
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous processing of organic semiconductors enabled by stable nanoparticles with built-in surfactants†
Raymundo
Marcial-Hernandez
 *a,
Sofia
Giacalone
a,
William G.
Neal
a,
Chang-Seuk
Lee
a,
Peter A.
Gilhooly-Finn
a,
Giulia
Mastroianni
b,
Dilara
Meli
c,
Ruiheng
Wu
d,
Jonathan
Rivnay
ef,
Matteo
Palma
a and
Christian B.
Nielsen
*a
*a,
Sofia
Giacalone
a,
William G.
Neal
a,
Chang-Seuk
Lee
a,
Peter A.
Gilhooly-Finn
a,
Giulia
Mastroianni
b,
Dilara
Meli
c,
Ruiheng
Wu
d,
Jonathan
Rivnay
ef,
Matteo
Palma
a and
Christian B.
Nielsen
*a
aDepartment of Chemistry, Queen Mary University of London, Mile End Road, London E1 4NS, UK. E-mail: r.hernandez@qmul.ac.uk; c.b.nielsen@qmul.ac.uk
bSchool of Biological and Behavioural Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, UK
cDepartment of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA
dDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA
eDepartment of Biomedical Engineering, Northwestern University, Evanston, Illinois 60208, USA
fSimpson Querrey Institute, Northwestern University, Chicago, Illinois 60611, USA
First published on 17th March 2023
Abstract
The introduction of oligoether side chains onto a polymer backbone can help to stabilise polymeric dispersions in water without the necessity of surfactants or additives when conjugated polymer nanoparticles are prepared. A series of poly(3-hexylthiophene) (P3HT) derivatives with different content of a polar thiophene derivative 3-((2-methoxyethoxy)methyl)thiophene was interrogated to find the effect of the polar chains on the stability of the formed nanoparticles, as well as their structural, optical, electrochemical, and electrical properties. Findings indicated that incorporation of 10–20 percent of the polar side chain led to particles that are stable over a period of 42 days, with constant particle size and polydispersity, however the particles from the polymer with 30 percent polar side chain showed aggregation effects. The polymer dispersions showed a stronger solid-like behaviour in water with decreasing polar side chain content, while thin film deposition from water was found to afford globular morphologies and crystallites with more isotropic orientation compared to conventional solution-processed films. As a proof-of-principle, field-effect transistors were fabricated directly from the aqueous dispersions demonstrating that polymers with hydrophilic moieties can be processed in water without the requirement of surfactants.
Introduction
Organic semiconductors (OSCs), which can be divided into small molecules1 and conjugated polymers, are widely investigated for their potential applications in a variety of research fields, including solar energy harvesting,2,3 display technologies,4,5 thermoelectric devices,6,7 bioelectronic systems,8,9 chemical sensors,1,10,11 thin-film transistors12–14 and as bioimaging probes.15,16 In the field of electronic manufacturing, OSCs hold an advantage over their inorganic counterparts because of their mechanical properties (flexibility, stretchability), relatively low cost, tuneability via chemical synthesis, and their solubility in organic solvents for their controlled processability into devices using scalable methods for industrial manufacture.17 The most common solvents used to process OSCs during the fabrication of organic electronic devices are mainly chlorinated and highly volatile solvents.18 These solvents are toxic, harmful for the environment and suspected of causing cancer,19 because of these reasons of concern, investigations into developing “greener” processing alternatives have increased in recent years.18Polymeric dispersions in aqueous media are an attractive alternative to avoid the use of halogenated solvents, especially if these dispersions can be processed using the same solution processing methods employed for conventional semiconductor inks.3,20 In a few cases, polymer dispersions have already been widely applied in the fabrication of thin-film organic electronics, for example in the case of poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS), where PSS works as a counter ion stabilising the dispersion.21,22 Conjugated polymer nanoparticles (CPNs) are polymeric dispersions typically used in bioimaging applications.16,23,24 These CPNs are a good alternative to dyes because often the latter are employed in silica matrices prone to colour leaching. The use of CPNs can overcome the leaching because the polymer itself is the probe and does not require an external matrix.25 The intrinsic hydrophobic nature of most OSCs require the use of surfactants to stabilise the aqueous dispersions and prevent agglomeration, and in some cases, the aid of co-stabilisers is necessary to generate the corresponding dispersion with appropriate particle uniformity and stability. Furthermore, the polymer solubility in a given solvent used for creating the dispersion determines the final polymer loading in the dispersion26 which is important for the subsequent solution processing step.
Surfactants and other additives can also act as charge carrier traps in the semiconductor layer which is one of the main reasons to avoid the use of CPNs to fabricate organic electronic devices along with the low polymeric content often achieved with dispersions.20,27 To avoid the use of surfactants, OSCs can be chemically modified by the incorporation of hydrophilic side chains that facilitate solubility in polar solvents and formation of the corresponding aqueous dispersion.9,28–31 Although these polymers have proven their potential as probes for biomarkers,32 and have overcome the use of surfactants, the implementation of CPNs in organic electronics has been limited due to the low content of the polymer in the corresponding dispersion.33 These polymer dispersions can be prepared using several methodologies, for example, nanoprecipitation and miniemulsion.34 Nanoprecipitation methodology requires the use of a water-miscible organic solvent that usually carries the polymer, once the addition of the polymer solution into the aqueous phase is made, the organic solvent is evaporated and the polymer is precipitated in small particles.35 Miniemulsion is a method where two non-miscible solvents form a stable emulsion using high energy methods, usually ultrasonication. Both emulsion methodologies can either use surfactants and co-stabilisers or be surfactant-free if the polymer contains the corresponding hydrophilic moieties.34 Herein, we have studied a series of poly(3-hexylthiophene) (P3HT) derivatives with different content of a polar thiophene derivative 3-((2-methoxyethoxy)methyl)thiophene incorporated in a random fashion to give statistical copolymers with varying ratios of hydrophilic and hydrophobic side chains (see Fig. 1). The emulsification of the polymers was systematically studied to generate a protocol for the corresponding polymer dispersions, along with the characterisation of their structural, optical, and electrochemical properties. Thin films were prepared via spray-coating from aqueous dispersion and compared to their conventionally solution-processed counterparts and studied in detail to assess their suitability for the fabrication of organic electronic devices exemplified by an organic field-effect transistor (OFET). Our work shows that the control of polar side chain content in P3HT based polymers can be used to tune the applicability of CPNs for organic electronics and that relatively short hydrophilic motifs can enable stable and surfactant-free semiconductor dispersions.
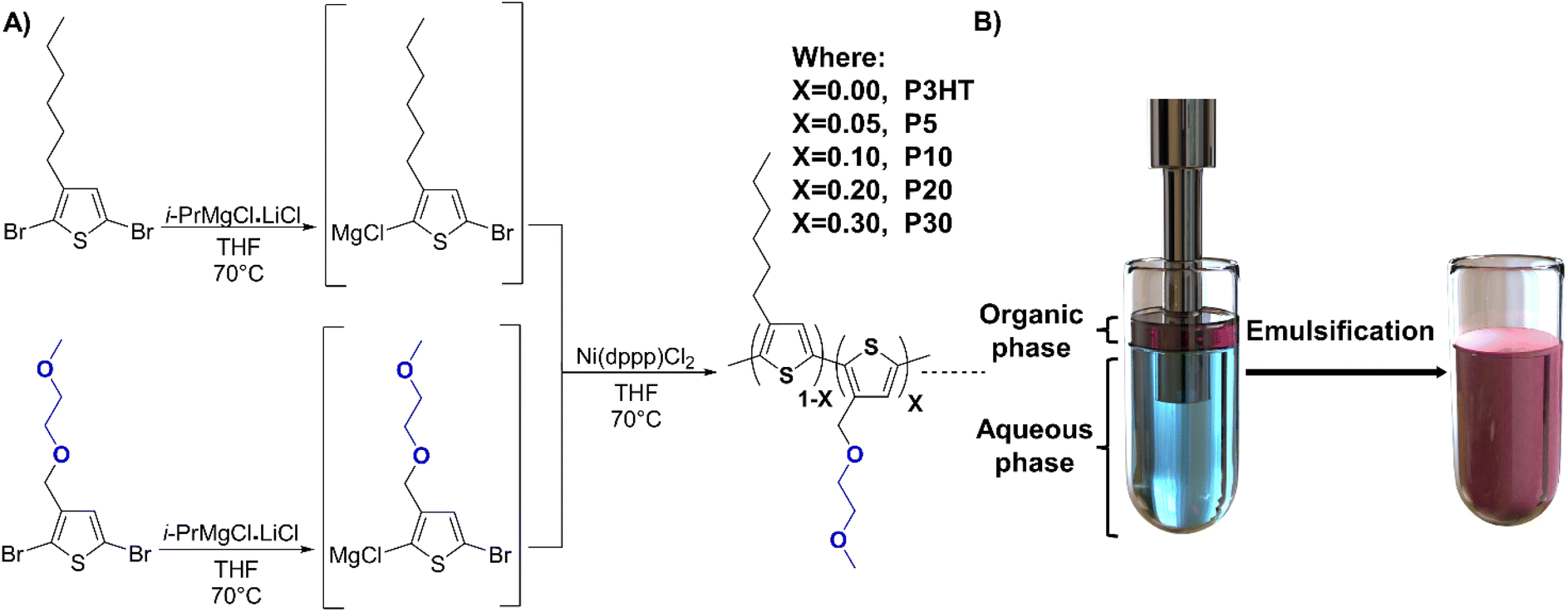 | ||
| Fig. 1 (A) Schematic representation of the random copolymer synthesis. (B) Representation of the ultrasonication process to generate the corresponding surfactant-free polymer dispersion. | ||
Results and discussion
Polymer synthesis
The statistical thiophene copolymers used in this study were synthesised according to the literature using the Grignard metathesis method.36,37 The procedure started with the synthesis of 2,5-dibromo-3-hexylthiophene and 2,5-dibromo-3-((2-methoxyethoxy)methyl)thiophene.37 Following lithiation of the corresponding brominated monomers to generate the magnesium chloride complexes via transmetallation, use of Ni(dppp)Cl2 as catalyst lead to the generation of the corresponding copolymers by controlling the ratio between the two thiophene species. Using molar ratios of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 90:10, 80:20 and 70:30 between the reactive 3-hexylthiophene and 3-((2-methoxyethoxy)methyl)thiophene species, we obtained polymers P5, P10, P20, and P30, respectively, where the number denotes the hydrophilic side chain content in percent (see Fig. 1). The crude polymers were purified by Soxhlet extraction using methanol, acetone, and hexane after which chloroform extraction afforded P5 (76% yield), P10 (77% yield), P20 (75% yield) and P30 (55% yield), respectively. The use of metal scavenging agents was omitted because most soluble salts are removed during the methanol and acetone washing steps, and the insoluble salts remain in the Soxhlet thimble, as suggested in the literature.38 Further details of the synthesis can be found in the ESI.† The obtained polymers were characterised via nuclear magnetic resonance (NMR) spectroscopy to confirm the ratio between the polar and the non-polar thiophene moieties (see Fig. S1†). The molecular weights obtained by gel-permeation chromatography (GPC) were similar to the previously reported ones, albeit with considerably lower molecular weight for the polymer P30 as detailed in Table 1.
:5, 90:10, 80:20 and 70:30 between the reactive 3-hexylthiophene and 3-((2-methoxyethoxy)methyl)thiophene species, we obtained polymers P5, P10, P20, and P30, respectively, where the number denotes the hydrophilic side chain content in percent (see Fig. 1). The crude polymers were purified by Soxhlet extraction using methanol, acetone, and hexane after which chloroform extraction afforded P5 (76% yield), P10 (77% yield), P20 (75% yield) and P30 (55% yield), respectively. The use of metal scavenging agents was omitted because most soluble salts are removed during the methanol and acetone washing steps, and the insoluble salts remain in the Soxhlet thimble, as suggested in the literature.38 Further details of the synthesis can be found in the ESI.† The obtained polymers were characterised via nuclear magnetic resonance (NMR) spectroscopy to confirm the ratio between the polar and the non-polar thiophene moieties (see Fig. S1†). The molecular weights obtained by gel-permeation chromatography (GPC) were similar to the previously reported ones, albeit with considerably lower molecular weight for the polymer P30 as detailed in Table 1.
| Polymer | M n (kDa) | Đ M | λ max solution | λ max thin film | λ max aqueous dispersion | λ max spray-coated film |
|---|---|---|---|---|---|---|
| Molecular weights were determined by GPC in chlorobenzene at 80 °C. Solution UV-vis spectra were recorded in chloroform from stock solutions (1 mg mL−1), thin films were spin-cast from polymer solutions in chloroform (5 mg mL−1). Aqueous dispersions were recorded in DI water and deposited onto glass substrates via spray-coating. | ||||||
| P3HT | 45 | 1.4 | 448 | 526 | — | — |
| P10 | 39 | 1.4 | 446 | 516 | 509 | 505 |
| P20 | 41 | 1.4 | 442 | 510 | 497 | 508 |
| P30 | 13 | 1.6 | 446 | 516 | 480 | 492 |
Preparation of polymer dispersions
The solubility of the polymers was investigated to determine the optimal solvent to employ during the dispersion preparation. The solubility in toluene was limited for all polymers and decreased when the polar side chain content increased, meanwhile the opposite effect was observed with chloroform. Lastly tetrahydrofuran (THF) was tested as solvent, affording a good solubility for all polymers. As THF is miscible with water it could also aid in the formation of the corresponding dispersions. Therefore, THF was selected as the organic phase to prepare the polymer dispersions. First, the polymer solutions in degassed THF were prepared, using a polymer loading of 10 mg mL−1 for a consistent analysis. The polymer solution was subsequently added to 10 mL of degassed deionised (DI) water. The mixture was placed in an ice bath and sonicated with an ultrasonic homogenizer using a 6 mm probe at 50 watts of power with a programmed time lapse of 4 minutes with intervals of 4 seconds of sonication and pause (see Fig. 1). The 6 mm probe diameter was selected according to fabricant suggestions when volumes of 10–100 mL are sonicated. Lower power rates than 50 watts afforded non-successful emulsifications, and the intervals of 4 seconds were selected to avoid a rapid increase of temperature during sonication. A non-controlled increase in temperature during the sonication could lead to premature precipitation of the polymer samples due to early evaporation of the organic phase. The formed emulsions were stirred at 50 °C under nitrogen to allow for the evaporation of the residual organic solvent. The resulting dispersions were filtered using 1.0 μm glass filters to remove precipitated polymer or impurities like dust. No further purification process is required for these surfactant-free polymer dispersions, compared with dispersions prepared using surfactants and co-stabilisers that often require the use of techniques like dialysis or precipitation to remove the excess of surfactants and other materials.Polymer dispersions were successfully prepared using P10, P20, and P30 polymers, on the other hand, P3HT and P5 precipitated during the sonication. The actual concentration of the polymer dispersions can differ from the expected ones based on loading, because of the unavoidable precipitation during the sonication and the evaporation of the organic phase. Therefore, to establish the concentration of each dispersion, a calibration curve of absorbance at the wavelength of maximum absorbance versus known concentration was generated from reference emulsions prepared using a surfactant (Fig. S5†). The dispersions used for the calibration curves were prepared by dissolving 10 mg of each polymer (P10, P20, and P30) in 1 mL of degassed THF, after which the solution was injected into a vial containing 10 mL of a sodium dodecyl sulphate (SDS) aqueous solution (7.5 mg mL−1). The mixture was sonicated using the same parameters as for the surfactant-free emulsification, and then the organic phase was evaporated.
Particle size and stability over time
The hydrodynamic diameter or particle size and polydispersity of each formed polymeric dispersion was investigated using dynamic light scattering (DLS) measurements. Aliquots of 5 μL of each aqueous dispersion were diluted in 1 mL of DI water prior to the DLS measurements which were carried out at 25 °C. The presented particle sizes and polydispersities (PDI) are average results of three measurements. As illustrated in Fig. 2, we observe that the particle size of the obtained dispersions decreases with increasing polar content in the polymers; 140 nm, 111 nm, and 64.8 nm for P30, P20, and P10, respectively, with a polymer loading of 10 mg mL−1. The PDI values, on the other hand, do not follow the same trend being 0.31, 0.16, and 0.22 for P30, P20, and P10, respectively.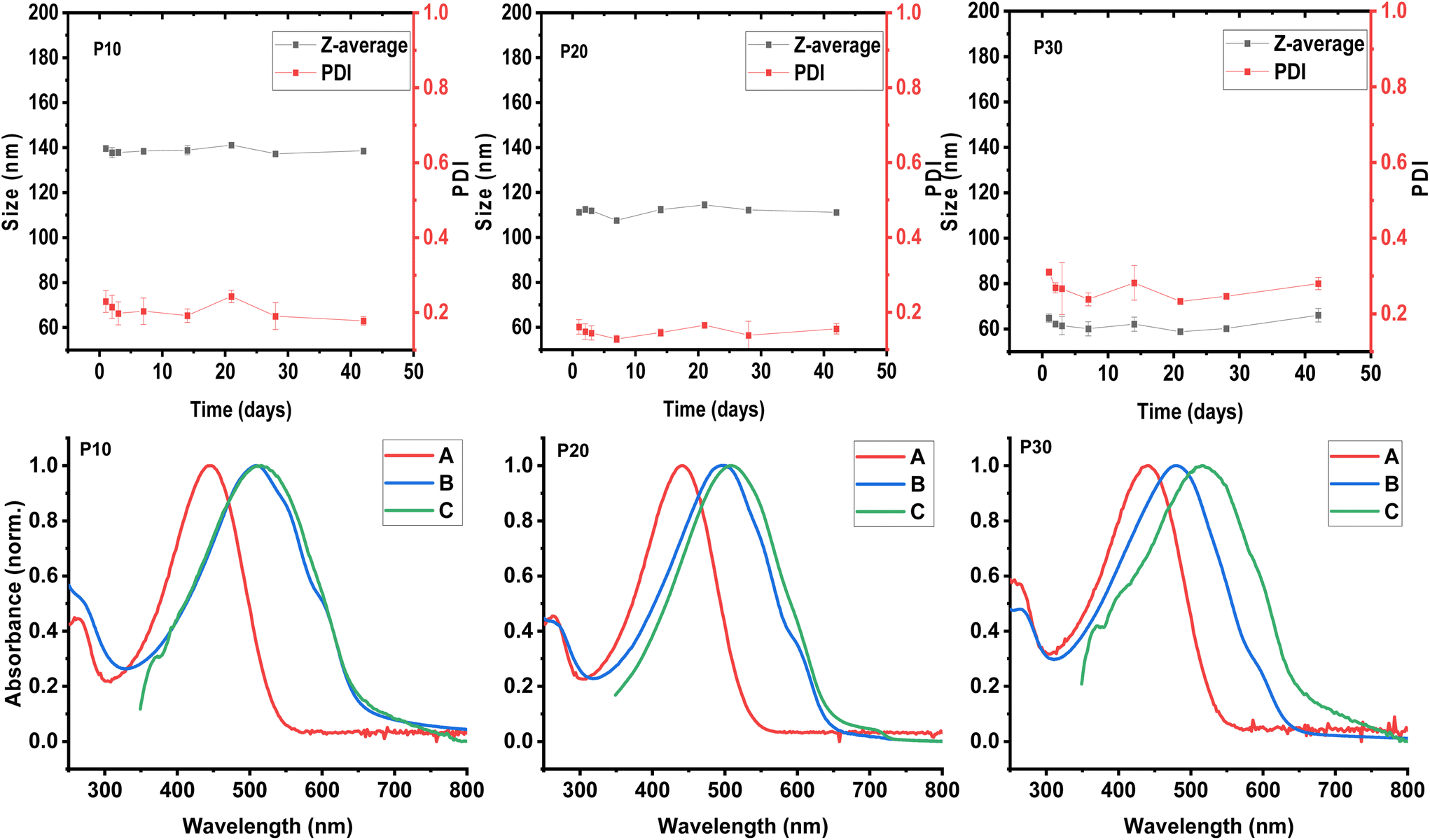 | ||
| Fig. 2 (Top) Analysis of particle stability over time via DLS measurements from P10 (left), P20 (centre), and P30 (right) dispersions in water. (Bottom) UV-vis spectra of (A) polymer solutions in chloroform, (B) polymer dispersions in water and (C) thin films spin-cast from chloroform solutions, for P10 (left), P20 (centre), and P30 (right). | ||
For a better understanding of the effects of the polar side chain content of the copolymers in the formed dispersions, the particle size and polydispersity were systematically studied over time for each formed dispersion. After 42 days for the three systems, no significant change in particle size was observed (see Fig. 2). Although the polydispersity was constant for P10 and P20, the P30 dispersion showed some variation over time with values above 0.3. These values represent the distribution of size populations of the corresponding sample. To the best of our knowledge, the influence of PDI of polymer nanoparticles have not been studied in the context of organic electronic applications. The particle size distribution may affect the uniformity of the generated polymer thin film, with broader distributions affording less uniform film morphologies to the detriment of electronic properties. In the case of the P30 dispersion, an increase in PDI value from ∼0.25 to values approaching 0.3 was observed during time, although the particle size remained relatively constant. The variation in PDI indicates that the nanoparticle populations are changing, probably due to the adherence between particles, also known as aggregation or flocculation.39 The aggregation of particles could lead to an eventual precipitation of the polymeric material; this is supported by the bimodal distribution observed in the particle size distribution measurement by intensity (see Fig. S2†), indicating at least two different populations of particles.
Optical and electrochemical properties
The aqueous polymeric dispersions were characterised by UV-vis spectroscopy to probe the solid-like behaviour that is typically observed in CPNs. Firstly, the solution and solid-state UV-vis spectra were recorded for each copolymer (P5, P10, P20, and P30) as well as hydrophobic and hydrophilic homopolymers P3HT and P3MEMT as reference (see Fig. S3†). In accordance with the literature, the polymer series show a gradual blue-shift in the absorption maximum (λmax) with increasing polar content both in solution and for thin films spin-cast from chloroform solutions (5 mg mL−1) onto glass substrates. In the solid state, the spectra of P3HT and P5 displayed a clear shoulder around 595 nm; this shoulder is ascribed to the 0–0 vibronic transition and decreases in intensity with increasing polar content and completely vanishes for P3MEMT, suggesting increasing structural disorder in the thin films with higher polar content, as also reported previously.37The UV-vis spectra for the aqueous CPN dispersions were prepared by taking aliquots of each dispersion and diluting in DI water. The λmax of these spectra followed the blue-shift trend that was observed for solution and thin-film spectra with λmax values of 509 nm, 497 nm, and 480 nm for P10, P20 and P30 respectively (see Fig. 2). The UV-vis spectrum of the P10 dispersion shows two low-energy shoulders at 591 and 543 nm, suggesting an increase in the interactions of π-orbitals (vibronic coupling) indicative of a higher degree of polymer backbone planarity. The λmax of the P10 dispersion shows a minor blue-shift of 6 nm relative to the thin film processed from chloroform solution and a red-shift of 65 nm when compared with the chloroform solution spectrum, corroborating the solid-like behaviour of the polymer nanoparticles dispersed in water. The spectrum of the P20 dispersion shows a defined shoulder at 596 nm from the 0–0 vibronic transition; the λmax represents a blue-shift of 12 nm when compared to the λmax of the corresponding thin film processed from chloroform solution, and a red shift of 57 nm when compared to the chloroform solution. The spectrum of the P30 dispersion, on the other hand, shows a small shoulder at 590 nm indicating some degree of planarity, however, the λmax is blue-shifted 35 nm when compared to the solid-state spectrum and red-shifted 40 nm when compared to the solution spectra. It is noticeable how the solid-like behaviour of the polymeric dispersions decreases with increasing polar content, which could be related to a better interaction with the aqueous medium and/or the previously observed increase in disorder of the polymer when the polar content increases.
The previously mentioned calibration curves for the reference dispersions using SDS as surfactant was generated by taking aliquots of 20 μL and measured by UV-vis spectroscopy, subsequently the linear fitting obtained from the absorbance at λmax at different concentrations allowed the determination of the corresponding molar extinction coefficient (εm) of each polymer. Assuming a neglectable effect of the surfactant on the absorption, the concentrations of the surfactant-free dispersions were determined by applying the Beer–Lambert law (see Fig. S5†).
The deposition of thin films to study the potential application of these CNP dispersions to fabricate organic electronics was subsequently investigated. Early attempts to generate thin films from the polymeric dispersions were unsuccessful due to the low concentration when spin-coating and slot-die coating were tested. This led us to explore the spray-coating technique to generate the corresponding thin-films; parameters like the spray distance and substrate temperature were optimised to generate the corresponding thin films. The ideal parameters found to deposit a thin layer are: 15 cm of distance from the tip nozzle to the substrate which was heated at 90 °C, with a slow spray rate of 0.5 mL of each dispersion.
The thin films obtained via spray-coating from the polymer dispersions showed λmax values of 505 nm, 508 nm, and 492 nm for P10, P20, and P30, respectively. These values represent a blue-shift when compared with the thin films prepared by spin-coating from organic solutions as depicted in Fig. 3. The spectra for the thin films obtained from the P10 and P20 dispersions show a similar pattern; in both cases the presence of two shoulders at 550 nm and 594 nm was observed, the shoulders are better defined for the films obtained from the dispersions than for those obtained from chloroform solution, indicating stronger intermolecular interactions for the former. The film obtained from the P30 dispersion displays a larger blue-shift of over 20 nm compared to the spin-cast film and a less-pronounced low-energy shoulder, suggesting a more disordered structure for the P30 spray-coated film compared to the film spin-cast from solution.
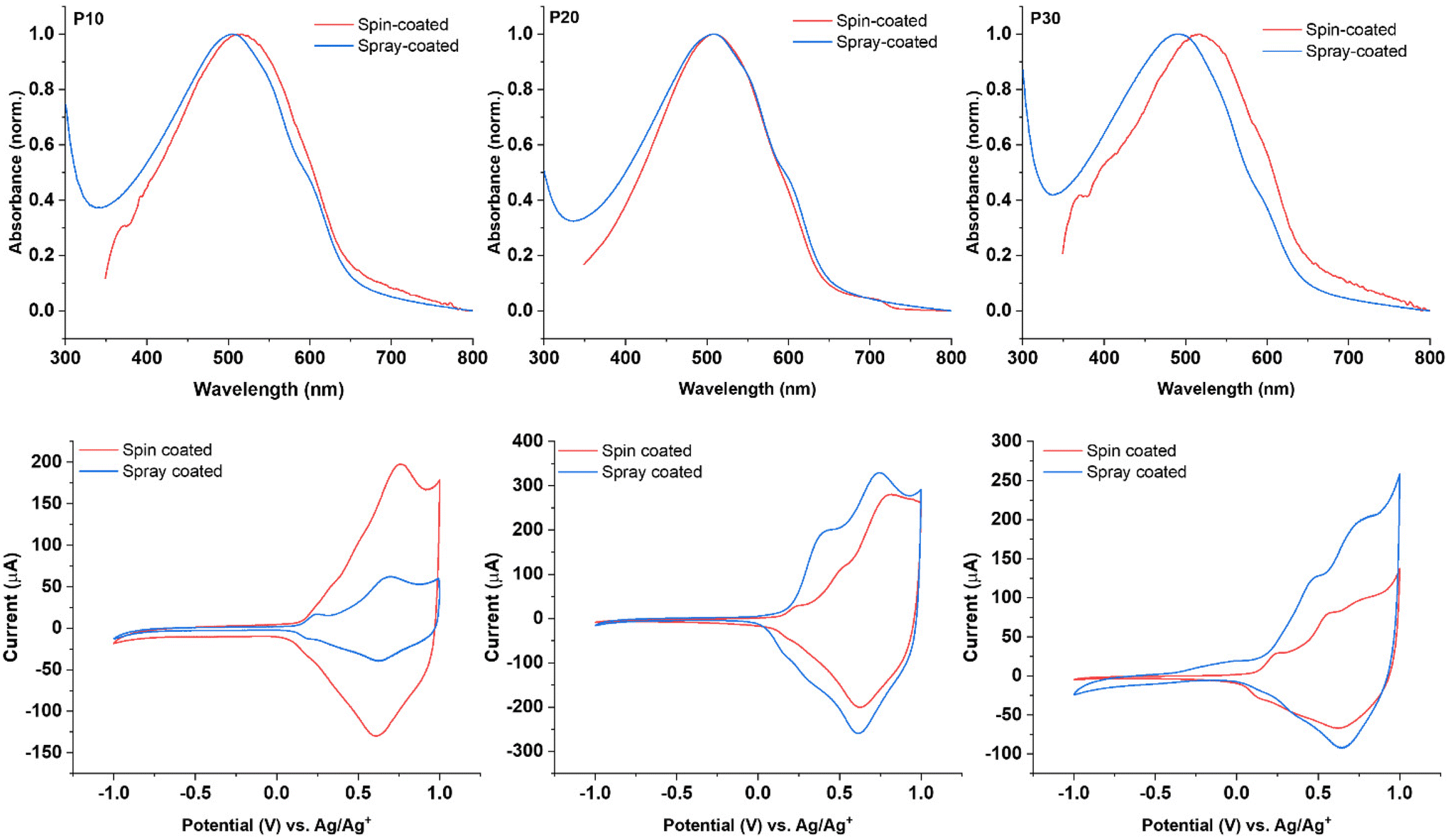 | ||
| Fig. 3 (Top) Solid state UV-vis spectroscopy of thin films deposited via spin-coating from organic solutions in chloroform (5 mg mL−1) and via spray-coating from polymer dispersions (1 mg mL−1). (Bottom) Cyclic voltammograms for thin films deposited onto indium tin oxide (ITO) coated glass via spin-coating from organic solutions in chloroform (5 mg mL−1) and via spray-coating from polymer dispersions (1 mg mL−1) in 0.1 M solution of tetrabutylammonium hexafluorophosphate in acetonitrile at a scan rate of 100 mV s−1, plots correspond to the second scan of each measurement of P10 (left), P20 (centre), and P30 (right). | ||
The electrochemical properties of the thin films obtained from the polymer dispersions were examined by cyclic voltammetry (CV) and compared to the corresponding polymer films spin-cast from solution. All films showed a reversible oxidative process irrespective of deposition technique with comparable onsets of oxidation when comparing the films deposited from solution and dispersion for each polymer (see Fig. 3). These results are in accordance with the literature,37 demonstrating that the polymer films deposited from dispersion broadly retain the electrochemical properties when compared with common solution processing techniques.
Solid state structure and surface morphology
First attempts of deposition by spray-coating carried out with substrates at 65 °C led to non-uniform films due to the aggregation of the droplets before the water evaporation, however, the obtained films were characterised by atomic force microscopy (AFM) to determinate the surface morphology of the generated particles as illustrated in Fig. 4. The films obtained from the P10 and P20 dispersions showed a granular or globular morphology, derived from the spherical shape of the polymeric particles. The spherical morphology has been commonly reported when different materials undergo ultrasonication treatment. The film obtained from the P30 dispersion shows an amorphous surface morphology; although some spherical particles can be observed, the characteristic granular morphology observed for P10 and P20 is not prevailing. We hypothesise that this is related to the increased polar content, which can promote a better interaction with the water, leading to less packed and organised polymer domains in the generated particles. This more amorphous morphology for P30 is in good agreement with the blue-shifted absorption spectra discussed above.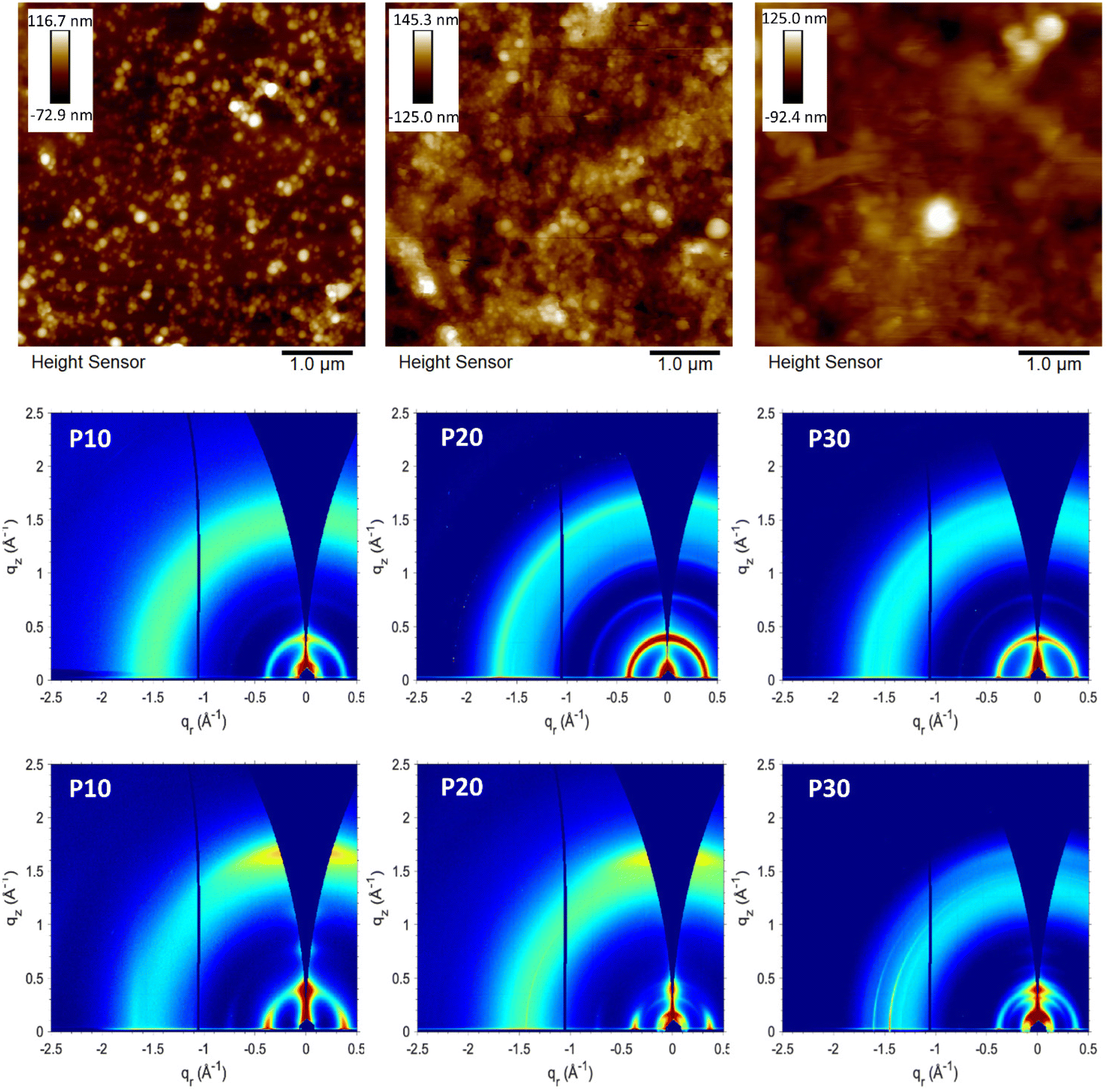 | ||
| Fig. 4 (Top) Atomic force micrographs of the polymer dispersions deposited via spray-coating onto glass substrates at 65 °C; P10 (left), P20 (centre), and P30 (right). Scale bars (insets) are in nm units. (Middle) 2D GIWAXS patterns of the polymer films deposited onto silicon substrates from aqueous dispersions via spray-coating at 90 °C. (Bottom) 2D GIWAXS patterns of the polymer films deposited onto silicon substrates from chloroform solutions via spin-coating. | ||
The films obtained when the dispersions were deposited onto substrates heated to 90 °C (Fig. S8†) presented a better large-area uniformity and showed the same nano- and microscale characteristics as the films deposited at 65 °C (Fig. 4 and Fig. S9†) with defined spherical particles for P10 and P20 with a globular morphology, and an amorphous morphology with some spherical particles for P30. We infer from the AFM data that the temperature of the substrate does not have a significant impact on the nanoscale morphology of the particles, however, as mentioned the higher temperature is required to avoid aggregation of the aqueous droplets during deposition and drying which will impair film uniformity. To further characterise the nanoparticles obtained, transmission electron microscopy (TEM) was carried out for each sample. Following density gradient centrifugation to remove larger aggregates formed during aging, the supernatants were collected and analysed by TEM and DLS. TEM images (Fig. S10†) confirmed the spherical morphology of the nanoparticles obtained, for P10 and P20 particle sizes between 40 and 110 nm where observed, whereas P30 showed particles ranging in size from 17 to 82 nm. Qualitative agreement with DLS measurements was confirmed by the obtained DLS particle sizes of 97 nm, 109 and 48 nm for P10, P20 and P30, respectively. For all samples, the formation of clusters that can lead to larger particles can be observed.
The crystalline domains of the polymer films were investigated with grazing incidence wide-angle X-ray scattering (GIWAXS) (see Fig. 4, Fig. S11 and S12†) and compared with the literature.37 Organic solutions and aqueous dispersions of the polymers were deposited via spin-coating and spray-coating respectively onto silicon substrates for the corresponding GIWAXS measurements. In general, all GIWAXS patterns displayed an in-plane d(100) lamellar peak regardless of the deposition method with little variation in the lamellar spacing (16–17 Å, see Table S1†). Compared to the films deposited by spin-coating from solution, the films deposited from the aqueous dispersions showed more distinct ring patterns indicative of an isotropic orientation of the crystallites relative to the substrate.40 Isotropic orientation was expected for the films from polymer dispersions if the particle shape is retained after deposition, because of the random orientation that the particles adopt during the deposition. This contrasts with the spin-coated films from solution that show a preferential edge-on orientation relative to the substrate with minor populations of face-on oriented crystallites. The AFM micrographs showed how the spherical or globular shape is retained for P10 and P20 films, explaining the isotropic behaviour observed in the GIWAXS patterns. Coherence lengths extracted from peak fitting of the lamellar (100) peaks were found to be 107 Å (∼6 repeat units), 144 Å (∼8 repeat units), and 114 Å (∼7 repeat units) for P10, P20, and P30, respectively, when spin-coated from chloroform solution, meanwhile values of 101 Å (∼6 repeat units), 133 Å (∼8 repeat units), and 159 Å (∼10 repeat units) are obtained for the corresponding spray-coated films from aqueous dispersion of P10, P20 and P30 respectively (see Table S1†). Apart from the more isotropic orientation of the crystallites in the films from the aqueous dispersions, as was expected, the GIWAXS data generally indicates that the solid-state packing is not markedly different when comparing the two deposition methods.
Finally, as a proof of concept, we investigated the possibility of fabricating organic electronic devices using these aqueous dispersions directly for the processing of the active layer. According to the literature,37 a decrease in electrical performance with increasing polar content was anticipated, hence, only P10 representing the lowest polar side chain content was tested in an organic field-effect transistor configuration as further detailed in the ESI (Fig. S13†). A bottom-gate bottom-contact OFET was fabricated by spray-coating the P10 aqueous dispersion at 90 °C onto a Si/SiO2 substrate with gold electrodes; to increase the wettability of the substrate, a 15-minute plasma treatment was performed. This un-optimised device was operational with an extracted hole mobility around 6 × 10−3 cm2 Vs−1. Albeit displaying lower charge carrier mobility compared with a reference device processed via spin-coating from a chloroform solution (5 × 10−2 cm2 Vs−1), this initial device work highlights the potential of this approach for fabricating active semiconductor layers via purely aqueous processing.
Conclusions
In summary, we have demonstrated that stable semiconducting polymer dispersions can be obtained by the introduction of non-ionic polar side chains onto the polymer backbone, here exemplified by an ethylene glycol motif and the well-studied poly(3-hexylthiophene) polymer. The presence of the polar side chains stabilises the aqueous dispersions without the use of surfactants or additives over a period of 42 days. Increasing the polar side chain content in the polymer can act in detriment to the dispersion stability as shown by an increase in polydispersity index over time for the P30 dispersions with 30% polar side chain content. The polymeric dispersions show a decrease in particle size with increasing polar content while blue-shifted absorption profiles likewise indicate a gradual loss of solid-state properties of the dispersions with increasing polar content. The dispersions were successfully deposited over different substrates via spray-coating at 90 °C. The isotropic distribution of crystallites observed by GIWAXS can be justified by the retention of the spherical shape of the particles once deposited, as corroborated by AFM and TEM measurements. Although a granular morphology was observed for P10 and P20 dispersions, P30 showed a more amorphous behaviour, indicating an aggregation effect taking place with higher polar side chain content. To assess the applicability of these aqueous dispersions for organic electronic device fabrication, the P10 dispersion with 10% polar side chain content was deposited onto a silicon substrate with patterned electrodes and characterised in a transistor configuration. A current response confirmed that the electrical properties are preserved, although the performance was lower when compared to devices where the semiconductor layer was spin-coated from organic solution.37 Further work to optimise the deposition process and subsequent post-processing steps such as thermal annealing is likely to bridge that gap in electrical performance. Moreover, the process could be particularly attractive in emerging fields such as organic thermoelectrics and bioelectronics where semiconducting polymers with polar side chain content are frequently observed to have much enhanced device performance compared to their non-polar counterparts.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge funding from the Engineering and Physical Sciences Research Council (EP/R032025/1) and the European Commission Horizon 2020 Future and Emerging Technologies (FET) project MITICS (964677). R. W. and J. R. acknowledge support from the National Science Foundation, NSF award DMR-1751308. C.-S. L. and M. P. acknowledge support from the Air Force Office of Scientific Research under award FA8655-21-1-7003. This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.References
- C. J. Kousseff, R. Halaksa, Z. S. Parr and C. B. Nielsen, Chem. Rev., 2022, 122, 4397–4419 CrossRef CAS PubMed.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- S. Yao, Z. Chen, F. Li, B. Xu, J. Song, L. Yan, G. Jin, S. Wen, C. Wang, B. Yang and W. Tian, ACS Appl. Mater. Interfaces, 2015, 7, 7146–7152 CrossRef CAS PubMed.
- C. Gu, A.-B. Jia, Y.-M. Zhang and S. X.-A. Zhang, Chem. Rev., 2022, 122, 14679–14721 CrossRef CAS PubMed.
- C.-C. Cheng, Y.-L. Chu, P.-H. Huang, Y.-C. Yen, C.-W. Chu, A. C. M. Yang, F.-H. Ko, J.-K. Chen and F.-C. Chang, J. Mater. Chem., 2012, 22, 18127–18131 RSC.
- S. Wang, G. Zuo, J. Kim and H. Sirringhaus, Prog. Polym. Sci., 2022, 129, 101548 CrossRef CAS.
- T. L. D. Tam and J. Xu, J. Mater. Chem. A, 2021, 9, 5149–5163 RSC.
- I. B. Dimov, M. Moser, G. G. Malliaras and I. McCulloch, Chem. Rev., 2022, 122, 4356–4396 CrossRef CAS PubMed.
- Z. S. Parr, R. B. Rashid, B. D. Paulsen, B. Poggi, E. Tan, M. Freeley, M. Palma, I. Abrahams, J. Rivnay and C. B. Nielsen, Adv. Electron. Mater., 2020, 6, 2000215 CrossRef CAS.
- A. Koklu, D. Ohayon, S. Wustoni, V. Druet, A. Saleh and S. Inal, Chem. Rev., 2022, 122, 4581–4635 CrossRef CAS PubMed.
- H. J. Kim, K. Perera, Z. Liang, B. Bowen, J. Mei and B. W. Boudouris, ACS Macro Lett., 2022, 11, 243–250 CrossRef CAS PubMed.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 CAS.
- P.-S. Lin, Y. Shoji, S. N. Afraj, M. Ueda, C.-H. Lin, S. Inagaki, T. Endo, S.-H. Tung, M.-C. Chen, C.-L. Liu and T. Higashihara, ACS Appl. Mater. Interfaces, 2021, 13, 31898–31909 CrossRef CAS PubMed.
- B. Kumar, B. K. Kaushik and Y. S. Negi, Polym. Rev., 2014, 54, 33–111 CrossRef CAS.
- T. F. Abelha, C. A. Dreiss, M. A. Green and L. A. Dailey, J. Mater. Chem. B, 2020, 8, 592–606 RSC.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- K. Kim, H. Yoo and E. K. Lee, Polymers, 2022, 14, 2960 CrossRef CAS PubMed.
- F. Campana, C. Kim, A. Marrocchi and L. Vaccaro, J. Mater. Chem. C, 2020, 8, 15027–15047 RSC.
- C. McDermott and J. J. A. Heffron, Int. J. Toxicol., 2013, 32, 136–145 CrossRef CAS PubMed.
- A. Rahmanudin, R. Marcial-Hernandez, A. Zamhuri, A. S. Walton, D. J. Tate, R. U. Khan, S. Aphichatpanichakul, A. B. Foster, S. Broll and M. L. Turner, Adv. Sci., 2020, 7, 2002010 CrossRef CAS PubMed.
- S. Carli, M. Di Lauro, M. Bianchi, M. Murgia, A. De Salvo, M. Prato, L. Fadiga and F. Biscarini, ACS Appl. Mater. Interfaces, 2020, 12, 29807–29817 CAS.
- X. Feng, X. Wang, M. Wang, S. Zhou, C. Dang, C. Zhang, Y. Chen and H. Qi, Chem. Eng. J., 2021, 418, 129533 CrossRef CAS.
- D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484–494 RSC.
- X. Lu, P. Yuan, W. Zhang, Q. Wu, X. Wang, M. Zhao, P. Sun, W. Huang and Q. Fan, Polym. Chem., 2018, 9, 3118–3126 RSC.
- Y. Braeken, S. Cheruku, A. Ethirajan and W. Maes, Materials, 2017, 10, 1420 CrossRef PubMed.
- R. Jenjob, T. Phakkeeree, F. Seidi, M. Theerasilp and D. Crespy, Macromol. Biosci., 2019, 19, 1900063 CrossRef PubMed.
- J. Cho, S. Yoon, K. M. Sim, Y. Jin Jeong, C. E. Park, S.-K. Kwon, Y.-H. Kim and D. S. Chung, Energy Environ. Sci., 2017, 10, 2324–2333 RSC.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- B. Schmatz, Z. Yuan, A. W. Lang, J. L. Hernandez, E. Reichmanis and J. R. Reynolds, ACS Cent. Sci., 2017, 3, 961–967 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- H. Duan, C. Guan, J. Xue, T. Malesky, Y. Luo, Y. Lin, Y. Qin and J. He, iScience, 2022, 25, 104220 CrossRef CAS PubMed.
- M. A. Chanu, S. Mondal, N. Zehra, A. S. Tanwar and P. K. Iyer, ACS Appl. Polym. Mater., 2022, 4, 3491–3497 CrossRef CAS.
- J. E. Millstone, D. F. J. Kavulak, C. H. Woo, T. W. Holcombe, E. J. Westling, A. L. Briseno, M. F. Toney and J. M. J. Fréchet, Langmuir, 2010, 26, 13056–13061 CrossRef CAS PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- P. A. Finn, I. E. Jacobs, J. Armitage, R. Wu, B. D. Paulsen, M. Freeley, M. Palma, J. Rivnay, H. Sirringhaus and C. B. Nielsen, J. Mater. Chem. C, 2020, 8, 16216–16223 RSC.
- J. H. Bannock, N. D. Treat, M. Chabinyc, N. Stingelin, M. Heeney and J. C. de Mello, Sci. Rep., 2016, 6, 23651 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, J. Am. Chem. Soc., 2006, 128, 968–975 CrossRef CAS PubMed.
- D. Liu, Y. Zhang and G. Li, J. Energy Chem., 2019, 35, 104–123 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06024h |
| This journal is © The Royal Society of Chemistry 2023 |