
Achieving photostability in dye-sensitized upconverting nanoparticles and their use in Fenton type photocatalysis†
Mannu
Kaur
 ,
Steven L.
Maurizio
,
Gabrielle A.
Mandl
and
John A.
Capobianco
*
,
Steven L.
Maurizio
,
Gabrielle A.
Mandl
and
John A.
Capobianco
*
Department of Chemistry and Biochemistry and Centre for NanoScience Research, Concordia University, 7141 Sherbrooke St W., Montreal, QC H4B 1R6, Canada. E-mail: John.Capobianco@concordia.ca
First published on 3rd August 2023
Abstract
Dye sensitization is a promising approach to enhance the luminescence of lanthanide-doped upconverting nanoparticles. However, the poor photostability of near-infrared dyes hampers their use in practical applications. To address this, commercial IR820 was modified for improved photostability and covalently bonded to amine-functionalized silica-coated LnUCNPs. Two methods of covalent linking were investigated: linking the dye to the surface of the silica shell, and embedding the dye within the silica shell. The photostability of the dyes when embedded in the silica shell exhibited upconversion emissions from NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ nanoparticles for over four hours of continuous excitation with no change in intensity. To highlight this improvement, the photostable dye-embedded system was successfully utilized for Fenton-type photocatalysis, emphasizing its potential for practical applications. Overall, this study presents a facile strategy to circumvent the overlooked limitations associated with photodegradation, opening up new possibilities for the use of dye-sensitized lanthanide-doped upconverting nanoparticles in a range of fields.
Introduction
The long-lived intermediate excited states of trivalent lanthanides have garnered considerable attention in the study of upconversion, where near-infrared (NIR) irradiation is converted to UV, visible, or higher energy NIR emissions.1–3 Due to their narrow absorption and emission bands,3 high chemical and colloidal stability,4 and low toxicity,5 lanthanide-doped upconverting nanoparticles (LnUCNPs) are used in many diverse applications such as photodynamic therapy,6 drug delivery,7 optical encoding,8 biosensing,9 photovoltaics,10 and bioimaging.11 However, due to the forbidden nature of their intraconfigurational 4f–4f transitions, they have low absorption coefficients, leading to low upconversion efficiencies.12 Consequently, the emission of light from LnUCNPs is commonly enhanced through an indirect sensitization technique involving an organic dye, known as the “antenna effect”,13–16 proposed by Weissman in 1942 to transfer energy from organic ligands to lanthanide ions.17Organic dyes possess considerably higher absorption cross-sections (3–4 orders of magnitude higher than that of lanthanide ions) and feature broad and tunable absorption bands, making them ideal light-harvesting agents.15,16,18,19 Moreover, the shift in absorbance relative to conventional Yb3+ (from 976 nm to around 800 nm) also reduces the absorption of NIR light by water in biological tissues and prevents laser-induced heating effects.20 This type of system has the advantage of protecting the activators from solvent-mediated quenching, as well as increasing the light absorption cross-section through the antenna effect.21 In recent years, numerous investigations have demonstrated the efficacy of dye sensitization in increasing the intensity of upconversion luminescence (UCL). This technique involves either electrostatically,22–25 or covalently26 linking an organic NIR-emitting dye with a high absorption cross-section (on the order of 10−16 to 10−17 cm2) to the surface of a nanoparticle. Alternatively, the dyes can be encapsulated in a polymer or mesoporous silica shell.27–30 The photostability of dyes has also been successfully achieved through incorporation within silica nanoparticles to generate fluorescent silica nanoparticles.31–34
The concept of dye-sensitized LnUCNPs was first introduced in 2012 by Zou et al. using carboxylic acid-modified cyanine dyes to enhance the UCL of NaYF4:Yb,Er LnUCNPs.35 Further, Prasad and co-workers demonstrated an energy-cascaded upconversion process where the energy is transferred from IR-808 to Nd3+, and then to Yb3+ and Tm3+, resulting in improved upconversion quantum yields.15 In 2019, Ju et al. designed core/shell/shell UCNPs with a core of NaYF4:Gd3+, an energy-concentrating shell of NaYF4:Yb3+,Er3+ and an outer shell of NaYF4:Nd3+,Yb3+ to interact with a NIR dye and facilitate energy transfer.36
Unfortunately, while dye-sensitized LnUCNPs exhibited more intense UCL, the release of the dyes from the surface37,38 and the limited photostability of NIR dyes still remains a challenge.39–42 Reports on the covalent linking of NIR dyes to silica-modified nanoparticles, or their encapsulation in polymers have alleviated the detachment issues.16,26,43,44 However, the photostability of NIR dyes still remains an obstacle since they are known to undergo bimolecular reactions with in situ generated singlet oxygen, resulting in degradation.39–42,45 While various steric and functional modifications have been explored to inhibit these bimolecular reactions,46–48 this has rarely been addressed with respect to dye-sensitized LnUCNPs. Our previous work took advantage of the steric modification of IR820, showing that the IR820-NO2-functionalized LnUCNPs had brighter and considerably longer UCL compared to IR820-sensitized LnUCNPs.45 However, the issue of photostability was not fully resolved.
For the first time, to our knowledge, we have demonstrated a means of generating photostable dye-sensitized LnUCNPs. To achieve this, a modified version of commercial IR820 was covalently embedded in silica-coated NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ active-core/active-shell LnUCNPs. To assess the photostability and effectiveness of this system, spectroscopic comparisons were made between electrostatic and covalently linked dye-LnUCNP systems. Additionally, quantum yield measurements and luminescence lifetimes were conducted to compare this system with Nd3+-doped core/shell nanoparticles, a commonly employed alternative for 808 nm-sensitized UCL. Finally, we demonstrate the applicability of this photostable dye-sensitized system for Fenton-type photocatalysis.
Experimental
All reagents were used without further purification. Gd2O3 (99.999%), Yb2O3 (99.999%), and Er2O3 (99.999%) were purchased from Chemicals 101 Corp. Sodium trifluoroacetate (98%) was purchased from Alfa Aesar. Oleic acid (90%), 1-octadecene (90%), IR820 (80%), 4-mercaptobenzoic acid (99%), tetraethylorthosilicate (99%), ammonium hydroxide solution (28–30%), 3-aminopropyltrimethoxysilane (97%), IGEPAL CO-520 (99%), N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide (EDC, 97%), N-hydroxysuccinimide (NHS, 98%), (3-aminopropyl)trimethoxysilane (97%), mesitylene (98%), L-arginine (≥98%), iron(III) chloride hexahydrate (≥98%), sodium acetate (≥99.0%), and polyvinylpyrrolidone (PVP, average Mw ∼55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Sigma Aldrich. Trace metal grade hydrochloric acid (99.999%) was purchased from Fisher Scientific. Hexadecyltrimethylammonium chloride (CTAC, >95.0%) was purchased from TCI Chemicals. For MALDI-MS analysis, 2,5-dihydroxybenzoic acid (DHB), was purchased from Sigma-Aldrich and used without further purification.
000) were purchased from Sigma Aldrich. Trace metal grade hydrochloric acid (99.999%) was purchased from Fisher Scientific. Hexadecyltrimethylammonium chloride (CTAC, >95.0%) was purchased from TCI Chemicals. For MALDI-MS analysis, 2,5-dihydroxybenzoic acid (DHB), was purchased from Sigma-Aldrich and used without further purification.
Synthesis of NaGdF4:Er3+(2%),20%Yb3+/NaGdF4:20%Yb3+ active core/active shell LnUCNPs
Core nanoparticles were synthesized using a previously established thermal decomposition method.49 1.25 mmol of lanthanide oxides (78 mol% Gd2O3, 20 mol% Yb2O3, and 2 mol% Er2O3) were dissolved in 10 mL of 50% v/v aqueous trifluoroacetic acid under reflux conditions, generating the trifluoroacetate precursors. Once dried, the precursors were combined with 2.5 mmol of sodium trifluoroacetate, all dissolved in 7.5 mL oleic acid and 7.5 mL 1-octadecene and degassed under vacuum at 120 °C for 30 min. In a separate flask, 12.5 mL of oleic acid and 12.5 mL 1-octadecene were degassed under vacuum at 120 °C for 30 min, followed by a temperature increase to 310 °C, at a rate of 10 °C min−1, under argon atmosphere. The precursor solution was then injected into the solvents at a rate of 1.5 mL min−1. This solution was left stirring at 300 rpm for 60 min. To synthesize the core/shell nanoparticles, 1.25 mmol of lanthanide oxides (80 mol% Gd2O3 and 20 mol% Yb2O3) were dissolved in 50% aqueous trifluoroacetic acid under reflux conditions, generating the shell precursor. Once dried, 2.5 mmol of sodium trifluoroacetate was added to the shell precursors and dissolved in 2.5 mL of oleic acid and 2.5 mL of 1-octadecene. This solution was degassed under vacuum at 120 °C for 30 min. Once the full 60 min core synthesis was complete, the shell precursor solution was then injected into the reaction vessel, at a rate of 0.5 mL min−1. This solution was maintained at 310 °C for 45 min. Once the oleate-capped nanoparticles were synthesized, the solution was cooled to room temperature in ambient conditions and precipitated in 40 mL of ethanol. The resulting pellet was centrifuged at 3900 rpm for 15 min, and washed three times via dispersion in hexane and precipitation in ethanol, centrifuging between washes. Samples were stored as a solid pellet covered with ethanol.Synthesis of NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+/NaGdF4:(10–20%)Nd3+
In addition to the active-core/active-shell nanoparticles, a shell of Nd3+ was synthesized using the same protocol as the Yb3+ active shell. 1.25 mmol lanthanide oxides were dissolved in trifluoroacetic acid (90–80 mol% Gd2O3 and 10–20 mol% Nd2O3) to include the intended Nd3+ dopant concentration. Degassing was completed using the same methods as mentioned previously, and this solution was injected into the reaction vessel after the active-shell synthesis (t = 105 min) at a rate of 0.5 mL min−1. This full solution was then left at 310 °C for 30 min. Once complete, the solution was cooled to room temperature under ambient conditions and precipitated/washed as mentioned previously.Synthesis of IR820-COOH
A nucleophilic substitution reaction was used to functionalize the IR820 dye with 4-mercaptobenzoic acid.81 0.11 mmol of dye and 0.58 mmol acid were dissolved in 5 mL anhydrous DMF, under an argon atmosphere at room temperature. This solution was allowed to react for 12 h. Once complete, the DMF was evaporated under reduced pressure. The crude product was filtered through a PTFE syringe filter and precipitated with anhydrous diethyl ether, yielding 93.7 mg (92%) of IR820-COOH.Synthesis of IR820-APTMS
The IR820-COOH dye (0.01 μmol) was dissolved in 10 mL of anhydrous DMSO, with 0.01 μmol EDC and 0.025 μmol NHS. This solution was stirred at room temperature, under nitrogen atmosphere for 8 h. 0.005 μmol APTMS was then added to the mixture and stirred for an additional 5 h. The crude product was filtered through a PTFE syringe filter and precipitated with anhydrous diethyl ether.Synthesis of the SL system
0.8 mL of IGEPAL CO-520 was mixed with 30 mg active-core/active-shell nanoparticles in 6 mL of hexane. After stirring for 30 min, 0.08 mL of ammonium hydroxide solution was added and sonicated for 20 min. Subsequently, 18 μmol of TEOS was added at a rate of 2.5 μL per 15 min, followed by stirring for 48 h. 4 μmol of APTMS was then added at the same rate and stirred for an additional 24 h. Once complete, the newly amine-functionalized nanoparticles were precipitated with ethanol, collected by centrifugation at 8000 rpm for 10 min, and washed three times with 10 mL ethanol.The covalent linking of IR820-COOH to the amine-functionalized surface of the nanoparticles was achieved using EDC/NHS amide coupling. IR820-COOH was dissolved in 20 mL of anhydrous DMSO, followed by the addition of varying amounts of EDC and NHS (Table S1†). The mixture was stirred at 200 rpm at room temperature for 8 h. 25 mg of the amine-functionalized nanoparticles dispersed in 2 mL DMSO was then added to the IR820-COOH solution, and the mixture was stirred for an additional 5 h at room temperature. The completed SL system was isolated using centrifugation at 13000 rpm for 10 min, followed by three washes with 10 mL ethanol.
Synthesis of the SE system
To synthesize the dye-embedded silica shell (with a thickness between 4–5 nm), 0.8 mL of IGEPAL CO-520 and 30 mg of oleate-capped nanoparticles were dispersed in 6 mL of hexane. After stirring for 30 min, 0.08 mL of ammonium hydroxide solution (28 wt% in water) is added and sonicated for 20 min. Subsequently, TEOS (according to Table S2†) was added at a rate of 2.5 μL per 15 min and stirred for 48 h. Following this, IR820-APTMS (according to Table S2†) was added and stirred for an additional 24 h. Finally, the nanoparticles were precipitated with ethanol, collected by centrifugation at 8000 rpm for 10 min, and washed three times with 10 mL ethanol.Transmission electron microscopy (TEM)
10 μL of 1 mg mL−1 solutions of nanoparticles (in either hexane or methanol, depending on the hydrophilicity of the sample) were dropped on 300 mesh copper grids (3 mm, formvar/carbon film supported). Micrographs were recorded using a Talos L120C scanning transmission electron microscope operating at an accelerating voltage of 120 kV.Energy dispersive X-ray spectroscopy (EDX)
EDX measurements were performed on a Jeol-JEM-2100F transmission electron microscope, operating at 200 kV. Scanning transmission electron microscopy (STEM) was used to record high angle annular dark field (HAADF) micrographs, which could then be used to acquire elemental mapping with an Oxford EDS detector (model Xplore).Powder X-ray diffraction (XRD)
Diffractograms of dried upconverting nanoparticles were collected using a Bruker AXS D2 Phaser (30 kV, 10 mA), and dried hematite nanoparticles were collected using a Rigaku MiniFlex (40 kV, 15 mA). Both instruments are equipped with a Cu-Kα source, and diffractograms were recorded with a resolution of 0.2°2θ.Upconversion emission spectroscopy
Nanoparticle solutions in methanol (5 mg mL−1) were excited using either 976 nm (Sky-laser, 0.8 W, 13 W cm−2) or 808 nm (SLOC Model IRM800T3-2500FC, 1.3 W, 5 W cm−2) irradiation. Upconverted emissions were collected at a right angle to the incident beam, using an optical fiber (Ocean Optics Inc, 600 μm) coupled to a visible bandpass filter (Thorlabs, 400–750 nm), connected to a Princeton Instruments FERGIE BRX-VR UV-NIR spectrograph fitted with a 1200 grooves per mm grating blazed at 290 nm, with a 50 μm entrance slit.Upconversion lifetimes
Lifetime measurements were recorded on the same solution samples of nanoparticles as the emission spectra, excited using a 976 nm diode laser (Coherent 6-pin 15 fiber-coupled F6 series laser diode, 500 μs pulse width, 1.5 W cm−2) or 808 nm diode laser (SLOC Model IRM800T3-2500FC, 500 μs pulse width, 5 W cm−2). Emissions were collected using a Jarrell-Ash Czerny-Turner 25-102 1 m double monochromator (1180 grooves per mm), coupled to a Peltier-cooled Hamamatsu R943-02 photomultiplier tube. Signals were processed through an SR440 Stanford Research Systems preamplifier and interpreted using an SR400 Stanford Research Systems gated photon counter, varying the 50 μs gate window to produce the lifetime profiles.Upconversion quantum yields
Quantum yield measurements were completed on the same nanoparticle solutions used for the emission spectra and lifetime measurements. Samples were irradiated using the same excitation sources as the emission spectra (976 and 808 nm). Emissions were collected using an Avantes AvaSphere-30-REFL integrating sphere, fiber-coupled to a Thorlabs FOFMS/M-UV filter mount that was fitted with either a 400–750 nm bandpass filter or a 60% neutral density filter to record the emissions or absorptions, respectively. The filter mount was fiber-coupled to an Avantes AvaSpec-ULS2048L spectrometer. Emission and absorption intensities were recorded in units of watts using a NIST calibrated AvaLight-DH-CAL-ISP30 lamp. These intensities were then converted to number of photons using the photon energy equation. Undoped NaGdF4 nanoparticles were used as a control for Nd3+-doped nanoparticles, while undoped NaGdF4 with an inert silica shell was used as a control for the SE system. Errors were calculated using triplicate measurements as well as the reported instrumental error.Synthesis of wide-pored silica shell
0.480 g of CTAC was mixed with 110 mL of water and 1.2 mL of mesitylene. 25 mg of SE nanoparticles were dispersed in 10 mL water and was added dropwise to the surfactant mixture. This solution was then sonicated for 30 min and stirred for an additional 30 min. 1.4 mL of butanol and 160 μL of TEOS was added to the solution at a rate of 0.5 mL h−1. Afterward, 24 mg of L-arginine was dissolved in 1 mL water and added to the solution. This was allowed to stir for 17 h at 50 °C under reflux.Synthesis of mesoporous silica shell was completed using the same method, omitting the addition of mesitylene.72
Synthesis and incorporation of α-Fe2O3 (hematite) nanoparticles
A previously reported hydrothermal method was used to synthesize the hematite nanoparticles.71 1.5 mmol of FeCl3·6H2O, 45 mmol of sodium acetate, and 1.5 g of PVP were mixed together with 30 mL of water and stirred for 2 h at 40 °C. The solution was then placed in a Teflon-lined stainless steel autoclave and heated at 200 °C for 18 h. After the reaction, the autoclave was cooled in ambient conditions, and the red solid product was collected by centrifugation and washed three times with water and ethanol. Finally, the red product was dried under vacuum at 70 °C for 12 h.30 mg of wide-pore-functionalized SE nanoparticles were dispersed in 2 mL ethanol. 2, 3, or 5 mL of a hematite nanoparticle solution (5 mg mL−1 in ethanol) was then added dropwise, and this solution was stirred for 24 h at 2000 rpm. The resulting nanocomposite (SE-WP-Fe) was collected by centrifugation and washed three times with ethanol.
Diffuse reflectance spectroscopy
Reflectance data was collected for dried hematite nanoparticles using a Cary 5000 Series UV-Vis spectrophotometer (Agilent Technologies) with a single beam range between 360–800 nm using an EasiDiff reflectance accessory (PIKE Technologies, Inc.).Electron paramagnetic resonance
Electron paramagnetic resonance (EPR) spectroscopy was performed at the McGill Chemistry Characterization facility (MC2) at McGill University in Montreal, Canada. Spectra were acquired on a Bruker Elexsys E580 X band EPR instrument using a high sensitivity resonator. The sample was analyzed in a flat cell (Suprasil, 150 μL, 0.3 mm wide) to minimize signal loss. A 1 G modulation amplitude and 20 mW power were used for the acquisition. A polynomial baseline correction was applied to each spectrum before fitting. Fitting was performed using WinSim2002 version 0.98 developed by the National Institute of Environmental Health Sciences at the National Institutes of Health. 50 mM solutions of 5,5-dimethyl-pyrroline N-oxide (DMPO) in methanol were used within 1 h of their preparation. In all cases, 2 mg mL−1 colloidal solutions of nanoparticles were used. In all samples involving H2O2, 10 μL of 30% H2O2 were added to a 2 mL dispersion of 2 mg mL−1 nanoparticles immediately before irradiation and/or signal acquisition. 808 nm irradiation was performed using the same laser previously discussed. 532 nm excitation was performed using a handheld diode laser (500 mW). For the irradiation experiments, samples were irradiated while inside the flat cell and the flat cell was immediately inserted into the EPR instrument for analysis after 15 min of irradiation. Control samples were treated identically to the irradiated samples.Evaluation of the photocatalytic activity of SE-WP-Fe system
The photocatalytic activity of the SE-WP-Fe nanocomposites was evaluated by observing the degradation of rhodamine B when exposed to 808 nm near-infrared laser irradiation. A mixture of 30 mg of the nanoparticles, 50 μL of 30% H2O2, and 10 mL of rhodamine B solution (4 × 10−6 M) was stirred for 1 h to reach the adsorption–desorption equilibrium of the dye with the catalyst. The stirred solution was then exposed to 808 nm irradiation and 1.5 mL aliquots were collected every 10 min. After removing any solid precipitate by centrifugation, an absorption spectrum (Agilent Technologies, Cary 5 Series UV–Vis–NIR Spectrophotometer) of each aliquot was recorded to evaluate the absorbance of the rhodamine B.Results and discussion
The NaGdF4:2%Er3+,20%Yb3+/NaGdF4:20%Yb3+ active-core/active-shell nanoparticles used throughout this work were synthesized using a double-injection thermal decomposition method.49 The as-synthesized core and core/shell nanoparticles were characterized by transmission electron microscopy (TEM) and X-ray diffraction (XRD). TEM micrographs display uniform size distributions, with an average core diameter of 19.8 ± 1.5 nm and core/shell diameter of 30.2 ± 3.0 nm, yielding a shell thickness of approximately 5.2 nm (Fig. S1a and b†). The XRD diffractograms are consistent with the hexagonal NaGdF4 structure (Fig. S1c†).50 UCL spectra upon 976 nm excitation, are displayed in Fig. S1d.† The characteristic green emissions of Er3+ from the 2H11/2/4S3/2 → 4I15/2 transitions are centered at 525 and 550 nm, respectively. Additionally, the red emission at 660 nm from the 4F9/2 → 4I15/2 transition is also observed. As expected, a nine-times enhancement in the UCL intensity for the core/shell LnUCNPs is observed relative to the core-only LnUCNPs.Commercial IR820 dye was modified with 4-mercaptobenzoic acid at the central aryl ring of the dye (herein referred to as IR820-COOH), which is ideal for this study since it can be excited at wavelengths ranging from 780–830 nm (Fig. S2a†), with a broad emission band that overlaps with the absorption of Yb3+ (Fig. S2b†).51 The structure of IR820-COOH was confirmed using absorption spectroscopy, 1H-NMR spectroscopy, and MALDI-TOF mass spectrometry (Fig. S3–S4†).
In this study, two methods of covalent linking of the IR820-COOH dye to the LnUCNPs were investigated: surface silica linking (SL) and silica embedded linking (SE) (Scheme 1). Although the method of linking the dye to the surface of a silica shell has been previously reported, it helps us to establish a benchmark to assess the photostability and the enhancement factor for the covalent embedded system.26,52 The SL method employs covalently linking the carboxylic acid-functionalized dye onto the surface of amine-functionalized silica-coated LnUCNPs by EDC/NHS amide coupling reaction, after the silica shell has been formed (Scheme 1a and Table S1†). The SE method involves first modifying IR820-COOH through EDC/NHS amide coupling with APTMS to produce IR820-APTMS, which results in the dye being embedded in the silica shell during polymerization on the LnUCNP surface (Scheme 1b). Silica shells with a thickness of 5 ± 1.5 nm and 4 ± 1.0 nm were obtained for the SL and SE systems, respectively (Fig. 1). The formation of the SL system was also characterized by Fourier-transform infrared (FTIR) spectroscopy (Fig. S5†) confirming the presence of the amide bond between the dye and silica shell at the surface of the LnUCNPs.
 | ||
| Scheme 1 Scheme of the two types of nanoparticle systems investigated in this work, where IR820 is functionalized at the central aryl ring to generate IR820-COOH to generate (a) NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ coated with silica and IR820-COOH linked to the surface via the EDC/NHS amide coupling reaction (SL) and (b) the NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+ coated with silica and IR820-APTMS embedded into the silica shell (SE). | ||
 | ||
| Fig. 1 TEM micrographs of (a) LnUCNPs coated with amine functionalized-SiO2 for the SL system. (b) LnUCNPs coated with IR820-CONH-APTMS embedded in the silica matrix in the SE system. | ||
Since it is essential to obtain a dye-sensitized system with maximal brightness to enable high sensitivity and lower detection limits for any application, various concentrations of the dye were studied in both methods to establish the optimum concentration of dye for improved UCL under 808 nm excitation (Fig. S6 and S7†). It is well-recognized that elevated concentrations of dye can lead to self-quenching, while low concentrations will not absorb sufficient NIR light.22,35,53 For the SL system, this was achieved by adding various concentrations of IR820-COOH to the EDC/NHS reaction. In contrast, modulating the dye content in the SE system is difficult because of the distance-dependent nature of the energy transfer between the dye and the LnUCNP. Reagent quantities had to be carefully tailored (Table S2, ESI†) to obtain similar-sized silica shells around the LnUCNPs in the range of 4–5 nm (Fig. S8†) such that a comparison can be made.
The upconversion emission spectra were recorded for both systems upon 808 nm excitation, showing characteristic Er3+ green and red emission bands (Fig. 2). It is evident for both methods that as the concentration of the dye increases, the Er3+ emission intensity increases, however beyond 340 and 390 dye molecules per nanoparticle (for the SL and SE systems respectively), any further increase in dye concentration leads to quenching of the UCL. Since NIR dyes have a small Stokes shift, two of the same dye molecules that are in close proximity can experience Förster resonance energy transfer (FRET), resulting in the self-quenching observed, known as homo-FRET.54,55 Moreover, π-stacking could also contribute to quenching, though this is unlikely in the SE system since the dyes are spatially separated and their movement is restricted within the silica framework.
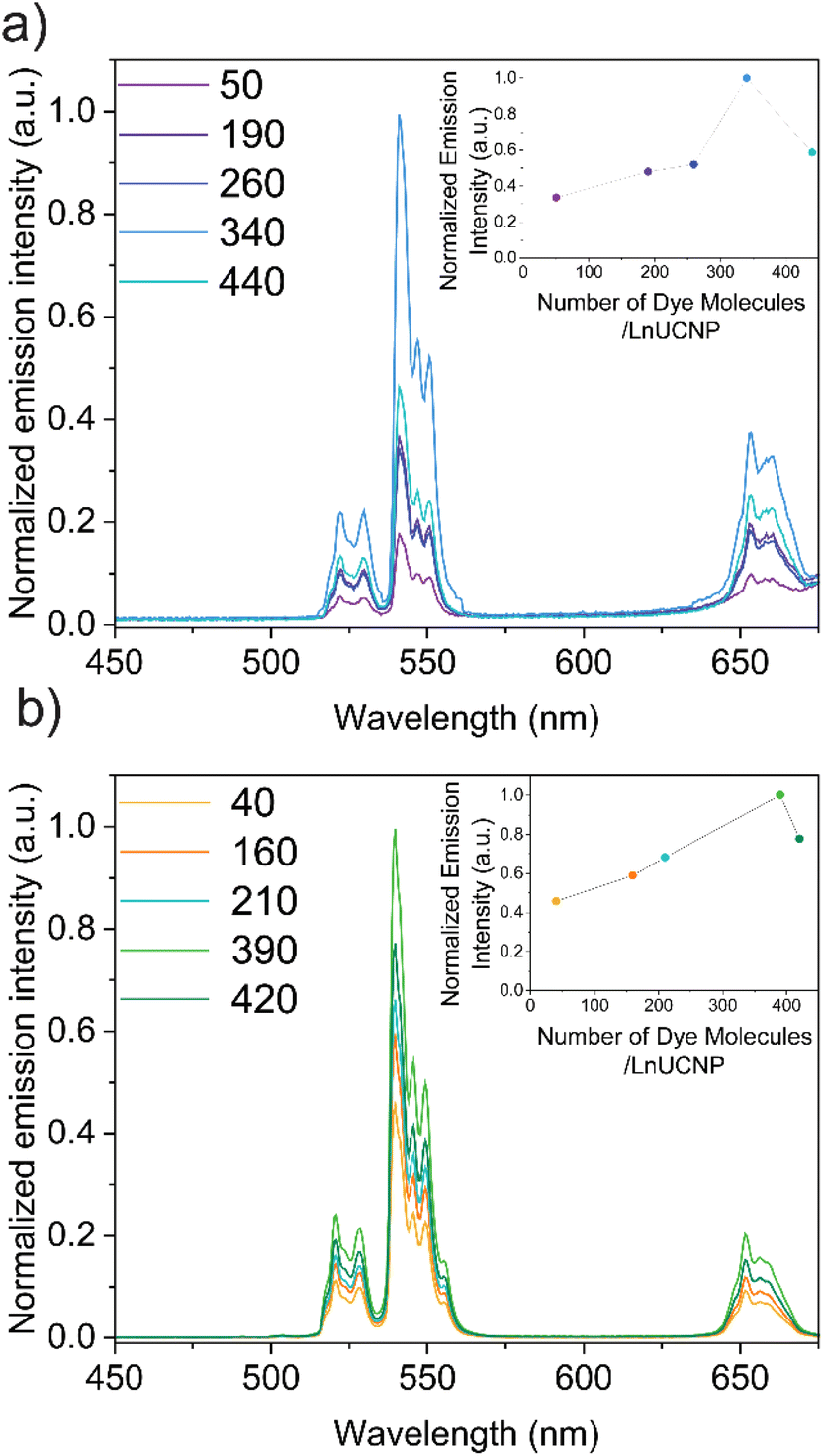 | ||
| Fig. 2 (a) Upconversion emission spectra of the SL system with different numbers of dye molecules per LnUCNP. (b) Upconversion emission spectra of the SE system with different numbers of dye molecules embedded into the silica sell per LnUCNP. | ||
Comparing the UCL intensities between the SL and SE system at their respective optimal dye concentrations are shown in Fig. 3. As observed in Fig. 3a and Fig. S9,† the SE system is 50% more intense than the SL system. The major difference between the two systems is the distance between the Yb3+ ions in the LnUCNP and the dye molecules. In the SL system, the dye molecules are spatially separated from the LnUCNPs due to the silica shell with a minimum distance of 5 nm as per the silica shell thickness. In contrast, in the SE system, the dye molecules are within the silica shell and therefore in closer proximity to the Yb3+ ions in the LnUCNP, with a maximum distance of 4 nm (from the LnUCNP surface to the edge of the 4 nm thick silica shell). As such, there should be a more efficient energy transfer between the dye molecules in the SE system and the Yb3+ ions.
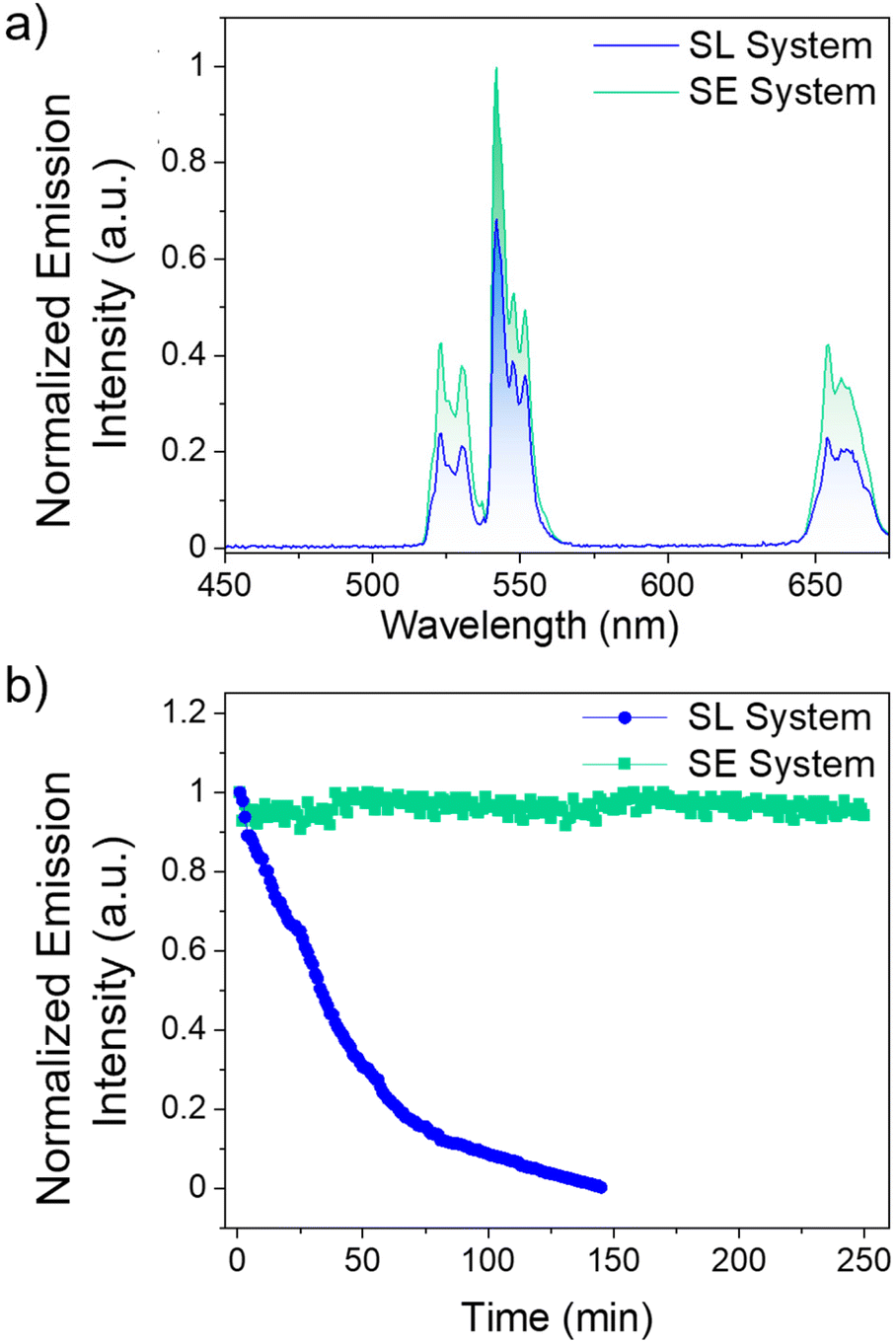 | ||
| Fig. 3 (a) Normalized upconversion emission spectra of the SL and SE systems under 800 nm excitation. (b) Emission intensity of the 4S3/2, 2H11/2 → 4I15/2 transition of Er3+ in the SL (blue) and SE (green) systems as a function of time under 800 nm excitation. | ||
For further comparison, the SL and SE systems were compared with a control sample where IR820-COOH was electrostatically linked to the surface of the LnUCNPs. Absorbance studies on both systems reveal no leakage of the dye even after 15 days, confirming this is not an issue for the covalently linked systems, whereas when the IR820-COOH dye is electrostatically linked there is substantial release from the LnUCNP surface (Fig. S10†). The electrostatically-linked system also showed the least efficient UCL and complete degradation of the dye after 80 minutes of irradiation (Fig. S11†). Kinetic studies were then completed on both systems (Fig. 3b) to establish the photostability of the dye by evaluating the UCL intensity of the 4S3/2 → 4I15/2 transition of Er3+ as a function of irradiation time under 808 nm excitation. After 2.5 hours, the emission of the SL system was no longer observed, indicating that although leaching from the surface is not an issue, the dye molecules bound to the surface of the LnUCNPs were still susceptible to photodegradation. Finally, the emission of the SE system remained constant over four hours of irradiation, suggesting upconversion could be harnessed for extended periods of time. We attribute this improvement in photostability to the likelihood that covalent encapsulation of IR820-APTMS prevents oxygen from reaching the dye molecules, thus no singlet oxygen can be formed to cause degradation. Moreover, the dye in the SE system is fully isolated from the environment and has restricted rotational freedom, while the SL system, notwithstanding its structural stability, is still susceptible to the environment and to in situ generated singlet oxygen. Therefore, owing to its encapsulation, the photostability of the dye molecules is considerably enhanced, which is a major advancement in terms of photostability over the past work in the dye-sensitized LnUCNPs.54–58 In summary, by embedding the IR820-APTMS dye into a silica network, it became possible to alleviate dye leaching and photodegradation, thereby generating a photostable dye-sensitized LnUCNP system capable of constant UCL emission intensity for over four hours of excitation.
In addition to NIR dyes, the use of Nd3+ is common to facilitate upconversion using 808 nm excitation, due to the 4I9/2 → 4F5/2 absorption band of Nd3+ which can transfer energy to Yb3+.59,60 While both NIR dyes and Nd3+ can be excited by the same wavelength, the NIR dyes have the additional benefit of having a higher absorption cross-section. In particular, the absorption cross-section of IR820 at 808 nm excitation is approximately 10−16 cm2,19 while that for the Nd3+ ion is about 4 × 10−20 cm2.61
To explore the potential differences in Nd3+- and dye-sensitized LnUCNPs, we conducted a systematic evaluation of a group of similarly-sized NaGdF4:Er3+,Yb3+/NaGdF4:Yb3+/NaGdF4:10–20%Nd3+ nanoparticles. TEM micrographs for 10, 15, and 20% Nd3+ show all compositions have uniform particle sizes and morphology, and nanoparticle sizes and shell thicknesses that are comparable to the SE system (Fig. S12†). XRD analysis confirms the hexagonal crystal structure for all samples (Fig. S13†). The UCL spectra of the core, core/shell, and core/shell/shell LnUCNPs were recorded under 976 nm excitation to ensure their luminescent properties were similar to each other and with the corresponding SE system (Fig. S14†). Emission spectra under 808 nm excitation were recorded to determine the optimal concentration of Nd3+, showing that 15% Nd3+ exhibited the most intense UCL (Fig. S15a†). Furthermore, on comparing the UCL of the optimized core/shell/NaGdF4:15%Nd3+ and the optimized SE system, under 808 nm excitation, the SE system exhibits 50% more intense emission (Fig. S15b†).
To evaluate the difference in energy transfer dynamics from Nd3+ or IR820-APTMS to Yb3+, upconversion lifetime measurements of Er3+ were recorded under 976 nm excitation (green 4S3/2 → 4I15/2 transition shown in Fig. 4, red 4F9/2 → 4I15/2 transition shown in Fig. S16a and b†). Using this excitation wavelength avoids absorption by Nd3+ or IR820-APTMS, exciting Yb3+ directly and allowing the study of potential “back-transfer” from the Er3+ ions in the core to the Nd3+ or IR820-APTMS in the outer shells. The addition of Nd3+ results in shortening of the Er3+ decay times, indicative of FRET between these two species. A possible mechanism is Er3+:4F9/2 + Nd3+:4I9/2 → Er3+:4I15/2 + Nd3+:4F9/2, which results in quenching of the Er3+ UCL, either directly or through a shift in the equilibrium of states.62 In contrast, the addition of the IR820-APTMS dye results in an increase in lifetime. This, in all likelihood, is a consequence of the formation of the intermediate TEOS shell, which has been reported to encapsulate water more efficiently than APTMS, resulting in greater phonon quenching within the TEOS shell.63 Regardless, no FRET was observed between Er3+ and IR820-APTMS, which is likely a contributing factor to why the SE system exhibits brighter UCL than Nd3+-doped LnUCNPs.
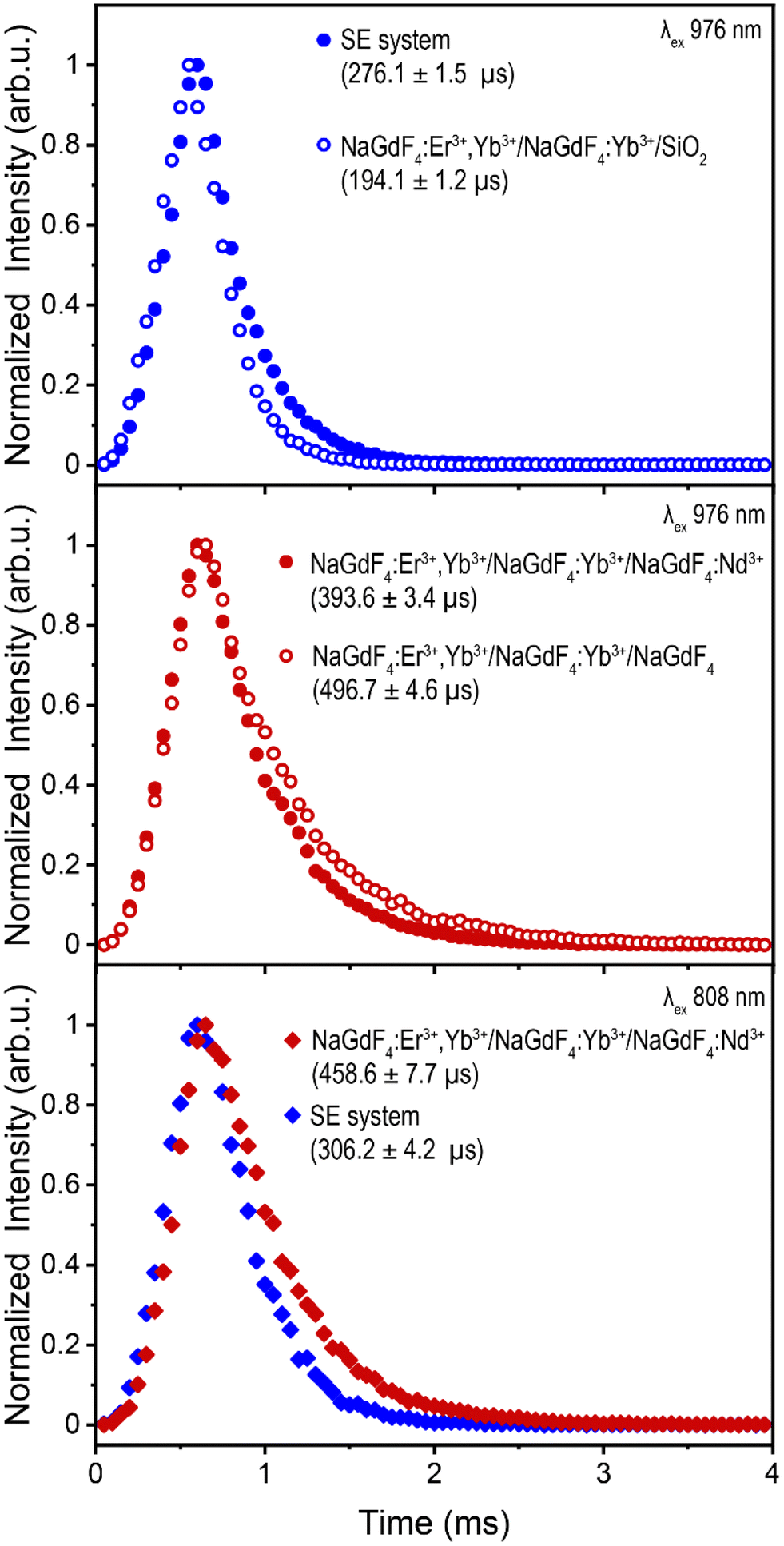 | ||
| Fig. 4 Upconversion lifetimes of the 2H11/2 → 4I15/2 transition of Er3+ in (a) the SE system (filled circles) and NaGdF4:Yb3+,Er3+/NaGdF4:Yb3+ LnUCNPs coated with unfunctionalized silica (outlined circles) under 976 nm excitation. (b) NaGdF4:Yb3+,Er3+/NaGdF4:Yb3+/NaGdF4:15% Nd3+ LnUCNPs (filled circles) and NaGdF4:Yb3+,Er3+/NaGdF4:Yb3+/NaGdF4 LnUCNPs (outlined circles) under 976 nm excitation. (c) The SE system (blue) and NaGdF4:Yb3+,Er3+/NaGdF4:Yb3+/NaGdF4:15%Nd3+ LnUCNPs (red) under 808 nm excitation. | ||
Lifetime measurements under 808 nm excitation, shown in Fig. 4, were undertaken to assess the dynamics when either Nd3+ or IR820-APTMS are excited directly. The resulting decay times are slightly longer than their 976 nm-excited counterparts, due to the additional energy transfer step from Nd3+ or IR820-APTMS to Yb3+. The decay time for Er3+ in the SE system is slightly shorter than in the Nd3+-doped LnUCNPs. This could be due to the vibrational modes of silica, resulting in phonon quenching that is not present in the Nd3+-doped NaGdF4 shell. Moreover, there is likely a contribution from the longer sensitizer lifetime, since the forbidden electronic transitions of Nd3+ have considerably slower decay times than the allowed singlet transition of IR820-APTMS, also resulting in the SE system decaying faster. However, since the decay time of the IR820-APTMS dye is shorter than Nd3+, questions arise regarding its effectiveness as a sensitizer, since shorter decay times are inefficient for FRET.64 Therefore, quantum yield measurements were performed to evaluate the FRET efficiencies from either Nd3+ or IR820-APTMS to sensitize Er3+.
Interestingly, both systems exhibit quantum yields of the same magnitude under 808 nm excitation (Nd3+-doped LnUCNPs: (7.3 ± 0.87) × 10−4%, SE system: (9.2 ± 2.9) × 10−4%), indicating the energy transfer efficiency from excited Nd3+ ions or IR820-APTMS molecules to Yb3+ is similar, despite their drastically different decay times. While Nd3+ → Yb3+ was expected to have a greater FRET efficiency, owing to its longer decay time, the spectral overlap between these two species is quite weak, indicating that the energy transfer is phonon-assisted.51,65,66 In the case of the SE system, while the decay time is much shorter, the spectral overlap between the dye and Yb3+ is much greater, resulting in comparable FRET efficiencies from both these sensitizers. Therefore, considering the higher absorption cross-section of the NIR dye, the SE system exhibits more intense UCL emissions.
There are several reports that utilize Ln3+-doped nanocrystals as efficient photocatalysts.67–69 However, due to the lack of structural architecture with dye-functionalized LnUCNPs, and the limited thermal and photostability of the NIR dyes, their incorporation with semiconducting photocatalysts has never been explored to our knowledge. In this work, we applied the SE system to facilitate Fenton-type photocatalysis, where 808 nm-sensitized UCL can transfer energy to hematite nanoparticles and generate reactive oxygen species (ROS) in the presence of hydrogen peroxide. This was accomplished by synthesizing a wide-pore silica shell around the SE system and incorporating the hematite nanoparticles into the pores. We evaluated the photocatalytic activity by examining the degradation of rhodamine B, a known carcinogen and water pollutant discharged from textile industries.70
The hematite (α-Fe2O3) nanoparticles were synthesized via a hydrothermal method and characterized by TEM, XRD, and diffuse reflectance.71 The α-Fe2O3 nanoparticle sizes were measured to be 15.5 ± 5.7 nm by TEM (Fig. S17a†), with the expected rhombohedral crystal phase as confirmed by XRD analysis (Fig. S17b†).71 Diffuse reflectance confirms a bandgap energy of approximately 2.2 eV, in agreement with the reported value71 (Fig. 5a and b). A wide-pore silica shell was coated around the SE system (SE-WP) through a typical sol–gel method that uses mesitylene as a pore swelling agent (Fig. 5c).72 This method renders a pore size of about 20–25 nm.73,74 The hematite nanoparticles were inserted into the pores via the self-absorption method.73 The insertion of hematite nanoparticles was confirmed using TEM, high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM), XRD, and energy dispersive X-ray spectroscopy (EDX). TEM micrographs demonstrated the random incorporation of hematite nanoparticles into the pores (Fig. 5d), XRD pattern for the composite nanoconstruct indicates that the diffraction peaks correspond to two distinct crystalline phases: β-NaGdF4 and α-Fe2O3 (Fig. 5e). HAADF-STEM imaging and EDX mapping of Gd, Si, and Fe provided confirmation of the LnUCNP core, silica shell, and hematite nanoparticles in the pores of the SE-WP system (Fig. 5f).
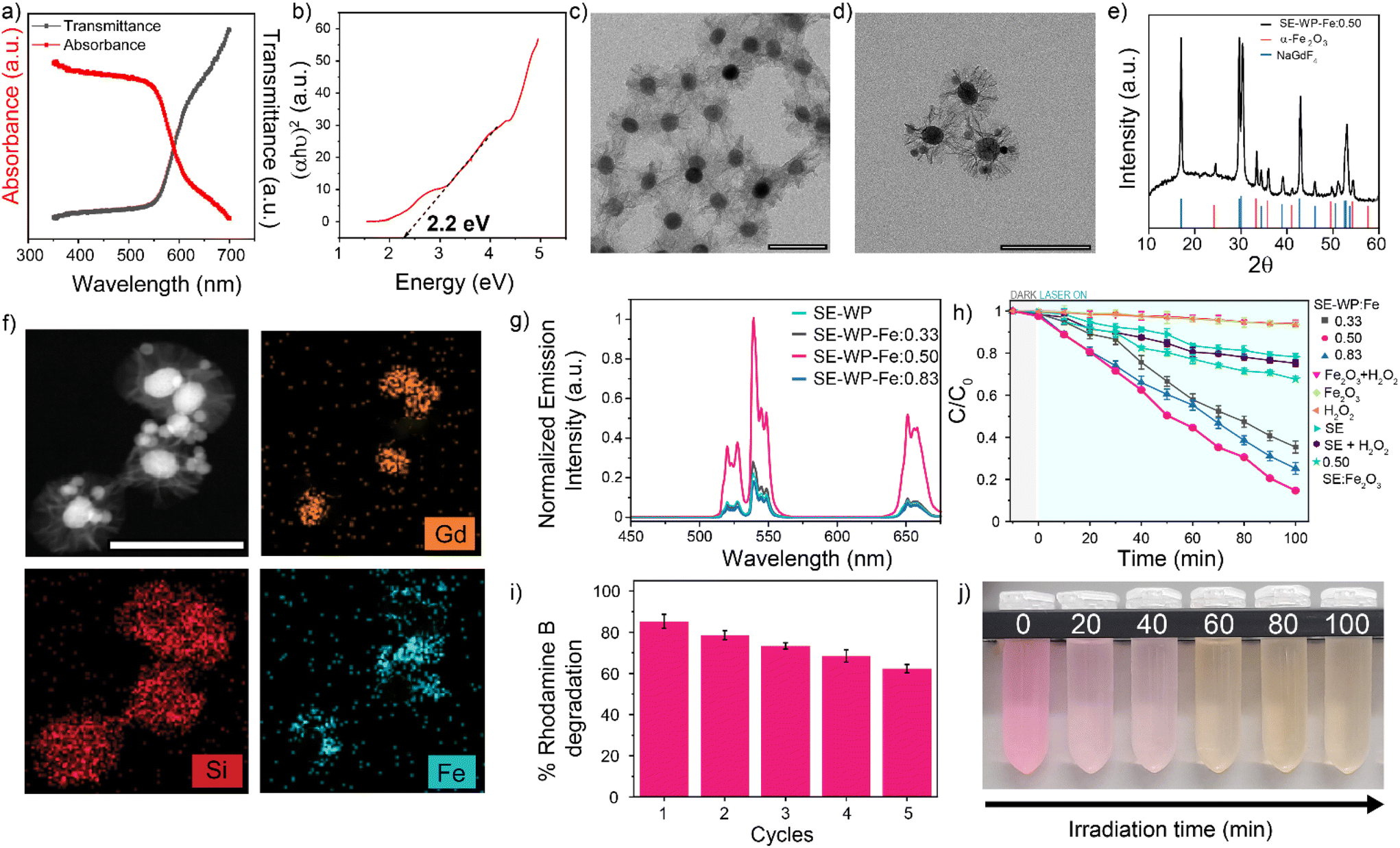 | ||
| Fig. 5 (a) Diffuse reflectance spectra for synthesized hematite nanoparticles and (b) the corresponding Tauc plot to determine the band gap energy. TEM micrographs for the wide-pored silica shell synthesized on the SE nanoparticles (c) before and (d) after the incorporation of hematite nanoparticles. (e) Powder XRD diffractogram and (f) HAADF and EDS mapping of the completed SE-WP-Fe system. (g) Upconversion emission spectra of the completed SE-WP-Fe system with varying concentrations of hematite nanoparticles. (h) Rhodamine B degradation measured from the relative absorbance spectra. (i) Repeated cycles of rhodamine B photodegradation after 100 min, recycling the SE-WP-Fe system. (j) Photographs of the rhodamine B solutions, illustrating the degradation of the dye after 100 min. All scale bars are 100 nm. | ||
In this system, the emissions from Er3+ under 808 nm excitation correspond to the band gap of hematite and thus are absorbed by the α-Fe2O3 nanoparticles such that the Fe3+ ions should photocatalytically decompose H2O2 to ROS, which degrades rhodamine B. Varied ratios of α-Fe2O3 to SE-WP (w/w) were studied (0.33, 0.50, and 0.83) to evaluate the effect of concentration on photocatalytic activity, herein the Fe2O3-loaded SE particles are referred to as SE-WP-Fe. The actual concentration of hematite nanoparticles incorporated in the LnUCNPs was established by UV-vis calibration curve (Fig. S18a and b†). As observed in Fig. 5g, the decrease in UCL upon increasing ratios of Fe2O3 to SE-WP indicates increased energy transfer accompanied by scattering, as expected.74
To confirm the potential for ROS production via the Fenton reaction from this system, electron paramagnetic resonance spectroscopy (EPR) was performed. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO), a well-established spin-trapping agent, was used to capture the ROS and enable the identification of the produced species. Shown in Fig. S19,† EPR spectra were obtained for each species in the presence and absence of H2O2, and the presence and absence of 808 nm excitation. As expected, in the absence of H2O2, in all cases weak or no EPR signal was obtained, confirming negligible ROS production, even under 808 nm excitation. A positive control was performed under direct excitation at 532 nm to excite Fe2O3 directly, from which the DMPO-X radical was generated; this is a well-established byproduct of substantial hydroxyl radical production.75 This was the only species observed when exciting the Fe2O3 directly. In contrast, under 808 nm excitation, four ROS were identified from the activity of the full SE-WP-Fe system (Table S4†). Specifically, hydroxyl radicals, superoxide radicals, and methylcarbonyl radicals were identified and their values are in agreement with what is observed for DMPO spin adducts in methanol and other Fenton-based systems.76,77 Notably, hydroxyl radicals are implicated in Fenton-reaction mediated degradation of rhodamine B as described by Frindy et al.78 The methylcarbonyl radicals are likely due to the decomposition and oxidation of methanol generated in the solution.76
In summary, the EPR spectral data suggest the Fenton reaction can occur from excitation at 808 nm in this system. The photocatalytic activities of the SE-WP-Fe system when excited with NIR light, were evaluated by monitoring the degradation of rhodamine B in methanol. It is known that the photogenerated electrons and holes in the semiconductor will travel to the surface, but many electron–hole pairs recombine during this process and do not contribute to the photocatalytic reaction.68,69,79,80 Thus, although increasing the α-Fe2O3 nanoparticles improves the absorption rate of upconverted emission, it also aggravates the recombination of photogenerated charge carriers and thus affects the activity of the photocatalyst.68 For this reason, several different ratios of Fe2O3 to LnUCNP (w/w) were evaluated for their photocatalytic activity. As shown in Fig. 5h, no detectable change in the rhodamine B concentration was observed when the α-Fe2O3 nanoparticles were exposed to 808 nm light alone, confirming this low-energy wavelength does not excite the hematite. Similar results were observed when the SE system was studied in the absence of hematite in the pores (Fig. 5h). The SE-WP-Fe nanoconstruct caused the successful degradation of rhodamine B under NIR irradiation for 100 minutes, with degradation of rhodamine B reaching 65%, 85%, and 75% when the hematite to SE-WP system mass ratios were 0.33, 0.50 and 0.83, respectively (Fig. 5h). It was found that the ratio of 0.50 had the most efficient photocatalytic activity, suggesting that the photocatalytic efficiency is dependent on the ratio of the LnUCNP and Fe2O3 nanoparticles.
To assess the reusability of the photocatalyst, the 0.50 SE-WP-Fe system was recycled and used for repeated degradation experiments. After five consecutive cycles, 62% of the initial rhodamine B concentration could still be degraded under the same conditions, indicating the relatively high stability and recyclability of the photocatalytic system (Fig. 5i and j). The decrease in photocatalytic efficiency may be attributed to the dissociation of hematite nanoparticles from the pores during the reaction and/or the recycling process.
To confirm the decrease in photocatalytic activity was not due to the degradation of the dye in the presence of H2O2 and hematite nanoparticles, we performed a time-dependent study on the UCL of the SE-WP-Fe system under 808 nm excitation in the presence and absence of H2O2. As observed in Fig. S20,† there was no change in the UCL intensity in the SE-WP-Fe system, even after 4 hours of 808 nm irradiation, indicating there is no dye degradation contributing to the loss of photocatalytic activity. In summary, the robust nature of the system presented herein has enabled dye-sensitized LnUCNPs for photocatalysis for the first time, to our knowledge. Embedding the dye within a silica framework prevents deleterious photodegradation, facilitating the use of these systems for long-term, repeated use under prolonged excitation.
Conclusion
This work addressed the photostability of dye-sensitized LnUCNPs, towards their use in solution-phase applications requiring prolonged and/or repeated irradiation. This was achieved by covalently embedding a modified IR820-APTMS dye into a silica matrix around the nanoparticles, to prevent interactions between the dye and oxygen present in the environment. Kinetic studies were conducted under 808 nm excitation, exhibiting no change in the UCL intensity of the dye-embedded system after four hours of irradiation. This enabled the use of the established SE system as a promising candidate to study the photocatalytic capability of dye-sensitized LnUCNPs. A wide-pored silica shell was synthesized around the SE system to encapsulate hematite nanoparticles, which have Fenton-type photocatalytic activity. Degradation of rhodamine B was observed under 808 nm irradiation, which was found to be efficient and reproducible across five cycles. As a result, the potential uses for dye-sensitized LnUCNPs can be expanded upon now that the limitation of dye photodegradation has been resolved.Author contributions
MK and JAC conceptualized the project. MK is credited with methodology, investigation, data acquisition, formal analysis, validation and visualization of the work. SLM is credited with data curation and data acquisition. GAM is credited with data acquisition and data visualization. MK, SLM and GAM wrote the original manuscript. All authors reviewed and edited the final manuscript. JAC is credited with funding acquisition, supervision, project administration and resource acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JAC is grateful to the Natural Sciences, Engineering and Research Council of Canada (NSERC) for sustained support of his research (no. RGPIN-2022-04704). JAC is a Concordia University Research Chair in Nanoscience and is grateful to Concordia University for financial support. M. K. is grateful to Concordia University for financial support through the Tuition Award of Excellence. SLM and GAM are grateful to NSERC for support from the Alexander Graham Bell CGS-D scholarships. SLM is grateful for financial support from Concordia University's doctoral incentive fellowship.References
- B. Chen and F. Wang, Trends Chem., 2020, 2, 427–439 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- Y. Wang, K. Zheng, S. Song, D. Fan, H. Zhang and X. Liu, Chem. Soc. Rev., 2018, 47, 6473–6485 RSC.
- S. Wilhelm, M. Kaiser, C. Würth, J. Heiland, C. Carrillo-Carrion, V. Muhr, O. S. Wolfbeis, W. J. Parak, U. Resch-Genger and T. Hirsch, Nanoscale, 2015, 7, 1403–1410 RSC.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC.
- F. Lu, L. Yang, Y. Ding and J. J. Zhu, Adv. Funct. Mater., 2016, 26, 4778–4785 CrossRef CAS.
- P. Lederhose, Z. Chen, R. Müller, J. P. Blinco, S. Wu and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 12195–12199 CrossRef CAS PubMed.
- S. Shikha, T. Salafi, J. Cheng and Y. Zhang, Chem. Soc. Rev., 2017, 46, 7054–7093 RSC.
- H. Yang, C. Han, X. Zhu, Y. Liu, K. Y. Zhang, S. Liu, Q. Zhao, F. Li and W. Huang, Adv. Funct. Mater., 2016, 26, 1945–1953 CrossRef CAS.
- R. Naccache, F. Vetrone and J. A. Capobianco, ChemSusChem, 2013, 6, 1308–1311 CrossRef CAS PubMed.
- H. Liu, M. K. G. Jayakumar, K. Huang, Z. Wang, X. Zheng, H. Ågren and Y. Zhang, Nanoscale, 2017, 9, 1676–1686 RSC.
- S. Banerjee, J. Koerner, M. Siebold, Q. Yang, K. Ertel, P. D. Mason, P. J. Phillips, M. Loeser, H. Zhang, S. Lu, J. Hein, U. Schramm, M. C. Kaluza and J. L. Collier, Opt. Express, 2013, 21, A726–A724 CrossRef CAS PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- X. Wu, H. Lee, O. Bilsel, Y. Zhang, Z. Li, T. Chen, Y. Liu, C. Duan, J. Shen, A. Punjabi and G. Han, Nanoscale, 2015, 7, 18424–18428 RSC.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS PubMed.
- T. Liang, Q. Wang, Z. Li, P. Wang, J. Wu, M. Zuo and Z. Liu, Adv. Funct. Mater., 2020, 30, 1910765–1910773 CrossRef CAS.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- H. R. M. Silva, W. M. Faustino, E. E. S. Teotonio, H. F. Brito, O. L. Malta and M. C. F. C. Felinto, J. Lumin., 2018, 207, 182–187 CrossRef.
- X. Wang, R. R. Valiev, T. Y. Ohulchanskyy, H. Ågren, C. Yang and G. Chen, Chem. Soc. Rev., 2017, 46, 4150–4167 RSC.
- L. M. Wiesholler, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and T. Hirsch, Nanoscale, 2019, 11, 13440–13449 RSC.
- G. Bao, S. Wen, G. Lin, J. Yuan, J. Lin, K. L. Wong, J. C. G. Bünzli and D. Jin, Coord. Chem. Rev., 2020, 429, 213642–213672 CrossRef.
- D. J. Garfield, N. J. Borys, S. M. Hamed, N. A. Torquato, C. A. Tajon, B. Tian, B. Shevitski, E. S. Barnard, Y. D. Suh, S. Aloni, J. B. Neaton, E. M. Chan, B. E. Cohen and P. J. Schuck, Nat. Photonics, 2018, 12, 402–407 CrossRef CAS.
- G. Chen, W. Shao, R. R. Valiev, T. Y. Ohulchanskyy, G. S. He, H. Ågren and P. N. Prasad, Adv. Opt. Mater., 2016, 4, 1760–1766 CrossRef CAS.
- S. Hao, Y. Shang, D. Li, H. Ågren, C. Yang and G. Chen, Nanoscale, 2017, 9, 6711–6715 RSC.
- C. Hazra, S. Ullah, Y. E. S. Correales, L. G. Caetano and S. J. L. Ribeiro, J. Mater. Chem. C, 2018, 6, 4777–4785 RSC.
- S. Wang, L. Liu, Y. Fan, A. M. El-Toni, M. S. Alhoshan, D. Li and F. Zhang, Nano Lett., 2019, 19, 2418–2427 CrossRef CAS PubMed.
- R. Lv, D. Wang, L. Xiao, G. Chen, J. Xia and P. N. Prasad, Sci. Rep., 2017, 7, 15753–15764 CrossRef PubMed.
- S. L. Lin, Z. R. Chen and C. A. Chang, Nanotheranostics, 2018, 2, 243–257 CrossRef PubMed.
- M. Zhao, B. Li, Y. Wu, H. He, X. Zhu, H. Zhang, C. Dou, L. Feng, Y. Fan and F. Zhang, Adv. Mater., 2020, 32, 2001172–2001179 CrossRef CAS PubMed.
- Q. Chen, C. Wang, L. Cheng, W. He, Z. Cheng and Z. Liu, Biomaterials, 2014, 35, 2915–2923 CrossRef CAS PubMed.
- V. Gubala, G. Giovannini, F. Kunc, M. P. Monopoli and C. J. Moore, Cancer Nanotechnol., 2020, 11, 1–43 CrossRef CAS.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- L. Jiao, F. Song, B. Zhang, H. Ning, J. Cui and X. Peng, J. Mater. Chem. B, 2017, 5, 5278–5283 RSC.
- T. Ribeiro, S. Raja, A. S. Rodrigues, F. Fernandes, C. Baleizão and J. P. S. Farinha, Dyes Pigm., 2014, 110, 227–234 CrossRef CAS.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- X. Zhang, W. Chen, X. Xie, Y. Li, D. Chen, Z. Chao, C. Liu, H. Ma, Y. Liu and H. Ju, Angew. Chem., Int. Ed., 2019, 58, 12245–12250 CrossRef.
- S. Wilhelm, ACS Nano, 2017, 11, 10644–10653 CrossRef CAS PubMed.
- S. Lu, J. Ke, X. Li, D. Tu and X. Chen, Aggregate, 2021, 2, e59–e76 CAS.
- A. Samanta, M. Vendrell, R. Das and Y. T. Chang, Chem. Commun., 2010, 46, 7406–7408 RSC.
- E. Engel, R. Schraml, T. Maisch, K. Kobuch, B. Konig, R. M. Szeimies, J. Hillenkamp, W. Baümler and R. Vasold, Invest. Ophthalmol. Visual Sci., 2008, 49, 1777–1783 CrossRef PubMed.
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Org. Biomol. Chem., 2015, 13, 7584–7598 RSC.
- R. R. Nani, J. A. Kelley, J. Ivanic and M. J. Schnermann, Chem. Sci., 2015, 6, 6556–6563 RSC.
- D. Wang, D. Wang, A. Kuzmin, A. Pliss, W. Shao, J. Xia, J. Qu and P. N. Prasad, Adv. Opt. Mater., 2018, 6, 1701142–1701151 CrossRef.
- Z. Liu, F. Ren, H. Zhang, Q. Yuan, Z. Jiang, H. Liu, Q. Sun and Z. Li, Biomaterials, 2019, 219, 119364–119375 CrossRef CAS PubMed.
- M. Kaur, G. A. Mandl, S. L. Maurizio, G. Tessitore and J. A. Capobianco, Nanoscale Adv., 2022, 4, 608–618 RSC.
- D. H. Li, C. L. Schreiber and B. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 12154–12161 CrossRef CAS PubMed.
- J. Cha, R. R. Nani, M. P. Luciano, G. Kline, A. Broch, K. Kim, J. M. Namgoong, R. A. Kulkarni, J. L. Meier, P. Kim and M. J. Schnermann, Bioorg. Med. Chem. Lett., 2018, 28, 2741–2745 CrossRef CAS PubMed.
- L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Org. Biomol. Chem., 2020, 18, 9385–9397 RSC.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- A. Li, T. Guan and Z. Sun, EPL, 2014, 106, 48001–48008 CrossRef.
- A. Aebischer, S. Heer, D. Biner, K. Krämer, M. Haase and H. U. Güdel, Chem. Phys. Lett., 2005, 407, 124–128 CrossRef CAS.
- H. Hu, L. Xiong, J. Zhou, F. Li, T. Cao and C. Huang, Chem. – Eur. J., 2009, 15, 3577–3584 CrossRef CAS PubMed.
- F. Zhao, D. Yin, C. Wu, B. Liu, T. Chen, M. Guo, K. Huang, Z. Chen and Y. Zhang, Dalton Trans., 2017, 46, 16180–16189 RSC.
- I. Miletto, E. Bottinelli, G. Caputo, S. Coluccia and E. Gianotti, Phys. Chem. Chem. Phys., 2012, 14, 10015–10021 RSC.
- G. Battistelli, A. Cantelli, G. Guidetti, J. Manzi and M. Montalti, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 139–151 CAS.
- R. I. Nooney, C. M. N. McCahey, O. Stranik, X. Le Guevel, C. McDonagh and B. D. MacCraith, Anal. Bioanal. Chem., 2009, 393, 1143–1149 CrossRef CAS PubMed.
- D. R. Larson, H. Ow, H. D. Vishwasrao, A. A. Heikal, U. Wiesner and W. W. Webb, Chem. Mater., 2008, 20, 2677–2684 CrossRef CAS.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesnert, Nano Lett., 2005, 5, 113–117 CrossRef CAS PubMed.
- X. Xie, Z. Li, Y. Zhang, S. Guo, A. I. Pendharkar, M. Lu, L. Huang, W. Huang and G. Han, Small, 2017, 13, 1602843–1602858 CrossRef PubMed.
- N. Song, B. Zhou, L. Yan, J. Huang and Q. Zhang, Front. Chem., 2019, 6, 1–8 RSC.
- B. M. Walsh, J. M. McMahon, W. C. Edwards, N. P. Barnes, R. W. Equall and R. L. Hutcheson, J. Opt. Soc. Am. B, 2002, 19, 2893–2903 CrossRef CAS.
- L. Tian, Z. Xu, S. Zhao, Y. Cui, Z. Liang, J. Zhang and X. Xu, Materials, 2014, 7, 7289–7303 CrossRef PubMed.
- W. Y. D. Yong, Z. Zhang, G. Cristobal and W. S. Chin, Colloids Surf., A, 2014, 460, 151–157 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- E. van der Kolk, P. Dorenbos, K. Krämer, D. Biner and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125110 CrossRef.
- X. Li, R. Wang, F. Zhang, L. Zhou, D. Shen, C. Yao and D. Zhao, Sci. Rep., 2013, 3, 3536–3543 CrossRef PubMed.
- S. Ullah, E. P. Ferreira-Neto, C. Hazra, R. Parveen, H. D. Rojas-Mantilla, M. L. Calegaro, Y. E. Serge-Correales, U. P. Rodrigues-Filho and S. J. L. Ribeiro, Appl. Catal., B, 2019, 243, 121–135 CrossRef CAS.
- L. Tan, D. Li, L. Zhang, L. Xu, Y. Zhao, L. Zhu and R. Qiao, J. Phys. Chem. C, 2020, 124, 18081–18090 CrossRef CAS.
- X. Han, L. Zhou, H. Zhuang, P. Wei, F. Li, L. Jiang and T. Yi, ACS Appl. Mater. Interfaces, 2022, 14, 18031–18042 CrossRef CAS PubMed.
- L. Liang, L. Cheng, Y. Zhang, Q. Wang, Q. Wu, Y. Xue and X. Meng, RSC Adv., 2020, 10, 28509–28515 RSC.
- M. Zhu, Y. Wang, D. Meng, X. Qin and G. Diao, J. Phys. Chem. C, 2012, 116, 16276–16285 CrossRef CAS.
- B. Ding, S. Shao, C. Yu, B. Teng, M. Wang, Z. Cheng, K. L. Wong, P. Ma and J. Lin, Adv. Mater., 2018, 30, 1802479–1802489 CrossRef PubMed.
- Y. Dai, D. Yang, D. Yu, S. Xie, B. Wang, J. Bu, B. Shen, W. Feng and F. Li, Nanoscale, 2020, 12, 5075–5083 RSC.
- C. Xu, C. Lei, Y. Wang and C. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112752–e202112774 CAS.
- A. Nsubuga, G. A. Mandl and J. A. Capobianco, Nanoscale Adv., 2021, 3, 1375–1381 RSC.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- K. M. Schaich and D. C. Borg, Lipids, 1988, 23, 570–579 CrossRef CAS PubMed.
- S. Frindy and M. Sillanpää, Mater. Des., 2020, 188, 108461–108473 CrossRef CAS.
- S. Liang, Y. Chang, Y. Wang, D. Zhang and X. Pu, Catal. Sci. Technol., 2019, 9, 2103–2110 RSC.
- W. C. Oh, S. Ye, J. G. Park, S. H. Kang and C. S. Lim, J. Ind. Eng. Chem., 2015, 32, 225–232 CrossRef CAS.
- L. Strekowski, C. J. Mason, H. Lee, R. Gupta, J. Sowell and G. Patonay, J. Heterocycl. Chem., 2003, 40, 913–916 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02845c |
| This journal is © The Royal Society of Chemistry 2023 |