
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reducing the pH dependence of hydrogen evolution kinetics via surface reactivity diversity in medium-entropy alloys†
Bao
Zhang‡
 *a,
Jia
Yao‡
bc,
Jia
Liu
d,
Tao
Zhang
a,
Houzhao
Wan
*bc and
Hao
Wang
bc
*a,
Jia
Yao‡
bc,
Jia
Liu
d,
Tao
Zhang
a,
Houzhao
Wan
*bc and
Hao
Wang
bc
aSchool of Physical and Mathematical Sciences, Nanyang Technological University, Singapore, 637371, Singapore. E-mail: bao.zhang@ntu.edu.sg
bHubei Yangtze Memory Laboratories, Wuhan 430205, China
cHubei Key Laboratory of Micro-Nanoelectronic Materials and Devices, School of Microelectronics, Hubei University, Wuhan, 430062, P. R. China. E-mail: houzhaow@hubu.edu.cn
dDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore, 117543, Singapore
First published on 30th August 2023
Abstract
The water dissociation step of the hydrogen evolution reaction is a well-known pH-dependent process, which makes sustainable hydrogen production suffer from sluggish kinetics. Herein, we demonstrate a surface reactivity diversity approach to reduce the pH dependence of HER kinetics in medium-entropy alloys. Grand canonical potential based calculation, CO-oxidation and potential of zero charge results showed that shifts in the Fermi level in neutral electrolytes lead to stronger M–H bonding (M = Ni, Pt, etc.) compared to those in basic solutions. These pH-dependent binding energies disrupt the optimized adsorption strength of advanced alkaline HER catalysts. By introducing a combination of a high surface reactivity metal (Mo) and a low surface reactivity metal (Cu/Zn) into Ni alloys, this surface reactivity diversity approach can significantly accelerate HER kinetics and allows for favorable adsorption of hydrogen and hydroxyl species at different pH levels. The resulting NiCuMo medium-entropy alloy exhibited impressive HER performance, with an overpotential of 63 mV at a current density of 100 mA cm−2 in alkaline electrolyte and 115 mV in neutral electrolyte. The intrinsic neutral HER activity of this NiCuMo is 3.65 times that of the benchmark alkaline HER catalyst. Furthermore, the NiCuMo-based membrane electrode assembly water electrolyzer can be stably operated for at least 200 h at a larger current density of 1.5 A cm−2. This surface reactivity diversity approach presents a promising design framework for less pH-dependent electrocatalysis.
Broader contextThe pH-dependent intermediate adsorption strength poses a challenge for pH universality of electrocatalysis. Effective approaches to this pH dependence problem have been rarely reported. Herein, we demonstrate a surface reactivity diversity strategy to medium-entropy alloys for reducing the pH dependence of the HER. By introducing a combination of a high surface reactivity metal (such as Mo) and a low surface reactivity metal (such as Cu/Zn) into Ni alloys, this surface reactivity diversity approach can significantly accelerate HER kinetics and also allows for favorable adsorption of hydrogen and hydroxyl species at different pH levels, resulting in a higher pH universality. Grand canonical potential based calculation, CO-oxidation and potential of zero charge results suggest that the shifts in the Fermi level at different pH levels are responsible for the pH-dependent binding energies, and these shifts can be compensated by the higher surface reactivity diversity of the medium-entropy alloy. This surface reactivity diversity strategy might open up new avenues for reasonable design of alloy catalysts and beyond. |
Renewable and sustainable pathways for producing fuels and chemicals could be a key part of our energy future.1–3 Hydrogen (H2) fuel is widely considered as a big player for sustainable energy.4–7 Among various hydrogen production approaches, electrocatalytic water splitting is expected to be the most promising candidate for green and large-scale hydrogen production.8–10 The cost factor is currently the main obstacle limiting the production of hydrogen from electrocatalytic water splitting. During electrocatalytic water splitting, hydrogen can be produced through water or proton reduction depending on the pH of the electrolyte. It is generally accepted that water reduction kinetics in basic and neutral media are much slower than those in acidic media, which mainly come from the sluggish kinetics of water dissociation.11–13
Substantial effort has been devoted to developing cost-effective HER electrocatalysts in basic14–16 and neutral media.17,18 Introduction of a water dissociation promoter is an important breakthrough in accelerating the water dissociation step, which was firstly reported by Markovic et al.11 In this approach, transition metal hydroxides are usually introduced as a water dissociation promoter and form a composite catalyst with the acidic HER electrocatalyst having suitable H* adsorption. The unsaturated transition metal atoms at the edges of the hydroxide usually have highly reactive d electrons. This can help cleave HO–H bonds and provides protons for the acidic HER electrocatalyst, leading to accelerated water dissociation kinetics. However, this high activity is only present near the interface. In addition to the difference between water and protons due to pH, the interfacial water and OH− may also affect the HER activity.19,20 This can result in an electrocatalyst surface that exhibits different hydrogen binding energies (HBE) in different pH environments.21,22 For example, Pt exhibits an optimal HBE under acidic conditions, but shows less satisfactory HBE under basic conditions. In this way, the optimal HBE under various pH values requires a wide range of surface reactivities.
Among various electrocatalysts, Ni based alloys and compounds are promising HER electrocatalysts with high activity, high stability and low cost. Commercially alkaline water electrolysis catalysts often involve porous nickel alloys, such as RANEY® nickel. NiMo alloys typically exhibit excellent alkaline hydrogen evolution activity.23 Besides, Ni based compounds, such as oxides24 and nitrides,25 also demonstrate notable HER catalytic activity. Herein, we present a surface reactivity diversity strategy for Ni alloy based HER electrocatalysts in non-acidic media. Specifically, we have constructed a NiCuMo medium-entropy alloy electrocatalyst that exhibits impressive performance. This electrocatalyst consists of Ni with medium surface reactivity towards the HER process, Mo with high surface reactivity that promotes water adsorption and dissociation, and Cu with low surface reactivity that accelerates hydrogen adsorption/desorption. The resulting electrocatalyst shows impressive HER electrocatalytic performance with ∼63 mV and ∼115 mV overpotentials at a current density of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mA cm−2 in 1 M KOH and pH7 buffer electrolyte, respectively.
mA cm−2 in 1 M KOH and pH7 buffer electrolyte, respectively.
We began with the theoretical calculations of the surface reactivity properties and HER electrocatalytic process of nickel and its binary alloys. The specific density functional theory (DFT) models of these alloys are shown in Fig. S1 (ESI†). As displayed in Fig. 1b, Ni has strong hydrogen adsorption with a free energy of hydrogen adsorption (GH) of −0.36 eV. However, water adsorption on the nickel surface is very weak, leading to a high energy barrier for water dissociation. In other words, hydrogen adsorption and water dissociation processes require different surface reactivities, with water dissociation requiring a much stronger surface reactivity. The water dissociation barriers (GTS) of Ni can reach over 1 eV, which indicates that Ni does not meet these requirements, resulting in unsatisfactory HER catalytic activity.
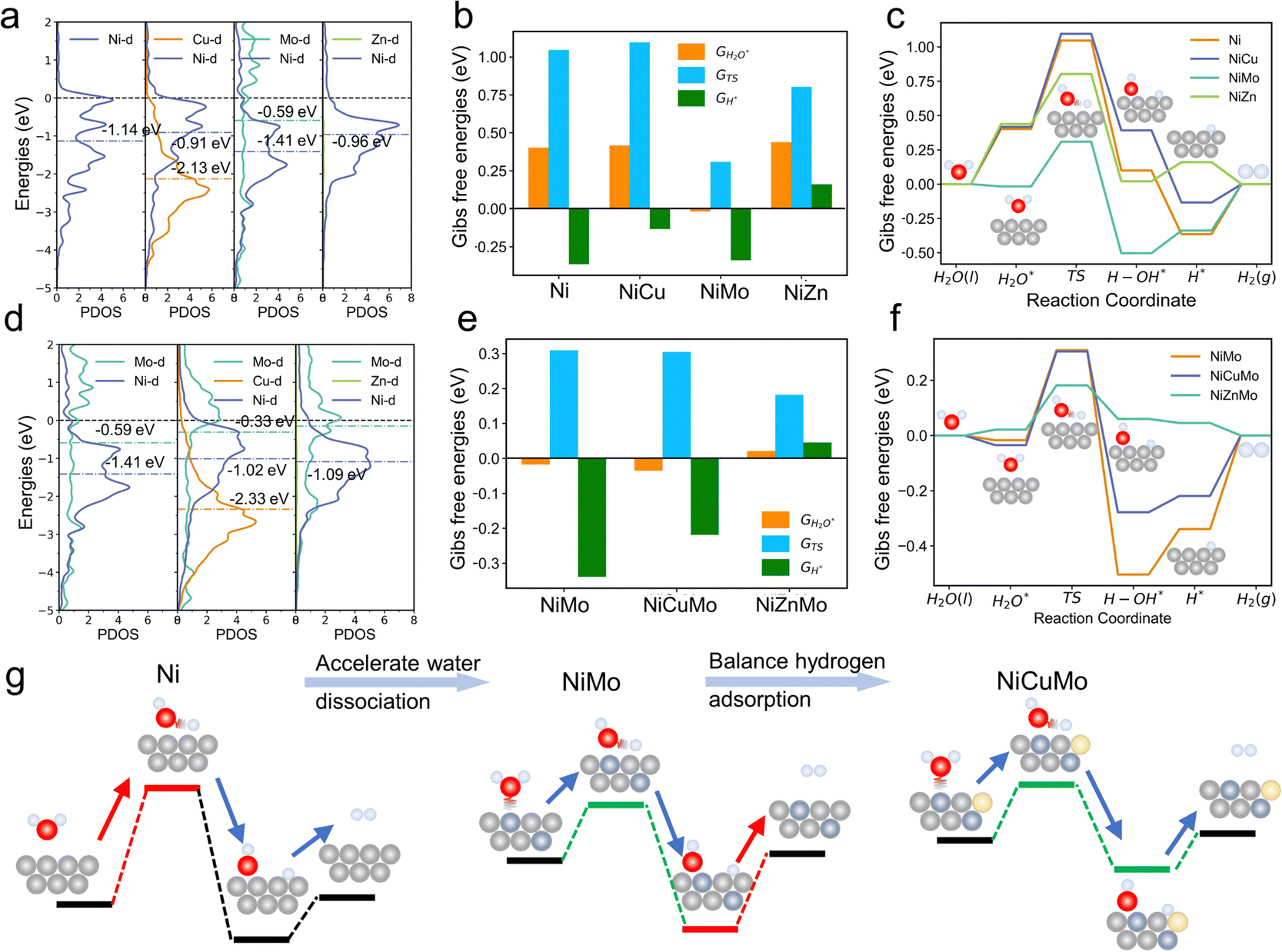 | ||
| Fig. 1 Theoretical design of Ni-based alloy. Projected density of states for the d orbitals of Ni and (a) Ni-based binary alloy, (d) NiMo alloy and NiMo-based ternary alloy. The free energy diagram of (b) and (c) Ni and Ni-based binary alloy, (e) and (f) NiMo alloy and Ni-based ternary alloy. (g) The schematic representation of the surface reactivity diversity enhanced HER mechanism for the NiCuMo electrocatalyst. Ni, Mo, Cu, O and H atoms are shown as light-grey, blue-grey, light-yellow, red and white balls, respectively. | ||
The d-band center (εd) is commonly used to evaluate the surface reactivity of metal and metal-like catalysts,26 with higher values indicating higher reactivity. As demonstrated in Fig. 1a, the εd of the Ni (111) surface is approximately −1.14 eV (vs. the Fermi level). When Cu is introduced, its lower εd (outside the lower boundaries) causes the εd of Ni to shift up to −0.91 eV. Although the weak surface reactivity of Cu can promote the GH of Ni to more optimal values, it does not assist with the water dissociation process. The synergistic effect between Ni and Cu has been utilized to improve the electrocatalytic activity of the catalysts for the HER,27 and we attribute this effect to the reactivity difference between Ni and Cu. This is analogous to the introduction of metal Zn with weak surface reactivity. In contrast to Cu and Zn, the introduction of Mo results in a higher εd and a shift in the εd down to −1.41 eV. Due to its high surface reactivity, Mo can interact strongly with water, leading to a much lower water dissociation energy barrier (∼0.31 eV). The dilemma remains that excessive H adsorption strength may be a major challenge for the HER activity of NiMo alloys.
To address the issue of inconsistent surface reactivity required for both hydrogen adsorption and water dissociation, a surface reaction diversity approach is proposed. This approach involves introducing both high surface reactivity metals and low surface reactivity metals into Ni alloys. As shown in Fig. 1d, the NiCuMo alloy has a diverse range of surface reactivity sites due to the wide variation of metal d-band centers, which range from −2.33 eV to −0.33 eV. As a result, the challenge of excessive hydrogen adsorption in NiMo alloys is improved by the presence of Cu, while the water dissociation energy barrier remains low (Fig. 1e and f). The introduction of weakly reactive Zn can also achieve accelerated kinetics. In the surface reactivity diversity strategy to medium-entropy alloy electrocatalysts (Fig. 1g), high surface reactivity metals are used to assist Ni and accelerate the water dissociation process, while low surface reactivity metals are used to improve the hydrogen adsorption strength of Ni. This allows for fast HER kinetics involving water to be achieved.
Encouraged by these results, Ni-based alloys were constructed by electrodeposition. As shown in Fig. S2 (ESI†), the alloy compositions were optimized, and uniform distributions of metal elements were found in these alloys (Fig. S3 and S4, ESI†). As illustrated in Fig. 2a and Fig. S5 (ESI†), the synthesized NiCuMo alloy features a pyramidal fractal structure, which maintains good contact with the metal foam while maintaining a robust microstructure under high currents.28,29 The XRD patterns of the NiCuMo alloy are shown in Fig. S6 (ESI†), and the typical peaks at 43.7° and 51.0° can be attributed NiCu(111) and NiCu(200) (PDF 03-065-9048). The NiCu crystalline phase has been previously reported in the co-electrodeposition process.30Fig. 2d shows the high-resolution TEM image of the NiCuMo alloy, demonstrating well-resolved lattice fringes. A lattice spacing of 0.207 nm is assigned to the (111) planes of the NiCu phase, in the middle range of pure Ni (0.202 nm) or Cu (0.209 nm). The corresponding TEM mappings (Fig. 2e) confirm the existence of Ni, Cu, and Mo. It is worth noting that the introduction of Cu inhibits the deposition of Mo, resulting in a dramatic decrease in the proportion of Mo. As shown in Table S2 (ESI†), the inductively coupled plasma optical emission spectroscopy (ICP-OES) test confirms low levels of Mo content (∼6.3 wt%). Mo is a rare metal and can be twice as expensive as Ni. This low Mo content is beneficial for reducing material costs. For comparison, Ni, NiCu and NiMo are also electrodeposited. As shown in Fig. S7 (ESI†), Ni and NiMo both exhibit smoother surface structures than NiCu, indicating the pyramidal fractal structure is mainly due to the introduction of Cu. The large electroactive area induced by Cu was consistent with previous reports.30,31 The NiZnMo was also electrodeposited with a much smoother surface than NiCuMo. The composition ratios of these alloys were evaluated by EDS shown in Fig. S8 and Table S1 (ESI†).
 | ||
| Fig. 2 Structure characterization of NiCuMo alloy. (a) Schematic of the preparation of the NiCuMo alloy by hydrogen bubble dynamic templated electrodeposition. (b) and (c) The SEM and (d) TEM images of NiCuMo alloy. (e) EDS elemental mapping images. | ||
To understand the electronic structure and the local environment of Mo atoms, X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) were used to characterize the NiMo and NiCuMo samples. As shown in Fig. 3a, b and Fig. S9 (ESI†), the Ni 2p and Mo 3d XPS spectra reveal that the surface Ni exists in the chemical states of metallic and divalent states, while the surface Mo is in mixed chemical states of Mo0 and Mo6+. This finding is similar to that of NiZnMo alloy (Fig. S10, ESI†). Additionally, positive shifts in binding energies were found for metallic Ni and Mo. The Mo K-edge spectra indicate that the electronic states of Mo in NiMo and NiCuMo samples become electron deficient states between the metallic and oxidized states owing to partial Mo6+ in the alloy.32 According to the k2-weighted FT of EXAFS spectra at the Mo K-edge in R space (Fig. 3d), the bond coordination of Mo atom is rearranged in the NiCuMo sample with a broadened peak around 2.1 Å. This bond is shorter than the Mo–Mo bond and is assumed to be the Ni/Cu–Mo bond. Besides, the coordination number of the Mo–Mo bond in the NiCuMo sample is much smaller than that of Mo foil. The EXAFS signal wavelet transforms of the NiCuMo sample also confirmed this rearranged bond coordination (Fig. 3e–g). These XAS results illustrated that the Mo atoms in the NiCuMo sample are highly dispersed.
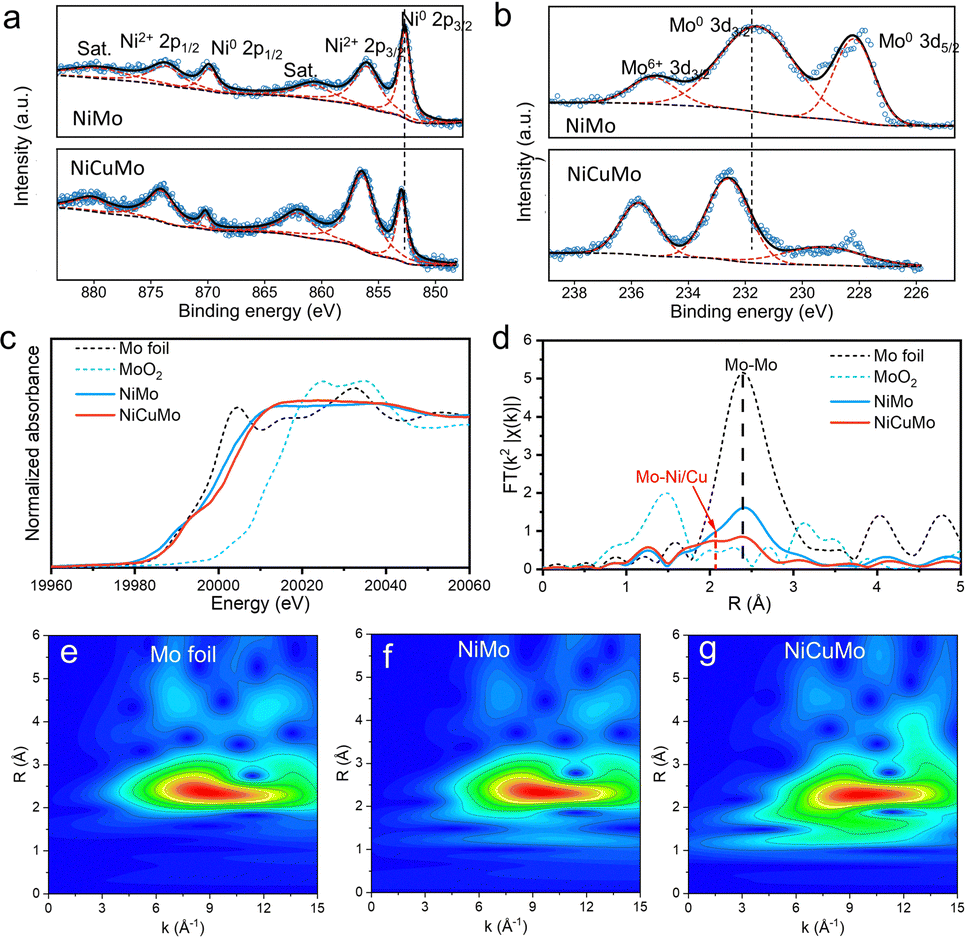 | ||
| Fig. 3 XPS and XAS spectral characterization. (a) Ni 2p and (b) Mo 3d XPS spectra. (c) Mo K-edge XAS and (d) the corresponding k2-weighted FT of EXAFS spectra in R space. (e)–(g) Wavelet transforms for the k3-weighted EXAFS signals at the Mo K-edge of Mo foil, NiMo and NiCuMo, respectively. | ||
The alkaline and neutral HER electrocatalytic performances of Ni-based alloys and commercial 20% Pt/C were evaluated in 1 M KOH and 1 M PBS (pH = 7) by using the Ag/AgCl reference electrode, and the performance in alkaline solutions was confirmed by the Hg/HgO reference electrode (Fig. S11, ESI†). The iR-corrected and pristine linear sweep voltammetry (LSV) curves in 1 M KOH are presented in Fig. 4a and Fig. S12 (ESI†). The overpotentials at a current density of 100mA cm−2 are 90, 248, and 263 mV for 20% Pt/C, NiCu, and Ni, respectively. The Tafel slopes for 20% Pt/C, NiCu, and Ni correspond to 116, 96, and 94 mV dec−1, respectively (Fig. 4c). These unsatisfied alkaline HER activities are due to the sluggish kinetics of water dissociation and restrict their alkaline HER performance.11–13 Benefiting from the synergism between Ni–Mo,30,31 NiMo exhibits an improved overpotential of 83 mV at a current density of 100mA cm−2. It further decreases to 63 mV by Cu introduction. The corresponding Tafel slopes also improved to 70 and 61 mV dec−1 for NiMo and NiCuMo, respectively. This demonstrates that the water reduction kinetics of Ni in alkaline media can be significantly enhanced using the surface reactivity diversity approach.
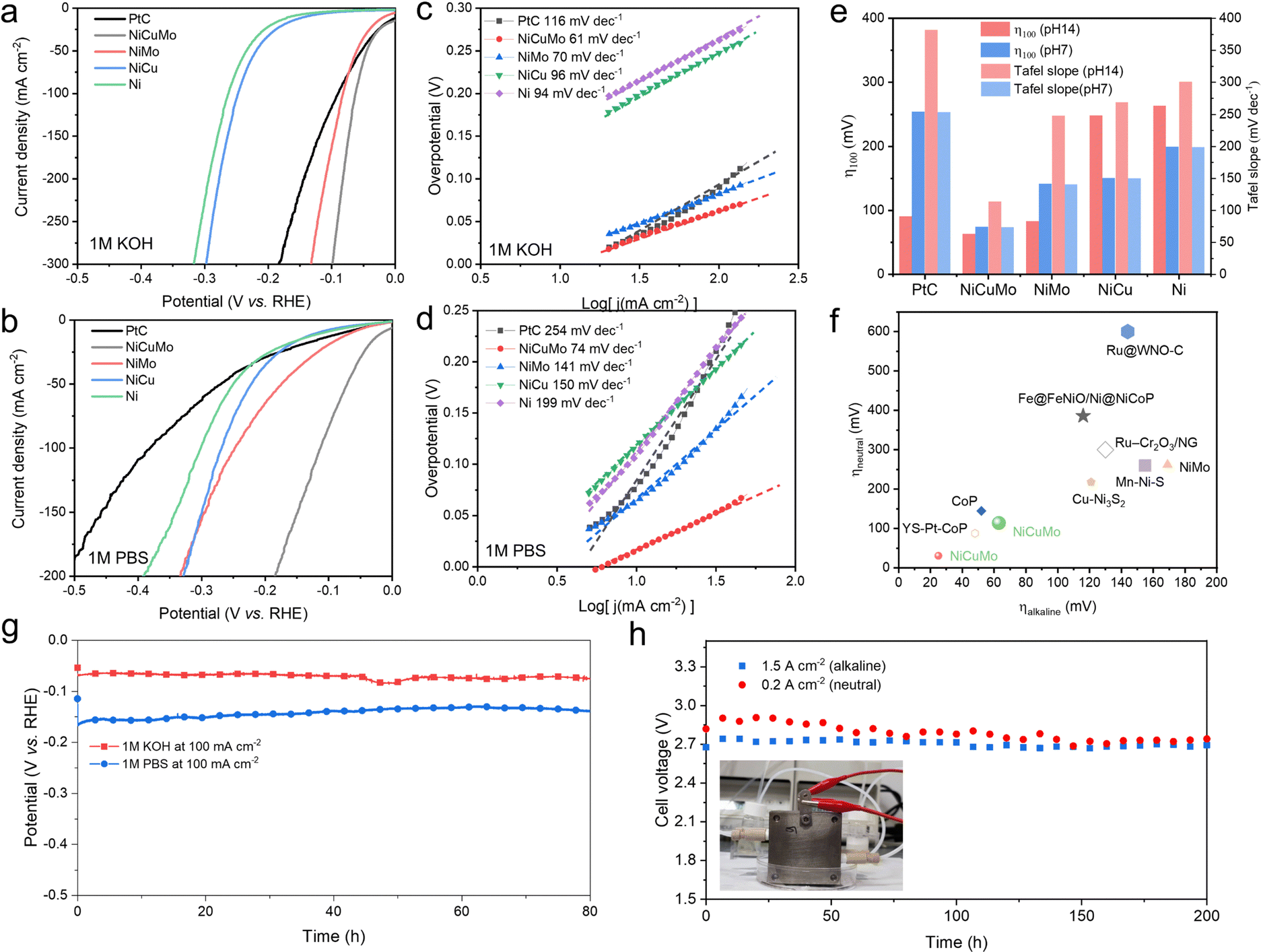 | ||
| Fig. 4 Hydrogen evolution reaction catalytic performance. Linear sweep voltammetry curves in (a) 1 M KOH and (b) 1 M PBS. Corresponding Tafel slopes for various electrocatalysts in (c) 1 M KOH and (d) 1 M PBS. (e) Comparison of overpotentials at 100 mA cm−2 and Tafel slopes. (f) Comparison of various HER electrocatalysts (marker size represents current density). (g) The chronopotentiometry curves of NiCuMo in alkaline and neutral media at a constant current density of −100 mA cm−2. (h) Membrane electrode assembly water electrolysis device stability tests at large current densities (20 °C ± 3 °C). | ||
In a neutral environment, different degrees of deterioration in HER electrocatalytic activity are observed. For 20% Pt/C, NiCu, and Ni catalysts, the required overpotentials at a current density of 100mA cm−2 increase to 382, 269, and 301 mV, respectively (Fig. 4b). The corresponding Tafel slopes also increase to 254, 150, and 199 mV dec−1, respectively. It is worth noting that, although NiMo exhibits high alkaline HER electrocatalytic activity, its neutral HER activity deteriorates significantly (Fig. 4e). NiMo exhibits a high overpotential of 248 mV to reach a current density of 100mA cm−2 in 1 M PBS medium, which is much higher than that in 1 M KOH (83 mV). The Tafel slope of NiMo in neutral HER also increases to 141 mv dec−1, indicating sluggish water reduction kinetics. This is likely due to the pH changes leading to less favorable intermediate species adsorption thermodynamics under neutral conditions. In contrast, NiCuMo ternary alloy has a very diverse range of surface reactivity sites and exhibits superior HER activity in both alkaline and neutral media. As expected from the DFT calculations, the NiZnMo medium-entropy alloy also exhibits superior HER activity in both alkaline and neutral media (Fig. S13, ESI†). Due to the well-known corrosion of Zn in alkaline solution, we focused our main efforts on the NiCuMo alloy.
The high HER catalytic activity of this NiCuMo ternary alloy was further confirmed by comparison with some superior HER catalysts (Fig. 2f, Tables S2 and S3, ESI†), such as YS–Pt–CoP,33 CoP,34 Ru@WNO–C,35 Mn–Ni–S,36 NiMo,37 Fe@FeNiO/Ni@NiCoP,38 Ru–Cr2O3/NG,39 and Cu–Ni3S2.40 This enhanced catalytic activity can also be achieved by replacing Cu with other weakly reactive metals, such as Zn, indicating the universality of the proposed surface reactivity diversity strategy. Besides, catalytic stability tests were conducted at 100 mA cm−2 in 1 M KOH and 1 M PBS to assess the electrochemical stability of this NiCuMo electrocatalyst. As shown in Fig. 4h, there is no noticeable increase in potential during the cycling test under both alkaline and neutral environments. The crystal structure and fractal structure of the NiCuMo electrocatalyst were both maintained in both alkaline and neutral HER stability tests (Fig. S14 and S15, ESI†), and no hydroxides were detected in the alkaline environment (Fig. S16, ESI†). EDS results indicated a significant increase in surface Mo content in neutral media (Fig. S17, ESI†), contributing to a slight activity enhancement. These results demonstrate that the NiCuMo electrocatalyst has strong long-term electrochemical stability.
To demonstrate the practical significance of the surface reactivity diversity for the pH-universal HER, we assembled a membrane electrode assembly (MEA) water electrolysis device. The cathode material of MEA was NiCuMo, and the anode materials were NiCoFe layered triple hydroxide41 and commercial IrO2 in alkaline and neutral electrolyzers, respectively. As shown in Fig. 4h and Fig. S18 and S19 (ESI†), the current density of the MEA composed of NiCuMo is higher than that of commercial Pt/C under the same cell voltage. The NiCuMo based MEA can be stably operated for at least 200 h at a larger current density in both neutral and alkaline electrolytes.
Distribution of relaxation times (DRT) analysis42 of electrochemical impedance spectroscopy (EIS) was employed to investigate the interfacial HER kinetics. Fig. 5 illustrates the decoupling of three interfacial processes with distinct frequencies from DRT peaks. These three interfacial processes were associated to the Volmer step (ASRVol), Heyrovsky step (ASRHey), and post-diffusion related to H2 recombination (ASRPD) from high frequency to low.43 The Volmer step (ASRVol) was identified as the rate-determining step (RDS) for both Ni and NiCu in neutral and alkaline HERs. Introducing Mo into Ni significantly reduced the ASRVol, making ASRHey the dominant process. This observation aligns with our DFT calculations, indicating that Mo, with its high surface reactivity, promotes water dissociation dynamics. Furthermore, the addition of Cu further reduces the dominant ASRHey, particularly in the neutral HER. The decrease in ASRHey was attributed to the more optimal hydrogen adsorption facilitated by Cu.
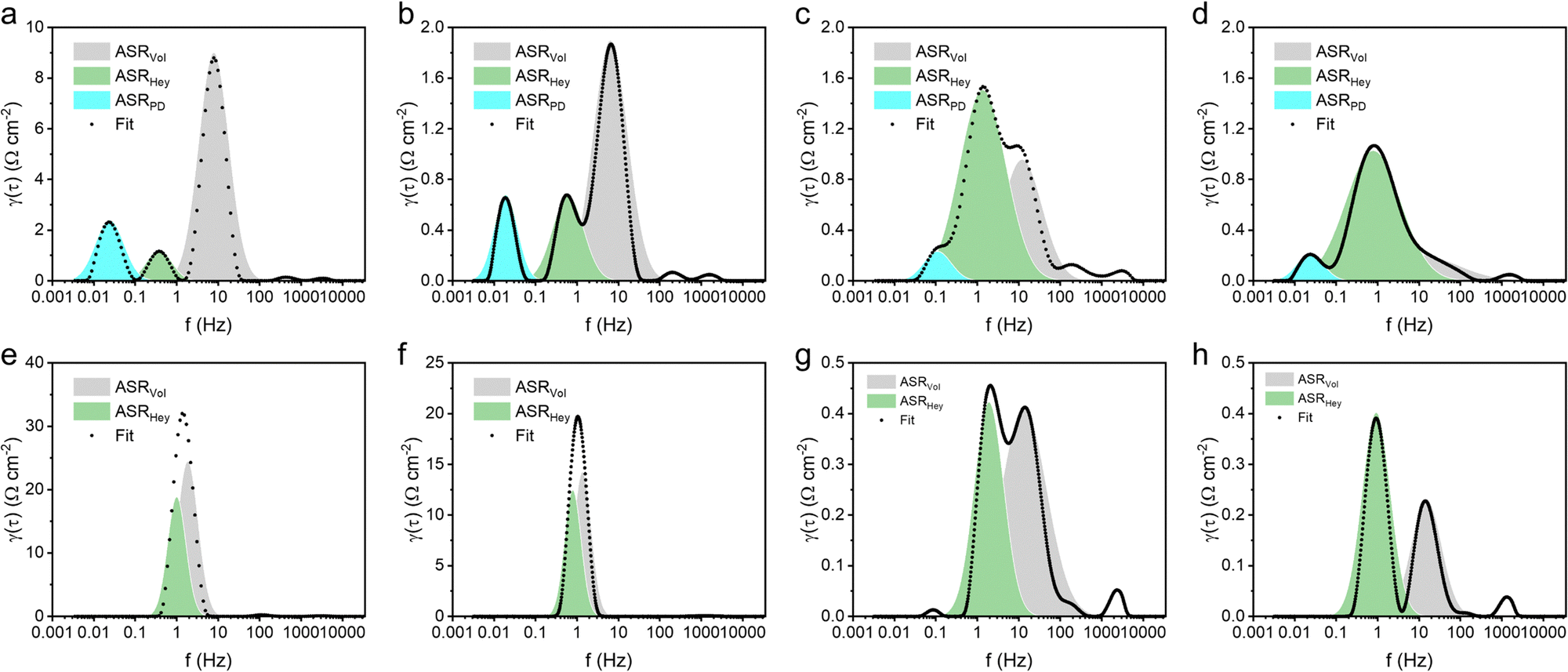 | ||
| Fig. 5 Interfacial HER kinetics. DRT plots of (a) Ni, (b) NiCu, (c) NiMo, and (d) NiCuMo at an overpotential of 100 mV in 1 M PBS solution. DRT plots in 1 M KOH solution of (e) Ni, (f) NiCu, (g) NiMo, and (h) NiCuMo at an overpotential of 100 mV. | ||
Furthermore, the intrinsic HER electrocatalytic activity of NiMo and NiCuMo was obtained via normalizing the HER current by their electrochemical surface areas (ECSA). The fractal structure of NiCuMo leads to a very high electrical double layer capacitance (280 mF cm−2) (Fig. S21, ESI†), which exhibits high intrinsic activity similar to that of NiMo in alkaline media (Fig. 6a). The intrinsic activities of NiMo and NiCuMo are significantly higher than those of Ni and NiCu (Fig. S22, ESI†). Interestingly, a clear difference between alkaline and neutral electrolytes was observed. Furthermore, this gap is significantly narrowed in NiCuMo (Fig. 6a and b). For instance, the η10 gap, which measures the overpotential required to reach a current density of 10 mA cm−2ECSA in alkaline and neutral electrolytes, is 105 mV for NiCu, but only about 36 mV for NiCuMo. The Tafel slope gap shows a similar trend. Remarkably, the intrinsic neutral HER activity of NiCuMo at an overpotential of 100 mV is 3.65 times that of NiMo.
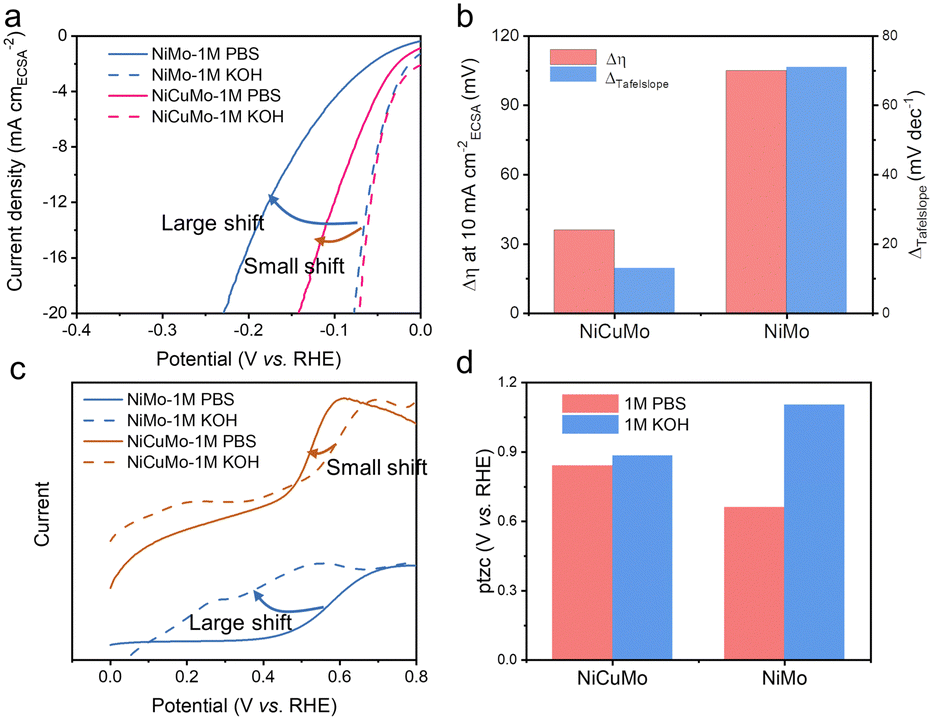 | ||
| Fig. 6 The activity gap between alkaline and neutral hydrogen evolution reactions. (a) ECSA normalized LSV curves of NiMo and NiCuMo. (b) Current density gap and Tafel slope gap. (c) LSV curves in CO-saturated electrolytes. (d) Potential of total zero charges in 1 M KOH and 1 M PBS. | ||
The activity gap between alkaline and neutral electrolytes can be attributed to less favorable intermediate species adsorption thermodynamics under neutral conditions. The CO-stripping measurement was used to evaluate the ability of the electrocatalysts to adsorb hydroxyl species, as the CO-oxidation process is initiated by reactive hydroxyl species.44 The onset potential shift of CO oxidation on NiCuMo was found to be much smaller compared to that of NiMo when measuring the LSV curves in CO-saturated electrolyte (Fig. 6c). The pH-dependent binding energies were the synergistic consequence from the altered electric field and interfacial electrolyte structures.45–47 The potential of total zero charge (ptzc) was used to account for these interfacial structures. Herein, ptzc values were evaluated by immersion i–t curves (Fig. S23–S27, ESI†).48,49 As shown in Fig. 6d, the ptzc of both NiMo and NiCuMo decreases with the electrolyte pH decrease, especially in the case of NiMo. This large shift of ptzc on NiMo indicates a significant electrochemical interface change under different pH environments, which leads to a dramatic change in the binding energies of intermediates. The pH-dependent ptzc change on NiCuMo is more moderate, allowing for the favorable adsorption of hydrogen and hydroxyl species at different pH levels. Noteworthily, the HER catalytic activity of our NiCuMo alloy is still pH-dependent and there is a long way to go to achieve a pH-independent HER.
To shed more light on the pH-dependent binding energies, grand canonical potential (GCP) based free energies of hydrogen adsorption on the Ni surface were analyzed. As shown in Fig. 7a, the hydrogen adsorption strength is enhanced as the solution pH decreases. This effect is attributed to the downward shift of the Fermi level and the increase in surface net charge at the potential of zero versus RHE as the solution pH decreases. The integrated crystal orbital Hamilton populations of the Ni–H bond also monotonously increase, indicating an enhanced Ni–H bond in neutral electrolyte (Fig. 7b). This enhanced M–H bond is also observed on other metal surfaces such as Ag, Au, Co, Pt, Ru, and Mo (Fig. 7c). This pH-dependent hydrogen binding energy has also been observed in previous experiments,21,22 and thus the optimal hydrogen binding energy under various pH values requires a wide range of surface reactivities.
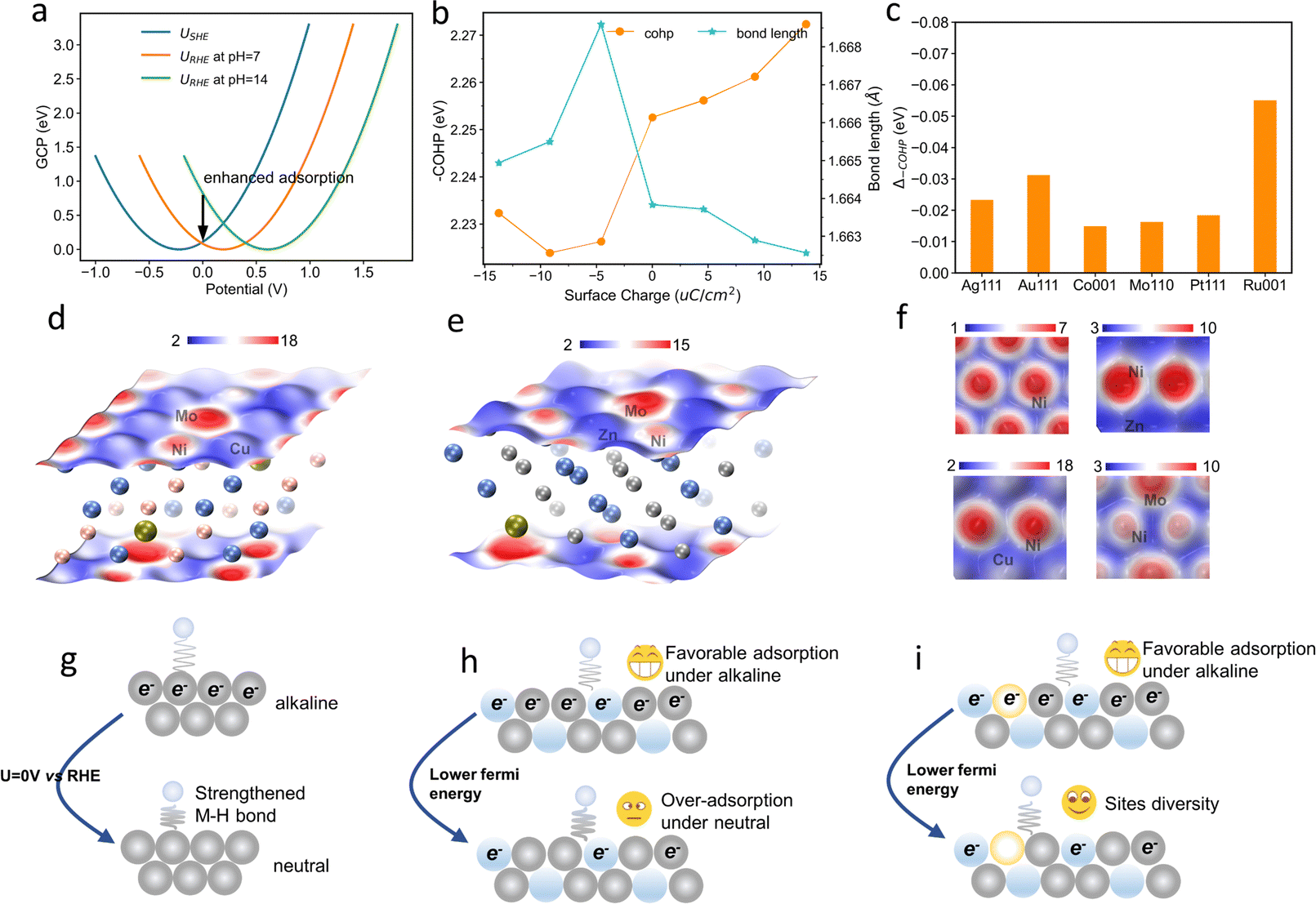 | ||
| Fig. 7 Surface reactivity diversity and pH-dependent hydrogen adsorption. (a) GCP based hydrogen adsorption free energies. (b) COHP and bond length of the Ni–H bond. (c) ICOHP changes as the surface charge becomes more negative. (d)–(f) Fermi softness of various surfaces. (g) The schematic representation of the surface reactivity diversity enabled pH-universal HER. Ni, Mo, Cu, and H atoms are shown as light-grey, blue-grey, light-yellow, and white balls, respectively. | ||
The spatial distribution of the surface reactivity of various catalysts was further visualized by the Fermi softness.50 The Fermi softness is the sum of all the weighted density of states values, in which the closer the state is to the Fermi level, the greater its contribution is. As shown in Fig. 7d–f, the averaged Fermi softness of a surface is integrated over the space of the first atomic layer of the surface via vertical projection. The Mo atoms show higher Fermi softness values than Ni in NiMo alloy, meaning higher surface reactivity of Mo. The Cu atoms show much lower Fermi softness values, suggesting the lower surface reactivity of Cu. The NiCuMo alloy has significantly different Fermi softness values on the surface, confirming its surface reactivity diversity. Under such a mechanism, the net surface charge of the catalyst under neutral hydrogen evolution is less than that under basic hydrogen evolution, strengthening hydrogen bonding (Fig. 7g). As a result, the favorable adsorption of hydrogen and hydroxyl species on NiMo in alkaline electrolytes will deteriorate as the electrolyte pH becomes more neutral. However, the surface reactivity diversity of the NiCuMo alloy allows for favorable adsorption strength of reaction intermediates in different pH environments, resulting in a higher pH universality.
Conclusions
In summary, a surface reactivity diversity strategy has been demonstrated for highly effective hydrogen evolution. The resulting NiCuMo medium-entropy alloy, which consists of three metals with different reactivities, exhibits a wide range of surface reactivities, promoting water adsorption and dissociation as well as hydrogen adsorption and desorption. This surface reactivity diversity promotes the HER kinetics as well as the pH universality. The resulting NiCuMo electrocatalyst exhibits ∼63 mV and ∼115 mV overpotentials at a current density of 100mA cm−2 in 1 M KOH and pH7 buffer electrolyte, respectively. This approach presents a solution for developing less pH-dependent electrocatalysis. It also holds the promise of a highly efficient design of alloy catalysts such as high entropy alloys, which may be useful in reactivity-based design of thermocatalysis and electrocatalysis.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
B. Z. and J. Y. contributed equally to this work. This research was supported by the National Natural Science Foundation of China (No. 52272198) and the Project funded by China Postdoctoral Science Foundation (No. 2020M682400 and 2021M690947).References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- A. Ahmed, A. Q. Al-Amin, A. F. Ambrose and R. Saidur, Int. J. Hydrogen Energy, 2016, 41, 1369–1380 CrossRef CAS.
- B. Tanç, H. T. Arat, E. Baltacıoğlu and K. Aydın, Int. J. Hydrogen Energy, 2019, 44, 10120–10128 CrossRef.
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- P. Da, Y. Zheng, Y. Hu, Z. Wu, H. Zhao, Y. Wei, L. Guo, J. Wang, Y. Wei, S. Xi, C.-H. Yan and P. Xi, Angew. Chem., 2023, 135, e202301802 CrossRef.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- Y. Zhou and H. J. Fan, ACS Mater. Lett., 2021, 3, 136–147 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- B. Zhang, J. Liu, J. Wang, Y. Ruan, X. Ji, K. Xu, C. Chen, H. Wan, L. Miao and J. Jiang, Nano Energy, 2017, 37, 74–80 CrossRef CAS.
- T. Zhang, Y. Liu, J. Yu, Q. Ye, L. Yang, Y. Li and H. J. Fan, Adv. Mater., 2022, 34, 2202195 CrossRef CAS PubMed.
- K. Xu, H. Ding, M. Zhang, M. Chen, Z. Hao, L. Zhang, C. Wu and Y. Xie, Adv. Mater., 2017, 29, 1606980 CrossRef PubMed.
- K. Xu, Y. Sun, Y. Sun, Y. Zhang, G. Jia, Q. Zhang, L. Gu, S. Li, Y. Li and H. J. Fan, ACS Energy Lett., 2018, 3, 2750–2756 CrossRef CAS.
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS.
- L. Zhai, X. She, L. Zhuang, Y. Li, R. Ding, X. Guo, Y. Zhang, Y. Zhu, K. Xu, H. J. Fan and S. P. Lau, Angew. Chem., Int. Ed., 2022, 61, e202116057 CrossRef CAS PubMed.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- X. Wang, C. Xu, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Nat. Commun., 2019, 10, 4876 CrossRef CAS PubMed.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- J. Jin, J. Yin, H. Liu, B. Huang, Y. Hu, H. Zhang, M. Sun, Y. Peng, P. Xi and C.-H. Yan, Angew. Chem., 2021, 133, 14236–14242 CrossRef.
- B. Zhang, L. Zhang, Q. Tan, J. Wang, J. Liu, H. Wan, L. Miao and J. Jiang, Energy Environ. Sci., 2020, 13, 3007–3013 RSC.
- J. Greeley, J. K. Nørskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- Y. Lu, S. Geng, S. Wang, S. Rao, Y. Huang, X. Zou, Y. Zhang, Q. Xu and X. Lu, Int. J. Hydrogen Energy, 2019, 44, 5704–5716 CrossRef CAS.
- Y. Li, W.-Z. Jia, Y.-Y. Song and X.-H. Xia, Chem. Mater., 2007, 19, 5758–5764 CrossRef CAS.
- H. Qiu, T. Tang, M. Asif, X. Huang and Y. Hou, Adv. Funct. Mater., 2019, 29, 1808468 CrossRef.
- H. L. S. Santos, P. G. Corradini, M. Medina, J. A. Dias and L. H. Mascaro, ACS Appl. Mater. Interfaces, 2020, 12, 17492–17501 CrossRef CAS PubMed.
- Z. Chang, L. Zhu, J. Zhao, P. Chen, D. Chen and H. Gao, Int. J. Hydrogen Energy, 2021, 46, 3493–3503 CrossRef CAS.
- K. Hu, M. Wu, S. Hinokuma, T. Ohto, M. Wakisaka, J. Fujita and Y. Ito, J. Mater. Chem. A, 2019, 7, 2156–2164 RSC.
- Z. Jiang, J. Ren, Y. Li, X. Zhang, P. Zhang, J. Huang, C. Du and J. Chen, Dalton Trans., 2019, 48, 8920–8930 RSC.
- T. Liu, L. Xie, J. Yang, R. Kong, G. Du, A. M. Asiri, X. Sun and L. Chen, ChemElectroChem, 2017, 4, 1840–1845 CrossRef CAS.
- G. Meng, H. Tian, L. Peng, Z. Ma, Y. Chen, C. Chen, Z. Chang, X. Cui and J. Shi, Nano Energy, 2021, 80, 105531 CrossRef CAS.
- L. Zeng, Z. Liu, K. Sun, Y. Chen, J. Zhao, Y. Chen, Y. Pan, Y. Lu, Y. Liu and C. Liu, J. Mater. Chem. A, 2019, 7, 25628–25640 RSC.
- F. Bao, E. Kemppainen, I. Dorbandt, R. Bors, F. Xi, R. Schlatmann, R. van de Krol and S. Calnan, ChemElectroChem, 2021, 8, 195–208 CrossRef CAS.
- Q. Che, X. Zhou, Q. Liu, Y. Tan and Q. Li, J. Mater. Chem. A, 2021, 9, 5833–5847 RSC.
- L. Tang, J. Yu, Y. Zhang, Z. Tang and Y. Qin, RSC Adv., 2021, 11, 6107–6113 RSC.
- L. Zhang, X. Gao, Y. Zhu, A. Liu, H. Dong, D. Wu, Z. Han, W. Wang, Y. Fang, J. Zhang, Z. Kou, B. Qian and T.-T. Wang, Nanoscale, 2021, 13, 2456–2464 RSC.
- J. Lee, H. Jung, Y. S. Park, N. Kwon, S. Woo, N. C. S. Selvam, G. S. Han, H. S. Jung, P. J. Yoo, S. M. Choi, J. W. Han and B. Lim, Appl. Catal., B, 2021, 294, 120246 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- T. Zhang, J. Jin, J. Chen, Y. Fang, X. Han, J. Chen, Y. Li, Y. Wang, J. Liu and L. Wang, Nat. Commun., 2022, 13, 6875 CrossRef CAS PubMed.
- J. Zhang, L. Zhang, J. Liu, C. Zhong, Y. Tu, P. Li, L. Du, S. Chen and Z. Cui, Nat. Commun., 2022, 13, 5497 CrossRef CAS PubMed.
- I. Hamada and Y. Morikawa, J. Phys. Chem. C, 2008, 112, 10889–10898 CrossRef CAS.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- S. Zhu, X. Qin, Y. Yao and M. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef PubMed.
- U. W. Hamm, D. Kramer, R. S. Zhai and D. M. Kolb, J. Electroanal. Chem., 1996, 414, 85–89 CrossRef.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- B. Huang, L. Xiao, J. Lu and L. Zhuang, Angew. Chem., Int. Ed., 2016, 55, 6239–6243 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00157a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |