
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale heterogeneity of arsenic and selenium species in coal fly ash particles: analysis using enhanced spectroscopic imaging and speciation techniques†
Nelson A.
Rivera
Jr
 *a,
Florence T.
Ling
bd,
Zehao
Jin
a,
Ajith
Pattammattel
c,
Hanfei
Yan
c,
Yong S.
Chu
c,
Catherine A.
Peters
b and
Heileen
Hsu-Kim
*a
*a,
Florence T.
Ling
bd,
Zehao
Jin
a,
Ajith
Pattammattel
c,
Hanfei
Yan
c,
Yong S.
Chu
c,
Catherine A.
Peters
b and
Heileen
Hsu-Kim
*a
aDepartment of Civil and Environmental Engineering, Duke University, Box 90287, Durham, North Carolina 27708, USA. E-mail: nelson.rivera@duke.edu; hsukim@duke.edu
bDepartment of Civil and Environmental Engineering, Princeton University, Princeton, New Jersey 08544, USA
cNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY 11973, USA
dEnvironmental Science Program, Department of Chemistry and Biochemistry, La Salle University, Philadelphia, PA 19141, USA
First published on 24th May 2023
Abstract
Coal combustion byproducts are known to be enriched in arsenic (As) and selenium (Se). This enrichment is a concern during the handling, disposal, and reuse of the ash as both elements can be harmful to wildlife and humans if mobilized into water and soils. The leaching potential and bioaccessibility of As and Se in coal fly ash depends on the chemical forms of these elements and their association with the large variety of particles that comprise coal fly ash. The overall goal of this research was to determine nanoscale and microscale solid phase mineral associations and oxidation states of As and Se in fly ash. We utilized nanoscale 2D imaging (30–50 nm spot size) with the Hard X-ray Nanoprobe (HXN) in combination with microprobe X-ray capabilities (∼5 μm resolution) to determine the As and Se elemental associations. Speciation of As and Se was also measured at the nano- to microscale with X-ray absorption spectroscopy. The enhanced resolution of HXN showed As and Se as either diffusely located around or comingled with Ca- and Fe-rich particles. The results also showed nanoparticles of Se attached to the surface of fly ash grains. Overall, a comparison of As and Se species across scales highlights the heterogeneity and complexity of chemical associations for these trace elements of concern in coal fly ash.
Environmental significanceFly ashes are the waste residuals collected during coal energy production and represent one of the largest industrial waste streams worldwide. The disposal of these residues is a significant challenge due the large volumes produced annually and the enrichment of toxic elements such as arsenic (As) and selenium (Se) in fly ash. This study describes the nanoscale heterogeneity of As and Se, both in distribution and speciation within individual fly ash grains. These insights help explain why the leaching potential of these elements are not easily predicted by elemental content. Rather, the heterogeneous distribution of As and Se in fly ash signifies that both intrinsic properties of the fly ash as well as extrinsic variables related to the leaching environment contribute to As and Se mobilization. |
Introduction
Coal fly ash and other coal combustion residues are known to be enriched in arsenic (As) and selenium (Se).1 This enrichment is a concern primarily during the handling, disposal, and reuse of the ash as both elements can be harmful to wildlife and humans if mobilized at concentrated quantities into water and soils. The extent of As and Se mobilization depends on the amount in the original ash as well as the chemical form of these elements and environmental conditions (e.g., water chemistry, redox conditions, soil surface chemistry, hydraulic conductivity).2,3The amounts and forms of As and Se in coal fly ash derive both from the original feed coal composition and from combustion conditions and other technologies to control flue gas emissions.1 Furthermore, coal-fired power plants are rapidly evolving in their operations in response to changes in the energy market as well as technological improvements for air pollution controls. For example, many coal power stations now employ air pollution control technologies, such as selective catalytic reduction and dry SOx capture, which require an injection of chemicals (e.g., ammonia, lime) into the flue gas, altering the composition of the fly ash products.4 In the U.S., coal-fired power plants are shifting operations from base-load to seasonal or peak-load generation,5 a shift that could influence the composition of residuals.
The concentrations, geochemical forms, and mobilization potentials of Se and As from coal fly ash disposal impoundments have been documented previously.2,3,6,7 For example, our prior survey7 of more than 100 coal fly ash samples showed that the Se and As contents vary by multiple orders of magnitude, depending on the coal feed stock. The leachable concentrations of As and Se are related, in part, to the total amounts of As and Se in the ash. However, our previous work2 demonstrates that leachable quantities do not always correlate to total element abundance. These results highlight the need to consider the chemical forms of As and Se as well as mass distribution within individual ash grains. In this respect, knowledge of the phase associations of As and Se in the fly ash matrix could offer clues to their mobilization potential in water.
For example, As and Se species hosted by Fe-rich particles are vulnerable to mobilization under reductive dissolution of the host phase.8,9 Changes in pH will also influence desorption potential of adsorbed As and Se ions.10 In contrast, Ca–arsenate and Ca–selenite minerals are low solubility phases and should be recalcitrant to such changes in environmental conditions.11,12
Many studies have attempted to evaluate As and Se speciation in fly ash,2,3,13–16 typically with spectroscopy- and microscopy-based tools. For example, several studies using synchrotron X-ray absorption spectroscopy (XAS) have shown that the arsenic exists primarily as As(V) while the selenium occurs in multiple oxidation states as Se(IV) and Se(0) in fly ash.3,13,17,18 These elements are thought to be associated with Fe phases (e.g., arsenate ions bound to iron oxides) or Ca phases (e.g., calcium arsenate and selenite).
Despite the breadth of work, knowledge gaps continue to exist, particularly in relating this information to leaching potential. Such gaps could be due to speciation methods that failed to capture molecular-level associations of these elements in fly ash, mass distribution of As and Se within individual ash particles, and heterogeneity between ash particles. For example, XAS methods rely on appropriate selection of reference As and Se phases, an uncertainty given the complexity of the fly ash matrix. Spatially resolved methods such as micro-X-ray fluorescence (μ-XRF) imaging can show elemental associations of the As and Se. However, this technique has resolution limitations as the beam size (∼5 μm) is similar to or larger than individual fly ash particles. During the coal combustion process, As and Se tend to volatilize in the flue gas.1,4 The deposition of these elements on fly ash surfaces occurs as the flue gas temperature reduces, resulting in the complexity of distribution within single ash particles that is difficult to ascertain with conventional microprobe and microscopy techniques.
Newly developed nanoscale X-ray imaging and spectroscopy offers additional insights for the determination of As and Se speciation, particularly if the As and Se are associated with nanoparticles. The Hard X-ray Nanoprobe (HXN) Beamline at the National Synchrotron Light Source II (NSLS-II) is a unique beamline enabling both nano-resolution imaging and X-ray absorption spectroscopy.19–21 Such information could inform both the mobility of the As and Se as well as leaching potential.
The objective of this research was to characterize fly ash particles at multiple length scales (nanometer to micrometer) to determine if elemental associations differ as a function of the resolution of characterization. Here, we utilized established synchrotron microprobe analyses combined with newly developed nanoscale synchrotron analyses (30–50 nm spot size). We sought to combine these analyses to improve our understanding of As and Se forms in fly ash matrices and to distinguish whether the As and Se species occur as discrete nanosized particles or are dispersed among a variety of host phases.
Materials
The fly ash samples selected for this study were collected in 2012–2015 and have been characterized in previous studies for major and minor element contents.7,22 Briefly, the fly ash samples were obtained from pulverized coal combustion units that were burning coals primarily originating from the central Appalachian (App), Powder River (PRB), and Illinois (IL) coal basins of the United States (Table 1). The total As and Se contents (mg kg−1) shown in Table 1 were determined previously by heated nitric acid extraction followed by inductively coupled plasma mass spectrometry.7,22 In our prior analyses with this method,7 we observed less than 10% relative standard deviation of the mean values for triplicate digestions of samples, indicating that the ash samples were well homogenized. Major elements Si, Al, Fe, and Ca contents (expressed in their wt% oxide form in Table 1) were determined by benchtop X-ray fluorescence spectroscopy (XRF).7,22 These fly ash samples were previously assessed for leachable As and Se contents (Table 2) through multiple extraction methods: 1) overnight leaching in deionized water (modeled after the Leaching Environmental Assessment Framework);23 2) leaching according to the Toxicity Characteristic Leaching Protocol (TCLP);23 and 3) modified BCR sequential extraction,22 as detailed in prior studies.22,23| Sample ID | Year sampled | As | Se | SiO2 | Al2O3 | Fe2O3 | CaO |
|---|---|---|---|---|---|---|---|
| mg kg−1 | mg kg−1 | wt% | wt% | wt% | wt% | ||
| App #1 | 2015 | 139 | 12 | 52.1 | 26.5 | 14.1 | 2.1 |
| App #2 | 2015 | 132 | 16 | 54.1 | 28.4 | 10.9 | 1.3 |
| PRB #1 | 2015 | 51 | 18 | 38.3 | 22.5 | 5.21 | 22.9 |
| IL #1 | 2012 | 137 | 10 | 45.7 | 21.2 | 26.4 | 1.9 |
| Deionized watera | TCLPa | Sequential extractionb | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %As leached | %Se leached | pH | %As leached | %Se leached | pH | %As in fraction | %Se in fraction | |||||||||
| F1 | F2 | F3 | F4 | Residual | F1 | F2 | F3 | F4 | Residual | |||||||
| a From Jin et al. (2023).23 b From Taggart et al. (2018);22 sequential extractions corresponded to: F1 deionized water soluble, F2 acid soluble (0.11 M acetic acid), F3 reducible (0.5 M NH3OH·HCl pH 2), F4 oxidizable (30% H2O2 followed by 1.0 M ammonium acetate at pH 2). | ||||||||||||||||
| App #1 | 1.6 | 27.3 | 10.1 | 11.8 | 15.5 | 5.2 | — | — | — | — | — | — | — | — | — | — |
| App #2 | — | — | — | — | — | — | 0.8 | 1.7 | 73.1 | 7.0 | 17.4 | 6.6 | 2.3 | 26.8 | 35.2 | 29.1 |
| PRB #1 | <0.01% | 0.72 | 11.8 | 5.3 | 29.5 | 9.6 | 0.2 | 1.8 | 5.3 | 9.4 | 83.4 | 6.8 | 2.9 | 3.7 | 69.6 | 17.1 |
| IL #1 | 0.67 | 8.3 | 10.0 | 0.41 | 2.1 | 5.2 | 0.0 | 2.3 | 45.0 | 6.4 | 46.3 | 8.5 | 0.5 | 13.4 | 45.9 | 31.6 |
Methods
Multiple solid-phase characterization techniques were performed on the fly ash samples to ascertain morphology, elemental associations, and oxidation states for As and Se. Random aliquots of fly ash from the various bulk ashes were collected for the analyses below.Imaging of the fly ash particles was performed using a Hitachi FlexSEM1000 scanning electron microscope (SEM). Samples were prepared by mixing a fly ash sample with 70% ethanol and letting the mixture settle. A 5 μL aliquot of the solution was then applied to a silicon chip with platinum fiducial markers (Norcada). SEM imaging was performed in high-vacuum mode at an accelerating voltage of 10 kV and a working distance of 5–6 mm. Results of the SEM imaging were used to determine areas of interest for the nano-XRF data collection.
The elemental associations of the fly ash were probed using μ-XRF and nano-XRF synchrotron capabilities. Thin sections of fly ash samples were prepared by Spectrum Petrographics (Vancouver, WA) for μ-XRF analysis at the Stanford Synchrotron Radiation Laboratory (SSRL) on Beamline 2–3. The samples were impregnated in Epotek 301 epoxy, cut to a thickness of 30 μm with a dimension of 27 mm × 46 mm, and mounted on a quartz slide. SSRL Beamline 2–3 is equipped with a vortex silicon drift detector with a nominal spot size of 5 μm × 5 μm. The maps of μ-XRF spectra were collected at 13 keV with a 100 ms dwell time and a 5 μm step size at ambient conditions. Intensity maps for As and Se were produced in the program SMAK.24 We also focused on Fe and Ca in the analysis of μ-XRF spectra due to prior work suggesting that As and Se are bound to host phases of these two major elements (as described in the Introduction) and also because of our prior leaching studies demonstrating selective release of As in a reducing extraction fluid (F3 fraction in Table 2). Areas of As and Se signal intensity on the XRF maps were selected for X-ray absorption near edge structure (XANES) spot analysis. The first derivative of a gold foil (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 919 eV) was used to calibrate the energy scale for both As and Se.
919 eV) was used to calibrate the energy scale for both As and Se.
The coal ash samples (App #1, App #2, and PRB #1) were also analyzed at the HXN Beamline at NSLS-II. Nano-XRF imaging of individual particles was conducted using a focused X-ray beam (∼30–50 nm) with a Fresnel X-ray zone plate. A three-element silicon drift detector (SDD, Vortex), positioned perpendicular to the X-ray beam, was used for collecting nano-XRF spectra. For nano-XRF imaging, raster scanning was used by continuous fly-scanning across regions of interest at 30–50 nm steps with dwell times from 100 to 250 ms, depending on the features of the samples. Nano-XRF spectra were collected at 12.7 keV just above the Se K-edge absorption. Spectral fitting of nano-XRF data was performed using PyXRF25 to create intensity maps for elements of interest (As, Se, Fe, Ca). Nano-XANES mapping was performed by collecting 60 maps over the energy range of 11845–11900 eV (energy stack). Additionally, XANES data was extracted from the energy stack using the software ImageJ. Details of the experimental procedure can be found in a prior publication.21
As and Se speciation at the bulk-scale was evaluated by synchrotron XAS. The data were collected at SSRL Beamline 11-2, running under dedicated conditions (3 GeV, 500 mA) using an unfocused beam. The beamline was configured with a rhodium mirror to reject higher order harmonics, a channel-cut Si(220) phi = 90° monochromator (beam size = 1 mm vertical × 10 mm horizontal), and a 100-element Ge detector. The energy scale for both As and Se was calibrated to the derivative maxima (11919 eV) detected with an Au metal foil reference standard. The Au calibrant was measured multiple times throughout the course of analysis to verify energy stability of the beamline. Fly ash samples were ground using an agate mortar and pestle and loaded into aluminum holders with Kapton tape windows. Multiple scans (from 6 to 9) were collected for each sample as they were held at 77 K in a liquid nitrogen cryostat. The IFEFFIT12 graphical user interface Athena was used for analyzing the extracted XAS data.26 The linear combination fitting module of Athena was used to determine the relative abundances of arsenic and selenium oxidation states for the XANES data (approximately 30 eV below and above the absorption edge).26 The samples were fit to As and Se standards. For As, the standards orpiment (As2S3), As(III) adsorbed to ferrihydrite (from the collection of P. O'Day27), and As(V) incorporated into a glass phase (from the collection of D. Hesterberg16) were used in the LCF fits. The main phases used for the Se LCF fits were FeSe, Se(0), Se(IV) associated with an aluminosilicate glass (obtained from collection of D. Hesterberg), and sodium selenate. Due to the variety of solid host phases in the samples, As and Se species of the same oxidation state with the same white line intensity could occur as multiple phases, both crystalline and amorphous. Thus, the XANES data analysis was primarily used to identify oxidation states of As and Se, rather than mineralogical associations of these elements.
Results
The multi-scale approach to characterizing fly ash revealed heterogeneities both with the sizes of the particles as well as heterogeneities in the speciation and spatial distribution of As and Se within these particles. SEM images of fly ash particles showed particles with spherical morphologies and a wide range of size. For example, particles of the PRB #1 sample (Fig. 1A) range in size from <500 nm to 5 μm, with smaller spherical particles mostly attached to larger ones. In a magnified view (Fig. 1B) of a ∼4 μm spherical fly ash particle in Fig. 1A, we observed multiple 500 nm particles attached to the surface. Fig. 1C and D show fly ash particles from App #1 and App #2. Similar to PRB #1, the App #1 and App #2 samples are comprised of spherical particles. We observed both smaller spherical particles attached to larger particles (Fig. 1C) as well as spherical particles dispersed on the holder (Fig. 1D). | ||
| Fig. 1 SEM images of: A) fly ash sample PRB #1 showing spherical particles of varying sizes; B) a magnified view of PRB #1 showing multiple sized particles clustered together, including <1 μm particles attached to a 4 μm particle; C) fly ash App #1; D) fly ash App #2. | ||
These results are similar to previous studies reporting a wide range in particle sizes from <500 nm to hundreds of μm.28–30 Note that the samples in our analysis and shown in Fig. 1 might be weighted towards the smaller-sized spheres due to the preparation methods for HXN beamline analyses, which involved droplet addition of an ethanol suspension of fly ash particles. Aggregation of the particles would be expected to occur during the drying process. Nevertheless, the SEM images highlight the importance of scale for the interpretation of X-ray-based analysis methods.
Arsenic distribution in fly ash grains
Both μ-XRF and nano-XRF maps show uneven distribution of As within individual ash grains (Fig. 2). For μ-XRF analyses, As was observed on multiple types of particles ranging in size from ∼50 μm to a single spot (5 × 5 μm pixel) for sample PRB #1 (Fig. 2A). Particles with As were also enriched with Fe, resulting in yellow shading spots in Fig. 2A. Single pixels of As (e.g., marked with an arrow in Fig. 2A) were also observed and did not appear to have associations with other elements under the imaging conditions. Arsenic is also known to associate with Ca-rich particles in fly ash.15 However, for the PRB #1 sample, the enrichment of As was not observed to co-occur in areas of high Ca counts for the μ-XRF map (Fig. 2A). We note that the Ca signal (and other lighter mass elements such as Al, Si, and S) could be decreased due to ambient air space between the sample and the detector. This has the potential to attenuate Ca fluorescence intensity and change the relative signals of As and Ca to each other masking the Ca–As association as well as other associations with trace elements (e.g., Se). This signal attenuation does not dampen Fe fluorescence intensity to the same degree. The nano-XRF data does show a more overlap with the Ca signal as these experiments were performed under a He environment at 250 torr. Additional elemental maps are for each nano-XRF sample are presented in ESI† Fig. S1–S3.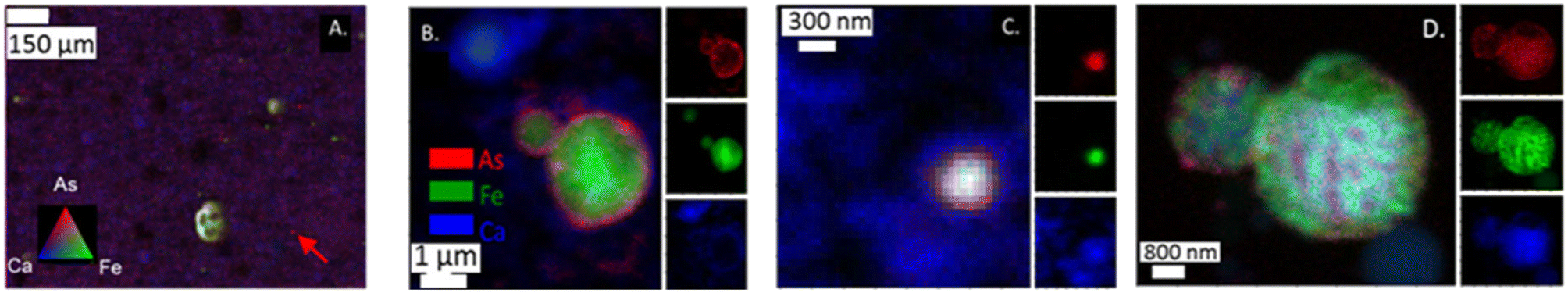 | ||
| Fig. 2 (A) μ-XRF analysis of PRB #1 fly ash showing As associated with a Fe-rich particle (resulting in yellow color) as well as spots which appear to have only an intense As (red) signal (see arrow); (B and C) nano-XRF analysis of App #1 fly ash. (D) Nano-XRF analysis of PRB #1 arsenic is dispersed across a particle with Fe and Ca. XRF intensity corresponding to As (red), Fe (green), and Ca (blue) show. At the nano-scale, particles were observed with As around a spherical particle associated with Fe (panel B) and a particle that appears to have a mixture of As,Fe, and Ca association (panel C). | ||
When nano-XRF mapping (30–50 nm spot size) was employed for arsenic analyses, As was observed to be heterogeneously distributed within single ash grains. For example, in the analysis of the App #1 fly ash (Fig. 2B and C), we observed multiple modes of distribution such as a coating of As on a fly ash grain (Fig. 2B) with a Fe core and diameter of 1 μm. This particle appeared to overlay a Ca layer or a different Ca-rich particle. In another fly ash particle (∼200 nm) in the App #1 sample (Fig. 2C), As was co-located with Fe and Ca, as indicated by the white core over the blue area. Fig. 2D shows a nano-XRF image of PRB #1. The fly ash particles have As dispersed across the particles but are not spatially concentrated as seen in Fig. 2B and C. Regardless, arsenic in PRB #1 was also observed in particles that also contained Fe and Ca.
Altogether, nanoscale mapping highlighted multiple As associations that include both As located at the boundary of Ca and Fe rich particles as well as As species co-located with Fe- and Ca-bearing particles. Note that the particles observed by nanoprobe analyses are smaller than the single pixel size for the μ-XRF data. Therefore, microprobe analyses could lead to the erroneous conclusion that “hot-spots” are discrete particles of arsenic rather than heterogeneous mixtures of multiple phases.
Selenium distribution in fly ash grains
Like As, Se was observed in the App #2 fly ash on both large fly ash grains (Fig. 3A) as well as smaller spots encompassing single to multiple pixels (5 × 10 μm2 area per pixel). For example, Se was co-located with Fe (yellow shaded area in Fig. 3A), similar to observations of As–Fe collocations (Fig. 2A). Additionally, a Se signal was overlaying a Ca signal (Fig. 3A) whereas this was not evident with the As (Fig. 2A). A Se hot-spot (Fig. 3A, arrow) shows Se surrounded by Fe (green) and Ca (blue) areas. A finer mapping of these smaller spots was not possible to determine whether these As and Se hotspots are separate chemical phases (e.g., As2O5) or distributed on fly ash grains <5 μm as seen in the SEM image (Fig. 1).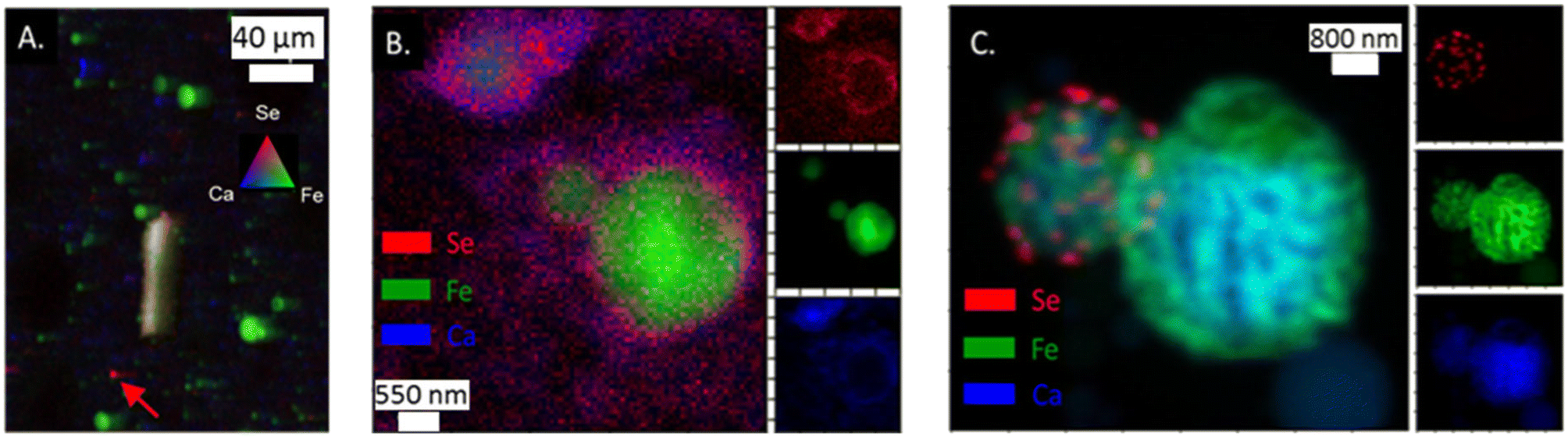 | ||
| Fig. 3 (A) μ-XRF analysis of App #2 fly ash showing Se associated with Fe-rich fly ash particle (yielding yellow color) as well as single spots with intense Se red signal (see arrow); (B) nano-XRF analysis of App #1 (B) showing diffuse distribution of Se. This image shows the same portion of the sample as mapped in Fig. 2B; (C) nano-XRF analysis of PRB #1 fly ash showing discrete Se particles about 100–500 nm on a Fe-coated Ca-rich sphere. | ||
Selenium in the fly ash was also observed to be concentrated on the surface boundary of individual fly ash grains in the nano-XRF data. For App #1 (Fig. 3B), Se was widely dispersed in the same areas as the As-bearing rind on the Fe-rich particle shown in Fig. 2B. Diffuse Se coatings surrounding a ∼600 nm Ca-rich particle (Fig. 3B) were identified where no Fe or As was observed. The nanoscale associations of Se with Fe- and Ca-rich zones are consistent with μ-XRF mapping where Se is associated with either Fe zones or Ca zones (Fig. 3).
While Se may be diffusely distributed on the surface of Fe- and Ca-rich particles (as shown in Fig. 3B), it can also appear as discrete nanoparticles. In Fig. 3C showing two spherical fly ash particles of 1 and 3 μm diameter, we observed discrete 50 nm hotspots of Se on the surface of the smaller particle for PRB #1. We did not observe other elements (e.g., Fe, Ca, Al, Si) that shared this morphology (Fig. S3†). Thus, these hotspots suggest the presence of Se nanoparticles on the fly ash grain, rather than Se diffusely distributed on the coatings. Selenium is capable of existing as nanoparticles in the environment31,32 and similar phases could be present in this sample although through a different formation mechanism.33 Unfortunately, we were not able to collect spectroscopic speciation data to confirm the oxidation state of Se in these particles.
While this study focused on As and Se, our data also indicated multiscale heterogeneity of major elements such as Fe and Ca. Some fly ash particles were distinctly rich in Fe or Ca (Fig. 3A and B). Images of other samples showed co-location of Fe and Ca, such as the 1 to 3 μm particles that appeared to have Fe encapsulating a particle with a Ca-rich core (Fig. 3C). These results are consistent with previous work showing the same type of iron coating where iron precipitates encapsulated a fly ash particle.28–30 The iron coating on the particle has similarities of magnetospheres found in fly ash. These iron coatings can range in composition and will be associated with Ca, Al, and Si.29,30
Bulk and spatially resolved speciation of As and Se
Bulk and μ-XANES data were collected on fly ash samples for both As and Se. Discrete spots were chosen to collect μ-XANES spectra based on the μ-XRF imaging to determine the microscale As and Se oxidation states. These data were compared to the bulk sample XANES data with the results demonstrating differences between the two for both As and Se (Fig. 4 and 5). Additionally, nano-XANES mapping of a fly ash grain for As was collected to determine nano-scale As speciation.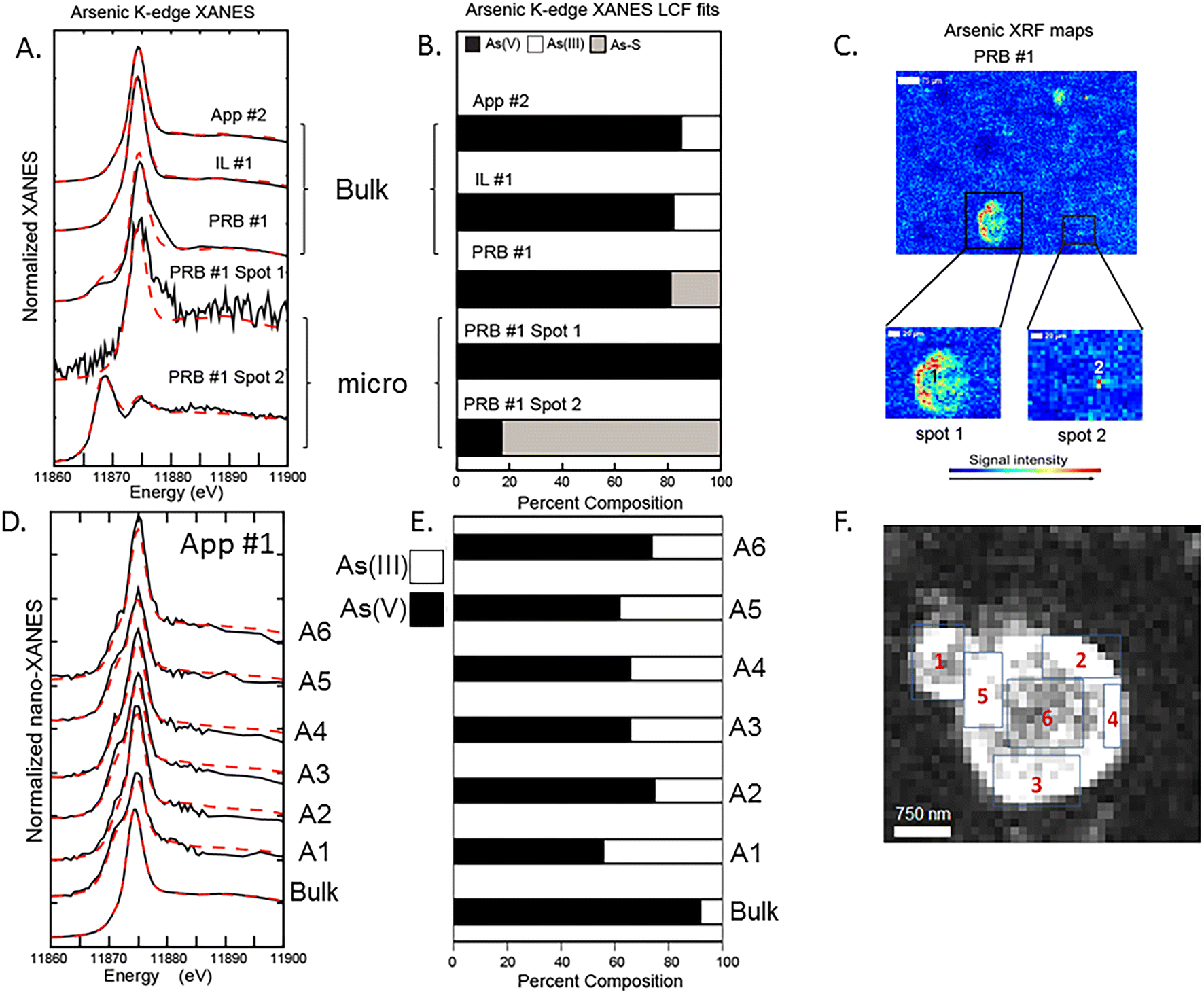 | ||
| Fig. 4 Arsenic speciation in fly ash samples analyzed with bulk, μ-, and nano XANES. A) As-XANES data (solid black lines) and respective model fits (red dotted lines) for selected fly ash samples analyzed at the bulk sample scale and the microprobe scale. B) Percent composition of the As for bulk and μ-XANES from linear combination fitting (LCF) with spectra of arsenic reference materials. C) μ-Focus XRF data showing spots for the μ-XANES. D) nano-XANES spectra for App #1 bulk and nano-XANES areas (A1–A6). E) LCF results for the nano-XANES indicating As(III) + As(V) oxidation states on the rind of a single particle. F) Nano-XRF map outlining individual areas analyzed for nano-XANES. | ||
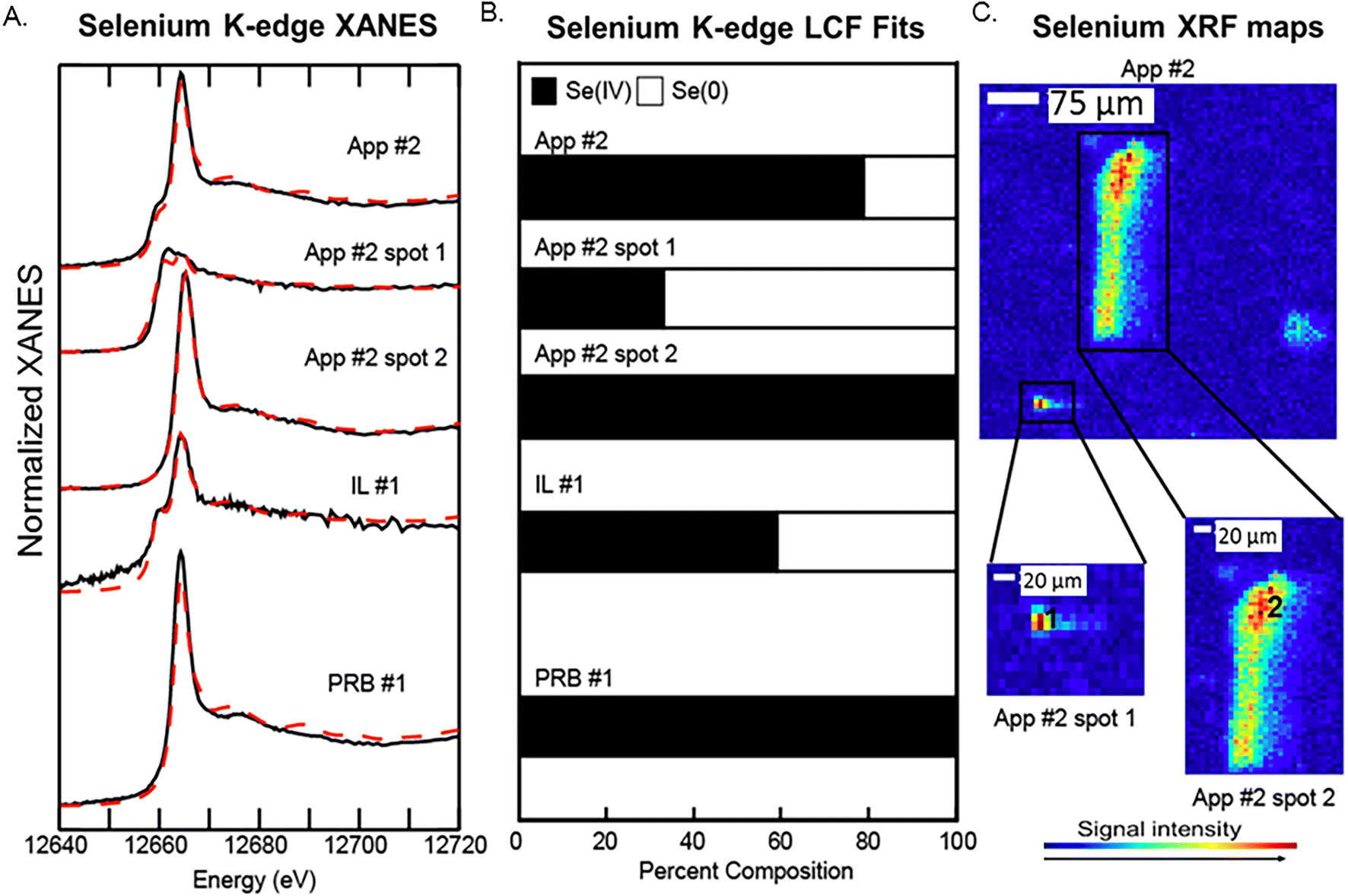 | ||
| Fig. 5 Selenium speciation in fly ash samples analyzed by bulk and μ-XANES. A) Se-XANES spectra data (solid black lines) and model fits (red dotted lines) for bulk and microprobe analysis. B) LCF model results showing percent composition of Se for spectra collected by bulk and μ-XANES. C) μ-XRF maps of Se showing spots analyzed by μ-XANES. | ||
Bulk As XANES (App #2, IL #1, and PRB #1) showed primarily As(V) (based on white line peaks at 11874–11875 eV) representing 80–95% of the total fit (Fig. 4A and B). Minor components were in the As(III) form (11871 eV) and another reduced As oxidation state (11869 eV) was fit with a sulfide phase. Both App #1 and IL #1 samples showed similar fitting with regards to As(V) and As(III) despite IL #1 having greater iron content. For example, the PRB #1 bulk XANES analysis showed a predominant As(V) peak (80% of total) and a secondary peak indicating the presence of a reduced As species (20%) (Fig. 4B). The PRB #1 spot 1 had As(V) as the main oxidation state similar to the bulk fit with no reduced As species. However, spot #2 (5 × 5 μm) showed reduced As fit with a sulfide phase (80%) and a minor amount of As(V) (20%). This potential for an arsenic sulfide phase could be a residual from the original coal feed stock in which trace elements such as arsenic are present in coal within pyrite and other sulfidic minerals. The μ-XRF and μ-XANES data indicate that heterogeneity can exist in the fly ash at the micron-scale. While the XANES fitting characterizes the oxidation state of the As and the mapping shows collocation of As with Fe- and Ca-bearing particles, this analysis does not necessarily provide confirmation of As-bearing molecular structures. For example for both PRB #1 (bulk sample) and PRB #1 spot1, LCF models are not overlapping the XANES data at spectra immediately after the white line energy of 11880 eV. Small deviations in the model fits at this spectral region of the data suggests that the reference materials, which represent idealized structures, are not fully reflecting the As-bearing phases and molecular coordination states in fly ash.
We attempted to discern the speciation of the As-rich rind shown in Fig. 2B (areas 2–5) at a finer resolution with nano-XANES mapping (Fig. 4D and E). These data indicated a mixture of As(III) and As(V) species in the rind (Fig. 4F), a mixture of oxidation states that was different than bulk XANES analysis, which indicated a predominance of As(V) in the App #1 sample. However, we also recognize that X-ray analyses have the potential to alter the sample by either reducing As(V) to As(III) or oxidizing a sample depending on the sample matrix.34 Further work should evaluate the importance of beam damage to the samples analyzed under these conditions.
Bulk Se XANES showed primarily Se(IV) (based on white line peaks at 12664 eV) and representing 60–100% of the total fit (Fig. 5A and B). A secondary peak as indicated by white line peak at ∼12658 eV corresponded to a reduced Se oxidation state and was best fit with the Se(0) reference spectra (0–40% of the Se spectra). The white line peaks for elemental Se(0) and Se-sulfide are relatively close; thus, we were not able to resolve the signals for the reduced Se forms because they were a minor component of the total Se. As such, we interpret the LCF fit with Se(0) to indicate the presence of both Se(0) and Se-sulfide. The μ-XANES analyses showed a heterogeneous distribution of Se species at the micrometer scale. For example, the LCF fits for spot 1 in App #2 showed a mixture of 40% Se(IV) and 60% reduced Se form, while a second spot showed only Se(IV) without any reduced Se (Fig. 5). These proportions differ from the bulk scale analysis of App #2 indicating an average proportion of 80% Se(IV) and 20% reduced Se.
Discussion
This work examined the composition of fly ash at multiple spatial scales to determine elemental associations and speciation of As and Se. Various modes of association of As and Se were observed, including As and Se species diffusely distributed across ash particle aggregates, concentrated at the edge or surface of particles, and discrete particles. Calcium and Fe were two major elements of fly ash that appeared to be prevalent with As and Se. The enhanced resolution at HXN showed As and Se species that were either diffusely located around or comingled with Ca- and Fe-bearing particles (Fig. 2 and 3).Arsenic was often co-located with Fe in the ash; however, such collocations were not universal (e.g., As spots without Fe signal PRB #1 in Fig. 2A). Such differences could explain the sequential extraction data that showed variable proportions of As from the fly ash was leached in the F3 NH3OH·HCl fluid (a fluid that can reductively dissolve Fe-oxide particles) (Table 2). Likewise, arsenic was sometimes concentrated at the surface of particles (APP #1, Fig. 2B) or mixed throughout individual ash particles (APP #1, PRB #1 in Fig. 2C and D), two modes of association that are expected to yield different rates of dissolution during leaching. Consistent with observations of surface-bound As in App #1, the proportion of leached As was 1.6% when this fly ash was leached in deionized water, while PRB #1 exhibited a smaller the leachable As percentage (<0.01%) for the same leaching assay (Table 2).
We also observed discrete Se-rich nanoparticles in the PRB #1 fly ash (Fig. 3C) while the Se signals in App #1 appeared to be more diffusively distributed at the nanoscale. Coincidently, App #1 contains larger proportion of water-leachable Se (27.3%) compared to the respective leachable Se value for PRB #1 (0.72%) (Table 2). The differences in Se distribution between the two fly ash samples can explain why such wide variation in Se leachability has been observed.23 These observations that would not have been possible to determine with the resolution of μ-XRF. The spot size for μ-XRF measurements in this study is approximately 5 μm × 5 μm. Any particles less than 5 μm would be within a single pixel during data collection, thus losing information on heterogeneity at the nanoscale.
These results demonstrate the nanoscale complexity of As and Se distribution within single grains of fly ash samples. Further analyses with a wider array of samples could inform the representation of the associations we observed in our samples. Nevertheless, data in this study illustrate why the release of these elements may not be easily discerned from bulk sample characterization such as oxidation state as determined by a bulk-beam XAS measurement or even by μ-XRF and μ-XAS. Such insights are relevant for understanding and predicting the chemical reactivity of these trace elements when fly ash is released into the environment. Arsenic species concentrated on the surface of particles particularly with Fe (as shown in Fig. 2B) could leach into the solution at environmentally relevant pH and redox conditions.8,35,36 In contrast, the leachability of As species distributed within other solid phases (e.g., Fig. 2D) would depend on conditions that influence mineral phase stability. The type of mineral that hosts As and Se species is also relevant to the dissolution and desorption potential of these elements. For example, surface-bound Se was observed in both Fe- and Ca-rich particles (Fig. 3) that are likely to require different environmental conditions (e.g., pH, redox potential) that lead to their dissolution and release of selenium ions. For example, a prior study by Schwartz et al. (2016)3 showed that the redox conditions in sediments strongly influence the release of aqueous As and Se from fly ash. Arsenic had greater release under anaerobic sediment conditions while selenium had the greatest release under aerobic conditions.
The presence of nanoparticulate Se (Fig. 3C) is particularly notable. The high specific surface area of nanoparticles can lead to greater reactivity when compared to micron-scale particles and surface coatings. Also, evaluations of the mobilization potential of selenium during the processing and disposal of fly ash should consider conditions for particle detachment in addition to mineral dissolution and ion desorption. The analysis of Se speciation at the nanoscale would provide further insight in this respect, and unfortunately was not available due to limited access to the HXN beamline. However, nano-XANES analyses of Se species is a feasible capability at this beamline and should be a focus for future work.
Altogether, these findings highlight the high degree of spatial and chemical heterogeneity of As and Se in coal fly ash. Multiscale analyses (from nano to micro-scale) are needed to establish binding mechanisms that are relevant to the reactivity and mobilization potential of toxic elements that are enriched in coal fly ash.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Fossil Energy under award #DE-FE0031748. NAR and HH were supported in part by the Human Health and Environmental Analysis Resource funded by the National Institute of Environmental Health Sciences (5U2C-ES030851). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. This research used resources Hard X-ray Nanoprobe (HXN) Beamline at 3-ID of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- F. Vejahati, Z. Xu and R. Gupta, Trace elements in coal: Associations with coal and minerals and their behavior during coal utilization – A review, Fuel, 2010, 89(4), 904–911, DOI:10.1016/j.fuel.2009.06.013.
- G. E. Schwartz, J. C. Hower, A. L. Phillips, N. Rivera, A. Vengosh and H. Hsu-Kim, Ranking Coal Ash Materials for Their Potential to Leach Arsenic and Selenium: Relative Importance of Ash Chemistry and Site Biogeochemistry, Environ. Eng. Sci., 2018, 35(7), 728–738, DOI:10.1089/ees.2017.0347 , (accessed 2022/10/11).
- G. E. Schwartz, N. Rivera, S.-W. Lee, J. M. Harrington, J. C. Hower, K. E. Levine, A. Vengosh and H. Hsu-Kim, Leaching potential and redox transformations of arsenic and selenium in sediment microcosms with fly ash, Appl. Geochem., 2016, 67, 177–185, DOI:10.1016/j.apgeochem.2016.02.013.
- R. Meij, Trace element behavior in coal-fired power plants, Fuel Process. Technol., 1994, 39(1), 199–217, DOI:10.1016/0378-3820(94)90180-5.
- M. Morey and A. Gorski, As U.S. coal-fired capacity and utilization decline, operators consider seasonal operation, US Energy Information Administration, 2020, https://www.eia.gov/todayinenergy/detail.php?id=44976# Search PubMed.
- Y.-T. Liu, T.-Y. Chen, W. G. Mackebee, L. Ruhl, A. Vengosh and H. Hsu-Kim, Selenium Speciation in Coal Ash Spilled at the Tennessee Valley Authority Kingston Site, Environ. Sci. Technol., 2013, 47(24), 14001–14009, DOI:10.1021/es4041557.
- R. K. Taggart, J. C. Hower, G. S. Dwyer and H. Hsu-Kim, Trends in the Rare Earth Element Content of U.S.-Based Coal Combustion Fly Ashes, Environ. Sci. Technol., 2016, 50(11), 5919–5926, DOI:10.1021/acs.est.6b00085.
- E. C. Gillispie, T. D. Sowers, O. W. Duckworth and M. L. Polizzotto, Soil Pollution Due to Irrigation with Arsenic-Contaminated Groundwater: Current State of Science, Curr. Pollut. Rep., 2015, 1(1), 1–12, DOI:10.1007/s40726-015-0001-5.
- B. D. Kocar and S. Fendorf, Thermodynamic Constraints on Reductive Reactions Influencing the Biogeochemistry of Arsenic in Soils and Sediments, Environ. Sci. Technol., 2009, 43(13), 4871–4877, DOI:10.1021/es8035384.
- N. Börsig, A. C. Scheinost, S. Shaw, D. Schild and T. Neumann, Uptake mechanisms of selenium oxyanions during the ferrihydrite-hematite recrystallization, Geochim. Cosmochim. Acta, 2017, 206, 236–253, DOI:10.1016/j.gca.2017.03.004.
- I. Baur and C. A. Johnson, Sorption of Selenite and Selenate to Cement Minerals, Environ. Sci. Technol., 2003, 37(15), 3442–3447, DOI:10.1021/es020148d.
- D. H. Moon, D. Dermatas and N. Menounou, Arsenic immobilization by calcium-arsenic precipitates in lime treated soils, Sci. Total Environ., 2004, 330(1–3), 171–185, DOI:10.1016/j.scitotenv.2004.03.016.
- A. Deonarine, A. Kolker, A. L. Foster, M. W. Doughten, J. T. Holland and J. D. Bailoo, Arsenic Speciation in Bituminous Coal Fly Ash and Transformations in Response to Redox Conditions, Environ. Sci. Technol., 2016, 50(11), 6099–6106, DOI:10.1021/acs.est.6b00957.
- J. C. Hower, U. M. Graham, A. Dozier, M. T. Tseng and R. A. Khatri, Association of the Sites of Heavy Metals with Nanoscale Carbon in a Kentucky Electrostatic Precipitator Fly Ash, Environ. Sci. Technol., 2008, 42(22), 8471–8477, DOI:10.1021/es801193y.
- Y. Luo, D. E. Giammar, B. L. Huhmann and J. G. Catalano, Speciation of Selenium, Arsenic, and Zinc in Class C Fly Ash, Energy Fuels, 2011, 25(7), 2980–2987, DOI:10.1021/ef2005496.
- N. Rivera, D. Hesterberg, N. Kaur and O. W. Duckworth, Chemical Speciation of Potentially Toxic Trace Metals in Coal Fly Ash Associated with the Kingston Fly Ash Spill, Energy Fuels, 2017, 31(9), 9652–9659, DOI:10.1021/acs.energyfuels.7b00020.
- B. Fu, J. C. Hower, S. Dai, S. M. Mardon and G. Liu, Determination of Chemical Speciation of Arsenic and Selenium in High-As Coal Combustion Ash by X-ray Photoelectron Spectroscopy: Examples from a Kentucky Stoker Ash, ACS Omega, 2018, 3(12), 17637–17645, DOI:10.1021/acsomega.8b02929.
- C. Senior, E. Granite, W. Linak and W. Seames, Chemistry of Trace Inorganic Elements in Coal Combustion Systems: A Century of Discovery, Energy Fuels, 2020, 34(12), 15141–15168, DOI:10.1021/acs.energyfuels.0c02375.
- H. A. Hunter, F. T. Ling and C. A. Peters, Coprecipitation of Heavy Metals in Calcium Carbonate from Coal Fly Ash Leachate, ACS ES&T Water, 2021, 1(2), 339–345, DOI:10.1021/acsestwater.0c00109.
- F. T. Ling, H. A. Hunter, J. P. Fitts, C. A. Peters, A. S. Acerbo, X. Huang, H. Yan, E. Nazaretski and Y. S. Chu, Nanospectroscopy Captures Nanoscale Compositional Zonation in Barite Solid Solutions, Sci. Rep., 2018, 8(1), 13041, DOI:10.1038/s41598-018-31335-3.
- A. Pattammattel, R. Tappero, M. Ge, Y. S. Chu, X. Huang, Y. Gao and H. Yan, High-sensitivity nanoscale chemical imaging with hard x-ray nano-XANES, Sci. Adv., 2020, 6(37) DOI:10.1126/sciadv.abb3615.
- R. K. Taggart, N. A. Rivera, C. Levard, J.-P. Ambrosi, D. Borschneck, J. C. Hower and H. Hsu-Kim, Differences in bulk and microscale yttrium speciation in coal combustion fly ash, Environ. Sci.: Processes Impacts, 2018, 20, 1390–1403, 10.1039/C8EM00264A.
- Z. Jin, J. Ren, N. A. Rivera, J. C. Hower and H. Hsu-Kim, Functional Predictor Variables for the Leaching Potential of Arsenic and Selenium from Coal Fly Ash, ACS ES&T Water, 2023, 3(4), 1105–1115, DOI:10.1021/acsestwater.2c00568.
- S. M. Webb, The MicroAnalysis Toolkit: X-ray Fluorescence Image Processing Software, AIP Conf. Proc., 2011, 1365(1), 196–199, DOI:10.1063/1.3625338 , (accessed 2022/07/05).
- L. Li, Y. Hanfei, X. Wei, Y. Dantong, H. Annie, L. Wah-Keat, I. C. Stuart and S. C. Yong, PyXRF: Python-based X-ray fluorescence analysis package, Proc. SPIE, 2017, 10389, 103890U, DOI:10.1117/12.2272585.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541 CrossRef CAS PubMed.
- P. A. O'Day, D. Vlassopoulos, R. Root and N. Rivera, The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(38), 13703–13708, DOI:10.1073/pnas.0402775101 , (accessed 2022/08/30).
- B. G. Kutchko and A. G. Kim, Fly ash characterization by SEM–EDS, Fuel, 2006, 85(17), 2537–2544, DOI:10.1016/j.fuel.2006.05.016.
- J. Yang, Y. Zhao, V. Zyryanov, J. Zhang and C. Zheng, Physical–chemical characteristics and elements enrichment of magnetospheres from coal fly ashes, Fuel, 2014, 135, 15–26, DOI:10.1016/j.fuel.2014.06.033.
- T. Czech, Morphology and Chemical Composition of Magnetic Particles Separated from Coal Fly Ash, Materials, 2022, 15(2), 528 CrossRef CAS PubMed.
- R. Jain, N. Jordan, S. Weiss, H. Foerstendorf, K. Heim, R. Kacker, R. Hübner, H. Kramer, E. D. van Hullebusch and F. Farges, et al. Extracellular Polymeric Substances Govern the Surface Charge of Biogenic Elemental Selenium Nanoparticles, Environ. Sci. Technol., 2015, 49(3), 1713–1720, DOI:10.1021/es5043063.
- N. S. Khoei, S. Lampis, E. Zonaro, K. Yrjälä, P. Bernardi and G. Vallini, Insights into selenite reduction and biogenesis of elemental selenium nanoparticles by two environmental isolates of Burkholderia fungorum, New Biotechnol., 2017, 34, 1–11, DOI:10.1016/j.nbt.2016.10.002.
- B. Yuan, H. Hu, Y. Huang, B. Fu, H. Liu, G. Luo, Y. Zhao and H. Yao, Condensation and adsorption characteristics of gaseous selenium on coal-fired fly ash at low temperatures, Chemosphere, 2022, 287, 132127, DOI:10.1016/j.chemosphere.2021.132127.
- Y.-S. Han, H. Y. Jeong, S. P. Hyun, K. F. Hayes and C.-M. Chon, Beam-induced redox transformation of arsenic during As K-edge XAS measurements: availability of reducing or oxidizing agents and As speciation, J. Synchrotron Radiat., 2018, 25(3), 763–770, DOI:10.1107/S1600577518002576.
- Y. Masue, R. H. Loeppert and T. A. Kramer, Arsenate and Arsenite Adsorption and Desorption Behavior on Coprecipitated Aluminum:Iron Hydroxides, Environ. Sci. Technol., 2007, 41(3), 837–842, DOI:10.1021/es061160z.
- Y. Wu, W. Li and D. L. Sparks, Effect of Iron(II) on Arsenic Sequestration by δ-MnO2: Desorption Studies Using Stirred-Flow Experiments and X-Ray Absorption Fine-Structure Spectroscopy, Environ. Sci. Technol., 2015, 49(22), 13360–13368, DOI:10.1021/acs.est.5b04087.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2en01056a |
| This journal is © The Royal Society of Chemistry 2023 |