
Highly stable and differentially arranged hexanuclear lanthanide clusters: structure, assembly mechanism, and magnetic resonance imaging†
Wen-Wen
Qin
a,
Yun-Lan
Li
a,
Zhong-Hong
Zhu
 *a,
Fu-Pei
Liang
a,
Qiong
Hu
*b and
Hua-Hong
Zou
*a
*a,
Fu-Pei
Liang
a,
Qiong
Hu
*b and
Hua-Hong
Zou
*a
aSchool of Chemistry and Pharmaceutical Sciences, State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Guangxi Normal University, Guilin 541004, P. R. China. E-mail: 18317725515@163.com; gxnuchem@foxmail.com
bGuangxi Key Laboratory of Agricultural Resources Chemistry and Biotechnology, College of Chemistry and Food Science, Yulin Normal University, Yulin 537000, P. R. China. E-mail: huqiongscut@163.com
First published on 18th September 2023
Abstract
The directed and controllable synthesis of lanthanide clusters with precise structures has received considerable research attention, however, progress in such remains sluggish. The steps performed in a reaction system under “black-box” conditions are unpredictable and have very low controllability. The precise customization of lanthanide clusters with the same number of cores but different arrangements is particularly difficult. Using bis-acylhydrazone-derived multidentate chelating ligands with different substituents, differentially arranged hexanuclear lanthanide clusters (Dy6 and HNP-Dy6) with identical core connections but different template-motif arrangements were constructed herein for the first time using a multidentate chelating coordination method. Specifically, Dy6 with face-to-face and dislocation-arrangement template motifs was obtained using –N(Et)2-substituted bis-acylhydrazone ligands with a strong steric hindrance effect. Changing –N(Et)2 to a benzene ring with a strong π–π interaction yielded HNP-Dy6 with inverted and coplanar arrangements of template motifs. The controllable construction of these two hexanuclear dysprosium clusters represented great progress in the precise synthesis of lanthanide clusters. High-resolution electrospray ionization–mass spectrometry (HRESI–MS) with different ion-source energies demonstrated the high stabilities of Dy6 and HNP-Dy6 in solutions. Time-dependent HRESI–MS tracked the formation processes of Dy6 and HNP-Dy6 and led to the following possible self-assembly mechanisms: L1 + 2Dy → Dy2L1 → Dy3L1 → Dy6(L1)2 and L2 + 2Dy → Dy2L2 → Dy3L2 → Dy5(L2)2/Dy6(L2)2 → Dy6(L2)2. At 1-T magnetic field, the longitudinal and transverse relaxation rates of Gd6 were 12.06 and 24.10 mM−1 s−1, respectively. Gd6 with highly aggregated Gd(III) exhibited high relaxation rates, indicating its great potential as a T1-weighted magnetic resonance imaging contrast agent. This work provides an example of the design and synthesis of lanthanide clusters with high stabilities and relaxation rates, taking a big step toward the precise and controllable synthesis of lanthanide clusters.
Introduction
Lanthanide clusters with a precise structure, a uniform nanoscale size, and rich functions have attracted widespread attention and have shown great application prospects in the fields of molecular magnetism, magnetocaloric effect, bio-imaging, solid-state luminescence, etc.1–11 In particular, gadolinium(III) clusters with a nanometer size and highly concentrated metal centers break through the limitations of conventional magnetic resonance imaging (MRI) contrast agents (CAs) and exhibit a high imaging contrast.12,13 According to the Solomon–Bloembergen Morgan (SBM) paramagnetic relaxation theory, it can be known that CAs with high relaxation, high resolution and high contrast can be obtained by connecting multiple single-nuclear or low-nuclear gadolinium complexes through multi-component integration. High-nuclear gadolinium clusters with highly aggregated Gd(III) undoubtedly show attractive application prospects in the field of MRI. In 2020, Sun et al. used 2,6-pyridine bistetrazolium-derived ligands with different bending angles to construct a water-soluble molecular cage Ln2nL3n, which showed high relaxation rates and excellent in vivo MRI imaging effects in mice.13 In 2023, Tong et al. used solvothermal synthesis technology to construct spherical nanoclusters Gd32 with high stability, low toxicity and excellent water solubility. The highly aggregated Gd(III) in the structure leads to its excellent MRI imaging contrast both in vivo and in vitro.12 Although substantial progress has been made in the application and expansion of lanthanide clusters, progress with regard to their controllable and precise synthesis remains unsatisfactory.14 Previously, lanthanide clusters primarily relied on hydrolysis and the anion template method to guide their structural synthesis. Various lanthanide clusters with beautiful shapes and diverse topologies, such as wheel-shaped Gd140, caged Gd60, hamburger-shaped Dy76, and tubular Dy72, have been constructed based on the abovementioned methods.15–19 In 2018, Bu et al. induced the synthesis of a rare hamburger-shaped dysprosium cluster Dy76 using a mixed-anion template that can be regarded as an assembly of two Dy48 clusters. In 2016, Zheng et al. synthesized a tubular dysprosium cluster Dy72 by controlling the hydrolysis of Dy(III) ions with N-methyldiethanolamine.19 In 2022, Zheng et al. synthesized multinuclear lanthanide hydroxide clusters under the guidance of combined I− and CO32− as mixed templates.20 The complex reaction and the extremely low controllability of the reaction system that lead to directional construction of lanthanide clusters remain to be very vague.The tracking of the self-assembly process and the study of the self-assembly mechanism have accelerated the pace of the directional and rational construction of lanthanide clusters.21–23 Since 2018, we have been using crystallography combined with high-resolution electrospray ionization–mass spectrometry (HRESI-MS) to explore and analyze the formation process and the self-assembly mechanism of several lanthanide clusters. We also proposed the out-to-in, linear, and annular growth mechanisms to guide the directional synthesis of lanthanide clusters.24–28 In 2020, our research group successfully constructed a double-cage dysprosium cluster Dy60 using a multidentate chelating coordination method, tracked the Dy60 formation process using HRESI-MS, and proposed the possible self-assembly mechanism of Dy60.29 In 2021, we first proposed that the out-to-in growth mechanism guides the directional construction of 16 nuclear discotic lanthanide clusters.25,26,30 In 2022, also for the first time, we guided the directional construction of chiral lanthanide clusters with different connection modes and shapes by manipulating the annular growth mechanism.27 We then successfully constructed the largest planar disk-shaped lanthanide cluster Dy19 under solvothermal conditions and proposed its possible assembly mechanism (i.e., the planar epitaxial or planar internal growth mechanism).31 The bulky bis-acylhydrazone and Schiff base-derived ligands with multidentate chelation coordination sites can quickly capture the lanthanide metal ions in solutions to form template units, thereby further assembling to obtain the target lanthanide clusters.24,32,33 These template units formed by linking lanthanide ions with bulky chelating ligands have a higher stability in solution than hydrolyzed products and anion templates, making the self-assembly process of lanthanide clusters more regular and promoting their designability.14 Based on the abovementioned foundations, we focus herein on the precise construction of lanthanide clusters with the same core connections but different template-motif arrangements, which has been difficult to achieve until now.
In this work, we use the –N(Et)2-substituted bis-acylhydrazone-derived ligand H2L1 (N′2,N′6-bis(4-(diethylamino)-2-hydroxybenzylidene)pyridine-2,6-dicarbohydrazide) and Dy(OAc)3·4H2O to perform a reaction under solvothermal conditions and acquire a Dy6 sample, which has face-to-face and dislocation-arrangement template motifs (Scheme 1). Changing –N(Et)2 to a benzene ring with a strong π–π interaction yields HNP-Dy6 with an inverted and coplanar arrangement of template motifs (Scheme 1). We employ HRESI-MS to evaluate the stabilities of the two hexanuclear clusters prepared in this study in a solution. Under the condition of an increasing ion-source energy from 0 to 100 eV, the molecular-ion peaks of Dy6 and HNP-Dy6 in the solution are found to be related to their structural skeletons, indicating that they both maintain a high stability in the solution. We implement time-dependent HRESI-MS to track the species changes during the Dy6 and HNP-Dy6 formations under solvothermal “black-box” reaction conditions. The intermediates of various reactions are identified. Their possible self-assembly mechanisms are proposed as follows: L1 + 2Dy → Dy2L1 → Dy3L1 → Dy6(L1)2 and L2 + 2Dy → Dy2L2 → Dy3L2 → Dy5(L2)2/Dy6(L2)2 → Dy6(L2)2. To the best of our knowledge, this is the first time that multidentate chelation coordination is manipulated through substitution effects to construct heterogeneous hexanuclear lanthanide clusters with the same cluster core connections but different template-motif arrangements. The high cluster stability in the solution promotes the expansion of its application in the solution. Notably, low Gd6 concentration exhibits high longitudinal and transverse relaxation rates (r1 and r2, respectively) at 1-T magnetic field, indicating its great potential as a T1-weighted MRI CA. This work paves a way for the directed and controllable synthesis of lanthanide clusters with precise structures and promotes the progress of lanthanide-cluster crystal engineering.
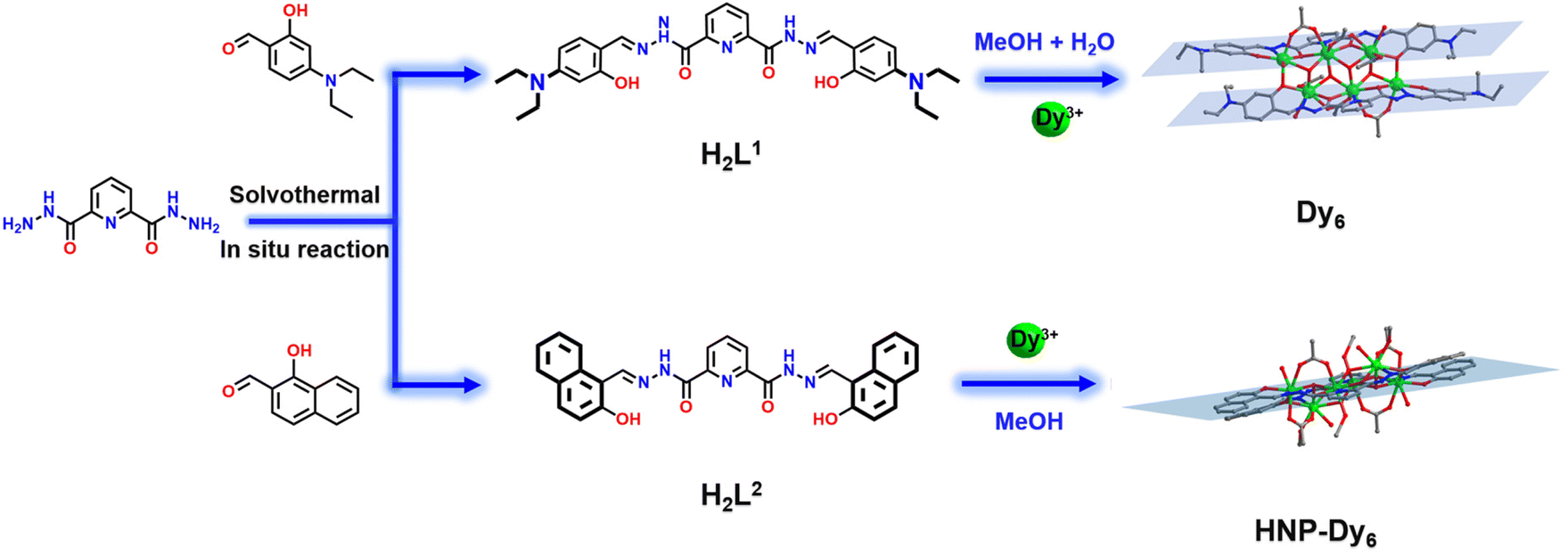 | ||
| Scheme 1 Schematic of the Dy6 and HNP-Dy6 synthesis. | ||
Results and discussion
Synthesis and structural analysis of Ln6 clusters (Ln = Dy and Gd)
Approximately 0.4 mmol of Dy(OAc)3·4H2O and 0.05 mmol of the organic ligand H2L1 (N′2,N′6-bis(4-(diethylamino)-2-hydroxybenzylidene)pyridine-2,6-dicarbohydrazide) were accurately weighed and dissolved in a mixed solvent of MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1.0 mL:0.3 mL) and added with 100 μL of triethylamine. After an even mixing, the reaction was derived under solvothermal conditions at 80 °C for 48 h to obtain an orange blocky crystal of Dy6 (Fig. 1). The results of single-crystal X-ray diffraction (SCXRD) showed that Dy6 crystallized in the P
:H2O (1.0 mL:0.3 mL) and added with 100 μL of triethylamine. After an even mixing, the reaction was derived under solvothermal conditions at 80 °C for 48 h to obtain an orange blocky crystal of Dy6 (Fig. 1). The results of single-crystal X-ray diffraction (SCXRD) showed that Dy6 crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group of the triclinic system (Tables S1–S4†). Dy6 comprised six Dy(III) ions, two multidentate chelating ligands (L1)2−, four μ3-O2−, six OAc−, and four terminally coordinated H2O molecules and had the following molecular formula: [Dy6(L1)2(μ3-O)4(OAc)6(H2O)4] (Fig. 1a). The six metal-centered Dy(III) ions of Dy6 were distributed on the equatorial plane and connected by oxygen atoms in the ligands and μ3-O2− bridges to form the {Dy/O} cluster nucleus (Fig. 1b). The multidentate chelating ligand (L1)2− chelated three Dy(III) ions to form the Dy3(L1) template motif. Notably, in the Dy6 structure, two Dy3(L1) were arranged in face-to-face and up-and-down dislocation arrangements (Fig. 1c and d, respectively). The two ligands at the cluster core periphery tightly wrapped the cluster core to resist the attack of foreign solvent molecules, further ensuring the Dy6 stability. Each ligand chelated three Dy(III) ions and had the following coordination modes: μ3–η1:η1:η2:η1:η2:η1:η1 (Fig. 1e). The six metal-centered Dy(III) ions in the Dy6 structure had three coordination environments. The metal-centered Dy1 had an O7N coordination environment provided by the (L1)2− ligand, μ3-O2−, OAc−, and H2O molecule (Fig. 1f). The SHAPE calculations showed that the coordination configuration was a Johnson biaugmented trigonal prism (J50) with a C2v symmetric environment (Table S4†). The metal-centered Dy2 had an O7N coordination environment provided by the (L1)2− ligand, μ3-O2−, and OAc− (Fig. 1f). The SHAPE calculations demonstrated that the coordination configuration was a biaugmented trigonal prism (J50) with a C2v symmetric environment (Table S4†). Meanwhile, the metal-centered Dy3 had an O6N2 coordination environment provided by the (L1)2− ligand, μ3-O2−, OAc−, and H2O molecule (Fig. 1f). The SHAPE calculations for this indicated that the coordination configuration was a Johnson pentagonal bipyramid (J13) with a D5h symmetric environment (Table S4†). According to further weak interaction analysis and stacking diagrams, the strong steric hindrance of –N(Et)2 caused the template units in the Dy6 structure to form face-to-face and dislocation arrangements (Fig. 1g and f). The structural analysis demonstrated that all the Dy–O/N bond lengths were within the normal range (Table S2†). We changed the metal salt to Gd(OAc)3·H2O and obtained the Gd6 homologue of Dy6 under the same conditions.
space group of the triclinic system (Tables S1–S4†). Dy6 comprised six Dy(III) ions, two multidentate chelating ligands (L1)2−, four μ3-O2−, six OAc−, and four terminally coordinated H2O molecules and had the following molecular formula: [Dy6(L1)2(μ3-O)4(OAc)6(H2O)4] (Fig. 1a). The six metal-centered Dy(III) ions of Dy6 were distributed on the equatorial plane and connected by oxygen atoms in the ligands and μ3-O2− bridges to form the {Dy/O} cluster nucleus (Fig. 1b). The multidentate chelating ligand (L1)2− chelated three Dy(III) ions to form the Dy3(L1) template motif. Notably, in the Dy6 structure, two Dy3(L1) were arranged in face-to-face and up-and-down dislocation arrangements (Fig. 1c and d, respectively). The two ligands at the cluster core periphery tightly wrapped the cluster core to resist the attack of foreign solvent molecules, further ensuring the Dy6 stability. Each ligand chelated three Dy(III) ions and had the following coordination modes: μ3–η1:η1:η2:η1:η2:η1:η1 (Fig. 1e). The six metal-centered Dy(III) ions in the Dy6 structure had three coordination environments. The metal-centered Dy1 had an O7N coordination environment provided by the (L1)2− ligand, μ3-O2−, OAc−, and H2O molecule (Fig. 1f). The SHAPE calculations showed that the coordination configuration was a Johnson biaugmented trigonal prism (J50) with a C2v symmetric environment (Table S4†). The metal-centered Dy2 had an O7N coordination environment provided by the (L1)2− ligand, μ3-O2−, and OAc− (Fig. 1f). The SHAPE calculations demonstrated that the coordination configuration was a biaugmented trigonal prism (J50) with a C2v symmetric environment (Table S4†). Meanwhile, the metal-centered Dy3 had an O6N2 coordination environment provided by the (L1)2− ligand, μ3-O2−, OAc−, and H2O molecule (Fig. 1f). The SHAPE calculations for this indicated that the coordination configuration was a Johnson pentagonal bipyramid (J13) with a D5h symmetric environment (Table S4†). According to further weak interaction analysis and stacking diagrams, the strong steric hindrance of –N(Et)2 caused the template units in the Dy6 structure to form face-to-face and dislocation arrangements (Fig. 1g and f). The structural analysis demonstrated that all the Dy–O/N bond lengths were within the normal range (Table S2†). We changed the metal salt to Gd(OAc)3·H2O and obtained the Gd6 homologue of Dy6 under the same conditions.
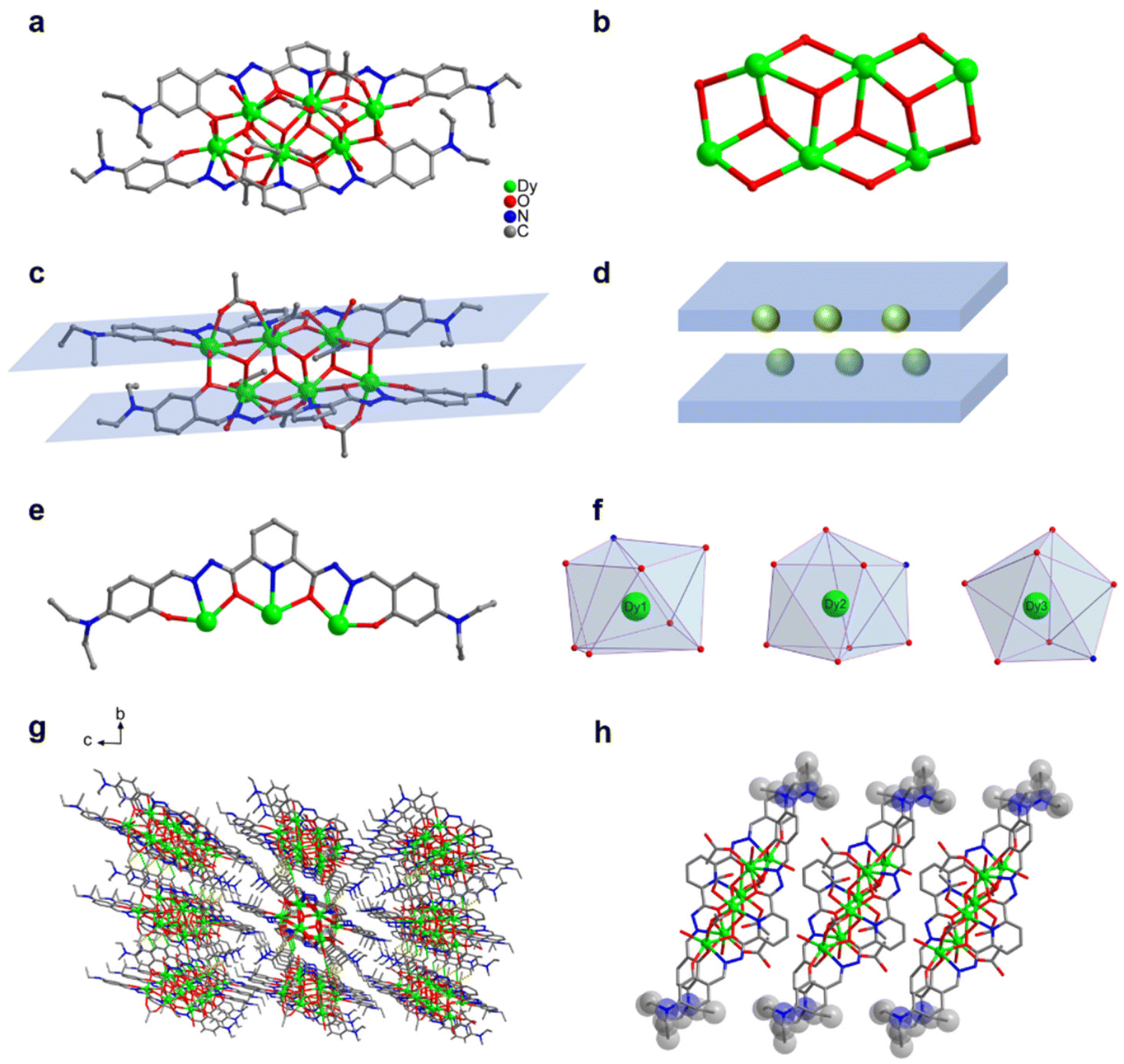 | ||
| Fig. 1 (a) Dy6 structure, (b) cluster core connection, (c) face-to-face and up-and-down dislocation arrangements of the Dy3(L1) template units, (d) schematic of the template-unit arrangement in Dy6, (e) coordination mode of ligand (L1)2−, (f) coordination configurations of the metal-centered ions Dy1–Dy3 (g) three-dimensional Dy6 accumulation, and (h) arrangement among the three independent units of Dy6. | ||
We changed 4-(diethylamino)salicylaldehyde to 2-hydroxy-1-naphthaldehyde to construct a diacylhydrazone-derived ligand, H2L2 (N′2,N′6-bis((2-hydroxynaphthalen-1-yl)methylene)pyridine-2,6-dicarbohydrazide). The yellow blocky crystals of HNP-Dy6 in Scheme 1 were obtained when the abovementioned solvothermal reaction was realized using H2L2 and the solvent was changed to 2.0 mL of anhydrous methanol. The SCXRD results showed that HNP-Dy6 crystallized in the P space group of the triclinic system (Tables S1–S4†). HNP-Dy6 comprised six Dy(III) ions, two multidentate chelating ligands (L2)2−, four μ3-O2−, four OAc−, four CH3O−, and four terminally coordinated H2O molecules with the following molecular formula: [Dy6(L2)2(μ3-O)4(OAc)4(CH3O)4(H2O)4]·3CH3OH (Fig. 2a). Similarly, the six metal-centered Dy(III) ions in the HNP-Dy6 structure were also distributed on the equatorial plane and connected by oxygen atoms in the ligands and μ3-O2− bridges to form the {Dy/O} cluster nucleus (Fig. 2b). The HNP-Dy6 structure comprised a multidentate chelating ligand that chelated three Dy(III) ions to form the Dy3(L2) template unit. Surprisingly, the two Dy3(L2) template units were found in the same plane and arranged in face-to-face and parallel arrangements, which was considerably different from Dy6 (Fig. 2c and d). The coordination mode of the multidentate chelating ligand (L2)2− was μ3–η1:η1:η2:η1:η2:η1:η1 (Fig. 2e). The six metal-centered Dy(III) ions in the HNP-Dy6 structure all had an O7N coordination environment. Their coordination configurations were Johnson biaugmented trigonal prism (J50) (Dy1), biaugmented trigonal prism (Dy2), and triangular dodecahedron (Dy3) (Fig. 1f and Table S5†). An obvious π–π stacking (3.820 Å) was observed in the HNP-Dy6 structure. The strong π–π interaction led to the reverse and planar arrangement of the template units (Fig. 2g and h). The structural analysis demonstrated that all the Dy–O/N bond lengths were within the normal range (Table S3†).
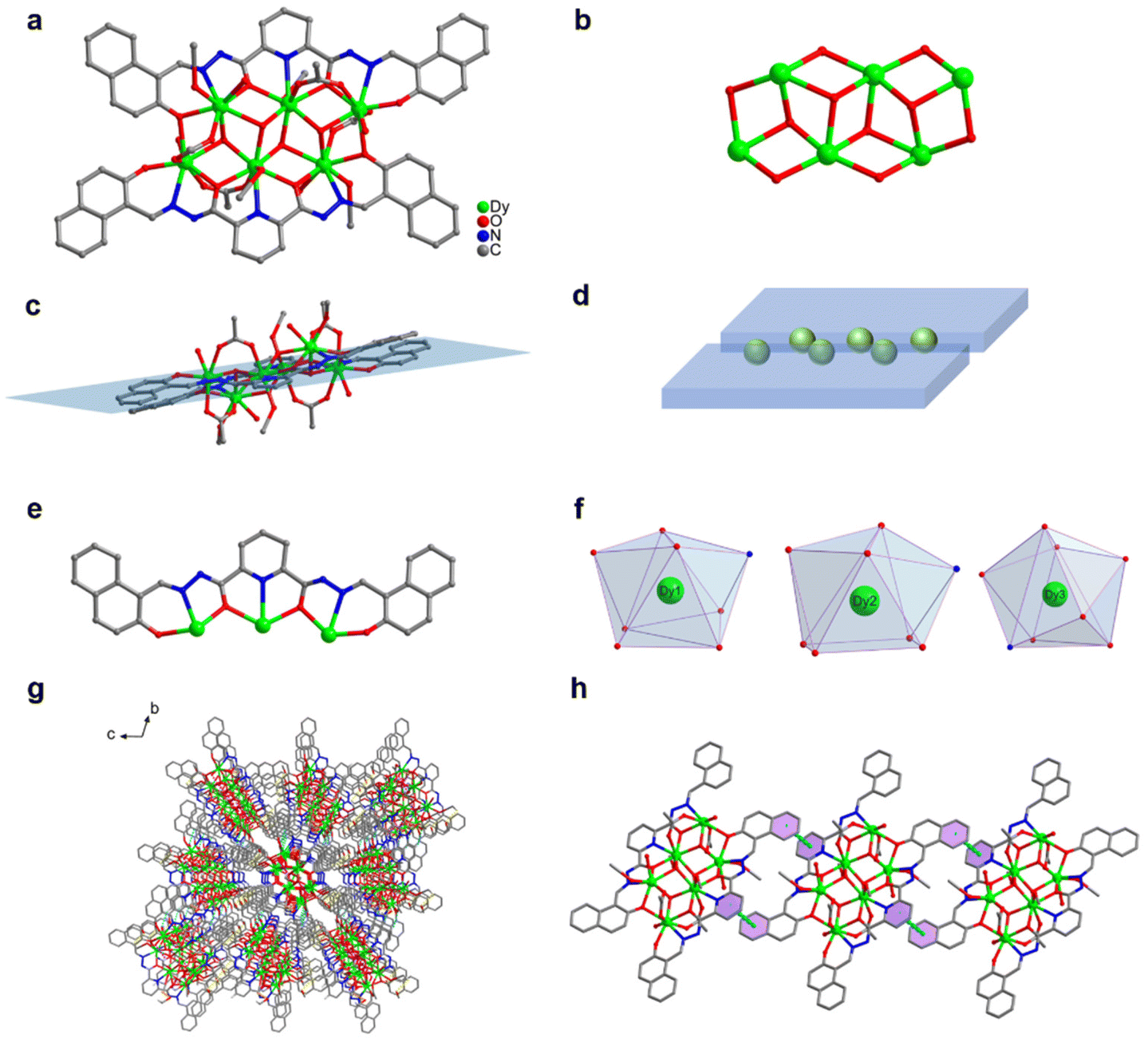 | ||
| Fig. 2 (a) HNP-Dy6 structure, (b) cluster core connection, (c) face-to-face arrangement of the Dy3(L2) template units in the same plane, (d) schematic of the template-unit arrangement in HNP-Dy6, (e) ligand coordination mode, (f) metal-centered coordination configuration, (g) three-dimensional HNP-Dy6 accumulation, and (h) π–π stacking of the three independent units of HNP-Dy6. | ||
The controllable construction of the two distinct hexanuclear dysprosium clusters marked a great progress in the precise lanthanide-cluster synthesis. We initially proposed the multidentate chelating coordination (MCC) method for the rational construction of lanthanide clusters. For the first time, we selected herein a diacylhydrazone-derived ligand with multiple coordination sites and manipulated the MCC method to realize the precise synthesis of planar and dislocation-parallel differential hexanuclear dysprosium clusters. Specifically, when the substituent –N(Et)2 with a large steric hindrance was selected, Dy6 was obtained with template units having face-to-face and upper and lower dislocation arrangements. Further increasing the conjugated ring on the ligand yielded HNP-Dy6 with template units located in the same plane and arranged face-to-face. To the best of our knowledge, most of the reported hexanuclear lanthanide clusters tend to be in the second arrangement. Precisely controlling and obtaining lanthanide clusters with the same cluster nuclei but different arrangements is difficult. Adjusting the multidentate chelating-ligand substituents enabled us to realize the directional assembly of heterogeneous hexanuclear lanthanide clusters with different arrangements. We were also able to reveal the controllability of the structural design of the hypernuclear lanthanide clusters, which is a big step toward a precise synthesis.
The thermal stability analysis of Dy6 and HNP-Dy6 was performed under a flowing N2 atmosphere. The temperature was slowly increased from 35 °C to 1000 °C at a 5 °C min−1 rate (Fig. S2†). As the temperature was gradually increased from 35 °C to 140 °C, we obtained a Dy6 weight loss rate of 5.18% that corresponded to the loss of four terminally coordinated water molecules and one OAc−. The theoretical value for this was 5.13%. The Dy6 weight loss rate was 11.60% when the temperature was increased from 310 °C to 405 °C. This process corresponded to the loss of five OAc−. The theoretical value for this was 11.55%. Finally, Dy6 was rapidly decomposed at a temperature above 480 °C (Fig. S2a†). Similarly, the HNP-Dy6 weight loss rate was 6.53% when the temperature was gradually increased from 35 °C to 115 °C. This corresponded to the loss of four terminally coordinated water molecules and three methanol molecules. The theoretical value for this was 6.78%. The HNP-Dy6 weight loss rate was 11.14% when the temperature was gradually increased from 32 °C to 390 °C. This rate corresponded to the loss of four terminally coordinated CH3O− and three OAc−. The theoretical value for this was 11.70%. Finally, the HNP-Dy6 structure was rapidly decomposed at a temperature of >500 °C (Fig. S2b†). The powder X-ray diffraction experimental and simulated values for Dy6, Gd6, and HNP-Dy6 were compared at 35 °C. The results showed their pure phases (Fig. S3†).
Stabilities of Dy6 and HNP-Dy6
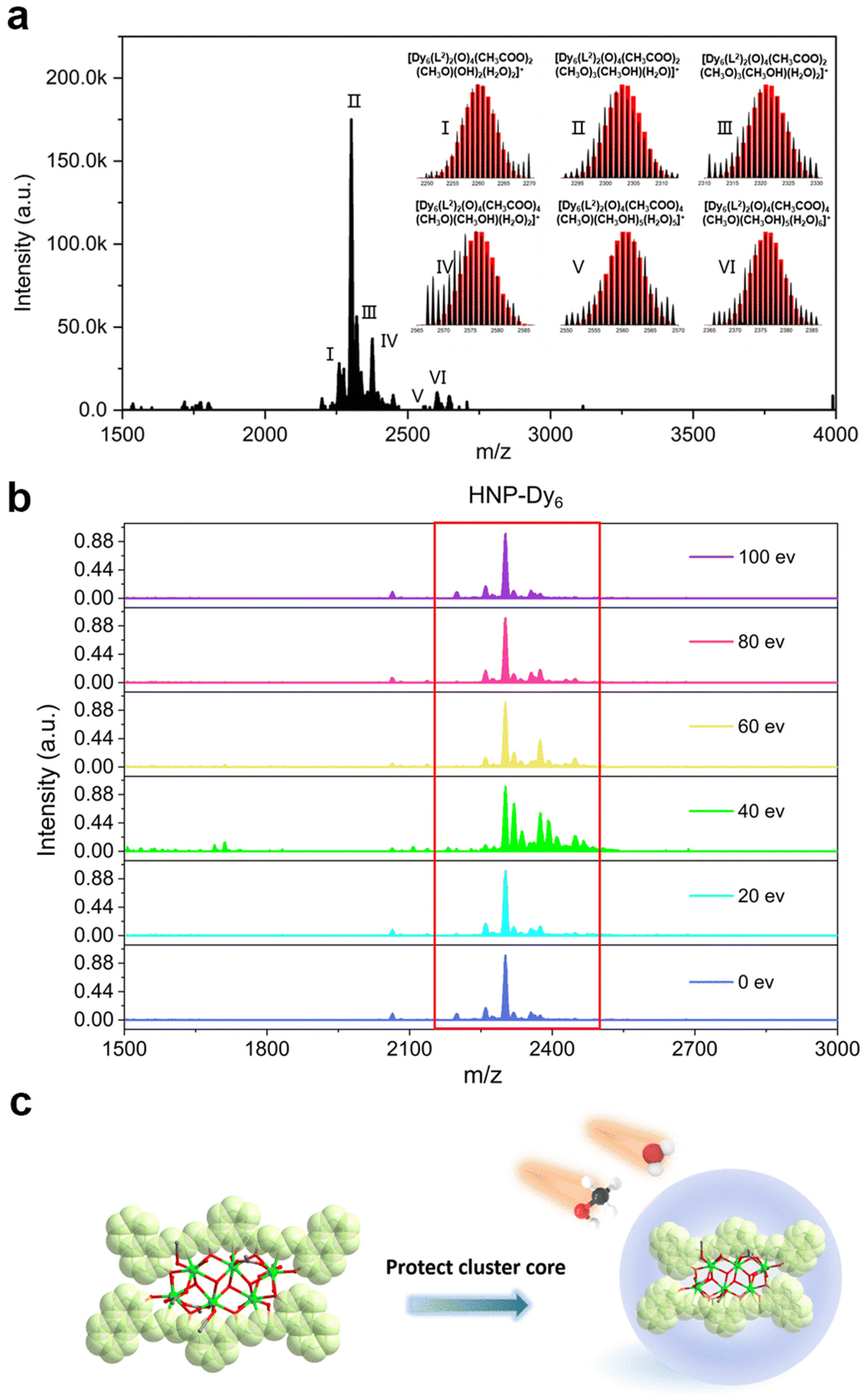
In recent years, HRESI-MS had been extensively used to evaluate the stability, solubility, and ionization degree of the lanthanide clusters in solutions.6,12,29,34 In this work, to further explore the stabilities of Dy6 and HNP-Dy6 in solution, we used HRESI-MS with different ion-source energies to monitor the molecular-ion peak fragments of Dy6 and HNP-Dy6. We specifically dissolved small amounts of pure Dy6 and HNP-Dy6 in chromatographic N,N-dimethylformamide (DMF) and performed HRESI-MS testing after extensive dilution using chromatographic methanol. The yielded data were collected within the m/z = 400–4000 range (Fig. 3a and 4a and Tables S6 and S7†). The results showed that Dy6 can maintain a high stability under the HRESI-MS conditions. The captured molecular-ion peaks of different m/z were related to the structural framework that can be assigned to [Dy6(L1)2(O)4(CH3COO)x(OH)(solv.)] (x ≤ 5; solv. = DMF, CH3OH, and H2O; +1, +2 valence). The abovementioned molecular-ion peak fragments were specifically resolved as [Dy6(L1)2(O)4(OH)4(CH3OH)3(H2O)2]2+ (calc. 1163.59, exp. 1163.54), [Dy6(L1)2(O)4(CH3COO)2(OH)2(CH3OH)2]2+ (calc. 1172.58, exp. 1172.55), [Dy6(L1)2(O)4(CH3COO)2(OH)2(CH3OH)2(H2O)]2+ (calc. 1181.09, exp. 1181.08), [Dy6(L1)2(O)4(OH)4(DMF)2(CH3OH)]2+ (calc. 1188.10, exp. 1188.08), [Dy6(L1)2(O)4(CH3COO)4(OH)(H2O)3]+ (calc. 2435.17, exp. 2435.16), [Dy6(L1)2(O)4(CH3COO)5(DMF)(H2O)]+ (calc. 2513.20, exp. 2513.18), and [Dy6(L1)2(O)4(CH3COO)4(OH)(DMF)(CH3OH)2(H2O)3]+ (calc. 2572.26, exp. 2572.22) (Fig. S4 and Table S6†). Similarly, HNP-Dy6 presented a +1 valent molecular-ion peak fragment related to the structural framework in the m/z = 2000–3000 range, which can be attributed to [Dy6(L2)2(O)4(CH3COO)x(CH3O)y (solv.)] (x ≤ 4; y ≤ 4; solv. = DMF, CH3OH, and H2O). The molecular-ion peak fragments were assigned to [Dy6(L2)2(O)4(CH3COO)2(CH3O)(OH)2(H2O)2]+ (calc. 2261.91, exp. 2261.87), [Dy6(L2)2(O)4(CH3COO)2(CH3O)3(CH3OH)(H2O)]+ (calc. 2302.96, exp. 2302.89), [Dy6(L2)2(O)4(CH3COO)2(CH3O)3(CH3OH)(H2O)2]+ (calc. 2321.97, exp. 2321.89), [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)(H2O)2]+ (calc. 2375.96, exp. 2375.94), [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)5(H2O)5]+ (calc. 2560.09, exp. 2560.02), and [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)5(H2O)6]+ (calc. 2576.10, exp. 2576.04) (Fig. S5 and Table S7†).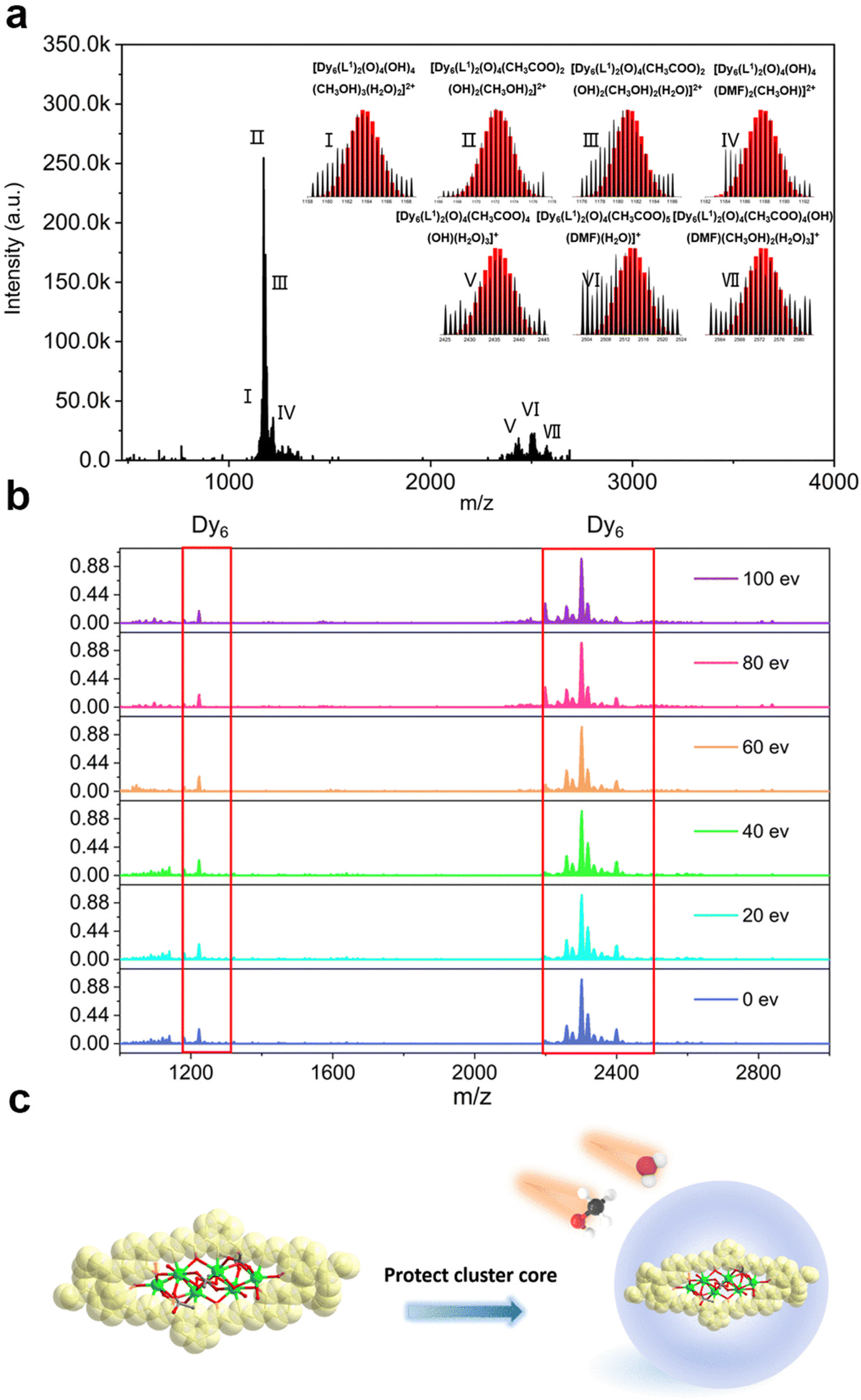 | ||
| Fig. 3 (a) HRESI-MS spectrum of Dy6 dissolved in DMF in the positive-ion mode, (b) HRESI-MS spectrum of Dy6 dissolved in DMF under different ion-source voltages (in-source CID), and (c) schematic of the chelating ligands effectively protecting the cluster core. | ||
 | ||
| Fig. 4 (a) HRESI-MS spectrum of HNP-Dy6 dissolved in DMF in the positive-ion mode, (b) HRESI-MS spectrum of HNP-Dy6 dissolved in DMF under different ion-source voltages (in-source CID), and (c) schematic of the chelating ligands effectively protecting the cluster core. | ||
Only the molecular-ion peaks [Dy6(L1)2(O)4(OH)4(CH3OH)3(H2O)2]2+ (calc. 1163.59, exp. 1163.56) and [Dy6(L1)2(O)4(CH3COO)4(OH)(H2O)3]+ (calc. 2435.16, exp. 2435.17), which were related to the complete structural skeleton, were detected in the Dy6 solution as the ion-source voltage was gradually increased from 0 to 100 eV (Fig. S6 and Table S8†). HNP-Dy6 also only depicted the molecular-ion peak fragment of [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)5(H2O)5]+ (calc. 2560.09, exp. 2560.04), which was consistent with the structural skeleton (Fig. S7 and Table S9†). These results indicate that both Dy6 and HNP-Dy6 exhibited high stabilities in solution (Fig. 3b and 4b). Based on the structural analysis results of Dy6 and HNP-Dy6, their stabilities can be attributed to the two large-volume chelating ligands on the cluster core periphery tightly wrapping around the cluster core, effectively resisting the attack of foreign solvent molecules on the cluster core (Fig. 3c and 4c).
Assembly mechanism of Dy6 and HNP-Dy6
We further explored the self-assembly mechanism of Dy6 and HNP-Dy6 by performing time-dependent HRESI-MS to track the species changes separately during their formation (Fig. 5a and 6a and Tables S10 and S11†). The HRESI-MS data suggested that Dy6 was formed through a step-by-step assembly. Fig. 5b depicts the time-dependent change trend of the species in the solution during the self-assembly process of Dy6. First, the reaction system was stirred at room temperature for 5 min (0 min), during which the ligand H2L immediately reacted with the metal Dy(III) ions to generate multiple [Dy2L1] fragments. The ligand fragment with the highest intensity appeared in the m/z = 877.49 position, which can be assigned to [Dy2(L1)(OH)2(DMF)8(CH3OH)6(H2O)4]2+ (Fig. 5a and S8†). The other molecular-ion peak fragments related to [Dy2L1] appeared at positions m/z = 878.48 and 891.50. By fitting, the molecular formulas of these fragments were found as [Dy2(L1)(OH)2(DMF)8(CH3OH)5(H2O)6]2+ (calc. 878.37, exp. 878.48) and [Dy2(L1)(OH)2(DMF)8(CH3OH)8(H2O)2]2+ (calc. 891.40, exp. 891.50) (Fig. 5a and S8†). The experimental results showed that the H2L1 ligand removed two protons and presented −2 valence under triethylamine regulation, while the (L1)2− ligand removed two protons combined with two metal-centered Dy(III) ions to form the [Dy2L1] fragments. The intensity of the [Dy2L1] fragments gradually decreased with increasing reaction time. These fragments disappeared after heating for 12 h and were transformed into a small amount of [Dy3L1] fragments with the increasing heating time. They began to appear at 5 min, formed in large quantities at 1 h, reached the maximum intensity at 6 h, and weakened after 12 h. Their molecular-ion peak appeared in the m/z = 1242.03–1416.14 range, and the molecular formulas obtained by fitting were [Dy3(L1)(OH)6(DMF)(H2O)2]+ (calc. 1242.13, exp. 1242.03), [Dy3(L1)(OH)6(CH3OH)2(H2O)2]+ (calc. 1265.16, exp. 1265.08), [Dy3(L1)(OH)6(DMF)2(CH3OH)2]+ (calc. 1345.07, exp. 1345.23), and [Dy3(L1)(OH)6(DMF)2(CH3OH)2(H2O)4]+ (calc. 1416.26, exp. 1416.14) (Fig. 5a and S8†). The [Dy6L12] fragments also began to appear at 5 min from the beginning of the reaction, formed in large quantities at 6 h, and showed a gradually increasing intensity. The molecular-ion peaks related to [Dy6L12] were observed in the m/z = 1151.53–2572.22 range. The molecular formulas of these fragments obtained by fitting were [Dy6(L1)2(O)4(CH3COO)(OH)3(CH3OH)2]2+ (calc. 1151.58, exp. 1151.53), [Dy6(L1)2(O)4(OH)4(CH3OH)3(H2O)2]2+ (calc. 1163.59, exp. 1163.54), [Dy6(L1)2(O)4(CH3COO)2(OH)2(CH3OH)2]2+ (calc. 1172.58, exp. 1172.55), [Dy6(L1)2(O)4(CH3COO)2(OH)2(CH3OH)2(H2O)]2+ (calc. 1181.09, exp. 1181.08), [Dy6(L1)2(O)4(OH)4(DMF)2(CH3OH)]2+ (calc. 1188.10, exp. 1188.08), [Dy6(L1)2(O)4(CH3COO)5(DMF)]+ (calc. 2496.21, exp. 2496.20), [Dy6(L1)2(O)4(CH3COO)5(DMF)(H2O)]+ (calc. 2513.20, exp. 2513.18), and [Dy6(L1)2(O)4(CH3COO)4(OH)(DMF)(CH3OH)2(H2O)3]+ (calc. 2572.26, exp. 2572.22) (Fig. 5a and S8). Surprisingly, the [Dy6L12] fragments also began appearing at the same time as the [Dy3L1] fragments were detected at 5 min from the start of the reaction. The results indicate that the trinuclear fragment [Dy3L1] was quickly used as a template motif after its formation to rapidly react to form the [Dy6L12] fragment. Therefore, the formation process of Dy6 underwent a stepwise assembly and had the possible assembly mechanism of L1 + 2Dy → Dy2L1 → Dy3L1 → 2Dy3L1 → Dy6L12 (Fig. 5c).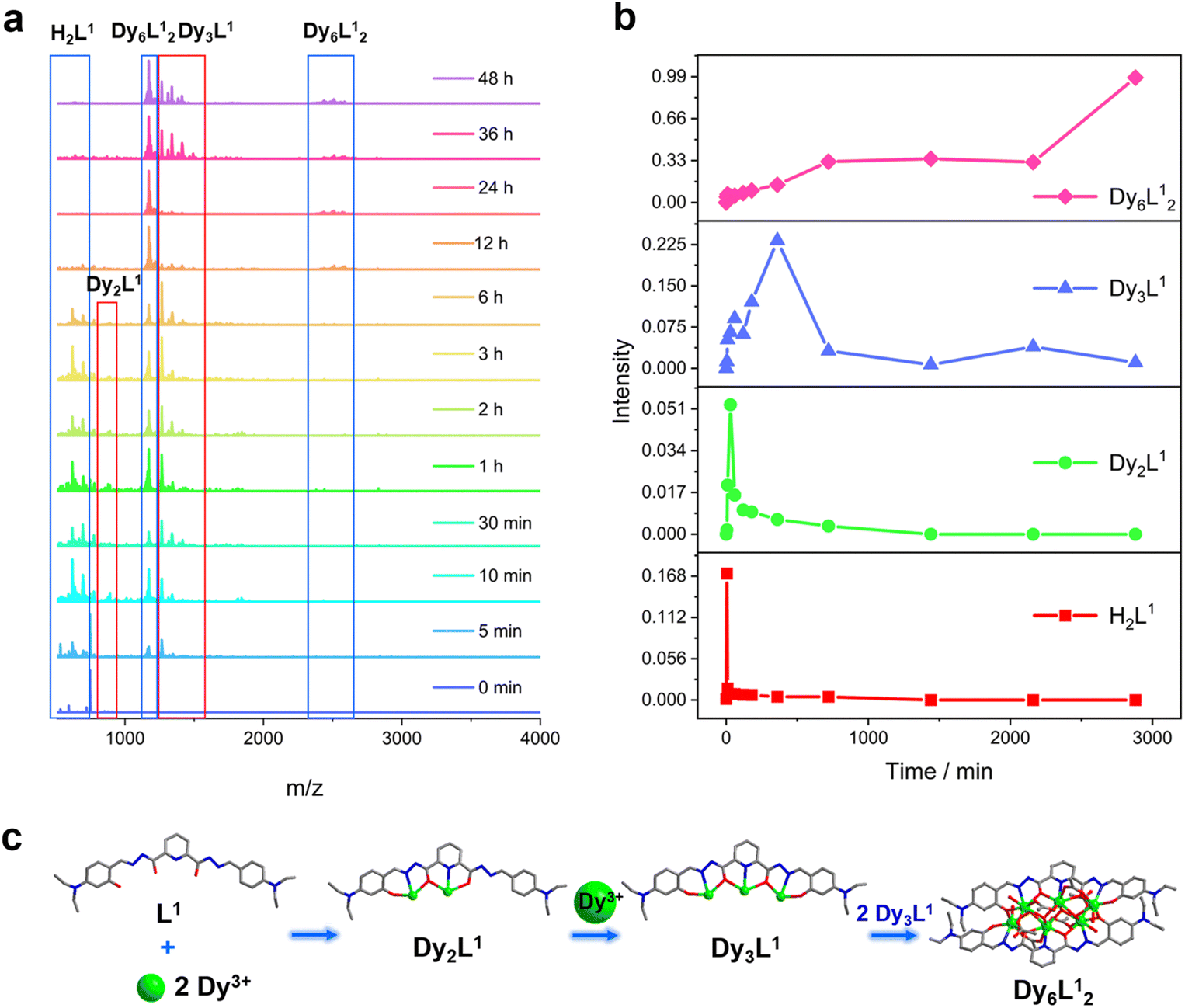 | ||
| Fig. 5 (a) Time-dependent HRESI-MS tracking of the Dy6 formation process, (b) intensity–time curves of the molecular-ion peaks of different species during the reaction, and (c) schematic of the possible formation process and the self-assembly mechanism of Dy6. | ||
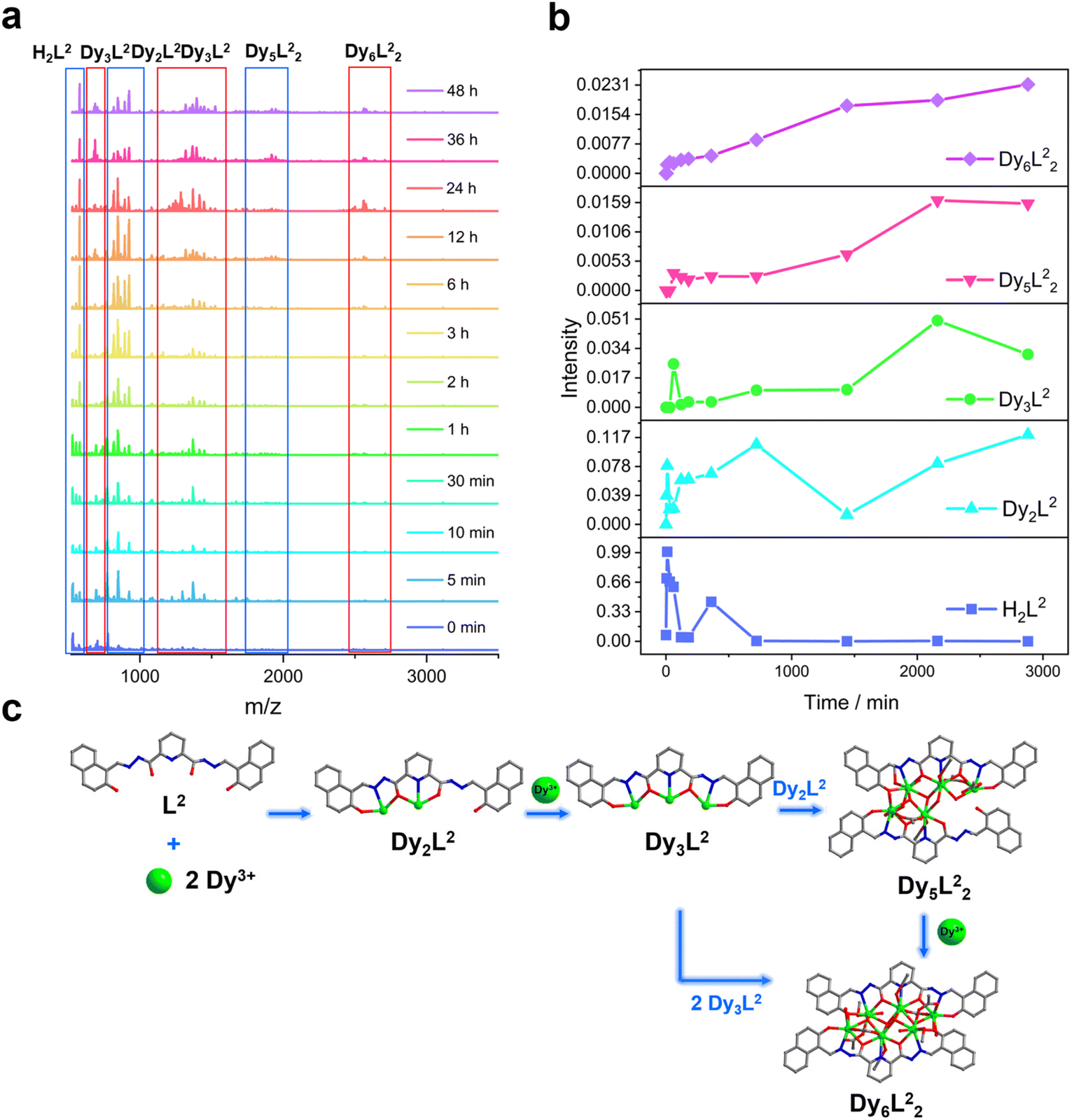 | ||
| Fig. 6 (a) Time-dependent HRESI-MS tracking of the HNP-Dy6 formation process, (b) HRESI-MS spectra intensity–time profiles of the species, and (c) schematic of the possible assembly mechanism of HNP-Dy6. | ||
Similarly, we tracked the HNP-Dy6 formation using time-dependent HRESI-MS and proposed its possible self-assembly mechanism. First, the reaction system was stirred at room temperature for 5 min (the label in the figure is 0 min), during which the ligand H2L immediately reacted with the metal-centered Dy(III) ions to generate multiple [Dy2L2] fragments. The ligand fragment with the highest intensity appeared in the m/z = 925.03 position that can be assigned to [Dy2(L2)(CH3O)2(OH)(H2O)]+ (Fig. 6a and S9†). The other molecular-ion peak fragments related to [Dy2L2] appeared at positions 1008.93 and 1070.96. The molecular formulas of these fragments obtained by fitting were [Dy2(L2)(CH3O)2(OH)(CH3OH)(H2O)4]+ (calc. 1009.10, exp. 1008.93) and [Dy2(L2)(OH)3(DMF)2(CH3OH)(H2O)]+ (calc. 1071.14, exp. 1070.96). We also observed that a small amount of [Dy2L2] fragments chelated with the metal-centered Dy(III) ions to form the [Dy3L2] fragments, which began appearing at 5 min. Their molecular-ion peaks appeared at the m/z = 1342.08–1524.10 position. The molecular formulas of these fragments obtained by fitting were [Dy3(L2)(OH)6(DMF)3(CH3OH)]+ (calc. 1342.13, exp. 1342.08), [Dy3(L2)(OH)6(DMF)(CH3OH)3(H2O)6]+ (calc. 1370.16, exp. 1370.07), [Dy3(L2)(OH)6(DMF)(CH3OH)4(H2O)2]+ (calc. 1448.22, exp. 1448.08), and [Dy3(L2)(OH)6(DMF)2(CH3OH)5(H2O)7]+ (calc. 1524.26, exp. 1524.10) (Fig. 6a and S9†). With the continuous increase of the heating time, the [Dy5L22] fragments started to appear at 1 h and formed by chelating a [Dy2L2] fragment with a [Dy3L2] fragment. The molecular-ion peak of the [Dy5L22] fragment was observed at the m/z = 1919.98–1967.96 position. The molecular formulas of these fragments acquired by fitting were [Dy5(L2)2(O)4(OH)2]+ (calc. 1919.95, exp. 1919.98), [Dy5(L2)2(O)4(OH)2(CH3OH)]+ (calc. 1946.94, exp. 1946.96), and [Dy5(L2)2(O)4(OH)2(CH3OH)(H2O)]+ (calc. 1967.98, exp. 1967.96). The [Dy3L2] and [Dy5L22] fragments gradually disappeared with up 48 h of heating time. The [Dy6L22] fragments also began to appear at 5 min from the beginning of the reaction, formed in large quantities at 6 h, and showed a gradually increasing intensity. The molecular-ion peaks related to the [Dy6L22] fragments appeared in the m/z = 2261.87–2576.04 range. The molecular formulas of these fragments obtained by fitting were [Dy6(L2)2(O)4(CH3COO)2(CH3O)(OH)2(H2O)2]+ (calc. 2261.91, exp. 2261.87), [Dy6(L2)2(O)4(CH3COO)2(CH3O)3(CH3OH)(H2O)]+ (calc. 2302.96, exp. 2302.87), [Dy6(L2)2(O)4(CH3COO)2(CH3O)3(CH3OH)(H2O)2]+ (calc. 2321.97, exp. 2321.89), [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)(H2O)2]+ (calc. 2375.96, exp. 2375.94), [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)5(H2O)5]+ (calc. 2560.09, exp. 2560.04), and [Dy6(L2)2(O)4(CH3COO)4(CH3O)(CH3OH)5(H2O)6]+ (calc. 2576.10, exp. 2576.04) (Fig. 6a and S9†). Therefore, the formation process of HNP-Dy6 was a combination of the stepwise and template assemblies with two possible self-assembly mechanisms: L2 + 2Dy → Dy2L2 → Dy3L2 → Dy5L22 → Dy6L22 or L2 + 2Dy → Dy2L2 → Dy3L2 → 2Dy3L2 → Dy6L22 (Fig. 6c).
MRI performance of Gd6
As one of the most mature medical imaging techniques available, MRI has been extensively used in clinical medical diagnosis in the recent years. However, CAs are usually required to improve the imaging contrast and clarity of MRI.12,13,35–38 Therefore, CAs with high resolution and clear imaging effects must be designed and developed. Gadolinium-based chelates are currently extensively used as a T1-weighted MRI CA for clinical diagnosis, and they usually require higher doses to achieve excellent imaging results.39–41 To explore the feasibility of Gd6 as a T1-weighted MRI CA, we tested the relaxation time with the Gd(III) concentration changes at 1-T magnetic field strength and obtained the longitudinal and transverse relaxation rates (r1 and r2, respectively). The results showed the Gd6r1 and r2 values of 12.06 and 24.10 mM−1 s−1, respectively, at 1-T magnetic field strength (Fig. 7a). Gd6 with a highly aggregated Gd(III) exhibited higher relaxation rates compared to the conventional Gd chelates. r2/r1 = 1.99 (r2/r1 < 2) indicated that Gd6 is a potential T1-weighted MRI CA candidate.12,42,43 Under the same magnetic field, the r1 and r2 of gadopentetate dimeglumine injection (Gd-DTPA), a currently used clinical MRI contrast agent, are 4.55 and 5.77 mM−1 s−1, respectively.12 In comparison, Gd6 shows higher relaxation values. With the gradual increase of the Gd(III) concentration of Gd6 in the aqueous solution, the T1-weighted image gradually became brighter under 1-T magnetic field. By contrast, the T2-weighted image gradually turned darker from bright, indicating that Gd6 exhibited a good T1 imaging effect in the solution. The T1 mapping images displayed by different Gd6 concentrations at 1-T magnetic field also depicted its great potential as an MRI CA in biomedical diagnosis (Fig. 7b). In addition, we dissolved Gd6 crystals in DMSO and dispersed them in H2O and phosphate buffered saline (PBS), respectively, and recorded the Gd6 dispersed in the above solutions in 1–5 days by UV-visible absorption spectroscopy, which was the absorbance (OD) change at 422 nm. We found that there was no significant change in the absorbance of Gd6 at 422 nm from the 1–5 days. The results show that Gd6 has excellent stability (Fig. S10†).5,12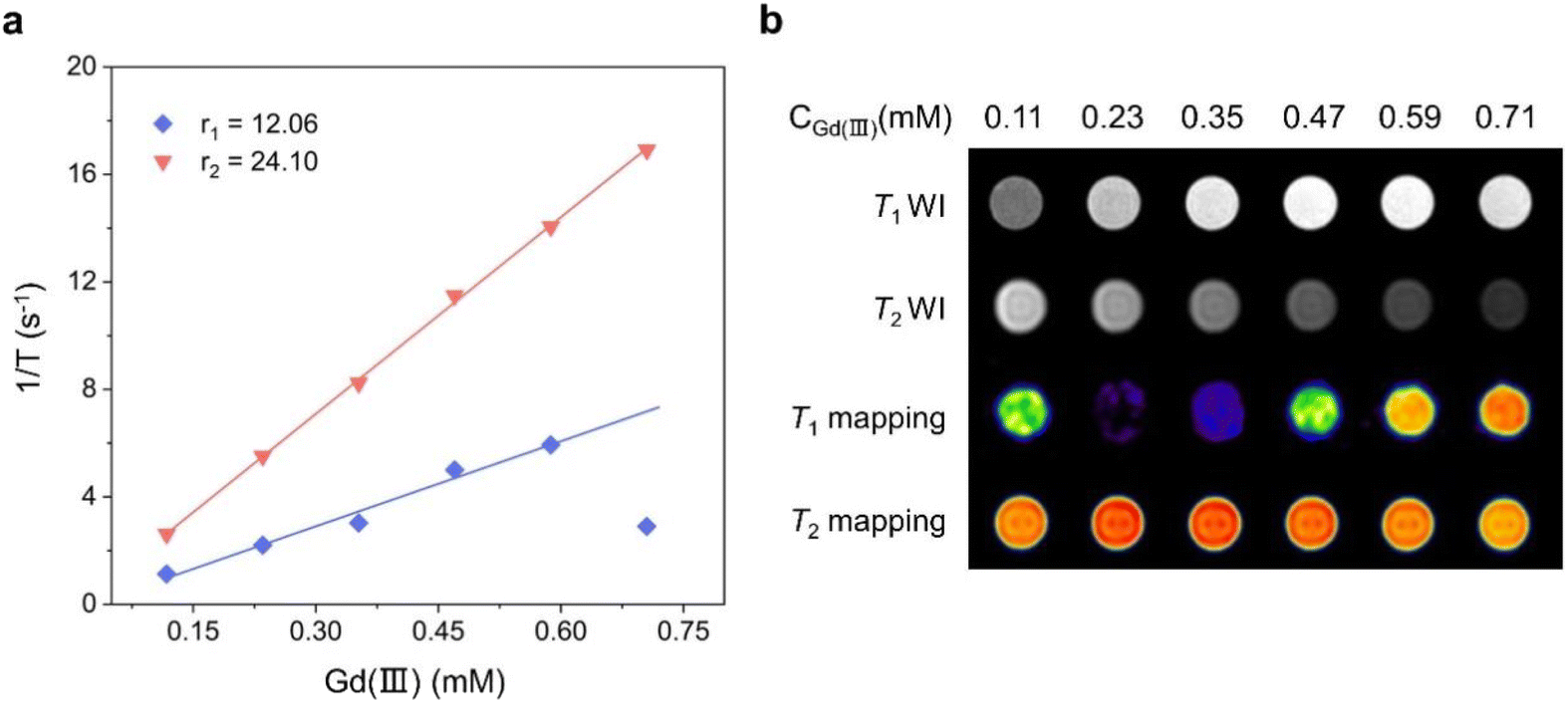 | ||
| Fig. 7 (a) Comparison of the r1 and r2 of Gd6 under 1-T magnetic field and (b) comparison of the solution imaging of different Gd6 concentrations under 1-T magnetic field. | ||
Magnetic analysis of Ln6
Lanthanide complexes have been extensively explored as candidate materials for single-molecule magnets (SMMs) and magnetic refrigerants. Especially the dysprosium complexes constructed by Dy(III) ions are mainly attributed to their significant magnetic anisotropy.34 Therefore, we investigated the magnetic behavior of Dy6 and HNP-Dy6. The temperature-varying molar susceptibilities of the Dy6 and HNP-Dy6 pure phases were tested under an external DC magnetic field of 1000 Oe in the 2–300 K temperature range (Fig. S11a and 11c†).44–49 The χmT values of Dy6 and HNP-Dy6 at 300 K were 83.65 and 82.13 cm3 K mol−1, respectively (6H15/2, S = 5/2, g = 4/3, J = 15/2, and L = 5). The χmT values of both Dy6 and HNP-Dy6 were slightly lower than the theoretical value (i.e., 85.02 cm3 K mol−1) of the six Dy(III) ions. As the temperature decreased, the χmT value of Dy6 continued to decrease, and when the temperature reached 2 K, the χmT of Dy6 droped rapidly to 68.15 cm3 K mol−1. However, when the temperature decreased from 300 K to 2 K, the χmT value of HNP-Dy6 first increased slowly and then decreased, and its χmT value reached 82.13 cm3 K mol−1 at 100 K, while its χmT decreased rapidly to 63.18 cm3 K mol−1 when the temperature reached 2 K.49,50 These findings showed that the temperature-decreasing χmT value may be caused by a decrease in the Stark sublevel of the Dy(III) excited state and the induced crystal field effect. Subsequently, we tested the field-dependent magnetization of Dy6 and HNP-Dy6 at different temperatures (2–5 K) and applied fields of 0–7 T. The M vs. H/T curves were plotted in Fig. S11b and 11d.† Their measured magnetizations at 2 K rapidly increased in the low-field region, but gradually became flat in the high-field region. Dy6 reached a maximum value of 75.98Nβ, while HNP-Dy6 reached 36.90Nβ at a 13 kOe static field. The M vs. H/T curves of Dy6 and HNP-Dy6 at different temperatures did not significantly overlap. The test results indicated that the Dy(III) ions may have low excited states and/or a large magnetic anisotropy. The hysteresis loops of Dy6 and HNP-Dy6 in Fig. S12† were not obvious at 2 K, which may be due to the existence of the crystal field and strong quantum tunneling effects. We further tested the alternating-current susceptibility of Dy6 and HNP-Dy6 to explore their dynamic magnetic behavior. We found that neither of the Dy6 nor HNP-Dy6 out-of-phase (χ′′) signals showed an obvious frequency-dependent behavior under the zero DC field (Fig. S13†). Generally speaking, multinuclear gadolinium clusters have better MCE properties and can be used as an excellent magnetocaloric material.51,52 We tested the magnetocaloric effect of Gd6 at 2–8 K and 0–7 T. As shown in Fig. S14,† when T = 2 K and ΔH = 7 T, the maximum experimental entropy change value of Gd6 is 25.76 J kg−1 K−1, which is smaller than the theoretical −ΔSm value (40.06 J kg−1 K−1), calculated based on the formula: −ΔSm = nRln(2S + 1) (R = 8.3145 J mol−1 K−1, S = 7/2). This can be attributed to the antiferromagnetic interaction between Gd(III) ions.17 In addition, its −ΔSm value is higher than most of the reported hexanuclear gadolinium clusters, showing better MCE performance.26,51 Therefore, magnetic studies show that Gd6 is a potential magnetic refrigeration material.
Conclusions
In conclusion, diacylhydrazone-derived ligands are used to enable controllable construction of differentiated hexanuclear lanthanide clusters with identical cores but different template-motif arrangements. Different substituents are used to regulate the template-motif arrangements formed by multidentate chelating ligands, taking a big step toward the controllable and precise synthesis of lanthanide clusters. HRESI-MS results prove that the two hexanuclear lanthanide clusters obtained herein exhibit high stabilities in solutions. Time-dependent HRESI-MS is employed to track the formation of these hexanuclear lanthanide clusters and propose their possible formation mechanisms. The findings reveal a novel strategy for the rational construction of lanthanide clusters with specific shapes and template-motif arrangements. Notably, the aggregation of Gd(III) ions leads to considerably enhanced relaxation rates of the Gd6 clusters and an excellent MRI resolution. Magnetic studies show that Gd6 exhibits remarkable MCE performance, which can be used as a potential magnetic refrigeration material. This work presents a new idea for the controllable synthesis of complex clusters and promotes progress in the crystal engineering of clusters with precise structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22061005, 22271068, and 22075058), and the Basic Ability Improvement Project of Young and Middle-Aged Teachers in Guangxi Colleges (2022KY0573), PhD Start-up Fund of Yulin Normal University (G2022ZK08).References
- Z. Zhu, M. Guo, X. L. Li and J. Tang, Molecular magnetism of lanthanide: Advances and perspectives, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef.
- X. Z. Li, C. B. Tian and Q. F. Sun, Coordination-Directed Self-Assembly of Functional Polynuclear Lanthanide Supramolecular Architectures, Chem. Rev., 2022, 122, 6374–6458 CrossRef CAS PubMed.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Lanthanide-directed synthesis of luminescent self-assembly supramolecular structures and mechanically bonded systems from acyclic coordinating organic ligands Dawn, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC.
- J. C. G. Bünzli, Lanthanide Luminescence for Biomedical Analyses and Imaging, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- H. L. Wang, Z. Ni, Z. Y. Ruan, Z. H. Zhu, P. Y. Liao, G. Feng, J. H. Jia and M. L. Tong, Spherical lanthanide nanoclusters toward white-light emission and cell membrane imaging, Nano Res., 2023, 16, 11495–11502 CrossRef CAS.
- M. Liu, H. Li, L. Bai, K. Zheng, Z. Zhao, Z. Chen, S. W. Ng, L. Ding and C. Zeng, Real-time and visual sensing devices based on pH-control assembled lanthanide-barium nano-cluster, J. Hazard. Mater., 2021, 413, 125291 CrossRef CAS PubMed.
- Y. Jiang, K. Zheng, Z. Liu, X. Yu, Q. Yang, T. Tang and C. Zeng, Synthesis of Two New Dinuclear Lanthanide Clusters and Visual Bifunctional Sensing Devices Based on the Eu Cluster, Adv. Opt. Mater., 2022, 10, 2102267 CrossRef CAS.
- D. Shao, W. J. Tang, Z. Ruan, X. Yang, L. Shi, X. Q. Wei, Z. Tian, K. Kumari and S. K. Singh, Water-driven reversible switching of single-ion magnetism and proton conduction in a dysprosium sulfonate, Inorg. Chem. Front., 2022, 9, 6147–6157 RSC.
- D. Shao, P. P. Sahu, W. J. Tang, Y. L. Zhang, Y. Zhou, F. X. Xu, X. Q. Wei, Z. Tian, S. K. Singh and X. Y. Wang, A single-ion magnet building block strategy toward Dy2 single-molecule magnets with enhanced magnetic performance, Dalton Trans., 2022, 51, 18610–18621 RSC.
- N. Qiao, X. Y. Xin, X. F. Guan, C. X. Zhang and W. M. Wang, Self-Assembly Bifunctional Tetranuclear Ln2Ni2 Clusters: Magnetic Behaviors and Highly Efficient Conversion of CO2 under Mild Conditions, Inorg. Chem., 2022, 61, 15098–15107 CrossRef CAS PubMed.
- X. L. Li, L. Zhao, J. Wu, W. Shi, N. Struch, A. Lützen, A. K. Powell, P. Cheng and J. Tang, Subcomponent self-assembly of circular helical Dy6(L)6 and bipyramid Dy12(L)8 architectures directed via second-order template effects, Chem. Sci., 2022, 13, 10048–10056 RSC.
- H. L. Wang, D. L. Liu, J. H. Jia, J. L. Liu, Z. Y. Ruan, W. Deng, S. P. Yang, S. G. Wu and M. L. Tong, High-Stability Spherical Lanthanide Nanoclusters for Magnetic Resonance Imaging, Natl. Sci. Rev., 2023, 10, nwad036 CrossRef PubMed.
- Z. Wang, L. He, B. Liu, L. P. Zhou, L. X. Cai, S. J. Hu, X. Z. Li, Z. Li, T. Chen, X. Li and Q. F. Sun, Coordination-Assembled Water-Soluble Anionic Lanthanide Organic Polyhedra for Luminescent Labeling and Magnetic Resonance Imaging, J. Am. Chem. Soc., 2020, 142, 16409–16419 CrossRef CAS PubMed.
- Y. L. Li, H. L. Wang, Z. H. Zhu, F. P. Liang and H. H. Zou, Recent advances in the structural design and regulation of lanthanide clusters: Formation and self-assembly mechanisms, Coord. Chem. Rev., 2023, 493, 215322 CrossRef CAS.
- X. Y. Zheng, J. Xie, X. J. Kong, L. S. Long and L. S. Zheng, Recent advances in the assembly of high-nuclearity lanthanide clusters, Coord. Chem. Rev., 2019, 378, 222–236 CrossRef CAS.
- X. Y. Zheng, Y. H. Jiang, G. L. Zhuang, D. P. Liu, H. G. Liao, X. J. Kong, L. S. Long and L. S. Zheng, A Gigantic Molecular Wheel of {Gd140}: A New Member of the Molecular Wheel Family, J. Am. Chem. Soc., 2017, 139, 18178–18181 CrossRef CAS PubMed.
- X. M. Luo, Z. B. Hu, Q. F. Lin, W. Cheng, J. P. Cao, C. H. Cui, H. Mei, Y. Song and Y. Xu, Exploring the Performance Improvement of Magnetocaloric Effect Based Gd-Exclusive Cluster Gd60, J. Am. Chem. Soc., 2018, 140, 11219–11222 CrossRef CAS PubMed.
- X. Y. Li, H. F. Su, Q. W. Li, R. Feng, H. Y. Bai, H. Y. Chen, J. Xu and X. H. Bu, A Giant Dy76 Cluster: A Fused Bi-Nanopillar Structural Model for Lanthanide Clusters, Angew. Chem., Int. Ed., 2019, 58, 10184–10188 CrossRef CAS PubMed.
- L. Qin, Y. Z. Yu, P. Q. Liao, W. Xue, Z. Zheng, X. M. Chen and Y. Z. Zheng, A “Molecular Water Pipe”: A Giant Tubular Cluster {Dy72} Exhibits Fast Proton Transport and Slow Magnetic Relaxation, Adv. Mater., 2016, 28, 10772–10779 CrossRef CAS PubMed.
- W. Huang, W. Chen, Q. Bai, Z. Zhang, M. Feng and Z. Zheng, Anion-Guided Stepwise Assembly of High-Nuclearity Lanthanide Hydroxide Clusters, Angew. Chem., Int. Ed., 2022, 61, e2022053 Search PubMed.
- Z. H. Zhu, X. F. Ma, H. L. Wang, H. H. Zou, K. Q. Mo, Y. Q. Zhang, Q. Z. Yang, B. Li and F. P. Liang, A triangular Dy3 single-molecule toroic with high inversion energy barrier: magnetic properties and multiple-step assembly mechanism, Inorg. Chem. Front., 2018, 5, 3155–3162 RSC.
- H. L. Wang, X. F. Ma, J. M. Peng, Z. H. Zhu, B. Li, H. H. Zou and F. P. Liang, Tracking the Stepwise Formation of the Dysprosium Cluster (Dy10) with Multiple Relaxation Behavior, Inorg. Chem., 2019, 58, 9169–9174 CrossRef CAS PubMed.
- Z. H. Zhu, J. M. Peng, H. L. Wang, H. H. Zou and F. P. Liang, Assembly Mechanism and Heavy Metal Ion Sensing of Cage-Shaped Lanthanide Nanoclusters, Cell Rep. Phys. Sci., 2020, 1, 100165 CrossRef CAS.
- H. L. Wang, T. Liu, Z. H. Zhu, J. M. Peng, H. H. Zou and F. P. Liang, A series of dysprosium clusters assembled by a substitution effect-driven out-to-in growth mechanism, Inorg. Chem. Front., 2021, 8, 2136–2143 RSC.
- Y. L. Li, H. L. Wang, Z. H. Zhu, J. Li, H. H. Zou, J. M. Peng and F. P. Liang, Truncation reaction regulates the out-to-in growth mechanism to decrypt the formation of brucite-like dysprosium clusters, Dalton Trans., 2022, 51, 197–202 RSC.
- Y. L. Li, H. L. Wang, Z. H. Zhu, J. Li, H. H. Zou and F. P. Liang, A Series of High-Nuclear Gadolinium Cluster Aggregates with a Magnetocaloric Effect Constructed through Two-Component Manipulation, Inorg. Chem., 2021, 60, 16794–16802 CrossRef CAS PubMed.
- B. F. Long, S. Yu, Z. H. Zhu, Y. L. Li, F. F. Liang and H. H. Zou, Coordination site manipulation of the annular growth mechanism to assemble chiral lanthanide clusters with different shapes and magnetic properties, Inorg. Chem. Front., 2022, 9, 5950–5959 RSC.
- B. F. Long, Y. L. Li, Z. H. Zhu, H. L. Wang, F. P. Liang and H. H. Zou, Assembly of pinwheel/twist-shaped chiral lanthanide clusters with rotor structures by an annular/linear growth mechanism and their magnetic properties, Dalton Trans., 2022, 416, 17040–17049 RSC.
- Z. R. Luo, H. L. Wang, Z. H. Zhu, T. Liu, X. F. Ma, H. F. Wang, H. H. Zou and F. P. Liang, Assembly of Dy60 and Dy30 cage-shaped nanoclusters, Commun. Chem., 2020, 3, 1–9 CrossRef PubMed.
- H. L. Wang, T. Liu, Z. H. Zhu, J. M. Peng, H. H. Zou and F. P. Liang, A series of dysprosium clusters assembled by a substitution effect-driven out-to-in growth mechanism, Inorg. Chem. Front., 2021, 8, 2136–2143 RSC.
- J. M. Peng, H. L. Wang, Z. H. Zhu, J. Bai, F. P. Liang and H. H. Zou, Series of the Largest Dish-Shaped Dysprosium Nanoclusters Formed by In Situ Reactions, Inorg. Chem., 2022, 61, 6094–6100 CrossRef CAS PubMed.
- H. L. Wang, Y. L. Li, Z. H. Zhu, X. L. Lu, F. P. Liang and H. H. Zou, Anion-Manipulated Hydrolysis Process Assembles of Giant High-Nucleation Lanthanide-Oxo Cluster, Inorg. Chem., 2022, 61, 20169–20176 CrossRef CAS PubMed.
- H. L. Wang, T. Liu, Z. H. Zhu, J. M. Peng, H. H. Zou and F. P. Liang, pH manipulates the assembly of a series of dysprosium clusters with subtle differences, Inorg. Chem. Front., 2021, 8, 3134–3140 RSC.
- Y. L. Li, W. W. Qin, L. H. L. Wang, Z. Z. Zhu, F. P. Liang and H. H. Zou, Highly Stable Drone-shaped Lanthanide Clusters: Structure, Assembly Mechanism, and Crystalline-Amorphous Transitions, Inorg. Chem. Front., 2023, 10, 5337–5346 RSC.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- D. Ni, W. Bu, E. B. Ehlerding, W. Cai and J. Shi, Engineering of inorganic nanoparticles as magnetic resonance imaging contrast agents, Chem. Soc. Rev., 2017, 46, 7438–7468 RSC.
- E. Debroye and T. N. Parac-Vogt, Towards polymetallic lanthanide complexes as dual contrast agents for magnetic resonance and optical imaging, Chem. Soc. Rev., 2014, 43, 8178–8192 RSC.
- H. Li and T. J. Meade, Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances, J. Am. Chem. Soc., 2019, 141, 17025–17041 CrossRef CAS PubMed.
- P. Verwilst, S. Park, B. Yoon and J. S. Kim, Recent advances in Gd-chelate based bimodal optical/MRI contrast agents, Chem. Soc. Rev., 2015, 44, 1791–1806 RSC.
- C. Sun, H. Lin, X. Gong, Z. Yang, Y. Mo, X. Chen and J. Gao, DOTA-Branched Organic Frameworks as Giant and Potent Metal Chelators, J. Am. Chem. Soc., 2020, 142, 198–206 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- C. He, X. Wu, J. Kong, T. Liu, X. Zhang and C. Duan, A hexanuclear gadolinium–organic octahedron as a sensitive MRI contrast agent for selectively imaging glucosamine in aqueous media, Chem. Commun., 2012, 48, 9290–9292 RSC.
- R. Wang, L. An, J. He, M. Li, J. Jiao and S. Yang, A class of water-soluble Fe(III) coordination complexes as T1-weighted MRI contrast agents, J. Mater. Chem. B, 2021, 9, 1787–1791 RSC.
- J. Liu, Y. C. Chen, J. L. Liu, V. Vieru, L. Ungur, J. H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X. M. Chen and M. L. Tong, A Stable Pentagonal Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization Reversal Barrier over 1000 K, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Z. Zhu, C. Zhao, T. Feng, X. Liu, X. Ying, X. L. Li, Y. Q. Zhang and J. Tang, Air-Stable Chiral Single-Molecule Magnets with Record Anisotropy Barrier Exceeding 1800 K, J. Am. Chem. Soc., 2021, 143, 10077–10082 CrossRef CAS PubMed.
- P. Zhang, L. Zhang, C. Wang, S. Xue, S. Y. Lin and J. Tang, Equatorially Coordinated Lanthanide Single Ion Magnets, J. Am. Chem. Soc., 2014, 136, 4484–4487 CrossRef CAS PubMed.
- B. Zhang, Z. Cheng, Y. Wu, L. Chen, R. Jing, X. Cai, C. Jiang, Y. Q. Zhang, A. Yuan, H. H. Cui and Z. Y. Li, Pseudo-mono-axial ligand fields that support high energy barriers in triangular dodecahedral Dy(III) single-ion magnets, Chem. Sci., 2022, 13, 13231–13240 RSC.
- H. S. Wang, P. F. Zhou, J. Wang, Q. Q. Long, Z. Hu, Y. Chen, J. Li, Y. Song and Y. Q. Zhang, Significantly Enhancing the Single-Molecule-Magnet Performance of a Dinuclear Dy(III) Complex by Utilizing an Asymmetric Auxiliary Organic Ligand, Inorg. Chem., 2021, 60, 18739–18752 CrossRef CAS PubMed.
- J. L. Liu, Y. C. Chen and M. L. Tong, Symmetry strategies for high performance lanthanide-based single-molecule magnets, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- Y. C. Chen, J. L. Liu, L. Ungur, J. Liu, Q. W. Li, L. F. Wang, Z. P. Ni, L. F. Chibotaru, X. M. Chen and M. L. Tong, Symmetry-Supported Magnetic Blocking at 20 K in Pentagonal Bipyramidal Dy(III) Single-Ion Magnets, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- J. Lu, V. Montigaud, O. Cador, J. Wu, L. Zhao, X. L. Li, M. Guo, B. Le Guennic and J. Tang, Lanthanide(III) Hexanuclear Circular Helicates: Slow Magnetic Relaxation, Toroidal Arrangement of Magnetic Moments, and Magnetocaloric Effect, Inorg. Chem., 2019, 58, 11903–11911 CrossRef CAS PubMed.
- Y. P. Qu, X. D. Huang, K. Xu, S. S. Bao and L. M. Zheng, Octahedral lanthanide clusters containing a central PO43− anion: structural, luminescent, magnetic and relaxometric properties, Dalton Trans., 2023, 52, 10489–10498 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2283643 and 2283644. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01490h |
| This journal is © the Partner Organisations 2023 |