking-Induced Chiral Self-Recognition of Hexabenzocoronene-Based [6]Helicenes, Eur. J. Org. Chem., 2022, e202200690 CAS.
- V. Pelliccioli, F. Cardano, G. Renno, F. Vasile, C. Graiff, G. Mazzeo, A. Fin, G. Longhi, S. Abbate, A. Rosetti, C. Villani, G. Viscardi, E. Licandro and S. Cauteruccio, Synthesis, Stereochemical and Photophysical Properties of Functionalized Thiahelicenes, Catalysts, 2022, 12, 366 CrossRef CAS.
- M. Baudillon, T. Cauchy, N. Vanthuyne, N. Avarvari and F. Pop, Configurationally stable dithia[7]helicene and dithia-quasi[8]circulene fused dithiolones, Org. Chem. Front., 2022, 9, 4260–4270 RSC.
- R. J. Malone, J. Spengler, R. A. Carmichael, K. Ngo, F. Würthner and W. A. Chalifoux, Synthesis and Properties of Achiral and Chiral Dipyrenoheteroles and Related Compounds, Org. Lett., 2023, 25, 226–230 CrossRef CAS PubMed.
- T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, Azaboradibenzo[6]helicene: Carrier Inversion Induced by Helical Homochirality, J. Am. Chem. Soc., 2012, 134, 19600–19603 CrossRef CAS PubMed.
- T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Enantioselective Synthesis of 1,12-Disubstituted [4]Helicenes, Angew. Chem., Int. Ed., 2020, 59, 5660–5664 CrossRef CAS PubMed.
- L. D. M. Nicholls, M. Marx, T. Hartung, E. González-Fernández, C. Golz and M. Alcarazo, TADDOL-Derived Cationic Phosphonites: Toward an Effective Enantioselective Synthesis of [6]Helicenes via Au-Catalyzed Alkyne Hydroarylation, ACS Catal., 2018, 8, 6079–6085 CrossRef CAS.
- P. Redero, T. Hartung, J. Zhang, L. D. M. Nicholls, G. Zichen, M. Simon, C. Golz and M. Alcarazo, Enantioselective Synthesis of 1-Aryl Benzo[5]helicenes Using BINOL-Derived Cationic Phosphonites as Ancillary Ligands, Angew. Chem., Int. Ed., 2020, 59, 23527–23531 CrossRef CAS PubMed.
- J. Zhang, M. Simon, C. Golz and M. Alcarazo, Gold-Catalyzed Atroposelective Synthesis of 1,1′-Binaphthalene-2,3′-diols, Angew. Chem., Int. Ed., 2020, 59, 5647–5650 CrossRef CAS PubMed.
- V. Pelliccioli, T. Hartung, M. Simon, C. Golz, E. Licandro, S. Cauteruccio and M. Alcarazo, Enantioselective Synthesis of Dithia[5]helicenes and their Postsynthetic Functionalization to Access Dithia[9]helicenes, Angew. Chem., Int. Ed., 2022, 61, e202114577 CrossRef CAS PubMed.
- F. Chen, W. Gu, A. Saeki, M. Melle-Franco and A. Mateo-Alonso, A Sterically Congested Nitrogenated Benzodipentaphene with a Double π-Expanded Helicene Structure, Org. Lett., 2020, 22, 3706–3711 CrossRef CAS PubMed.
- D. I. Tonkoglazova, A. V. Gulevskaya, K. A. Chistyakov and O. I. Askalepova, Synthesis, crystal structures and properties of carbazole-based [6]helicenes fused with an azine ring, Beilstein J. Org. Chem., 2021, 17, 11–21 CrossRef CAS PubMed.
- Z. Xie and F. Würthner, Perylene Bisimides with Rigid 2,2′-Biphenol Bridges at Bay Area as Conjugated Chiral Platforms, Org. Lett., 2010, 12, 3204–3207 CrossRef CAS PubMed.
- F. Würthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C.-C. You, P. Jonkheijm, J. v. Herrikhuyzen, A. P. H. J. Schenning, P. P. A. M. van der Schoot, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, Supramolecular p−n-Heterojunctions by Co-Self-Organization of Oligo(p-phenylene Vinylene) and Perylene Bisimide Dyes, J. Am. Chem. Soc., 2004, 126, 10611–10618 CrossRef PubMed.
- C. Lütke Eversloh, Z. Liu, B. Müller, M. Stangl, C. Li and K. Müllen, Core-extended terrylene tetracarboxdiimide: Synthesis and chiroptical characterization, Org. Lett., 2011, 13, 5528–5531 CrossRef PubMed.
- F. Saal and P. Ravat, Imide-Functionalized Helical PAHs: A Step towards New Chiral Functional Materials, Synlett, 2021, 32, 1879–1890 CrossRef CAS.
- J. Li, P. Li, M. Fan, X. Zheng, J. Guan and M. Yin, Chirality of Perylene Diimides: Design Strategies and Applications, Angew. Chem., Int. Ed., 2022, 61, e202202532 CAS.
- L. Zhang, S. Chen, J. Jiang, X. Dong, Y. Cai, H.-J. Zhang, J. Lin and Y.-B. Jiang, C- and S-Shaped Perylene Diimide Heterohelicenes: Modular Synthesis and Spiral-Stair-Like π-Stacking, Org. Lett., 2022, 24, 3179–3183 CrossRef CAS PubMed.
- D. Meng, G. Liu, C. Xiao, Y. Shi, L. Zhang, L. Jiang, K. K. Baldridge, Y. Li, J. S. Siegel and Z. Wang, Corannurylene Pentapetalae, J. Am. Chem. Soc., 2019, 141, 5402–5408 CrossRef CAS PubMed.
- G. Liu, Y. Liu, C. Zhao, Y. Li, Z. Wang and H. Tian, Stereoselective Chiral Molecular Carbon Imides Featuring 12-Fold [5]helicenes Around Four Cores, Angew. Chem., Int. Ed., 2023, 62, e202214769 CAS.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, Three-Bladed Rylene Propellers with Three-Dimensional Network Assembly for Organic Electronics, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS PubMed.
- M. Żyła-Karwowska, H. Zhylitskaya, J. Cybińska, T. Lis, P. J. Chmielewski and M. Stępień, An Electron-Deficient Azacoronene Obtained by Radial π Extension, Angew. Chem., Int. Ed., 2016, 55, 14658–14662 CrossRef PubMed.
- M. Navakouski, H. Zhylitskaya, P. J. Chmielewski, T. Lis, J. Cybińska and M. Stępień, Stereocontrolled Synthesis of Chiral Heteroaromatic Propellers with Small Optical Bandgaps, Angew. Chem., Int. Ed., 2019, 58, 4929–4933 CrossRef CAS PubMed.
- B. Liu, M. Böckmann, W. Jiang, N. L. Doltsinis and Z. Wang, Perylene Diimide-Embedded Double [8]Helicenes, J. Am. Chem. Soc., 2020, 142, 7092–7099 CrossRef CAS PubMed.
- C. Schaack, A. M. Evans, F. Ng, M. L. Steigerwald and C. Nuckolls, High-Performance Organic Electronic Materials by Contorting Perylene Diimides, J. Am. Chem. Soc., 2022, 144, 42–51 CrossRef CAS PubMed.
- X. Xiao, S. K. Pedersen, D. Aranda, J. Yang, R. A. Wiscons, M. Pittelkow, M. L. Steigerwald, F. Santoro, N. J. Schuster and C. Nuckolls, Chirality Amplified: Long, Discrete Helicene Nanoribbons, J. Am. Chem. Soc., 2021, 143, 983–991 CrossRef CAS PubMed.
- P. Osswald and F. Würthner, Effects of Bay Substituents on the Racemization Barriers of Perylene Bisimides: Resolution of Atropo-Enantiomers, J. Am. Chem. Soc., 2007, 129, 14319–14326 CrossRef CAS PubMed.
- L. Zhang, I. Song, J. Ahn, M. Han, M. Linares, M. Surin, H.-J. Zhang, J. H. Oh and J. Lin, π-Extended perylene diimide double-heterohelicenes as ambipolar organic semiconductors for broadband circularly polarized light detection, Nat. Commun., 2021, 12, 142 CrossRef CAS PubMed.
- B. Mahlmeister, R. Renner, O. Anhalt, M. Stolte and F. Würthner, Axially chiral bay-tetraarylated perylene bisimide dyes as non-fullerene acceptors in organic solar cells, J. Mater. Chem. C, 2022, 10, 2581–2591 RSC.
- R. Renner, B. Mahlmeister, O. Anhalt, M. Stolte and F. Würthner, Chiral Perylene Bisimide Dyes by Interlocked Arene Substituents in the Bay Area, Chem. – Eur. J., 2021, 27, 11997–12006 CrossRef CAS PubMed.
- B. Mahlmeister, M. Mahl, H. Reichelt, K. Shoyama, M. Stolte and F. Würthner, Helically Twisted Nanoribbons Based on Emissive Near-Infrared Responsive Quaterrylene Bisimides, J. Am. Chem. Soc., 2022, 144, 10507–10514 CrossRef CAS PubMed.
- I. V. Alabugin and E. Gonzalez-Rodriguez, Alkyne Origami: Folding Oligoalkynes into Polyaromatics, Acc. Chem. Res., 2018, 51, 1206–1219 CrossRef CAS PubMed.
- K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, Traceless Directing Groups in Radical Cascades: From Oligoalkynes to Fused Helicenes without Tethered Initiators, J. Am. Chem. Soc., 2015, 137, 1165–1180 CrossRef CAS PubMed.
- J. K. Li, X. Y. Chen, Y. L. Guo, X. C. Wang, A. C. H. Sue, X. Y. Cao and X. Y. Wang, B,N-Embedded Double Hetero[7]helicenes with Strong Chiroptical Responses in the Visible Light Region, J. Am. Chem. Soc., 2021, 143, 17958–17963 CrossRef CAS PubMed.
- W. Zheng, T. Ikai, K. Oki and E. Yashima, Consecutively fused single-, double-, and triple-expanded helicenes, Nat. Sci., 2022, 2, e20210047 CAS.
- S. H. Pun, K. M. Cheung, D. Yang, H. Chen, Y. Wang, S. V. Kershaw and Q. Miao, A Near-Infrared Absorbing and Emissive Quadruple Helicene Enabled by the Scholl Reaction of Perylene, Angew. Chem., Int. Ed., 2022, 61, e202113203 CrossRef CAS PubMed.
- T. Ikai, S. Yamakawa, N. Suzuki and E. Yashima, One-Step Simultaneous Synthesis of Circularly Polarized Luminescent Multiple Helicenes Using a Chrysene Framework, Chem. – Asian J., 2021, 16, 769–774 CrossRef CAS PubMed.
- J. Hu, Q. Xiang, J. Xu, Z. Xu, G. Chen and Z. Sun, Stable and twisted 5,6:12,13-dinaphthozethrene from angular π-extension, Chem. Commun., 2021, 57, 9712–9715 RSC.
- T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, Synthesis, Structures, and Properties of Hexapole Helicenes: Assembling Six [5]Helicene Substructures into Highly Twisted Aromatic Systems, J. Am. Chem. Soc., 2017, 139, 18512–18521 CrossRef CAS PubMed.
- V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, Chiral Nanographene Propeller Embedding Six Enantiomerically Stable [5]Helicene Units, J. Am. Chem. Soc., 2017, 139, 18508–18511 CrossRef CAS PubMed.
- F. Zhang, E. Michail, F. Saal, A. M. Krause and P. Ravat, Stereospecific Synthesis and Photophysical Properties of Propeller-Shaped C90H48 PAH, Chem. – Eur. J., 2019, 25, 16241–16245 CrossRef CAS PubMed.
- Y. Nakai, T. Mori and Y. Inoue, Theoretical and Experimental Studies on Circular Dichroism of Carbo[n]helicenes, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS PubMed.
- R. Yamano, Y. Shibata and K. Tanaka, Synthesis of Single and Double Dibenzohelicenes by Rhodium–Catalyzed Intramolecular [2 + 2 + 2] and [2 + 1 + 2 + 1] Cycloaddition, Chem. – Eur. J., 2018, 24, 6364–6370 CrossRef CAS PubMed.
- Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Müllen and A. Narita, Benzo-Fused Double [7]Carbohelicene: Synthesis, Structures, and Physicochemical Properties, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef CAS PubMed.
- Y. Hu, G. M. Paternò, X. Y. Wang, X. C. Wang, M. Guizzardi, Q. Chen, D. Schollmeyer, X. Y. Cao, G. Cerullo, F. Scotognella, K. Müllen and A. Narita, π-Extended Pyrene-Fused Double [7]Carbohelicene as a Chiral Polycyclic Aromatic Hydrocarbon, J. Am. Chem. Soc., 2019, 141, 12797–12803 CrossRef CAS PubMed.
- J. Tan, X. Xu, J. Liu, S. Vasylevskyi, Z. Lin, R. Kabe, Y. Zou, K. Müllen, A. Narita and Y. Hu, Synthesis of a π-Extended Double [9]Helicene, Angew. Chem., Int. Ed., 2023, 62, e202218494 CrossRef CAS PubMed.
- Z. Sun, C. Yu, N. Zhang, L. Li, Y. Jiao, P. Thiruvengadam, D. Wu and F. Zhang, Divergent Synthesis of Double Heterohelicenes as Strong Chiral Double Hydrogen-Bonding Donors, Org. Lett., 2022, 24, 6670–6675 CrossRef CAS PubMed.
- A. Konishi and M. Yasuda, Breathing New Life into Nonalternant Hydrocarbon Chemistry: Syntheses and Properties of Polycyclic Hydrocarbons Containing Azulene, Pentalene, and Heptalene Frameworks, Chem. Lett., 2021, 50, 195–212 CrossRef CAS.
- Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Construction of Heptagon-Containing Molecular Nanocarbons, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef CAS PubMed.
- S. H. Pun and Q. Miao, Toward Negatively Curved Carbons, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed.
- S. Liu, L. M. Roch, O. Allemann, J. Xu, N. Vanthuyne, K. K. Baldridge and J. S. Siegel, 1,2,3- versus 1,2-Indeno Ring Fusions Influence Structure Property and Chirality of Corannulene Bowls, J. Org. Chem., 2018, 83, 3979–3986 CrossRef CAS PubMed.
- S. Wang, M. Tang, L. Wu, L. Bian, L. Jiang, J. Liu, Z.-B. Tang, Y. Liang and Z. Liu, Linear Nonalternant Isomers of Acenes Fusing Multiple Azulene Units, Angew. Chem., Int. Ed., 2022, 61, e202205658 CAS.
- Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K. H. Low, S. Zhou, D. L. Phillips, X. Feng and J. Liu, Defective Nanographenes Containing Seven-Five-Seven (7-5-7)-Membered Rings, J. Am. Chem. Soc., 2021, 143, 2353–2360 CrossRef CAS PubMed.
- M. Schnitzlein, C. Mützel, K. Shoyama, J. M. Farrell and F. Würthner, PAHs Containing both Heptagon and Pentagon: Corannulene Extension by [5 + 2] Annulation, Eur. J. Org. Chem., 2022, 2022, e202101273 CrossRef CAS.
- T. Kirschbaum, F. Rominger and M. Mastalerz, A Chiral Polycyclic Aromatic Hydrocarbon Monkey Saddle, Angew. Chem., 2020, 132, 276–280 CrossRef.
- X. Yang, F. Rominger and M. Mastalerz, Contorted Polycyclic Aromatic Hydrocarbons with Two Embedded Azulene Units, Angew. Chem., Int. Ed., 2019, 58, 17577–17582 CrossRef CAS PubMed.
- T. Kirschbaum, F. Rominger and M. Mastalerz, Synthesis of a Benzotrisazulene via Trioxobenzotrisazulene, Chem. – Eur. J., 2023, e202301470 CrossRef PubMed.
- D. Popp, S. M. Elbert, C. Barwig, J. Petry, F. Rominger and M. Mastalerz, Palladium-Catalyzed Cyclization of a Pyryne Precursor to Higher Pyrenylenes, Angew. Chem., Int. Ed., 2023, 62, e202219277 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2023 |