
Developing electrochemical hydrogenation towards industrial application
Julian T.
Kleinhaus
 a,
Jonas
Wolf
ab,
Kevinjeorjios
Pellumbi
ab,
Leon
Wickert
ab,
Sangita C.
Viswanathan
ab,
Kai
junge Puring
b,
Daniel
Siegmund
*ab and
Ulf-Peter
Apfel
*ab
a,
Jonas
Wolf
ab,
Kevinjeorjios
Pellumbi
ab,
Leon
Wickert
ab,
Sangita C.
Viswanathan
ab,
Kai
junge Puring
b,
Daniel
Siegmund
*ab and
Ulf-Peter
Apfel
*ab
aInorganic Chemistry I, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: daniel.siegmund@umsicht.fraunhofer.de; ulf.apfel@rub.de
bFraunhofer Institute for Environmental, Safety and Energy Technology UMSICHT, Osterfelder Str. 3, 46047 Oberhausen, Germany
First published on 10th October 2023
Abstract
Electrochemical hydrogenation reactions gained significant attention as a sustainable and efficient alternative to conventional thermocatalytic hydrogenations. This tutorial review provides a comprehensive overview of the basic principles, the practical application, and recent advances of electrochemical hydrogenation reactions, with a particular emphasis on the translation of these reactions from lab-scale to industrial applications. Giving an overview on the vast amount of conceivable organic substrates and tested catalysts, we highlight the challenges associated with upscaling electrochemical hydrogenations, such as mass transfer limitations and reactor design. Strategies and techniques for addressing these challenges are discussed, including the development of novel catalysts and the implementation of scalable and innovative cell concepts. We furthermore present an outlook on current challenges, future prospects, and research directions for achieving widespread industrial implementation of electrochemical hydrogenation reactions. This work aims to provide beginners as well as experienced electrochemists with a starting point into the potential future transformation of electrochemical hydrogenations from a laboratory curiosity to a viable technology for sustainable chemical synthesis on an industrial scale.
 From left to right: Julian T. Kleinhaus, Jonas Wolf, Kevinjeorjios Pellumbi, and Leon Wickert | Julian T. Kleinhaus, Jonas Wolf, Kevinjeorjios Pellumbi, and Leon Wickert all received their MSc in Chemistry from the Ruhr University Bochum and are currently doctoral students at Ruhr University Bochum and Fraunhofer UMSICHT. JTK investigates electrochemical hydrogenation reactions with molecular as well as heterogenous catalysts within the Cluster of Excellence RESOLV. His work focuses on stereoselective electrosynthetic processes. JW develops innovative electrode and cell concepts for the electrocatalytic hydrogenation of organic substrates. He is funded by the Studienstiftung des deutschen Volkes. KP aims to establish holistic pathways for the transfer of novel catalysts and materials into scalable electrolyzers for CO2 electroreduction and electrochemical hydrogenations. He is currently a recipient of the Kekulé-Fellowship of the Fonds of the Chemical Industry. The research of LW focusses on the development of electrochemical systems for the sustainable hydrogenation of organic molecules. He specializes on the conversion of water-insoluble substrates and the design of high-throughput electrochemical cells. |
 Sangita C. Viswanathan | Sangita Viswanathan obtained her BSc in Chemistry from the Stella Maris College in Chennai, India in 2019 before she moved to Germany for pursuing her Master studies at Ruhr University Bochum. In 2022, she joined the group of Prof. Dr Apfel where she is working on the development of optimization of electrochemical organic hydrogenation reactions. |
 Kai junge Puring | Kai junge Puring obtained his PhD at Fraunhofer Institute for Environmental, Safety and Energy Technology (UMSICHT), Oberhausen and Ruhr University Bochum for his work on the development of catalysts and gas diffusion electrodes for the electrochemical CO2 reduction with Prof. Dr U.-P. Apfel. After finishing his PhD he proceeded research in the fields of water and CO2 electrolysis technologies at Fraunhofer UMSICHT, where he is currently Group Manager for Electrolysis Technologies. His research interests focus on the ‘Lab-to-Pilot’ development of next generation membrane electrolysis technologies from materials research to process design. |
 Daniel Siegmund | Daniel Siegmund obtained his PhD from the Ruhr University Bochum for his work on the development of novel organometallic anti-biotics with Prof. N. Metzler-Nolte. In 2018, he joined the Fraunhofer Institute for Environmental, Safety and Energy Technology (UMSICHT) in Oberhausen where he is currently a group manager for electrocatalysis in the department of electrosynthesis and leading the NanoMatFutur research group H2Organic. His research interests focus on the development of sustainable electrocatalytic synthesis processes for organic commodity and fine-chemicals. |
 Ulf-Peter Apfel | Ulf Apfel received his PhD from the Friedrich-Schiller University Jena. After a postdoctoral stay at MIT (2011/2012), he started his independent career at the Ruhr University Bochum funded by the ‘‘Fonds der Chemischen Industrie’’ and the DFG as an Emmy Noether group leader. He holds a professorship at the Ruhr University Bochum since 2019 and is leading the department Electrosynthesis at Fraunhofer UMSICHT. His research interests are in the field of technical electrochemistry with a special emphasis on the electrochemical reactor and catalyst design. |
Key learning points(1) Electrochemical hydrogenations (EChH) offer enormous potential to sustainably replace classic thermocatalytic hydrogenations.(2) Scalability and efficient process design of EChH is crucially dependent on the employed reactor design. Currently employed systems with increasing scalability include H-type cells for basic evaluation, whereas flow cells and zero-gap electrolyzers offer the potential for industrial application. (3) Although EChH is known for all relevant substrate classes, only for specific substrates, e.g., toluene or alkynes, efficient processes at elevated current densities (>100 mA cm−2) are known. (4) Material and component development need to be accelerated to obtain cheap and efficient electrocatalysts, durable membranes and optimized reactor parts (e.g., flow-fields) to achieve meaningful process optimizations. (5) Techno-economic and life-cycle assessments of EChH processes are a crucial, but currently rarely considered factors in evaluating both environmental and profitability aspects to prove the industrial relevance of this electrolysis process. |
1. Introduction
Hydrogenation reactions, i.e., the reductive addition of hydrogen to an inorganic or organic substrate, can safely be considered as one of the most important transformation classes in the chemical industry. It is estimated that about a quarter of all synthetic chemical processes contain at least one hydrogenation step.1 The range of substrates hereby includes edible oils and fats,2 petrochemical products3 such as paraffins, food supplements4 and even fine chemicals such as pharmaceuticals.5 The enormous range of conceivable starting materials and products implies that hydrogenation reactions are important on almost any scale imaginable. In classical (thermo)chemical hydrogenation approaches, gaseous hydrogen is reacted with the respective substrates under increased pressure, possibly increased temperatures and, depending on the exact application, a metal-catalyst. Currently, a large part of the available hydrogen is produced from fossil fuels with the release of climate-damaging carbon dioxide, so that hydrogenation operations are also indirectly subject to considerable CO2 emissions.6 Electrochemical hydrogenations (EChH) and likewise a large number of other electroorganic syntheses have the potential to overcome the severe disadvantages of their traditional counterpart. The clear advantages of electrochemical approaches include generally good controllability, the possibility of implementation under mild conditions as well as the elimination of gaseous hydrogen and the associated need for a dedicated hydrogen infrastructure.7 Furthermore, even stoichiometric hydridic reducing agents, commonly applied in reducing especially carbonyl species can be replaced by an electrocatalytic process, avoiding the formation of surplus waste. Clearly, these advantages of electrochemistry were already directly apparent to the pioneers of electrocatalytic hydrogenation and it is no surprise that a large number of compounds have already been successfully converted by electrolysis. It is therefore surprising that hardly any industrial process, to the best of our knowledge, uses dedicated electrochemical or electrocatalytic hydrogenation as of 2023.8–11 On closer analysis, several reasons can be given for this fact: firstly, an industrial process requires scaled electrochemical reactors. In contrast, however, the majority of academic test reactions are carried out in small, less scalable cells, so that an effective transfer to the actual application is considerably more difficult. Here, it must be noted that reactor configurations have a considerable influence on decisive target variables of the electrochemical reaction (Faraday efficiency, cell potential, achievable current densities). Secondly, electro-organic reactions in general often require organic solvents with low conductivity. These solvent limit achievable currents in cells with a liquid electrolyte and at the same time make high demands on the used materials.11,12 Thirdly, many approaches still require expensive or complex components, e.g., catalysts, which are unsuitable for a cost-competitive scaled target process. Lastly, the knowledge on reaction mechanisms and working principles is limited and cannot be easily transferred between different substrates and catalysts hampering efficient process development.13Yet, we believe that hydrogenation reactions can be one of the most promising candidates for direct positive impact on the carbon footprint of the chemical industry in the course of electrification. This potential of electrocatalytic hydrogenation is very well demonstrated by the largest electrosynthetic process established in industry, the hydrodimerization of acrylonitrile to adiponitrile. Adiponitrile is an important precursor for Nylon and therefore produced in Mt scale, a significant portion (>500 kt) of which is generated electrochemically by Ascend.13 It is estimated, that by the use of renewable electricity for this single step, the total CO2 emissions of Nylon production could be reduced by around 20%.12 At the same time, industrial electrochemical hydrogenations become relevant in industrial research,14–18 where, e.g., the ENEOS Corporation aims at implementing methylcyclohexane produced by electrochemical hydrogenation as a liquid organic hydrogen carrier. In February 2023, a 150 kW-electrolyzer was scheduled to go into operation to demonstrate the feasibility of the technology.18
In this tutorial review, we therefore present not only the latest developments in the field, but in particular the added value of individual results for next steps towards industrial application. In doing so, newcomers to the field will receive an informed overview of the basics as well as currently developing trends. Finally, as an active group in the field of EChH, we provide insights into pitfalls in the practical implementation that we encountered in the past.
2. Fundamentals
2.1 Mechanistic considerations
When considering the cathodic transformation of organic compounds to hydrogenated products, two general pathways have to be distinguished which in turn are not necessarily distinguished by the pure nature of the obtained products (Fig. 1).19,20 There is (1) the electrocatalytic hydrogenation (denoted as ECH, Fig. 1), which involves the intermediate generation of adsorbed or chemisorbed hydrogen on the electrocatalyst surface (EHads). This surface hydrogen species in turn can react with a chosen organic substrate (Org) to form the desired hydrogenated product (OrgH2). For this to occur, the organic substrate is required to interact with the catalyst surface in a favorable manner, i.e. in the majority of cases needs to be adsorbed on the catalyst surface as well (EOrgads), however bound too strongly to prevent any mobility on the surface or even prohibit the desorption of the final product.21,22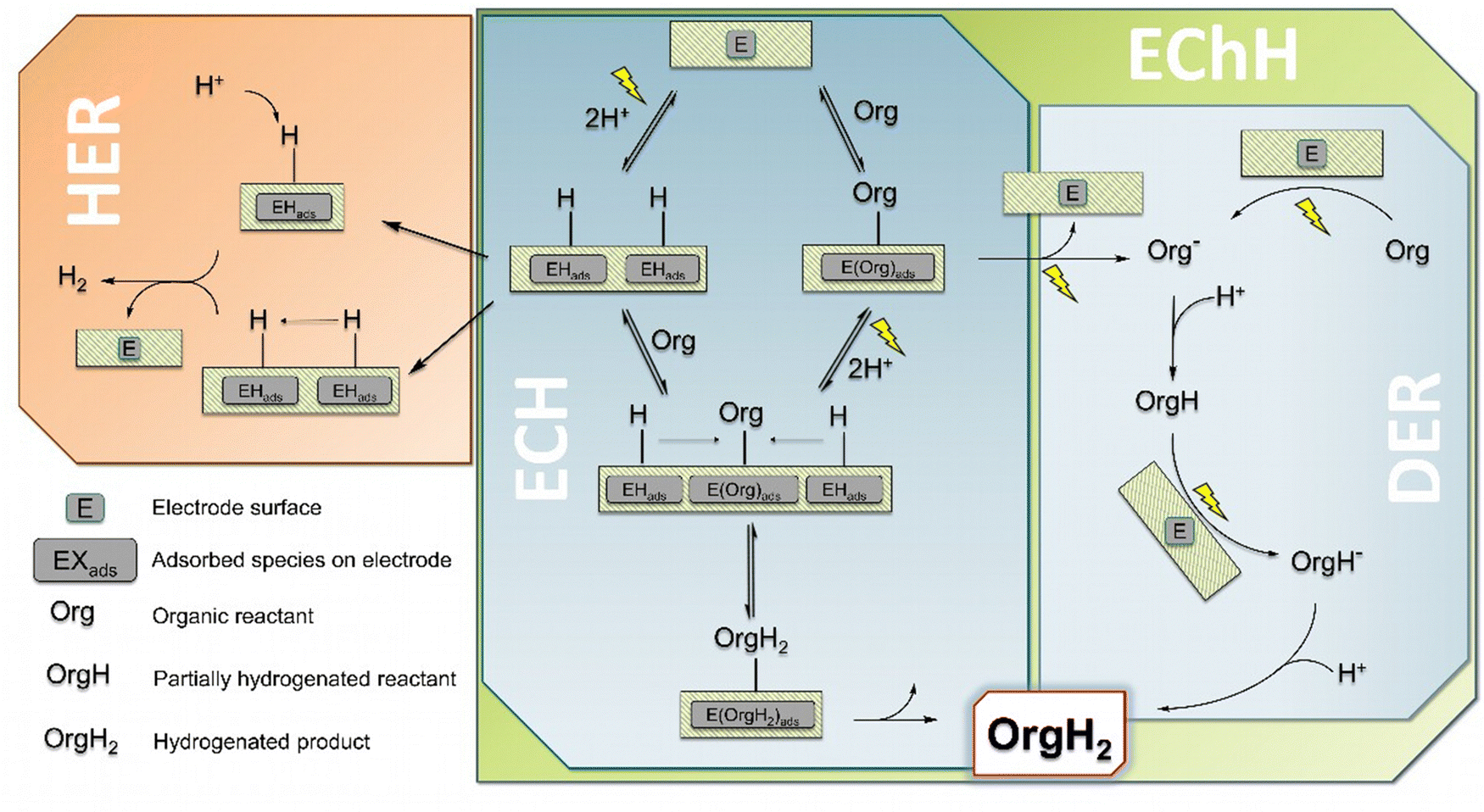 | ||
| Fig. 1 Overview of the main reaction mechanisms for electrochemical hydrogenations (EChH): ECH: electrocatalytic hydrogenation, DER: direct electroreduction. HER (left) describes the well-known hydrogen evolution reaction as major side reaction. | ||
(2) There is also the possibility for a (multiple) direct electroreduction of the organic substrate (Org−, Fig. 1 right) which is followed by protonation from a proton source in the electrolyte to obtain the very same product as in case (1) (first mono-hydrogenated OrgH, finally OrgH2). This pathway can be referred to as direct electroreduction (DER).23 However, the terms electrochemical as well as electrocatalytic hydrogenation are often used synonymously, with either EChH or ECH used as generic term. In our view EChH should be used to describe the general reaction class and hence will be used throughout this work if the exact mechanism (with ECH/DER being boundary cases) is of minor importance. One reason for this apparent inconsistency could be a focus on a successful synthesis, whereby the exact elucidation of the underlying mechanism is of secondary importance. Furthermore, the described idealized mechanisms can simultaneously contribute to a real-world electrolysis application.
The sole availability of both mechanisms equips the synthetic organic chemist with the possibility to stir the hydrogenation reaction in specific directions in order to increase the chemoselective outcome, i.e., if the molecule of interest contains multiple functional groups to be potentially hydrogenated or an over-hydrogenation needs to be prevented. The major handles to control the underlying mechanism and thus to influence the product selectivity are the chosen electrode surfaces (catalysts), the applied electrode potential/current density (which in turn strongly depends on the catalyst) and the composition of the electrolyte (chemical nature, pH).24 Generally, catalysts favoring ECH reactions are chosen from rather good hydrogen evolution (HER) catalysts for the reason that the first reaction step of ECH, formation of EHads, is actually equivalent to the Volmer-reaction step in hydrogen production.25 However, for the ECH to proceed favorably, the reactive collisions of EHads with adsorbed reactants Orgads needs to outcompete the recombination of two EHads (Tafel-step) or the protonation of EHads (Heyrovsky-step), both leading to subsequent desorption of H2. From these considerations it is immediately evident that hydrogen formation is the major side reaction of the ECH and sophisticated fine tuning of reaction conditions (T, pH) as well as carefully chosen electrode materials are needed. In case of DER-mechanisms additional side reactions may become more dominant, such as downstream reactions of electrochemically reduced organic substrates aside of simple protonation, e.g. the recombination of two reduced species in a coupling process or the hydrogenolysis of polarized bonds.7,26 On the contrary, with DER it is conceivable to selectively address certain functional groups in organic molecules in the presence of multiple reducible parts by controlled potential electrolysis.
2.2 Benchmarking
In order to compare different technological approaches to the EChH and also derive techno-economical evaluations, key performance indicators (KPI) have to be defined. While the thermocatalytic hydrogenation is well known and thus these KPIs exist and target values are defined,27 currently no universal benchmarks for EChH have been established. In this respect, we herein discuss important key performance indicators for the EChH. In general, it is advisable to adopt well-established nomenclature and metrics from related electrochemical processes, here especially water electrolysis and carbon dioxide reduction, that could help to define KPIs for the EChH in the future.28For thermochemical hydrogenation reactions the key performance indicators are typically the turnover or productivity of the process, the conversion of the substrate to the target product either over a process runtime (batch) or a single pass of the catalyst bed (continuous) depending on batch or continuous process design and the selectivity for a certain product within a product spectrum.29 In electrochemical reactions the turnover of a reaction with a molar product flow (ṅ) is directly tied to the electrical current supplied to the EChH unit by Faraday's law (eqn (1)). Herein i denotes the electric current, F the Faraday constant, t the duration of electrolysis and z the number of transferred electrons for a single given species to its corresponding product.
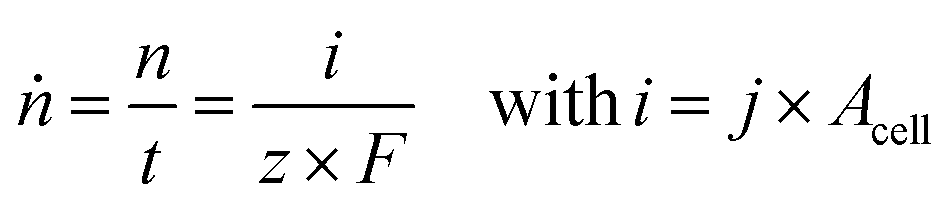 | (1) |
Similar to thermochemical hydrogenations, principles of (single pass) conversion (SPC, eqn (2)) apply to measure the capability of the process to achieve high conversion by dividing the molar product flow (ṅp) with the molar reactant flow (ṅr), which will reduce the required purification expenditure.
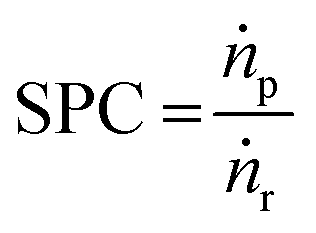 | (2) |
The selectivity of an electrochemical process is typically defined by the Faraday or current efficiency (eqn (3)) which expresses the percentage of the current that is used for a certain reaction product p by evaluating the theoretically possible molar product flow (ṅtheor) against the practically observed one (ṅp). We want to highlight that for deep understanding of electrochemical processes in general, it is important to determine the Faraday efficiencies for all products and side-products as gaps in the observed faradaic efficiency to 100% may result not only from the main side-reactions (in the case of EChH the parasitic hydrogen evolution) but could also arise from phenomena such as product cross-over or parasitic currents. Hence, product analysis should not only be limited to the cathode compartment but rather also anodic compartments which is useful for pinpointing deficiencies of the applied cell design.
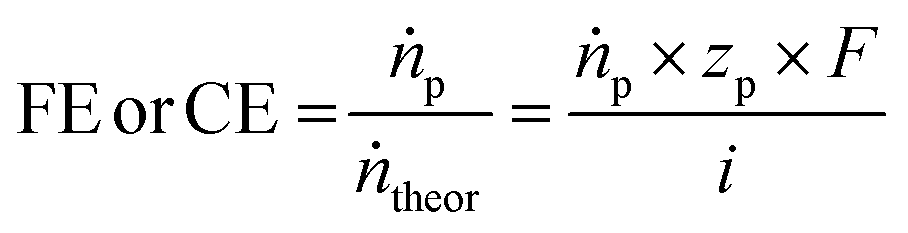 | (3) |
| UCell = E0 + ηiR + ηCat,A + ηCat,C + ηMTL | (4) |
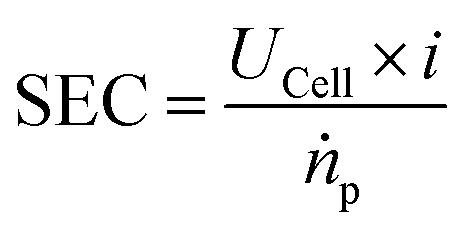 | (5) |
2.3 Performance reporting and guidelines
The wide variety of reported performance indicators like Faraday efficiency, conversion, and conversion rate already makes the comparison between different electrochemical experiments difficult. Moreover, all these indicators can only be assessed in relation to others as, e.g., a high conversion does not necessarily indicate an efficient process if the obtained current efficiency is low. In a similar fashion, high initial Faraday efficiencies that drop at elevated conversion constitute a significant problem. On the contrary, different philosophies for reporting electrochemical data exist, which somewhat hinder the detailed comparison of key performance indicators such as current (density) and transferred charge across publications. Considering different cell and electrode geometries and concepts, the broad variety of used catalyst and substrate classes, and the individual combinations of those variables, a comparison and analysis of the reported data is extremely challenging. For intensely studied substrates like furfural derivatives such analyses are available, but for the broad spectrum of other hydrogenation substrates this is not the case.19,32,33 This issue restricts the assessment of own results in comparison with the available literature but also hampers fundamental understanding of the ongoing processes and development of theoretical models. Recently, ACS has published guidelines for unifying the reporting of electrochemical data.34 The authors encourage researchers in this rising field to take these considerations seriously and actively participate in standardizing the field by carefully reporting all relevant parameters.3. Reactors for electrochemical hydrogenations
As for all electrolytic processes, the employed reactor type and the associated design play a critical role in controlling the product spectrum, as well as for optimizing the overall process efficiency. In this chapter, we explore the different types of reactors currently employed in the field of EChH, presenting their advantages, disadvantages, and limitations. Overall, electrochemical reactors for EChH can be separated into three types: H-type reactors, flow cells and zero-gap electrolyzers (Fig. 2).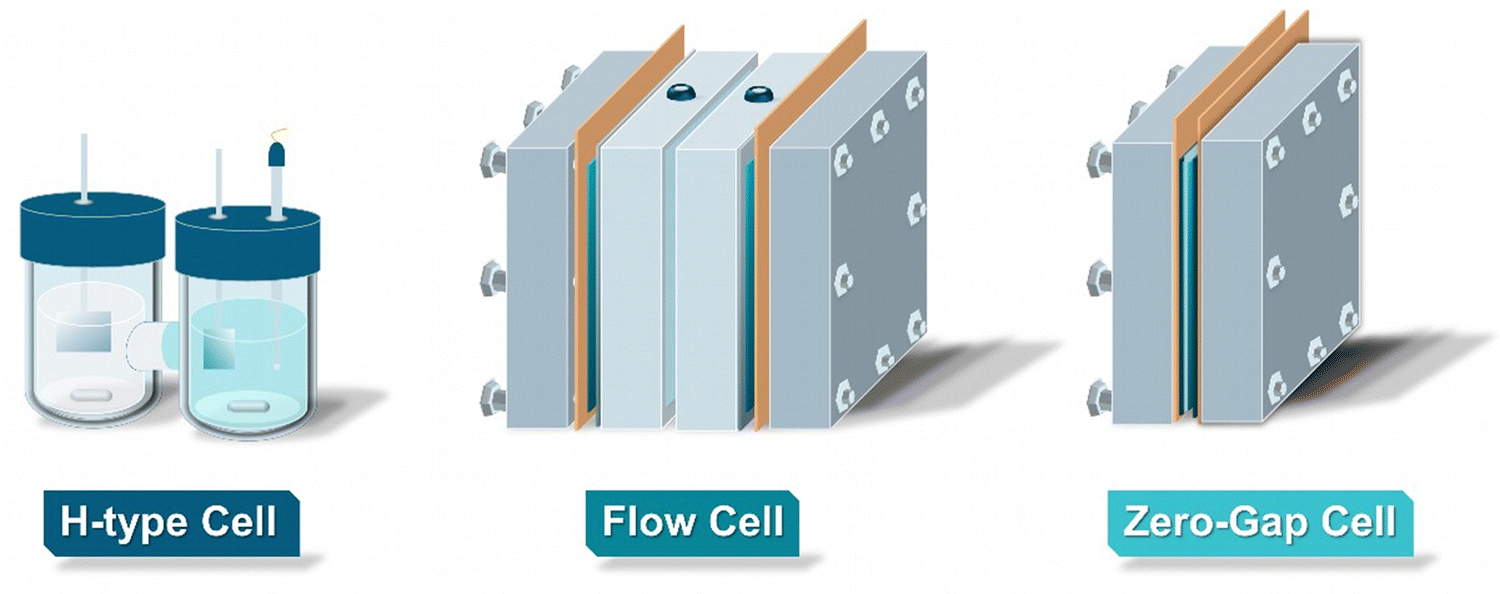 | ||
| Fig. 2 Schematic representation of the three major reactor types employed for the EChH. | ||
3.1 H-type reactors & lab-scale concepts
The primary workhorses of emerging electrochemical technologies such as the EChH are often H-type cells. Typically consisting of two half-cells either made of glass or durable polymers such as PTFE or PEEK, such designs are ideal for lab-scale experiments, due to their modular and inexpensive character and the possibility to physically separate the cathode and anode sides.35 Characteristically, H-type cells are able to operate with low electrolyte volumes, in both aqueous and organic environments, at different temperature ranges, while in-flow gas-sampling can be easily performed to quantify the product spectrum. Through this plethora of advantages, such designs provide important information on the activities and selectivity of different electrocatalysts, influence of electrolyte species and electrode architecture on the EChH. Nevertheless, limitations exist with low current densities leading to low overall product conversions. While H-type cells are omnipresent across all electrocatalytic fields, we like to steer the attention of the interested reader to novel lab-scale designs that appear to be unique to the ECH and which could accelerate catalytic screening and mechanistic understanding (Fig. 3).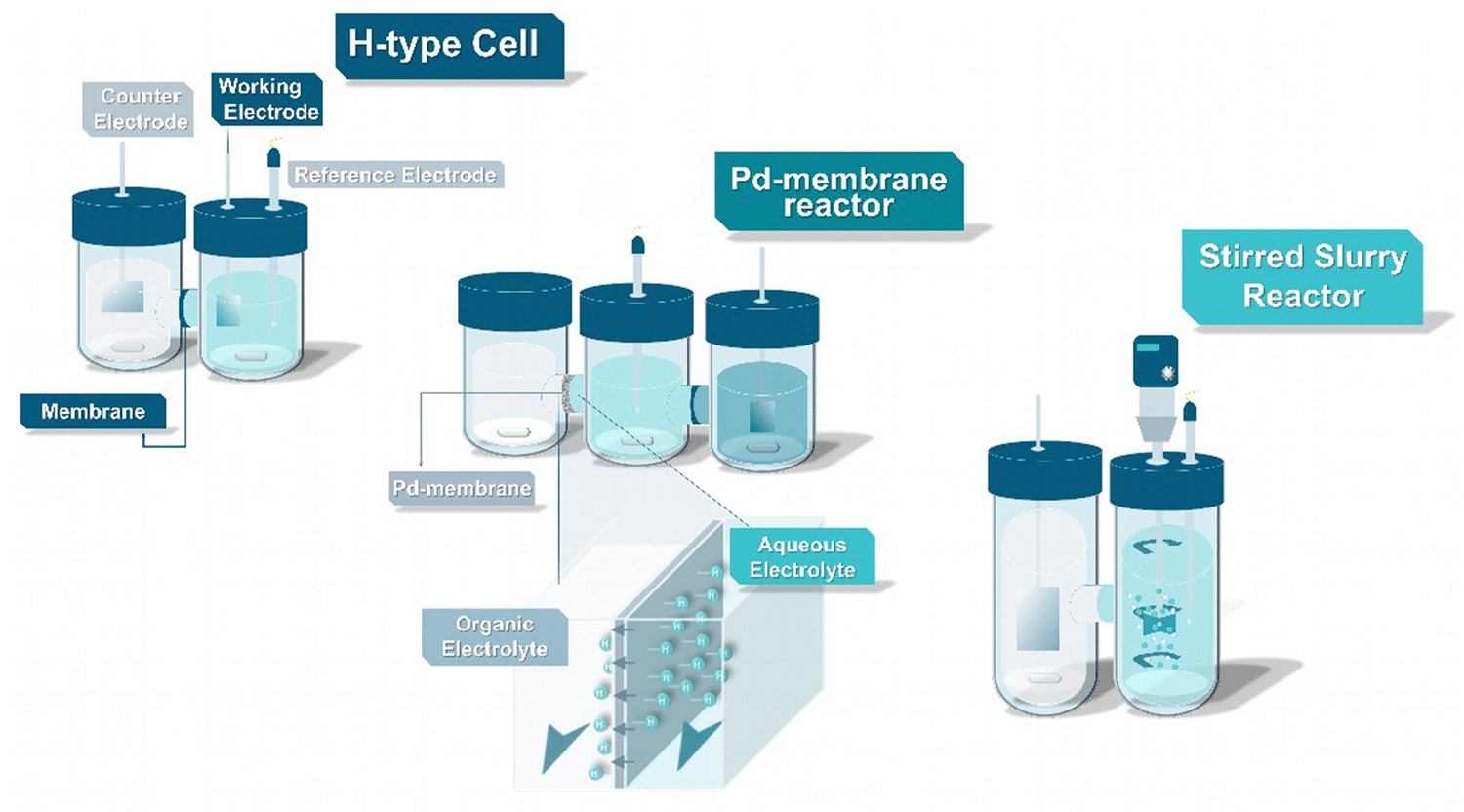 | ||
| Fig. 3 Advanced reactor concepts derived from an initial H-type approach for EChH. | ||
In addition, to using supported electrocatalysts and foil-based electrodes, Berlinguette and coworkers have driven the development of palladium membrane-based reactors for ECH as advanced specialized types of H-type cells.36–41 Specifically, hydrogen species bound to the cathode during electrolysis in aqueous environments can permeate through a Pd-lattice in Pd-membranes towards a separate compartment where the organic substrate is hydrogenated. This approach does not only take advantage of the well-known ability of Pd to perform hydrogenation reactions, but also its ability to transport hydrogen atoms through the Pd lattice, to effectively separate an aqueous environment providing protons from the organic hydrogenation compartment, which enables the use of organic solvents for the hydrogenation of hydrophobic organic substrates. This separation also facilitates the investigation of underlying mechanisms and electrocatalytic screenings via metal sputtering methods,38,42,43 permits the efficient deuteration of organic compounds44 as well as the regeneration of enzymatic cofactors.40 Further groups have adopted this design, with Zhang et al. expanding the approach for the conversion of acetonitrile to ammonia,45 Xu et al. applying this reactor design for reductive treatment of halogen-containing contaminants in water,46 and Sun and coworkers even enabled a highly efficient dual hydrogenation by applying Pd-membranes not only as the cathode but also as the anode for formaldehyde oxidation.47
Notably, Wijaya et al. demonstrated the promising perspective of mechanically stirred H-type slurry reactors to reach higher current densities (up to about 250 mA cm−2).48 Herein, a Nafion-separated slurry reactor, employing 5 wt% of well dispersed Pt/C as the catalyst in the cathode compartment, was used for the hydrogenation of guaiacol. The study also sheds light on the effect of catholyte-anolyte combinations, temperature, and the role of galvanostatic vs. potentiostatic control for the hydrogenation of guaiacol, highlighting the intricate interdependencies within efficient ECH reactions. Employing a similar reactor design, Han et al. took advantage of the ability of Pd to efficiently create reactive hydride species on its surface by immobilizing Pd-particles on carbon nanotubes (CNTs).49 Here, CNTs act not only as a conductivity support, but assist in the creation of oil–water pickering emulsions in a stirred reactor, containing the target substrate, here styrene, within them and hence increasing the mass activity of the used Pd. The advantage of bi-phasic systems has also been demonstrated by Jiang et al. separating the less water–soluble products of furfural hydrogenation by the addition of organic oils to the catholyte compartment.50 Overall, such novel concepts point out the vast and still unexplored research opportunities within the field of EChH, towards generating tailored electrode architectures, coupled with reactor design towards increasing mass activity of hydrogenation electrocatalysts. We would therefore urge the community to not limit itself to simple H-type concepts, but further explore and innovate towards improved reactor designs, possibly influencing the overall field of electrosynthesis. Yet, the transferability to scaled systems alongside a conservation of performance remains to be explored. Contrary, two reactor designs exist that would allow the transition from H-type cells to fully built large-scale reactors, namely flow and zero-gap electrolyzers.
3.2 Flow cell electrolyzers
The general objective of flow electrolyzers (Fig. 4) is to minimize the inter-electrode distances to decrease ohmic losses while maximizing the mass transport of reactants.35 The employed electrodes are usually either plates or foils of metal electrocatalysts or materials deposited on conductive metal supports. Thin polymeric spacers allow for the transport of electrolyte through the cell compartments, while the addition of turbulence promotors within these frames aims to meliorate reactant diffusion. The electrolytes are commonly continuously circulated through the cathode and anode compartment, either in batch or single-pass mode, while both divided and undivided variants exist. Divided variants often employ ion-exchange membranes (IEMs) to control the ionic transport in solution. In addition, membrane separated configurations allow for the complete separation of the cathode and anode compartments, minimizing crossover of reactants and products, while, however, adding an additional resistance factor. Three main types of IEMs exist, cation-, anion-exchange membranes (CEMs & AEMs) as well as bipolar membranes (BPMs).35 In more detail, CEMs transport protons or cationic species, depending on the anolyte composition, alongside water from the anode to the cathode, whereas AEMs transport anionic species, e.g., OH− generated during the EChH, to the anode alongside water through electro-osmosis. BPMs traditionally consist of laminated CEMs and AEMs, either operating in a so-called forward-bias leading to the recombination of H+/OH− at the membrane junction, or in reverse-bias, where water is dissociated into H+ and OH− being transferred to the differently charged electrodes, respectively.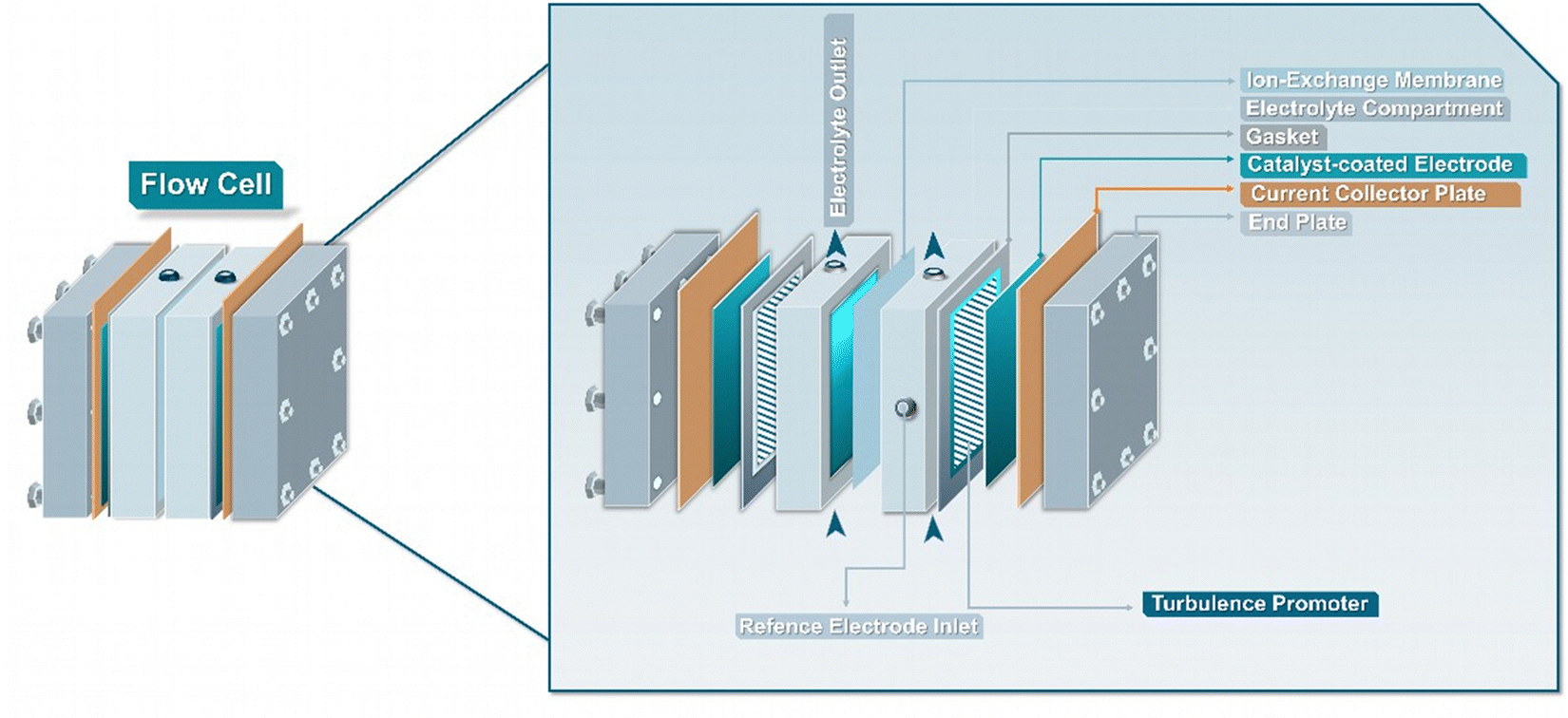 | ||
| Fig. 4 Schematic representation of a typical flow-reactor configuration for electrolysis. | ||
Taking advantage of the increased elevated mass transport provided by flow electrolyzers, Zhu et al. and Bu et al. were able to severely shorten the required conversion time as well as to increase the achievable partial current density to 750 mA cm−2 for the EChH of the vitamin synthon 2-methyl-3-butin-2-ol (MBY) to its alkenol counterpart (2-methyl-3-butene-2-ol (MBE)) directly opening a promising route for the adoption of flow cells in industrially-relevant applications.51,52 Moreover, the Berlinguette group expanded the use of the above mentioned Pd-membrane reactor to a flow configuration, accompanied by significant increases in reaction rates (15-fold).53 Simultaneously, the group demonstrated that sputtering Pd on porous PTFE-membranes yields similar activities, presenting a more cost-effective alternative to their previously reported concept.54 Compared to their divided counterpart, undivided flow reactors on the other hand largely simplify the reactor architecture, allowing for more robust and simpler electrolyzer scale-up. Dell’Anna et al. investigated the hydrogenation of cis,cis-muconic acid in both undivided and Nafion-divided electrolyzers.55 The influence between the use of a Nafion-membrane to separate the two cell compartments and non-divided variant was investigated, showing minimal differences. However, it is important to note, that the generated product and employed reactants were not prone to oxidation on the anode, which could become an issue for other investigations aiming to implement such designs.
An interesting sub-category of flow electrolyzers are micro-fluidic electrochemical reactors. Their design involves the use of micro-structured electrodes separated only by a thin gap. Both divided and undivided variants exist. Overall, such cells provide a pathway towards continuous operation, achieving faster conversions and higher yields for the target product.56–58 Furthermore, compared to typical batch cells, micro-flow electrolyzers present added advantages of providing a more uniform heat and mass transport as well as a homogeneous current distribution. While micro-fluidic cells have in recent years become an important tool of organic synthesis laboratories,59 their use in the case of EChH remains relatively rare. Cao et al. investigated the electrochemical hydrogenation of furfural in a micro-flow reactor, showing how this reactor architecture can allow for lower dwell times and elevated conversion of bio-mass derivates.60 Paired with computational fluid dynamics (CFD) simulations, Noel and co-workers were able to demonstrate how Taylor Flow reactors, characterized by gaseous and liquid saturated regions, lead to local increases of the cell resistance limiting the efficiency of such reactors.61 Further designs focusing on electrochemical synthesis have in recent years worked on resolving such issues, offering interesting reactor alternatives for EChH researchers. Interestingly, Atobe and co-workers showed how micro-flow reactors could be employed to performed so-called paired reactions of anodic and cathodic species.62–64 Specifically for the EChH, benzylamine was dimerized to the respective imine at the anode compartment prior to diffusing to a graphite cathode electrode through the micro-fluidic channel for the performance of the EChH. This approach circumvented issues of crossover but also of the use of a membrane. By employing ionic liquid electrolytes, Atobe and coworkers were able to ensure the non-mixing of the two electrolytes in a microfluidic cell via careful tuning of reactor architecture to maintain a stable liquid–liquid interface. Overall, multiple investigations involving flow based electrolyzers demonstrate the promise of EChH to rapidly scale-up promising reactions. Notably, for this reactor architecture microfluidic cells could possibly open novel pathways towards continuous operation against the commonly performed batch mode operation.
3.3 Zero-gap electrolyzers
Across the field of EChH, alongside H-type cells, the zero-gap electrolyzer constitutes the most-frequently employed electrolyzer class, with substrate classes ranging from bio-oils, biomass derivates, unsaturated carbon bonds, carbonyls and aromatics, among others (Fig. 5).65–69 The term ‘zero-gap’ here refers to the direct contact of anode and cathode with the ion-conductive membrane, which functions as a solid electrolyte. This operation mode has two main benefits. First, the direct contact of the electrode with the membrane reduces the electrode distance and with it the inner resistance of the cell, thereby enabling operation at high current densities above 1 A cm−2.70 Second, the presence of the solid electrolyte makes a liquid electrolyte superfluous. In CO2 electrolysis, this enables the use of a gaseous substrate stream,35,71–73 a concept that could also be adapted in EChH, but more importantly it also enables substrate solutions in organic solvents with low conductivity.65–69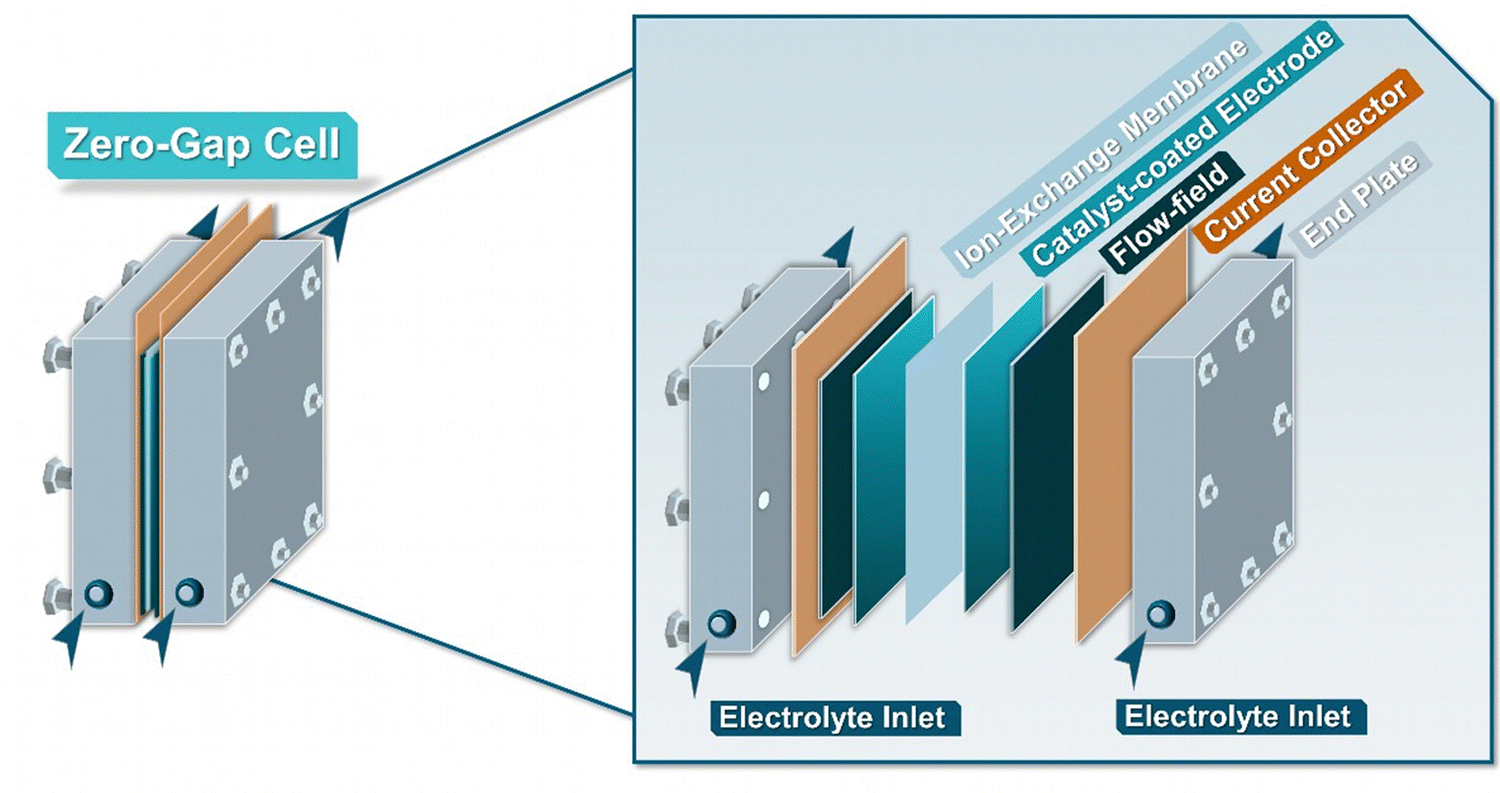 | ||
| Fig. 5 Schematic representation of a typical zero-gap electrolyzer. | ||
The substrate solutions are transported through a flow field structure to a conductive porous substrate, ensuring the diffusion of reactants and conduction of current to the catalytic layers, located in direct proximity to the membrane. Although in the case of flow-cell electrolyzers, the catalytic particles can only be deposited onto the electrode surface, for ZGEs the catalytic layer can additionally be directly applied on the surface of the membrane.74,75 The so-called catalyst coated substrate (CCS) configuration can be directly achieved by spray-coating, dip-coating or doctor blading, while catalyst-coated membrane (CCM) configurations can additionally be performed through the use of the decal process, involving the transfer of catalytic layer to conductive membranes via hot-pressing (Fig. 6). Comparatively, the CCS approach is known to be more robust and minimize membrane swelling, though the CCM approach is currently preferred, delivering higher catalyst utilization and better ionic-conductivity within the membrane-catalyst interface.
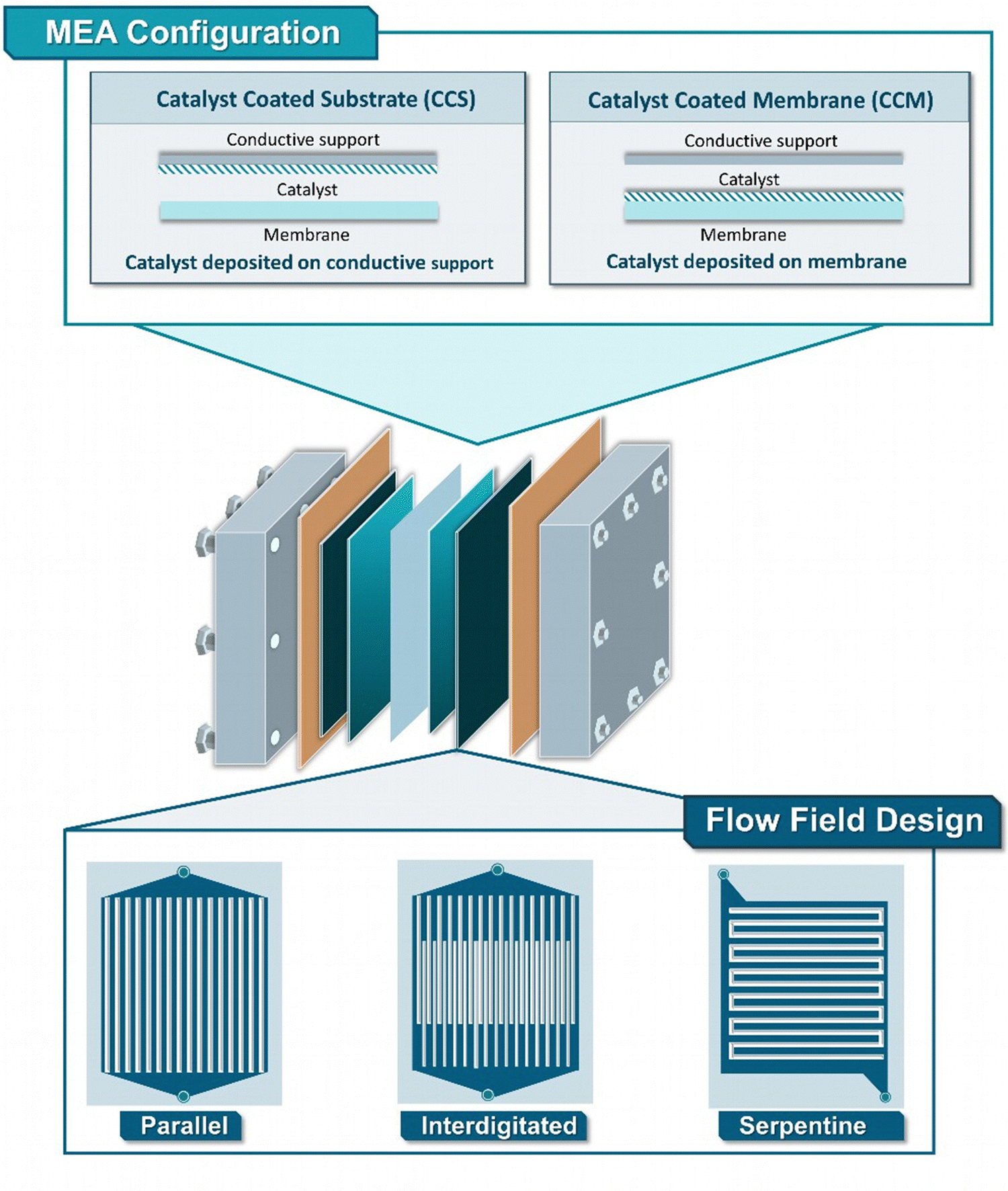 | ||
| Fig. 6 Detailed schematic representation showing the different techniques towards the generation of a MEA (CCS and CCM) as well as widely used flow field designs (parallel, interdigitated, serpentine).76 | ||
Compared to a flow electrolyzer, zero-gap designs allow for minimization of the electrolyzer resistance and better control of the local environment depending on the employed membrane type and process conditions. Overall, the previously mentioned membrane types, CEMs and AEMs and BPMs can also be employed in ZGEs, with the transferred ionic species being shown in Fig. 8. Despite of these advantages, the efficiency of this reactor type is strongly dependent on the performance and degree of optimization of the individual components, such as the membrane type and thickness and electrode architecture of both the anode and the cathode.77 It is thus evident that an efficient zero-gap electrolyzer requires the interdisciplinary effort of chemical sciences and engineering to understand and tune the interplay between different approaches/parameters.
Commonly, hydrogen oxidation has been one of the most employed anode reactions in the case of zero-gap EChH electrolyzers.66–69,78 Specifically, hydrogen gas is oxidized on the surface of a Pt catalyst to protons, which may be transported to the cathode side through the membrane. The operation mode, of these so-called ‘proton-pumps’ (Fig. 7) has been mainly used for the hydrogenation of bio-oils and aromatic compounds, being recently expanded to stereo- and diastereoselective and asymmetric hydrogenation reactions.68,78 Although the perspective towards real-life applicability of this anode reaction is currently unclear and heavily dependent on the local hydrogen supply, the main application of this operating mode lies not on the electro-synthetic but the hydrogen-storage side of the EChH, so-called LOHCs (liquid organic hydrogen carriers). Numerous supports have shown how this reaction pairing can be employed to efficiently store hydrogen in such LOHCs which mainly consist of aromatic compounds, with toluene being one of the most investigated carriers.79 Interestingly, since hydrogen oxidation on Pt is highly efficient, with the required overpotential lying close to 0 V,80 the use of proton-pumps could become an interesting platform for a better testing and understanding of electrocatalysts and different membranes in zero-gap electrolyzers, since the obtained cell voltage corresponds closely to the cathode performance.
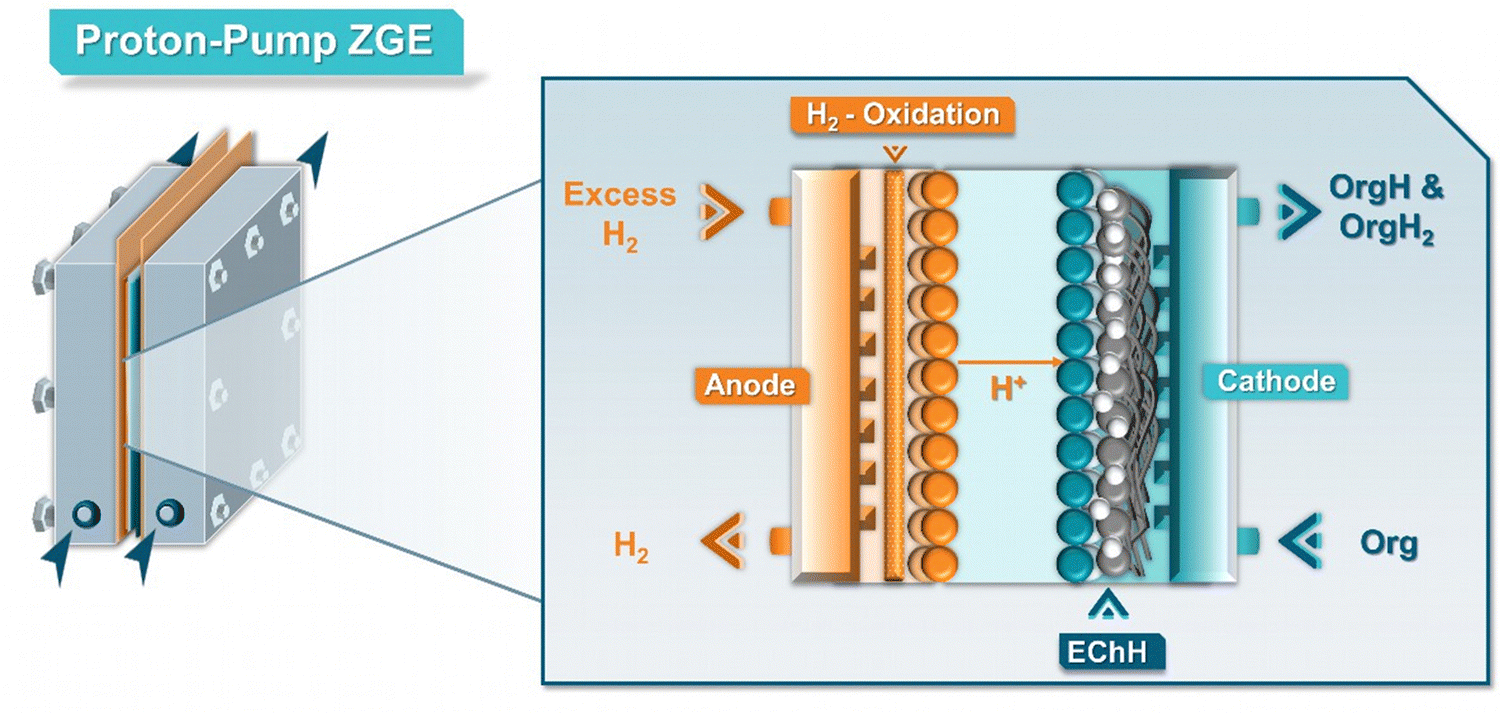 | ||
| Fig. 7 Detailed schematic representation of a ‘proton-pump’ zero-gap electrolyzer (ZGE) and its working–principle. | ||
Moreover, zero-gap electrolyzers have in specific cases demonstrated accelerated scalability across different cell sizes. Montiel and coworkers initially demonstrated the ECH of acetophenone to 1-phenyethanol in a 10 cm2 zero-gap electrolyzer and then transitioned to a 100 cm2 pre-pilot scale, using a Pd-based cathode and a Pt-coated proton-pump.81–83 Similarly, Weber and co-workers demonstrated a pathway for the scalable implementation of hydrogenation reaction in three different cell scales from 10 cm2 to 200 cm2.84
Nevertheless, although electrolytic stacks have been presented in multiple electro-synthetic approaches, examples distinctly for EChH reactions remain scarce.17,18 Here, inspiration could be drawn from more established electrolysis technologies such as H2 production. The EChH could in this case be performed either in large electrolytic stacks with electrodes in the m2 range, or in smaller stack bundles for a more tailored and decentralized production.85
Moreover, despite their omnipresence and success in the field of EChH, systematic studies on the influence of reactor components, such as the employed support, binder nature and content, membrane, and flow field structures, for a broad range of substrates are currently limited.75,76,86–88 Regarding the interplay between the employed support and substrate type, Chen et al. investigated the hydrogenation of maleic acid and butanone.86 Their study also highlights issues with the stability of the commonly used Nafion membranes and binder under elevated organic substrate concentrations, leading to cracking and peeling of the catalytic layer. Similarly, we demonstrated that a conductive carbon felt used in the redox-flow battery field significantly outcompetes conventional conductive supports such as carbon cloths and carbon papers, showing that the field of EChH can draw inspiration from multiple electrochemical fields.87
Regarding the role of the key component of zero-gap electrolyzers, the ion-exchange membrane (Fig. 8), Diaz et al. compared AEM and CEM membranes for the hydrogenation of furfural. Their results demonstrate that AEM-separated zero-gap electrolyzers outcompete their CEM-based counterparts at current densities of 50 mA cm−2.89 These results are in accordance with results from our group showing that AEMs lead to higher selectivity for the hydrogenation of MBY on pentlandite-based, (Fe,Ni)9S8, electrodes.75 Specifically, the comparison of CCM and CCS-based electrodes, and different porous transport layer showed that: (a) CCM-deposited electrodes outperformed CCS ones in terms of achieved cell efficiency and (b) the compression generated between the two PTLs plays a critical role regarding the obtained EChH activity, possibly affected by current and fluid distribution (Fig. 6). Regarding the role of the flow-field structure, Nagasawa et al. rather unconventionally demonstrated that in the case of toluene EChH, a porous-carbon material outcompeted commonly used flow-field structures (parallel, interdigitated, serpentine),76 subsequently showing that impregnation of the mentioned carbon-based flow field with Pt outperformed a Pt-loaded carbon paper support generated via doctor-blading.90
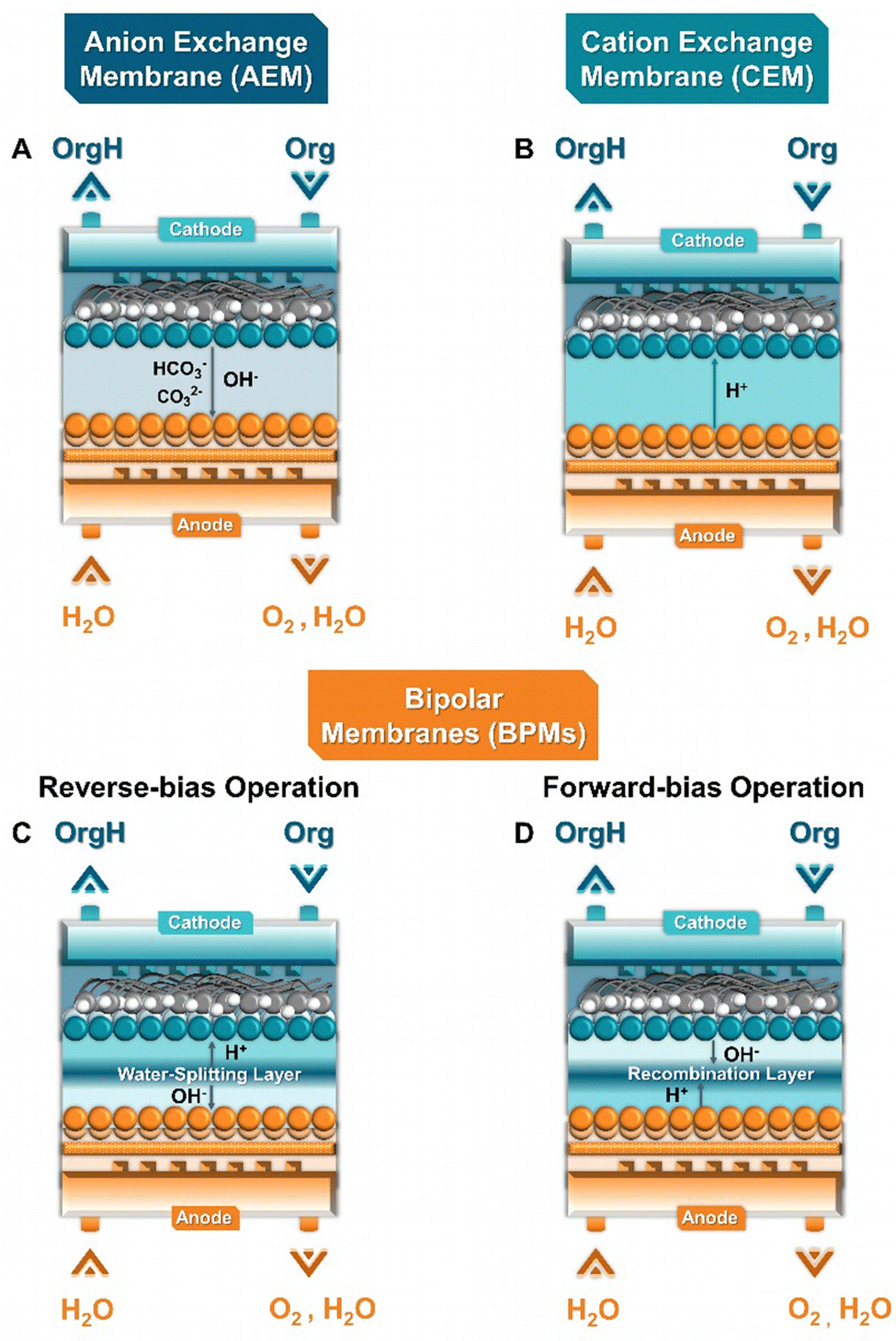 | ||
| Fig. 8 Detailed schematic representation of the mainly transported species within different membrane types in zero-gap electrolyzers. | ||
Importantly, performing electrochemical hydrogenations does not exclude the simultaneous valorization of organic compounds at the anode. Exemplarily, Liu et al. demonstrated the parallel oxidation and reduction of HMF in a zero-gap electrolyzer, employing a Nafion-membrane.91,92 Overall, such parallel electro-synthetic routes, could significantly increase the overall electrosynthetic output of the electrolyzer and concurrently its applicability, though clear systematic studies on product crossover between the two cell compartments over long-periods of operation are currently missing. Notably, the crossover of liquid products is not an issue that only EChH electrolyzers currently face.93–97 Specifically, liquid products, such as acids and alcohols, generated during the electrocatalytic reduction of CO2 readily diffuse through the employed AEMs, due to electro-osmotic pressure. In addition to using BPMs, one additional solution to this problem has been the development of the three-compartment ZGEs. Here, both electrodes are separated with the help of ion-exchange membranes from a middle compartment, aiming towards the complete separation of liquid products prior to landing on the anode side. With our review aiming to spark innovative designs for the EChH, we envision that coupling the important know-how of microfluidic cells and flow chemistry with such reactor designs could lead to their further improvement.
At this point, we would like to emphasize that a reactor that fits all thinkable target reactions does not exist. Specifically, the reactor and architecture must be critically tailored to the production requirements and complexity of the target product. As a rough comparison, for small-scale fine-chemicals, even simple undivided batch cells could be an efficient approach, since only small amounts of product are produced, and accordingly only low currents are needed. In contrast for target substrates being currently converted in the ton-scale annually such as MBY4 or the hydrogenation of toluene as a potential LOHC,79 optimized cells and electrodes aiming at maximizing cell efficiency are required. Systematic studies between the interplay of the cost of reactor scale-up and product value will surely create a clearer picture in the future.
All in all, the field of EChH has experienced major innovation leaps, enriching the pool of hydrogenated reactants. Nevertheless, despite these advancements, most EChH reactions are being performed in batch mode, under low current densities (<100 mA cm−2), with substrate concentrations in the range of 100–1000 mmol or even lower. While this operation mode is completely valid for fine-chemicals, it becomes an obstacle for the application of EChH at industrially relevant scales with respect to the conversion of substrates in the ton-scale, such as biomass. Evidently, to achieve rapid transition between small and large-scale EChH applications, more systematic studies on the influence of the process parameters, design of components and reactor architecture on the EChH are required. Such investigations would not only allow to find optimal conditions for each substrate class but also to establish continuously operated EChH reactions. Possible sources of inspiration could be CO2- and CO-electrolyzers, redox flow batteries, as well as fuel cells and H2-generating electrolyzers. Similarly, although our review focuses mainly on low-temperature based EChH systems, recent advances in the field of high-temperature electrolytic systems, operating at temperatures (500–1000 °C), could also provide an additional inspiration source.98,99 Notably, instead of employing an ion-conductive polymer membrane, a solid-oxide layer between the cathode and anode ensures under such elevated temperatures the necessary ionic-conductivity. Though the stability of organic substrates under such temperatures is a crucial subject, we believe that such concepts could present interesting coupling opportunities between current chemical plants and electrolyzers, either via employing residual heat in a direct or thermo-electrically driven manner or towards introducing inorganic separation layers in the field of EChH. Here, we would like to guide interested readers to current literature reviews that better describe the mechanism and applications of solid-oxide electrolysis.98,100–105
4. Substrates
Since the EChH is already known for decades, a large variety of compounds has already been tested (Fig. 9).106 The EChH of C–C, C–N– and N–O-unsaturated compounds might contribute to the building of simple as well as highly complex fine chemicals in the pharmaceutical or specialty chemicals sector. Meanwhile, aromatic compounds such as dibenzyl toluene are widely debated candidates for future large-scale hydrogen storage with their hydrogenation being a crucial process step that should be decoupled from the necessity of the intermediate production and storage of hydrogen gas. The reductive workup of pyrolysis oil from biomass feedstocks constitutes a further future-proof EChH-application potentially opening the pathway for the replacement of fossil fuels in multiple sectors.19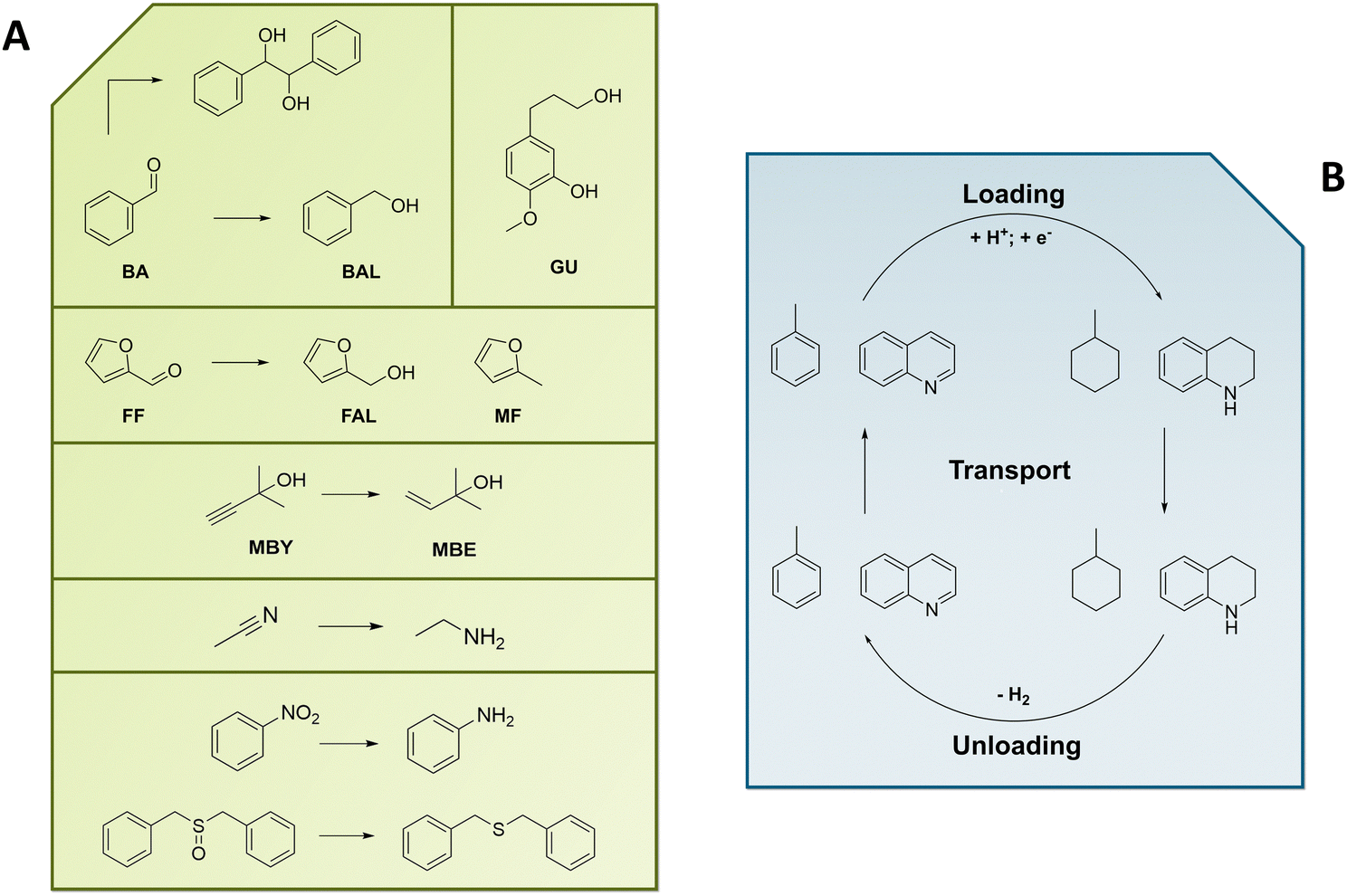 | ||
| Fig. 9 Selected prominent examples for organic substrates in hydrogenation reactions. (A) Biomass model aldehydes and their hydrogenation, deoxygenation and dimerization; hydrogenation of exemplary C–C-, C–N–, N–O– and S–O-unsaturated substrates (B) Loading and Unloading of potential LOHCs. | ||
In this chapter an overview of the broad variety of substrates discussed in literature is provided. At the same time, we want to highlight two examples with industrial relevance. While furfural (FF) and hydroxymethylfurfural (HMF) as biomass derivatives surely would fit this category, we herein focus on toluene as a potential LOHC (model) and 2-methylbut-3-yn-2-ol (MBY), an important building block for the vitamin and fragrance industry, providing insight from our own research.
4.1 C–O-unsaturated compounds
Due to their accessibility from biomass and derived compound mixtures as well as the large synthetic value of their downstream products for pharmacological, specialty chemical-, fuel- or polymer-related applications107 and many more, oxygenated organic compounds are among the most intensely researched and established substrate classes for electrocatalytic hydrogenations.108–111 As compounds commonly found in pyrolyzed biomass feedstocks, benzaldehyde (BA)32,33,112–116 and FF33,109,110,117 are widespread model substrates for diverse EChH-related investigations, that may yield benzyl alcohol (BAL) or furyl alcohol (FA) and 2-methylfuran (MF), respectively. While other aldehydes,84,118,119 ketones such as acetophenone81,120,121 or acetone,122,123 α-ketoesters124,125 and α-ketoacids126 are also widely represented as substrates in literature, the EChH of non-activated carboxylic acids remains challenging.19,127 This is mainly due to the low electrophilicity of the O–C–O-unit caused by conjugation which is lowered even further upon deprotonation of the acid functionality so that the EChH of carboxylic acids often requires elevated temperatures (60–150 °C).126,128 The reduction of dimethyl formamide as a model amide to the corresponding amine with relevance for the pharmaceutic industry on a copper catalyst was also reported.129 Next to C–O-double bond hydrogenation, C–O-single bond cleavage is of large interest from a starting material-perspective since, e.g., waste lignin moieties bear methoxy groups.108 Furthermore, from a product-perspective the reduction of carbohydrates may yield valuable products for the food industry.130 As catalysts for C–O-hydrogenation, palladium is to be considered the benchmark.81,110,112,117,131–133 While palladium and other noble metals like platinum,115,122,134–137 ruthenium117,128,138,139 and rhodium117,140,141 dominate the field, there is an increasing number of protocols featuring abundant metal catalysts107,108,113,117,119,126,130,142 with copper as the most prominent example.109,110,117,143 Most reported protocols operate at relatively low current densities and electrode areas of <30 mA cm−2 and <10 cm2, respectively, while allowing faradaic efficiencies of >90% in many cases are achieved depending on the substrate.33,84,107,109,116 Nonetheless, industrial applicability has received growing attention by researchers so that conversions in gram-scale116 and even successful EChH of crude biomass-derived pyrolysis oil are described.126As mentioned above, aldehydes are most frequently represented in literature as C–O-unsaturated substrates for EChH reactions and are highly abundant in biomass feedstocks. Owing to the large reactivity of aldehyde molecules adsorbed on a catalyst surface,112 the prevalent issue in literature for rendering EChH protocols of C–O-unsaturated substrates more industrially applicable is steering the selectivity towards certain desired organic products opposed to the hydrogen evolution reaction (HER) and the formation of products from side reactions. Next to EChH and dehydration, the main reaction paths are dimerization and polymerization resulting from coupling of multiple substrate radical species formed by DER-processes on the catalyst surface.33,144,145 A main factor that promotes the undesired HER are highly cathodic half-cell potentials that are often required for the formation of intermediate radical anions.33,109,146 Moreover, the often used acidic working conditions may increase the reactivity of aldehydes.122 The selectivity of the EChH of furfural towards highly reduced products such as 2-methylfuran may be improved by increasing the active catalyst surface,109 by catalyst doping,109,110,121 introducing surface vacancies to the catalyst84 or fine-tuning the accessible crystal planes of the catalyst surface.134
On the other hand, interactions between catalyst support and substrate might play a role to, e.g., favor the formation of aromatic pinacol dimers over the corresponding alcohol.116 Such interactions were also shown to play a role in enhancing the overall EChH efficiency of benzaldehyde by participation of Brønsted-acid sites of electrode supports in the hydrogenation.112 Thus, a more holistic view on the electrolysis setup rather than focusing merely on the catalyst is not only beneficial for maximizing energy efficiency but also for the chemical selectivity of EChH processes. Moreover, base metal catalysts showed enhanced dimer formation compared to noble metals.33 Tuning of further parameters such as substrate concentration and temperature were also shown to play a role in steering EChH selectivity with, e.g., higher process temperatures favoring dimerization over EChH in the conversion of furfural.146 Transferring the state of the art of process parameters in alkaline water electrolyzers, such as the current density, at which efficient operation is possible, to a similar reactor for the EChH of biomass-derived organics, product streams of 2–15 t d−1 per m3 cell volume were shown to be realistic production volumes of hydrogenated pyrolysis oil assuming an uptake of four electrons per molecule and an average molecular mass of 150 g mol−1.19
4.2 C–C-unsaturated compounds
Among C–C-unsaturated substrates, EChH especially provides the possibility to mildly hydrogenate molecules sensitive to typical thermocatalytic conditions such as enlarged temperatures and pressures, e.g., edible oils. The ECH of oils is known for decades and even allows the conversion of the pure starting material in a glass frit-reactor.106,147 A further substrate class, whose common hydrogenation products, Z-alkenes, are highly desirable for the synthesis of pharmacological and natural products and organic functional materials, are alkynes.37,52,148–150 Unsaturated esters and acids on the other hand may be accessible from sugar or lignin and yield products usable for polyesters and plastics.130,151,152 Next to palladium37,67,148,153,154 as the benchmark catalyst material and other noble metals like platinum153,155,156 and ruthenium,151,157 abundant metal catalysts130,149,151,158 are increasingly represented in recent research with nickel150,151,159 as one of the most reported examples.A challenge rather often addressed in recent literature for C–C-unsaturated substrates is the electrocatalytic deuteration with partially very selective and efficient protocols that allow the enhancement of the lifetime and efficacy of drugs in a physiological context.44,149 As an attempt to make the EChH of this substrate class more industrially feasible, the conversion of crude biotechnological feeds containing α,β-unsaturated acids is also reported with only minor efficiency drops compared to the pure substrates.151 As a special representative of C–C-unsaturated substrates, aromatic compounds, are also well established in EChH literature ranging from oxygenated derivatives such as methoxylated lignin monomers160 and benzoic acid69 to neat aromatics like benzene,161 toluene162–164 or xylene.165 While platinum is by far the most used catalyst material for aromatic compounds.69,160,162–164 Some recently developed systems focus on cooperative multi-metal catalysts to enhance reaction efficiency and to steer the selectivity towards C–C-hydrogenation in the presence of sensitive reducible functional groups.69,160 Regarding the loading and unloading of liquid organic hydrogen carriers (LOHCs) as the most promising application of the EChH of aromatic compounds in the future, diverse protocols for highly concentrated or even pure benzene derivatives exist that are able to maintain cell voltages of 2 V at current densities of up to 400 mA cm−2.76,163–165 In this regard, for the thermodynamically rather challenging hydrogenation of aromatic compounds, current densities and cell concepts are notably most ready for application compared to other substrate classes. If the LOHC-technology should establish itself as a key storage concept in the upcoming hydrogen economy, the EChH of such may potentially join the refinement of biomass-derived pyrolysis oil in becoming the largest-scale EChH-application mid-term underlining the importance of research on system scale-up for the corresponding substrate classes.
Researchers are fully aware of the potential use of TL/MCH as a hydrogen storage system leading to TL-related EChH-protocols to be by far the closest to industrial applicability, as mentioned above.79 Generally, toluene is subject of a relatively high number of studies conducted in application-oriented flow reactors rather than in H-type cells, in contrast to the vast majority of substrates in EChH-research.76,90,164,168–170
The focus of TL-EChH-studies partially lies on similar topics as for other substrate classes such as the optimization of catalyst systems towards lower loadings, especially of PGM-components.162–164,171–173 Exemplarily, Yamanaka et al. could show a similar EChH-activity for a combined Ru- and Ir-catalyst with a fifth of the loading of a corresponding pure Pt-catalyst. The system could then be optimized towards a Ru-Ir-alloy that with a proven synergistic effect due to favored TL- and hydride-adsorption for Ru and Ir, respectively, and lastly to Ir-doped Ru-nanoparticles that showed an even higher activity and Ir-material efficiency than the previous two systems.88,163,171
In contrast to other substrate classes however, the field of TL-EChH is majorly concerned with optimizing the electrode and cell architecture as well as the process parameters of membrane reactors towards large-scale applicability. While, e.g., the studies by Yamanaka et al. and Mitsushima et al., respectively, underline the tremendous influence of the utilized porous transport layer, Mitsushima et al. conducted a broad investigation of short- and long-term TL-crossover through a variety of polymer membranes as an utterly important quantity for system scale up.88,168,174 Furthermore, Mitsushima et al. carried out an extended study on the influence of TL concentration and current density on the efficiency of membrane reactors with varying cathode flow field structures. Fortunately, TL-EChH proceeded more efficiently at large TL-concentrations for elevated current densities, presumably due to the avoidance of an excessively high hydride coverage of the catalyst, while a porous carbon electrode support without an additional flow field structure was shown to favor the EChH at those conditions.76 This system was optimized later on by incorporating remarkably low loadings of 0.02 mg cm−2 of Pt as the active catalyst.90
Considering that crucial hurdles towards industrial application of TL-EChH have already been mastered, including the maximization of starting material concentration in the feed and of current densities whilst maintaining low cell voltages and catalyst loadings, EChH has the potential to at least partially account for the probably steeply rising demand of MCH as a hydrogen carrier in the future. This will especially hold true at sites where limitations in plant architecture do not allow the intermediate formation and storage of gaseous hydrogen, which must be an object of respective technoeconomic assessments in the future.
According to estimations by the IEA,175 the transport sector alone, which has a comparatively good compatibility with LOHC technology due to the existing liquid fuel infrastructure, is expected to feature an annual demand of 35 Mt H2 by 2040. Assuming that this entire amount will be stored in the TL/MCH-system, it would be equivalent to an annual production volume of 568.2 Mt of MCH. The ECH-based production of this amount would require 497![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 134 electrolyzers consisting of 50 cells with 1 m2 electrode surface, respectively, under uninterrupted operation assuming the currently best performing systems by Nishiki and coworkers164 as the basis of this calculation. Each electrolyzer would have an annual production volume of 1143 t in this case. Indeed, an electrolyser of similar scale already exists as a demonstration plant built by the Japanese ENEOS Corporation, set up in Australia and operated since February 2023, featuring a surface area of 3 m2 and a performance input of 150 kW from an adjacent solar power plant.18
134 electrolyzers consisting of 50 cells with 1 m2 electrode surface, respectively, under uninterrupted operation assuming the currently best performing systems by Nishiki and coworkers164 as the basis of this calculation. Each electrolyzer would have an annual production volume of 1143 t in this case. Indeed, an electrolyser of similar scale already exists as a demonstration plant built by the Japanese ENEOS Corporation, set up in Australia and operated since February 2023, featuring a surface area of 3 m2 and a performance input of 150 kW from an adjacent solar power plant.18
The continuous stability and operability of the electrolyzers over such extended durations will be the main hurdle for area-wide EChH-application in this field and are also investigated in the realm of the abovementioned project regarding influences of climate and fluctuations in current supply. The economic feasibility of an EChH-based production of this order of magnitude against conventional MCH-production methods and even other hydrogen storage methodologies must be evaluated in detailed future assessments.
By using copper or copper phosphide nanoarrays on copper foam as a catalyst in a flow cell setup, Zhang and coworkers52,176 could achieve partial current densities of over 750 mA cm−2 for MBE, while maintaining a high selectivity for the alkene and a current efficiency of 58%. It was shown that with 357 g gcat−1 h−1 (batch, 1 cm2) and 169 g gcat−1 h−1 (single pass, 25 cm2) the mass activity of the copper was significantly higher than that of state-of-the-art thermocatalysts. With increasing MBY concentrations the observed Faraday efficiency dropped, probably due to a too high coverage of the active sites. It was also shown that the achieved current density should be sufficient for an economic implementation of the reaction in industry.
We recently demonstrated that the electrochemical hydrogenation of MBY can also be efficiently performed by metal sulfides of the pentlandite class (M9S8; M = Co, Ni, Fe) maintaining the activity for >100 h at 160 mA cm−2. While the metal composition influences the activity and selectivity, we could show that, on the way to a functional electrolyzer process, parameters and components like the electrode support and membrane strongly influence the observed performance.75,87 Notably, the cell potential and also the hydrogenation activity are sensitive towards the design and processing of the anode.75 Taken together, the industrial transformation of the thermocatalytic MBY hydrogenation to an electrocatalytic one seems everything but implausible. The promising holistic approach for the hydrogenation of MBY includes scalable and energy efficient cells, high current densities (>100 mA cm−2), optimization procedures for the whole cell performance and a techno-economic assessment. At the same time, both approaches still suffer from limitations of the available anion-exchange membranes that are prone to substrate crossover. From our experience these limitations become even stronger when trying to enlarge the substrate scope to non-polar organic substrates as membranes, binders, and tubes can be easily attacked by the required organic solvents. These and further challenges for application oriented EChH research will be addressed in the concluding chapter.
4.3 C–N-unsaturated compounds
One major advantage of electrocatalytic protocols is the opening of new reaction pathways with finely tuned selectivities compared to conventional (thermal or thermocatalytic) conversions. Thus, e.g. primary and secondary amines can be easily obtained by the electrocatalytic reduction of the respective nitriles or imines circumventing the use of stoichiometric reducing agents as well as the need for overstoichiometric provision of reactants which are the main drawbacks of conventional protocols.177However, records for the EChH of C–N-unsaturated substrates remain still rare compared to C–C or C–O unsaturated compounds.177–179 Certainly, one of the most prominent examples is the hydrogenation of adiponitrile to 1,6-diaminohexane178 with high relevance for the polymer industry. As soon as the substrate-independent challenges of reactor scale-up such as avoidance of noble metal catalysts and maintaining high reaction efficiencies at enhanced current densities are addressed, C–N-unsaturated substances may also gain importance as LOHC-candidates, e.g. in the form of N-heterocycles.159
4.4 NOx- and SOx-containing compounds
A large variety of research projects in recent years targeted the EChH of nitrogen180 and sulfur oxides,181 the latter primarily considered as atmospheric pollutants. While literature on the hydrogenation of organic NOx-containing compounds is relatively abundant, reports on the EChH of SOx-containing organics remain rather rare. The EChH of organic nitrogen oxides is particularly interesting for the formation of amines with relevance for the pharmaceutic industry with the corresponding nitro-substituted starting materials mostly being easily accessible, especially in the case of aromatic substances.182–190 A dominant current challenge in the EChH of organic nitrogen oxides is associated with the acidic pH that is often required for a relatively efficient conversion, similarly to the EChH of oxygenated compounds.191 Organic sulfoxides on the other hand are contained as pollutants in fossil fuels so that their economical and easy reduction by electrocatalytic hydrogenation is desirable.192 Next to nitrobenzene and nitrophenol,182–188 azo- and azoxy-aromatics193 as well as broad ranges of aromatic sulfoxides194 as model substrates, even the desulfurization of pure Diesel by continuously electrocatalytically regenerated sodium borohydride is known.192 With this application in mind, the importance of sulfoxide-EChH may be expected to increase short- and mid-term due to the still inclining global use of fossil fuels.While the previously discussed challenges regarding efficiency and selectivity apparently become increasingly important at larger reactor scales and resource expenditure for all substrate classes, the reaction order might also change with increasing scale.131 To ensure economically feasible efficiencies and selectivities of EChH-processes in scaled reactors, the focus in these research areas must and is expected to experience a shift from catalytic materials towards the interplay of more technical parameters such as the electrode architecture and the cell configuration, which was shown to be of utmost importance in some of the studies discussed above. Regarding the reactor feeds an acceleration of the development of larger-scale solvent-free protocols, potentially even operating with crude starting material mixtures, is conceivable next to a general scale-up in reactor dimensions.
5. Catalysts
5.1 Overview
The vast majority of hydrogenation catalysts consists of metals that are either known to catalyse thermal hydrogenations such as palladium,32,33,37,42,81,82,84,112,195,196 ruthenium,32,88,118,191,197–201 and nickel33,108,178,199,202–209 or known to easily form active hydrogen species on their surface under reductive conditions like platinum.21,22,24,48,122,134,136,152,165,182,210–215 The other frequently reported catalyst is copper, which is mainly applied for the hydrogenation of furfural derivatives26,60,109,110,216,217 and nitrobenzene derivatives.182–187 Still, other metals like silver,84,209,218–222 gold,143,161,209,223,224 and other late transition metals32,33,107,142,209,225–230 have also been evaluated for EChH. Metals with high overpotentials for the hydrogen evolution like lead, bismuth or cadmium55,142,209,229,231,232 and also carbon materials124,191,233–235 were reported to enable hydrogenation reactions but the limitation to activated substrates indicates in these cases the reaction proceed via the direct reduction of the substrates (DER, see Section 2.1).Typically, the metals are employed as carbon supported catalysts, nanoparticles or directly as plates or foils. Expectedly, small particle sizes are usually preferred due to the increased surface area and as for other electrocatalytic reactions and nanostructuring as well as the incorporation of defects can severely improve the observed catalyst activity.52,134,136,150,199,236,237 In a similar fashion, single atom catalysts238 are well known for their high (electro)catalytic activity and have been reported for several electrochemical reductions, e.g., of protons,239–241 carbon dioxide,239–241 nitrogen,240,241 and nitrate.242–247 Likewise, applications of organic hydrogenations and reductions have been reported.109,137,248–250
For PGMs, that show underpotential deposition of hydrogen, i.e. the formation of reduced hydrogen species on the metal surface at potentials more positive than the standard potential of H+/H2, electrochemical hydrogenations can occur at positive potentials vs. RHE.22,134 However, the current densities under these conditions are usually limited to a few mA cm−2 and hence this phenomenon is not relevant for preparative electrolysis. Still, PGMs are often the catalyst material of choice,154,163,251 although, especially for platinum, the competing HER has to be considered at elevated current densities and increasingly cathodic potentials.252 For almost all substrate classes competitive PGM-free catalysts were reported including alkynes and alkenes,52,87,149 carbonyls,32,33,236 furfural derivatives26,60,107,109,110,216,217 and aromatic nitro compounds.182–188 Even for the hydrogenation of phenol derivatives to cyclohexanols nickel based catalysts are known although operating at low current densities (<10 mA cm−2) and at elevated temperatures (60–75 °C).108,208,237 On the other hand, one relevant substrate class discussed in literature that still requires PGMs for efficient catalysis are aromatic compounds like toluene. This fact is especially interesting since the hydrogenation of toluene and similar compounds is one of the only types of EChH, that is well known to efficiently operate at current densities of >400 mA cm−2 in polymer electrolyte membrane reactors.76,163–165
5.2 Recent developments
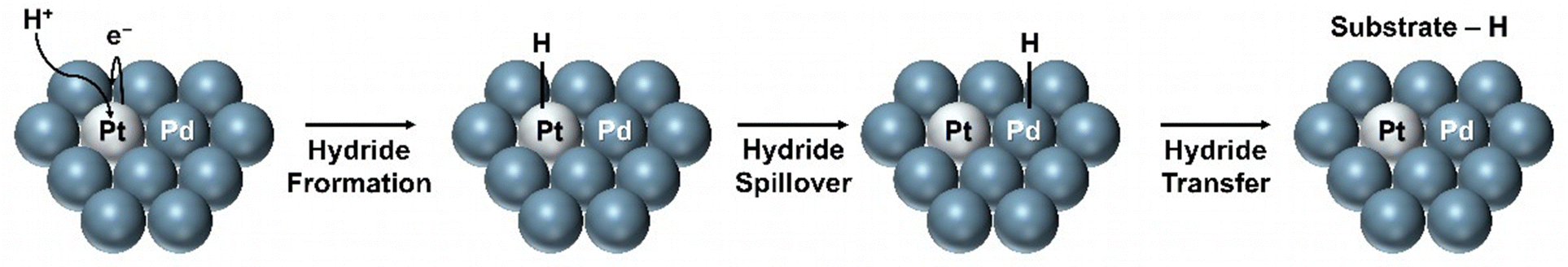 | ||
| Fig. 10 Hydride spillover on bimetallic catalyst surfaces. | ||
In 2020, the field regained interest, when a bimetallic manganese complex 1 for the hydrogenation of carbonyl compounds was reported (Fig. 11).283 This complex, initially designed for carbon dioxide reduction, features a phenolic hydroxide that functions as a proton relay and is crucial for high activity. Both, 1 and the subsequently reported, monometallic complex 2284 were reported to selectively reduce ketones and aldehydes in the presence of non-conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. However, 2 was reported to fully reduce an α,β-unsaturated ketone to the saturated alcohol. Also in 2020, Armstrong and Waymouth reported on the molybdenum complex 3 for the electro-hydrogenation of benzaldehyde, which is enabled by the formation of a stable metal hydride under reductive conditions.114
C double bonds. However, 2 was reported to fully reduce an α,β-unsaturated ketone to the saturated alcohol. Also in 2020, Armstrong and Waymouth reported on the molybdenum complex 3 for the electro-hydrogenation of benzaldehyde, which is enabled by the formation of a stable metal hydride under reductive conditions.114
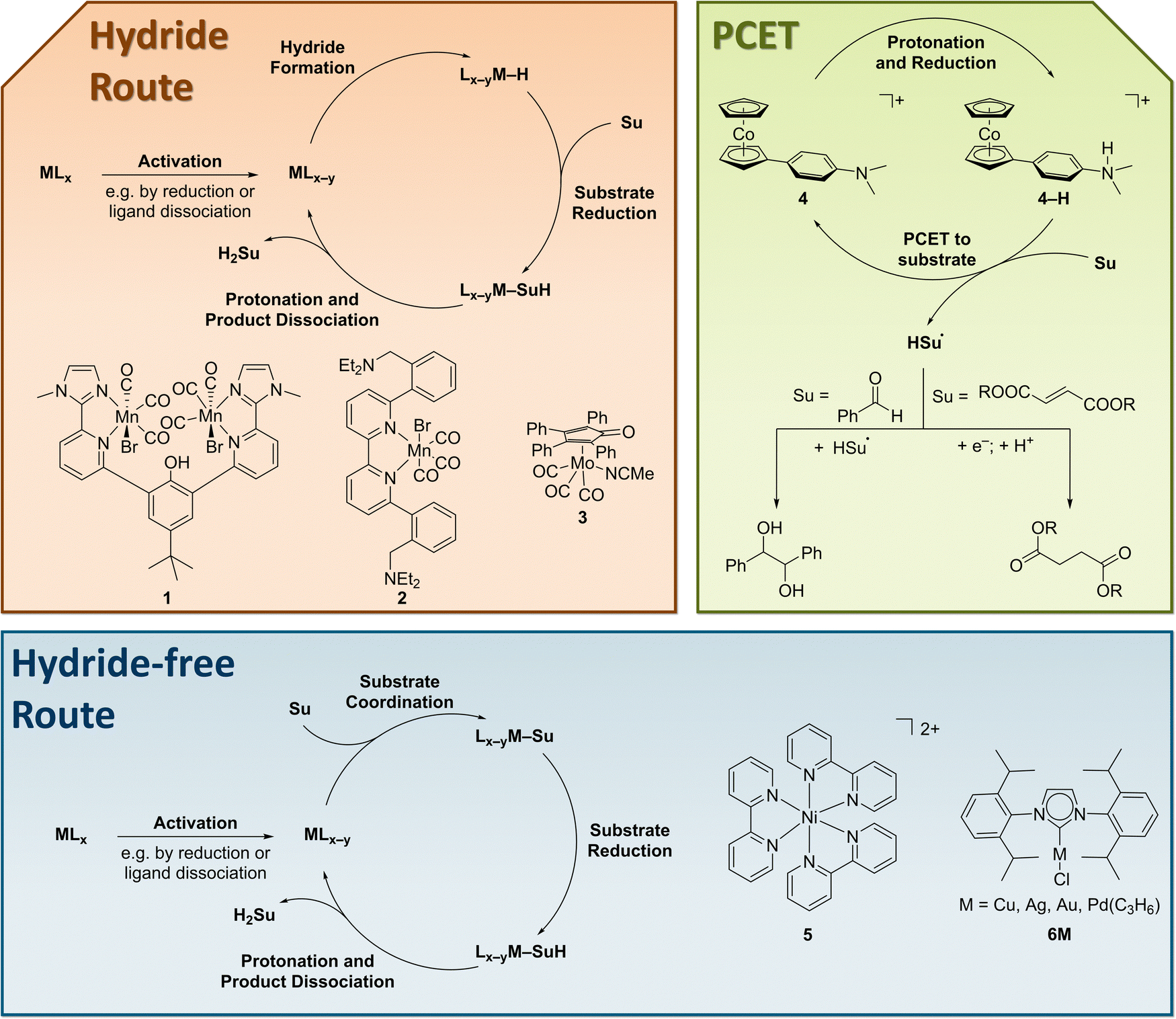 | ||
| Fig. 11 Selected molecular complexes for hydrogenation reactions and their different mechanisms of action. | ||
A different reactivity was demonstrated for complex 4, which functions as a PCET mediator.285 Due to the radical reactivity the complex does not support the hydrogenation of benzaldehyde but rather enables a reductive C–C coupling to the pinacol coupling product. In contrast, the electron-deficient carbon–carbon double bond in fumarate esters could be hydrogenated.286 In this case, the radical intermediate does not dimerize. Similar reactivity under reductive conditions was also reported for other alkenes earlier always accompanied by dimerization.287–290 In combination with molecular nickel phosphine hydrogenation catalysts, 4 mediates the hydrogenation of alkynes and enones.291
The application of the simple complex Ni(bpy)32+5 (bpy = 2,2′-bipyridine) enabled the semihydrogenation of alkynes via molecular electrosynthesis.292 In contrast to the examples above, this complex relies on the strong coordination of the substrate to the metal centre and avoids hydride and radical intermediates.293 Similarly, a series of NHC complexes (6M) was reported to efficiently reduce acetylene to ethylene. This report constitutes the first incorporation of molecular EChH catalyst into a functional electrode and highlights the potential of this approach.294 These recent reports highlight the potential of molecular electrocatalysis for the hydrogenation of organic molecules. Still, the prevailing efficiency-gap to heterogenous electrocatalytic materials and the lack of application in advanced cell concepts needs to be overcome in the future.
6. Current challenges and outlook
Even though great effort has been made to develop innovative catalyst and reactor designs for the electrochemical hydrogenation of a variety of different functional groups, several obstacles remain to be surmounted especially with an efficient industrial application of this technology in mind.6.1 Product purification
To circumvent energy intensive purification of the target product from the electrolyte mixture, conversion towards a desired hydrogenation should ideally be close to 100% (conversion & selectivity).130 Even though, the work-up of a reaction mixture is of major importance in chemical industry, only little attention is paid to this subject in EChH research. Especially in electrochemical conversions, researchers face the challenge of separating the product from a rather complex mixture of solvent, reactants, supporting electrolyte and sometimes more additives. Additionally, when operating under industrially preferred continuous flow conditions, the purification should follow in a continuous manner as well. Besides the liquid electrolyte mixture, hydrogen gas is, in most cases, observed as a side product of the EChH due to the parasitic HER. Despite of its value as an energy carrier, hydrogen gas could be recycled into the same electrocatalytic process to minimize its net accumulation299 or it can be employed for downstream utilization for a coupled thermocatalytic hydrogenation processes using gaseous H2.300In the conventional thermocatalytic hydrogenation process, pure substrates or highly concentrated solutions of substrate are hydrogenated to minimize the post-processing costs and increase the production rate. Likewise, simplifying the electrolyte mixture of the EChH towards high substrate concentrations or, more preferably, to a pure substrate solution would greatly reduce the financial post processing effort.301–303 To address these issues, it is necessary to develop new reactor and electrode designs that enable the conversion of ideally pure substrates. In the past, concepts were introduced that reduce the required solvent volume (e.g. zero-gap reactor87,154,172) or avoid the mixing of electrolytes and substrate in the first place (e.g. the palladium membrane reactor).36,37,47,170,304–307 Albeit the challenging and cost intensive scale-up of this approach, similar optimized concepts could pave the way to introduce a green and cost-efficient EChH for large-scale industrial processes.
6.2 Electrolyte and component optimization
Besides the previously mentioned catalysts described in Chapter 5, there are several other essential parameters that must be considered for optimization for efficient large-scale electrochemical hydrogenation (Fig. 12). These include the solvent/electrolyte mixture, ion-conducting membrane, catalyst binders, porous catalyst support structures, additives as well as current collectors and flow fields.58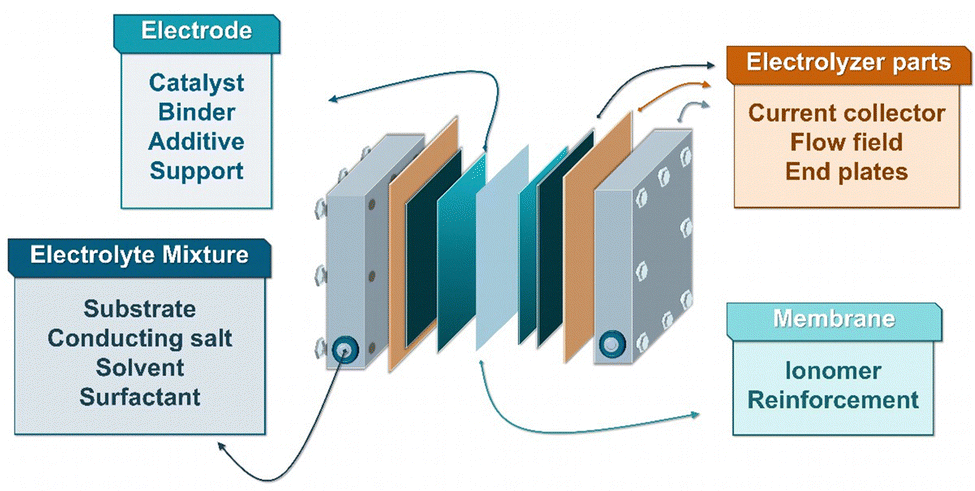 | ||
| Fig. 12 Electrolyte and cell components to be considered for process optimization. | ||
The role of the electrolyte in the EChH depends on the employed electrolysis cell. For H-type setups, the ion transport between the electrodes is the most important task.35 Regarding more advanced setups, such as zero-gap systems, the membrane takes over this responsibility. Therefore, in these systems, the electrolyte solution, or more precisely, the substrate solution, mainly defines the reaction environment at the catalyst surface and facilitates the substrate transport having a major impact on the selectivity and yield of the hydrogenation.65,87 Moreover, the stability of all cell components must be sufficient in the solvent of choice to withstand ideally thousands of ours of electrolysis. Multiple electrolyte-related factors influence the EChH such as solvent type, supporting electrolyte, substrate concentration and pH that have been described in the past. For example, the selectivity of an EChH can be, among other parameters, controlled by the substrate concentration, influencing the reactant flux to the electrode surface.178 Mathanker et al. pointed out that the activity of catalyst sites, interfacial water layer structure, transition state energy of reactants and the local pH is influenced by the choice of electrolyte salt, e.g., due to ion interaction with electrode surface and blocking of active sites.308 Additionally, multiple groups have pointed out the importance of pH adjustment to control side reactions and component decomposition.60,207,309 It has been shown in these works that, while low pH is a major contributor to an increased hydrogen production, alkaline pH can facilitate decomposition of less durable substrates. Therefore, yields can be maximized by a fine-tuning of the electrolyte pH for a given hydrogenation reaction. Additionally, the solvent of the electrolyte plays a major role in controlling a systems performance.60
Fluoropolymeric compounds like PTFE, PVDF or other vinyl polymers like PVP or PVA as well as ionomers like Nafion may be used to immobilize catalyst particles on a conductive support and provide suitable ion exchange properties at the catalyst surface.87,170,310 Popular examples for materials enhancing the conductivity between the catalyst particles are carbon-based compounds like carbon black170,222 or CNTs,311 among others. Nafion is among the most frequently used separators for divided PEM-EChH,68,81,89,161,165,312–314 while a broader range of anion-exchange ionomers is employed such as Fumion, among others.75,87 Despite their frequent use in EChH, ionomers and membranes require significant optimization for a reliable use in the presence of organic solvents and substrates. From our experience, the application of (an)ion-exchanging membranes is often limited by their poor stability in organic media and swelling effects that can significantly decrease the systems stability or increase the necessary cell potential. Therefore, the role of the membrane is of significant importance in EChH since many of the commercially available membrane materials are not compatible with commonly used organic solvents.
Modestino and coworkers observed a particularly pronounced swelling effect in Nafion-membranes for short-chain alcohol solvents, opposed to alkanes.315 In this context, our group recently proved the importance of a sufficiently tight and homogeneous interface between membrane and electrode to ensure an efficient EChH of MBY especially in PEM-based cells.75 This points out the necessity to develop means to tackle uncontrolled membrane swelling as a challenge. Swelling may also result in increased crossover of solvents to the adjacent cell compartment, which can be mitigated by thicker membranes. Yet, this procedure does not automatically lead to improved electrolyser efficiency.316 Due to these reasons and since swelling also significantly alters the structure–property relationships of the respective polymers, it especially makes the reliable EChH of substrates that require such solvents more challenging. Additionally, there is a lack of literature for evaluating other membrane properties and their effects on the EChH such as reinforcement with materials like PEEK. Also, inorganic filler particles, e.g. layered zirconium phosphate were reported to increase mechanical and chemical membrane stability, especially at enhanced temperatures and mitigate swelling. The same studies present promising properties of ionomer-treated PTFE membranes, which allow a more controlled tuning of membrane thickness, porosity and morphology, whilst maintaining ionic conductivity and ensuring superior stability to conventional ionomer membranes.316 Furthermore, besides the chemical environment of the membrane, the mode of electrolyte flow was shown to influence the water content of the membrane having distinctive effects on conductivity and thus electrolyzer efficiency. In this context, contrarious flow of anolyte and catholyte was more beneficial than flow into the same direction.317
Apart from challenges regarding mechanical stability, it is noteworthy to mention that the major membrane decomposition mechanism includes the reaction with radical intermediates; an issue that becomes even more decisive when considering that common EChH catalysts such as Pt/C may display Fenton reactivity.318 Targeted molecular ionomer design can address this issue, e.g., by varying amount and distribution of sulfonyl groups in proton-exchanging polymers.316 A similar approach may be taken to create ionomers with enhanced stability in strongly alkaline conditions, that may be required for catalyst stability or sufficiently fast OER kinetics, as done for AEM polymers.319
In our opinion, the development of suitable ionomers as binding and membrane materials and the elucidation of effects regarding the fabrication methods, that largely influence membrane properties as well,320 is crucial to successfully apply the EChH for a broader range of hydrogenation reactions.
While the membrane is facilitating the ion-exchange between anolyte and catholyte, the conductive catalyst support combined with a suitable flow field design plays a crucial role for simultaneously controlling transport of electrons and substrate molecules to the catalytic surface of the electrode. In the past, large scale electrochemical processes have employed capillary gap reactors321 or plate electrodes with flow channels8 to ensure sufficient conversion. In lab-scales, catalyst supports from other fields of high conversion electrocatalysis such as HER, CO2RR, fuel cells, or battery systems, e.g., graphite felts, carbon cloth or carbon paper provide a high surface area combined with an outstanding electrical conductivity that is necessary for EChH. Some of these catalyst supports, especially highly porous materials have shown great promise in the hydrogenation of organic molecules, recently.75,84,87 Yet, similar to the development of suitable ionomers, researchers in the field of EChH are currently limited to catalyst supports and flow channel designs from other application fields. In order to develop tailor-made supports for the EChH, future research must focus on understanding diffusion and flow characteristics in order to optimize porosity, flow rates, as well as substrate convection and distribution on the electrode. A suitable pathway to develop catalyst supports and flow fields to maximize the conversion by tuning the above-mentioned parameters can be modelling and simulation of fluid dynamics.
6.3 Techno-economic and life cycle assessment considerations
Renewable energy costs have been drastically decreasing in recent years, enabling to directly power the EChH by the most affordable and sustainable energy sources.322 Beyond this decrease, the extremely fast development of electrolyzers and their rapidly decreasing cost contribute to the affordability of electrochemical solutions in organic synthesis.323 However, prior to application, performance targets have to be determined, to evaluate the profitability of the whole process. Amongst the few techno-economic analyses (TEA) that have been conducted for electrocatalytic hydrogenation processes, the majority covers the field of biomass conversion, among others.130,160,324–327 It was found from these reports that electrifying the EChH of organic compounds to high value-added chemicals can be more profitable compared to conventional syntheses in the case of producing methoxycyclohexanes, 4-tert-butylcyclohexanol, cinnamylalcohol, 2-methoxycyclohexanol, phenol and 2-methyl-3-butene-2-ol (Fig. 13), while the cost advantage increases with higher current density and Faraday efficiency.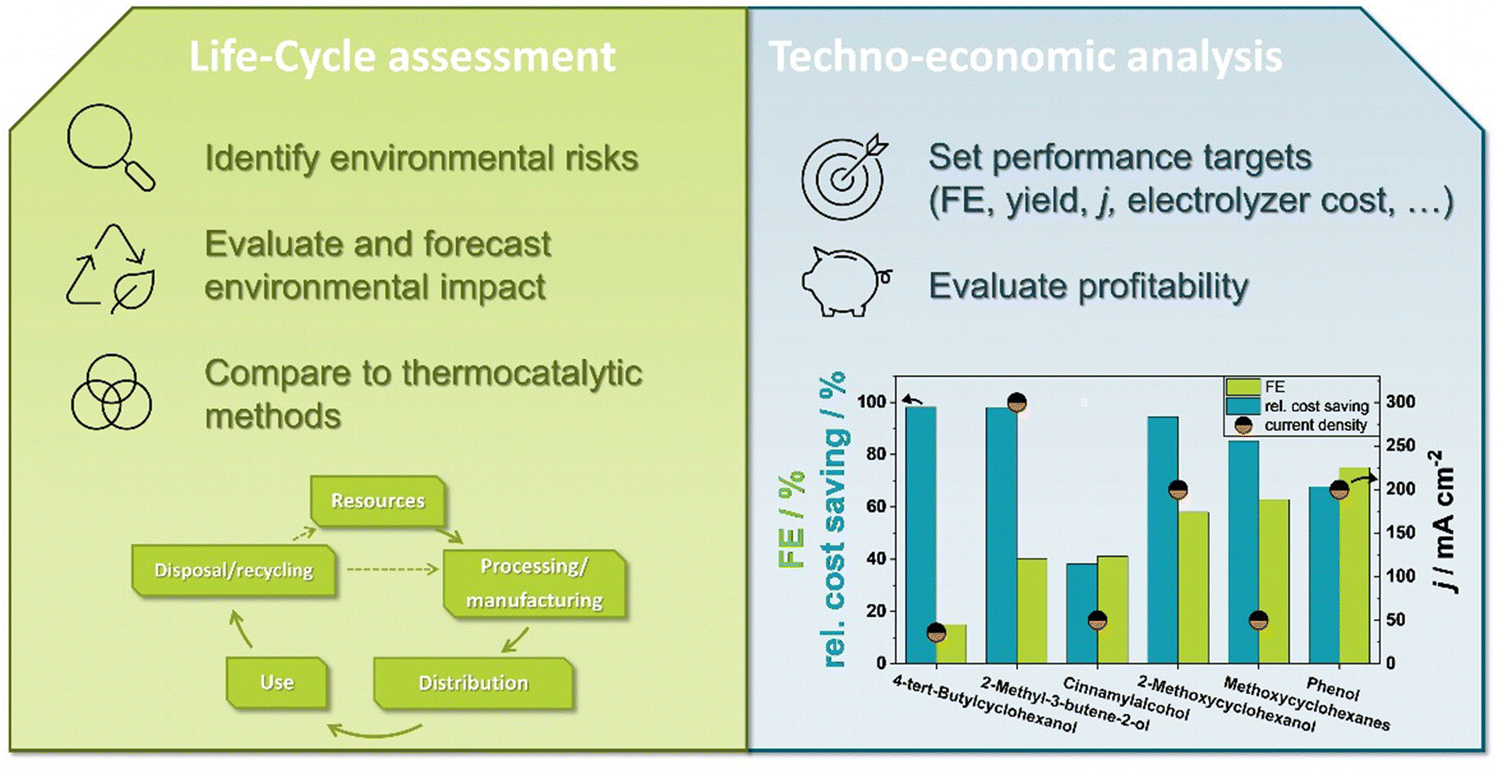 | ||
| Fig. 13 Overview over lifecycle assessment and techno-economic analysis. Diagram: Relative cost saving and Faraday efficiency with respect to the applied current density for the electrified hydrogenation of different organic compounds derived from the respective techno-economic analysis. | ||
Additionally, the assessments demonstrate that the most significant contributions to the total process cost are strongly dependent on the Faraday efficiency of the product generation and the applied current density along with the cell voltages. Depending on the target product, even at moderate Faraday efficiencies of >60% and small current densities of 5 mA cm−2, the EChH process can be more affordable than conventional methods, as Peng et al.160 have reported for the case of 2-methoxycyclohexanol. However, the amount of product to be produced and the subsequent purification steps have to be critically considered. Nevertheless, for the same product, increasing the process efficiency further to Faraday efficiencies of >95% and current densities above 200 mA cm−2 could further reduce production costs by a at least a factor of two. The above-mentioned techno-economic assessments additionally indicate that several other factors, e.g., full cell voltage, electrolyte composition, product separation, (noble metal) catalyst costs and lifetime, catalyst loading, and the corresponding anodic oxidation reaction must be considered for an accurate and complete picture. However, there are additional factors that TEA in literature often not account for. These include electronic losses for voltage/current generators, ohmic losses in terms of heating and correspondingly required cooling, cost of recirculation, cost of binders/membranes and their lifetime, separation of salt/electrolyte from product mixture and challenges regarding scalability. Also, often neglected in studied EChH reactions is the long-term behaviour of the electrochemical cell, which plays a crucial role in cost estimations as continuous electrolysis in large scale should operate efficiently for thousands of hours. Besides the hydrogenation process itself, the financial burdens can be drastically decreased by coupling the EChH with an organic anodic oxidation process in the same facility.36,195,222 This way, the electrosynthetic output can be increased to >100% while a single electrolyzer can produce more than one product at the same time.222
In addition to the profitability aspect, environmental considerations such as cradle to grave life cycle assessments are a crucial part of investigating future perspectives of the EChH. The goal of these assessments is the evaluation of the environmental impact by identification of environmental risks and a comparison to the prevalent thermochemical processes. Thus, critical issues can be identified, and the process can be adapted accordingly. While there have been sophisticated LCAs of related and more mature processes such as CO2RR and HER,328–332 this topic is scarcely elucidated for EChH. Yet, even for intermediate technology readiness levels that EChH can be classified as, LCA should be conducted as they can give an essential forecast of the environmental impacts of a process in the future.333 Thus far, mostly general overviews of environmental advantages of EChH compared to their thermocatalytic counterparts are described in literature. However, detailed studies with standardized methodologies for the environmental impact in comparison to the currently used hydrogenation technologies are urgently required to offer a more complete picture of the EChH process for chemical industry and to provide target values for process development. Taken together, it must be stated that meaningful technological breakthrough in the field of electrosynthesis nowadays can only be achieved when chemical sciences, (reactor/cell) engineering, material sciences as well as life-cycle and techno-economic assessments work together in a synergistic fashion. Furthermore, critical input through dialogues with a potential operator of this technology (industry) has to be considered and taken even into early development stages.334
7. Conclusion
Besides decades of research in EChH and the obvious advantages of electroorganic reactions over traditional chemical transformations, hardly any industrial EChH is realized yet and the well-established thermocatalytic hydrogenation is still the technique of choice for industrial hydrogenation in different scales. Herein, we outlined the major achievements of recent years in material/catalyst development, reactor design, and process optimization and identified the apparent shortcomings on the way of industrialization. Furthermore, we outlined potential routes for future investigations, which we hope will inspire young researchers new to the field as well as experienced organic (electro)chemists to work with us on future electrochemical solutions in hydrogenation. A close cooperation of researchers, industrial partners and politicians would significantly accelerate the process of developing next generations electrochemical systems for a variety of organic compounds to finally electrify the hydrogenation industry.Author contributions
This tutorial review was conceptualized and written by all authors.Abbreviations
| AEM | Anion-exchange membrane |
| BA | Benzaldehyde |
| BAL | Benzyl alcohol |
| BPM | Bipolar membrane |
| CCM | Catalyst coated membrane |
| CCS | Catalyst coated substrate |
| CE | Current efficiency |
| CEM | Cation-exchange membrane |
| CFD | Computational fluid dynamics |
| CNT | Carbon nanotubes |
| DER | Direct electroreduction |
| ECH | Electrocatalytic hydrogenation |
| EChH | Electrochemical hydrogenation |
| FE | Faradaic efficiency |
| FF | Furfural |
| HER | Hydrogen evolution reaction |
| HMF | Hydroxymethylfurfural |
| IEM | Ion-exchange membrane |
| KPI | Key performance indicator |
| LCA | Life cycle assessment |
| LOHC | Liquid organic hydrogen carrier |
| MBA | 2-Methyl-2-butan-2-ol |
| MBE | 2-Methyl-3-butene-2-ol |
| MBY | 2-Methyl-3-butin-2-ol |
| MCH | Methylcyclohexane |
| MEA | Membrane electrode assembly |
| NHC | N-heterocyclic carbon |
| PCET | Proton-coupled electron transfer |
| PEEK | Polyether ether ketone |
| PGM | Platinum group metal |
| PTFE | Polytetrafluoroethylene |
| PVA | Polyvinyl alcohol |
| PVDF | Polyvinylidene fluoride |
| PVP | Polyvinylpyrrolidone |
| RHE | Reversible hydrogen electrode |
| SEC | Specific electricity consumption |
| SPC | Single-pass conversion |
| TEA | Techno-economic analysis |
| TL | Toluene |
| TOF | Turnover frequency |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. T. K. and U.-P. A. acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy-EXC2033-390677874-RESOLV as well as support by the Mercator Research Center Ruhr (MERCUR.Exzellenz, “DIMENSION” Ex-2021-0023 and “KataSign” Ko-2021-0016). U.-P. A., D. S. and K. j. P. acknowledge funding from the Fraunhofer Internal Programs under Grant no. Attract 097-602175 and the Fraunhofer Lighthouse project “ShaPID”. D. S. is grateful for financial support by the BMBF (Federal ministry of education and research) within the NanoMatFutur Project “H2Organic” (no. 03XP0421) as well as by the DFG within the Priority Program “Nitroconversion” (no. 501491300). J. W. and K. P. gratefully acknowledge PhD fellowships granted by the Studienstiftung des deutschen Volkes and the Fonds of the Chemical Industry, respectively.References
- M. Zaarour, J. Cazemier and J. Ruiz-Martínez, Catal. Sci. Technol., 2020, 10, 8140–8172 RSC
.
- U. P. Laverdura, L. Rossi, F. Ferella, C. Courson, A. Zarli, R. Alhajyoussef and K. Gallucci, ACS Omega, 2020, 5, 22901–22913 CrossRef CAS PubMed
- M. D. Garba and S. D. Jackson, Appl. Petrochem. Res., 2017, 7, 1–8 CrossRef CAS
-
W. Bonrath, J. Medlock, J. Schütz and B. Wüstenberg, in Hydrogenation, ed. I. Karamé, IntechOpen, London, 2012 Search PubMed
- E. Masson, E. M. Maciejewski, K. M. P. Wheelhouse and L. J. Edwards, Org. Process Res. Dev., 2022, 26, 2190–2223 CrossRef CAS
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Energy Fuels, 2021, 35, 16403–16415 CrossRef
- A. S. May and E. J. Biddinger, ACS Catal., 2020, 10, 3212–3221 CrossRef CAS
- D. E. Danly, J. Electrochem. Soc., 1984, 131, 435C–442C CrossRef
- C. a C. Sequeira and D. M. F. Santos, J. Braz. Chem. Soc., 2009, 20, 387–406 CrossRef CAS
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC
- D. E. Blanco and M. A. Modestino, Trends Chem., 2019, 1, 8–10 CrossRef CAS
- M. J. Orella, Y. Román-Leshkov and F. R. Brushett, Curr. Opin. Chem. Eng., 2018, 20, 159–167 CrossRef
- DSM IP Assets B.V., International Application, WO2022238349A1, 2022
- SIEMENS AG, German Pat., DE102016218230A1, 2016
- SIEMENS AG, German Pat., DE102018201287A1, 2018
- ENEOS Corporation, Chiyoda Corporation and Queensland University of Technolog, ENEOS, Chiyoda, and QUT Successfully Scaled Up an Australian CO2-Free Hydrogen Supply Chain Demonstration using Direct MCH® ∼Filling a FCV with hydrogen derived from renewable energy∼, https://www.eneos.co.jp/english/newsrelease/2021/pdf/20211102_01.pdf, (accessed 14 September 2023).
- ENEOS Corporation, Australian Demonstration Plant Begins Operation toward the Development of a CO2-free Hydrogen Supply Chain, https://www.eneos.co.jp/english/newsrelease/2022/pdf/20230130_01.pdf, (accessed 14 September 2023).
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed
- P. Zhang and L. Sun, Chin. J. Chem., 2020, 38, 996–1004 CrossRef CAS
- S. Guan, G. A. Attard and A. J. Wain, ACS Catal., 2020, 10, 10999–11010 CrossRef CAS PubMed
- C. J. Bondue and M. T. M. Koper, J. Am. Chem. Soc., 2019, 141, 12071–12078 CrossRef CAS PubMed
- J. Yang, H. Qin, K. Yan, X. Cheng and J. Wen, Adv. Synth. Catal., 2021, 363, 5407–5416 CrossRef CAS
- M. A. Villalba and M. T. M. Koper, Electrochim. Acta, 2022, 417, 140264 CrossRef CAS
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC
- X. Yuan, K. Lee, M. T. Bender, J. R. Schmidt and K. Choi, ChemSusChem, 2022, 15, e202200952 CrossRef CAS PubMed
-
C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, John Wiley & Sons, New York, NY, 2., Auflage., 2011 Search PubMed
-
P. Millet, Electrochemical Power Sources: Fundamentals, Systems, and Applications, Elsevier, 2022, pp. 37–62 Search PubMed
- M. Zhang, M. Wang, B. Xu and D. Ma, Joule, 2019, 3, 2876–2883 CrossRef
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS
- European Comission, Chemicals Strategy for Sustainability Towards a Toxic-Free Environment, European Commission, Brussels, 2020.
- J. A. Lopez-Ruiz, E. Andrews, S. A. Akhade, M.-S. Lee, K. Koh, U. Sanyal, S. F. Yuk, A. J. Karkamkar, M. A. Derewinski, J. Holladay, V.-A. Glezakou, R. Rousseau, O. Y. Gutiérrez and J. D. Holladay, ACS Catal., 2019, 9, 9964–9972 CrossRef CAS
- E. Andrews, J. A. Lopez-Ruiz, J. D. Egbert, K. Koh, U. Sanyal, M. Song, D. Li, A. J. Karkamkar, M. A. Derewinski, J. Holladay, O. Y. Gutiérrez and J. D. Holladay, ACS Sustainable Chem. Eng., 2020, 8, 4407–4418 CrossRef CAS
- S. Minteer, J. Chen, S. Lin, C. Crudden, S. Dehnen, P. V. Kamat, M. Kozlowski, G. Masson and S. J. Miller, ACS Energy Lett., 2023, 8, 1793–1794 CrossRef CAS
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed
- R. S. Sherbo, R. S. Delima, V. A. Chiykowski, B. P. MacLeod and C. P. Berlinguette, Nat. Catal., 2018, 1, 501–507 CrossRef CAS
- R. S. Sherbo, A. Kurimoto, C. M. Brown and C. P. Berlinguette, J. Am. Chem. Soc., 2019, 141, 7815–7821 CrossRef CAS PubMed
- A. Huang, Y. Cao, R. S. Delima, T. Ji, R. P. Jansonius, N. J. J. Johnson, C. Hunt, J. He, A. Kurimoto, Z. Zhang and C. P. Berlinguette, JACS Au, 2021, 1, 336–343 CrossRef CAS PubMed
- R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215–224 RSC
- A. Kurimoto, S. A. Nasseri, C. Hunt, M. Rooney, D. J. Dvorak, N. E. LeSage, R. P. Jansonius, S. G. Withers and C. P. Berlinguette, Nat. Commun., 2023, 14, 1814 CrossRef CAS PubMed
- M. D. Stankovic, J. F. Sperryn, R. S. Delima, C. C. Rupnow, M. B. Rooney, M. Stolar and C. P. Berlinguette, Energy Environ. Sci., 2023, 16, 3453–3461 RSC
- A. Kurimoto, R. P. Jansonius, A. Huang, A. M. Marelli, D. J. Dvorak, C. Hunt and C. P. Berlinguette, Angew. Chem., Int. Ed., 2021, 60, 11937–11942 CrossRef CAS PubMed
- B. Y. Tang, R. P. Bisbey, K. M. Lodaya, W. L. Toh and Y. Surendranath, Nat. Catal., 2023, 6, 339–350 CrossRef CAS
- A. Kurimoto, R. S. Sherbo, Y. Cao, N. W. X. Loo and C. P. Berlinguette, Nat. Catal., 2020, 3, 719–726 CrossRef CAS
- Y. Zhang and N. Kornienko, Chem Catal., 2022, 2, 499–507 CrossRef CAS
- Y. Xu, Z. Mao, R. Qu, J. Wang, J. Yu, X. Luo, M. Shi, X. Mao, J. Ding and B. Liu, Nat. Water, 2023, 1, 95–103 CrossRef
- G. Han, G. Li and Y. Sun, Nat. Catal., 2023, 6, 224–233 CrossRef CAS
- Y. P. Wijaya, T. Grossmann-Neuhaeusler, R. D. Dhewangga Putra, K. J. Smith, C. S. Kim and E. L. Gyenge, ChemSusChem, 2020, 13, 629–639 CrossRef CAS PubMed
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS
- M. Jiang, J. Tan, Y. Chen, W. Zhang, P. Chen, Y. Tang and Q. Gao, Chem. Commun., 2023, 59, 3103–3106 RSC
- K. Zhu, J. Ma, L. Chen, F. Wu, X. Xu, M. Xu, W. Ye, Y. Wang, P. Gao and Y. Xiong, ACS Catal., 2022, 12, 4840–4847 CrossRef CAS
- J. Bu, S. Chang, J. Li, S. Yang, W. Ma, Z. Liu, S. An, Y. Wang, Z. Li and J. Zhang, Nat. Commun., 2023, 14, 1533 CrossRef CAS PubMed
- R. P. Jansonius, A. Kurimoto, A. M. Marelli, A. Huang, R. S. Sherbo and C. P. Berlinguette, Cell Rep. Phys. Sci., 2020, 1, 100105 CrossRef
- R. S. Delima, R. S. Sherbo, D. J. Dvorak, A. Kurimoto and C. P. Berlinguette, J. Mater. Chem. A, 2019, 7, 26586–26595 RSC
- M. N. Dell’Anna, M. Laureano, H. Bateni, J. E. Matthiesen, L. Zaza, M. P. Zembrzuski, T. J. Paskach and J.-P. Tessonnier, Green Chem., 2021, 23, 6456–6468 RSC
- M. Atobe, Curr. Opin. Electrochem., 2017, 2, 1–6 CrossRef CAS
- S. I. Fletcher, F. B. Sillars, R. C. Carter, A. J. Cruden, M. Mirzaeian, N. E. Hudson, J. A. Parkinson and P. J. Hall, J. Power Sources, 2010, 195, 7484–7488 CrossRef CAS
- M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2018, 118, 4541–4572 CrossRef CAS PubMed
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC
- Y. Cao and T. Noël, Org. Process Res. Dev., 2019, 23, 403–408 CrossRef CAS PubMed
- Y. Cao, C. Soares, N. Padoin and T. Noël, Chem. Eng. J., 2021, 406, 126811 CrossRef CAS
- F. Amemiya, K. Fuse, T. Fuchigami and M. Atobe, Chem. Commun., 2010, 46, 2730 RSC
- F. Amemiya, H. Matsumoto, K. Fuse, T. Kashiwagi, C. Kuroda, T. Fuchigami and M. Atobe, Org. Biomol. Chem., 2011, 9, 4256 RSC
- F. Amemiya, T. Kashiwagi, T. Fuchigami and M. Atobe, Chem. Lett., 2011, 40, 606–608 CrossRef CAS
- N. Itoh, W. C. Xu, S. Hara and K. Sakaki, Catal. Today, 2000, 56, 307–314 CrossRef CAS
- K. Takano, H. Tateno, Y. Matsumura, A. Fukazawa, T. Kashiwagi, K. Nakabayashi, K. Nagasawa, S. Mitsushima and M. Atobe, Chem. Lett., 2016, 45, 1437–1439 CrossRef CAS
- A. Fukazawa, J. Minoshima, K. Tanaka, Y. Hashimoto, Y. Kobori, Y. Sato and M. Atobe, ACS Sustainable Chem. Eng., 2019, 7, 11050–11055 CrossRef CAS
- A. Fukazawa, K. Tanaka, Y. Hashimoto, Y. Sato and M. Atobe, Electrochem. Commun., 2020, 115, 106734 CrossRef CAS
- A. Fukazawa, Y. Shimizu, N. Shida and M. Atobe, Org. Biomol. Chem., 2021, 19, 7363–7368 RSC
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable. Energy Fuels, 2020, 4, 2114–2133 RSC
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS
- R. Phillips and C. W. Dunnill, RSC Adv., 2016, 6, 100643–100651 RSC
- K. Pellumbi, J. Wolf, S. C. Viswanathan, L. Wickert, M.-A. Kräenbring, J. T. Kleinhaus, K. junge Puring, F. Özcan, D. Segets, U.-P. Apfel and D. Siegmund, RSC Sustainable, 2023, 1, 631–639 RSC
- K. Nagasawa, A. Kato, Y. Nishiki, Y. Matsumura, M. Atobe and S. Mitsushima, Electrochim. Acta, 2017, 246, 459–465 CrossRef CAS
- K. Shigemasa, A. Atienza-Márquez, K. Inoue, S. Jang, F. I. R. Peña, T. Araki, T. Terao, K. Nagasawa and S. Mitsushima, J. Power Sources, 2023, 554, 232304 CrossRef CAS
- Y. Shimizu, J. Harada, A. Fukazawa, T. Suzuki, J. N. Kondo, N. Shida and M. Atobe, ACS Energy Lett., 2023, 8, 1010–1017 CrossRef CAS
- M. Niermann, A. Beckendorff, M. Kaltschmitt and K. Bonhoff, Int. J. Hydrog. Energy, 2019, 44, 6631–6654 CrossRef CAS
- X. Jiao, C. Lin, N. P. Young, C. Batchelor-McAuley and R. G. Compton, J. Phys. Chem. C, 2016, 120, 13148–13158 CrossRef CAS
- A. Sáez, V. García-García, J. Solla-Gullón, A. Aldaz and V. Montiel, Electrochim. Acta, 2013, 91, 69–74 CrossRef
- A. Sáez, V. García-García, J. Solla-Gullón, A. Aldaz and V. Montiel, Electrochem. Commun., 2013, 34, 316–319 CrossRef
- C. Mozo Mulero, A. Sáez, J. Iniesta and V. Montiel, Beilstein J. Org. Chem., 2018, 14, 537–546 CrossRef PubMed
- J. D. Egbert, E. C. Thomsen, S. A. O’Neill-Slawecki, D. M. Mans, D. C. Leitch, L. J. Edwards, C. E. Wade and R. S. Weber, Org. Process Res. Dev., 2019, 23, 1803–1812 CrossRef CAS
- P. Shirvanian, A. Loh, S. Sluijter and X. Li, Electrochem. Commun., 2021, 132, 107140 CrossRef CAS
- W. Chen, G. He, F. Ge, W. Xiao, J. Benziger and X. Wu, ChemSusChem, 2015, 8, 288–300 CrossRef CAS PubMed
- K. Pellumbi, L. Wickert, J. T. Kleinhaus, J. Wolf, A. Leonard, D. Tetzlaff, R. Goy, J. A. Medlock, K. junge Puring, R. Cao, D. Siegmund and U.-P. Apfel, Chem. Sci., 2022, 13, 12461–12468 RSC
- Y. Inami, H. Ogihara and I. Yamanaka, Electrocatalysis, 2018, 9, 204–211 CrossRef CAS
- L. A. Diaz, T. E. Lister, C. Rae and N. D. Wood, ACS Sustainable Chem. Eng., 2018, 6, 8458–8467 CrossRef CAS
- K. Nagasawa, Y. Sawaguchi, A. Kato, Y. Nishiki and S. Mitsushima, Electrochemistry, 2018, 86, 339–344 CrossRef CAS
- H. Liu, T.-H. Lee, Y. Chen, E. W. Cochran and W. Li, Green Chem., 2021, 23, 5056–5063 RSC
- H. Liu, T. Lee, Y. Chen, E. W. Cochran and W. Li, ChemElectroChem, 2021, 8, 2817–2824 CrossRef CAS
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS
- N. Wang, R. K. Miao, G. Lee, A. Vomiero, D. Sinton, A. H. Ip, H. Liang and E. H. Sargent, SmartMat, 2021, 2, 12–16 CrossRef CAS
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed
- Y. Kuang, H. Rabiee, L. Ge, T. E. Rufford, Z. Yuan, J. Bell and H. Wang, Energy Environ. Mater., 2023, e12596 CrossRef
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed
- F. Kosaka, T. Nakamura, A. Oikawa and J. Otomo, ACS Sustainable Chem. Eng., 2017, 5, 10439–10446 CrossRef CAS
- N. Minh, Solid State Ion., 2004, 174, 271–277 CrossRef CAS
- A. Brisse, J. Schefold and M. Zahid, Int. J. Hydrog. Energy, 2008, 33, 5375–5382 CrossRef CAS
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS
- V. Venkataraman, M. Pérez-Fortes, L. Wang, Y. S. Hajimolana, C. Boigues-Muñoz, A. Agostini, S. J. McPhail, F. Maréchal, J. Van Herle and P. V. Aravind, J. Energy Storage, 2019, 24, 100782 CrossRef
- Y. Jiang, F. Chen and C. Xia, J. Power Sources, 2021, 493, 229713 CrossRef CAS
- J. Zamudio-García, L. Caizán-Juanarena, J. M. Porras-Vázquez, E. R. Losilla and D. Marrero-López, J. Power Sources, 2022, 520, 230852 CrossRef
- S. Fokin, Z. Für Elektrotechnik Elektrochem., 1906, 12, 749–768 CrossRef
- M. S. Dhawan, G. D. Yadav and S. Calabrese Barton, Sustainable Energy Fuels, 2021, 5, 2972–2984 RSC
- Y. Zhou, G. E. Klinger, E. L. Hegg, C. M. Saffron and J. E. Jackson, J. Am. Chem. Soc., 2020, 142, 4037–4050 CrossRef CAS PubMed
- P. Zhou, Y. Chen, P. Luan, X. Zhang, Z. Yuan, S.-X. Guo, Q. Gu, B. Johannessen, M. Mollah, A. L. Chaffee, D. R. Turner and J. Zhang, Green Chem., 2021, 23, 3028–3038 RSC
- P. Zhou, L. Li, V. S. S. Mosali, Y. Chen, P. Luan, Q. Gu, D. R. Turner, L. Huang and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202117809 CrossRef CAS PubMed
- N. Wolff, O. Rivada-Wheelaghan and D. Tocqueville, ChemElectroChem, 2021, 8, 4019–4027 CrossRef
- K. Koh, U. Sanyal, M. Lee, G. Cheng, M. Song, V. Glezakou, Y. Liu, D. Li, R. Rousseau, O. Y. Gutiérrez, A. Karkamkar, M. Derewinski and J. A. Lercher, Angew. Chem., Int. Ed., 2020, 59, 1501–1505 CrossRef CAS PubMed
- S. Calado Galvão De Melo, M. E. P. Da Silva, M. E. B. Da Silva, J. A. Da Paz, C. M. B. De Menezes Barbosa, F. D. De Menezes, R. N. A. Loureiro, M. Navarro, J. Â. P. Da Costa, G. F. Da Silva, A. A. O. Villa and M. Vilar, Int. J. Hydrog. Energy, 2020, 45, 22855–22872 CrossRef
- K. C. Armstrong and R. M. Waymouth, Organometallics, 2020, 39, 4415–4419 CrossRef CAS
- N. Singh, U. Sanyal, G. Ruehl, K. A. Stoerzinger, O. Y. Gutiérrez, D. M. Camaioni, J. L. Fulton, J. A. Lercher and C. T. Campbell, J. Catal., 2020, 382, 372–384 CrossRef CAS
- C. Liu, R. Li, W. Zhou, Y. Liang, Y. Shi, R.-L. Li, Y. Ling, Y. Yu, J. Li and B. Zhang, ACS Catal., 2021, 11, 8958–8967 CrossRef CAS
- U. Sanyal, K. Koh, L. C. Meyer, A. Karkamkar and O. Y. Gutiérrez, J. Appl. Electrochem., 2021, 51, 27–36 CrossRef CAS
- X. Huang, L. Zhang, C. Li, L. Tan and Z. Wei, ACS Catal., 2019, 9, 11307–11316 CrossRef CAS
- S. Han, Y. Shi, C. Wang, C. Liu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100337 CrossRef CAS
- M. Villalba, M. del Pozo and E. J. Calvo, Electrochim. Acta, 2015, 164, 125–131 CrossRef CAS
- M. A. Villalba and M. T. M. Koper, J. Phys. Chem. C, 2020, 124, 25884–25891 CrossRef CAS
- X. Wang, Y. Jiao, L. Li, Y. Zheng and S. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202114253 CrossRef CAS PubMed
- C. J. Bondue and M. T. M. Koper, J. Catal., 2019, 369, 302–311 CrossRef CAS
- D. B. Nemez, B. K. Sidhu, P. K. Giesbrecht, J. D. Braun and D. E. Herbert, Org. Chem. Front., 2021, 8, 549–554 RSC
- N. V. Rees, R. J. Taylor, Y. X. Jiang, I. R. Morgan, D. W. Knight and G. A. Attard, J. Phys. Chem. C, 2011, 115, 1163–1170 CrossRef CAS
- M. Sadakiyo, S. Hata, T. Fukushima, G. Juhász and M. Yamauchi, Phys. Chem. Chem. Phys., 2019, 21, 5882–5889 RSC
- S. Ono and T. Yamauchi, Bull. Chem. Soc. Jpn., 1952, 25, 404–407 CrossRef CAS
- T. Dalavoy, J. Jackson, G. Swain, D. Miller, J. Li and J. Lipkowski, J. Catal., 2007, 246, 15–28 CrossRef CAS
- R. Jia, L. Haixin, L. Shanshan, L. Yujiao, D. Yan and W. Yingli, Int. J. Electrochem. Sci., 2020, 15, 3914–3921 CrossRef CAS
- J. E. Matthiesen, M. Suástegui, Y. Wu, M. Viswanathan, Y. Qu, M. Cao, N. Rodriguez-Quiroz, A. Okerlund, G. Kraus, D. R. Raman, Z. Shao and J.-P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 7098–7109 CrossRef CAS
- J. A. Lopez-Ruiz, U. Sanyal, J. Egbert, O. Y. Gutiérrez and J. Holladay, ACS Sustainable Chem. Eng., 2018, 6, 16073–16085 CrossRef CAS
- F. Laplante, L. Brossard and H. Ménard, Can. J. Chem., 2003, 81, 258–264 CrossRef CAS
- N. Takano and C. Seki, Electrochemistry, 2006, 74, 596–598 CrossRef CAS
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Nat. Catal., 2019, 2, 243–250 CrossRef CAS
- R. C. Z. Lofrano, J. M. Madurro, L. M. Abrantes and J. R. Romero, J. Mol. Catal. Chem., 2004, 218, 73–79 CrossRef CAS
- C. Kim and H. Lee, Catal. Commun., 2009, 11, 7–10 CrossRef CAS
- S. Zhao, Y. Wen, X. Peng, Y. Mi, X. Liu, Y. Liu, L. Zhuo, G. Hu, J. Luo and X. Tang, J. Mater. Chem. A, 2020, 8, 8913–8919 RSC
- Z. Li, M. Garedew, C. H. Lam, J. E. Jackson, D. J. Miller and C. M. Saffron, Green Chem., 2012, 14, 2540 RSC
- B. Zhang, J. Zhang and Z. Zhong, ACS Appl. Energy Mater., 2018, 1, 6758–6763 CrossRef CAS
- J.-C. Moutet, C. Duboc-Toia, S. MØnage and S. Tingry, Adv. Mater., 1998, 665–667 CrossRef CAS
- J.-C. Moutet, L. Yao Cho, C. Duboc-Toia, S. Menage, E. C. Riesgo and R. P. Thummel, New J. Chem., 1999, 23, 939–944 RSC
- R. J. M. Bisselink, M. Crockatt, M. Zijlstra, I. J. Bakker, E. Goetheer, T. M. Slaghek and D. S. van Es, ChemElectroChem, 2019, 6, 3285–3290 CrossRef CAS
- D. C. Cantu, A. B. Padmaperuma, M.-T. Nguyen, S. A. Akhade, Y. Yoon, Y.-G. Wang, M.-S. Lee, V.-A. Glezakou, R. Rousseau and M. A. Lilga, ACS Catal., 2018, 8, 7645–7658 CrossRef CAS
- S. Huang, B. Gong, Y. Jin, P. H.-L. Sit and J. C.-H. Lam, ACS Catal., 2022, 12, 11340–11354 CrossRef CAS
- S. Huang, Y. Jin, M. Zhang, K. Yan, S.-P. Feng and J. C.-H. Lam, Green Chem., 2022, 24, 7974–7987 RSC
- B. Zhao, M. Chen, Q. Guo and Y. Fu, Electrochim. Acta, 2014, 135, 139–146 CrossRef CAS
- G. J. Yusem and P. N. Pintauro, J. Am. Oil Chem. Soc., 1992, 69, 399–404 CrossRef CAS
- Y. Wu, C. Liu, C. Wang, S. Lu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 21170–21175 CrossRef CAS PubMed
- Y. Wu, C. Liu, C. Wang, Y. Yu, Y. Shi and B. Zhang, Nat. Commun., 2021, 12, 3881 CrossRef CAS PubMed
- Y. Ling, Y. Wu, C. Wang, C. Liu, S. Lu and B. Zhang, ACS Catal., 2021, 11, 9471–9478 CrossRef CAS
- F. J. Holzhäuser, J. Artz, S. Palkovits, D. Kreyenschulte, J. Büchs and R. Palkovits, Green Chem., 2017, 19, 2390–2397 RSC
- E. Lamy-Pitara, S. El Mouahid, S. Kerkeni and J. Barbier, Electrochim. Acta, 2003, 48, 4311–4316 CrossRef CAS
- B. Li and H. Ge, Sci. Adv., 2019, 5, eaaw2774 CrossRef CAS PubMed
- S. Nogami, N. Shida, S. Iguchi, K. Nagasawa, H. Inoue, I. Yamanaka, S. Mitsushima and M. Atobe, ACS Catal., 2022, 12, 5430–5440 CrossRef CAS
- K. A. San, V. Chen and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2017, 9, 9823–9832 CrossRef CAS PubMed
- Z. Wang, A. Garg, L. Wang, H. He, A. Dasgupta, D. Zanchet, M. J. Janik, R. M. Rioux and Y. Román-Leshkov, ACS Catal., 2020, 10, 6763–6770 CrossRef CAS
- F. Zhang, J. Fang, L. Huang, W. Sun, Z. Lin, Z. Shi, X. Kang and S. Chen, ACS Catal., 2019, 9, 98–104 CrossRef CAS
- C. Xing, Y. Xue, X. Zheng, Y. Gao, S. Chen and Y. Li, Angew. Chem., 2023, e202310722 CAS
- M. Li, C. Liu and B. Zhang, Sci. Bull., 2021, 66, 1047–1049 CrossRef CAS PubMed
- T. Peng, T. Zhuang, Y. Yan, J. Qian, G. R. Dick, J. Behaghel De Bueren, S.-F. Hung, Y. Zhang, Z. Wang, J. Wicks, F. P. Garcia De Arquer, J. Abed, N. Wang, A. Sedighian Rasouli, G. Lee, M. Wang, D. He, Z. Wang, Z. Liang, L. Song, X. Wang, B. Chen, A. Ozden, Y. Lum, W. R. Leow, M. Luo, D. M. Meira, A. H. Ip, J. S. Luterbacher, W. Zhao and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 17226–17235 CrossRef CAS PubMed
- H. Huang, Y. Yu and K. H. Chung, Int. J. Hydrog. Energy, 2014, 39, 13832–13837 CrossRef CAS
- T. Imada, M. Chiku, E. Higuchi and H. Inoue, ACS Catal., 2020, 10, 13718–13728 CrossRef CAS
- Y. Inami, H. Ogihara, S. Nagamatsu, K. Asakura and I. Yamanaka, ACS Catal., 2019, 9, 2448–2457 CrossRef CAS
- S. Mitsushima, Y. Takakuwa, K. Nagasawa, Y. Sawaguchi, Y. Kohno, K. Matsuzawa, Z. Awaludin, A. Kato and Y. Nishiki, Electrocatalysis, 2016, 7, 127–131 CrossRef CAS
- R. Fonocho, C. L. Gardner and M. Ternan, Electrochim. Acta, 2012, 75, 171–178 CrossRef CAS
- D. Klvana, J. Chaouki, D. Kusohorsky, C. Chavarie and G. M. Pajonk, Appl. Catal., 1988, 42, 121–130 CrossRef CAS
- M. Markiewicz, Y.-Q. Zhang, M. T. Empl, M. Lykaki, J. Thöming, P. Steinberg and S. Stolte, Energy Environ. Sci., 2019, 12, 366–383 RSC
- K. Nagasawa, K. Tanimoto, J. Koike, K. Ikegami and S. Mitsushima, J. Power Sources, 2019, 439, 227070 CrossRef CAS
- K. Nagasawa, Y. Sawaguchi, A. Kato, Y. Nishiki and S. Mitsushima, Electrocatalysis, 2017, 8, 164–169 CrossRef CAS
- K. Takano, H. Tateno, Y. Matsumura, A. Fukazawa, T. Kashiwagi, K. Nakabayashi, K. Nagasawa, S. Mitsushima and M. Atobe, Bull. Chem. Soc. Jpn., 2016, 89, 1178–1183 CrossRef CAS
- Y. Inami, S. Iguchi, S. Nagamatsu, K. Asakura and I. Yamanaka, ACS Omega, 2020, 5, 1221–1228 CrossRef CAS PubMed
- A. Fukazawa, K. Takano, Y. Matsumura, K. Nagasawa, S. Mitsushima and M. Atobe, Bull. Chem. Soc. Jpn., 2018, 91, 897–899 CrossRef CAS
- T. Imada, Y. Iida, Y. Ueda, M. Chiku, E. Higuchi and H. Inoue, Catalysts, 2021, 11, 318 CrossRef CAS
- Y. Bao, T. W. Napporn, K. Nagasawa and S. Mitsushima, Electrocatalysis, 2019, 10, 184–194 CrossRef CAS
- International Energy Agency, Global demand for hydrogen from electrolysis and fossil fuels with CCUS by scenario, 2030 and 2040, https://www.iea.org/data-and-statistics/charts/global-demand-for-hydrogen-from-electrolysis-and-fossil-fuels-with-ccus-by-scenario-2030-and-2040, (accessed 31 May 2023).
- S. Yang, J. Bu, R. Bai, J. Lin, S. An, Y. Wu, Y. Guo, J. Gao and J. Zhang, Chin. J. Chem., 2023, 41, 3618–3624 Search PubMed
- R. Xia, D. Tian, S. Kattel, B. Hasa, H. Shin, X. Ma, J. G. Chen and F. Jiao, Nat. Commun., 2021, 12, 1949 CrossRef CAS PubMed
- D. E. Blanco, A. Z. Dookhith and M. A. Modestino, ACS Sustainable Chem. Eng., 2020, 8, 9027–9034 CrossRef CAS
- C. Wei, Y. Fang, B. Liu, C. Tang, B. Dong, X. Yin, Z. Bian, Z. Wang, J. Liu, Y. Qian and G. Wang, Nat. Commun., 2023, 14, 3847 CrossRef CAS PubMed
- Y. Alvarez-Gallego, X. Dominguez-Benetton, D. Pant, L. Diels, K. Vanbroekhoven, I. Genné and P. Vermeiren, Electrochim. Acta, 2012, 82, 415–426 CrossRef CAS
- L. E. Barrosse-Antle, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2008, 112, 3398–3404 CrossRef CAS
- B. Wouters, X. Sheng, A. Boschin, T. Breugelmans, E. Ahlberg, I. F. J. Vankelecom, P. P. Pescarmona and A. Hubin, Electrochim. Acta, 2013, 111, 405–410 CrossRef CAS
- Z. Li, M. He, Y. Wen, X. Zhang, M. Hu, R. Li, J. Liu, J. Chu, Z. Ma, X. Xing, C. Yu, Z. Wei and Y. Li, Inorg. Chem., 2020, 59, 1522–1531 CrossRef CAS PubMed
- M. Jin, Y. Liu, X. Zhang, J. Wang, S. Zhang, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, Appl. Catal., B, 2021, 298, 120545 CrossRef CAS
- W. Xu, C. Yu, J. Chen and Z. Liu, Appl. Catal., B, 2022, 305, 121062 CrossRef CAS
- Y. Gao, Q. Xue, J. Li, M. Zhang, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2022, 14, 14202–14209 CrossRef CAS PubMed
- D. Carvajal, R. Arcas, C. A. Mesa, S. Giménez, F. Fabregat-Santiago and E. Mas-Marzá, Adv. Sustainable Syst., 2022, 6, 2100367 CrossRef CAS
- S. Wu, X. Huang, H. Zhang, Z. Wei and M. Wang, ACS Catal., 2022, 12, 58–65 CrossRef CAS
- C. Tan, F. Yao, C. Ma, L. He, X. Yang, T. Wang, L. Xiang, G. Yu, Z. Yu, Z. Zeng and C. Zhang, Chem. Eng. J., 2023, 467, 143448 CrossRef CAS
- Y. Wang, Y. Liu, Z. Zhao, Z. Zheng, A. M. Balu, R. Luque and K. Yan, Mater. Today, 2023, 66, 84–91 CrossRef CAS
- A. Moya, J. Creus, N. Romero, J. Alemán, X. Solans-Monfort, K. Philippot, J. García-Antón, X. Sala and R. Mas-Ballesté, Dalton Trans., 2020, 49, 6446–6456 RSC
- C. Shu, T. Sun, J. Jia and Z. Lou, Ind. Eng. Chem. Res., 2013, 52, 7660–7667 CrossRef CAS
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Natl. Sci. Rev., 2020, 7, 285–295 CrossRef CAS PubMed
- Z. Kong, C. Pan, M. Li, L. Wen and W. Guo, Green Chem., 2021, 23, 2773–2777 RSC
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS
- L. C. Meyer, U. Sanyal, K. A. Stoerzinger, K. Koh, J. L. Fulton, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2022, 12, 11910–11917 CrossRef CAS
- L. Coche, B. Ehui, D. Limosin and J. C. Moutet, J. Org. Chem., 1990, 55, 5905–5910 CrossRef CAS
- Z. Li, S. Kelkar, L. Raycraft, M. Garedew, J. E. Jackson, D. J. Miller and C. M. Saffron, Green Chem., 2014, 16, 844–852 RSC
- K. J. Carroll, T. Burger, L. Langenegger, S. Chavez, S. T. Hunt, Y. Román-Leshkov and F. R. Brushett, ChemSusChem, 2016, 9, 1904–1910 CrossRef CAS PubMed
- M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS
- G. Bharath and F. Banat, ACS Appl. Mater. Interfaces, 2021, 13, 24643–24653 CrossRef CAS PubMed
- M. Fujihira, A. Yokozawa, H. Kinoshita and T. Osa, Chem. Lett., 1982, 1089–1092 CrossRef CAS
- D. Robin, M. Comtois, A. Martel, R. Lemieux, A. K. Cheong, G. Belot and J. Lessard, Can. J. Chem., 1990, 68, 1218–1227 CrossRef CAS
- D. S. Santana, G. O. Melo, M. V. F. Lima, J. R. R. Daniel, M. C. C. Areias and M. Navarro, J. Electroanal. Chem., 2004, 569, 71–78 CrossRef CAS
- M. V. F. Lima, F. D. Menezes, B. de Barros Neto and M. Navarro, J. Electroanal. Chem., 2008, 613, 58–66 CrossRef CAS
- M. Vilar, J. L. Oliveira and M. Navarro, Appl. Catal. Gen., 2010, 372, 1–7 CrossRef CAS
- Z. Li, S. Kelkar, C. H. Lam, K. Luczek, J. E. Jackson, D. J. Miller and C. M. Saffron, Electrochim. Acta, 2012, 64, 87–93 CrossRef CAS
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron, D. J. Miller and J. E. Jackson, Green Chem., 2015, 17, 601–609 RSC
- Y. Kwon, Y. Y. Birdja, S. Raoufmoghaddam and M. T. M. Koper, ChemSusChem, 2015, 8, 1745–1751 CrossRef CAS PubMed
- A. Wieckowski, S. D. Rosasco, G. N. Salaita, A. Hubbard, B. E. Bent, F. Zaera and G. A. Somorjai, J. Am. Chem. Soc., 1985, 107, 5910–5920 CrossRef CAS
- K. Amouzegar and O. Savadogo, Electrochim. Acta, 1994, 39, 557–559 CrossRef CAS
- A. J. Wain, M. A. O’Connell and G. A. Attard, ACS Catal., 2018, 8, 3561–3570 CrossRef CAS
- Y. P. Wijaya, R. D. D. Putra, K. J. Smith, C. S. Kim and E. L. Gyenge, ACS Sustainable Chem. Eng., 2021, 9, 13164–13175 CrossRef CAS
- Z. Liang, M. A. Villalba and M. T. M. Koper, Electrochim. Acta, 2021, 391, 138911 CrossRef CAS
- C. J. Bondue, Z. Liang and M. T. M. Koper, J. Phys. Chem. C, 2021, 125, 6643–6649 CrossRef CAS PubMed
- A. S. May, S. M. Watt and E. J. Biddinger, React. Chem. Eng., 2021, 6, 2075–2086 RSC
- R. J. Dixit, K. Bhattacharyya, V. K. Ramani and S. Basu, Green Chem., 2021, 23, 4201–4212 RSC
- B.-L. Chen, Y. Xiao, X.-M. Xu, H.-P. Yang, H. Wang and J.-X. Lu, Electrochim. Acta, 2013, 107, 320–326 CrossRef CAS
- H. Yang, T. Lv, W. Sun, Y. Du, H. Wang and J. Lu, RSC Adv., 2014, 4, 30584–30586 RSC
- H. Yang, D. Chi, Q. Sun, W. Sun, H. Wang and J. Lu, Chem. Commun., 2014, 50, 8868–8870 RSC
- J. J. Roylance, T. W. Kim and K.-S. Choi, ACS Catal., 2016, 6, 1840–1847 CrossRef CAS
- X. H. Chadderdon, D. J. Chadderdon, T. Pfennig, B. H. Shanks and W. Li, Green Chem., 2019, 21, 6210–6219 RSC
- M. Byrne and A. T. Kuhn, J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases, 1972, 68, 1898 CAS
- J. P. Oña, R.-M. Latonen, N. Kumar, M. Peurla, I. Angervo and H. Grénman, Electrochim. Acta, 2023, 437, 141536 CrossRef
- U. Sanyal, J. Lopez-Ruiz, A. B. Padmaperuma, J. Holladay and O. Y. Gutiérrez, Org. Process Res. Dev., 2018, 22, 1590–1598 CrossRef CAS
- A. Brisach-Wittmeyer, N.-A. Bouchard, R. Breault and H. Ménard, Can. J. Chem., 2006, 84, 1640–1647 CrossRef
- J.-C. Moutet, Y. Ouennoughi, A. Ourari and S. Hamar-Thibault, Electrochim. Acta, 1995, 40, 1827–1833 CrossRef CAS
- P. Dabo, A. Cyr, J. Lessard, L. Brossard and H. Ménard, Can. J. Chem., 1999, 77, 1225–1229 CrossRef CAS
- T. R. Dos Santos, P. Nilges, W. Sauter, F. Harnisch and U. Schröder, RSC Adv., 2015, 5, 26634–26643 RSC
- L. Liu, H. Liu, W. Huang, Y. He, W. Zhang, C. Wang and H. Lin, J. Electroanal. Chem., 2017, 804, 248–253 CrossRef CAS
- D. E. Blanco, R. Atwi, S. Sethuraman, A. Lasri, J. Morales, N. N. Rajput and M. A. Modestino, J. Electrochem. Soc., 2020, 167, 155526 CrossRef CAS
- J. R. Ochoa-Gómez, F. Fernández-Carretero, F. Río-Pérez, A. García-Luis, T. Roncal and E. J. García-Suárez, Green Chem., 2019, 21, 164–177 RSC
- P. K. Giesbrecht, D. B. Nemez and D. E. Herbert, Chem. Commun., 2018, 54, 338–341 RSC
- J. Li, L. He, X. Liu, X. Cheng and G. Li, Angew. Chem., 2019, 131, 1773–1777 CrossRef
- Y. Qin, J. Lu, Z. Zou, H. Hong, Y. Li, Y. Li, L. Chen, J. Hu and Y. Huang, Org. Chem. Front., 2020, 7, 1817–1822 RSC
- C. M. B. Holt, S. Murphy, M. R. Gray and D. Mitlin, Catal. Commun., 2010, 12, 314–317 CrossRef CAS
- P. Zhou, S. Guo, L. Li, T. Ueda, Y. Nishiwaki, L. Huang, Z. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202214881 CrossRef CAS PubMed
- V. B. Saptal, V. Ruta, M. A. Bajada and G. Vilé, Angew. Chem., Int. Ed., 2023, 62, e2022193 CrossRef PubMed
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J. Luo and X. Sun, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS
- L. Zou, Y. Wei, C. Hou, C. Li and Q. Xu, Small, 2021, 17, 2004809 CrossRef CAS PubMed
- T. Zhu, Q. Chen, P. Liao, W. Duan, S. Liang, Z. Yan and C. Feng, Small, 2020, 16, 2004526 CrossRef CAS PubMed
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed
- X. Zhao, X. Jia, Y. He, H. Zhang, X. Zhou, H. Zhang, S. Zhang, Y. Dong, X. Hu, A. V. Kuklin, G. V. Baryshnikov, H. Ågren and G. Hu, Appl. Mater. Today, 2021, 25, 101206 CrossRef
- J. Li, M. Li, N. An, S. Zhang, Q. Song, Y. Yang and X. Liu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2105628118 CrossRef CAS PubMed
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208215 CrossRef CAS PubMed
- J. Zhao, X. Ren, X. Liu, X. Kuang, H. Wang, C. Zhang, Q. Wei and D. Wu, Chem. Eng. J., 2023, 452, 139533 CrossRef CAS
- N. Li, X. Song, L. Wang, X. Geng, H. Wang, H. Tang and Z. Bian, ACS Appl. Mater. Interfaces, 2020, 12, 24019–24029 CrossRef CAS PubMed
- M. A. Bajada, J. Sanjosé-Orduna, G. Di Liberto, S. Tosoni, G. Pacchioni, T. Noël and G. Vilé, Chem. Soc. Rev., 2022, 51, 3898–3925 RSC
- Z. Mukadam, S. Liu, A. Pedersen, J. Barrio, S. Fearn, S. Ch Sarma, M.-M. Titirici, S. B. Scott, I. E. L. Stephens, K. Chan and S. Mezzavilla, Energy Environ. Sci., 2023, 16, 2934–2944 RSC
- J. Ma, Z. Wang, T. Majima and G. Zhao, ACS Catal., 2022, 12, 14062–14071 CrossRef CAS
- S. Nogami, K. Nagasawa, A. Fukazawa, K. Tanaka, S. Mitsushima and M. Atobe, J. Electrochem. Soc., 2020, 167, 155506 CrossRef CAS
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed
- J. Jiang, R. Zhai and X. Bao, J. Alloys Compd., 2003, 354, 248–258 CrossRef CAS
- Y.-N. Yue, D. Wu, S. Zeng, M.-P. Yang, H. Wang and J.-X. Lu, New J. Chem., 2017, 41, 7853–7856 RSC
- L. Li, M. Yang, L. Cheng, J. Shi, J. Gou and J. Xu, React. Chem. Eng., 2023 10.1039/10.1039.D3RE00312D
- V. Márquez, J. S. Santos, J. G. Buijnsters, S. Praserthdam and P. Praserthdam, Electrochim. Acta, 2022, 410, 139972 CrossRef
- Y.-R. Zhang, B.-X. Wang, L. Qin, Q. Li and Y.-M. Fan, Green Chem., 2019, 21, 1108–1113 RSC
- G. Cheng, Z. Zhai, J. Sun, Y. Ran, W. Yang, F. Tan and Z. Zhang, Chem. Eng. J., 2023, 474, 145631 CrossRef CAS
- K. Zhu, X. Xu, M. Xu, P. Deng, W. Wu, W. Ye, Z. Weng, Y. Su, H. Wang, F. Xiao, Z. Fang and P. Gao, ChemElectroChem, 2021, 8, 3855–3862 CrossRef CAS
- X. Xu, J. Ma, F. Wu, K. Zhu, H. Zhou, Y. Zhang, X. Li, Y. Zhou, G. Jia, D. Liu, P. Gao and W. Ye, Inorg. Chem. Front., 2022, 9, 3444–3452 RSC
- D. Siegmund, N. Blanc, M. Smialkowski, K. Tschulik and U. Apfel, ChemElectroChem, 2020, 7, 1514–1527 CrossRef CAS
- A. Mukherjee, M. Abdinejad, S. S. Mahapatra and B. C. Ruidas, J. Mater. Chem. A, 2023, 11, 9300–9332 RSC
- H. G. Shiraz, X. Crispin and M. Berggren, Int. J. Hydrog. Energy, 2021, 46, 24060–24077 CrossRef CAS
- R. Watanabe, M. Yamauchi, M. Sadakiyo, R. Abe and T. Takeguchi, Energy Environ. Sci., 2015, 8, 1456–1462 RSC
- M. Sadakiyo, S. Hata, X. Cui and M. Yamauchi, Sci. Rep., 2017, 7, 17032 CrossRef PubMed
- M. Yamauchi, S. Hata, H. Eguchi, S. Kitano, T. Fukushima, M. Higashi, M. Sadakiyo and K. Kato, Catal. Sci. Technol., 2019, 9, 6561–6565 RSC
- T. Fukushima and M. Yamauchi, J. Appl. Electrochem., 2021, 51, 99–106 CrossRef CAS
- T. Fukushima and M. Yamauchi, Chem. Commun., 2019, 55, 14721–14724 RSC
- X.-Q. Pan, X.-Y. Zhang, G.-X. Huang, S.-C. Mei, J.-W. Huang, J.-J. Chen, W.-J. Liu and H.-Q. Yu, Appl. Catal., B, 2023, 323, 122191 CrossRef CAS
- J. Tan, J. Shao, Y. Shi, W. Zhang and Q. Gao, ACS Sustainable Chem. Eng., 2022, 10, 13525–13533 CrossRef CAS
- H. Li, Y. Gao, Y. Wu, C. Liu, C. Cheng, F. Chen, Y. Shi and B. Zhang, J. Am. Chem. Soc., 2022, 144, 19456–19465 CrossRef CAS PubMed
- J. Tan, W. Zhang, Y. Shu, H. Lu, Y. Tang and Q. Gao, Sci. Bull., 2021, 66, 1003–1012 CrossRef CAS PubMed
- W. S. Knowles, J. Chem. Educ., 1986, 63, 222 CrossRef CAS
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS
- H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385–1393 CrossRef CAS PubMed
- G. Durin, N. Kaeffer and W. Leitner, Curr. Opin. Electrochem., 2023, 41, 101371 CrossRef CAS
- R. Ruppert, S. Herrmann and E. Steckhan, Tetrahedron Lett., 1987, 28, 6583–6586 CrossRef CAS
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS
- S. Chardon-Noblat, J. Mol. Catal. Chem., 1995, 99, 13–21 CrossRef CAS
- C. Caix, S. Chardon-Noblat, A. Deronzier, J.-C. Moutet and S. Tingry, J. Organomet. Chem., 1997, 540, 105–111 CrossRef CAS
- Z. Chen, C. R. K. Glasson, P. L. Holland and T. J. Meyer, Phys. Chem. Chem. Phys., 2013, 15, 9503 RSC
- I. Fokin and I. Siewert, Chem. – Eur. J., 2020, 26, 14137–14143 CrossRef CAS PubMed
- I. Fokin, K.-T. Kuessner and I. Siewert, ACS Catal., 2022, 12, 8632–8640 CrossRef CAS
- M. J. Chalkley, P. Garrido-Barros and J. C. Peters, Science, 2020, 369, 850–854 CrossRef CAS PubMed
- J. Derosa, P. Garrido-Barros and J. C. Peters, J. Am. Chem. Soc., 2021, 143, 9303–9307 CrossRef CAS PubMed
- H. Rasnoshik, A. Masarwa, H. Cohen, I. Zilbermann, E. Maimon and D. Meyerstein, Dalton Trans., 2010, 39, 823–833 RSC
- O. Schutz, A. Masarwa, I. Zilbermann, E. Maimon, H. Cohen and D. Meyerstein, J. Coord. Chem., 2010, 63, 2528–2541 CrossRef CAS
- O. Schutz, A. Masarwa, I. Zilbermann, E. Maimon, H. Cohen and D. Meyerstein, Eur. J. Inorg. Chem., 2014, 932–940 CrossRef CAS
- H. Shimakoshi, Z. Luo, K. Tomita and Y. Hisaeda, J. Organomet. Chem., 2017, 839, 71–77 CrossRef CAS
- J. Derosa, P. Garrido-Barros, M. Li and J. C. Peters, J. Am. Chem. Soc., 2022, 144, 20118–20125 CrossRef CAS PubMed
- M.-Y. Lee, C. Kahl, N. Kaeffer and W. Leitner, JACS Au, 2022, 2, 573–578 CrossRef CAS PubMed
- G. Durin, M.-Y. Lee, M. A. Pogany, T. Weyhermüller, N. Kaeffer and W. Leitner, J. Am. Chem. Soc., 2023, 145, 17103–17111 CrossRef CAS PubMed
- L. Zhang, Z. Chen, Z. Liu, J. Bu, W. Ma, C. Yan, R. Bai, J. Lin, Q. Zhang, J. Liu, T. Wang and J. Zhang, Nat. Commun., 2021, 12, 6574 CrossRef CAS PubMed
- H.-P. Yang, H. Wang and J.-X. Lu, Electrochem. Commun., 2015, 55, 18–21 CrossRef CAS
- H.-P. Yang, Q. Fen, H. Wang and J.-X. Lu, Electrochem. Commun., 2016, 71, 38–42 CrossRef CAS
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed
- S. K. Green, J. Lee, H. J. Kim, G. A. Tompsett, W. B. Kim and G. W. Huber, Green Chem., 2013, 15, 1869 RSC
- T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., Int. Ed., 2019, 58, 3562–3565 CrossRef CAS PubMed
- Z. Ogumi, K. Nishio and S. Yoshizawa, Electrochim. Acta, 1981, 26, 1779–1782 CrossRef CAS
- Z. Ogumi, M. Inaba, S. Ohashi, M. Uchida and Z. Takehara, Electrochim. Acta, 1988, 33, 365–369 CrossRef CAS
- M. Paidar, V. Fateev and K. Bouzek, Electrochim. Acta, 2016, 209, 737–756 CrossRef CAS
- G. Li, G. Han, L. Wang, X. Cui, N. K. Moehring, P. R. Kidambi, D. Jiang and Y. Sun, Nat. Commun., 2023, 14, 525 CrossRef CAS PubMed
- C. Iwakura, T. Abe and H. Inoue, J. Electrochem. Soc., 1996, 143, L71–L72 CrossRef CAS
- C. Iwakura, Y. Yoshida and H. Inoue, J. Electroanal. Chem., 1997, 431, 43–45 CrossRef CAS
- H. Inoue, T. Abe and C. Iwakura, Chem. Commun., 1996, 55 RSC
- A. Mathanker, W. Yu, N. Singh and B. R. Goldsmith, Curr. Opin. Electrochem., 2023, 40, 101347 CrossRef CAS
- P. N. Pintauro and J. R. Bontha, J. Appl. Electrochem., 1991, 21, 799–804 CrossRef CAS
- W. An, J. K. Hong and P. N. Pintauro, J. Appl. Electrochem., 1998, 28, 947–954 CrossRef CAS
- V. Lordi, N. Yao and J. Wei, Chem. Mater., 2001, 13, 733–737 CrossRef CAS
- P. N. Pintauro, M. P. Gil, K. Warner, G. List and W. Neff, Ind. Eng. Chem. Res., 2005, 44, 6188–6195 CrossRef CAS
- G. R. List, K. Warner, P. Pintauro and M. Gil, J. Am. Oil Chem. Soc., 2007, 84, 497–501 CrossRef CAS
- M. J. Torres, P. Sánchez, A. De Lucas-Consuegra and A. R. De La Osa, Mol. Catal., 2020, 492, 110936 CrossRef CAS
- A. Katzenberg, A. Angulo, A. Kusoglu and M. A. Modestino, Macromolecules, 2021, 54, 5187–5195 CrossRef CAS
- S. Mitov, B. Vogel, E. Roduner, H. Zhang, X. Zhu, V. Gogel, L. Jörissen, M. Hein, D. Xing, F. Schönberger and J. Kerres, Fuel Cells, 2006, 6, 413–424 CrossRef CAS
- W. Yang, D. Cha and Y. Kim, Energy, 2019, 185, 386–395 CrossRef
- H. Liu, H. A. Gasteiger, A. Laconti and J. Zhang, ECS Trans., 2006, 1, 283–293 CrossRef CAS
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., 2009, 121, 6621–6624 CrossRef
- R. F. Silva, M. De Francesco and A. Pozio, Electrochim. Acta, 2004, 49, 3211–3219 CrossRef CAS
-
N. Aust, in Encyclopedia of Applied Electrochemistry, ed. G. Kreysa, K. Ota and R. F. Savinell, Springer New York, New York, NY, 2014, pp. 1392–1397 Search PubMed
- B. S. Crandall, T. Brix, R. S. Weber and F. Jiao, Energy Fuels, 2023, 37, 1441–1450 CrossRef CAS
- R. Way, M. C. Ives, P. Mealy and J. D. Farmer, Joule, 2022, 6, 2057–2082 CrossRef
- Q. Tu, A. Parvatker, M. Garedew, C. Harris, M. Eckelman, J. B. Zimmerman, P. T. Anastas and C. H. Lam, Environ. Sci. Technol., 2021, 55, 3240–3249 CrossRef CAS PubMed
- M. Wang, T. Peng, C. Yang, B. Liang, H. Chen, M. Kumar, Y. Zhang and W. Zhao, Green Chem., 2022, 24, 142–146 RSC
- H. Chen, T. Peng, B. Liang, D. Zhang, G. Lian, C. Yang, Y. Zhang and W. Zhao, Green Chem., 2022, 24, 3655–3661 RSC
- M. J. Orella, S. M. Brown, M. E. Leonard, Y. Román-Leshkov and F. R. Brushett, Energy Technol., 2020, 8, 1900994 CrossRef
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed
- K. Bareiß, C. De La Rua, M. Möckl and T. Hamacher, Appl. Energy, 2019, 237, 862–872 CrossRef
- L. J. Müller, A. Kätelhön, M. Bachmann, A. Zimmermann, A. Sternberg and A. Bardow, Front. Energy Res., 2020, 8, 15 CrossRef
- G. Zhao, M. R. Kraglund, H. L. Frandsen, A. C. Wulff, S. H. Jensen, M. Chen and C. R. Graves, Int. J. Hydrog. Energy, 2020, 45, 23765–23781 CrossRef CAS
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B.-M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed
- N. Thonemann and A. Schulte, Environ. Sci. Technol., 2019, 53, 12320–12329 CrossRef CAS PubMed
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, JACS Au, 2021, 1, 527–535 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |