
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the formation mechanism of the biphenylene network†
Kaifeng
Niu
ab,
Qitang
Fan
c,
Lifeng
Chi
 *bd,
Johanna
Rosen
a,
J. Michael
Gottfried
*c and
Jonas
Björk
*a
*bd,
Johanna
Rosen
a,
J. Michael
Gottfried
*c and
Jonas
Björk
*a
aDepartment of Physics, Chemistry and Biology, IFM, Linköping University, 581 83 Linköping, Sweden. E-mail: Jonas.bjork@liu.se
bInstitute of Functional Nano & Soft Materials (FUNSOM) and Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou 215123, China. E-mail: chilf@suda.edu.cn
cDepartment of Chemistry, Philipps-Universität Marburg, 35032 Marburg, Germany. E-mail: michael.gottfried@chemie.uni-marburg.de
dDepartment of Materials Science and Engineering, Macau University of Science and Technology, Macau, 999078, China
First published on 3rd January 2023
Abstract
We have computationally studied the formation mechanism of the biphenylene network via the intermolecular HF zipping, as well as identified key intermediates experimentally, on the Au(111) surface. We elucidate that the zipping process consists of a series of defluorinations, dehydrogenations, and C–C coupling reactions. The Au substrate not only serves as the active site for defluorination and dehydrogenation, but also forms C–Au bonds that stabilize the defluorinated and dehydrogenated phenylene radicals, leading to “standing” benzyne groups. Despite that the C–C coupling between the “standing” benzyne groups is identified as the rate-limiting step, the limiting barrier can be reduced by the adjacent chemisorbed benzyne groups. The theoretically proposed mechanism is further supported by scanning tunneling microscopy experiments, in which the key intermediate state containing chemisorbed benzyne groups can be observed. This study provides a comprehensive understanding towards the on-surface intermolecular HF zipping, anticipated to be instructive for its future applications.
New conceptsOn-surface synthesis has presented a range of pioneering achievements for creating atomically precise low-dimensional carbon-based materials. Intermolecular HF zipping represents a conceptually new approach for fabricating nonbenzenoid carbon allotropes and was recently used to realize the two-dimensional biphenylene network on Au(111) (Q. Fan, et al., Science, 2021, 372, 852–856). However, the further application of this approach is hindered by the lack of insights of the formation mechanism. Here, by computationally analyzing the thermodynamics and reaction kinetics of the intermolecular HF zipping, we have demonstrated that the formation of the biphenylene network follows a novel intra-polymer mechanism consisting of defluorination, dehydrogenation, and C–C coupling stages. Our theoretical calculations have shown that the chemisorbed phenylene group on the surface serves as both the key intermediate states and facilitates subsequent C–C couplings, making the mechanism conceptually different from most other cascade reactions on surfaces. Importantly, we also present experimental support for such intermediate structures, by imaging chemisorbed phenylenes by scanning probe microscopy. Our work shows that the on-surface intermolecular HF zipping can be a powerful means for the design of new carbon allotropes and complements the toolbox of on-surface synthesis. |
Introduction
The fascinating properties of carbon allotropes, such as carbon nanotubes and graphene, have ignited the interests for the rational design of carbon-based nanostructures built from sp, sp2, and sp3 hybridized carbon atoms.1–3 The current progress for the synthesis of low-dimensional carbon materials by rational surface-assisted C–C coupling on metal surfaces or metal oxides has initiated the era of designing carbon allotropes with atomic precision.4,5 Such bottom-up approach has become a powerful methodology for the fabrication low-dimensional functional materials such as 2D covalent organic frameworks and graphene nanoribbons.6,7 In general, it consists of two main steps: (i) the design and synthesis of molecular building blocks, and (ii) thermally triggered transformation of precursors to target structure via C–C coupling.8,9 Generally, the quality of the target carbon nanostructure is determined by different factors such as appropriate molecular building blocks, choice of surface, coverage of molecules, and temperature. More importantly, the transformation from building blocks to desired carbon structures should be performed with exceptionally high efficiency and selectivity so that the polymers can be confined in the desired dimensions.10Extensive efforts have been made, demonstrating a plethora of various reactions, to construct C–C bonds, triggered on different single-crystal metal or metal oxide surfaces. Up to now, the studied reactions comprise of Ullmann-coupling,11 homo-coupling of terminal alkynes,12 decarboxylative polymerization,13 oligomerization of heterocyclic carbenes,14 dehydrogenative homocoupling,15 cyclodehydrogenation,16 cyclodehydrofluorination,17etc. These studies have demonstrated that both intermolecular and intramolecular reactions can be achieved via on-surface synthesis, and explored the possibility of fabricating large organic molecules in a controlled manner.16,18 In addition, density functional theory (DFT) calculations offer a strong tool for elucidating the mechanisms for various reactions, providing comprehensive understandings of the on-surface synthesis.19–23
Recently, a new methodology for the rational synthesis of extended sp2 carbon systems via intramolecular HF zipping has been developed.24 Nevertheless, the products of such approach have been limited into purely hexagonal net,25–27 while the formation of regular carbon nets with non-hexagons still remains challenging.28,29 In this regard, the development of intermolecular HF zipping is facilitating the fabrication of ordered non-benzenoid carbon allotropes. Fan et al. have synthesized two-dimensional biphenylene networks (BPN) with periodically arranged four-, six-, and eight-membered rings with atomic precision and high selectivity.30 The ultra-flat network is achieved by the intermolecular HF zipping between self-assembled poly(2,5-difluoro-para-phenylene) (PFPP) chains on the Au(111) surface after annealing to 700 K.30 The experimental observation and further theoretical investigations have demonstrated that the BPN exhibits uncommon metallic conduction, which is of great value in the development of carbon-based electronic materials.30–32 Despite the experimental success, the underlying mechanism for the intermolecular HF zipping still remains unclear. Therefore, a complete theoretical understanding of the intermolecular HF zipping on the Au(111) surface is crucial for the rational design of new on-surface synthesis protocols making use of such reactions.
Herein, we present a detailed analysis of the intermolecular HF zipping by density functional theory (DFT) calculations. The PFPP chains represented by oligomers consisting of four phenylene groups are employed as the precursor (I in Fig. 1a). The reaction steps of HF zipping reaction between two precursors are investigated via electronic structure theory-based transition state calculations, in which the formation of the first two four-membered carbon rings (I–III in Fig. 1a) is considered. By exploring a range of possible reaction pathways, we identified that the intermolecular HF zipping exhibits an “intra-polymer” mechanism, in which the defluorination and the dehydrogenation at the same phenylene group take place successively and are followed up by C–C couplings. The defluorinated and dehydrogenated phenylene group form C–Au bonds with the surface, resulting in the “standing” benzyne group. Such chemisorption configuration can effectively reduce the barrier of the following C–C coupling step, resulting in the formation of the four-membered rings. Importantly, the chemisorbed precursors with standing benzyne groups are not only identified by theoretical calculations but also observed by corroborating Scanning Tunnelling Microscopy (STM) experiments.
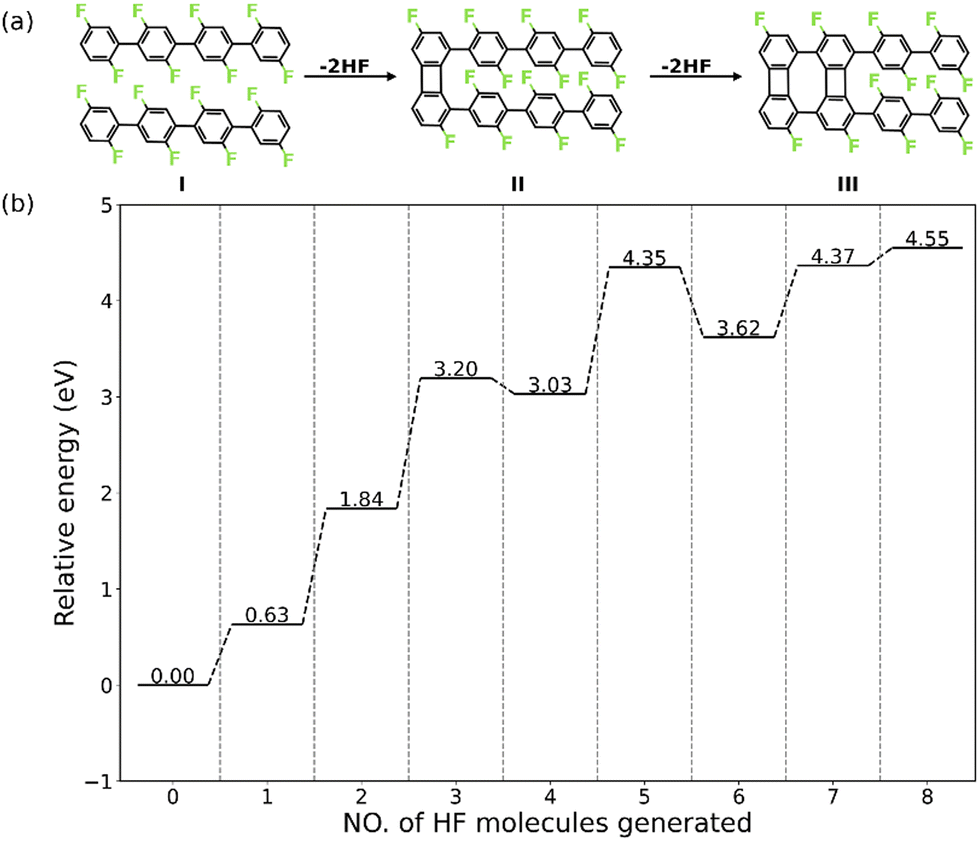 | ||
| Fig. 1 (a) Schematic illustration of the first four HF zipping reactions. (b) The stepwise energy profile of interchain HF zipping reactions on the Au(111) surface. | ||
Results and discussion
The intermolecular HF zipping reaction, between poly-difluorophenylene on Au(111), involves multiple dehydrofluorination steps and requires high temperatures (600 K to 700 K) to be triggered experimentally.30 In order to reduce the complexity of the system without compromising the integrity of the pathway, we model the intermolecular HF zipping using oligomers consisting of four difluorophenylene groups (valence bond structure I, Fig. 1a). To obtain a holistic knowledge of the reaction mechanism, we firstly investigate thermodynamic properties of each dehydrofluorination step. The intermolecular HF zipping is initiated from the physisorbed molecular precursors I. The lateral condensation starting from I therefore includes eight dehydrofluorinations, leading to the formation of four four-membered and three eight-membered carbon rings, together with eight HF molecules. At each step, the relative energy (ΔEn) is calculated by| ΔEn = En − E0 − n × EHF(g) | (1) |
Nevertheless, the dehydrofluorination steps are predicted to be thermodynamically favored (i.e., the Gibbs reaction free energy is negative) under the experimental conditions (T = 700 K and p = 1 × 10−10 mbar),30 in which the entropy of each generated HF molecule will contribute significantly with −3.25 eV to the free energy of each step (eqn (S6), ESI†), making the endothermic reaction highly exergonic (Fig. S1, ESI†).21 Such significant change of the reaction energy indicates that it is the reaction kinetics that is vital in each dehydrofluorination step and what hampers the reaction from proceeding at lower temperature. Furthermore, the stepwise energy profile suggests that the HF zipping reactions can be categorized into two types: scenario (i) the HF zipping at the terminal phenyl groups (n = 1, 2, 7, 8), in which each HF elimination reaction is endothermic, and scenario (ii) the HF zipping at the phenylene groups in the middle of molecular chains (n = 3, 4, 5, 6), in which the HF elimination exhibits endothermic characteristic when n is odd while the reaction is exothermic when n is an even number. The appropriateness of the model system has been verified by employing a molecular precursor consisting of six difluorophenylene groups. By comparing stepwise energy profiles (Fig. 1b and Fig. S2, ESI†) for different precursors, the reaction energies for the formation of first 2 four-membered rings exhibit good numerical convergence (1.84 eV vs. 1.92 eV at n = 2, and 3.03 eV vs. 3.12 eV at n = 4 for 4-ring precursor and 6-ring precursor, respectively). Therefore, the 4-ring precursor (I in Fig. 1a) is expected to model the formation of the two first four-membered rings of the biphenylene network accurately. The second four-membered ring formation for the 4-ring precursor mimics the behavior of the corresponding reaction between central units of the 6-ring precursor well and is expected to describe the reaction mechanism relevant for the real polymer system. For these reasons, investigations of the reaction mechanism are focused on the first 4 HF zipping steps, consisting of the formation of four-membered ring at terminal phenyl group (I to II, corresponding to the scenario (i) and at phenylene bridge in the middle of precursors (II to III, corresponding to the scenario (ii)).
Generally, one HF zipping reaction consists of the elimination of the HF molecule and the C–C coupling. The first question to answer is which reaction step takes place first. The physisorbed configuration of the precursor provides three alternatives to trigger the HF zipping, namely, concerted HF elimination and C–C coupling, dehydrogenation, and defluorination. However, our calculations have shown that the concerted pathway exhibits high energy barriers (larger than 4 eV, see Fig. S3, ESI†), while activation energies for the dehydrogenation and defluorination are significantly lower. In addition, the activation energy for the defluorination is 0.37 eV lower than that of the dehydrogenation (2.14 eV for defluorination vs. 2.51 eV for dehydrogenation, Fig. S4, ESI†). Therefore, the Au surface-catalyzed defluorination is identified as the first step of HF zipping reactions.
Fig. 2 summarizes the most favored mechanism for the formation of the 1st four-membered ring. As seen, two main stages are included in the pathway: the surface-catalyzed defluorinations and dehydrogenations (S0–S4), and the C–C coupling between chemisorbed precursors (S4–S5b). Detailed reaction pathways for the first stage (S0–S4) are depicted in Fig. 3, in which two defluorinations and two dehydrogenations are incorporated. Unlike the polarized C–F bond created by the γ-alumina catalyst,8 the C–F bond at the terminal phenyl group is completely activated with an energy barrier of 2.14 eV, resulting in the chemisorption of the defluorinated precursor on the Au(111) surface (S0–S1). Starting from S1, the HF elimination can be achieved by two mechanisms, namely, inter-polymer mechanism (the 1st HF is formed after the intermolecular C–C coupling) and the intra-polymer mechanism (the 1st HF is formed within the same precursor). The former mechanism has been proposed for the cyclodehydrofluorination on metal oxides, in which the C–F bond is catalyzed by metal atoms allowing the Friedel–Crafts–like arylation simultaneously.33 However, the intermolecular C–C coupling starting from S1 is not favored due to the relatively high energy barrier of 1.86 eV (Fig. S6, ESI†). Such high barrier can be ascribed to strong C–Au interactions which prohibit the C–C coupling.34 Furthermore, the activation energy for the dehydrogenation in the intra-polymer mechanism is found to be significantly reduced due to the close proximity of the C–H bond to the surface.35 As shown in Fig. 3, the C–H activation exhibits a smaller barrier than that of the defluorination in the first step (ETS2 – ES1 = 1.59 eV < ETS1 – ES0 = 2.14 eV). Such low C–H activation energy indicates that the defluorination and dehydrogenation at the same phenyl/phenylene group will always take place consecutively. Therefore, not only the intermolecular mechanism (Fig. S5–S7, ESI†) can be excluded, but also the further reactions can be predicted. Starting from S2, similar reactions can be identified for the second precursor molecule, in which defluorination and dehydrogenation take place sequentially (S2–S4) with all energy barriers lower than the first defluorination step. Accordingly, both precursors are chemisorbed on the Au surface with standing benzyne rings, together with 2 HF molecule released. It is noted that the benzyne-like groups generated by C–F/C–H activations can directly achieve homo-coupling to form four-membered rings, which is known as the benzyne mechanism in organic chemistry.36 However, the benzyne-like groups exhibit strong interactions with the surface by forming C–Au bonds, resulting in chemisorbed phenylene groups (S4). Such molecule–surface interactions agree well with the experimental observation of C–Au bonds by X-ray photoelectron spectroscopy (XPS).30 As a result, the existence of chemisorbed phenylene groups indicates that the benzyne mechanism is merged with the intra-polymer mechanism.
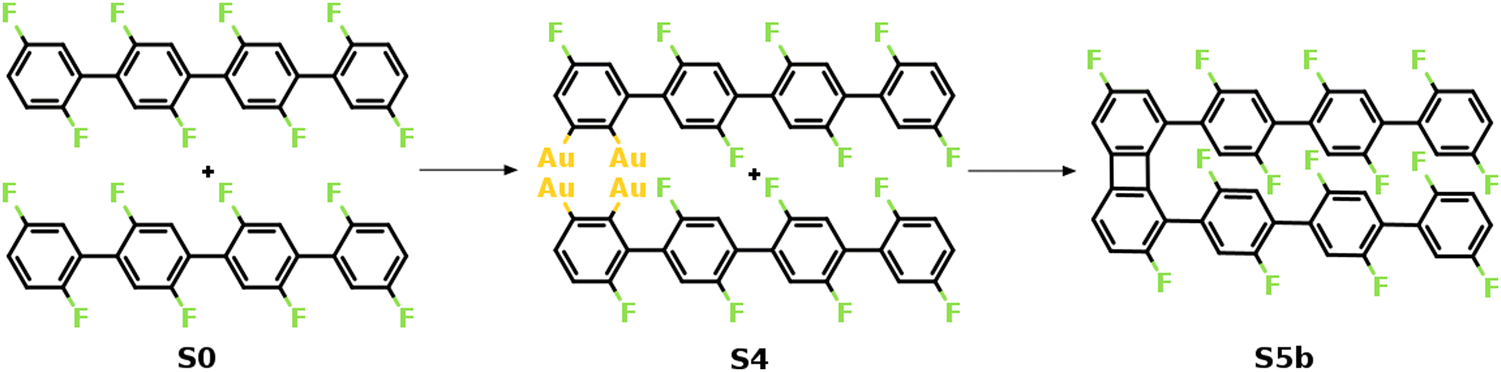 | ||
| Fig. 2 Mechanism of the formation of the 1st four-membered carbon ring. The valence structures are labeled consistently with the calculated reaction pathway. | ||
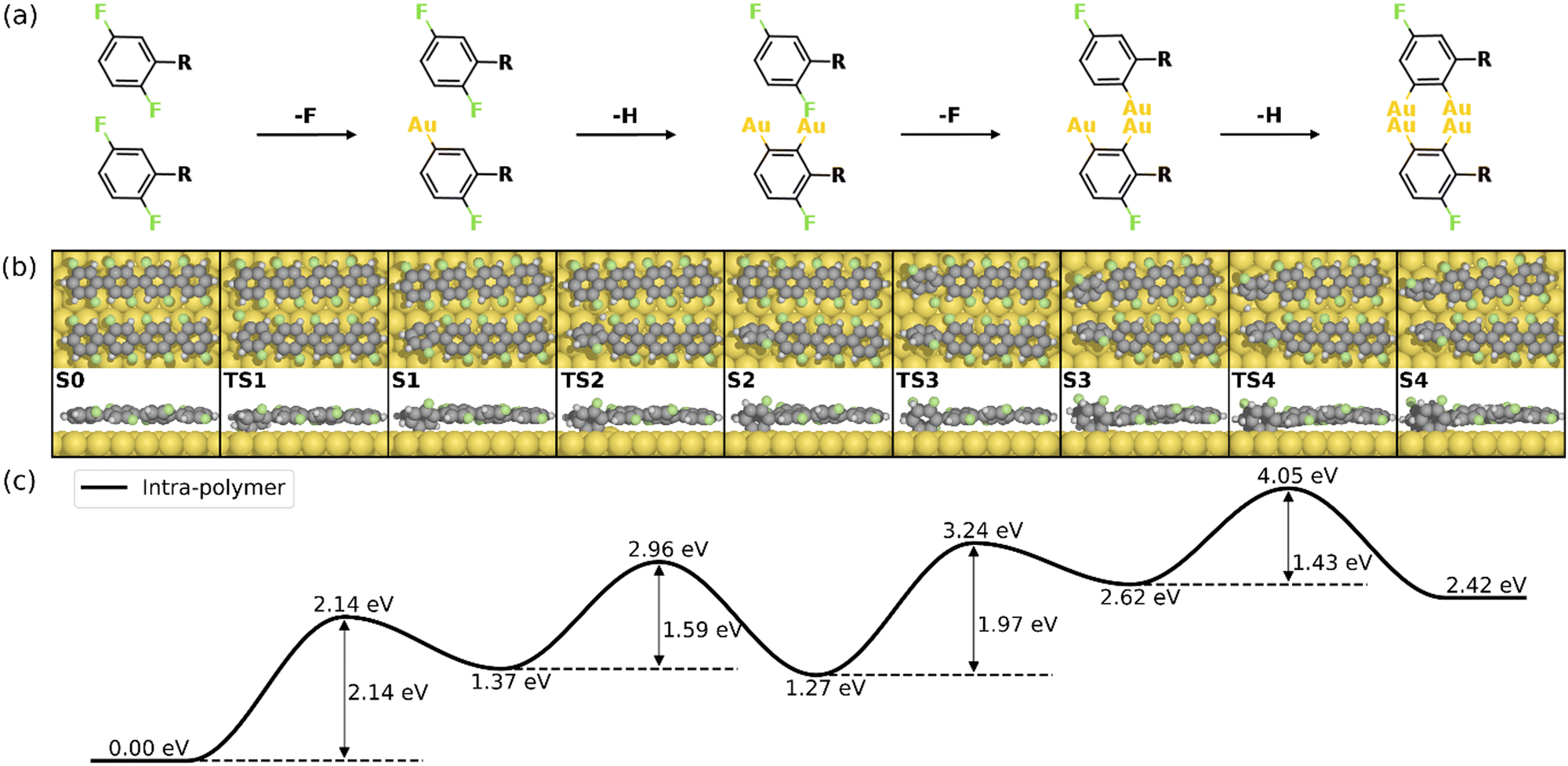 | ||
| Fig. 3 Reaction mechanism of the first four steps of the HF zipping reactions (S0–S4). (a) Valence bond structures for each intermediate state. (b) The reaction pathway and (c) the corresponding energy profile. Definition of energies is given in eqn (S3) (see ESI†). The C, H, F, and Au atoms are represented by the gray, white, green, and yellow circles, respectively. | ||
The formation of the four-membered ring starting from S4 is shown in Fig. 4. As seen, two intermolecular C–C bonds are formed consecutively. The first C–C coupling (S4–S5a) exhibits a barrier of 1.52 eV, resulting in one C–C bond while the other C atoms remain chemisorbed. The four-membered ring is finalized by the second C–C coupling with a barrier of 0.97 eV. Note that neither of the C–C coupling barriers exceed the C–F activation energy (2.14 eV), i.e. the formation of the 1st four-membered ring can be expected once the first defluorination has proceeded. It is noted that the formation of the six-membered ring can be initiated at the S4 simultaneously. However, the C–C coupling steps for the formation of a six-membered ring exhibit higher activation energies than the formation of a four-membered rings (1.93 eV for a six-membered ring and 1.52 eV for a four-membered ring, see Fig. S8, ESI†). This is probably due to the strong twist needed to make a six-membered ring, which is anticipated to be even more difficult between central units of the PFFP chains. Furthermore, following the formation of the first four-membered ring the positions of the PFPP chains will be locked with respect to each other favoring the further formation of four- and eight-membered rings. Consequently, the formation of the BPN is preferred over graphene, agreeing well with experiments.30
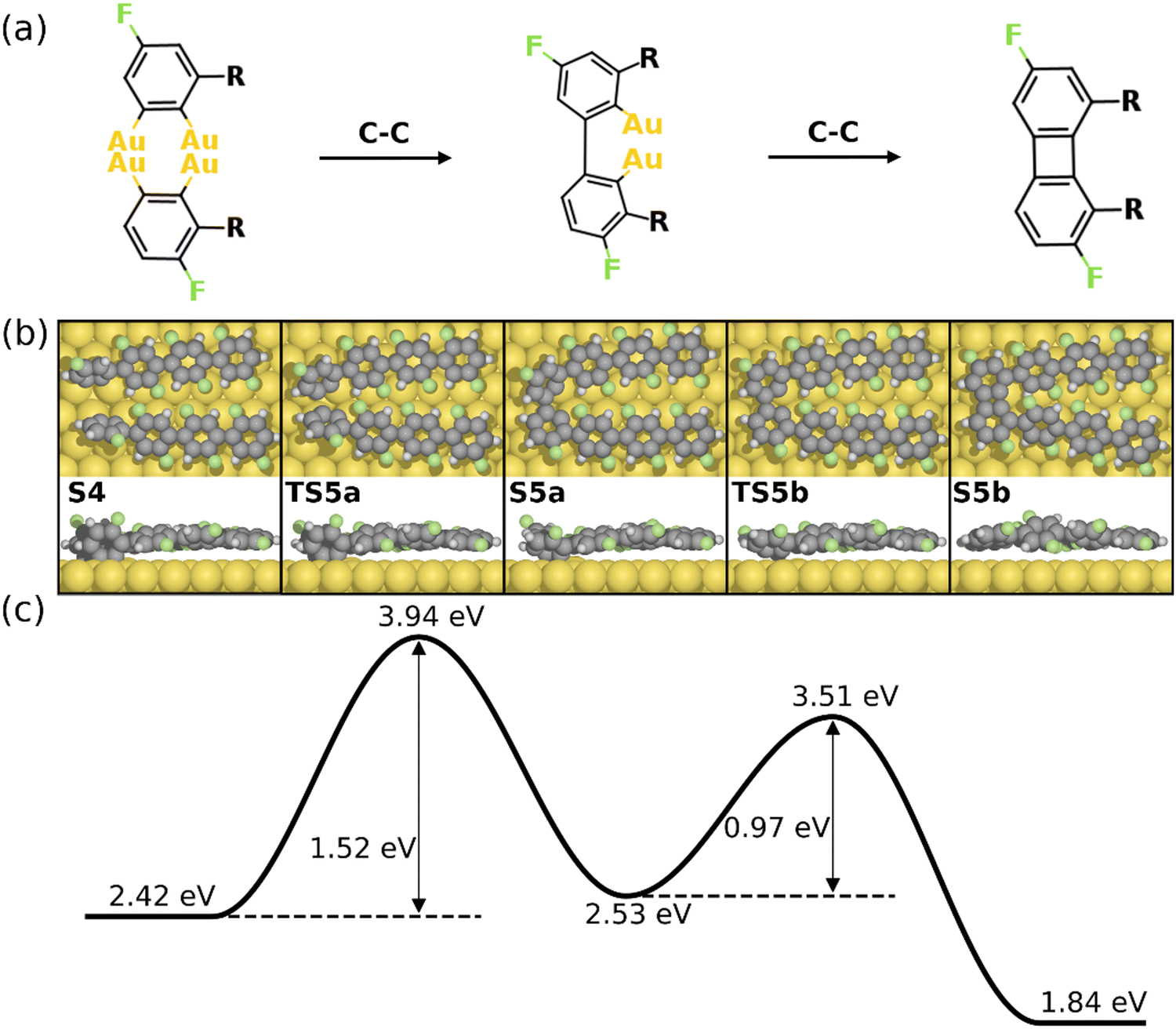 | ||
| Fig. 4 Reaction mechanism of C–C coupling steps for the formation of the 1st four-membered ring (from S4–S5b). (a) The valence bond structure of each intermediate states. (b) and (c) are the reaction pathway and the energy profile, respectively. Definition of energies are given in eqn (S3) (see ESI†). The C, H, F, and Au atoms are represented by the gray, white, green, and yellow circles, respectively. | ||
As previously shown, the formation of the four-membered ring at the phenylene groups in the middle of molecular chains possesses different thermodynamic characteristics compared to that at the terminal phenyl rings. To be specific, the 3rd C–C coupling is an endothermic reaction while the 4th C–C coupling is exothermic at 0 K, indicating that it is presumably easier to form the 2nd four-membered ring. However, the reaction pathway shows that the reaction is kinetically restricted. The most favored reaction pathway for the formation of the 2nd four-membered ring is summarized in Fig. 5, including four stages (S5b-S12). The first stage (S5b-S8) follows the same mechanism of the 1st four-membered ring formation, in which the defluorination and the dehydrogenation proceed successively at the same phenylene group. As shown in Fig. 6, the reaction initiates at S5b, in which two precursors are connected via the 1st four-membered ring. In order to achieve the close proximity of the target C–F bond to the surface, an extra rotation step of the phenylene group is required (S5b-S6). Starting from S6, the C–F bond scission can be catalyzed by the surface Au atom, generating a phenylene radical stabilized by the C–Au bond (S7). Although two steps are included, the overall barrier for the defluorination (S5b-S7) is similar to other defluorination steps (2.02 eV). Such barrier indicates that the non-planar geometry of phenylene groups will not significantly influence the activity of defluorinations. In addition, the chemisorption in S7 promotes reaction activity of the following dehydrogenation, leading to a low C–H activation energy of 1.29 eV (S7 and S8). Furthermore, the possibility of the C–C coupling directly after S7, following the inter-polymer mechanism, has been investigated (summarized in Fig. S9–S11, ESI†). However, such mechanism can be effectively prohibited due to the relatively high barrier of the direct C–C coupling (2.07 eV, Fig. S11, ESI†). Therefore, subsequent steps are considered starting from the chemisorbed phenylene group (S8).
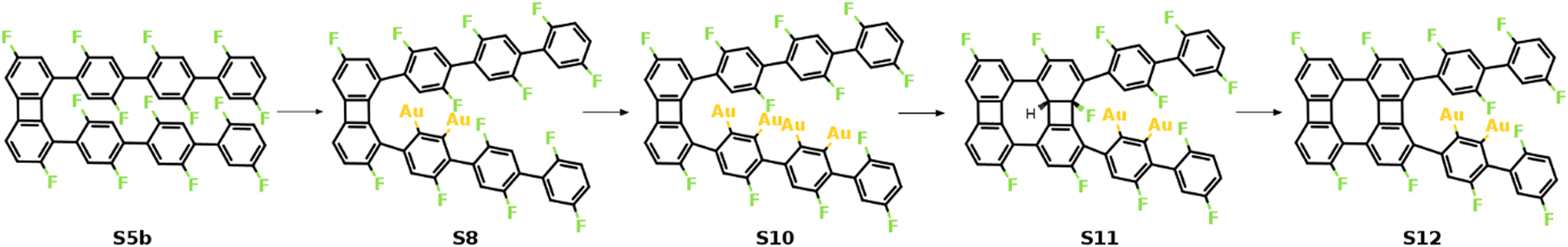 | ||
| Fig. 5 Schematic mechanism of the formation of the 2nd four-membered carbon ring. Valence structures are labeled consistently with the calculated reaction pathway. | ||
 | ||
| Fig. 6 Reaction mechanism of the formation of the 2nd four-membered ring (S5b–S8). (a) The valence bond structures for each intermediate state. Note that two precursors are now connected by the 1st four-membered ring, which is simplified to “R–R”. (b) The reaction pathway and (c) the energy profile for the defluorination and dehydrogenation reactions. Definition of energies is given in eqn (S3) (see ESI†). The C, H, F, and Au atoms are represented by the gray, white, green, and yellow circles, respectively. | ||
Starting from S8, it is crucial to clarify whether the formation of the 2nd four-membered ring follows the same mechanism as that of the 1st four-membered ring, i.e., whether the defluorination and dehydrogenation take place serially on the other molecular chain. However, DFT calculations show that the defluorination on the second precursor molecule exhibits an activation energy of 2.42 eV, which is higher than all the other defluorination steps (Fig. S12, ESI†). Furthermore, the consecutive dehydrogenation possesses a barrier of 1.89 eV, which is about 0.5 eV higher than other dehydrogenations. Such high activation energies can be ascribed to the mismatch between the phenylene group and the Au surface. Generally, the stoichiometry between the metal active sites and target C–F/C–H bond is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, i.e., one metal atom can only catalyze one C–F/C–H bond.37 However, due to the rigidity of the 1st four-membered ring, only one surface Au atom can serve as the active site for both the defluorination (S8–S9’) and dehydrogenation (S9’–S10’). As a result, the defluorination and dehydrogenation possess higher energy barriers. In addition, the standing benzyne group generated by defluorination and dehydrogenation prefers to form two C–Au bonds with separate surface Au atoms (e.g., S4 and S8) in a “di-σ” manner.38 However, such configuration leads to this benzyne group rotating further from the other molecule (S10’), providing additional steric hindrance. Therefore, a more difficult C–C coupling step with a barrier as high as 2.64 eV is identified (S10’–S11’), indicating that such pathway is unlikely to take place on the Au(111) surface.
:1, i.e., one metal atom can only catalyze one C–F/C–H bond.37 However, due to the rigidity of the 1st four-membered ring, only one surface Au atom can serve as the active site for both the defluorination (S8–S9’) and dehydrogenation (S9’–S10’). As a result, the defluorination and dehydrogenation possess higher energy barriers. In addition, the standing benzyne group generated by defluorination and dehydrogenation prefers to form two C–Au bonds with separate surface Au atoms (e.g., S4 and S8) in a “di-σ” manner.38 However, such configuration leads to this benzyne group rotating further from the other molecule (S10’), providing additional steric hindrance. Therefore, a more difficult C–C coupling step with a barrier as high as 2.64 eV is identified (S10’–S11’), indicating that such pathway is unlikely to take place on the Au(111) surface.
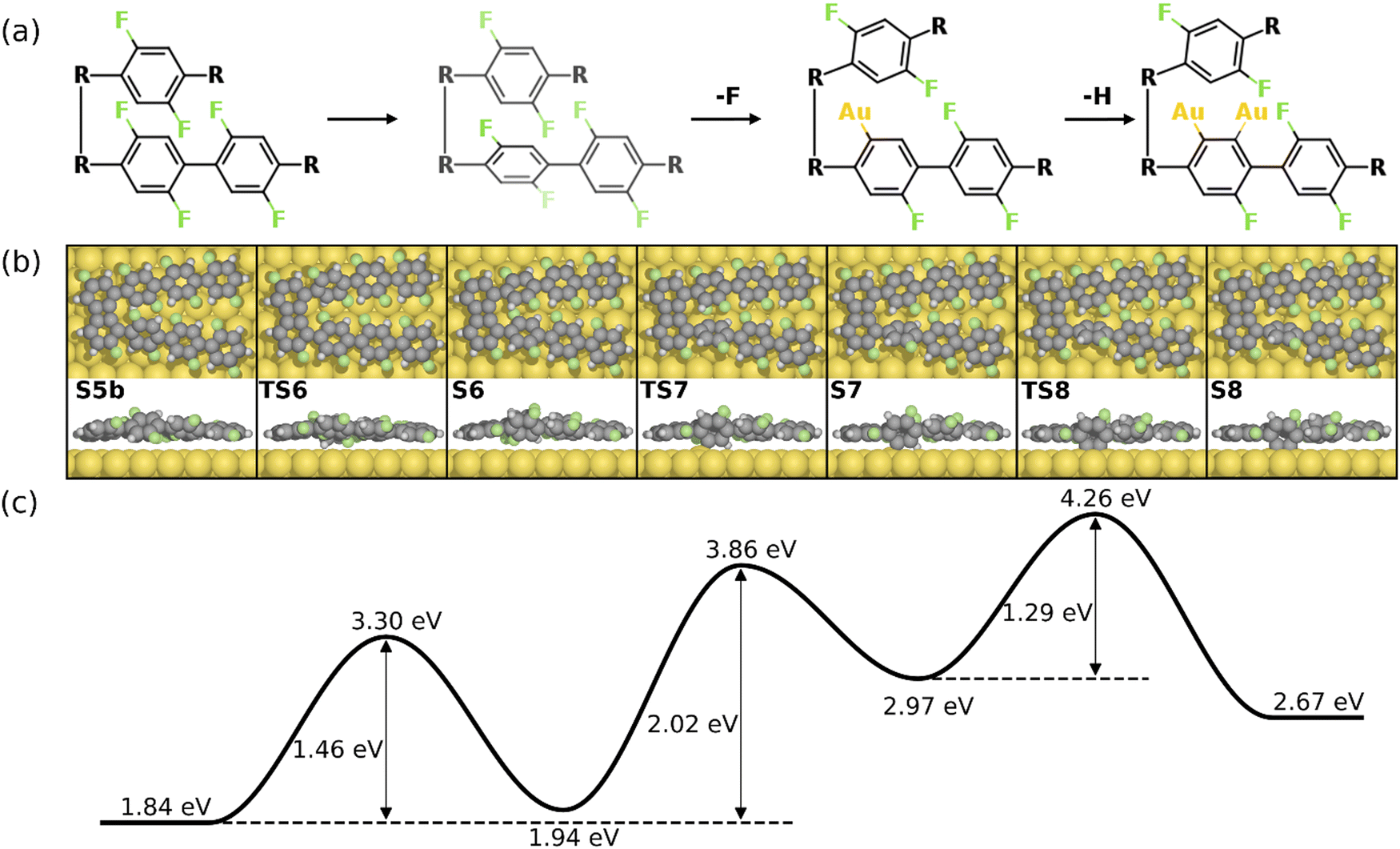
Alternatively, the defluorination can be proceeded on the same precursor with the chemisorbed standing benzyne group. Fig. 7 depicts the final four steps of the formation of the 2nd four-membered ring. As seen, the continued defluorination (S8 and S9) and dehydrogenation (S9 and S10) on the same precursor molecule exhibit lower activation energies than those on the other precursor molecule (2.06 eV vs. 2.42 eV and 1.60 eV vs. 1.89 eV). Passing through TS10, two phenylene groups in the same precursor form C–Au bonds in the “di-σ” configuration while the phenylene groups in the other precursor molecule remain unchanged (S10). The C–C coupling between one chemisorbed benzyne and the pristine phenylene can proceed in succession, leading to the formation of a four-membered carbon ring with H and F attached (S10 and S11). Taking into account that the C–C coupling between two standing benzyne groups require an energy barrier of 2.64 eV (S10’ and S11’ in Fig. S12, ESI†), the coupling between one benzyne group and one pristine group is more likely (ETS11 – ES10 = 2.41 eV). Such decreased barrier suggests that the adjacent chemisorbed benzyne group can effectively increase the activity of C–C coupling. In addition, the coupling step (S10 and S11) is also identified as the rate-limiting step as it exhibits the highest energy barrier in the overall intra-polymer mechanism. After the formation of the four-membered carbon ring, the dehydrofluorination (S11 and S12) can be proceeded with a relatively small activation energy (1.63 eV). This concludes the intra-polymer mechanism of HF zipping reactions, in which the building block (S12) containing four-, six-, and eight-membered rings are generated for the further synthesis of two-dimensional carbon allotropes.
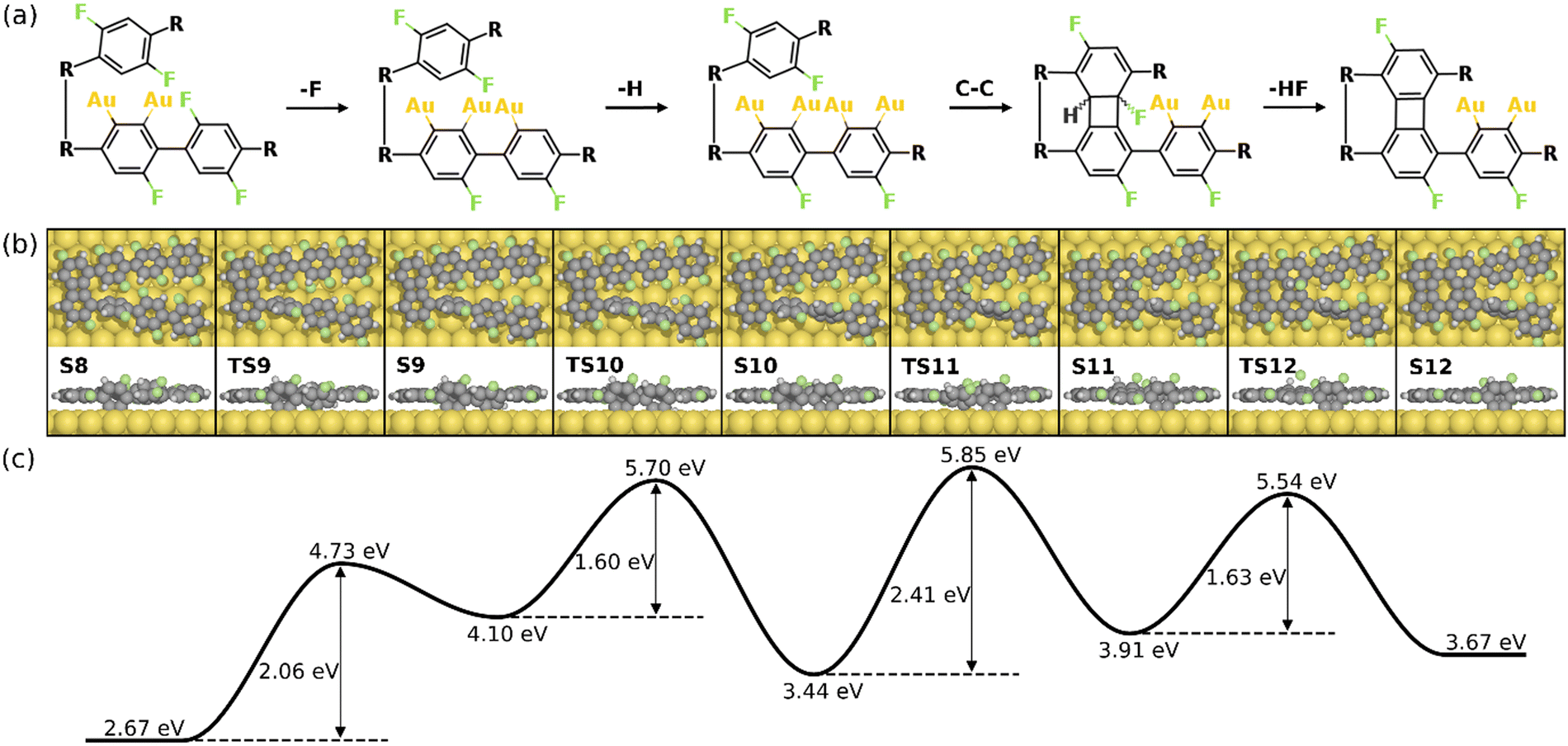 | ||
| Fig. 7 Reaction mechanism of the last four steps in the formation of the 2nd four-membered carbon ring (S8–S12). (a) The valence bond structures of each intermediate states. (b) The reaction pathway and (c) the corresponding energy profile. Definition of energies are given in eqn (S3) (see ESI†). The C, H, F, and Au atoms are represented by the gray, white, green, and yellow circles, respectively. | ||
As a final remark, the convergence of the energy profile has been investigated to obtain accurate energies. The relative energies for each intermediate state and corresponding reaction energies of each reaction step are summarized in Table S1 (ESI†). The overall energy profile increases due to the decreasing of the molecule–surface interactions in denser k-point sampling. Nevertheless, most reaction steps show good numerical convergence at the 2 × 2 k-point sampling (below 0.1 eV).39 However, the C–C coupling steps are demonstrated to be sensitive to the k-point sampling since they result in the most significant change in molecule–surface interaction (two C–Au bonds break simultaneously as C–C bonds form). We therefore also elucidate the transition states for key C–C coupling steps (S4–S5b and S10–S11) with 2 × 2 k-point sampling. The energy profiles for C–C coupling steps show that denser k-point sampling leads to decreased energy barriers (Fig. S13 and S14, ESI†). In particular, the rate-limiting step for the formation of the 2nd four-membered ring is reduced from 2.41 eV to 2.29 eV (Fig. S14, ESI†). Importantly, this does not influence the selectivity and the identification of the rate-limiting step, and the energy profile obtained with Γ point sampling are considered adequate to explain the reaction mechanism. Furthermore, the thermodynamics for rate-limiting steps are investigated by calculating the Gibbs free energy profiles (eqn S7–S9, ESI†) in order to investigate the effects of vibrational enthalpy and entropy at a temperature of 700 K. Our calculations have shown that the vibrational enthalpy and entropy due to high temperature exhibit limited contribution to the rate-limiting barriers (barriers decreased around 0.1 eV, see Fig. S15 and S16, ESI†). Thus, the calculated potential energy profiles are sufficient to understand the formation mechanism of BPN.
In the proposed intra-polymer mechanism, the C–Au bonds are inevitably generated after defluorination and dehydrogenation, yielding chemisorbed benzyne groups standing on the surface. Such bonding characteristics have been previously been proposed based on XPS measurements.30 More importantly, the intermediate states containing standing benzyne groups are observed by STM. Fig. 8a shows the formed poly(2,5-difluoro-para-phenylene) (PFPP) chains with intact C–F groups at 470 K (corresponding to S0). After annealing of PFPP chains to 600 K, C–F bonds and C–H bonds are partially activated, resulting in bright protrusions in the PFPP chains (Fig. 8b). Furthermore, apparent height profile line-scan shows that the height fluctuation of the PFPP chain is less than 10 pm, while bright protrusions are about 80 pm higher than the PFPP chain (Fig. 8c and d). It is worthy to note that the C–C coupling between chemisorbed benzyne groups is the rate-limiting step, the intermediate states containing standing benzyne groups are therefore expected to be stabilized on the surface. Consequently, the standing benzyne groups can be observed as bright protrusions as they exhibit higher apparent height with respect to the rest of the molecular chain, agreeing well to STM simulations where the standing phenylene groups appear brighter than the rest of molecular precursors (Fig. S17, ESI†). Such experimental observations and simulation results corroborate the reaction mechanism proposed by theoretical calculations.
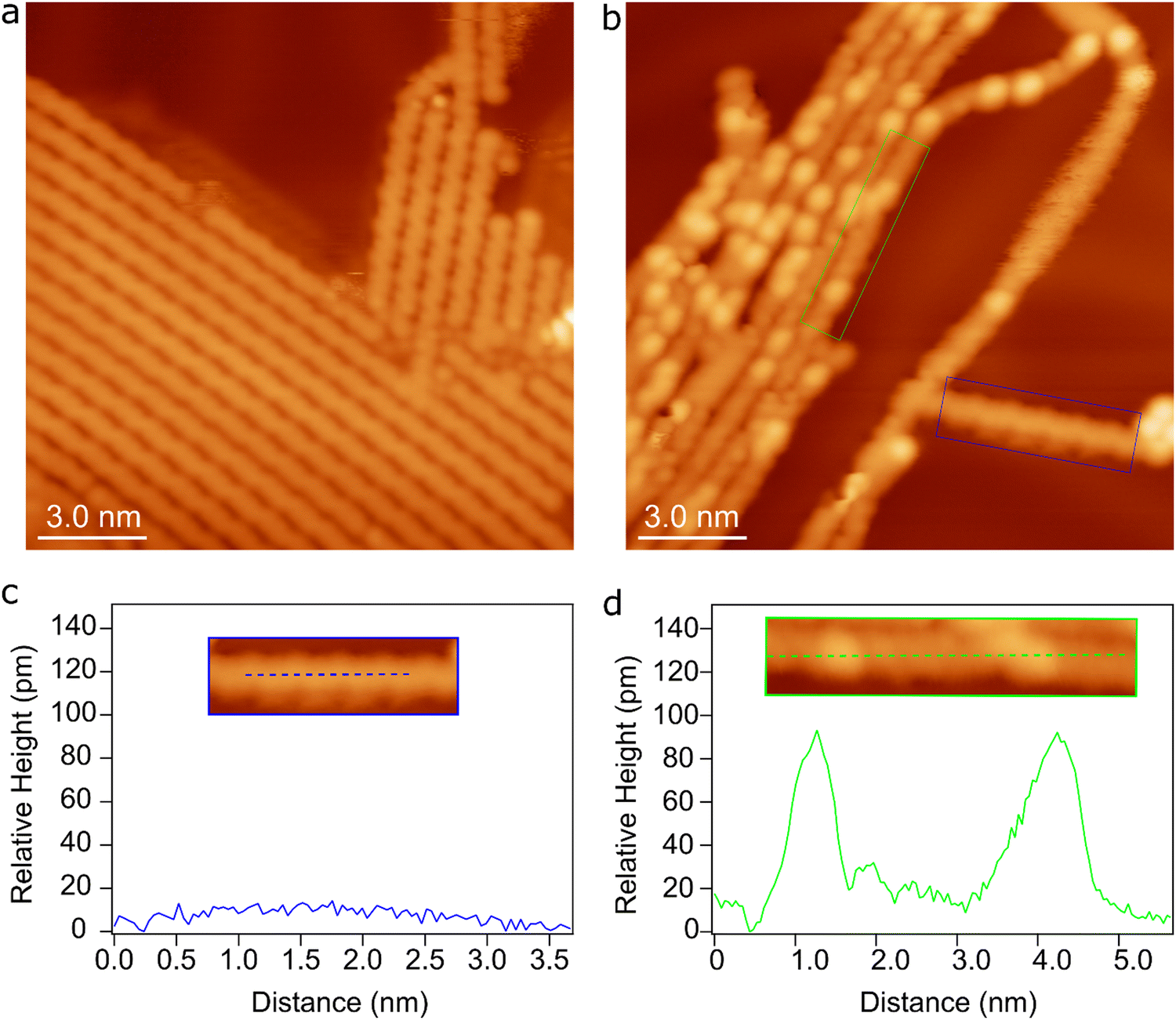 | ||
| Fig. 8 STM images taken after deposition of a submonolayer of 4,4′′-dibromo-2,2′,2′′,5,5′,5′′-hexafluoro-1,1′:4′,1′′-terphenyl onto Au(111) followed by annealing to (a) 473 K and (b) 600 K. Tunneling parameters: (a) U = −1.8 V, I = 0.17 nA; (b) U = −0.5 V, I = 0.25 nA. (c) and (d) show the apparent height profiles along the blue and green line in the insets, which are cut out from panel (b) as marked by the blue and green rectangles. | ||
Conclusions
In conclusion, we have investigated the mechanism for the intermolecular HF zipping reactions on the Au(111) surface. The formation of four-membered carbon rings follows the intra-polymer mechanism, in which the C–F and C–H activations take place consecutively, followed by the C–C coupling. Of importance, surface Au atoms not only serve as active sites to catalyze target C–F/C–H bonds, but also form C–Au bonds with the defluorinated and/or dehydrogenated phenylene groups, leading to intermediate states with standing benzyne groups. Such chemisorption configuration can effectively reduce the C–C coupling barriers, making HF zipping reactions possible on the surface. Of importance, the reduced C–C coupling barrier guarantees a higher selectivity of HF zipping reactions towards the biphenylene network over graphene, in which the formation of six-membered rings is kinetically suppressed. Furthermore, our experimental observation confirms the existence of the standing benzyne groups, validating the proposed intra-polymer mechanism. The insights provided in the present work contribute significantly to the understanding of this new on-surface synthesis approach. We anticipate such mechanism can broaden the application of on-surface HF zipping and the accelerate the rational design of low-dimensional carbon allotropes.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge funding by the Swedish Research council, the Deutsche Forschungsgemeinschaft (223848855-SFB1083), and the LOEWE Focus Group PriOSS. J. R. acknowledges support from the Knut and Alice Wallenberg (KAW) Foundation for a Fellowship/Scholar Grant, and from the Göran Gustafsson foundation. K. N. and L. C. acknowledge the Collaborative Innovation Centre of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the 111 Project. Q. F. thanks the Alexander von Humboldt-Foundation for a Research Fellowship for Postdoctoral Researchers. This work was supported by the National Natural Science Foundation of China (NSFC, Grant No. 21790053, and 51821002) and the Ministry of Science and Technology (2017YFA0205002). Computational resources were allocated at the National Supercomputer Centre, Sweden, and PDC Center for High Performance Computing, both allocated by SNIC, as well as at the LUMI supercomputer in CSC's data center in Finland.References
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- H. R. Karfunkel and T. Dressler, J. Am. Chem. Soc., 1992, 114, 2285–2288 CrossRef CAS.
- P. A. Held, H. Fuchs and A. Studer, Eur. J. Chem., 2017, 23, 5874–5892 CrossRef CAS PubMed.
- H. Zhang, H. Lin, K. Sun, L. Chen, Y. Zagranyarski, N. Aghdassi, S. Duhm, Q. Li, D. Zhong, Y. Li, K. Mullen, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- D. Sharapa, A.-K. Steiner and K. Amsharov, Phys. Status Solidi B, 2018, 255, 1800189 CrossRef.
- X. Zhou and G. Yu, Adv. Mater., 2020, 32, 1905957 CrossRef CAS PubMed.
- F. Diederich and M. Kivala, Adv. Mater., 2010, 22, 803–812 CrossRef CAS PubMed.
- D. F. Perepichka and F. Rosei, Science, 2009, 323, 216–217 CrossRef CAS PubMed.
- Y. Q. Zhang, N. Kepcija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- H. Y. Gao, P. A. Held, M. Knor, C. Muck-Lichtenfeld, J. Neugebauer, A. Studer and H. Fuchs, J. Am. Chem. Soc., 2014, 136, 9658–9663 CrossRef CAS PubMed.
- M. Matena, T. Riehm, M. Stöhr, T. A. Jung and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 2414–2417 CrossRef CAS PubMed.
- Y. He, M. Garnica, F. Bischoff, J. Ducke, M.-L. Bocquet, M. Batzill, W. Auwärter and J. V. Barth, Nat. Chem., 2017, 9, 33–38 CrossRef CAS PubMed.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61–67 CrossRef CAS PubMed.
- K. Amsharov, Phys. Status Solidi B, 2016, 253, 2473–2477 CrossRef CAS.
- G. Otero, G. Biddau, C. Sánchez-Sánchez, R. Caillard, M. F. López, C. Rogero, F. J. Palomares, N. Cabello, M. A. Basanta, J. Ortega, J. Méndez, A. M. Echavarren, R. Pérez, B. Gómez-Lor and J. A. Martín-Gago, Nature, 2008, 454, 865–868 CrossRef CAS PubMed.
- J. Björk, F. Hanke and S. Stafström, J. Am. Chem. Soc., 2013, 135, 5768–5775 CrossRef PubMed.
- J. Björk, Y. Q. Zhang, F. Klappenberger, J. V. Barth and S. Stafström, J. Phys. Chem. C, 2014, 118, 3181–3187 CrossRef.
- J. Björk, J. Phys. Chem. C, 2016, 120, 21716–21721 CrossRef.
- Z. Zhang, D. F. Perepichka and R. Z. Khaliullin, J. Phys. Chem. Lett., 2021, 12, 11061–11069 CrossRef CAS PubMed.
- S. Blankenburg, J. Cai, P. Ruffieux, R. Jaafar, D. Passerone, X. Feng, K. Müllen, R. Fasel and C. A. Pignedoli, ACS Nano, 2012, 6, 2020–2025 CrossRef CAS PubMed.
- A.-K. Steiner and K. Y. Amsharov, Angew. Chem., Int. Ed., 2017, 56, 14732–14736 CrossRef CAS PubMed.
- M. Kolmer, R. Zuzak, A. K. Steiner, L. Zajac, M. Engelund, S. Godlewski, M. Szymonski and K. Amsharov, Science, 2019, 363, 57–60 CrossRef CAS PubMed.
- C. Ashworth, Nat. Rev. Mater., 2019, 4, 81 CrossRef.
- M. Kolmer, A.-K. Steiner, I. Izydorczyk, W. Ko, M. Engelund, M. Szymonski, A.-P. Li and K. Amsharov, Science, 2020, 369, 571–575 CrossRef CAS PubMed.
- J. Hieulle, E. Carbonell-Sanromà, M. Vilas-Varela, A. Garcia-Lekue, E. Guitián, D. Peña and J. I. Pascual, Nano Lett., 2018, 18, 418–423 CrossRef CAS PubMed.
- S. Mishra, M. Krzeszewski, C. A. Pignedoli, P. Ruffieux, R. Fasel and D. T. Gryko, Nat. Commun., 2018, 9, 1714 CrossRef PubMed.
- Q. Fan, L. Yan, M. W. Tripp, O. Krejčí, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Liljeroth and J. M. Gottfried, Science, 2021, 372, 852–856 CrossRef CAS PubMed.
- I. Alcón, G. Calogero, N. Papior, A. Antidormi, K. Song, A. W. Cummings, M. Brandbyge and S. Roche, J. Am. Chem. Soc., 2022, 144, 8278–8285 CrossRef PubMed.
- Y. Liao, X. Shi, T. Ouyang, J. Li, C. Zhang, C. Tang, C. He and J. Zhong, J. Phys. Chem. Lett., 2021, 12, 8889–8896 CrossRef CAS PubMed.
- J. Ichikawa, M. Yokota, T. Kudo and S. Umezaki, Angew. Chem., Int. Ed., 2008, 47, 4870–4873 CrossRef CAS PubMed.
- Q. Zhong, K. Niu, L. Chen, H. Zhang, D. Ebeling, J. Björk, K. Müllen, A. Schirmeisen and L. Chi, J. Am. Chem. Soc., 2022, 144, 8214–8222 CrossRef CAS PubMed.
- Q. Fan, S. Werner, J. Tschakert, D. Ebeling, A. Schirmeisen, G. Hilt, W. Hieringer and J. M. Gottfried, J. Am. Chem. Soc., 2018, 140, 7526–7532 CrossRef CAS PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic chemistry, Oxford university press, 2nd edn, 2012 Search PubMed.
- Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS PubMed.
- P. S. Cremer, X. C. Su, Y. R. Shen and G. A. Somorjai, J. Phys. Chem., 1996, 100, 16302–16309 CrossRef CAS.
- C. Bronner, J. Björk and P. Tegeder, J. Phys. Chem. C, 2014, 119, 486–493 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The computational parameters and methods. The definition of relative energies of each intermediate state. The entropy contribution of the HF molecule to the reaction thermodynamics. The alternative first reaction step and the inter-polymer reaction pathway. The reaction pathway for the formation of the six-membered ring. The alternative coupling steps for the formation of the 2nd four-membered ring. The k-point sampling convergence of energies and the energy profile for the C–C coupling steps with 2 × 2 k-point sampling. The Gibbs free energy profiles for rate-limiting steps. STM simulations of the chemisorbed phenylene groups. See DOI: https://doi.org/10.1039/d2nh00528j |
| This journal is © The Royal Society of Chemistry 2023 |