
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterplay of the mechanical and structural properties of DNA nanostructures determines their electrostatic interactions with lipid membranes†
Diana
Morzy
 a,
Cem
Tekin
a,
Vincenzo
Caroprese
a,
Roger
Rubio-Sánchez
bcd,
Lorenzo
Di Michele
bc and
Maartje M. C.
Bastings
*ae
a,
Cem
Tekin
a,
Vincenzo
Caroprese
a,
Roger
Rubio-Sánchez
bcd,
Lorenzo
Di Michele
bc and
Maartje M. C.
Bastings
*ae
aProgrammable Biomaterials Laboratory, Institute of Materials, School of Engineering, Ecole Polytechnique Fédérale Lausanne, Lausanne, 1015, Switzerland. E-mail: maartje.bastings@epfl.ch
bDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, London W12 0BZ, UK
cfabriCELL, Molecular Sciences Research Hub, Imperial College London, London W12 0BZ, UK
dCavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB3 0HE, UK
eInterfaculty Bioengineering Institute, School of Engineering, Ecole Polytechnique Fédérale Lausanne, Lausanne, 1015, Switzerland
First published on 16th January 2023
Abstract
Nucleic acids and lipids function in close proximity in biological processes, as well as in nanoengineered constructs for therapeutic applications. As both molecules carry a rich charge profile, and frequently coexist in complex ionic solutions, the electrostatics surely play a pivotal role in interactions between them. Here we discuss how each component of a DNA/ion/lipid system determines its electrostatic attachment. We examine membrane binding of a library of DNA molecules varying from nanoengineered DNA origami through plasmids to short DNA domains, demonstrating the interplay between the molecular structure of the nucleic acid and the phase of lipid bilayers. Furthermore, the magnitude of DNA/lipid interactions is tuned by varying the concentration of magnesium ions in the physiologically relevant range. Notably, we observe that the structural and mechanical properties of DNA are critical in determining its attachment to lipid bilayers and demonstrate that binding is correlated positively with the size, and negatively with the flexibility of the nucleic acid. The findings are utilized in a proof-of-concept comparison of membrane interactions of two DNA origami designs – potential nanotherapeutic platforms – showing how the results can have a direct impact on the choice of DNA geometry for biotechnological applications.
Introduction
Nucleic acids and lipids are crucial components of every cell, and are abundant in every biological environment. Many research endeavours focus on exploring the relationship between them. Primarily, interactions between DNA and lipids are crucial in biomedical constructs, with lipofection-based delivery of DNA vaccines1–3 standing out as a particularly significant example. In such formulations, nucleic acids form complexes with cationic lipids,4,5 although their interactions with natural lipid mixtures have also been observed. Specifically, it has been suggested that DNA/lipid binding plays a role in vivo, being responsible for the formation of a nuclear pore.6,7 Finally, the understanding of these interactions contributes to the development of novel biomaterials, particularly in DNA nanotechnology, aiming to construct DNA-based mimics of membrane proteins8–11 or active membrane signalling platforms.12,13DNA/lipid interactions are most frequently studied where DNA is chemically modified either with a hydrophobic moiety, ensuring its attachment to lipid bilayers,8,14–16 or by incorporating cationic lipids to provide electrostatic attraction with negatively-charged nucleic acids.4,17 Neither of these scenarios is representative of DNA and lipids found in biological environments. Therefore, a complete understanding of the emergence and relevance of interactions between nucleic acids and lipids is still lacking.
In particular, such interactions are strongly guided by electrostatic phenomena, as DNA carries a negative charge in the form of phosphates (PO4−), and the known library of cellular lipids displays a wide range of charge profiles: from lipids with negatively charged headgroups18 to zwitterionic species.19 The latter are the most frequent components of cell membranes,20 and more specifically phosphatidylcholines are a major building block of cellular lipid bilayers,20,21 carrying a positively-charged choline and a negatively-charged phosphate group – the latter is identical to the one found in the DNA backbone. Particularly in view of complex ionic compositions of physiological fluids,22–24 understanding the interplay between this network of charges is crucial for fully unravelling the mechanisms behind biological functions of cells, and implementing DNA-based nanomaterials in biomedical research.
When unmodified DNA and phospholipids are incubated together in the presence of divalent cations, e.g., Mg2+, the ions provide electrostatic bridges between phosphate groups, leading to DNA/lipid binding.25,26 This bridging phenomenon is based on a multivalent effect, where multiple connections (cation bridges) allow for a stable attachment. Such multivalent processes are often defined through the valency (number of bridges),27,28 and also flexibility of the binding pair.29,30 The latter in particular is a relevant parameter to study, in view of the wide range of mechanical properties exhibited by both native31 and engineered32 DNA nanostructures. By analysing the electrostatics-based DNA/lipid interactions in the context of multivalent processes, we realize that parameters related to all three components, i.e., lipids, DNA and bridging ions, are responsible for the formation and stability of the attachment.
In this work, we particularly focus on the nucleic acid structure, using the unique tunability of the DNA structural and mechanical properties. With initial microscopy observations, we identify two DNA parameters whose interplay is crucial in guiding the electrostatic interactions with lipids: its flexibility and size. We combine the results from confocal microscopy imaging and zeta (ζ) potential measurements to further understand the effects of each component on the interaction balance of lipids/ions/DNA. By manipulating the mechanical properties and structural geometry, we can unravel their effects on the described interactions, better understand the relations between these molecules commonly found together in biological environments, and find novel control mechanisms for DNA-based nanotherapeutics interacting with cellular membranes.
Results
Attachment of DNA to lipid bilayers is most often achieved either by modifying nucleic acid strands with hydrophobic moieties8,14,15,33 or by creating formulations involving positively-charged – cationic – lipids.4,17 Here, we analysed binding between DNA and lipid bilayers as they are mostly found in biological environments: unmodified nucleic acids and zwitterionic lipids. In this scenario, interactions are dominated by the electrostatic properties of the molecules, where negatively-charged phosphates on DNA and on lipid headgroups are bridged by positively-charged, divalent ions (Fig. 1a). Regarding the binding as a multivalent process, we identified two key groups of parameters that affect this attachment: mechanical parameters, like the lipid bilayer phase or DNA flexibility, and valency parameters, determining the number of connections formed, like the size of the DNA construct or the number of bridging ions. Interplay between them determines local variations in binding kinetics and surface concentration, thus affecting the observed DNA attachment to lipid membranes.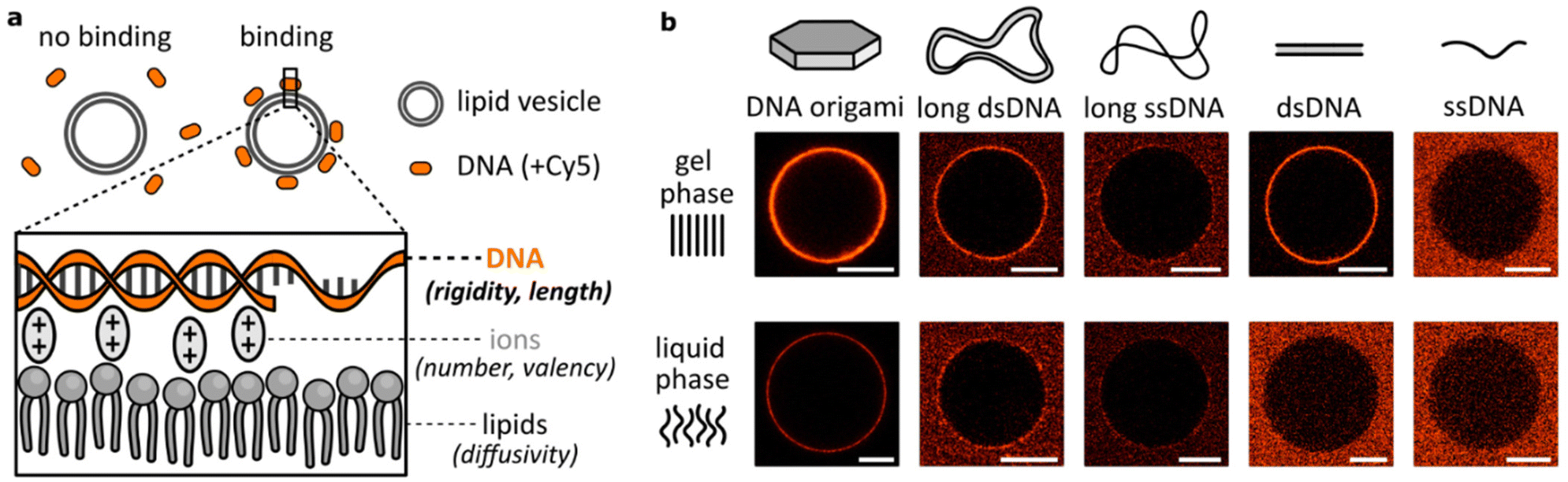 | ||
| Fig. 1 The effects of divalent cation bridging between DNA and zwitterionic lipids vary with the DNA design. (a) Negatively charged phosphates of the DNA backbone and lipid headgroups are electrostatically bridged with divalent cations, which manifests as DNA binding to the lipid membrane. Parameters related to each component (DNA, ions, lipids) will affect the binding process. (b) Representative micrographs of DNA structures interacting with DPPC (gel phase) and DOPC (liquid phase) lipid vesicles in the presence of 12 or 1 mM MgCl2, for DNA origami and the other structures, respectively. The panel represents qualitative comparison; only the intensities for large constructs (DNA origami, long ds/ssDNA) can be compared between the gel and liquid phases. Large constructs are modified with six, while small ones with one Cy5 label. Scale bars: 10 μm. | ||
Primarily, this work centres on studying the role of the DNA structural design in electrostatic interactions with zwitterionic lipids. Our initial microscopy observations of various fluorescently-labelled DNA structures coating the surface of lipid vesicles are shown in Fig. 1b. Lipid vesicles in either gel (DPPC) or liquid (DOPC) phase were incubated with three large constructs based on a long (7560 nt) scaffold: DNA origami, long double-stranded (ds)DNA and long single-stranded (ss)DNA, and two smaller structures: 20 bp dsDNA and 40 nt ssDNA. This first qualitative comparison demonstrated big disparities between DNA constructs. Noticeably, even though we previously observed no binding of dsDNA to lipids in the liquid phase,26 here we observed that large DNA structures provide coating of the DOPC vesicles’ surface – particularly in the case of relatively rigid DNA origami. Thus, high-valency constructs based on the long DNA scaffold all bind to liquid phase bilayers, although only a faint binding of long ssDNA is observed, in both lipid phases. On the other hand, short ssDNA showed no clear attachment even in the gel phase, indicating that its small size is an important factor determining the lipid interactions. Following these initial observations, we hypothesize that the structural (size) and mechanical (flexibility) properties of DNA are crucial in determining binding. The results presented in Fig. 1b clearly demonstrate not only the effects of DNA design, but also a major role played by the bilayer structure in guiding the electrostatic binding of DNA, agreeing with our previous report.26 We have also previously explored the effects of ion identity on this phenomenon. In particular, while monovalent cations are unable to bridge negative phosphates, they can compete with divalent cations and suppress bridging at sufficiently high concentrations.14,26,34,35 Here, we further expand our insights into the role of ions, particularly Mg2+, deepening our understanding on how cations influence DNA/lipid attachment in the context of structural variations.
Ions’ concentration and valency determine electrostatic DNA/lipid interactions
We have focused our research on the ion most often introduced when working with DNA-based systems: Mg2+, included at a concentration range between 0 and 4 mM, spanning the physiologically-relevant values in serum: [0.75, 1.25] mM.36 When zwitterionic DMPC vesicles were incubated with increasing concentrations of MgCl2, we observed their ζ potential increasing from negative values to positive ones (Fig. 2a). Having confirmed that positively charged Mg2+ ions interact with lipid bilayers, we incubated dsDNA (40 bp) with increasing magnesium concentrations (Fig. 2b). As the vesicles were cooled down below their transition temperature (Tm ≈ 24 °C, see Fig. S1†), a drop in their ζ potential was observed in the presence of DNA. We interpret this change as a result of the attachment of negatively charged duplexes to the lipid bilayer as it transitioned from the liquid to gel phase. Changes in the ζ potential of the vesicles (Δζ = ζliquid − ζgel) with increasing Mg2+ content are shown to be closely correlated with the observed attachment of DNA: at higher [Mg2+], a drop towards more negative ζ potential values is observed, suggesting that with a higher number of cations more DNA is able to bind to the vesicle's surface. Similarly, attachment of fluorescently-labelled dsDNA to DPPC bilayers was observed to increase as the structures were incubated with increasing MgCl2 concentrations. The distributions of coating intensities observed by confocal microscopy are shown in Fig. 2c.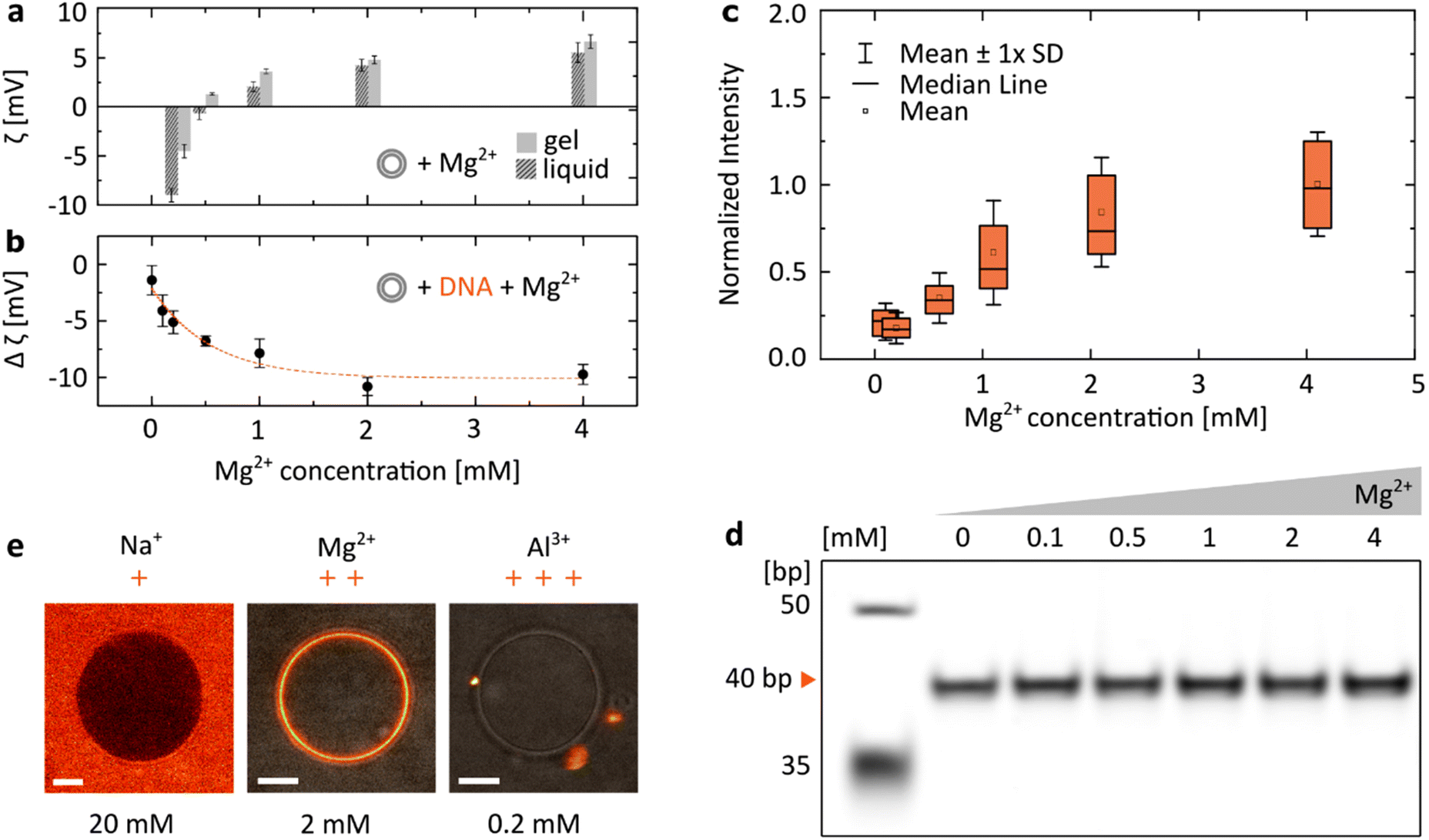 | ||
| Fig. 2 The electrostatic bridging between DNA (40 bp dsDNA) and lipids is determined by the concentration and valency of ions. (a) ζ potential measurements of DMPC vesicles as a function of the MgCl2 concentration, measured for the gel (15 °C) and liquid (35 °C) phases. (b) ζ potential measurements of DNA incubated with DMPC vesicles show an increasingly negative change when transitioning between the liquid and gel phases (Δζ = ζliquid − ζgel) at increasing Mg2+ concentrations, suggesting an increased attachment of negatively charged DNA. For all ζ measurements, error bars represent standard deviation from three measurements, each consisting of at least 15 runs. Dashed line represents an exponential fit. (c) Quantitative analysis of DNA attachment to DPPC lipid bilayers, observed as a fluorescence signal around vesicles (as in Fig. 1b), plotted against Mg2+ concentration. Box plots represent values measured from at least 80 vesicles (details in Table S2†). (d) PAGE analysis demonstrates that the effects of Mg2+ concentration on the yield of folding of a 40 bp DNA duplex are negligible (further analysis in Fig. S2†). (e) Representative micrographs demonstrate the effects of ion valency on the DNA/lipid binding; images show Cy5-labelled dsDNA incubated with DPPC vesicles at room temperature. Concentration of respective ions is stated below each image. Scale bars: 5 μm. | ||
As the structural integrity of dsDNA depends on the ionic composition of the surrounding medium, we wanted to exclude the possibility that DNA structures could unfold at lower [Mg2+], leaving solely non-binding single strands (Fig. 1b), which could account for the observed changes in the magnitude of binding. PAGE analysis of duplexes folded in respective MgCl2 buffers shows that their effects on the stability of the double strands are negligible (Fig. 2d and Fig. S2†). Thus, the observed changes in DNA/lipid attachment are the result of Mg2+-dependent electrostatic binding, rather than structural changes in the nucleic acid constructs.
In addition, the effects of ions on the behaviour of nucleic acids can be related not only to the changes in their concentration, but also to their valency (Fig. 2e). Since monovalent cations (Na+) do not provide electrostatic bridges, even at higher concentrations no DNA/lipid attachment is observed. On the other hand, the higher valency of Al3+ could provide stable binding. However, due to their ability to condense nucleic acids,37 only aggregates of fluorescently-labelled dsDNA were observed both attached and separated from the lipid vesicles.
DNA length and flexibility determine electrostatic DNA/lipid interactions
We have discussed the role of lipid phases and ions on the electrostatic DNA/lipid binding. In this section, we focus our studies on unravelling the effects of the remaining, third component of the analysed system: the DNA structural design. With the vastness of its potential in biomedical applications,38–42 the tunability offered by DNA nanotechnology makes it an exciting platform for understanding and controlling the interactions of biomaterials with cellular lipid membranes.As we can observe from micrographs in Fig. 1b, the lipid binding differs between dsDNA and ssDNA, as well as between large and small constructs. From these findings we identify two DNA design parameters determining the membrane attachment: (a) The size of the DNA structure, which is a proxy for the valency of binding; longer DNA strands carry more phosphates, thus providing more binding sites (assuming an excess of mediating cations). (b) The flexibility of the DNA construct, which influences the entropic penalty of binding; more flexibility of the structure in solution indicates higher loss of degrees of freedom during binding to the surface.29 Here, we aim to comprehensively unravel binding dependency on these two parameters, and better characterize the interplay between them. Which one, size or flexibility, should be prioritized when designing lipid-interacting DNA constructs?
With this goal in mind, we have designed a library of linear nanostructures, as presented schematically in Fig. 3a. The constructs vary in length (20, 40, and 60 bp), determining the valency of binding. In turn, flexibility was modulated by introducing single-stranded domains between 20 bp double-stranded segments in the structures based on a single long strand. With their lesser rigidity, the ssDNA parts act as flexible linkers for the rigid binding dsDNA domains, and we assume that their interactions with lipids are negligible compared with those of dsDNA (see Fig. 1b and Fig. S3†). In order to quantitatively represent the concept of conformational flexibility considered here, we collected end-to-end distances measured for each construct using coarse-grained oxDNA simulations, and observed the width of their distributions, presented in Fig. 3b, as an indication of the conformational flexibility of the structures. Fig. 3d–f additionally illustrate the flexibility of nanostructures, mapping their root mean square fluctuations (RMSF) on the coarse-grained model of linear DNA. The simulations confirm that with longer ssDNA linkers the constructs can be treated as more flexible.
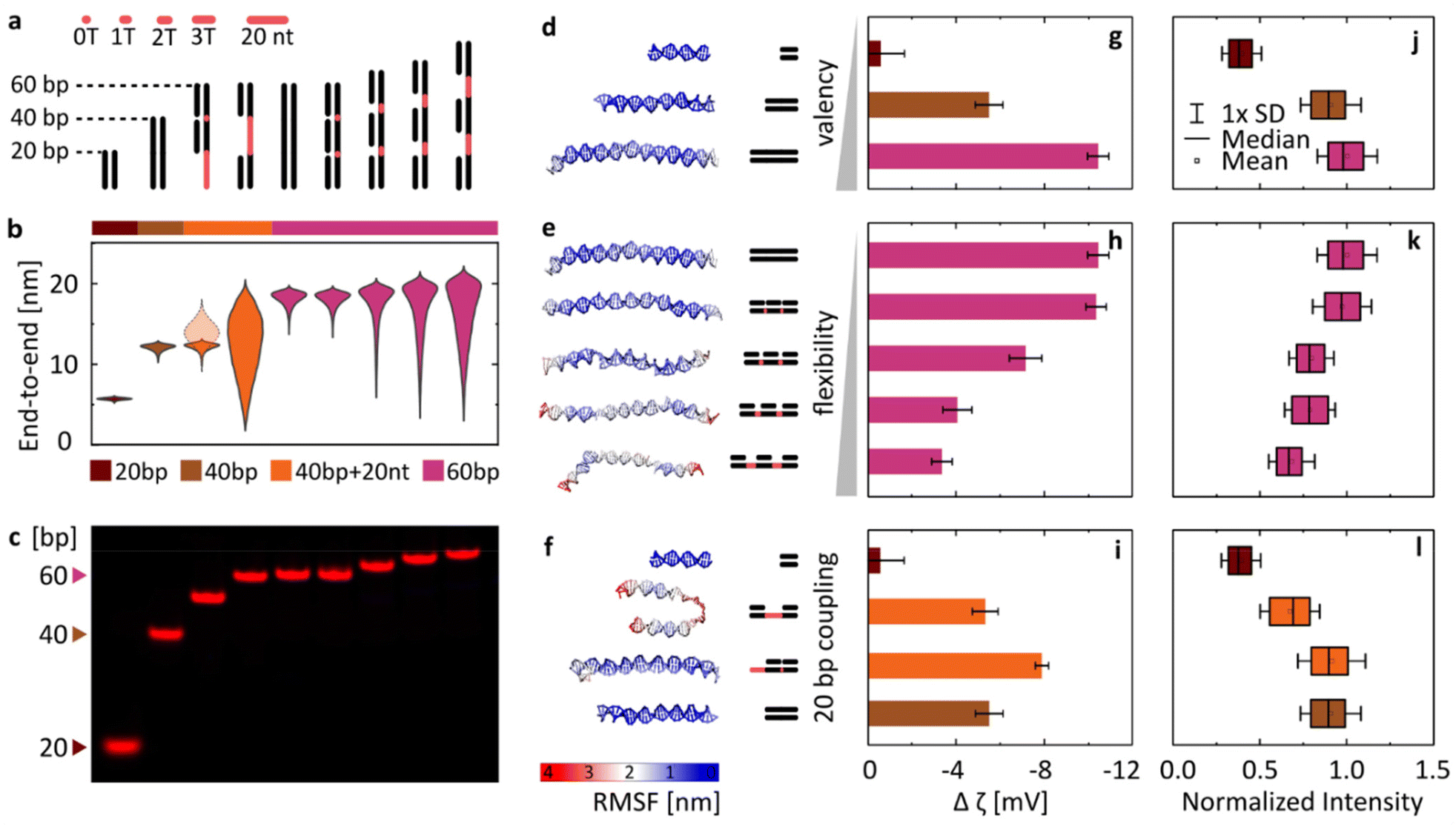 | ||
| Fig. 3 Varying the DNA design affects the magnitude of its binding to lipid bilayers (gel phase) in the presence of MgCl2. (a) Schematic overview of the library of linear DNA constructs. Single-stranded regions are highlighted in red. (b) Violin plots of end-to-end distances between dsDNA regions obtained from coarse-grained simulations. The semi-transparent plot for the third structure (==−) represents the distance between the double and single-stranded termini of the nanostructure. (c) PAGE analysis of the Cy5-labelled duplexes. (d–f) Last frames from coarse-grained simulations of DNA structures, mapping their root-mean-square-fluctuations (RMSF). (g–i) Changes in ζ potential when transitioning between the liquid and gel phases (defined as in Fig. 2b) for the library of duplexes. Measurements were performed in 2 mM Mg2+. Error bars represent standard deviation from at least three measurements, each consisting of at least 15 runs. (j–l) Fluorescence signal observed around DPPC vesicles under the confocal microscope, indicating attachment of Cy5-labelled duplexes. Imaging was performed in 1 mM Mg2+. Box plots represent values measured from at least 200 vesicles (details in Table S3†). | ||
The designed structures were folded and PAGE-analysed (Fig. 3c), and their Mg2+-mediated attachment to lipids was assessed with ζ potential measurements (Fig. 3g–i) and by confocal microscopy (Fig. 3j–l). Note that the ζ potential values are influenced by two factors: the number of bound duplexes and the charge they carry. For example, even though microscopy suggests similar attachment of duplexes of 40 bp (==) and 40 bp + 20 nt (==−), the additional charge of 20 nt carried by the latter results in a bigger ζ potential drop. Additionally, in ζ potential measurements, small unilamellar vesicles (SUVs) with a diameter of 100 nm are used, and the higher curvature that this vesicle size implies might pose an additional factor influencing changes in binding: the attachment of smaller constructs could be favoured, as they will induce less stress on the curvature of the membrane. Thus, microscopy imaging of structures labelled with the same number of fluorophores binding to giant unilamellar vesicles (GUVs) with an effectively “flat” membrane surface on the scale of the used linear constructs is a more direct way to make conclusions about DNA binding, while its good agreement with the trends observed with ζ potential measurements provides an additional confirmation of the observed behaviours.
Taking this notion into account, we conclude that the results from both experimental techniques are in good agreement. Furthermore, the initial analysis instantly confirms our hypothesis: DNA/lipid binding depends on the size and flexibility of DNA. As the length of the duplexes increases (Fig. 3d), more binding sites (more phosphates) lead to more stable and stronger binding, observed as a larger shift of ζ potential towards negative values (Fig. 3g) and a higher intensity of fluorescent coating on the vesicles observed under the confocal microscope (Fig. 3j). On the other hand, the structure of higher flexibility (Fig. 3e) loses more degrees of freedom as a result of surface binding, shifting the rebinding kinetics and affecting the local surface concentration of attached strands. Thus flexibility leads to less favourable and weaker attachment, observed as a smaller decrease of ζ potential values (Fig. 3h) and a lower intensity of vesicle coating (Fig. 3k). We can present this as a general concept, where the level of binding is correlated positively with size (valency) and negatively with the flexibility of the nucleic acid.
Often, the differences between designs are a combination of both these parameters. In Fig. 3f, we have shown a set of structures formed by various configurations of the 20 bp double-stranded domain, with the corresponding ζ potential measurements and the fluorescence imaging results summarized in panels Fig. 3i and l, respectively. The continuous duplex of 40 bp was compared with designs based on the 60 nt strand, with two 20 bp parts either adjacent to each other (==−)or separated by 20 nt-long ssDNA (=–=). While the obtained results, particularly the more reliable quantification of microscopy observations (Fig. 3l), suggest that the ==− structure binds similarly to the continuous 40 bp duplex, the more flexible =−= design shows reduced binding, near the values obtained for a single 20 bp domain. However, despite significant differences in the mechanical properties between them, the two designs do not reach the level of differences obtained when changing the length of constructs (e.g., 20 vs. 40 bp). We thus observe that valency plays a big role in determining DNA/lipid attachment, and is likely the more dominating one among the two factors in guiding the cation-mediated bridging.
The interplay between DNA size and flexibility determines lipid binding of DNA origami constructs
The results presented in the previous section suggest that while flexibility has a clear impact on the Mg2+-mediated binding of dsDNA to zwitterionic lipids, valency plays a more dominant role in determining the attachment. We examined whether we can apply these findings to more complex structures to program their relative binding efficiency.To this end, we introduced two DNA origami designs: a large, single-layer plate based on a square lattice43,44 (Fig. 4a) and a smaller disk built on a hexagonal lattice45 (Fig. 4b). Both structures have been published before, and their use as potential nanotherapeutics has been highlighted in the scientific literature.12,46 With similar molecular weight, the two main differences in their structures are: (I) the plate has a higher number of phosphates available for surface binding than the disk (≈3.5× more). This is caused not only by the surface area of the plate being larger than that of the disk, but also by the cross-sections that determine which DNA helices are in immediate contact with the surface. The square lattice of a plate results in a higher number of phosphates in the proximity to a lipid bilayer than the hexagonal-lattice of a disk, as is highlighted in Fig. 4a and b. (II) The single-layered, large plate is more flexible than the compact disk with a honeycomb cross-section, as indicated by the ranges of RMSF predicted computationally: [0.3–1.5] nm and [1.3–5.4] nm for the disk and the plate, respectively (see CanDo analysis in Fig. S4†).
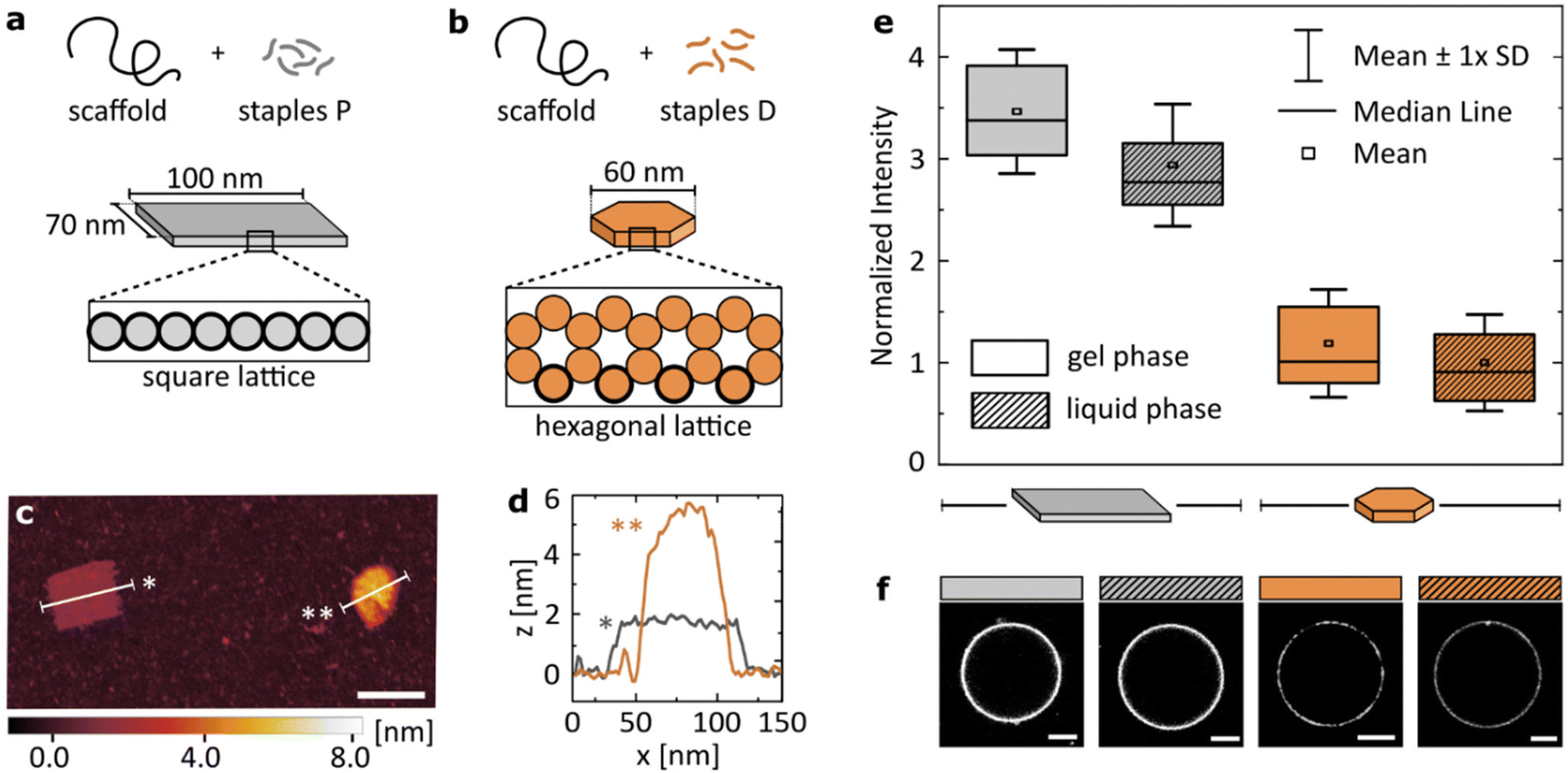 | ||
| Fig. 4 Structural details of the DNA origami design determine the magnitude of its binding to lipids. Geometric details of the two studied origami designs: (a) large, single-layered DNA plate and (b) more densely packed, smaller DNA disk. Cross-sections of the designs highlight helices in direct contact with the surface (bold outline). (c) Representative AFM image showing the two structures. Scale bar: 100 nm. (d) Plot profiles as marked on the image in (c). (e) Fluorescence intensity of Alexa488-labelled (6×) DNA origami, coating vesicles either in gel (DPPC) or liquid (DOPC) phase. Imaging was performed in 12 mM Mg2+. All values normalized to the mean value obtained for the disk binding to lipids in the liquid phase, are corrected by the yield of the fluorescent labelling of each structure. Box plots represent values measured from at least 50 vesicles (details in Table S4†). (f) Representative confocal images of the respective fluorescent signal, corrected by the yield of the fluorescent labelling of each structure. Scale bars: 5 μm. | ||
In short, we compared the cation-mediated lipid binding of a flexible structure with high valency (plate) with that of a more rigid one with lower valency (disk). We folded each design, and characterized them by agarose gel electrophoresis (AGE, Fig. S5†) and atomic force microscopy (AFM, Fig. S6†), verifying the correct folding and structure of the constructs. A representative AFM image of the two structures measured simultaneously is presented in Fig. 4c, alongside their profile plots (Fig. 4d) confirming the expected geometry of the designs. We quantitatively compare the binding of the two DNA origami structures to both gel (DPPC) and liquid (DOPC) phase bilayers. The box plots of the obtained signal distributions, measured for at least 50 individual vesicles in each sample, are shown in Fig. 4e, while the representative images of the fluorescent DNA coating of vesicles are presented in Fig. 4f.
Firstly, we confirm observations which are shown in Fig. 1b: both large origami designs bind to the liquid phase bilayers, which was never observed for smaller yet highly rigid dsDNA constructs. Thus, the high valency of binding, provided by the large surface area of origami, allows binding also to the less favourable surface of the liquid phase bilayers. However, this attachment is still weaker than that to gel phase membranes, although the difference is less pronounced than that with smaller dsDNA structures (Fig. 1b).
Secondly, the high valency plate exhibits stronger binding than the smaller disk. We suggest that this difference is caused by the dominance of valency in determining the strength of binding. This hypothesis agrees with our initial observations from studying linear constructs, although we note that studying a larger library of comparable origami nanostructures could bring deeper insight into the importance of design details in the interactions with the liquid phase bilayers.
In the next section we will further discuss the implications of these findings, both from the perspective of DNA/lipid interactions, and using a general model for multivalent binding. Here, we additionally emphasize the meaning of these results for researchers working with DNA origami and lipid-based constructs (including cell membranes): large DNA constructs can be found binding non-specifically to lipid bilayers, producing a strong background signal. We have shown how this electrostatic attachment can be tuned through the DNA design and the ionic composition of the media, and hope that these findings would help with the experimental design of future DNA-based therapeutics and sensors.
Conclusions
We have studied electrostatic, cation-mediated interactions between unmodified DNA and zwitterionic bilayers, and found that their binding is determined by each component of the molecular system: lipids, ions and DNA. Particularly the latter, DNA structural design, offers a wide range of tunable parameters that can be easily implemented as control mechanisms for guided membrane binding.Firstly, we experimentally observed differences in binding to bilayers in either the gel or liquid phase. We confirmed that binding to liquid phase bilayers is hindered compared with the gel phase ones, and also demonstrated that structures with a large surface area, and thus high valency of binding, can attach to bilayers in the liquid phase. This is an important observation for DNA nanotechnologists working with biological systems, as cellular membranes typically exhibit liquid phase lipid bilayers.47 One of the potential benefits of our observations is the changes to present techniques for the cellular delivery of large nucleic acids (DNA/RNA) either for transfection or vaccination purposes, where currently-used toxicity-inducing48 cationic lipids could potentially be substituted for DNA/RNA formulations with more natural zwitterionic lipids. Even though here we focused on understanding the interplay between the parameters of DNA design, we acknowledge that the structure and mobility of lipids – and the mobility of surface-bound receptors in general49 – are crucial factors determining binding processes, coupled with other conditions discussed here. The valency (origami vs. short dsDNA) determining the difference in binding to liquid and gel phases is one example of such coupling between parameters that can be harnessed to finetune DNA/lipid interactions and is thus a promising next step for further research.
Secondly, we showed how the concentration and type of ions determine the electrostatic binding. In particular, we demonstrated that ion valency can affect the bridging ability (monovalent Na+) and accessibility of DNA by inducing its clustering (trivalent Al3+). We are still in the process of understanding how ion types determine binding dependency on the DNA design. It has been previously reported that the interactions of nucleic acids with lipids via cation-mediated attachment are correlated with the degree to which the ions bind to the lipids,7 and the divalent-to-monovalent ion ratio was shown to have an impact on the DNA/lipid interactions.14,26,35 Thus, in the future studies, more precise mimicking of physiological conditions is a crucial next step in developing biological applications reliant on the presented phenomenon.
Finally, the major interest of this work was to unravel the importance of the DNA structural design in determining electrostatic binding to lipid bilayers. With the library of DNA structures, we showed that binding depends on the valency/flexibility interplay and we performed an initial analysis of the importance of the two parameters. Our findings allowed us to discuss the differences in binding between two distinct DNA origami designs: plate and disk. These and similar DNA-based constructs are often developed as potential therapeutics and drug delivery platforms50–52 – applications implying interactions with lipid-based membranes. Therefore, the analysis of design parameters discussed here is crucial for further improvement, and gaining new means of control over these nanotools.
The valency/flexibility interplay is important not only for electrostatic binding, and is not exclusive to DNA/lipid systems, but a similar analysis can also be applied to all multivalent processes. Multivalency is most often described through the combination of the affinity and valency of binding, yet we realize that the mechanical properties – flexibility – are a critical parameter as well. Importantly, the balance between the mechanical and structural parameters is bound to differ between systems, where e.g., stronger affinity of binding will render flexibility irrelevant. Further research work in the direction presented here will have a major relevance in two aspects of multivalent processes: (I) it will shed more light on various biological phenomena, helping us to understand why biological multivalent binders evolved in a certain way. For example, we will observe with more clarity the differences in binding between enveloped viruses carrying high numbers of flexible receptors53,54 and antibodies with a low valency, but well-defined geometry.55,56 (II) Better understanding of the valency/flexibility interplay in binding will help design cost-effective membrane-targeting therapeutics, guiding the choices of the design parameters. For example, one can ask whether a newly developed drug requires its high valency (= high costs) or could it instead achieve similar performance with an increased rigidity of the scaffold. Thus, we believe that our work opens paths towards deeper comprehension of fundamental biological phenomena, as well as towards guiding the application-based research in the biomedical sector.
Materials and methods
All chemical compounds were obtained from Sigma-Aldrich, unless stated otherwise. DNA oligonucleotides were purchased from Integrated DNA Technologies, Inc, while lipids were provided by Avanti® Polar Lipids.DNA design and folding
Each designed DNA oligonucleotide was analysed using the NUPACK suite,57 in order to prevent the formation of secondary structures and to ensure the sufficient yield of folding. All the strands were dissolved to a final concentration of 100 μM: unmodified ones in IDTE buffer (10 mM Tris, 0.1 mM EDTA (Ethylenediaminetetraacetic acid), pH 8.0) and labelled ones in Milli-Q purified water. Strands were then stored at 4 °C, except for dye-modified ones, which were stored in −20 °C.In order to fold the designed duplex structures, strands were dissolved to a final concentration of 1 μM in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0). Additionally, the structures were folded in a buffer with magnesium concentration stated for each experiment. The mixture was then annealed to 90 °C, and cooled down to room temperature for over 45 minutes. Folded structures were all stored at 4 °C. The sequences and details of modifications are listed in Table S1.†
In order to fold DNA origami structures, 10 nM ssDNA scaffold (p7560 for disk, p7249 for plate, tilibit nanosystems) was mixed with an excess of staples (7× for disk, 10× for plate) in a TE buffer containing 18 mM MgCl2. The disk mixture was annealed to 80 °C for 5 min, and then cooled from 60 °C to 20 °C at a cooling rate of −1 °C h−1. The plate mixture was annealed to 80 °C for 5 min, and then cooled from 60 °C to 40 °C at a rate of −18.75 °C h−1, upon which it was brought down to 20 °C. Folded structures were all stored at 4 °C. The structures were purified by PEG purification, as described previously.12 The sequences of staples and further characterization of the origami designs can be found in previous reports on the plate43,58 and the disk.12,45
Long dsDNA and ssDNA structures were obtained by mixing the p7560 scaffold (10 nM) with a 2× excess of either a full set of staples (dsDNA) or six of them (ssDNA), both in the TE buffer containing 1 mM MgCl2. The mixtures were annealed to 80 °C and then cooled down to 20 °C for over 14 h. Staples provided overhanging handles for the attachment of Cy5-labelled oligonucleotides, which were added in the next step (5× excess), by incubating for 1 h at 37 °C.
Coarse-grained simulations
The single structures of the double stranded domains were simulated using the oxDNA259 forcefield in the oxDNA simulation engine.60 The pipeline to generate topology, initial configuration, external force files, actual simulation and related analyses was identical for each of the structures. At first, we used the vHelix61 in Autodesk Maya ver. 2018b to generate a first CAD. We then converted in initial configuration and topology files using the tacoxDNA website.62 Afterwards we generated external force files that would prevent base-pair melting using the relative function in the oxView website.63,64 Relaxation of the structures was performed through a minimization in which the external forces were activated and the maximum backbone force was capped to ∼250 pN. We then deactivated the forces and proceeded with 4 relaxation runs in the Canonical ensemble, warming up the structure from 1 °C to 25 °C. A final production run still in the Canonical ensemble was then carried out for at least 1.5 μs. During this step we collected frames for every 15 ps. The final trajectory, the topology and configuration files were then converted using in-house python scripts to MDCRD, PSF and PDB files for treating of periodic boundary conditions and measuring in physical units using the software VMD.65 The end to end distances used as reference the centre of mass of the second base pair and the second but last base pair. Statistics were calculated over 3 independent replicas.For the 4 relaxing runs the temperature employed were respectively 1 °C, 10 °C, 20 °C, and 25 °C with the time step maintained at 1.5 fs. For the production run the time step was 3.03 fs.
When not stated otherwise, parameters were maintained in the standard values used in the production runs in the oxDNA web.66
Vesicle preparation
Giant unilamellar vesicles (GUVs) used in the microscopy imaging were prepared by electroformation. The lipids used were DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) and DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) dissolved to the final concentration of 5 mg ml−1 in chloroform. The lipid mixture was distributed over an ITO-coated glass slide (15–25 Ω sq−1, Sigma-Aldrich), chloroform evaporation was ensured by desiccating the lipid film for 30 min. A nitrile O-ring was placed on top of the dry film, 800 μl of 200 mM sucrose was used as a buffer to hydrate the lipid layer, and a second ITO glass was placed on top. The osmolality of the buffer was around 200 mOsm, with all the dilution buffers adjusted accordingly. All the buffers were adjusted to pH 7.5 (using sodium hydroxide and hydrochloride solutions). A sinusoidal waveform with an amplitude value of 2 V was applied, with a frequency of 10 Hz for the first 2 h and then 2 Hz for the next hour. In the case of electroformation of GUVs containing DPPC lipids, the process was performed at 60 °C to ensure bilayer fluidity. Obtained GUVs were stored at 4 °C and used within a week.Large unilamellar vesicles (LUVs) used for the ξ potential measurements were prepared with extrusion, using a commercially available extruder (Avanti® Polar Lipids, Avanti Mini Extruder), following the producer's protocol. Whatman® Filter Supports and Whatman® Nuclepore Track-Etched Membranes (100 nm) were obtained from Sigma Aldrich. The lipid used was DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine). The lipid layer, formed in a round-bottom flask, was hydrated in 200 mM sucrose. The extrusion was performed at 40 °C to ensure bilayer fluidity. DLS measurements after each extrusion protocol were used to confirm the correct size distribution of the obtained vesicles (Fig. S7†). LUVs were stored at 4 °C and used within a week.
Confocal microscopy imaging
The confocal microscopy images were acquired on a Leica SP8 inverted microscope using an HC PL APO CS2 63×/1.4 oil immersion objective. All imaging was performed at room temperature. Cy5 excitation was performed using a 638 nm solid state laser at 1% laser power for studying DNA duplexes, with emission recorded between 650 and 720 nm. Alexa488 excitation was performed with a 488 nm laser at 3% power for DNA origami, with emission recorded between 500 and 570 nm. These settings do not apply to qualitative comparison presented in Fig. 1b, where non-binding structures required a higher laser power to observe the DNA dispersed in a solution – the images presented there are processed using the Leica Application Suite (LAS) X. The FIJI67 was used to analyse all the remaining images, particularly to adjust the intensity of signals observed for the disk and plate samples, to reflect on their different fluorescence yield as observed from the gel electrophoresis. All reported values are after the subtraction of the background signal.In all the experiments, DNA structures were diluted in an osmotically balanced glucose buffer with Mg2+ added at the required concentration. In the experiments comparing DPPC and DOPC vesicles (Fig. 1b, 4e and f), the PC content of GUVs was compared using the commercially available fluorimetric assay (Sigma-Aldrich), following the calibration curve (Fig. S8†), and the volume of vesicle solutions were adjusted accordingly. Similarly, the concentration of DNA structures, particularly DNA origami that was recovered after the purification protocol, could differ, and thus was adjusted based on the SYBR signal on the gel (Fig. S9†). The intensity of DNA membrane coating was measured using the FIJI, and the numbers of vesicles measured in each experiment are listed in Tables S2–4.† The values listed in Fig. 4e and the images shown in Fig. 4f are corrected for the differences in the fluorescence of the two origami designs.
The FRAP measurements were performed on vesicles containing NBD-PC (0.5%, ex/em 464/531 nm) to confirm the presence of gel and liquid phases in the experiments, and the results are shown in Fig. S10.† Using the FRAP function of the microscope's software, a spot of ∅ 4 μm was bleached and the fluorescence recovery was observed. Five frames were collected pre-bleaching. Bleaching was performed for over 0.1 s with 99% laser power and the fluorescence recovery was recorded for 50 frames. The results were fitted and analysed as described previously.26
Dynamic and electrophoretic light scattering
The measurements were performed on a Zetasizer Nano ZSP (Malvern Panalytical) with an excitation wavelength of 633 nm and a scattering angle fixed at 173°. For assessing the hydrodynamic radius, 100 μl of 10× diluted vesicles were measured in a disposable ZEN0040 cuvette. The ζ potential measurements were performed in a DTS1070 cell, with [DNA] = 15 nM and 200× diluted LUVs in 800 μl of 200 mM sucrose solution. Different ratios of DNA to vesicle content were studied, as presented in Fig. S11.† For each experiment, the mixture was prepared at 35 °C, above the phase transition temperature Tt of DMPC (measured with differential scanning calorimetry (DSC) to be around 24 °C (Fig. S1†)), ensuring the liquid phase of the bilayer, and measured at this temperature. Next the same mixture was cooled down to 15 °C, equilibrated for 2 min and measured again. The results represent a change in the ζ potential between these two measurements (Δζ = ζliquid − ζgel). The experiments shown in Fig. 3c were performed at 2 mM Mg2+. At least 3 measurements were performed for each condition, each of at least 15 runs.Gel electrophoresis
Polyacrylamide gel electrophoresis (PAGE) was used to confirm the expected folding of DNA duplex designs. The gels were prepared with polyacrylamide at a concentration of 10%, 0.5× TBE (Tris, borate, EDTA) and either 1 mM MgCl2 (Fig. 3c) or 0 mM MgCl2 (Fig. 2d). The previously described protocol8 was adopted.Agarose gel electrophoresis (AGE) was used to confirm the expected folding of DNA origami and large constructs. The gels were prepared with agarose at a concentration of 2%, 0.5× TBE, 0.01% SYBR Safe and either 10 mM (Fig. S5†) or 1 mM MgCl2 (Fig. S9†). 3 μl of a DNA sample were mixed with 2 μl of its folding buffer and 1 μl of the 6× loading dye, and 5 μl of the mixture were loaded on the gel. The gel was run in 0.5× TBE with the respective concentration of MgCl2 at 60 mV for 90 min. The FIJI was used to analyse the images.
Atomic force microscopy
The AFM images were acquired in the tapping mode in liquid on a Cypher VRS (Asylum Research Inc.) with a BioLever mini cantilever (BL-AC40TS-C2, Olympus). To prevent the diffusion of DNA on mica, freshly cleaved mica was pretreated with 40 μl of 100 mM NiCl2. After 30–60 s incubation, mica was rinsed with 20 mL of Milli-Q water and dried by compressed air. Typically, 5 μL of DNA solution at approximately 1 nM concentration was deposited upon pretreated mica and incubated for 20–30 s. Meanwhile, the tip of the cantilever was immersed in 20 μL of 10 mM MgAc2 before engaging the surface to prevent bubble formation between the tip and the surface. During imaging, the setpoint was minimized while preserving good tracking and an integral gain of around 60 was used. The AFM images shown in this manuscript were processed by a scar correction followed by a first-order polynomial line-by-line correction while excluding an Otsu's mask.Data availability
All data are available free of charge under https://doi.org/10.5281/zenodo.6984753 (https://zenodo.org/record/6984753#.YvZd4xzP2Uk).Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. M. and M. M. C. B. acknowledge the support from the Volkswagen Stiftung “Life?” (Grant No. 98200). V. C. and M. M. C. B. acknowledge the support from the Swiss National Science Foundation (Eccellenza Grant PCEGP2_181137). R. R. S. acknowledges support from the EPSRC CDT in Nanoscience and Nanotechnology (NanoDTC, Grant No. EP/L015978/1), the Mexican National Council for Science and Technology and the Cambridge Trust. R. R. S. and L. D. M. acknowledge funding from the Royal Society Research Fellows Enhanced Research Expenses (RF/ERE/210029). L. D. M. acknowledges support from a Royal Society University Research Fellowship (UF160152) and from the European Research Council (ERC) under the Horizon 2020 Research and Innovation Programme (ERC-STG No. 851667 NANOCELL). The authors thank Dr Jorieke Weiden and Dr Eva Kursinkal for their help with DNA origami protocols.References
- J. E. Francis, I. Skakic and P. M. Smooker, Methods Mol. Biol., 2022, 2412, 355–366 CrossRef PubMed.
- M. Rak, A. Góra-Sochacka and Z. Madeja, Methods Mol. Biol., 2021, 2183, 391–404 CrossRef CAS PubMed.
- A. Tejeda-Mansir, A. García-Rendón and P. Guerrero-Germán, Biotechnol. Genet. Eng. Rev., 2019, 35, 46–68 CrossRef CAS PubMed.
- R. Marty, C. N. N′soukpoé-Kossi, D. Charbonneau, C. M. Weinert, L. Kreplak and H. A. Tajmir-Riahi, Nucleic Acids Res., 2009, 37, 849–857 CrossRef CAS PubMed.
- G. Hermanson, V. Whitlow, S. Parker, K. Tonsky, D. Rusalov, M. Ferrari, P. Lalor, M. Komai, R. Mere, M. Bell, K. Brenneman, A. Mateczun, T. Evans, D. Kaslow, D. Galloway and P. Hobart, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13601–13606 CrossRef CAS PubMed.
- V. Kuvichkin, Ann. Endocrinol. Metab., 2017, 1, 1–7 CrossRef.
- V. Kuvichkin, Bioelectrochemistry, 2002, 58, 3–12 CrossRef CAS PubMed.
- D. Morzy, H. Joshi, S. E. Sandler, A. Aksimentiev and U. F. Keyser, Nano Lett., 2021, 21, 9789–9796 CrossRef CAS PubMed.
- D. Sobota, H. Joshi, A. Ohmann, A. Aksimentiev and U. F. Keyser, Nano Lett., 2020, 20, 4306–4311 CrossRef CAS PubMed.
- J. R. Burns, A. Seifert, N. Fertig and S. Howorka, Nat. Nanotechnol., 2016, 11, 152–156 CrossRef CAS PubMed.
- R. Rubio-Sánchez, G. Fabrini, P. Cicuta and L. di Michele, Chem. Commun., 2021, 57, 12725 RSC.
- A. Comberlato, M. M. Koga, S. Nüssing, I. A. Parish and M. M. C. Bastings, Nano Lett., 2022, 22, 2506–2513 CrossRef CAS PubMed.
- R. M. L. Berger, J. M. Weck, S. M. Kempe, O. Hill, T. Liedl, J. O. Rädler, C. Monzel, A. Heuer-Jungemann, R. M. L. Berger, S. M. Kempe, T. Liedl, J. O. Rädler, J. M. Weck, A. Heuer-Jungemann, O. Hill and C. Monzel, Small, 2021, 17, 2101678 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, J. List and F. C. Simmel, Acc. Chem. Res., 2014, 47, 1807–1815 CrossRef CAS PubMed.
- R. Rubio-Sánchez, S. E. Barker, M. Walczak, P. Cicuta and L. Di Michele, Nano Lett., 2021, 21, 2800–2808 CrossRef PubMed.
- D. Morzy, M. Schaich and U. F. Keyser, Molecules, 2022, 27(2), 578 CrossRef CAS PubMed.
- D. Zhi, Y. Bai, J. Yang, S. Cui, Y. Zhao, H. Chen and S. Zhang, Adv. Colloid Interface Sci., 2018, 253, 117–140 CrossRef CAS PubMed.
- M. P. Platre and Y. Jaillais, Plant Signaling Behav., 2017, 12, e1282022 CrossRef PubMed.
- O. Szekely, A. Steiner, P. Szekely, E. Amit, R. Asor, C. Tamburu and U. Raviv, Langmuir, 2011, 27, 7419–7438 CrossRef CAS PubMed.
- J. H. Lorent, K. R. Levental, L. Ganesan, G. Rivera-Longsworth, E. Sezgin, M. Doktorova, E. Lyman and I. Levental, Nat. Chem. Biol., 2020, 16, 644–652 CrossRef CAS PubMed.
- S. Jackowski, J. Biol. Chem., 1996, 271, 20219–20222 CrossRef CAS PubMed.
- R. D. Moore and G. A. Morrill, Biophys. J., 1976, 16, 527–533 CrossRef CAS PubMed.
- H. Bischof, S. Burgstaller, M. Waldeck-Weiermair, T. Rauter, M. Schinagl, J. Ramadani-Muja, W. F. Graier and R. Malli, Cells, 2019, 8, 492 CrossRef CAS PubMed.
- A. Kaasik, D. Safiulina, A. Zharkovsky and V. Veksler, Am. J. Physiol., 2007, 292, C157–C163 CrossRef CAS PubMed.
- D. H. Mengistu, K. Bohinc and S. May, J. Phys. Chem. B, 2009, 113, 12277–12282 CrossRef CAS PubMed.
- D. Morzy, R. Rubio-Sánchez, H. Joshi, A. Aksimentiev, L. di Michele and U. F. Keyser, J. Am. Chem. Soc., 2021, 143, 15 CrossRef PubMed.
- Y. Liu, P. Wijesekara, S. Kumar, W. Wang, X. Ren and R. E. Taylor, Nanoscale, 2021, 13, 6819–6828 RSC.
- W. Hawkes, D. Huang, P. Reynolds, L. Hammond, M. Ward, N. Gadegaard, J. F. Marshall, T. Iskratsch and M. Palma, Faraday Discuss., 2019, 219, 203–219 RSC.
- F. J. Martinez-Veracoechea and M. E. Leunissen, Soft Matter, 2013, 9, 3213–3219 RSC.
- F. J. Martinez-Veracoechea and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10963–10968 CrossRef CAS PubMed.
- A. Basu, D. G. Bobrovnikov and T. Ha, J. Mol. Biol., 2021, 433, 166861 CrossRef CAS PubMed.
- J. Ji, D. Karna and H. Mao, Chem. Soc. Rev., 2021, 50, 11966–11978 RSC.
- D. Morzy, M. Schaich and U. F. Keyser, Molecules, 2022, 27(2), 578 CrossRef CAS PubMed.
- S. Kempter, A. Khmelinskaia, M. T. Strauss, P. Schwille, R. Jungmann, T. Liedl and W. Bae, ACS Nano, 2019, 13, 996–1002 CAS.
- Y. Xin, S. Martinez Rivadeneira, G. Grundmeier, M. Castro and A. Keller, Nano Res., 2020, 13(11), 3142–3150 CrossRef CAS.
- F. C. Mooren, M. Moreno Geada, J. Singh, R. Stoll, W. Beil and W. Domschke, Biochim. Biophys. Acta, Mol. Cell Res., 1997, 1358, 279–288 CrossRef CAS PubMed.
- C. Qin, F. Kang, W. Zhang, W. Shou, X. Hu and Y. Gao, Water Res., 2017, 123, 58–66 CrossRef CAS PubMed.
- M. Zahid, B. Kim, R. Hussain, R. Amin and S. H. Park, Nanoscale Res. Lett., 2013, 8, 119 CrossRef PubMed.
- A. Keller and V. Linko, Angew. Chem., Int. Ed., 2020, 59, 15818–15833 CrossRef CAS PubMed.
- H. Shen, Y. Wang, J. Wang, Z. Li and Q. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 13859–13873 CrossRef CAS PubMed.
- Z. Huang, L. Qiu, T. Zhang and W. Tan, Matter, 2021, 4, 461–489 CrossRef CAS.
- P. Chidchob and H. F. Sleiman, Curr. Opin. Chem. Biol., 2018, 46, 63–70 CrossRef CAS PubMed.
- J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Schueder and R. Jungmann, Nat. Protoc., 2017, 12(6), 1198–1228 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- A. S. Eklund, A. Comberlato, I. A. Parish, R. Jungmann and M. M. C. Bastings, ACS Nano, 2021, 15, 17668–17677 CrossRef CAS PubMed.
- M. Godonoga, T. Y. Lin, A. Oshima, K. Sumitomo, M. S. L. Tang, Y. W. Cheung, A. B. Kinghorn, R. M. Dirkzwager, C. Zhou, A. Kuzuya, J. A. Tanner and J. G. Heddle, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- T. Sych, C. O. Gurdap, L. Wedemann and E. Sezgin, Membranes, 2021, 11, 323 CrossRef CAS PubMed.
- S. Cui, Y. Wang, Y. Gong, X. Lin, Y. Zhao, D. Zhi, Q. Zhou and S. Zhang, Toxicol. Res., 2018, 7, 473 CrossRef CAS PubMed.
- D. Morzy and M. Bastings, Angew. Chem., Int. Ed., 2022, 61, e202114167 CrossRef CAS PubMed.
- E. Tasciotti, Nat. Biotechnol., 2018, 36, 234–235 CrossRef CAS PubMed.
- Q. Jiang, S. Liu, J. Liu, Z. Wang and B. Ding, Adv. Mater., 2019, 31, 1804785 CrossRef CAS PubMed.
- J. Weiden and M. M. C. Bastings, Curr. Opin. Colloid Interface Sci., 2021, 52, 101411 CrossRef CAS.
- M. Delguste, C. Zeippen, B. Machiels, J. Mast, L. Gillet and D. Alsteens, Sci. Adv., 2018, 4, 1273 CrossRef PubMed.
- J. L. Cuellar-Camacho, S. Bhatia, V. Reiter-Scherer, D. Lauster, S. Liese, J. P. Rabe, A. Herrmann and R. Haag, J. Am. Chem. Soc., 2020, 142, 12181–12192 CrossRef CAS PubMed.
- T. Li, M. B. Tracka, S. Uddin, J. Casas-Finet, D. J. Jacobs and D. R. Livesay, PLoS Comput. Biol., 2015, 11, 473–479 Search PubMed.
- J. Zimmermann, F. E. Romesberg, C. L. Brooks and I. F. Thorpe, J. Phys. Chem. B, 2010, 114, 7359–7370 CrossRef CAS PubMed.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS PubMed.
- A. H. Clowsley, W. T. Kaufhold, T. Lutz, A. Meletiou, L. Di Michele and C. Soeller, J. Am. Chem. Soc., 2020, 142, 12069–12078 CrossRef CAS PubMed.
- B. E. K. Snodin, F. Randisi, M. Mosayebi, P. Šulc, J. S. Schreck, F. Romano, T. E. Ouldridge, R. Tsukanov, E. Nir, A. A. Louis and J. P. K. Doye, J. Chem. Phys., 2015, 142, 234901 CrossRef PubMed.
- P. Šulc, F. Romano, T. E. Ouldridge, L. Rovigatti, J. P. K. Doye and A. A. Louis, J. Chem. Phys., 2012, 137, 135101 CrossRef PubMed.
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Högberg, Nature, 2015, 523, 441–444 CrossRef CAS PubMed.
- A. Suma, E. Poppleton, M. Matthies, P. Šulc, F. Romano, A. A. Louis, J. P. K. Doye, C. Micheletti and L. Rovigatti, J. Comput. Chem., 2019, 40, 2586–2595 CrossRef CAS PubMed.
- E. Poppleton, J. Bohlin, M. Matthies, S. Sharma, F. Zhang and P. Šulc, Nucleic Acids Res., 2020, 48, e72–e72 CrossRef CAS PubMed.
- J. Bohlin, M. Matthies, E. Poppleton, J. Procyk, A. Mallya, H. Yan and P. Šulc, Nat. Protoc., 2022, 17(8), 1762–1788 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- E. Poppleton, R. Romero, A. Mallya, L. Rovigatti and P. Šulc, Nucleic Acids Res., 2021, 49, W491–W498 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05368c |
| This journal is © The Royal Society of Chemistry 2023 |