
Regulation engineering of the surface and structure of perovskite-based electrocatalysts for the oxygen evolution reaction
Ning
Yu
,
Zhi-Jie
Zhang
,
Yong-Ming
Chai
* and
Bin
Dong
 *
*
State Key Laboratory of Heavy Oil Processing, College of Chemistry and Chemical Engineering, China University of Petroleum (East China), Qingdao 266580, P. R. China. E-mail: ymchai@upc.edu.cn; dongbin@upc.edu.cn; Fax: +86-532-86981156; Tel: +86-532-86981156
First published on 13th June 2023
Abstract
Perovskite oxides of low cost and with an adjustable structure have been widely used as electrocatalysts for the oxygen evolution reaction (OER). However, most perovskite oxides have inherent barriers of low catalytic activity and poor electrical conductivity. The reaction at the active site and the adsorption/desorption of the reactants are carried out more efficiently on the catalyst surface. In order to regulate the surface and structure of a catalyst to improve the catalytic activity of perovskite oxides in the OER, many strategies have been explored. In this paper, the descriptor of catalytic activity of perovskite oxides is described. The oxygen evolution mechanisms (the adsorbate evolution mechanism, AEM, and lattice oxygen evolution mechanism, LOM) and the regulation mode between AEM and LOM have been deeply analyzed to guide the design of high performing catalysts for the OER. The research progress of perovskite oxide catalysts in recent years in terms of surface composition, design morphology control and defect engineering is reviewed systematically. In particular, the Ir/Ru perovskite in acidic media that can be used in a proton exchange membrane water electrolyser (PEMWE) is discussed in detail. Finally, the existing problems of perovskite oxide based electrocatalysts and their prospects for practical applications in the OER are presented.
 Ning Yu | Ning Yu received her BS degree from Liaocheng University. She is pursuing her PhD under the supervision of Professor Bin Dong in the State Key Laboratory of Heavy Oil Processing, College of Chemistry and Chemical Engineering, China University of Petroleum (East China). Her research interests focus on the perovskite-based nanomaterials for electrocatalysis for water electrolysis. |
 Zhi-Jie Zhang | Zhijie Zhang received her BS degree from Liaocheng University. She is pursuing her master's degree under the supervision of Professor Bin Dong in the State Key Laboratory of Heavy Oil Processing, College of Chemistry and Chemical Engineering, China University of Petroleum (East China). Her research interests focus on transition metal-based nanostructures for electrocatalysis for water splitting. |
 Yong-Ming Chai | Prof. Yong-Ming Chai received his PhD from China University of Petroleum (East China) in 2008. Now he is a professor and the deputy director of State Key Laboratory of Heavy Oil Processing, College of Chemistry and Chemical Engineering, China University of Petroleum (East China). He carried out research as a visiting scholar in Marquette University from 2015.03 to 2016.03. His research interests focus on transition metal-based catalysts for the utilization and production of green hydrogen including the hydrodesulfurization process of heavy oil and water splitting. Prof. Chai is an associate editor of Nano-Micro Letters, a Young Editorial Board Member of Journal of Fuel Chemistry and Technology. |
 Bin Dong | Prof. Bin Dong received his BS and PhD from Lanzhou University in 2002 and 2008, respectively. He carried out research as a visiting scholar in Marquette University from March 2014 to March 2015. Now he is a Professor at the State Key Laboratory of Heavy Oil Processing, College of Chemistry and Chemical Engineering, China University of Petroleum (East China). His research interests mainly focus on transition metal-based functional materials for energy conversion and storage including electrocatalysis and photoelectrocatalysis. Prof. Dong has published more than 150 SCI papers including 7 ESI papers as the first-author or corresponding author with an H index 56. He was selected as a Highly Cited Chinese Researcher from Elsevier in 2022. Prof. Dong is an Editorial Board Member of Chinese Chemical Letters, an assistant editor of Nano-Micro Letters, a Young Editorial Board Member of Nano-Micro Letters, eScience, a Young Editorial Board Member of Chinese Journal of Structural Chemistry, and a member of the International Society of Electrochemisty. |
1. Introduction
Clean and renewable energy technologies have been rapidly developed to address serious environmental problems caused by excessive consumption of non-renewable resources such as natural gas, oil and coal.1,2 Hydrogen is an ideal renewable energy source with zero pollution. Electrochemical water decomposition hydrogen production (H2O → H2 + 1/2O2) is widely regarded as one of the most attractive sustainable energy conversion and storage technologies.3–5 Essentially, the existence of slow multistep proton-coupled electron transfer in the oxygen evolution reaction (OER) prevents the realization of hydrogen production for efficient water electrolysis. Noble metal catalysts, such as RuO2 and IrO2, show excellent OER performance, but their high cost in practical applications seriously hinders their widespread application.6,7 This challenge has ignited a surge of research to find cost-effective and durable non-precious transition metal oxides as electrocatalysts for OERs.In 1839, a mineral composed of calcium titanate (CaTiO3) was first discovered from skarn by the Prussian mineralogist Gustav Rose, who named it after the mineralogist L. A. Perovski. To date, significant progress has been made in the in-depth understanding of the reaction mechanism of perovskite-based catalysts and in the design of efficient catalysts (Fig. 1). The typical single perovskite Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) exhibits higher intrinsic activity in alkaline electrolytes than the IrO2 reference in some cases.8 A series of compelling dual perovskites with outstanding OER activity have been predicted and further evaluated by developing activity descriptor data-driven machine learning methods that can accelerate the design of highly active and low-cost electrocatalysts.9 Perovskite oxides usually represent a class of materials with the general formula ABO3 (A = alkaline, alkaline earth or rare earth metal, B = transition metal, and O = oxygen). Recently, perovskite oxides with ABO3 rich in earth elements have attracted great interest in OER catalysis due to their high intrinsic activity and large composition/structure adjustability.10–12 In the basic perovskite oxide structure, the B-site cations coordinate with the oxygen atom octahedron (BO6), whose shared angles in the three directions of the crystal are located in the center of the cube (Fig. 2a). The A-site cations exhibit 12-fold coordination in the octahedral interspace. The perovskite crystal structure shows incomparable flexibility, which can incorporate almost all metal ions in the periodic table (about 90% of the elements in the periodic table) into the perovskite structure (Fig. 2b). Many studies have developed catalysts with double perovskites (A2B2O6),13 triple perovskites (A3B3O9),14 quadruple perovskites (A4B4O12),15 and Ruddlesden–Popper perovskites (An+1BnO3n+1 (n = 1, 2, and 3)).16 The special structure and properties of perovskite offer several advantages in the OER: (1) Perovskite oxides have a robust crystalline structure that is highly stable under harsh reaction conditions, such as high temperatures and corrosive environments, making them suitable for use in the OER. (2) The electronic properties of perovskite oxides can be tuned by varying the composition of the A and B-site cations, allowing for the optimization of the electronic structure for OER catalysis. (3) The unique combination of metal cations in the perovskite structure can lead to synergistic effects that enhance the catalytic activity of the material, leading to improved OER performance. (4) Perovskite oxides can achieve high catalytic activity for the OER at low overpotentials, which is the extra energy needed to drive the reaction forward.
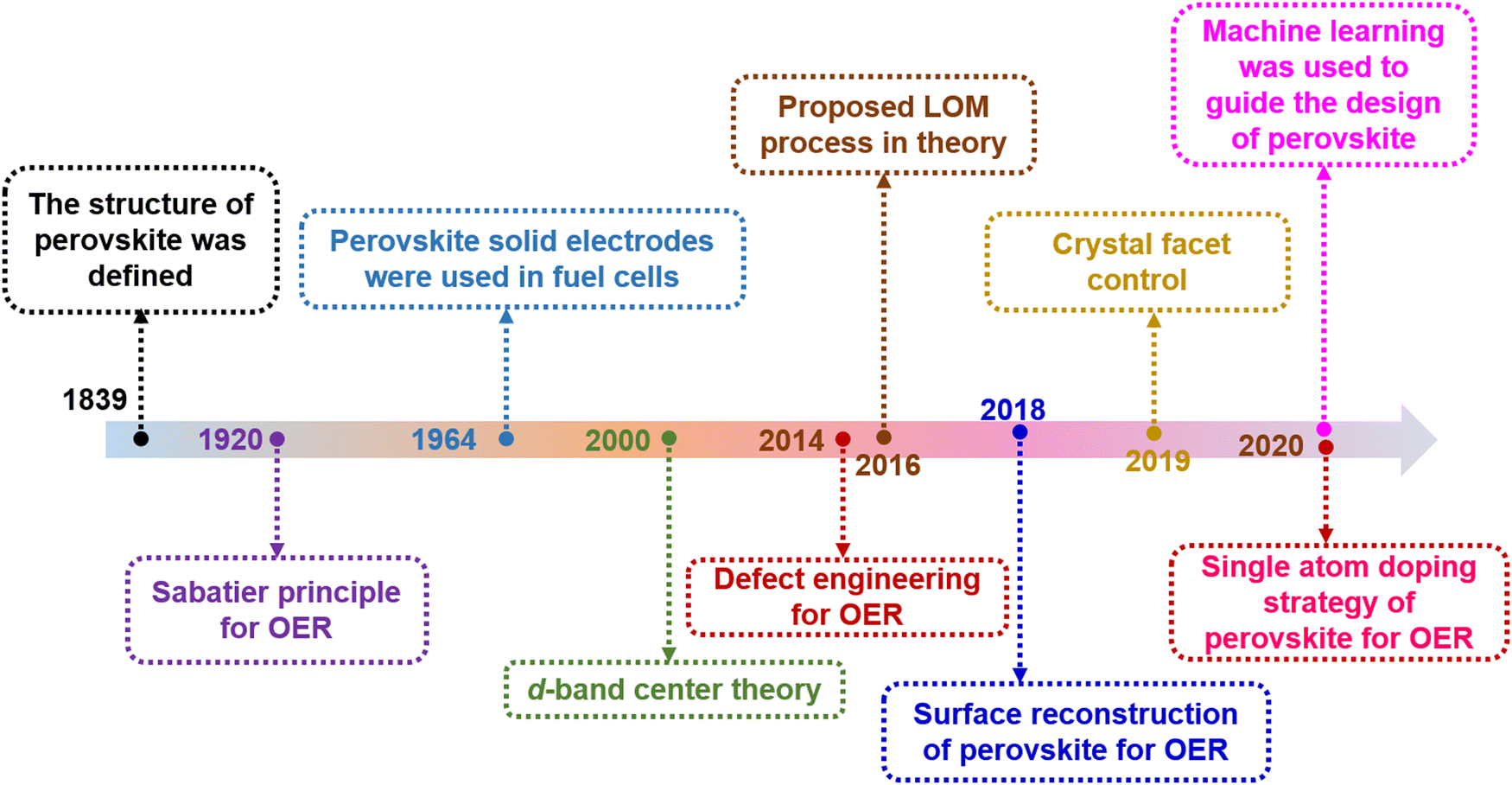 | ||
| Fig. 1 The timeline-based accomplishments in the exploration of perovskite oxides for the OER. All the data were searched for in the Web of Science Core Collection. | ||
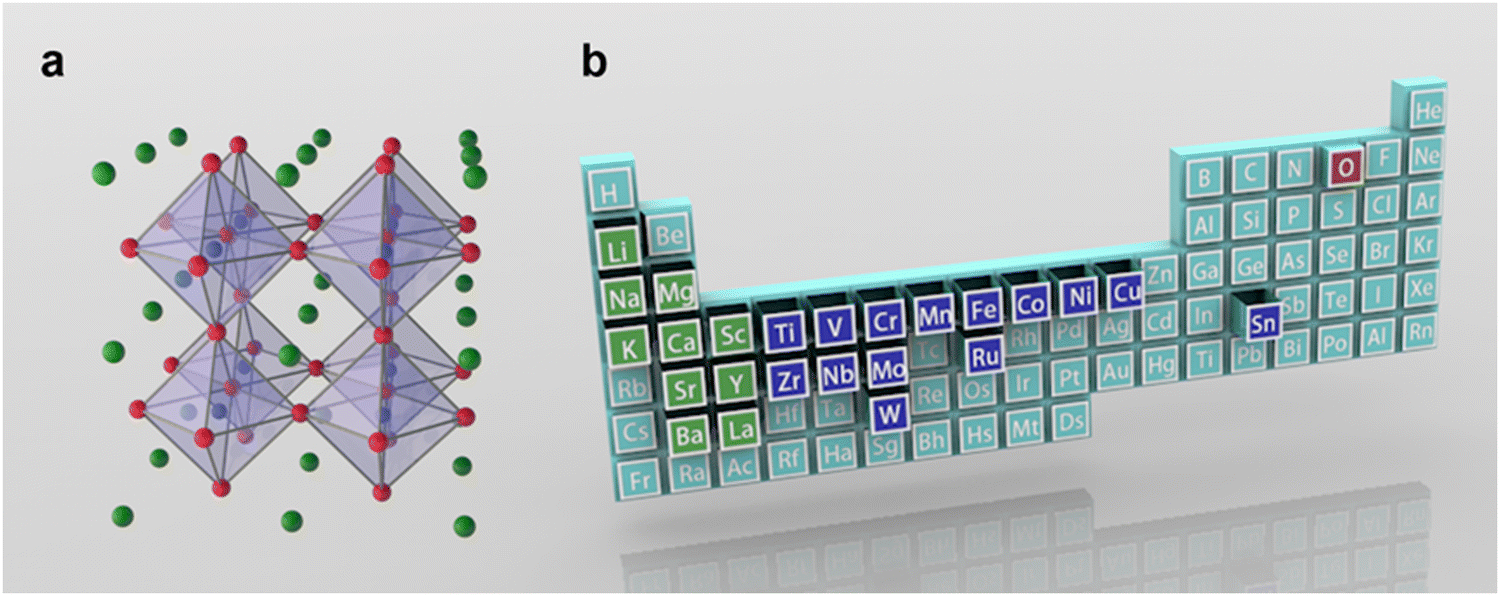 | ||
| Fig. 2 (a) Schematic illustration of the structure for perovskites (ABO3). (b) The commonly used element in perovskite oxides. Green is the A-site element, blue is the B-site element, and red is oxygen. | ||
The special structure allows perovskite to exhibit properties comparable to those of Ir/Ru base standards.17 Noble-metal base perovskite oxides in polymer electrolyte membrane (PEM) water electrolyzers have the advantages of a more stable structure and a lower noble metal content when running at a higher current density.12,18 However, pure perovskite oxides (for example, LaNiO3, LaCoO3, LaFeO3, etc.) as OER catalysts still have the disadvantages of poor conductivity and few active sites limiting their development in this field. Reducing the overpotential of the OER, or the energy level of the catalyst should be closer to the equilibrium potential of 1.23 V (2H2O(l) → O2(g) + 4H+(aq) + 4e−, E0 = 1.23 V), is frequently one of the criteria for measuring the catalytic activity in the design of catalysts.19,20 The reaction can be initiated thermodynamically because the electron exchange between the electrocatalyst and the reactants is more favorable. Many effective strategies, such as hybridization, crystal facet control, morphology control, and defect engineering, have been applied to improve the intrinsic activity and stability of perovskite oxides for the OER. Perovskite oxides are widely used in catalysis, electrocatalysis and photocatalysis. They are also employed as components of devices that facilitate new energy innovations, such as catalytic metal–air cells,21 water splitting,22 and solid oxide fuel cells.23
Herein, the descriptor of catalytic activity of perovskite oxides is introduced. The recent strategy for adjusting the physical and chemical characteristics of perovskite oxides is described for application in OER. The catalytic mechanisms of the OER, including AEM and LOM, are deeply understood, and the regulation of the oxygen evolution mechanism is summarized. Strategies for improving the catalytic activity and stability of OER perovskite oxides are systematically studied, including composition engineering, morphology modulation, defect engineering, etc. In particular, the development of Ir/Ru-based perovskite oxide catalysts for PEMWEs under acidic environments is discussed. Finally, the challenges with employing perovskite oxides in practical applications as OER electrocatalysts are prospected.
2. Reaction mechanisms of perovskite-based electrocatalysts
2.1. AEM
The OER on the anode is one of the half reactions in the water cracking process, which is a 4-electron process. There are two widely accepted OER mechanisms, identified as the adsorbate evolution mechanism (AEM) and lattice oxygen evolution mechanism (LOM). OER behaves differently under acidic and alkaline conditions.The AEM process occurs through multiple oxygen intermediates including OH*, O*, OOH*, and O2, in which a series of consistent electron–proton transfer steps occur between the metal band and the adsorbent oxygen intermediate.24–26 Because of this process, AEM is considered to be a metal redox catalytic reaction. For AEM, the OER is caused by water adsorption on the catalyst surface and subsequent hydrolytic dissociation. The important intermediates are M–O and M–OOH, which eventually produce oxygen.
Exploring the real active site in the catalytic process of OER catalysts is the focus of many researchers. In AEM, it is widely believed that the active site is a transition metal ion, at which water molecules are adsorbed and oxidation occurs (Fig. 3a). Many reports have shown that the presence of B-site metal ions in the octahedral structure of BO6 in perovskite-type OER catalysts plays an important role in the catalytic process.27,28 For example, Fe ions in perovskite oxides have been shown to be highly catalytic elements of the OER, and can be doped with Co to achieve higher catalytic performance.28 According to Man et al. 's theoretical analysis, the activity of perovskite oxides presents volcanic maps in the following order: SrCoO3 > LaNiO3 > SrNiO3 > SrFeO3 > LaCoO3 > LaFeO3 > LaMnO3.29 This trend was also verified through experiments by Bockris et al. and Y. Matsumoto et al.30,31 The perovskite oxide at the top of the volcano showed a better catalytic performance. Perovskite oxides on the left half of the volcano suggest a binding force that is too strong with the adsorbent, whereas perovskite oxides on the right half represent a binding force that is too weak. The best OER performance can only be attained at the active site with the appropriate adsorption intensity. In AEM, OER activity is independent of pH at the reversible hydrogen electrode (RHE) scale. Despite this progress, a number of issues remain to be resolved.
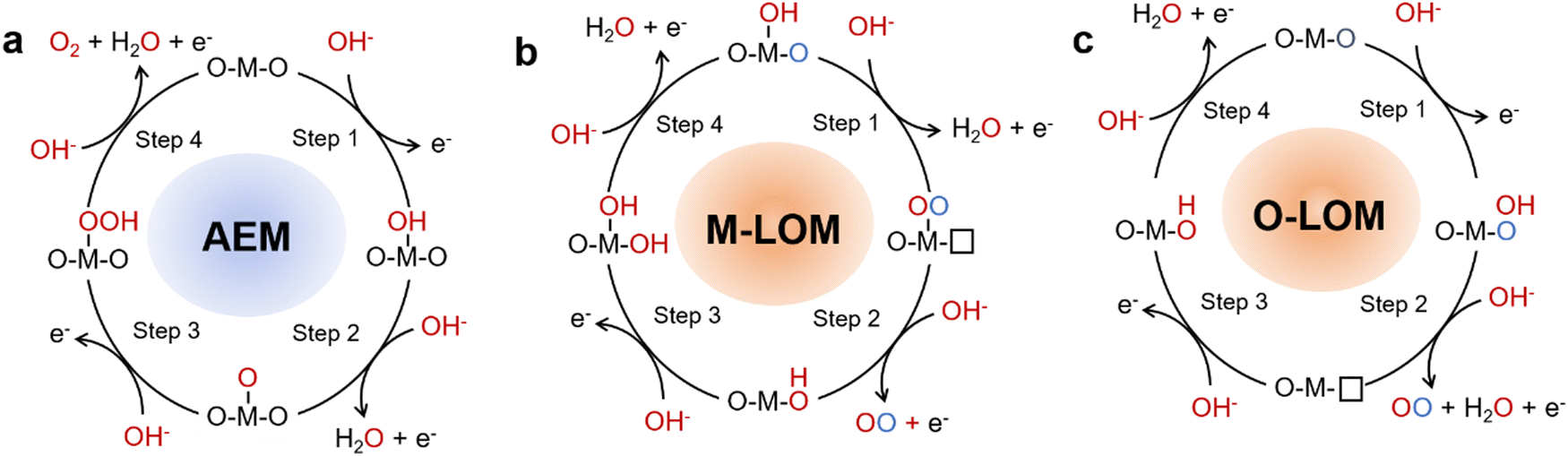 | ||
| Fig. 3 Schematic OER mechanisms on perovskite oxides: (a) AEM, (b) M–LOM, and (c) O–LOM. | ||
2.2. LOM
With the development of a large number of OER catalysts, it has been found that the OER catalytic activity of some materials does not follow the volcanic relationship. The active oxygen evolution activity of some catalysts depends on the pH in the electrolyte.32,33 One explanation for the discrepancy is that these catalysts do not actually follow the reaction mechanism described above. Inspired by this, the lattice oxygen oxidation mechanism (LOM) has been widely studied. Through theoretical calculation, Mefford et al. found that lattice oxygen was also involved in the OER process of La1−xSrxCoO3−δ, which may explain the reason for its high activity. This mechanism has also been verified by experiments.34 Shao-Horn et al. found that the oxygen produced in the OER process came from lattice oxygen on SrCoO3−δ, Pr0.5Ba0.5CoO3−δ and La0.5Sr0.5CoO3−δ by in situ18O isotope-labeled mass spectrometry.32 In the LOM, the metal-lattice oxygen oxidation mechanism (M–LOM) and oxygen-lattice oxygen oxidation mechanism (O–LOM) are divided according to whether the active site is metal or lattice oxygen (Fig. 3b and c).35 In the first step of M–LOM, –OO is formed by dehydrogenation at the transition metal site where –OH is adsorbed. On dehydrogenation, the O of the adsorbed –OH binds to the lattice O. In the third step of M–LOM, OO evolves into O2, forming an oxygen vacancy. The oxygen vacancy is then filled by OH−. Finally, deprotonation occurs and the initial surface is restored. There is decoupling of the proton–electron transfer step in the LOM due to oxygen vacancy generation, resulting in the pH-dependent OER properties previously observed. Shao-Horn et al. also proposed a new reaction mechanism (O–LOM, Fig. 3c), which is similar to M–LOM.32 The main difference between M–LOM and O–LOM is that M–LOM assumes that the active site of perovskite is the metal site, whereas O–LOM assumes that it is the lattice-oxygen site.36 Gao-Ren Li et al. established the relationship between lattice oxygen evolution reaction and dynamic surface structure.37 Through theoretical calculations and in situ characterization, such as in situ X-ray absorption near edge structures (XANES) and in situ Raman, it has been demonstrated that surface Fe sites are catalytic centers of LOM, and Sr vacancies can promote the LOM by raising the 2p level of oxygen. To verify whether LOM is dominant in the OER: first, it is possible to test whether the catalyst performance is strongly dependent on pH; Secondly, the performance of catalysts with LOM is generally improved after the CV test. Third, direct isotope labeling is used to observe whether the O in the production of O2 comes from lattice oxygen.2.3. Regulation of the reaction mechanism
In LOM, O–O radical coupling is achieved directly without the involvement of M–OOH intermediates.39 The process avoids the limitation of the adsorption energy scale, resulting in the catalyst working in LOM showing higher apparent activity than one working in AEM.39–42 However, the catalyst is usually accompanied by a large amount of surface reconstruction during the LOM process due to the redox and overflow of lattice oxygen, which is very unfavorable to the stability of the catalyst.38 Therefore, how to regulate the reaction path between AEM and LOM to achieve a coordinated symbiosis of catalyst activity and stability is a problem that needs to be considered for catalyst design.The way InIch the OER mechaIism is regulated can be differentiated according to whether or not the chemical composition of the catalyst is changed. In general, people can optimize the reaction mechanism by changing the material of catalyst to regulate the distribution state of electrons or the adsorption energy of reaction intermediates. In the Mott–Hubbard cationic redox environment, when U (the energy difference between the empty upper Hubbard band (UHB) and the full lower Hubbard band (LHB)) is much smaller than Δ (the charge-transfer gap), it causes the oxygen non-bonding 2p band to be located in the metal LHB, allowing electrons to only come from LHB during the OER process (Fig. 4a).43 Transition metal cations of perovskite oxidized species act as active sites and redox centers for intermediates in the adsorption reaction, thus following AEM in the OER process. WIen U is much larger than Δ, the gap is going to be a charge transfer type. The 2p band of oxygen provides electrons, and under applied potential conditions, the non-bonded 2p band of oxygen crosses the Fermi level. The oxygen site will act as a redox center and an LOM-based electron transport pathway may be triggered. For example, by doping with some foreign ion, either the transition metal ion at the B-site or an ion at the A-site.32,44,45 On the other hand, AEM and LOM can be realized in a class of materials by adjusting defect content or introducing lattice stress while keeping the chemical composition of materials unchanged. Perovskite oxides have high structural tolerance, resulting in a wide range of adjustment of metal 3d and oxygen 2p band structure. Shao-Horn, Stevenson and Xu and colleagues have successfully implemented AEM or LOM in different perovskite oxides. The disordered structure can be introduced into perovskite-type catalysts by ball milling and electrical activation to control oxygen defects (Fig. 4b).32,34,40 This process is usually accompanied by dynamic reconfiguration, and the operation of the LOM mechanism can be judged by whether it depends on the pH of the electrolyte. By cleverly controlling oxygen defect content, Chun-Hua Ya et al. found that the dominant OER mechanism on LaxSr1−xCoO3 can be arbitrarily converted between AEM–LOM–AEM, accompanied by the changing trend of volcanic activity (Fig. 4c–e).38 By doping other metal ions at the A-site, ball milling, electrical activation and other ways to activate lattice oxygen to adjust oxygen hole concentration, so as to achieve the purpose of mechanism conversion. The synergistic optimization of catalyst activity and stability can be realized by changing the mechanism. In conclusion, the difficulty of cationic and anionic redox reactions in the OER potential region determines which catalytic mechanism is used, thus affecting the performance and behavior of OER catalysts.
 | ||
| Fig. 4 (a) Schematic descriptions of two possible band structures of perovskite oxides under different oxygen evolution reaction mechanisms. (b) CV measurements from pH = 12.5 to 14 recorded at 10 mV s−1. (c) Effect of ball milling time on different oxygen evolution mechanisms. (d) The dependence of the dominant peak of the p band and the orbital center of the p band on VO concentration. (e) Transformation of the OER mechanism of oxygen deficiency control and explanation of the structure–activity relationship. Reproduced with permission.38 Copyright 2022, American Association for the Advancement of Science. | ||
3. Catalytic activity descriptors for perovskite oxides
Several OER activity descriptors have been developed over the past few decades, including free energy of adsorbent, eg occupancy, and metal–oxygen covalence which have the effects of the d-band center and O 2p-band center.The free energy of adsorbent (ΔGads*) obtained by theoretical calculation is also a very important descriptor. The catalytic activity of perovskite oxides is strongly influenced by their surface properties, which are determined by their ΔGads*. In the context of catalysis, it determines the ability of a material to bind and activate reactant molecules, such as H2O, and facilitate their transformation into product molecules, such as O2. In general, perovskite oxides with a higher ΔGads* of water molecules have been found to exhibit higher catalytic activity for the OER (Fig. 5a).46 However, excessively high ΔGads* can also result in strong binding of water molecules to the surface, leading to kinetic limitations and decreased catalytic activity. Man et al. considers 18 classes of perovskite and plots ηOER as a function of ΔGO*–ΔGOH*, thus obtaining the universal volcanic relationship.29 In summary, ΔGads* of water molecules on the surface of perovskite oxides is an important factor that determines their catalytic activity in the OER, with an optimal range that balances binding strength and kinetic accessibility for efficient conversion of water into oxygen.
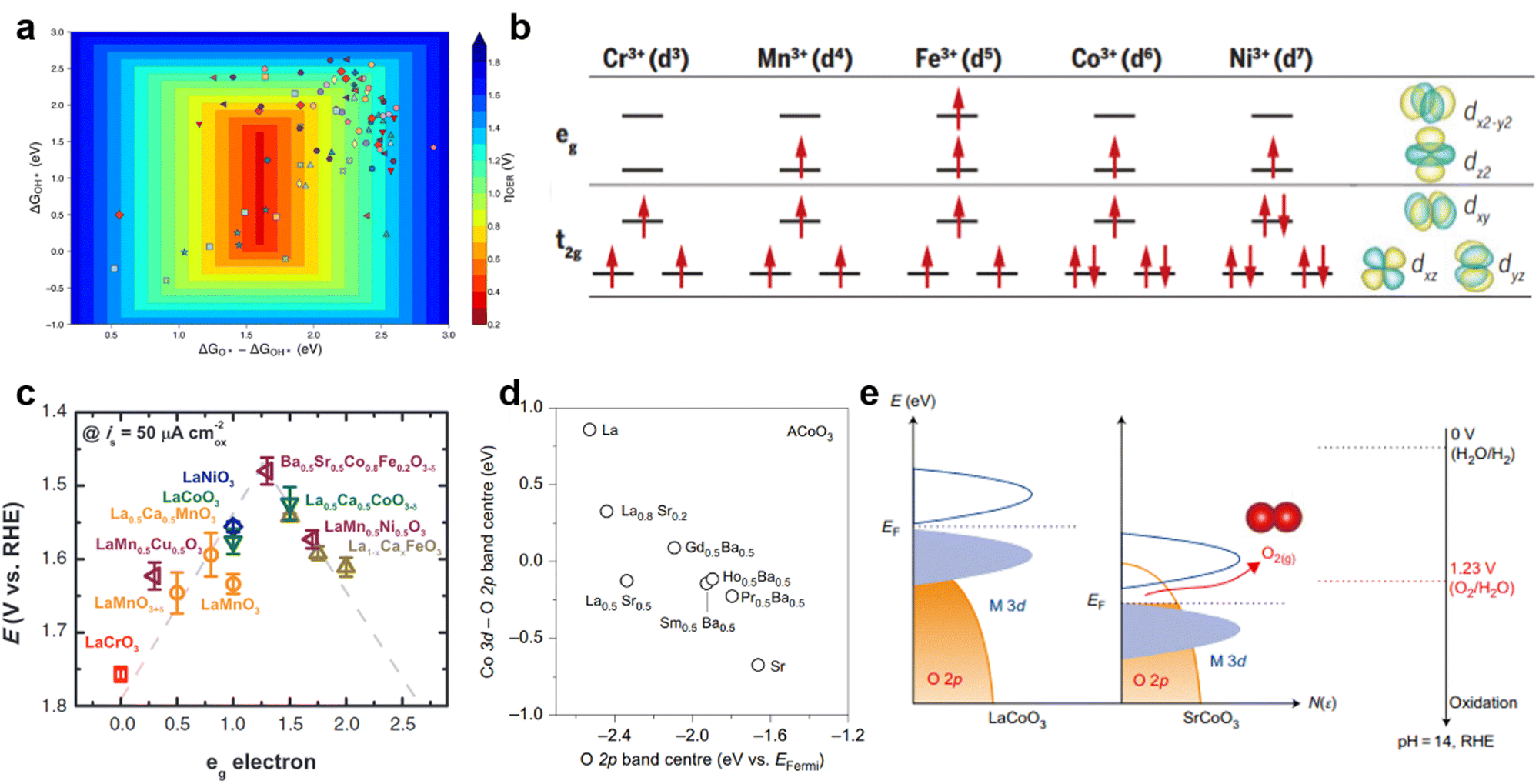 | ||
| Fig. 5 (a) Two-dimensional OER activity plot of theoretical overpotentials (ηOER) for all the materials explored in this study as a function of ΔGO*–ΔGOH* and ΔGOH*. Reproduced with permission.46 Copyright 2020, American Chemical Society. (b) The electronic configuration of some transition metals and related metal orbitals. Reproduced with permission.17 Copyright 2017, American Association for the Advancement of Science. (c) The relationship between the OER catalytic activity and the occupancy of the eg-symmetry electron of the transition metal (B in ABO3). Reproduced with permission.39 Copyright 2011, Science. (d) The difference between the Co 3d band center and O 2p band center and O 2p relative to the Fermi level in ACoO3 perovskites with different A-site cations. (e) Schematic representation of M 3d-O 2p overlap. Reproduced with permission.32 Copyright 2017, Nature Publishing Group. | ||
OER activity generally depends on oxygen binding strength. The Shao-Horn group proposed that eg filling of surface transition metal cations (B-site cations) can greatly affect the binding of the OER intermediate and oxide surface, thus affecting the activity of OER. The 3d orbital of the B-site ion in perovskite is hybridized with the 2p orbital of the O. There is a high spatial overlap between the dz2 and dx2−y2 orbitals in the transition metal and the 2p of oxygen to form σ orbitals. The dxy, dyz and dxz metal orbitals have low spatial overlap with oxygen to form π orbitals. The anti-bonding orbitals σ* and π* are called eg and t2g orbitals of the octahedral coordination field, respectively. The number and spin state of d-electrons of B-site metal ions affect the filling state of eg. For example, the filling state of eg for Cr3+, Mn3+, Fe3+, Co3+, and Ni3+ correspond to 0, 1, 2, 1, and 1, respectively (Fig. 5b).17 Electron transfer between surface cations and adsorption intermediates can be facilitated more directly because the eg orbital overlaps more strongly with the oxygen-associated adsorbent than the t2g orbital. Therefore, a Shao-Horn (SH) principle with eg track filling as the activity descriptor is proposed.39 According to the principle of Shao-Horn (SH), the performance of an oxygen evolution catalyst can be optimized by adjusting the eg orbital filling of the metal site. They found that the eg orbitals of Ba0.5Sr0.5Co0.8Fe0.32O3 (BSCF) surface transition metal ions will participate in bonding with surface adsorbed anions via σ bond, resulting in significantly higher OER intrinsic activity than IrO2. This strategy of doping other metal cations at the B-site to regulate eg orbital filling has been applied by others to achieve better oxygen evolution performance, including Ca2Mn2O5, CaCu3Fe4O12, SrNb0.1Co0.7Fe0.2O3−δ, and Ca0.9Yb0.1MnO3−δ. This descriptor captures the energy variation of the electrochemical OER from the perspective of molecular orbital theory, leading to the formulation of unique OER activity design principles (Fig. 5c). In other words, when surface transition metal ions such as eg orbital filling ≈1, the perovskite oxide catalyst has better catalytic performance.
The relative distance between the band center of the metal 3d (M 3d) and oxygen 2p (O 2p), the covalency of M–O, can be used as a descriptor of catalytic activity. The covalence of M–O bonds in perovskite-type oxides containing various metal ions has an important effect on their catalytic activity and stability.47 The smaller the relative distance between the M d-band center and O 2p-band center, the stronger the covalent bond of perovskite oxide. Suitable covalent strength can accelerate the charge transfer barrier between metal and oxygen, thus promoting OER properties. But too high covalence will lead to the instability of the catalyst.48 For AEM, first of all, the increase of covalence leads to an increase in electron density at the metal site and the decrease of binding energy between the metal site and the oxygen intermediate species.49 Moreover, the binding energy is adjusted by doping and the length of the M–O bond is changed. The covalency is enhanced and the bond length is reduced, which promotes the formation of oxygenophilic intermediates. In addition, the enhanced M–O covalence will increase the efficiency of charge transfer and reduce the electron charge transfer impedance of perovskite oxide catalysts in the OER process.50 For LOM, the intensity M–O covalency may cause the O 2p center to shift towards higher energies relative to the Fermi level (Fig. 5d). The location of the center of the O 2p band affects the energy required to form oxygen vacancies.45 Perovskite oxides with a higher O 2p band center are more likely to form oxygen holes and activate lattice oxygen, which is conducive to improving OER performance. It has also been proved that when M–O bond covalence is enhanced, the contribution of O 2p at the Fermi level is enhanced as a new catalytic center. Yang Shao-Horn et al. also proved by DFT that when La3+ is replaced by Sr2+ at the A-site, the Fermi level is closer to the calculated O 2p band center, which is accompanied by a decrease in the energy gap between the metal 3d and O 2p band center (Fig. 5e).32 Compared to LaCoO3, the O 2p state of the Fermi level of SrCoO3 is higher than the redox energy of the O2/H2O, making the oxidation of lattice oxygen in perovskite more thermodynamically favorable.
4. Engineering of the surface and structure of perovskite oxides
Due to the low conductivity, small number of active sites and slow reaction kinetics, the electrocatalytic activity of the original pure perovskite oxides is not ideal. Therefore, various strategies have been used to improve the electrocatalytic performance of perovskite oxide systems (Table 1). In order to compare the relative activity of the catalyst, the overpotential required to produce a current density of 10 mA cm−2 is usually used as the evaluation index (the current density of the solar water splitting device with an efficiency of 12.3%). When the current density is greater than or equal to 10 mA cm−2, the catalyst is considered to have the potential of industrialization.54| Electrocatalyst | Overpotential [mV vs. RHE] | Tafel slope [mV dec−1] | Electrolyte | Loading [mg cm−2] | Electrode | Ref. |
|---|---|---|---|---|---|---|
| La1−xCexNiO3 | 270 | 45 | 1 M KOH | 0.4 | Carbon paper | 22 |
| Ce doped LaCoO3 | 390 | 112 | 1 M KOH | 0.25 | Glassy carbon | 55 |
| La1−xSnxNiO3−δ | 345 | 74 | 1 M KOH | 0.367 | Glassy carbon | 59 |
| La1−xSrxCoO3 | 85 | 1 M KOH | 0.25 | Glassy carbon | 51 | |
| La2NiMnO6 | 350 | 58 | 1 M KOH | Glassy carbon | 53 | |
| La1−xFeO3−δ | 410 | 54 | 0.1 M KOH | 0.232 | Glassy carbon | 65 |
| PrBa0.5Sr0.5Co2−xFexO5+δ | 290 | 69 | 1 M KOH | 0.192 | Glassy carbon | 66 |
| SrCo0.4Fe0.2W0.4O3−δ | 300 | 1 M KOH | 0.232 | Glassy carbon | 67 | |
| LaFexNi1−xO3 | 302 | 50 | 1 M KOH | Glassy carbon | 52 | |
| F-Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 280 | 101.67 | 1 M KOH | 0.255 | Rotating disk | 68 |
| S-doped LaCoO3 | 364 | 126.7 | 1 M KOH | Glassy carbon | 69 | |
| LaFeO3−x−δClx | 59 | 0.1 M KOH | Rotating disk | 70 | ||
| Sr3NiFeMoO9−δ | 260 | 42.3 | 1 M KOH | 3 | Ni-foam | 44 |
| (La0.6Sr0.4) (Co0.2[FeMnNiMg]0.8)O3 | 320 | 45 | 1 M KOH | 0.196 | Glassy carbon | 71 |
| La(CrMnFeCo2Ni)O3 | 325 | 51.2 | 1 M KOH | 2.35 | Ni-foam | 72 |
4.1. Composition design
Due to the unique structural advantages of perovskite, component regulation can be conducted at the A, B, and O site respectively. Doping of elements generally optimizes the crystal structure parameters and adjusts the electronic structure to achieve the best performance of the OER.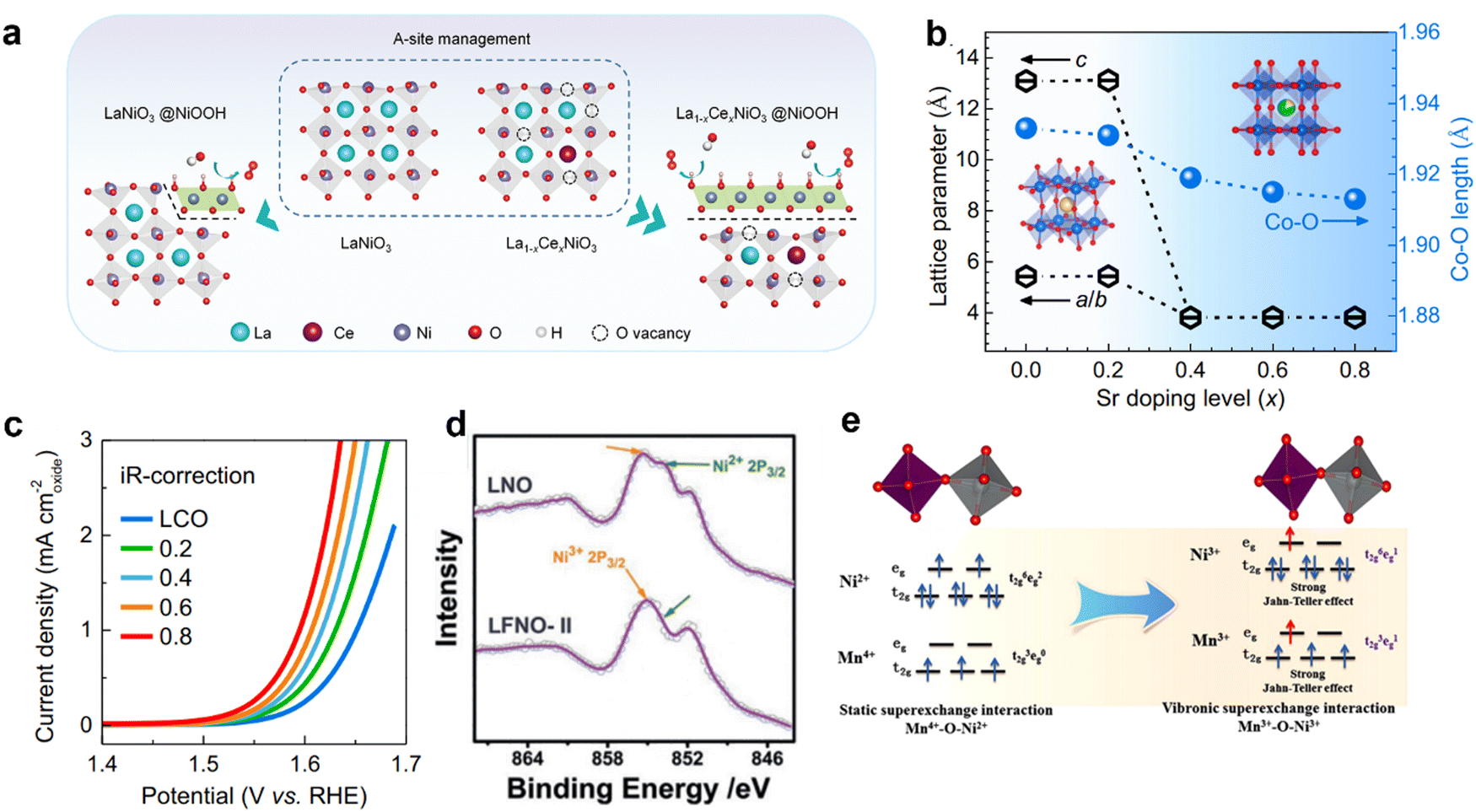 | ||
| Fig. 6 (a) Schematic diagram of Ce doping accelerated surface reconstruction at site A. Reproduced with permission.22 Copyright 2021, Wiley-VCH GmbH. (b) The lattice parameters of La1−xSrxCoO3 evolve and the length of the Co–O bond changes with the increase of Sr doped in the A-site. (c) LSV polarization curves with iR-correction of La1−xSrxCoO3 for the OER. Reproduced with permission.51 Copyright 2021, Elsevier. (d) XPS spectra of Ni 2p3/2 for LaFexNi1−xO3. Reproduced with permission.52 Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) The influence of electron super-exchange between Ni3+–O–Mn3+ induces strong Jahn–Teller distortion of MnO6 and NiO6. Reproduced with permission.53 Copyright 2018, American Chemical Society. | ||
The doping of other elements at the A-site also affects the reaction mechanism of the perovskite catalyst during the OER. For example, the mechanism of catalytic OER has been shown recently to shift from AEM to LOM, through adjusting the Sr in the La1−xSrxCoO3−δ system of doping.32,34,36 Zong-ping Shao et al. have provided direct evidence of lattice oxygen in the OER by design of the Si-incorporated SrCo1−ySiyO3−δ system.32 This strategy of regulating the reaction mechanism through A-site doping provides a new perspective for constructing high performance catalysts of the OER.
A-site doping also plays a role in improving the stability of catalysts, especially the OER under acidic conditions. Under acidic conditions, the cation in perovskite oxide can easily dissolve into the electrolyte during the OER process, leading to the collapse of the structure.60,61 The introduction of external cations at the A-site has been reported to improve the activity and stability of ruthenium oxides by preventing or delaying the dissolution of cations. It is found that doping Na+ in SrRuO3 improves the durability of SrRuO3 perovskite, and maintains 85% of the initial activity after 20 cycles.62 Higher activity and durability of Sr1−xKxRuO3 (x = 0.00, 0.05, 0.10 and 0.20) can be obtained by partially doped monovalent K+ cations.63 The distortion degree of RuO6 octahedron in perovskite is reduced by doping Na+/K+ in SrRuO3. After Na+/K+ doping, some Ru cations are oxidized to Ru5+, which reduces the Jahn–Teller effect of Ru4+ cations in RuO6 octahedron.64 By partially doping Na+/K+ at the Sr2+ site, SrRuO3 results in an oxide of Ru atoms with higher oxidation states and higher symmetry, thus improving stability. In other words, doping of A-site elements optimizes lattice parameters, leading to improved stability. Moreover, it can adjust the electron distribution state of B-site ions and adjust the covalence of metal - oxygen, thus enhancing the activity for the OER.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in LaFexNi1−xO3.52 It is confirmed by in situ testing that Ni2+, Ni3+, Fe3+ and Fe4+ exist in single crystal samples of LaFexNi1−xO3, and when Fe replaces Ni, the price of Ni (Fe) will decrease (increase). It can be seen from XPS that the strength of Ni2+ is reduced, indicating that the introduction of Fe into LaNiO3 can increase the oxidation state of Ni (Fig. 6d).52 DFT simulations show the introduction of an additional Fe 3d state near the Fermi level by B-site doping. Electron rearrangement in the Ni–O–Fe bridge strengthens the d-orbital coupling and increases the transition metal (TM) 3d bandwidth, thereby increasing the activity of the OER. In addition, the introduction of other metal ions into the B-site will also affect the lattice structure of the perovskite catalyst. It has been reported that Fe replaces Ni in epitaxial LaFexNi1−xO3 films (x = 0, 0.125, 0.25, 0.33, 0.375, 0.5, and 1.0), resulting in a monotone increase in the out-of-plane lattice parameter (c) with increasing x.
:2 in LaFexNi1−xO3.52 It is confirmed by in situ testing that Ni2+, Ni3+, Fe3+ and Fe4+ exist in single crystal samples of LaFexNi1−xO3, and when Fe replaces Ni, the price of Ni (Fe) will decrease (increase). It can be seen from XPS that the strength of Ni2+ is reduced, indicating that the introduction of Fe into LaNiO3 can increase the oxidation state of Ni (Fig. 6d).52 DFT simulations show the introduction of an additional Fe 3d state near the Fermi level by B-site doping. Electron rearrangement in the Ni–O–Fe bridge strengthens the d-orbital coupling and increases the transition metal (TM) 3d bandwidth, thereby increasing the activity of the OER. In addition, the introduction of other metal ions into the B-site will also affect the lattice structure of the perovskite catalyst. It has been reported that Fe replaces Ni in epitaxial LaFexNi1−xO3 films (x = 0, 0.125, 0.25, 0.33, 0.375, 0.5, and 1.0), resulting in a monotone increase in the out-of-plane lattice parameter (c) with increasing x.
Double perovskites (A2BB′O6) can also be synthesized by introducing metal ions at the B-site (where the B-site is occupied alternately by different cations B and B′), with the presence of oxygen bridges connecting each B and B′ atom pair.27,82,83 The coupling effect can be enhanced by adjusting the B–O–B′, providing a basis for tailoring their electronic structure. The ordered arrangement of B-site ions results in a superior physical correlation effect in double perovskites. The eg electron filling state of metal can be adjusted by optimizing B-site ions. On the other hand, theoretical and experimental studies have proved that doping other ions at the B-site of perovskite oxides can regulate the electron distribution of metal–oxygen bonds and accelerate the formation of active components. For example, the electron super-exchange of Ni3+–O–Mn3+ in La2NiMnO6 nanoparticles causes the electron filling states of eg of Mn and Ni ions to become uniform (Fig. 6e).53 In addition, the electron super-exchange interaction of Ni3+–O–Mn3+ induces strong Jahn–Teller distortion of MnO6 and NiO6 octahedrons, elongating M–O bonds and contributing to the formation of Mn/Ni hydroxide/oxide active components on the surface of La2NiMnO6.84
In particular, the electrocatalytic performance of perovskite materials can be improved by adding nonmetallic elements at the B-site. By adding the corresponding salt to the synthesis, P or B can be introduced at the B-site of the lattice.85 P. R. Slater and co-workers found that the conductivity of CaMn0.95P0.05O3−δ and CaMn0.95B0.05O3−δ was significantly improved compared with the original catalysts, which is attributed to the addition of O22−/O− and the optimization of electron filling in eg.86 Another phosphor-doped perovskite SrCo0.85Fe0.1P0.05O3−δ (SCFP) has been reported to stabilize the crystal structure of the oxide by introducing P. In addition, it has been reported that silicon-doped perovskite can stabilize the cubic structure and enhance the ionic conductivity of perovskite, such as SrM1−xSixO3 (M = Fe, Mn, Co), Ba2In2−xSixO5+x/2 (x = 0,0.1, 0.2, 0.3).87–89 Therefore, the addition of nonmetallic elements to the B-site of perovskite oxides opens up a new field for the design of high-performance and stable oxygen evolution catalysts.
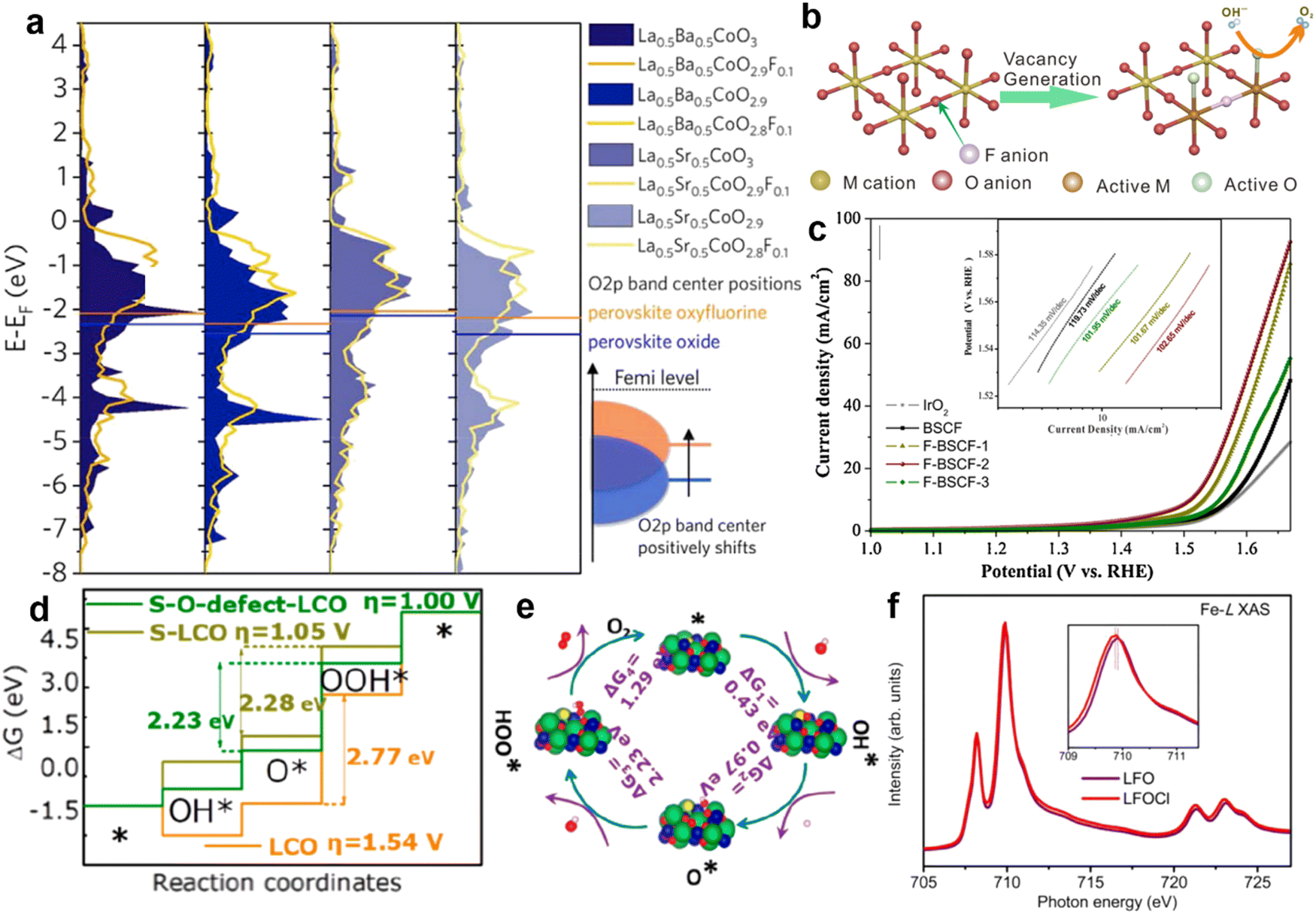 | ||
| Fig. 7 (a) The density of states (the sum of the upper and lower spin states) in the O 2p band of perovskite oxides with different F doping contents. Reproduced with permission.90 Copyright 2018, Elsevier. (b) The process by which F replaces O-site atoms to produce O2−/O− species. (c) LSV diagram regarding different contents of F substitution in Ba0.5Sr0.5Co0.8Fe0.2O3−δ. Reproduced with permission.68 Copyright 2019, Elsevier. (d) Gibbs free energy variation of LaCoO3 doped with S and oxygen defects in the four steps of the OER. (e) Reaction mechanism of S-doping leading to an oxygen defect of LCO for the OER. Reproduced with permission.69 Copyright 2020 American Chemical Society. (f) The sXAS spectra of Fe-L for LaFeO3 and LaFeO2.9−δCl0.1. Reproduced with permission.70 Copyright 2020, Elsevier. | ||
4.2. Morphology engineering
The morphology of perovskite oxide has an important effect on the OER performance. Structures with a larger electrochemically active surface area (ECSA) tend to have more catalytic active sites, which is conducive to enhancing the transport of substances. Morphological engineering has more influence on the catalytic surface. In recent years, perovskite oxides have been developed by researchers with a variety of nanostructures, such as: nanoparticles, nanowires, nanotubes, nanorods, nanospheres and so on. Meso-porous and macro-porous structures were found in SrNb0.1Co0.7Fe0.2O3−δ perovskite nanorods prepared by electrospinning (Fig. 8a). Compared with the bulk structure, the large pore size distribution range and the increase of pore number are conducive to the exposure of the catalytic active site and the mass transport associated with electrocatalysis, thus showing high efficiency OER activity in alkaline solution.91 The yolk–shell La0.9Sr0.1CoO3 perovskite microspheres were synthesized using a one pot method. The unique Yolk–Shell La0.9Sr0.1CoO3 perovskite microspheres showed strong catalytic activity for the OER (Fig. 8b).92 Its structure has advantages: (1) larger ECSA on the unit scale of coarse nanocrystals provides more active sites for the OER; (2) the shorter length is conducive to the diffusion of electrons and oxygen ions, thus speeding up the electrochemical kinetics; (3) the thin hollow structure on the three-dimensional (3D) space is conducive to maintaining the integrity of the structure and preventing aggregation or sintering, which is conducive to maintaining a stable structure during the long catalytic process. Hong Yang et al. prepared LaCoO3 with porous particles and hollow nanospheres using a hydrothermal method (Fig. 8c). By adding the organic substance glycine to the synthesis as a pore-forming agent, it will spill out in the structure due to high temperature calcination. This makes LaCoO3 with the amorphous surface structure exhibit a low initial potential in OERs (Fig. 8d). The OER performance of LaCoO3 hollow nanospheres (∼1.50 V) is the best compared with that of dense (∼1.55 V) and porous particles (∼1.51 V).93 Zong-ping Shao et al. impregnated a neatly arranged colloidal crystal template of polymethyl methacrylate (PMMA) with a mixed metallic solution (Fig. 8e).94 Perovskite oxide with a 3D ordered macro-porous structure was obtained by removing the tightly packed spherical template by calcination. Open interconnected 3D ordered macro-porous structures have uniformly distributed pores with an average diameter of about 200 nm, resulting in a high specific surface area, which is conducive to O2/OH− transport channels and electron migration (Fig. 8f, g). Yang's group also prepared sea urchin-like nanostructures with high specific surface area (48 m2 g−1), which followed the “nucleation-growth-assembly” route.96 Regulating the nanostructure of perovskite oxides can also affect the electron configuration of active sites. For example, by reducing the particle size, the regulation successfully increased the fill of eg of Co3+ in LaCoO3 from about 1.0 to an optimized configuration close to 1.2.95 The orbital hybridization of Co 3d-O 2p and the weakening of the Co–O bond favor the formation of –OOH species, thus increasing the intrinsic activity of LaCoO3.97,98 In addition, by normalized specific activity comparison in the Brunauer–Emmett–Teller (BET) region, the activity of the 80 nm sample is still 1.8 times that of the bulk sample, indicating that the significant enhancement of OER activity is mainly due to the increase of reactivity at the active site, that is, due to the spin state transition of Co3+ on the surface (Fig. 8h).95 The surface regulation of perovskite oxides is more complicated. It can not only increase the exposure of more active sites, but also regulate the internal electron distribution. The influence on oxygen evolution performance has many aspects that need to be further studied and explored, to provide more basis for the design of catalysts with high performance. | ||
| Fig. 8 (a) Scanning electron microscopy (SEM) image of nanorods SrNb0.1Co0.7Fe0.2O3−δ prepared by electrospinning technology. Reproduced with permission.91 Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinhei. (b) TEM image of Yolk–Shell La0.9Sr0.1CoO3. Reproduced with permission.92 Copyright 2015, The Royal Society of Chemistry. (c) SEM images of hollow spheres LaCoO3. (d) CV curves of LaCoO3 with different morphologies for the OER. Reproduced with permission.93 Copyright 2017, American Chemical Society. (e) Schematic diagram of the advantages of 3D ordered macro-porous electrocatalysts for the OER. (f) LSV curves of various catalysts for the OER. (g) TEM image for 3D ordered macro-porous electrocatalysts. Reproduced with permission.94 Copyright 2018, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (h) Mass and special activities for nanoparticle catalysts of different sizes at an overpotential = 0.49 V. Reproduced with permission.95 Copyright 2016, Springer Nature. | ||
4.3. Defect engineering
The introduction of anion or cation defects in the perovskite structure may regulate the electron distribution and optimize the reaction path, which contributes to the electrocatalytic capacity.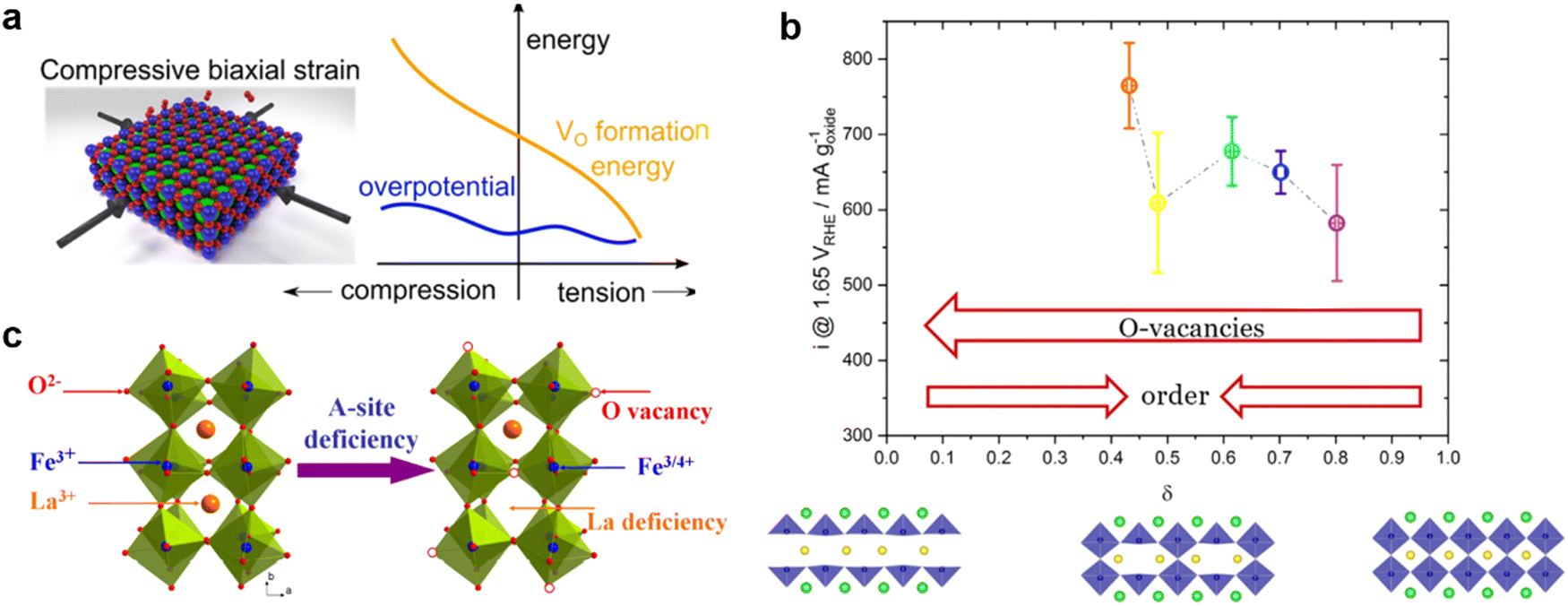 | ||
| Fig. 9 (a) Schematic diagram of the effect of compressive strain and tension strain on OER overpotential and oxygen vacancy formation energy. Reproduced with permission.99 Copyright 2016, American Chemical Society. (b) OER activity of PrBaCo2O5+δ in different oxidation stoichiometry. The structure diagrams below show the distribution of oxygen vacancy in the structure of PBCO with δ = 0, 0.5 and 1. Reproduced with permission.100 Copyright 2021, Wiley-VCHGmbH. (c) Schematic diagram of the formation of oxygen vacancy and Fe4+ in La1−xFeO3−δ perovskite with A-site deficiency. Reproduced with permission.65 Copyright 2016, American Chemical Society. | ||
Among the several mechanisms proposed for the LOER is the involvement of active sites for oxygen evolution by oxygen, resulting in the creation of surface oxygen vacancies. In other words, there is a perovskite catalyst with a high oxygen vacancy content and oxygen may occur through the LOM. The ability of PrBaCo2O5+δ to accommodate a wide range of oxygen vacancies makes it an excellent candidate for studying oxygen vacancies. Emiliana Fabbri et al. systematically studied the increase of OER activity in PrBaCo2O5+δ materials with the increase of lattice oxygen vacancy. However, the relationship between OER activity and oxidation stoichiometry is not linear (Fig. 9b). When δ ≈ 0.5, PrBaCo2O5+δ forms alternating [CoO6] octahedrons and [CoO5] square pyramids in the plane of Pr. This arrangement of oxygen vacancies causes significant changes in Co–O coordination and Co–O plane buckling, which is detrimental to the OER activity of perovskite.100 In general, the reduction atmosphere can be maintained during synthesis, physical treatment (Ar plasma treatment, biaxial compression/tension strain), or A/B site elements can be doped to form oxygen vacancies in perovskite oxides. The presence of an appropriate amount of oxygen vacancy regulates crystallographic characteristics, rearranges electron distribution and optimizes adsorption energy, thus achieving excellent performance for the OER.
5. Noble-perovskites for the OER in acidic media
Recently, a proton exchange membrane water electrolyser (PEMWE) with high current density (maximum 2–3 A cm−2) and ultrahigh gas purities has become a commercial or near-commercial electrochemical water cracking technology.113–115 However, PEMWEs require the use of noble metal based electrocatalysts in the anode, such as iridium (Ir), ruthenium (Ru) or their oxide forms. The high cost and scarcity of Ir and Ru limit the large-scale application of PEMWE, but the instability of some non-noble metal catalysts in strongly acidic solutions makes their electrocatalytic OER activity lower than that of noble-metal based materials. Therefore, it is necessary to develop noble metal OER electrocatalysts with high electrocatalytic activity and good stability in acidic solutions. One of the more direct approaches is to develop catalysts with lower noble metal content while ensuring high performance. Perovskite oxide is a candidate for acid catalysts because it can be doped with multiple metal elements at the A/B-site (Table 2). The OER process of Ir/Ru-based perovskite oxide is often accompanied by the dissolution of elements, which not only affects the structure, but also forms a new catalytic surface.| Electrocatalyst | Overpotential [mV vs. RHE] | Durability tests | Electrolyte | Loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|
| Ba2YIrO6 | 340 | 1 h @j = 10 mA cm−2 | 0.1 M HClO4 | 0.015 | 12 |
| IrOx/SrIrO3 | 270–290 | 30 h @j = 10 mA cm−2 | 0.5 M H2SO4 | 116 | |
| IrOx/9R-BaIrO3 | 230 | 48 h @j = 10 mA cm−2 | 0.5 M H2SO4 | 0.283 | 118 |
| 6H-SrIrO3 | 248 | 30 h @j = 10 mA cm−2 | 0.5 M H2SO4 | 0.90 | 117 |
| Sr0.9Na0.1RuO3 | 170 | 0.1 M HClO4 | 0.51 | 62 | |
| CaCu3Ru4O12 | 171 | 24 h @j = 10 mA cm−2 | 0.5 M H2SO4 | 0.25 | 121 |
| Cr0.6Ru0.4O2 | 175 | 10 h @j = 10 mA cm−2 | 0.5 M H2SO4 | 0.283 | 122 |
| W0.2Er0.1Ru0.7O2−δ | 167 | 500 h @j = 10 mA cm−2 | 0.5 M H2SO4 | 0.33 | 18 |
| SrTi1−xIrxO3 | 247 | 20 h @j = 10 mA cm−2 | 0.1 M HClO4 | 0.21 | 123 |
| Sr2IrO4 | 286 | 6 h @j = 10 mA cm−2 | 0.1 M HClO4 | 0.08 | 124 |
| Sr4IrO6 | 287 | 6 h @j = 10 mA cm−2 | 0.1 M HClO4 | 0.08 | 124 |
5.1. Ir-based perovskites
Jaramillo and co-workers prepared a catalyst composed of SrIrO3 epitaxial thin films by pulsed laser deposition.116 Sr was leach from the surface layer of SrIrO3 film to form a highly active surface layer IrOx, which improved the activity of the catalyst in the stability test held at 10 mA cmgeo−2 (current normalization to the geometric surface area of the electrode) in 0.5 M H2SO4. An overpotential of 340 mV is required for the initial SrIrO3 to reach 10 mA cmgeo−2. After the current density of 10 mA cmgeo−2 was maintained for 10 minutes, the overpotential decreased to 320 mV. After 2 hour constant-current tests, IrOx/SrIrO3 only required 270 mV of overpotential to reach 10 mA cmgeo−2. DFT calculations were performed to further understand the production of active species on the surface of SrIrO3 (Fig. 10a and b). The theoretical overpotential is calculated for the possible generation of various overburden structures, IrO2-anatase, anatase IrO2(001), IrO2-sheets, IrO3, rutile IrO2(110), rutile IrO2(100), on IrOx/SrIrO3 surfaces with Sr defects (Fig. 10b). Studies have shown that the formation of IrO3 or anatase IrO2 sites may be stable and active overlay structures, but the presence of other more active species cannot be excluded. In other words, dissolution of IR-based perovskite oxides tends to occur in acidic OERs. How to expose more active sites in a way that minimizes dissolution and reconstitution is the key question. Xiao-xin Zou et al. studied Sr2IrO4 of the Ruddlesden–Popper phase, which has unique structural advantages: (1) the interlayer Sr2+ can be exchanged with protons in a controlled manner while keeping the perovskite structure unchanged; (2) protonated layered perovskite can be stripped into nanosheets with a higher surface area and more active sites. Iridium nanosheet structures were obtained by liquid phase stripping with interlayer channels that prevented electron transfer between the inner and outer Ir atoms, thus reducing the charge density around the Ir sites outside the surface.120 It regulates the ability of the Ir site to bind oxygen intermediates and improves OER activity. Xiao-xin Zou et al. found that 6H-SrIrO3 had 27.1 wt% less iridium than IrO2, but 6H-SrIrO3 had almost 4 times the OER activity of IrO2 at 1.525 V. The crystal structure of 6H-SrIrO3 was still maintained after 48 days of exposure in strong acidic medium (0.5 M H2SO4).117 Moreover, Ir species were not detected in acidic solution, indicating that 6H-SrIrO3 has good durability in acidic environments. The results of DFT simulations show that the surface of 6H-SrIrO3 with Ir atoms in a face-sharing octahedral dimer has better catalytic activity than the surface of 6H-SrIrO3 with Ir atoms in an isolated horn-sharing IrO6 octahedron for the OER (Fig. 10c). Yan and co-workers studied 9R-BaIrO3 with 1nm surface IrOx nanoparticles (IrOx/9R-BaIrO3).118 Its iridium mass activity (168 A gIr−1) is about 16 times higher than that of IrO2 (10 A gIr−1) in acidic OERs (Fig. 10d). The structure of the active layer on the surface of IrOx/9R-BaIrO3 evolved during the acidic OER process. During the acidic OER process, the valence state of Ir changes, in which the final surface contains a high content of the high valence state Ir5+Ox. The crystal morphology of the catalyst also evolved from the initial nanoparticle crystal to amorphous octahedron (Fig. 10e). The willingness of IrOx/9R-BaIrO3 to have excellent acidic activity of the OER can be attributed to the formation of a large amount of high-valence amorphous iridium oxide on the surface and strong metal conductivity.118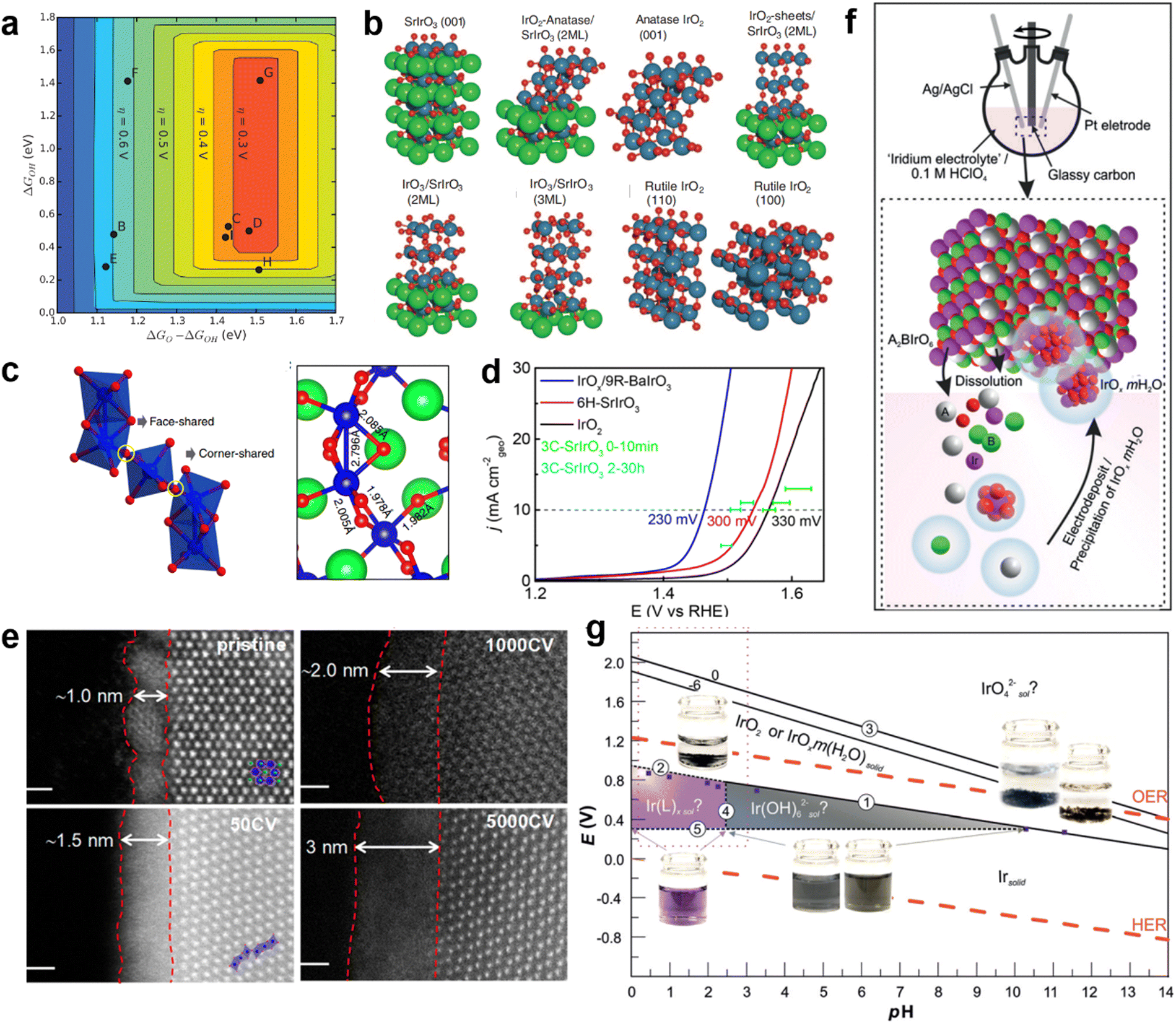 | ||
| Fig. 10 (a) The theoretical overpotential of IrOx with different structures exists on the surface with O* and OH* binding energies as the descriptor. (b) The model of the surface of IrOx with different structures. Reproduced with permission.116 Copyright 2016, American Association for the Advancement of Science. (c) Crystal structure of 6H-SrIrO3 and different connection modes of IrO6 octahedron. Reproduced with permission.117 Copyright 2018, Sprinter Nature. (d) LSV curves of IrOx/9R-BaIrO3, IrO2, and 6H-SrIrO3 in 0.5 M H2SO4. (e) HAADF-STEM image of the IrOx/9R-BaIrO3 surface after a CV test with different times. Reproduced with permission.118 Copyright 2021, American Chemical Society. (f) The proposed OER catalytic mechanism in Ir-based perovskite oxidation species involves iridium dissolution-electrodeposition. (g) Iridium EH–pH diagram. Reproduced with permission.119 Copyright 2019, Wiley-VCH. | ||
According to the research, a small amount of Ir will be dissolved in a strongly acidic environment, and then deposited on the surface of the catalyst in the form of oxides (IrOx·mH2O). The behavior of Ir-based perovskite oxides in the OER can be summarized as solution-electrodeposition of iridium species (Fig. 10f). The behavior of Sr2FeIr(V)O6 and Sr2Fe0.5Ir0.5(V)O4 in acidic OER was studied using CV. The estimated precipitation potential is Ep−IrOx = 0.92 V (vs. RHE). In other words, electrodeposition occurs when the applied external potential exceeds the precipitating potential of IrOx·mH2O. This re-deposition of Ir species from the perovskite structure onto the catalyst surface to form IrOx·mH2O is correlated with the applied potential, which follows the Pourbaix diagram of iridium.119 Moreover, the OER activity of Ir-based perovskite oxides is also constrained by a stable potential/pH domain.
Alexis Grimaud et al. reconstructed EH-pH maps of iridium based on their experimental results and the equation established by Pourbaix, thus further discussing the existence forms of Ir species in different potential and electrolyte environments (Fig. 10g).119Ep−IrOx was used as the critical potential for distinguishing soluble iridium in solids IrOx·mH2O and IrO2. Ir-based perovskites, such as Sr2CoIr(V)O6, La2LiIr(V)O6, Ba2PrIr(V)O6 and Ba2LaIr(V)O6, have higher open circuit potentials than Ep−IrOx. When exposed to low pH, these perovskite iridium species precipitate to IrOx·mH2O as OER active species under their open-circuit voltage.
5.2. Ru-based perovskites
Like Ir-based perovskite oxides, Ru-based perovskite oxides also dissolve or are more easily dissolved in acidic environments, which is the reason why Ru-based perovskite oxides have been less studied in acidic OER. Schmidt et al. prepared SrRuO3 nanoparticles using a simple flame spray synthesis method.61 The thermodynamically stable phases of Sr2Ru3O10, Sr4Ru2O9, Sr2RuO4 and SrRuO3 were explored by adjusting the different proportions of Sr and Ru during synthesis. In acidic environments, perovskite oxides undergo an OER process accompanied by dissolution at an oxidation potential (ABO3 + 2H+ ↔ A2+ + BO2 + H2O). RuO2 easily forms RuO2(OH)2 and further RuO4. Therefore, the high current response of Ru-based perovskite oxide materials in an acidic environment may be due to the OER and Ru corrosion. Studies of SrRuO3 films using a rotating ring disk electrode with ruthenium platinum rings, where the measured currents were attributed to the OER (90%) and Ru corrosion (10%). In the Pourbaix diagram of SrRuO3, it is found that SrRuO3 is easily decomposed by chemical dissolution in any aqueous solution (Fig. 11a).61 As a result, SrRuO3 has difficulty maintaining the perovskite structure in applications involving contact with water-based solutions (mainly involving the OER). The instability of the SrRuO3 structure is due to the dissolution of Sr2+ in most pH domains within the operating potential range of the acidic electrolyte.61 Even at its open circuit potential, Sr material is immediately released into the water phase. The second cause of instability is that Ru, which is the active substance of the OER, also dissolves during the whole oxidation process (Fig. 11b). The phase transition from RuO2 to RuO4 occurred only at about 1.3 (vs. RHE) of Ru dissolved in acidic medium.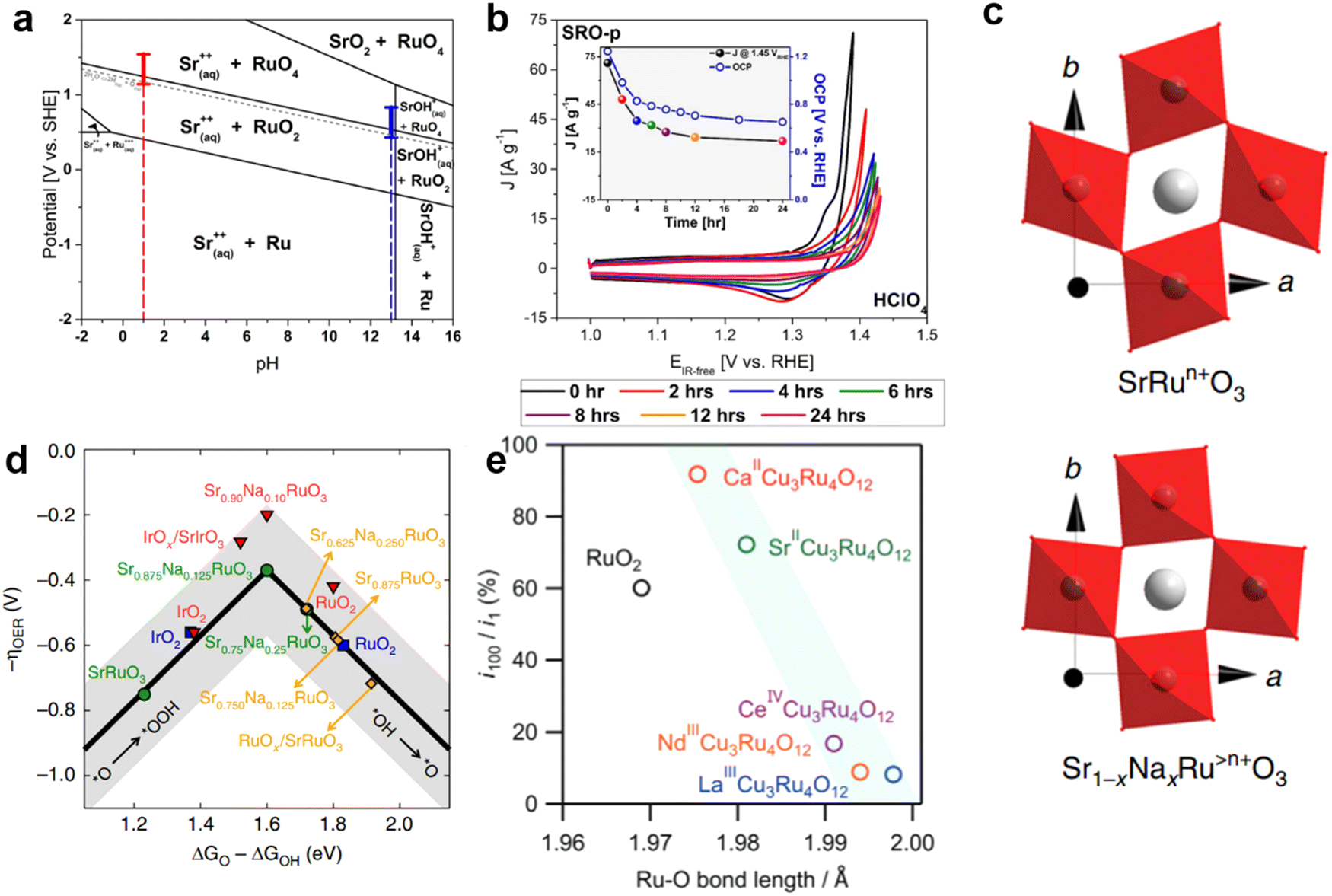 | ||
| Fig. 11 (a) Pourbaix diagrams of SrRuO3. (b) The CV diagram of 10 mV s−1 was performed at 0, 2, 4, 6, 8, 12, and 24 hours after initial contact of the SrRuO3 with the synthetic air saturated electrolyte in HClO4. Reproduced with permission.61 Copyright 2017, American Chemical Society (c) Diagram of the inhibition of octahedral distortion of SrRuO3 doped with Na+. (d) The plot of OER Volcano-type activity. Reproduced with permission.62 Copyright 2019, Sprinter Nature. (e) A graph showing the relationship between the length of the Ru–O bond and the ratio of current density i100/i1 at the 100th and 1st cycles. Reproduced with permission.125 Copyright 2022, Wiley-VCH GmbH. | ||
In order to further improve the catalytic OER durability of Ru-based perovskite oxides in acid, Na was doped at the A-site. When Na+ replaces Sr2+, the surface oxygen p-band center and Ru d-band center in SrRuO3 are positively shifted (Fig. 11c). The binding strength between SrRuO3 and the intermediate was optimized to make the activity of the OER close to the optimum. After the 20th cycle of CV, SrRuO3 lost more than 85% of its initial activity, while NaxSr1−xRuO3 lost only about 15% of its initial activity in 0.1 M HClO4.62 According to DFT, Na-doping reduces the dissolution rate of Sr and Ru, resulting in higher structural stability (Fig. 11d). On the one hand, Ru is known to oxidize to the unstable Ru>4+ at a potential above ∼1.4 V (vs. RHE). The incorporation of Na+ makes Ru>4+ stable in the lattice of SrRuO3, which can be used as the catalytic center to adjust its charge according to the different adsorbent. In addition, the addition of Na+ reduces the deformability of RuO6 octahedron, thus improving the structural stability in the OER. In general, the method of Na-doping can adjust the electron distribution state of Ru, improve the dissolution potential, and slow down the distortion of RuO6 octahedron, so as to achieve the purpose of enhancing the durability of Ru-based perovskite oxide in acidic media. García et al. optimized the doping of different contents of K+ in Sr1−xKxRuO3 (x = 0, 0.05, 0.10, 0.20) to improve the activity of the OER in O2-saturated 0.1 M HClO4.63 When the current density reaches 10 mA cm−2, the required voltage of SrRuO3 is about 1.4 V, while the voltage of Sr0.95K0.05RuO3 drops to about 1.35 V. After K+ doping, the B-site Ru cation is oxidized to the higher oxidation state Ru5+, thus alleviating the Jahn–Teller effect in the RuO6 octahedron. The influence of cations on the distortion degree of the perovskite structure is further discussed by introducing the Goldschmidt tolerance factor (t):  . Where 〈A–O〉 and 〈B–O〉 are the main distances of ABO3 molecular perovskite.126 When t = 1, perovskite oxide has great symmetry. The value of t, 0.994, 0.999, 1.004 and 1.013, increases with the increase of K+ content, indicating that the doping of K+ could adjust the distortion degree of the perovskite structure and improve the durability.
. Where 〈A–O〉 and 〈B–O〉 are the main distances of ABO3 molecular perovskite.126 When t = 1, perovskite oxide has great symmetry. The value of t, 0.994, 0.999, 1.004 and 1.013, increases with the increase of K+ content, indicating that the doping of K+ could adjust the distortion degree of the perovskite structure and improve the durability.
CaCu3Ru4O12 was reported to show an ultra-low overpotential of 171 mV at 10 mA cmgeo−2 in 0.5 M H2SO4.121 In addition, its mass activity (1942 A gRu−1) is 170 times that of commercial RuO2 at 1.50 V. In terms of crystal structure, the refined Ru–O bond length of CaCu3Ru4O12 (1.9808(7) Å) is longer than the average Ru–O bond length of RuO2 (∼1.97 Å). This indicates that the Ru–O bond strength of CaCu3Ru4O12 is weaker than that of RuO2. The theoretical OER overpotential was further evaluated from DFT calculations. Free energy calculated on relatively stable RuO2(110) and CaCu3Ru4O12(001) surfaces. The free energy difference between ΔGHOO* and ΔGO* in the rate-determining step of RuO2 is 2.08 eV, resulting in a high OER overpotential of 0.85V. For CaCu3Ru4O12, ΔGHOO*–ΔGO* is significantly reduced to 1.89 eV, corresponding to a theoretical overpotential of 0.66 V. This suggests that the unique quadruple structure of CaCu3Ru4O12 is associated with superior activity of the OER. Shunsuke Yagi et al. further investigated the effect of A-site cations on the catalytic activity and stability of ACu3Ru4O12 (A= Ca, Sr, La, Nd, Ce) in acidic aqueous solution.125 The different cations at A-sites change the average valence states of Cu and Ru and the bond length of Ru–O. The relationship between the activity of the OER and average Cu valence shows a volcanic type relationship (Fig. 11e). In addition, it is further proved that the stability of perovskite oxide in the OER increases with the increase of Ru–O covalence with the increase of Ru–O bond length. The adjustment of different ions in quadruple perovskite oxides provides a new idea for designing OER catalysts with high activity and stability in strongly acidic aqueous solutions.
6. Conclusions and perspectives
In this review, descriptors widely accepted for the catalytic activity of perovskite oxides are first discussed. The widely studied oxygen evolution mechanisms (AEM and LOM) were discussed and their differences were analyzed in terms of active sites and reaction pathways (Fig. 12). In particular, the method of regulating the mutual conversion of the AEM–LOM mechanism to obtain the best performance of the OER was introduced. On the basis of understanding these theories, the design and development of high performance OER catalysts are guided. Then, the design strategies of high efficiency perovskite oxide electrocatalysts adjusting the structure and surface in recent years, including component design, morphology control and defect engineering, were introduced. Finally, the research progress of Ir/Ru-based perovskite oxide catalysts in an acidic environment is introduced. Although perovskite oxides have made great progress in the field of the OER, there are still several directions that need further consideration in the future. | ||
| Fig. 12 A schematic representation of the strategies on the design of perovskite oxide electrocatalysts for the OER. | ||
6.1. Mechanism of perovskite oxide for the OER
In the proposed oxygen evolution reaction mechanism (AEM and LOM), metals or oxygen is mainly used for the adsorption of OH and oxygen production sites. The mechanism of the OER, especially lattice oxygen, should be better understood. For example, the catalyst performance depending on the pH value is used as one of the criteria to determine the occurrence of LOM. However, the adsorption process of intermediates is poorly matched with the catalyst in the interpretation of LOM. Another, as the LOM has a lower overpotential than the AEM, has been confirmed by the DFT calculation. But its physical origin has not been explored. In particular, perovskite oxides in acidic media are often associated with dissolution and structural collapse, where the extent of LOM contribution to the OER in acidity remains to be determined. A deep understanding of the oxygen evolution mechanism of perovskite oxide is of guiding significance for the design of high-performance catalysis, so we should further study the catalytic mechanism.Improvements can be made in two ways. On the one hand, more efficient and accurate in situ characterization techniques, such as in situ Raman spectroscopy, in situ X-ray diffraction and synchrotron radiation X-ray absorption spectroscopy, are developed to realize real-time monitoring of surface changes of perovskite oxides. On the other hand, the application of complex theoretical computing techniques, machine learning and artificial intelligence can more accurately and effectively study the intermediate steps in the catalytic process, so as to guide the design of OER electrocatalysts and their further optimization. In particular, machine learning can recombine information from published experimental data and open-source databases by obtaining large amounts of material information, as opposed to high-throughput DFT approaches that rely heavily on time-consuming and expensive computational simulations.
6.2. Interface engineering of perovskite oxides
The catalytic reaction occurs on the surface of the electrocatalyst. The reactants are first adsorbed on the surface of the catalyst, then converted to products, and finally the products are desorbed from the surface of the catalyst to the electrolyte.127 The OER reaction rate is determined by the chemical bond between the substrate molecule and the catalyst.128 Therefore, interface engineering is very critical to the catalytic activity of perovskite oxides. On the one hand, the interface between the catalyst and electrolyte can be adjusted. Introducing foreign ions into the electrolyte to achieve dynamic equilibrium on the catalyst surface is a fast and effective way.77,127 Alternatively, ultra-thin amorphous FeOOH nanosheets are constructed on the catalyst surface to decorate perovskite structures.129 On the other hand, the interface is constructed in the catalyst. Integration of metals and metal compounds has received increasing attention because the Mott–Schottky effect allows the transfer of electrons between metals and metal compounds.130 The surface of perovskite oxide can be modified to form an interface structure and achieve a strong synergistic effect.On the whole, interfacial regulation can efficiently control the atomic and electronic structures of perovskite oxides in order to optimize chemical bond strength, electronic interaction, or achieve a synergistic impact, as well as enhance the charge transfer ability.
6.3. Practical electrolyzer test and application
Currently, most tests of OER in research are performed on rotating disk electrodes in beakers, where catalyst tests have a small area and low voltage. Laboratory testing conditions do not meet industrial requirements. Moreover, there are more uncontrollable factors in industrial application, which leads to the existing evaluation system of catalysts not being complete and accurate. For industrial applications, electrocatalysis require better performance and stability under industry-related conditions such as high current density, long working hours, high pressure and temperature.131–133 The joint European Fuel Cell and Hydrogen Project has set a target for the industrialization of electrolytic water, with an alkaline hydrolysis current density of 800 mA cm−2 and a PEM hydrolysis current density of 2500 mA cm−2 by 2030.134 Therefore, the test phase in the laboratory should be carried out under simple simulation of industrial conditions (current density greater than 200 mA cm−2, temperature at 50–80 °C). It is necessary to design and study more stable and efficient electrolytic water perovskite oxides.The perovskite oxide catalyst assembled in proton exchange membrane water PEMWE for testing is also one of the keys to large-scale application. Although many transition metals have good oxygen evolution activity in acidic environments, it is tough to achieve the unity of high activity and stable structure. In order to reduce the cost, in addition to reducing the content of precious metals by utilizing the advantages of perovskite oxide structure which can accommodate a variety of elements mentioned above, it is also an effective way to maximize the catalytic efficiency of precious metals. Such as building single-atoms materials, heterogeneous structures, etc. single-atoms catalysts have been widely studied for their high catalytic efficiency. But it still faces obstacles to reuniting and shedding. So there is still a long way to go to develop perovskite oxide catalysts for practical application. In addition, PEMWE contains a number of membrane electrode components, including an electrocatalyst layer, a membrane/polymer and a porous transport layer.135,136 The assembly of these membranes also has a great influence on the catalytic performance. It is also important to develop membrane electrode assemblies with high corrosion resistance and high-quality transmission rates. It is inferred that the industrialization of water electrolysis based on perovskite oxide electrocatalyst can be realized faster and more accurately by combining the testing and the investment of electrolytic cell under industrial conditions.
6.4. Economic considerations of perovskite oxides
Economic considerations are also important conditions for the development of perovskite oxide catalysts in the future. In order to realize the large-scale application of perovskite oxide catalysts, the technical cost should be considered. The cost, on the one hand, comes from the cost of raw materials. On the other hand, the costs are those incurred in the actual production, assembly and application processes. More economical membrane electrode components should be developed to reduce the energy consumption in the electrochemical reaction and so on.In recent years, inorganic perovskite solar cells with excellent photoelectronic properties used as the active layer of inorganic perovskite solar cells in the field of new energy have been rapidly developed.137–139 Decommissioned inorganic perovskite materials are rich in transition metal resources and their high value recovery is beneficial to the metal resource cycle and environmental protection. Solid wastes containing transition metals can be used as potential raw materials. Transforming them into advanced functional materials through etching and other means is more in line with the requirements of a green economy. We believe that combined efforts to develop efficient electrochemical water decomposition catalysts will accelerate perovskite oxide catalysts to become participants in the realization of the ideal hydrogen energy society.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (52174283 and 52274308).References
- R. Lee, The Outlook for Population Growth, Science, 2011, 333, 569–573 CrossRef CAS PubMed
.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, A review on fundamentals for designing oxygen evolution electrocatalysts, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC
- B. Lu, D. Wang, C. Zhao, K. Zhu, J. Yan, G. Wang, D. Cao and K. Ye, The novel dual-category active sites of NiCoP/CoP as high-performance electrocatalyst for urea electrolysis and synergistic hydrogen production, Mater. Chem. Front., 2022, 6, 1681–1689 RSC
- R.-Y. Fan, J.-Y. Xie, H.-J. Liu, H.-Y. Wang, M.-X. Li, N. Yu, R.-N. Luan, Y.-M. Chai and B. Dong, Directional regulating dynamic equilibrium to continuously update electrocatalytic interface for oxygen evolution reaction, Chem. Eng. J., 2022, 431, 134040 CrossRef CAS
- T. Wang, X. Cao and L. Jiao, Ni2P/NiMoP heterostructure as a bifunctional electrocatalyst for energy-saving hydrogen production, eScience, 2021, 1, 69–74 CrossRef
- Y.-N. Zhou, F.-L. Wang, S.-Y. Dou, Z.-N. Shi, B. Dong, W.-L. Yu, H.-Y. Zhao, F.-G. Wang, J.-F. Yu and Y.-M. Chai, Motivating high-valence Nb doping by fast molten salt method for NiFe hydroxides toward efficient oxygen evolution reaction, Chem. Eng. J., 2022, 427, 131643 CrossRef CAS
- X. Li, Q. Hu, H. Yang, T. Ma, X. Chai and C. He, Bimetallic two-dimensional materials for electrocatalytic oxygen evolution, Chin. Chem. Lett., 2022, 33, 3657–3671 CrossRef CAS
- J.-I. Jung, H. Y. Jeong, M. G. Kim, G. Nam, J. Park and J. Cho, Fabrication of Ba0.5Sr0.5Co0.8Fe0.2O3–δ Catalysts with Enhanced Electrochemical Performance by Removing an Inherent Heterogeneous Surface Film Layer, Adv. Mater., 2015, 27, 266–271 CrossRef CAS PubMed
- Z. Song, X. Wang, F. Liu, Q. Zhou, W.-J. Yin, H. Wu, W. Deng and J. Wang, Distilling universal activity descriptors for perovskite catalysts from multiple data sources via multi-task symbolic regression, Mater. Horiz., 2023, 10, 1651–1660 RSC
- Y. Bu, S. Kim, O. Kwon, Q. Zhong and G. Kim, A Composite Catalyst Based on Perovskites for Overall Water Splitting in Alkaline Conditions, ChemElectroChem, 2019, 6, 1520–1524 CrossRef CAS
- J. Chen, J. Wu, Y. Liu, X. Hu and D. Geng, Assemblage of Perovskite LaNiO3 Connected With In Situ Grown Nitrogen-Doped Carbon Nanotubes as High-Performance Electrocatalyst for Oxygen Evolution Reaction, Phys. Status Solidi A, 2018, 215, 1800380 CrossRef
- O. Diaz-Morales, S. Raaijman, R. Kortlever, P. J. Kooyman, T. Wezendonk, J. Gascon, W. T. Fu and M. T. M. Koper, Iridium-based double perovskites for efficient water oxidation in acid media, Nat. Commun., 2016, 7, 12363 CrossRef CAS PubMed
- X. Xu, Y. Zhong and Z. Shao, Double Perovskites in Catalysis, Electrocatalysis, and Photo(electro)catalysis, Trends Chem., 2019, 1, 410–424 CrossRef CAS
- N.-I. Kim, Y. J. Sa, T. S. Yoo, S. R. Choi, R. A. Afzal, T. Choi, Y.-S. Seo, K.-S. Lee, J. Y. Hwang, W. S. Choi, S. H. Joo and J.-Y. Park, Oxygen-deficient triple perovskites as highly active and durable bifunctional electrocatalysts for oxygen electrode reactions, Sci. Adv., 2018, 4, eaap9360 CrossRef PubMed
- I. Yamada, H. Fujii, A. Takamatsu, H. Ikeno, K. Wada, H. Tsukasaki, S. Kawaguchi, S. Mori and S. Yagi, Bifunctional Oxygen Reaction Catalysis of Quadruple Manganese Perovskites, Adv. Mater., 2017, 29, 1603004 CrossRef PubMed
- X. Xu, Y. Pan, Y. Zhong, R. Ran and Z. Shao, Ruddlesden–Popper perovskites in electrocatalysis, Mater. Horiz., 2020, 7, 2519–2565 RSC
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Perovskites in catalysis and electrocatalysis, Science, 2017, 358, 751–756 CrossRef CAS PubMed
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Dopants fixation of Ruthenium for boosting acidic oxygen evolution stability and activity, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed
- C. R. Lhermitte and B. M. Bartlett, Advancing the Chemistry of CuWO4 for Photoelectrochemical Water Oxidation, Acc. Chem. Res., 2016, 49, 1121–1129 CrossRef CAS PubMed
- T. Liu, Q. Zhang and Q. Zhao, Theoretical insight into the anion vacancy healing process during the oxygen evolution reaction on TaON and Ta3N5, Phys. Chem. Chem. Phys., 2022, 24, 13999–14006 RSC
- V. F. Mattick, X. Jin, T. Yang, R. E. White and K. Huang, Unraveling Oxygen Electrocatalysis Mechanisms on a Thin-Film Oxygen-Deficient Perovskite La0.6Sr0.4CoO3−δ, ACS Appl. Energy Mater., 2018, 1, 3937–3946 CrossRef CAS
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, A-Site Management Prompts the Dynamic Reconstructed Active Phase of Perovskite Oxide OER Catalysts, Adv. Energy Mater., 2021, 11, 2003755 CrossRef CAS
- L. Zhao, K. Chen, Y. Liu and B. He, A novel layered perovskite as symmetric electrode for direct hydrocarbon solid oxide fuel cells, J. Power Sources, 2017, 342, 313–319 CrossRef CAS
- V. Tripkovic, H. A. Hansen, J. M. Garcia-Lastra and T. Vegge, Comparative DFT+U and HSE Study of the Oxygen Evolution Electrocatalysis on Perovskite Oxides, J. Phys. Chem. C, 2018, 122, 1135–1147 CrossRef CAS
- S. Lee, M. R. Ashwin Kishore, D. Kim, H. Kang, J. Chun, L. S. Oh, J. H. Park, H. J. Kim, J. S. Yoo and E. Lim, Direct O–O Coupling Promoted the Oxygen Evolution Reaction by Dual Active Sites from Ag/LaNiO3 Interfaces, ACS Appl. Energy Mater., 2022, 5, 14658–14668 CrossRef CAS
- M. T. M. Koper, Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis, Chem. Sci., 2013, 4, 2710–2723 RSC
- S. Vasala and M. Karppinen, A2B′B′′O6 perovskites: A review, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS
- Z. Wang, Z. Hao, F. Shi, K. Zhu, X. Zhu and W. Yang, Boosting the oxygen evolution reaction through migrating active sites from the bulk to surface of perovskite oxides, J. Energy Chem., 2022, 69, 434–441 CrossRef CAS
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS
- J. O. M. Bockris and T. Otagawa, The Electrocatalysis of Oxygen Evolution on Perovskites, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS
- Y. Matsumoto and E. Sato, Electrocatalytic
properties of transition metal oxides for oxygen evolution reaction, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed
- A. Grimaud, A. Demortière, M. Saubanère, W. Dachraoui, M. Duchamp, M.-L. Doublet and J.-M. Tarascon, Activation of surface oxygen sites on an iridium-based model catalyst for the oxygen evolution reaction, Nat. Energy, 2016, 2, 16189 CrossRef
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Water electrolysis on La1−xSrxCoO3−δ perovskite electrocatalysts, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J.-P. M. Veder, D. Guan, R. O’Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed
- X. Rong, J. Parolin and A. M. Kolpak, A Fundamental Relationship between Reaction Mechanism and Stability in Metal Oxide Catalysts for Oxygen Evolution, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS
- J.-W. Zhao, H. Zhang, C.-F. Li, X. Zhou, J.-Q. Wu, F. Zeng, J. Zhang and G.-R. Li, Key roles of surface Fe sites and Sr vacancies in the perovskite for an efficient oxygen evolution reaction via lattice oxygen oxidation, Energy Environ. Sci., 2022, 15, 3912–3922 RSC
- M. Lu, Y. Zheng, Y. Hu, B. Huang, D. Ji, M. Sun, J. Li, Y. Peng, R. Si, P. Xi and C.-H. Yan, Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites, Sci. Adv., 2022, 8, eabq3563 CrossRef CAS PubMed
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts, Nat. Energy, 2019, 4, 329–338 CrossRef CAS
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Iron-facilitated dynamic active-site generation on spinel CoAl2O4 with self-termination of surface reconstruction for water oxidation, Nat. Catal., 2019, 2, 763–772 CrossRef CAS
- Y. Duan, S. Sun, Y. Sun, S. Xi, X. Chi, Q. Zhang, X. Ren, J. Wang, S. J. H. Ong, Y. Du, L. Gu, A. Grimaud and Z. J. Xu, Mastering Surface Reconstruction of Metastable Spinel Oxides for Better Water Oxidation, Adv. Mater., 2019, 31, 1807898 CrossRef PubMed
- J. Zaanen, G. A. Sawatzky and J. W. Allen, Band gaps and electronic structure of transition-metal
compounds, Phys. Rev. Lett., 1985, 55, 418–421 CrossRef CAS PubMed
- A. K. Tomar, U. N. Pan, N. H. Kim and J. H. Lee, Enabling Lattice Oxygen Participation in a Triple Perovskite Oxide Electrocatalyst for the Oxygen Evolution Reaction, ACS Energy Lett., 2023, 8, 565–573 CrossRef CAS
- H. Chen, C. Lim, M. Zhou, Z. He, X. Sun, X. Li, Y. Ye, T. Tan, H. Zhang, C. Yang, J. W. Han and Y. Chen, Activating Lattice Oxygen in Perovskite Oxide by B-Site Cation Doping for Modulated Stability and Activity at Elevated Temperatures, Adv. Sci., 2021, 8, 2102713 CrossRef CAS PubMed
- G. T. K. K. Gunasooriya and J. K. Nørskov, Analysis of Acid-Stable and Active Oxides for the Oxygen Evolution Reaction, ACS Energy Lett., 2020, 5, 3778–3787 CrossRef CAS
- Z. Shi, X. Wang, J. Ge, C. Liu and W. Xing, Fundamental understanding of the acidic oxygen evolution reaction: mechanism study and state-of-the-art catalysts, Nanoscale, 2020, 12, 13249–13275 RSC
- J. Zhu, S. Li, Z. Zhuang, S. Gao, X. Hong, X. Pan, R. Yu, L. Zhou, L. V. Moskaleva and L. Mai, Ultrathin Metal Silicate Hydroxide Nanosheets with Moderate Metal–Oxygen Covalency Enables Efficient Oxygen Evolution, Energy Environ. Mater., 2022, 5, 231–237 CrossRef CAS
- L. Heymann, M. L. Weber, M. Wohlgemuth, M. Risch, R. Dittmann, C. Baeumer and F. Gunkel, Separating the Effects of Band Bending and Covalency in Hybrid Perovskite Oxide Electrocatalyst Bilayers for Water Electrolysis, ACS Appl. Mater. Interfaces, 2022, 14, 14129–14136 CrossRef CAS PubMed
- Z.-Y. Yu, Y. Duan, Y. Kong, X.-L. Zhang, X.-Y. Feng, Y. Chen, H. Wang, X. Yu, T. Ma, X. Zheng, J. Zhu, M.-R. Gao and S.-H. Yu, General Synthesis of Tube-like Nanostructured Perovskite Oxides with Tunable Transition Metal–Oxygen Covalency for Efficient Water Electrooxidation in Neutral Media, J. Am. Chem. Soc., 2022, 144, 13163–13173 CrossRef CAS PubMed
- Z. Shen, M. Qu, J. Shi, F. E. Oropeza, V. A. de la Peña O'Shea, G. Gorni, C. M. Tian, J. P. Hofmann, J. Cheng, J. Li and K. H. L. Zhang, Correlating the electronic structure of perovskite La1−xSrxCoO3 with activity for the oxygen evolution reaction: The critical role of Co 3d hole state, J. Energy Chem., 2022, 65, 637–645 CrossRef CAS
- H. Wang, J. Wang, Y. Pi, Q. Shao, Y. Tan and X. Huang, Double Perovskite LaFexNi1−xO3 Nanorods Enable Efficient Oxygen Evolution Electrocatalysis, Angew. Chem., Int. Ed., 2019, 58, 2316–2320 CrossRef CAS PubMed
- Y. Tong, J. Wu, P. Chen, H. Liu, W. Chu, C. Wu and Y. Xie, Vibronic Superexchange in Double Perovskite Electrocatalyst for Efficient Electrocatalytic Oxygen Evolution, J. Am. Chem. Soc., 2018, 140, 11165–11169 CrossRef CAS PubMed
- A. L. Hoang, S. Balakrishnan, A. Hodges, G. Tsekouras, A. Al-Musawi, K. Wagner, C.-Y. Lee, G. F. Swiegers and G. G. Wallace, High-performing catalysts for energy-efficient commercial alkaline water electrolysis, Sustainable Energy Fuels, 2023, 7, 31–60 RSC
- J. Qian, T. Wang, Z. Zhang, Y. Liu, J. Li and D. Gao, Engineered spin state in Ce doped LaCoO3 with enhanced electrocatalytic activity for rechargeable Zn–Air batteries, Nano Energy, 2020, 74, 104948 CrossRef CAS
- D. A. Kumar, S. Selvasekarapandian, H. Nithya, J. Leiro, Y. Masuda, S.-D. Kim and S.-K. Woo, Effect of calcium doping on LaCoO3 prepared by Pechini method, Powder Technol., 2013, 235, 140–147 CrossRef CAS
- J. Qian, J. Li, B. Xia, J. Zhang, Z. Zhang, C. Guan, D. Gao and W. Huang, Multi-stability modulating of alkaline-earth metal doped LaCoO3 for rechargeable Zn–air batteries, Energy Storage Mater., 2021, 42, 470–476 CrossRef
- L. He, Y. Zhang, Y. Zang, C. Liu, W. Wang, R. Han, N. Ji, S. Zhang and Q. Liu, Promotion of A-Site Ag-Doped Perovskites for the Catalytic Oxidation of Soot: Synergistic Catalytic Effect of Dual Active Sites, ACS Catal., 2021, 11, 14224–14236 CrossRef CAS
- C. Liu, D. Ji, H. Shi, Z. Wu, H. Huang, Z. Kang and Z. Chen, An A-site management and oxygen-deficient regulation strategy with a perovskite oxide electrocatalyst for the oxygen evolution reaction, J. Mater. Chem. A, 2022, 10, 1336–1342 RSC
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Functional links between stability and reactivity of strontium ruthenate single crystals during oxygen evolution, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed
- B.-J. Kim, D. F. Abbott, X. Cheng, E. Fabbri, M. Nachtegaal, F. Bozza, I. E. Castelli, D. Lebedev, R. Schäublin, C. Copéret, T. Graule, N. Marzari and T. J. Schmidt, Unraveling Thermodynamics, Stability, and Oxygen Evolution Activity of Strontium Ruthenium Perovskite Oxide, ACS Catal., 2017, 7, 3245–3256 CrossRef CAS
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, Á. García, J. Torrero, M. T. Fernández-Díaz, P. Bencok, M. A. Peña, J. L. G. Fierro and S. Rojas, Na-doped ruthenium perovskite electrocatalysts with improved oxygen evolution activity and durability in acidic media, Nat. Commun., 2019, 10, 2041 CrossRef PubMed
- I. Rodríguez-García, D. Galyamin, L. Pascual, P. Ferrer, M. A. Peña, D. Grinter, G. Held, M. Abdel Salam, M. Mokhtar, K. Narasimharao, M. Retuerto and S. Rojas, Enhanced stability of SrRuO3 mixed oxide via monovalent doping in Sr1−xKxRuO3 for the oxygen evolution reaction, J. Power Sources, 2022, 521, 230950 CrossRef
- H.-T. Jeng, S.-H. Lin and C.-S. Hsue, Orbital Ordering and Jahn–Teller Distortion in Perovskite Ruthenate SrRuO3, Phys. Rev. Lett., 2006, 97, 067002 CrossRef PubMed
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS
- H. Jo, Y. Yang, A. Seong, D. Jeong, J. Kim, S. H. Joo, Y. J. Kim, L. Zhang, Z. Liu, J.-Q. Wang, S. K. Kwak and G. Kim, Promotion of the oxygen evolution reaction via the reconstructed active phase of perovskite oxide, J. Mater. Chem. A, 2022, 10, 2271–2279 RSC
- G. Chen, Z. Hu, Y. Zhu, Z.-G. Chen, Y. Zhong, H.-J. Lin, C.-T. Chen, L. H. Tjeng, W. Zhou and Z. Shao, Ultrahigh-performance tungsten-doped perovskites for the oxygen evolution reaction, J. Mater. Chem. A, 2018, 6, 9854–9859 RSC
- J. Xiong, H. Zhong, J. Li, X. Zhang, J. Shi, W. Cai, K. Qu, C. Zhu, Z. Yang, S. P. Beckman and H. Cheng, Engineering highly active oxygen sites in perovskite oxides for stable and efficient oxygen evolution, Appl. Catal., B, 2019, 256, 117817 CrossRef CAS
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Modulation of Electronics of Oxide Perovskites by Sulfur Doping for Electrocatalysis in Rechargeable Zn–Air Batteries, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS
- Y. Zhu, Q. Lin, Z. Wang, D. Qi, Y. Yin, Y. Liu, X. Zhang, Z. Shao and H. Wang, Chlorine-anion doping induced multi-factor optimization in perovskties for boosting intrinsic oxygen evolution, J. Energy Chem., 2021, 52, 115–120 CrossRef CAS
- L. Tang, Y. Yang, H. Guo, Y. Wang, M. Wang, Z. Liu, G. Yang, X. Fu, Y. Luo, C. Jiang, Y. Zhao, Z. Shao and Y. Sun, High Configuration Entropy Activated Lattice Oxygen for O2 Formation on Perovskite Electrocatalyst, Adv. Funct. Mater., 2022, 32, 2112157 CrossRef CAS
- T. X. Nguyen, Y.-C. Liao, C.-C. Lin, Y.-H. Su and J.-M. Ting, Advanced High Entropy Perovskite Oxide Electrocatalyst for Oxygen Evolution Reaction, Adv. Funct. Mater., 2021, 31, 2101632 CrossRef CAS
- G. Valderrama, A. Kiennemann, C. U. de Navarro and M. R. Goldwasser, LaNi1−xMnxO3 perovskite-type oxides as catalysts precursors for dry reforming of methane, Appl. Catal., A, 2018, 565, 26–33 CrossRef CAS
- X. Shen, Y. Sun, Y. Wu, J. Wang, E. Jiang, X. Xu, J. Su and Z. Jia, The coupling of CH4 partial oxidation and CO2 splitting for syngas production via double perovskite-type oxides LaFexCo1−xO3, Fuel, 2020, 268, 117381 CrossRef CAS
- X. Cheng, B.-J. Kim, E. Fabbri and T. J. Schmidt, Co/Fe Oxyhydroxides Supported on Perovskite Oxides as Oxygen Evolution Reaction Catalyst Systems, ACS Appl. Mater. Interfaces, 2019, 11, 34787–34795 CrossRef CAS PubMed
- B. W. Kwon, J. H. Oh, G. S. Kim, S. P. Yoon, J. Han, S. W. Nam and H. C. Ham, The novel perovskite-type Ni-doped Sr0.92Y0.08TiO3 catalyst as a reforming biogas (CH4 + CO2) for H2 production, Appl. Energy, 2018, 227, 213–219 CrossRef CAS
- N. Yu, H.-J. Liu, Y.-N. Cao, Q.-Y. Wang, Y. Ma, J.-F. Yu, H. Hu, Y.-M. Chai and B. Dong, Dynamic equilibrium of external Fe3+ to effectively construct catalytic surface of perovskite LaNi1−xFexO3 for water oxidation, Colloids Surf., A, 2022, 654, 130042 CrossRef CAS
- N. Yu, X.-Y. Zhang, Q.-Y. Wang, H.-J. Liu, G. Han, F.-G. Wang, Y.-L. Zhou, Y.-M. Chai and B. Dong, Electrochemical activation construction of LaCo1−x−yNixFeyO3 promoted by optimized lattice Ni doping toward water oxidation, Int. J. Hydrogen Energy, 2022, 47, 39097–39107 CrossRef CAS
- B.-J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, Functional Role of Fe-Doping in Co-Based Perovskite Oxide Catalysts for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2019, 141, 5231–5240 CrossRef CAS PubMed
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution, Nat. Commun., 2013, 4, 2439 CrossRef PubMed
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Phosphorus-Doped Perovskite Oxide as Highly Efficient Water Oxidation Electrocatalyst in Alkaline Solution, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS
- K. I. Kobayashi, T. Kimura, H. Sawada, K. Terakura and Y. Tokura, Room-temperature magnetoresistance in an oxide material with an ordered double-perovskite structure, Nature, 1998, 395, 677–680 CrossRef CAS
- M. García-Hernández, J. L. Martínez, M. J. Martínez-Lope, M. T. Casais and J. A. Alonso, Finding Universal Correlations between Cationic Disorder and Low Field Magnetoresistance in FeMo Double Perovskite Series, Phys. Rev. Lett., 2001, 86, 2443–2446 CrossRef PubMed
- S. Zhao, L. Shi, S. Zhou, J. Zhao, H. Yang and Y. Guo, Size-dependent magnetic properties and Raman spectra of La2NiMnO6 nanoparticles, J. Appl. Phys., 2009, 106, 123901 CrossRef
- J. F. Shin, A. Orera, D. C. Apperley and P. R. Slater, Oxyanion doping strategies to enhance the ionic conductivity in Ba2In2O5, J. Mater. Chem., 2011, 21, 874–879 RSC
- J. M. Porras-Vazquez, T. F. Kemp, J. V. Hanna and P. R. Slater, Synthesis and characterisation of oxyanion-doped manganites for potential application as SOFC cathodes, J. Mater. Chem., 2012, 22, 8287–8293 RSC
- J. M. Porras-Vazquez, T. Pike, C. A. Hancock, J. F. Marco, F. J. Berry and P. R. Slater, Investigation into the effect of Si doping on the performance of SrFeO3−δ SOFC electrode materials, J. Mater. Chem. A, 2013, 1, 11834–11841 RSC
- H. Wang, Y.-F. Yu, Q.-W. Chen and K. Cheng, Carboxyl-functionalized nanoparticles with magnetic core and mesopore carbon shell as adsorbents for the removal of heavy metal ions from aqueous solution, Dalton Trans., 2011, 40, 559–563 RSC
- J. Li, S. Bernard, V. Salles, C. Gervais and P. Miele, Preparation of Polyborazylene-Derived Bulk Boron Nitride with Tunable Properties by Warm-Pressing and Pressureless Pyrolysis, Chem. Mater., 2010, 22, 2010–2019 CrossRef CAS
- B. Hua, M. Li, W. Pang, W. Tang, S. Zhao, Z. Jin, Y. Zeng, B. Shalchi Amirkhiz and J.-L. Luo, Activating p-Blocking Centers in Perovskite for Efficient Water Splitting, Chem, 2018, 4, 2902–2916 CAS
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, A Perovskite Nanorod as Bifunctional Electrocatalyst for Overall Water Splitting, Adv. Energy Mater., 2017, 7, 1602122 CrossRef
- S. Bie, Y. Zhu, J. Su, C. Jin, S. Liu, R. Yang and J. Wu, One-pot fabrication of yolk–shell structured La0.9Sr0.1CoO3 perovskite microspheres with enhanced catalytic activities for oxygen reduction and evolution reactions, J. Mater. Chem. A, 2015, 3, 22448–22453 RSC
- J. Kim, X. Chen, P.-C. Shih and H. Yang, Porous Perovskite-Type Lanthanum Cobaltite as Electrocatalysts toward Oxygen Evolution Reaction, ACS Sustainable Chem. Eng., 2017, 5, 10910–10917 CrossRef CAS
- J. Dai, Y. Zhu, Y. Zhong, J. Miao, B. Lin, W. Zhou and Z. Shao, Enabling High and Stable Electrocatalytic Activity of Iron-Based Perovskite Oxides for Water Splitting by Combined Bulk Doping and Morphology Designing, Adv. Mater. Interfaces, 2019, 6, 1801317 CrossRef
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Engineering electrocatalytic activity in nanosized perovskite cobaltite through surface spin-state transition, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed
- C. Jin, X. Cao, L. Zhang, C. Zhang and R. Yang, Preparation and electrochemical properties of urchin-like La0.8Sr0.2MnO3 perovskite oxide as a bifunctional catalyst for oxygen reduction and oxygen evolution reaction, J. Power Sources, 2013, 241, 225–230 CrossRef CAS
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, In Operando Identification of Geometrical-Site-Dependent Water Oxidation Activity of Spinel Co3O4, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed
- S. Malkhandi, P. Trinh, A. K. Manohar, A. Manivannan, M. Balasubramanian, G. K. S. Prakash and S. R. Narayanan, Design Insights for Tuning the Electrocatalytic Activity of Perovskite Oxides for the Oxygen Evolution Reaction, J. Phys. Chem. C, 2015, 119, 8004–8013 CrossRef CAS
- H. A. Tahini, X. Tan, U. Schwingenschlögl and S. C. Smith, Formation and Migration of Oxygen Vacancies in SrCoO3 and Their Effect on Oxygen Evolution Reactions, ACS Catal., 2016, 6, 5565–5570 CrossRef CAS
- E. Marelli, J. Gazquez, E. Poghosyan, E. Müller, D. J. Gawryluk, E. Pomjakushina, D. Sheptyakov, C. Piamonteze, D. Aegerter, T. J. Schmidt, M. Medarde and E. Fabbri, Correlation between Oxygen Vacancies and Oxygen Evolution Reaction Activity for a Model Electrode: PrBaCo2O5+δ, Angew. Chem., Int. Ed., 2021, 60, 14609–14619 CrossRef CAS PubMed
- J. Kim, X. Yin, K.-C. Tsao, S. Fang and H. Yang, Ca2Mn2O5 as Oxygen-Deficient Perovskite Electrocatalyst for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed
- Y. Lu, A. Ma, Y. Yu, R. Tan, C. Liu, P. Zhang, D. Liu and J. Gui, Engineering Oxygen Vacancies into LaCoO3 Perovskite for Efficient Electrocatalytic Oxygen Evolution, ACS Sustainable Chem. Eng., 2019, 7, 2906–2910 CrossRef CAS
- J. Bian, Z. Li, N. Li and C. Sun, Oxygen Deficient LaMn0.75Co0.25O3−δ Nanofibers as an Efficient Electrocatalyst for Oxygen Evolution Reaction and Zinc–Air Batteries, Inorg. Chem., 2019, 58, 8208–8214 CrossRef CAS PubMed
- L. Wang, K. A. Stoerzinger, L. Chang, X. Yin, Y. Li, C. S. Tang, E. Jia, M. E. Bowden, Z. Yang, A. Abdelsamie, L. You, R. Guo, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, Strain Effect on Oxygen Evolution Reaction Activity of Epitaxial NdNiO3 Thin Films, ACS Appl. Mater. Interfaces, 2019, 11, 12941–12947 CrossRef CAS PubMed
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, Enhancing Perovskite Electrocatalysis through Strain Tuning of the Oxygen Deficiency, J. Am. Chem. Soc., 2016, 138, 7252–7255 CrossRef CAS PubMed
- H. Wang, J. Qi, N. Yang, W. Cui, J. Wang, Q. Li, Q. Zhang, X. Yu, L. Gu, J. Li, R. Yu, K. Huang, S. Song, S. Feng and D. Wang, Dual-Defects Adjusted Crystal-Field Splitting of LaCo1−xNixO3−δ Hollow Multishelled Structures for Efficient Oxygen Evolution, Angew. Chem., Int. Ed., 2020, 59, 19691–19695 CrossRef CAS PubMed
- F. You, J. Wan, J. Qi, D. Mao, N. Yang, Q. Zhang, L. Gu and D. Wang, Lattice Distortion in Hollow Multi-Shelled Structures for Efficient Visible-Light CO2 Reduction with a SnS2/SnO2 Junction, Angew. Chem., Int. Ed., 2020, 59, 721–724 CrossRef CAS PubMed
- D. N. Mueller, M. L. Machala, H. Bluhm and W. C. Chueh, Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions, Nat. Commun., 2015, 6, 6097 CrossRef CAS PubMed
- L. Yan, Y. Lin, X. Yu, W. Xu, T. Salas, H. Smallidge, M. Zhou and H. Luo, La0.8Sr0.2MnO3-Based Perovskite Nanoparticles with the A-Site Deficiency as High Performance Bifunctional Oxygen Catalyst in Alkaline Solution, ACS Appl. Mater. Interfaces, 2017, 9, 23820–23827 CrossRef CAS PubMed
- Y. Xue, H. Miao, S. Sun, Q. Wang, S. Li and Z. Liu, (La1−xSrx)0.98MnO3 perovskite with A-site deficiencies toward oxygen reduction reaction in aluminum-air batteries, J. Power Sources, 2017, 342, 192–201 CrossRef CAS
- L. Tang, Y. Rao, L. Wei, H. Zheng, H. Liu, W. Zhang and K. Tang, A-site Cation Defects (Ba0.5Sr0.5)1–xCo0.8Fe0.2O3–δ Perovskites as Active Oxygen Evolution Reaction Catalyst in Alkaline Electrolyte†, Chin. J. Chem., 2021, 39, 2692–2698 CrossRef CAS
- N. Li, J. Guo, Y. Ding, Y. Hu, C. Zhao and C. Zhao, Direct Regulation of Double Cation Defects at the A1A2 Site for a High-Performance Oxygen Evolution Reaction Perovskite Catalyst, ACS Appl. Mater. Interfaces, 2021, 13, 332–340 CrossRef CAS PubMed
- Y. Shi, K. Huang, L. Shen, C. Ding, Z. Lu, H. Tan, C. Guo and C. Yan, Understanding the in-plane electron transport in low noble metal proton exchange membrane water electrolyser, J. Power Sources, 2022, 549, 232130 CrossRef CAS
- M. Upadhyay, A. Kim, S. S. Paramanantham, H. Kim, D. Lim, S. Lee, S. Moon and H. Lim, Three-dimensional CFD simulation of proton exchange membrane water electrolyser: Performance assessment under different condition, Appl. Energy, 2022, 306, 118016 CrossRef CAS
- F. Barbir, PEM electrolysis for production of hydrogen from renewable energy sources, Sol. Energy, 2005, 78, 661–669 CrossRef CAS
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, A highly active and stable IrOx SrIrO3 catalyst for the oxygen evolution reaction, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed
- L. Yang, G. Yu, X. Ai, W. Yan, H. Duan, W. Chen, X. Li, T. Wang, C. Zhang, X. Huang, J.-S. Chen and X. Zou, Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers, Nat. Commun., 2018, 9, 5236 CrossRef CAS PubMed
- N. Li, L. Cai, C. Wang, Y. Lin, J. Huang, H. Sheng, H. Pan, W. Zhang, Q. Ji, H. Duan, W. Hu, W. Zhang, F. Hu, H. Tan, Z. Sun, B. Song, S. Jin and W. Yan, Identification of the Active-Layer Structures for Acidic Oxygen Evolution from 9R-BaIrO3 Electrocatalyst with Enhanced Iridium Mass Activity, J. Am. Chem. Soc., 2021, 143, 18001–18009 CrossRef CAS PubMed
- R. Zhang, N. Dubouis, M. B. Osman, W. Yin, M. T. Sougrati, D. A. D. Corte, D. Giaume and A. Grimaud, A Dissolution/Precipitation Equilibrium on the Surface of Iridium-Based Perovskites Controls Their Activity as Oxygen Evolution Reaction Catalysts in Acidic Media, Angew. Chem., Int. Ed., 2019, 58, 4571–4575 CrossRef CAS PubMed
- H. Chen, L. Shi, K. Sun, K. Zhang, Q. Liu, J. Ge, X. Liang, B. Tian, Y. Huang, Z. Shi, Z. Wang, W. Zhang, M. Liu and X. Zou, Protonated Iridate Nanosheets with a Highly Active and Stable Layered Perovskite Framework for Acidic Oxygen Evolution, ACS Catal., 2022, 12, 8658–8666 CrossRef CAS
- X. Miao, L. Zhang, L. Wu, Z. Hu, L. Shi and S. Zhou, Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation, Nat. Commun., 2019, 10, 3809 CrossRef PubMed
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Chromium-ruthenium oxide solid solution electrocatalyst for highly efficient oxygen evolution reaction in acidic media, Nat. Commun., 2019, 10, 162 CrossRef PubMed
- X. Liang, L. Shi, Y. Liu, H. Chen, R. Si, W. Yan, Q. Zhang, G.-D. Li, L. Yang and X. Zou, Activating Inert, Nonprecious Perovskites with Iridium Dopants for Efficient Oxygen Evolution Reaction under Acidic Conditions, Angew. Chem., Int. Ed., 2019, 58, 7631–7635 CrossRef CAS PubMed
- A. L. Strickler, D. Higgins and T. F. Jaramillo, Crystalline Strontium Iridate Particle Catalysts for Enhanced Oxygen Evolution in Acid, ACS Appl. Energy Mater., 2019, 2, 5490–5498 CrossRef CAS
- W. Liu, K. Kawano, M. Kamiko, Y. Kato, Y. Okazaki, I. Yamada and S. Yagi, Effects of A-site Cations in Quadruple Perovskite Ruthenates on Oxygen Evolution Catalysis in Acidic Aqueous Solutions, Small, 2022, 18, 2202439 CrossRef CAS PubMed
- M. A. Peña and J. L. G. Fierro, Chemical Structures and Performance of Perovskite Oxides, Chem. Rev., 2001, 101, 1981–2018 CrossRef PubMed
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. Farinazzo Bergamo Dias Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, Dynamically Stable Active Sites from Surface Evolution of Perovskite Materials during the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2021, 143, 2741–2750 CrossRef CAS PubMed
- C. Hu, J. Hong, J. Huang, W. Chen, C. U. Segre, K. Suenaga, W. Zhao, F. Huang and J. Wang, Surface decoration accelerates the hydrogen evolution kinetics of a perovskite oxide in alkaline solution, Energy Environ. Sci., 2020, 13, 4249–4257 RSC
- Z. Zhang, B. He, L. Chen, H. Wang, R. Wang, L. Zhao and Y. Gong, Boosting Overall Water Splitting via FeOOH Nanoflake-Decorated PrBa0.5Sr0.5Co2O5+δ Nanorods, ACS Appl. Mater. Interfaces, 2018, 10, 38032–38041 CrossRef CAS PubMed
- X. Liang, L. Shi, R. Cao, G. Wan, W. Yan, H. Chen, Y. Liu and X. Zou, Perovskite-Type Solid Solution Nano-Electrocatalysts Enable Simultaneously Enhanced Activity and Stability for Oxygen Evolution, Adv. Mater., 2020, 32, 2001430 CrossRef CAS PubMed
- F. Yang, Y. Luo, Q. Yu, Z. Zhang, S. Zhang, Z. Liu, W. Ren, H.-M. Cheng, J. Li and B. Liu, A Durable and Efficient Electrocatalyst for Saline Water Splitting with Current Density Exceeding 2000 mA cm−2, Adv. Funct. Mater., 2021, 31, 2010367 CrossRef CAS
- L. A. King, M. A. Hubert, C. Capuano, J. Manco, N. Danilovic, E. Valle, T. R. Hellstern, K. Ayers and T. F. Jaramillo, A non-precious metal hydrogen catalyst in a commercial polymer electrolyte membrane electrolyser, Nat. Nanotechnol., 2019, 14, 1071–1074 CrossRef CAS PubMed
- Q. Yang, G. Li, K. Manna, F. Fan, C. Felser and Y. Sun, Topological Engineering of Pt-Group-Metal-Based Chiral Crystals toward High-Efficiency Hydrogen Evolution Catalysts, Adv. Mater., 2020, 32, 1908518 CrossRef CAS PubMed
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Recent Advances in Design of Electrocatalysts for High-Current-Density Water Splitting, Adv. Mater., 2022, 34, 2108133 CrossRef CAS PubMed
- G. S. Ogumerem and E. N. Pistikopoulos, Parametric optimization and control for a smart Proton Exchange Membrane Water Electrolysis (PEMWE) system, J. Power Sources, 2020, 91, 37–49 CAS
- P. Shirvanian and F. van Berkel, Novel components in Proton Exchange Membrane (PEM) Water Electrolyzers (PEMWE): Status, challenges and future needs. A mini review, Electrochem. Commun., 2020, 114, 106704 CrossRef CAS
- W. Xiang, S. Liu and W. Tress, Interfaces and Interfacial Layers in Inorganic Perovskite Solar Cells, Angew. Chem., Int. Ed., 2021, 60, 26440–26453 CrossRef CAS PubMed
- H. Zhang, W. Xiang, X. Zuo, X. Gu, S. Zhang, Y. Du, Z. Wang, Y. Liu, H. Wu, P. Wang, Q. Cui, H. Su, Q. Tian and S. Liu, Fluorine-Containing Passivation Layer via Surface Chelation for Inorganic Perovskite Solar Cells, Angew. Chem., Int. Ed., 2023, 62, e202216634 CrossRef CAS PubMed
- L. Zhang, B. Li, J. Yuan, M. Wang, T. Shen, F. Huang, W. Wen, G. Cao and J. Tian, High-Voltage-Efficiency Inorganic Perovskite Solar Cells in a Wide Solution-Processing Window, J. Phys. Chem. Lett., 2018, 9, 3646–3653 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2023 |