
An injectable supramolecular hydrogel as a self-drug-delivery system for local chemoimmunotherapy against melanoma†
Sourabh
Bera
,
Hemanta Kumar
Datta
and
Parthasarathi
Dastidar
 *
*
School of Chemical Sciences, Indian Association for the Cultivation of Science (IACS), 2A and 2B, Raja S. C. Mullick Road, Jadavpur, Kolkata-700032, West Bengal, India. E-mail: ocpd@iacs.res.in; ocpdastidar@gmail.com
First published on 1st July 2023
Abstract
Skin-cancer melanoma caused 57k death in 2020. Some of the available therapies are: topical application of a gel loaded with an anti-skin cancer drug and intravenous injection of immune cytokines; however, both the approaches have drawbacks such as inefficient internalization of the drug in cancer cells and a short half-life with severe side effects, respectively. Interestingly, we observed for the first time that a subcutaneously implanted hydrogel designed and synthesized by coordinating NSAIDs and 5-AP with Zn(II) can effectively combat melanoma cell (B16-F10)-induced tumors in C57BL/6 mice. Both in vitro and in vivo results show that it can effectively reduce PGE2 expression, consequently upregulating IFN-γ and IL-12 that eventually engage M1-macrophages for activating T cells (CD8+), triggering apoptosis. This unique all-in-one self-drug-delivery approach, wherein the hydrogel implant is made from the drug molecules itself providing both chemotherapy and immunotherapy in combating deadly melanoma, highlights the supramolecular chemistry-based bottom-up approach in cancer therapy.
1 Introduction
Cancer is the second leading cause of death worldwide.1 According to the report published by the World Health Organisation (WHO) in 2022, 57k people died worldwide in 2020 from melanoma only.2 Most often, anticancer drugs are administered via the intravenous route, triggering unwanted toxicity in healthy tissues and causing severe side effects. To minimize this, intravenous nanomedicines targeting tumor sites are being developed.3–5 However, local chemotherapy to the tumor site appears promising because of high local concentration, a longer retention time, and slow and sustained release of the drug to the tumor site which consequently increases the anticancer potential and minimizes the chance of affecting healthy tissues. Drug-encapsulated hydrogel formulations are promising because of the biocompatibility of the gelling solvent (water) and its easy administration to the tumor site via the topical or subcutaneous route.To avoid synthetic vehicle-related toxicities, a hydrogel matrix can be obtained from natural polymers, synthetic peptides, proteins, nucleic acids, etc.6–8 Boronic ester formation induced cross-linked polymer-based hydrogels to load and release erlotinib (anticancer drug)9 and aPDL1 (antibody of programmed death ligand 1),10,11 cross-linked nucleic acid nanogels for effective siRNA delivery,12 sequestering ATP by a designed peptide-based hydrogel to boost the activity of the anticancer drug doxorubicin (DOX),13 DNA gel nanoparticles for tumor therapy,14 protocatechualdehyde-based Fe(III) metallo-hydrogels for loading and release of DOX,15 gelatin-based hydrogels loaded with a NO donor for anti-tumor immunotherapy,16 supramolecular hydrogels releasing both DOX and an immunosuppressive agent for cancer therapy,17 and syringeable immunomodulatory multidomain nanogels for tumor immunotherapy18 were reported to be effective, as evident from both in vitro and in vivo experiments.
Designing such formulations requires careful choice of a hydrogel matrix that should be sustainable, biocompatible, biodegradable, etc. However, gaining access to such a hydrogel matrix often requires a nontrivial synthetic protocol (for example, a peptide-based hydrogel matrix); it must also be amenable for facile loading of drugs and suitable for slow and sustained release of drugs, avoiding burst release.
An alternative approach [known as self-drug-delivery (SDD)], wherein a drug molecule is converted to a supramolecular gel,19–34 is being investigated with much interest, because in here, one can avoid the challenges involved in the design and synthesis of a gel matrix and the drug can be delivered by itself to the target site;35–38 for example, a hydrogel obtained by sonicating the anticancer drug raltitrexed was found to be effective in treating post-surgical cancer.39 A plasma-activated thermosensitive biogel as an exogenous ROS carrier for post-surgical treatment of cancer was developed.40 An injectable and biodegradable DNA hydrogel was reported to be effective in tumor therapy.14 A trident peptide-based hydrogel functionalized with phosphotyrosine, camptothecin and hydroxychloroquine was demonstrated to be effective in enhanced chemotherapy and immunotherapy for spatial suppression of tumor metastasis.41
Most of these fascinating SDD systems, however, were dependent on one way or the other on covalent synthesis, which is relatively difficult compared to metal–ligand coordination chemistry. Drug delivery systems developed by exploiting metal–ligand coordination chemistry offer various advantages over covalent chemistry-based systems: they (a) are easily synthesized, (b) are amenable for atomic-level structure determination by single-crystal X-ray diffraction (SXRD), providing crucial insights for further development, (c) have tuneable physicochemical properties arising from virtually infinite combinations of ligands (drug molecules) and metal salts, and (d) are easily biodegradable owing to the relatively labile metal–ligand coordination bond compared to the covalent bond.42–46
We have developed a few metal–ligand coordination-based SDD systems that include non-steroidal anti-inflammatory drug (NSAID)-anchored Zn(II) coordination polymer-based metallogels effective in inhibiting cell migration of melanoma cells B16-F10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47,48 and multi-drug-derived Zn(II) coordination polymer-based nano-sized particles for killing both Mycobacterium tuberculosis and the human lung cancer cell line A549 with no in vivo toxicity.49
47,48 and multi-drug-derived Zn(II) coordination polymer-based nano-sized particles for killing both Mycobacterium tuberculosis and the human lung cancer cell line A549 with no in vivo toxicity.49
With this background in mind, we set out to develop an SDD system by exploiting simple coordination chemistry that would target melanoma skin cancer. Herein, we report the design, synthesis and single crystal structures of a series of Zn(II) coordination complexes (CCs) derived from 5-amino-1,10-phenanthroline (5-AP) and various NSAIDs (meclofenac – MEC, diclofenac – DIC, mefenamic acid – MEF and flufenamic acid – FLU). A majority of the CCs formed highly stable hydrogels at room temperature. MEC-derived CCs showed significant anticancer activity in both MTT and scratch assays against the melanoma cell line B16-F10. Further in vitro experiments (cell cycle, nuclear condensation, annexin V-FITC/PI, mitochondrial membrane potential (MMP), Hoechst efflux assays) suggested an apoptotic pathway. In vivo experiments by subcutaneous injection of an MEC-derived hydrogel (MHG-1) to the melanoma-induced tumor site in a C57BL/6 mouse model showed a remarkable anti-tumor effect; upregulation of pro-inflammatory cytokines (IFN-γ, TNF-α, IL-12, IL-2 and IL-6) observed in the hydrogel-treated tumor tissue further supported the apoptotic pathway and increased the cytotoxic T cell population, suggesting the role of the hydrogel in anti-tumor immunity (Scheme 1).
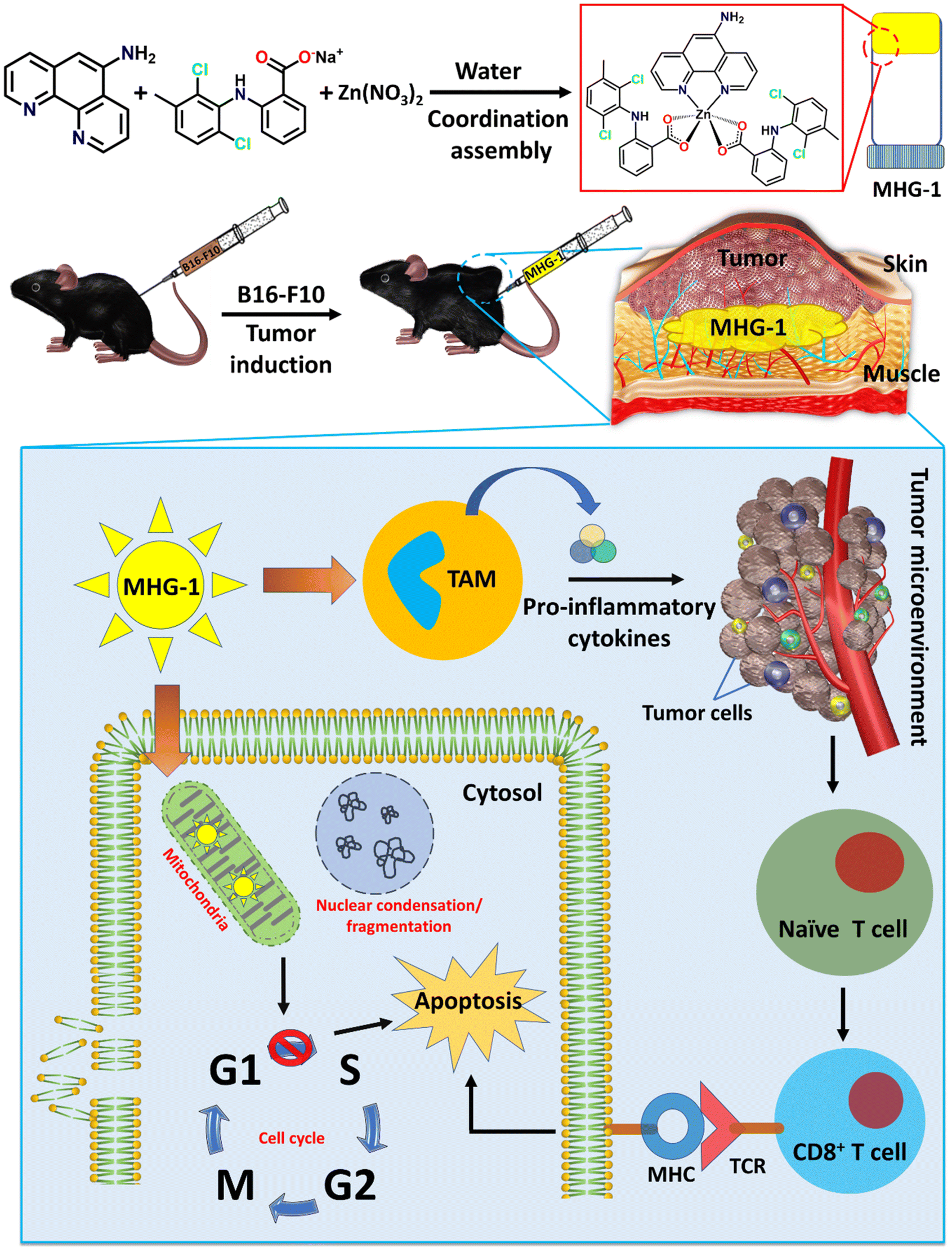 | ||
| Scheme 1 Schematic representation of the work done here (TAM: tumor associated macrophages; MHG-1: metallohydrogel-1; MHC: major histocompatibility complex; TCR: T cell receptor). | ||
2 Results and discussion
2.1 Design, synthesis and single crystal structures of the CCs
The main objective of the present work was to develop a hydrogel-based SDD system that can subcutaneously be implanted on the tumor site for plausible anti-tumor therapy. For this purpose, we exploited simple coordination chemistry, wherein, we employed a N donor chelating ligand, namely 5-AP, various Na-NSAIDs (MEC, DIC, MEF and FLU), and Zn(NO3)2 to synthesize a series of Zn(II) CCs as potential anticancer agents against melanoma-induced tumors (Fig. 1A). While 5-AP metal complexes are reported to have anticancer properties,50 NSAIDs are known for their immunomodulatory effects on tumor microenvironments;51 Zn salts, on the other hand, do possess anti-inflammatory properties,52 which is an advantage in the context of cancer therapy. It is well known that hydrogen bonding and hydrophobic/hydrophilic balance play a crucial role in the supramolecular hydrogelation process.53,54 Thus, a large π surface of 5-AP and hydrogen bonding functionalities (–NH2 of 5-AP and –COO and –NH of the NSAIDs) are expected to facilitate the hydrogelation process under suitable conditions.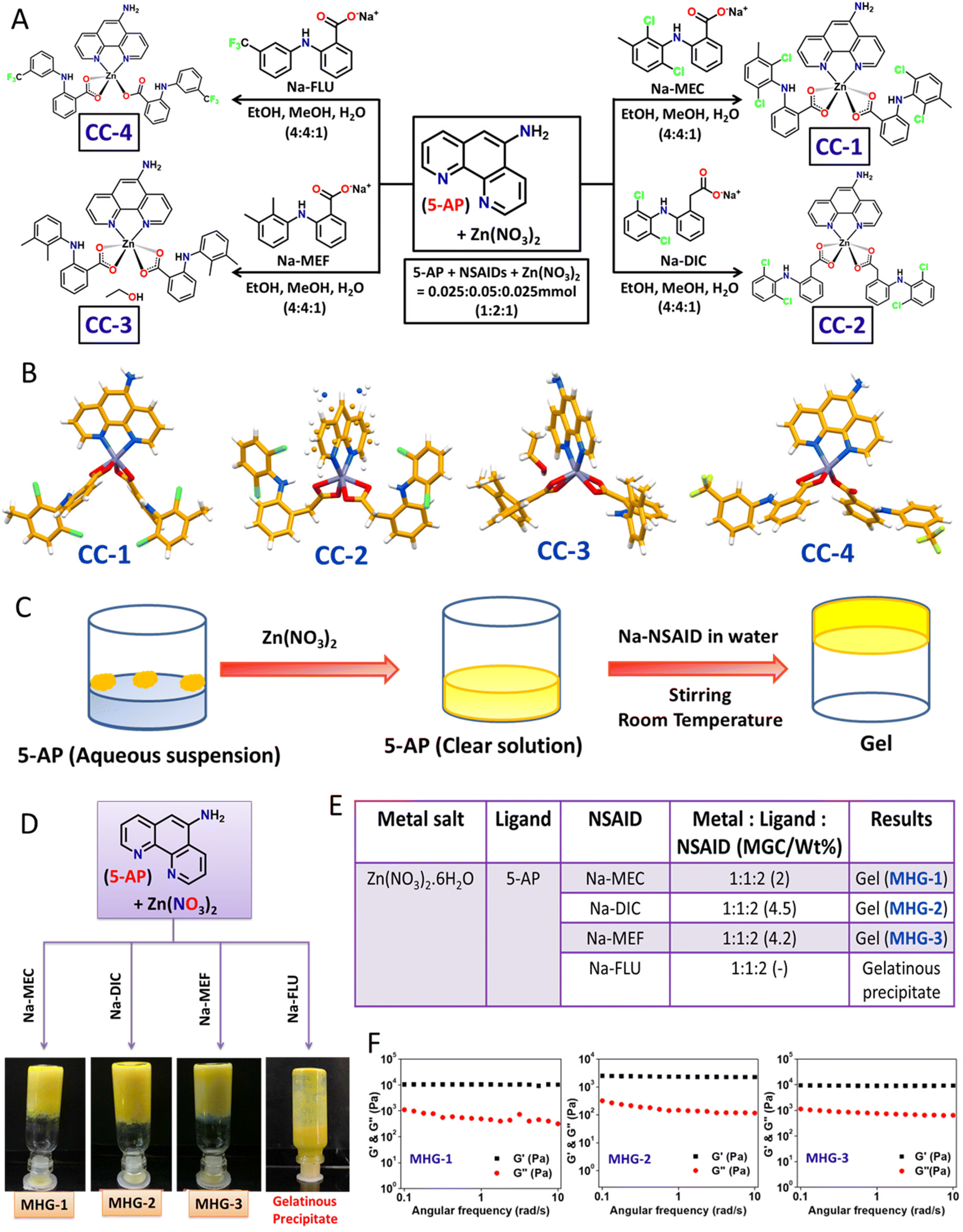 | ||
| Fig. 1 Synthesis of the CCs, hydrogel formation and characterization. (A) Synthetic route for the CCs. (B) Crystal structures of the CCs. (C) Schematic representation of the hydrogel preparation strategy. (D) Image of the inverted vials containing hydrogels. (E) Gelation data. (F) Dynamic rheology (frequency sweep, 8 wt% hydrogel). | ||
:2:1 molar ratio in EtOH/MeOH/water (4:4:1 v/v/v) at room temperature, followed by slow evaporation in a long tube yielded crystalline products after one week (Fig. 1A). Blue shift (Δν = 6–41 cm−1) of the carboxylate stretching with respect to that of the corresponding Na-NSAID in the FT-IR of these crystals strongly suggested carboxylate-Zn coordination (Fig. S1 and Table S1†). Elemental analysis clearly supported the stoichiometry, suggesting the formation of Zn(5-AP)(NSAID)2 complexes (see the Experimental section).
Single crystals of these complexes were subjected to X-ray diffraction (Table S2†). While CC-1 and CC-4 were isomorphous, displaying an identical space group (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and near identical cell dimensions, CC-2 and CC-3 belonged to a monoclinic crystal system with space groups P2/c and Pc, respectively. Except in CC-4, the metal centre Zn(II) was six coordinated, owing to the chelating coordination mode of both 5-AP and carboxylates of the corresponding NSAID moieties; mono-dentate coordination of one of the NSAID moieties in CC-4 resulted in five coordinated Zn(II) species, suggesting the formation of coordination complexes having a general formula Zn(5-AP)(NSAID)2 (Fig. 1B). Interestingly, only CC-3 contained a lattice occluded solvate molecule (EtOH) in its asymmetric unit. It may also be noted that half of the complex molecule was located in the asymmetric unit in CC-2 as the metal centre Zn(II) resided on a 2-fold axis consequently inducing a rotational disorder on 5-AP. Careful analyses revealed that the N atom of the disordered 5-AP moiety was involved in hydrogen bonding with the carboxylate O atoms of the neighbouring molecules (Fig. S3 and Table S4†). However, in the remaining cases, the complex molecules were located at general positions. Molecular self-assembly in all the crystal structures was mainly governed by hydrogen bonding and π–π stacking. Driven by strong intermolecular N–H⋯O hydrogen bonding involving the amino N and carboxylate O atoms and π–π stacking interactions involving the large π surface of 5-AP, both CC-1 and CC-4 formed centrosymmetric dimers, which were further packed in the crystal structures sustained by π–π stacking interactions involving 5-AP and F-containing phenyl moieties (Fig. S2 and S5 and Tables S3 and S6†). In CC-3, the coordination complex molecules formed a hydrogen bonded chain through N–H⋯O interactions involving the carboxylate O and amino N atoms. The lattice occluded solvate EtOH molecules connected these chains through O–H⋯O and N–H⋯O interactions, resulting in an overall 2D hydrogen bonded network, which was packed in a parallel manner along the a-axis (Fig. S4 and Table S5†). These results clearly suggested that both the ingredients (5-AP and NSAIDs) were successfully anchored to the Zn(II) metal centre with a precise stoichiometry of 1:2:1, and a pure crystalline product could be obtained. Powder X-ray diffraction (PXRD) data supported the crystalline phase purity of the synthesized CCs (Fig. S10†).
) and near identical cell dimensions, CC-2 and CC-3 belonged to a monoclinic crystal system with space groups P2/c and Pc, respectively. Except in CC-4, the metal centre Zn(II) was six coordinated, owing to the chelating coordination mode of both 5-AP and carboxylates of the corresponding NSAID moieties; mono-dentate coordination of one of the NSAID moieties in CC-4 resulted in five coordinated Zn(II) species, suggesting the formation of coordination complexes having a general formula Zn(5-AP)(NSAID)2 (Fig. 1B). Interestingly, only CC-3 contained a lattice occluded solvate molecule (EtOH) in its asymmetric unit. It may also be noted that half of the complex molecule was located in the asymmetric unit in CC-2 as the metal centre Zn(II) resided on a 2-fold axis consequently inducing a rotational disorder on 5-AP. Careful analyses revealed that the N atom of the disordered 5-AP moiety was involved in hydrogen bonding with the carboxylate O atoms of the neighbouring molecules (Fig. S3 and Table S4†). However, in the remaining cases, the complex molecules were located at general positions. Molecular self-assembly in all the crystal structures was mainly governed by hydrogen bonding and π–π stacking. Driven by strong intermolecular N–H⋯O hydrogen bonding involving the amino N and carboxylate O atoms and π–π stacking interactions involving the large π surface of 5-AP, both CC-1 and CC-4 formed centrosymmetric dimers, which were further packed in the crystal structures sustained by π–π stacking interactions involving 5-AP and F-containing phenyl moieties (Fig. S2 and S5 and Tables S3 and S6†). In CC-3, the coordination complex molecules formed a hydrogen bonded chain through N–H⋯O interactions involving the carboxylate O and amino N atoms. The lattice occluded solvate EtOH molecules connected these chains through O–H⋯O and N–H⋯O interactions, resulting in an overall 2D hydrogen bonded network, which was packed in a parallel manner along the a-axis (Fig. S4 and Table S5†). These results clearly suggested that both the ingredients (5-AP and NSAIDs) were successfully anchored to the Zn(II) metal centre with a precise stoichiometry of 1:2:1, and a pure crystalline product could be obtained. Powder X-ray diffraction (PXRD) data supported the crystalline phase purity of the synthesized CCs (Fig. S10†).
2.2 Hydrogelation
The self-assembly features involving both π–π stacking and hydrogen bonding present in these CCs raised hope of the possibility of hydrogel formation. However, none of the CCs were soluble in water at room temperature. Interestingly, addition of solid Zn(NO3)2 to an aqueous suspension of 5-AP resulted in a clear solution, suggesting effective coordination between 5-AP and Zn(II). To our delight, addition of aqueous solution of the corresponding Na-NSAID, followed by mechanical stirring, produced opaque yellow metallohydrogels (MHG-1, MHG-2 and MHG-3) at room temperature for CC-1, CC-2 and CC-3, as evident from the tube inversion test; a gelatinous precipitate was obtained for CC-4 (Fig. 1C and D). In these experiments, the molar ratio of the reactants was maintained as guided by the corresponding single crystal structures (Fig. 1E). It is noteworthy that instantaneous hydrogelation of the drug complexes at room temperature (contrary to the usual heat-cool treatment to obtain gels) is advantageous for SDD. Dynamic rheological studies were carried out to confirm the viscoelastic properties of the MHGs. Amplitude sweep experiments performed on the hydrogels (8 wt%) displayed the linear viscoelastic (LVE) region, which is typical of gel-like materials (Fig. S11†). Frequency sweep experiments, on the other hand, clearly established the viscoelastic properties of these hydrogels, as evident from the fact that the elastic modulus (G′) was more than the loss modulus (G′′) and remained frequency (ω s−1) invariant especially in the longer time scale (Fig. 1F). All the hydrogels were found to be rheologically strong, as evident from the remarkable tanδ values ranging 0.05–0.09 (Table S7†). The morphology of the dried hydrogels under TEM appeared to be colloidal aggregates (Fig. S12†). The absence of entangled fibrillar networks, i.e., self-assembled fibrillar networks (SAFiNs),55 typically observed for supramolecular gels, indicated that gelation did not take place via SAFiN formation in our case; rather, interactions involving the nanosized particles of the coordination complexes were responsible for the formation of gel networks.56 Interestingly, the stretching frequency of the carboxylate of the Na-NSAID experienced a blue shift (Δν = 5–78 cm−1) in the corresponding xerogels, suggesting carboxylate–Zn coordination in the xerogels as well (Fig. S14 and Table S8†).
2.3 Rheoreversibility, tube-squeezing-out ability, long-term stability and injectability make MHG-1 suitable for real-life applications
Low tanδ values from the frequency sweep experiments suggested that the gels were rheologically strong. Now, to use these hydrogels for local applications, they must possess good thixotropic properties. Interestingly, the hydrogels (8 wt%) showed good rheoreversibility over several low–high strain cycles during the thixotropic experiments (Fig. 2A). Not only that, during these studies, we noticed that the hydrogel MHG-1 (which had the least tanδ value) showed good adhering properties on a glass surface (Fig. 2B), which was also found when a small amount of it was taken on fingers and was randomly moved (Fig. 2C and ESI movie 1†). This property might be an advantage for its topical use (Fig. 2D). Another important fact was the long-time stability of the hydrogel. For that reason, we kept MHG-1 (4 wt%, 1 mL) for 60 days at room temperature and under light exposure and air-tight conditions (Fig. 2E), and after that, evaluated the viscoelastic properties by frequency sweep experiments (Fig. 2F). The data suggested that the hydrogel had good viscoelastic properties, showing a remarkable tanδ value (0.08) even after 60 days. We have also evaluated its squeezing property and injectability to use it in a more practical way. The data showed that the hydrogel could be squeezed out from a plastic tube and injection syringe without any phase separation, suggesting the hydrogel could become a potential candidate in topical as well as local applications for therapeutic use (Fig. 2G and H and ESI movies 2 and 3†).
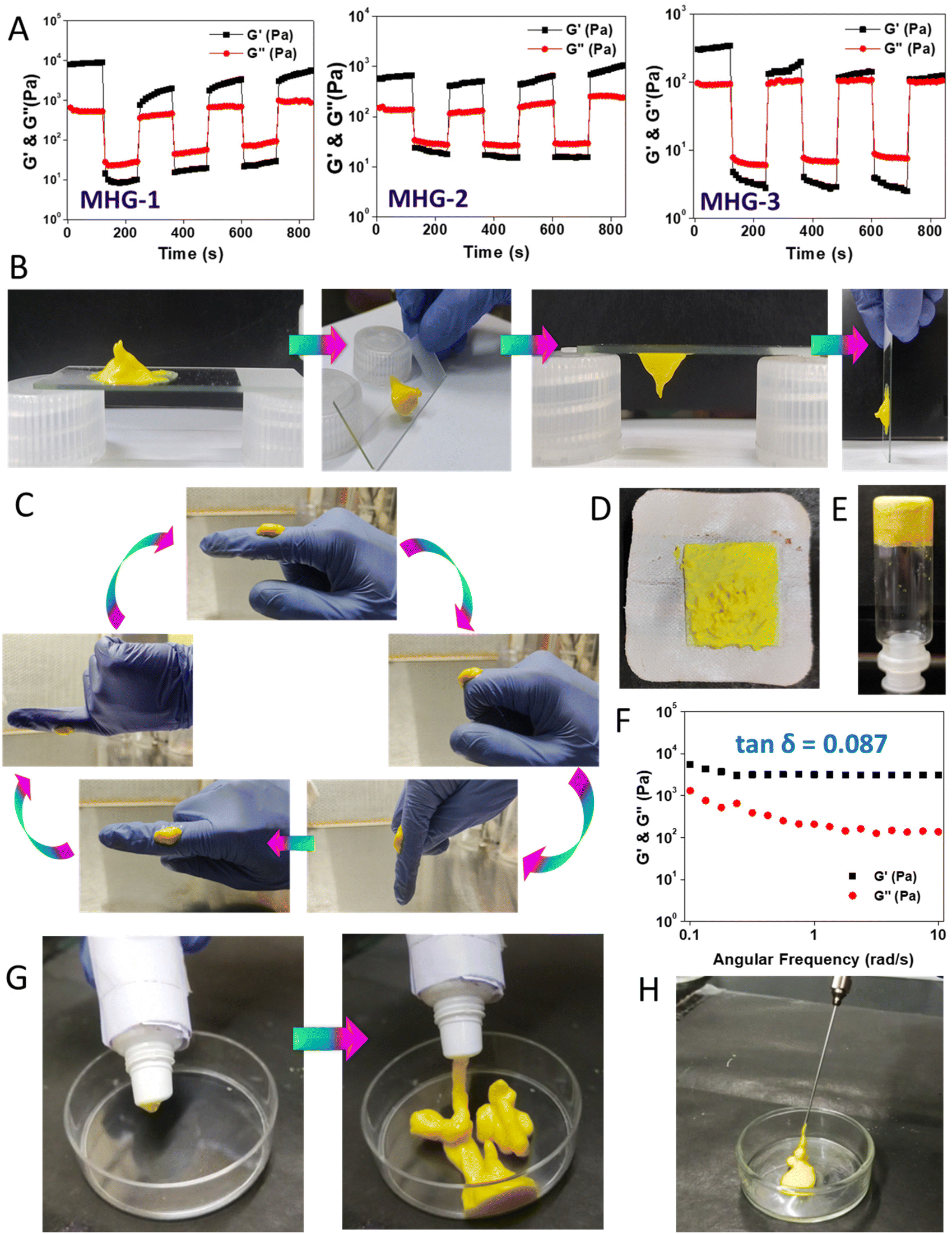 | ||
| Fig. 2 Material properties of the hydrogel. (A) Rheoreversibility of the hydrogels (MHG-1, MHG-2 and MHG-3). (B and C) Adhering properties of MHG-1 (4 wt%). (D) A bandage pad containing MHG-1. (E and F) Image of the inverted vial and dynamic rheology (frequency sweep, 4 wt% hydrogel) of 60-day-old MHG-1. (G and H) Squeezing-out properties and injectability of MHG-1. | ||
2.4 Biological studies
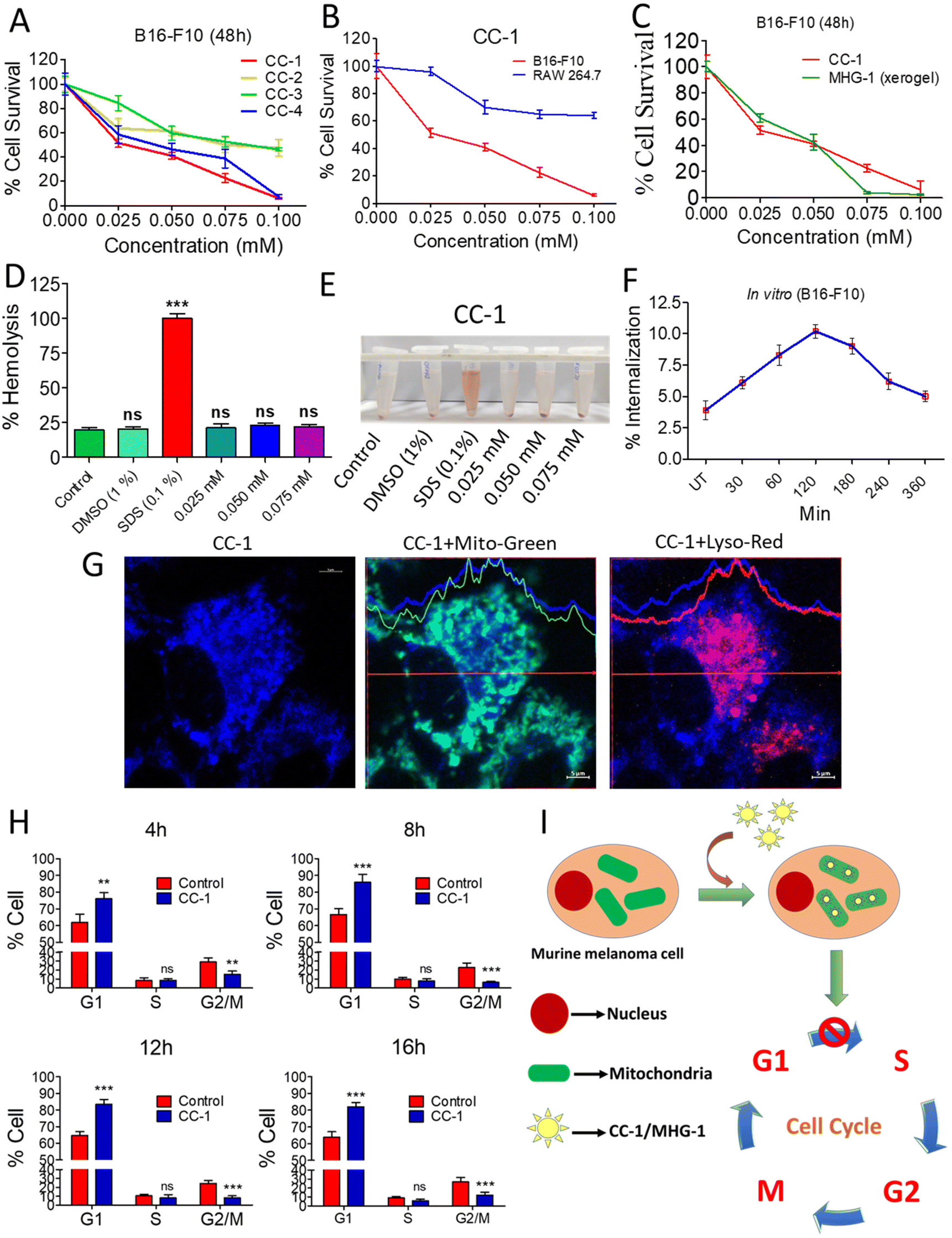 | ||
| Fig. 3 Anticancer activity of CC-1 on murine melanoma cells (B16-F10) under in vitro conditions. (A) MTT assay of the CCs against B16-F10 cells. (B) MTT assay of CC-1 against RAW 264.7 and B16-F10 cells after 48 h. (C) MTT assay of CC-1 and MHG-1 (xerogel) against B16-F10 cells after 48 h. (D) Hemolysis of CC-1 on murine blood (all calculations were carried out by considering that 100% hemolysis occurred in the SDS-treated set; DMSO was used as the control solvent). (E) Image of CC-1-, SDS- and DMSO-treated murine blood after 3 h incubation. (F) % Internalization of CC-1 (0.025 mM) in B16-F10 cells as per the flow cytometry data (see Fig. S23†) (UT – untreated cells). (G) LSCM images of CC-1 (0.025 mM)-treated B16-F10 cells after 120 min incubation. Significant overlap of blue and green fluorescence due to CC-1 internalization and MitoTracker™-stained mitochondria, respectively, and lack of that with red fluorescence due to the LysoTracker™-stained lysosome confirmed mitochondrial internalization of CC-1 (scale bar = 5 μm). (H) Cell cycle analysis of CC-1 (0.025 mM)-treated B16-F10 cells (see Fig. S28†). (I) Schematic representation of CC-1 internalization and cell cycle arrest. In the line graphs, data are represented as mean ± SD (n = 3); bar graphs are represented as mean + SD (n = 3), where **p < 0.01 and ***p < 0.001 and ns is non-significant. | ||
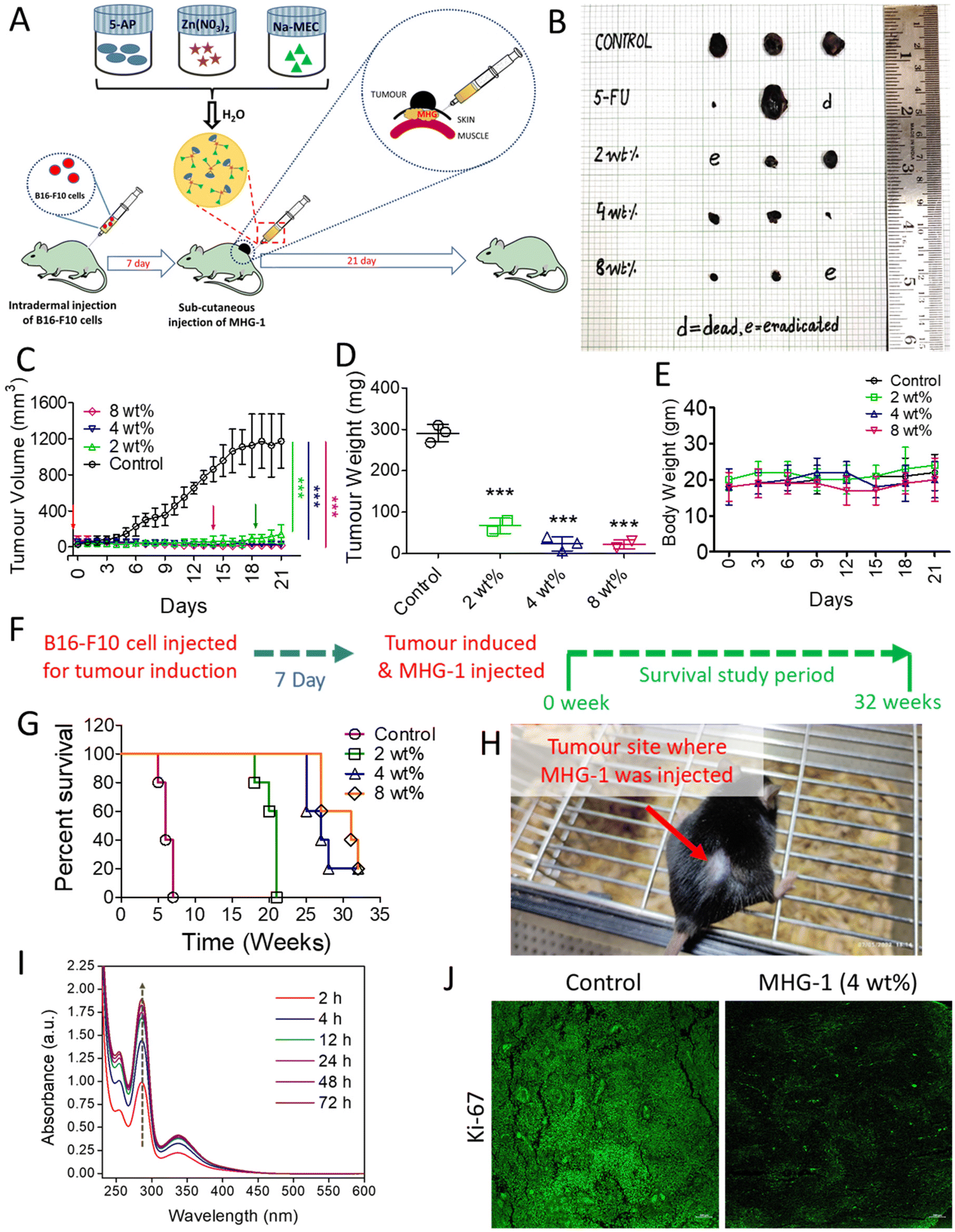 | ||
| Fig. 4 In vivo anticancer activity of MHG-1 and its in vitro release. (A) Schematic representation of the overall in vivo procedure. (B) The size of the tumors under various conditions. (C) Tumor volume (mm3) during the treatment period (21 days). Tumor eradication was noticed in 2 (green arrow) and 8 wt% (magenta arrow) MHG-1 treated mice after 18 and 14 days, respectively. Note that each group contained three mice and tumor eradication was observed in one mouse in both cases (2 and 8 wt%). The red arrow indicates the day at which the hydrogel was implanted. (D) Tumor weight after the 21-day MHG-1 treatment. (E) Body weight of the tumor bearing mice treated with MHG-1 during the treatment period (21 days). (F) Strategy for the survival study. (G) Survivability plot of the MHG-1 treated mice for 32 weeks. (H) Image of MHG-1 (4 wt%)-treated melanoma bearing mice after 32 weeks. The image shows complete eradication of the tumor after MHG-1 treatment. The red arrow indicates the tumor site, where MHG-1 was injected. (I) Time-dependent UV-vis spectrum of the released component from the MHG-1 bed (prepared in PBS) (the dashed arrow represents an increase in absorption of the leached out supernatant solution with time). (J) The LSCM image of the anti-Ki-67 monoclonal antibody-stained tumor tissue treated with MHG-1 (4 wt%). The tumor was excised after 21 days of MHG-1 treatment. Reduced Ki-67 expression (low-intensity green fluorescence compared to the control) was observed after MHG-1 treatment (scale bar = 100 μm). In the graphs, data are represented as mean ± SD (n = 3); where ***p < 0.001. | ||
In vitro cell migration and cell cycle data revealed the inhibition of cancer cell proliferation after the treatment. As Ki-67 is upregulated in any proliferative cells such as cancerous cells,62 we incubated the tumor tissue collected after 21 days of the treatment with 4 wt% hydrogel with an anti-Ki-67 monoclonal antibody. It was clear from the LSCM images that the expression of Ki-67 was greatly reduced in the treated tissues as compared to the control, thereby supporting the role of the hydrogel as an anticancer agent (Fig. 4J and Fig. S51†).
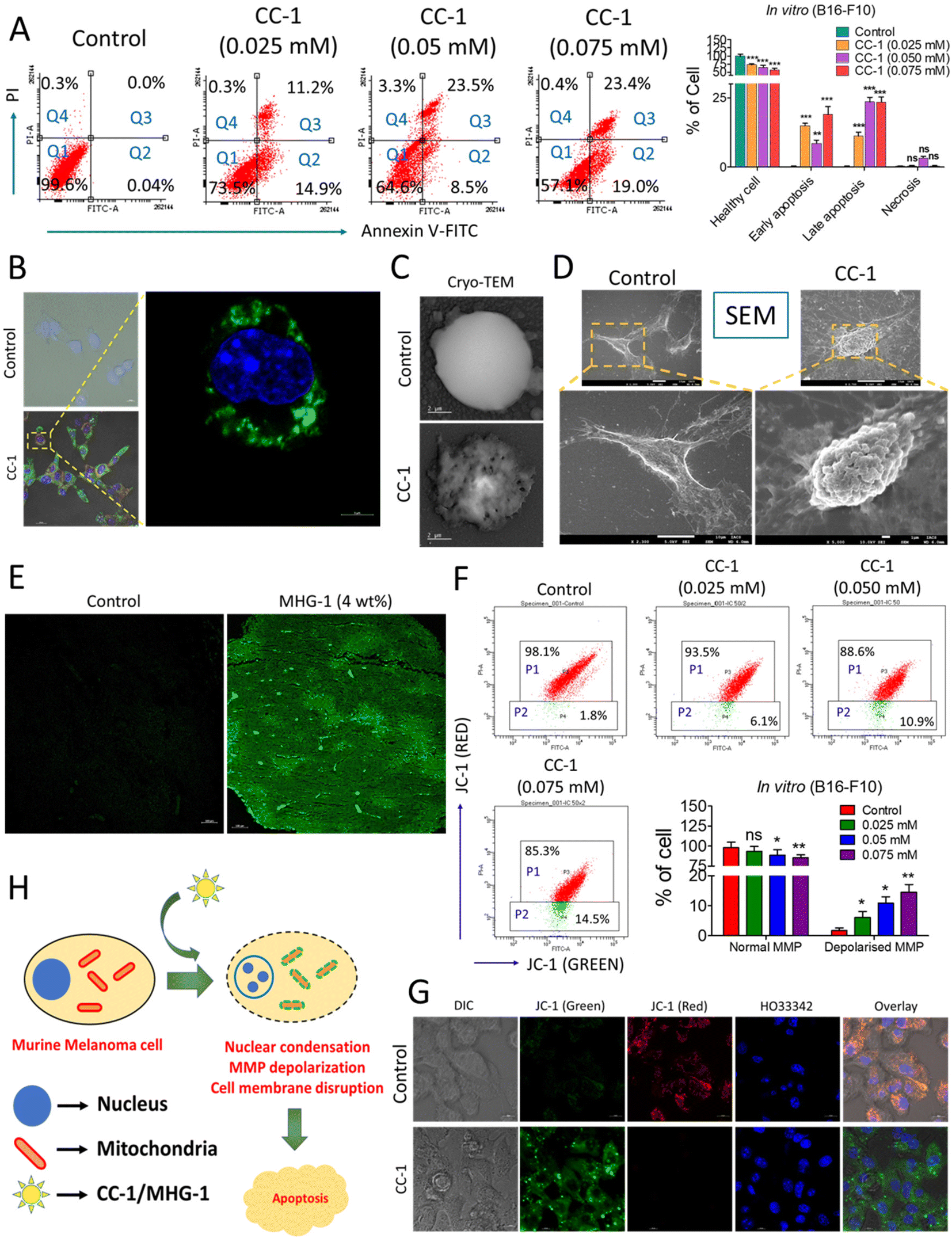 | ||
| Fig. 5 Probing the onset of apoptosis with CC-1/MHG-1 in murine melanoma cells (A) Flow cytometry of CC-1-treated B16-F10 cells, after 48 h incubation (Q1-healthy cells; Q2-early apoptosis; Q3-late apoptosis; Q4-necrosis). (B) Part of the LSCM images of CC-1 (0.025 mM)-treated B16-F10 cells depicted in Fig. S34,† highlighting the onset of apoptosis as revealed from the green fluorescence arising due to annexin V-FITC binding with the extracellular phosphatidyl serine (scale bar = 20 μm). (C and D) Cryo-TEM and SEM images of a single B16-F10 cell under treated (CC-1/0.025 mM) and untreated (control) conditions after 48 h (scale bar = 2 μm and 10 μm), respectively. (E) The LSCM image of untreated and MHG-1 (4 wt%)-treated tumor tissues stained with annexin V-FITC. The tumor was excised after 21 days of MHG-1 treatment. Increased green fluorescence arising due to annexin V-FITC binding with the extracellular phosphatidyl serine supported apoptosis in in vivo as well (scale bar = 100 μm). (F) Flow cytometry of CC-1-treated B16-F10 cells stained with JC-1 after 48 h. Increased cell population emitting green fluorescence (P2) indicated MMP depolarization, supporting apoptosis. (G) LSCM images of CC-1 (0.025 mM)-treated B16-F10 cells stained with JC-1 after 48 h. Distinct green fluorescence in the overlay image of the treated cells suggested MMP depolarization, supporting apoptosis (scale bar = 10 μm). (H) Schematic representation of the mechanism of cancer cell death. In the graphs, data are represented as mean + SD (n = 3), where *p < 0.05, **p < 0.01 and ***p < 0.001 and ns is non-significant. | ||
Apoptosis was also evident from the mitochondrial membrane potential (MMP) assay with JC-1 in B16-F10 cells treated with CC-1 in a dose-dependent manner; a gradual increase of green fluorescence with an increase in the concentration of CC-1 in flow cytometry clearly indicated depolarization of MMP, supporting apoptosis (Fig. 5F). It was also confirmed by the LSCM images of B16-F10 cells under various staining conditions (Fig. 5G). Flow cytometry data from a single cell suspension from the hydrogel-treated tumor tissues also supported apoptosis, as evident from the JC-1 assay (Fig. S38†). Thus, our findings from both in vitro and in vivo studies clearly suggested that CC-1 (in vitro)/MHG-1 (in vivo) triggers apoptosis on the cancer cells via MMP depolarisation (Fig. 5H).
Finally, we were able to record a video, where cells were triggering apoptosis after treatment with CC-1 (concentration: 10 times higher than IC50) using a confocal microscope. The video clearly demonstrated the internalization of CC-1-induced apoptotic bleb formation, with notable morphological changes (Fig. 6 and ESI movie 5†).
 | ||
| Fig. 6 Internalization of CC-1 (blue) triggering apoptosis. | ||
Hoechst efflux assay. Drug resistance to chemotherapy and tumor reoccurrence are common in cancer treatment, which is primarily due to side population (SP) cells having cancer stem cell-like properties.63,64 Efflux of chemotherapeutic drugs by SP cells can be monitored by the efflux of HO33342, which produces red fluorescence. Decreasing red fluorescence in the flow cytometry data from B16-F10 cells treated with verapamil (a well-known drug that kills SP cells, as the positive control) and CC-1 with varying concentrations clearly supported the ability of CC-1 in killing SP cells (Fig. S39†). Similar experiments performed on a single cell suspension of the hydrogel-treated tumor tissues also supported the in vitro data (Fig. S40†).
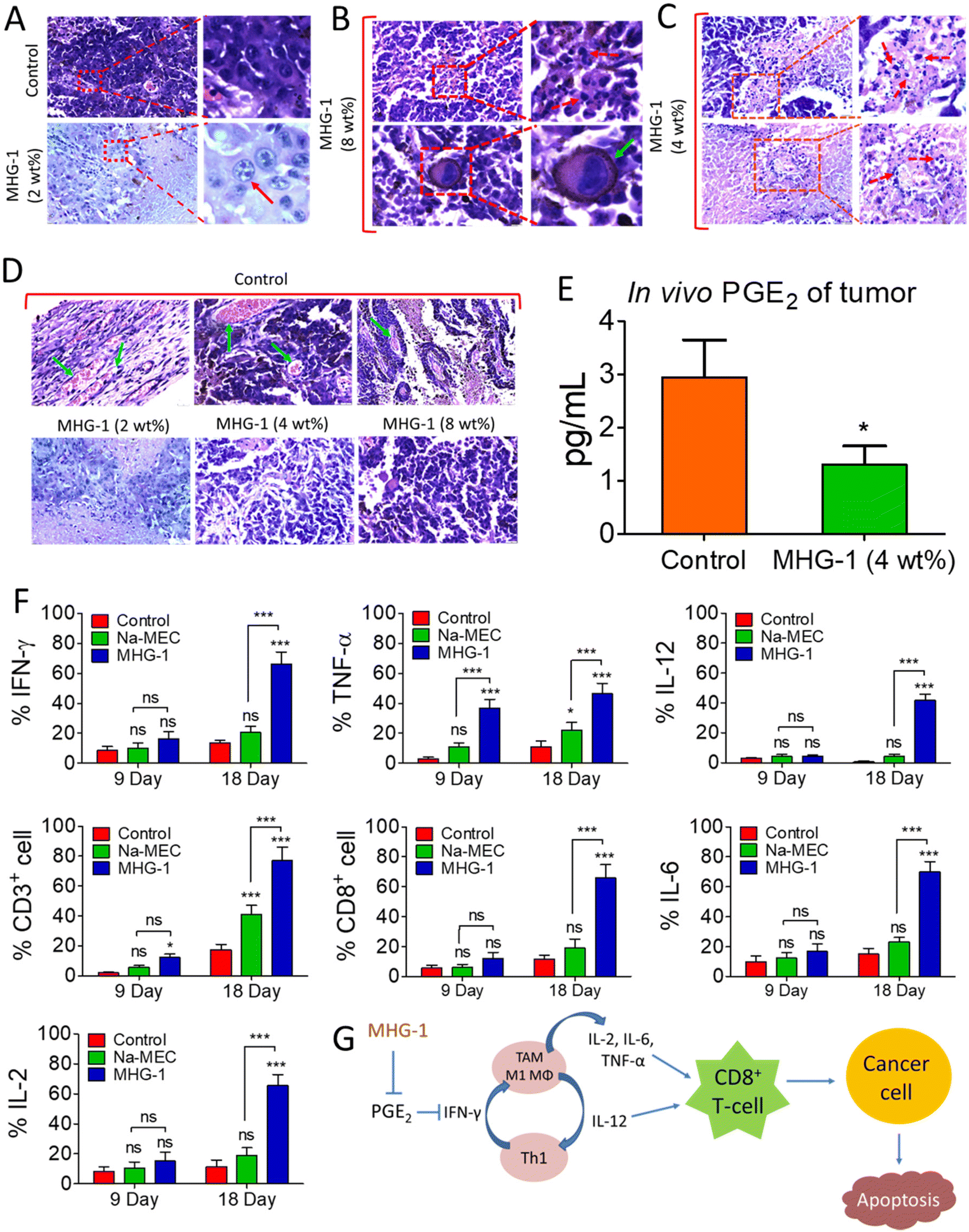 | ||
| Fig. 7 Optical microscopic images of the H&E-stained tumor tissue (the tumor was excised after the 21-day MHG-1 treatment) and immunomodulation in the tumor microenvironment with MHG-1. (A) The intense blue dots within the nuclei (red arrow) suggest nuclear condensation/fragmentation (scale bar = 20 μm). (B) The giant cell with lobular or kidney shaped nuclei (green arrow) suggests monocyte infiltration in the tumor (scale bar = 10 μm). (C) The blue dots (red arrow dashed line) within the cell suggest nuclear fragmentation (scale bar = 20 μm). (D) The light blue arrows suggest the presence of blood vessels in the control set. In contrast, in the MHG-1-treated tumor tissue, blood vessels were absent (scale bar = 20 μm). (E) PGE2 assay of the tumor tissue supernatant after 18 days of MHG-1 (4 wt%) treatment. (F) Graphical representation of pro-inflammatory cytokine expression and T-cell populations in the tumor microenvironment, after MHG-1 (4 wt%) treatment. Data were obtained from flow cytometry. (G) Schematic representation of the MHG-1-induced immunoediting mechanism in the tumor microenvironment, leading to cancer cell apoptosis. In the graphs, data are represented as mean + SD (n = 3), where *p < 0.05 and ***p < 0.001 and ns is non-significant. | ||
Since the hydrogel under study contained a NSAID component, namely MEC, and since it showed encouraging results as an anticancer agent triggering apoptosis as revealed by various in vitro and in vivo experiments including inhibition of angiogenesis and a sign of a pro-inflammatory immune response in the histopathological studies (see above), we decided to investigate the intracellular PGE2 level and the status of the immune system in the tumor microenvironment after the hydrogel treatment. The PGE2 assay carried out on inflamed RAW 264.7 cells (LPS treated) showed significant reduction of PGE2 concentration when treated with CC-1 and Na-MEC, indicating the well-known role of NSAIDs in PGE2 reduction present in CC-1 (Fig. S46†). Similar results were also obtained for the hydrogel-treated tumor and skin tissues, suggesting the effective role of reducing PGE2 due to subcutaneous administration of the hydrogel (Fig. 7E and S47†).
The reduction in PGE2 expression in the tumor microenvironment often changes the cytokine secretions, which could exert anti-tumor activity.78 Furthermore, it is well known that PGE2 inhibits IFN-γ production and opposes IL-12 signalling by proliferating Th2 lymphocytes.78 Thus, we investigated IFN-γ and IL-12 contents in the tumor microenvironment after the treatment. The flow cytometry data suggested induction in IFN-γ as well as IL-12 expression after 9 and 18 days of the hydrogel treatment with respect to that of the control (Fig. 7F, S48 and S49†). The overexpression of IFN-γ in the tumor microenvironment indicated an increase in M1 macrophage population.71 Furthermore, IFN-γ from Th1 cells recruits and maintains the M1 phenotype of macrophages; on the other hand, IL-12 from M1 macrophages recruits and maintains the Th1 phenotype.79 In our case also, simultaneous overexpression of IL-12 along with IFN-γ was observed. Thus, it may be an indication of maintenance and recruitment of Th1 lymphocytes and M1 macrophages. To further confirm this, we checked the expression of other pro-inflammatory cytokines (TNF-α, IL-2 and IL-6) related to Th1 lymphocytes and M1 macrophages. The data depict an increased cytokine expression after the hydrogel treatment (Fig. 7F, S48 and S49†). These results suggested an increased M1 population in the tumor microenvironment. It is noteworthy that IL-2 or its combination with drug/other cytokines is used as subcutaneous/intravenous injection in melanoma immune therapy.80,81 Furthermore, IL-2 not only showed regression of metastatic melanoma, but also could be a useful immunotherapy for tumor eradication. Due to the short serum half-life of IL-2, it faces several limitations.82 In our case, the hydrogel itself is able to induce IL-2 concentration locally without any notable side effects. In addition, in some of the cases, we have observed complete eradication of the tumors. It is well known that cytokines (IL-12, IL-6, TNF-α and IL-2) are directly/indirectly responsible for the maturation and activation of cytotoxic T cells (CD8+ cells), which interact with cancer cells, triggering apoptosis/necrosis.79 Thus, we investigated T cell populations in the hydrogel-treated tumor microenvironment. Our data suggested an increase in total T cell population (CD3+ cells), indicating expansion of tumor infiltrating T cells as well as cytotoxic T cell population (CD8+ cells) (Fig. 7F, S48 and S49†).
In summary, the subcutaneously injected hydrogel potentially reduces PGE2 expression, which in turn induces IFN-γ and IL-12. IFN-γ and IL-12 recruited M1 macrophages and Th1 lymphocytes, which secreted pro-inflammatory cytokines (IL-12, IL-6, TNF-α and IL-2). Further, the pro-inflammatory cytokines activated the cytotoxic T cells (CD8+ cells). Finally, the activated T cells (CD8+ cells) interacted with the cancer cells, triggering apoptosis (Fig. 7G).
3 Conclusion
A series of Zn(II) coordination complexes derived from NSAIDs and 5-AP were designed, synthesized and crystallographically characterized for developing a self-drug-delivery system as hydrogels for treating the most fatal type of skin cancer (melanoma). One such hydrogel having MEC as the NSAID component, when subcutaneously implanted as a single dose, showed promising results in inhibiting melanoma-induced tumor growth in C57BL/6 mice, including complete eradication of tumors in some cases. Both in vitro and in vivo results suggested that MEC being a NSAID effectively reduced the intracellular PGE2 concentration, thereby upregulating IFN-γ and IL-12, which in turn modulated the immune microenvironment of the tumor site by recruiting M1 macrophages and Th1 lymphocytes. Consequently, the secreted pro-inflammatory cytokines (IL-12, IL-6, TNF-α, IL-2) activated cytotoxic T cells (CD8+), which eventually triggered apoptosis. The results presented herein highlight the following merits:(a) Simple coordination chemistry and the concept of supramolecular chemistry have been employed to design hydrogels as a potential self-drug-delivery system for melanoma therapy. The absence of the need for a drug delivery vehicle usually required in a gel-based therapy understandably eliminates the concerns pertaining to drug loading and its effective release to the target site.
(b) The successful preparation of a stable and injectable hydrogel allows us to implant it to the tumor site via the subcutaneous route. The usual topical gel formulation in treating melanoma (e.g., 5-fluorouracil loaded gel formulation) suffers from inefficient delivery of the anticancer drug to the cancer cells due to spreading and drying issues of the applied gel. However, the subcutaneous route (as it is implanted below the tumor) prevents both spreading and drying, thereby helping in maintaining high local concentration of the drug and its sustained release to the target site.
(c) Both in vitro and in vivo data strongly support that the all-in-one self-drug-delivery system thus developed is capable of providing both chemo- and immunotherapy to combat melanoma, as evident from the decreased intracellular PGE2 level and up-regulation of IL-2. It is noteworthy that immunotherapy wherein the immune cytokine IL-2 is injected intravenously suffers from its short half-life and severe side effects.
The overall work reported in this paper is schematically depicted in Fig. S54.† The approach of developing a self-drug-delivery system by implanting a supramolecular drug-derived hydrogel to deliver chemotherapeutic agents along with an immunomodulator to combat melanoma is expected to open up new avenues in cancer therapy.
Author contributions
S. B. performed the synthesis, characterization, gelation, rheology, SXRD, PXRD, MTT, and scratch assay and performed all biological experiments along with and under the supervision of H. K. D. and P. D. conceptualized and supervised the project, analyzed the results, and co-wrote the manuscript with input from all the co-authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
P. D. thanks SERB (CRG/2019/000790) for the financial support. S. B. thanks UGC for SRF and H. K. D. thanks IACS for the RA fellowship. The authors thank Chanchal K. Das and Debapriya Ghatak for helping in flow cytometry and LSCM data collection. The authors also thank Dr Amitava Sengupta, Cancer Biology Division, CSIR-Indian Institute of Chemical Biology, Kolkata for gifting the PE-CF594 rat anti-mouse CD31 antibody. S. B. and H. K. D. thank Dr Poulami Chakraborty and Dr Protap Biswas for their continuous help and support. S. B and H. K. D. also thank Subhra Sankar Dutta for the help during LC-MS data collection. S. B. thanks Prosenjit Bera for the help in graphic designing. SXRD data were collected in the DBT-funded X-ray diffraction facility under the CEIB program at the School of Chemical Sciences, IACS, Kolkata. The animal experiments were performed under the ethical permission of Institutional Animal Ethics Committee (IAEC) having approval no. IACS/IAEC/2021-02 and were conducted following the IAEC guidelines.References
- https://www.who.int/health-topics/cancer#tab=tab_1 .
- https://www.iarc.who.int/news-events/global-burden-of-cutaneous-melanoma-in-2020-and-projections-to-2040/ .
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Strom, J. Controlled Release, 2012, 161, 175–187 CrossRef CAS PubMed.
- L. Cai, S. Liu, J. Guo and Y. G. Jia, Acta Biomater., 2020, 113, 84–100 CrossRef CAS PubMed.
- S. Khajouei, H. Ravan and A. Ebrahimi, Adv. Colloid Interface Sci., 2020, 275, 102060 CrossRef CAS PubMed.
- M. G. Raucci, U. D'Amora, A. Ronca and L. Ambrosio, Adv. Funct. Mater., 2020, 9, 2000349 CAS.
- S. Y. Lee, M. Yang, J. H. Seo, D. I. Jeong, C. Hwang, H. J. Kim, J. Lee, K. Lee, J. Park and H. J. Cho, ACS Appl. Mater. Interfaces, 2021, 13, 2189–2203 CrossRef CAS PubMed.
- J. Qi, T. Ding, T. Liu, X. Xia, S. Wu, J. Liu, Q. Chen, D. Zhang and H. Zhao, Adv. Funct. Mater., 2022, 32, 2204273 CrossRef CAS.
- F. Wang, H. Su, D. Xu, M. K. Monroe, C. F. Anderson, W. Zhang, R. Oh, Z. Wang, X. Sun, H. Wang, F. Wan and H. Cui, Biomaterials, 2021, 279, 121182 CrossRef CAS PubMed.
- F. Ding, Q. Mou, Y. Ma, G. Pan, Y. Guo, G. Tong, C. H. J. Choi, X. Zhu and C. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3064–3068 CrossRef CAS PubMed.
- H. Wang, Z. Feng, Y. Qin, J. Wang and B. Xu, Angew. Chem., Int. Ed., 2018, 57, 4931–4935 CrossRef CAS PubMed.
- L. Zhou, W. Pi, M. Hao, Y. Li, H. An, Q. Li, P. Zhang and Y. Wen, Biomater. Sci., 2021, 9, 4904 RSC.
- N. Liu, S. Wu, X. Tian and X. Li, J. Mater. Chem. B, 2022, 10, 5165 RSC.
- J. Kim, D. M. Francis, L. F. Sestito, P. A. Archer, M. P. Manspeaker, M. J. O'Melia and S. N. Thomas, Nat. Commun., 2022, 13, 1479 CrossRef CAS PubMed.
- B. Wang, J. Chen, J. S. Caserto, X. Wang and M. Ma, Nat. Commun., 2022, 13, 3821 CrossRef CAS PubMed.
- C. Song, H. Phuengkham, Y. S. Kim, V. V. Dinh, I. Lee, I. W. Shin, H. S. Shin, S. M. Jin, S. H. Um, H. Lee, K. S. Hong, S.-M. Jin, E. Lee, T. H. Kang, Y.-M. Park and Y. T. Lim, Nat. Commun., 2019, 10, 3745 CrossRef PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- P. Dastidar, Gels, 2019, 5(1), 15 CrossRef CAS PubMed.
- S. Bonardd, B. Maiti, S. Grijalvo, J. Rodríguez, H. Enshaei, G. Kortaberria, C. Alemán and D. Díaz Díaz, Soft Matter, 2022, 18, 4963–4972 RSC.
- M. Samateh, S. Marwaha, J. K. James, V. Nanda and G. John, Carbohydr. Res., 2022, 521, 108647 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS.
- D. Ghosh, M. Górecki, G. Pescitelli and K. K. Damodaran, Angew. Chem., Int. Ed., 2021, 60, 24406–24410 CrossRef CAS PubMed.
- R. Luboradzki, O. Gronwald, M. Ikeda, S. Shinkai and D. N. Reinhoudt, Tetrahedron, 2000, 56, 9595–9599 CrossRef CAS.
- A. R. Hirst, D. K. Smith, M. C. Feiters, H. P. M. Geurts and A. C. Wright, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef CAS PubMed.
- S. L. James, G. O. Lloyd and J. Zhang, CrystEngComm, 2015, 17, 7976–7977 RSC.
- O. Lebel, M. Perron, T. Maris, S. F. Zalzal, A. Nanci and J. D. Wuest, Chem. Mater., 2006, 18, 3616–3626 CrossRef CAS.
- J. A. Foster, M. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- D. Díaz Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41(2), 315–326 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115(24), 13165–13307 CrossRef CAS PubMed.
- F. Zhao, M. L. Ma and B. Xu, Chem. Soc. Rev., 2009, 38, 883–891 RSC.
- P. Dastidar, R. Roy, R. Parveen and K. Sarkar, Adv. Ther., 2019, 2, 1800061 CrossRef.
- Q. Qian, D. Wang, L. Shi, Z. Zhang, J. Qian, J. Shen, C. Yu and X. Zhu, Biomaterials, 2021, 265, 120403 CrossRef CAS PubMed.
- H. Zhang, S. Xu, J. Zhang, Z. Wang, D. Liu, L. Guo, C. Cheng, Y. Cheng, D. Xu, M. G. Kong, M. Rong and P. K. Chu, Biomaterials, 2021, 276, 121057 CrossRef CAS PubMed.
- G. Gao, Y.-W. Jiang, W. Zhan, X. Liu, R. Tang, X. Sun, Y. Deng, L. Xu and G. Liang, J. Am. Chem. Soc., 2022, 144(26), 11897–11910 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed.
- E. Bellido, T. Hidalgo, M. V. Lozano, M. Guillevic, R. Simón-Vázquez, M. J. Santander-Ortega, Á. González-Fernández, C. Serre, M. J. Alonso and P. Horcajada, Adv. Healthcare Mater., 2015, 4, 1246–1257 CrossRef CAS PubMed.
- D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaïm, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef CAS.
- W.-H. Chen, X. Yu, W.-C. Liao, Y. S. Sohn, A. Cecconello, A. Kozell, R. Nechushtai and I. Willner, Adv. Funct. Mater., 2017, 27, 1702102 CrossRef.
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed.
- P. Biswas and P. Dastidar, Inorg. Chem., 2021, 60(5), 3218–3231 CrossRef CAS PubMed.
- P. Biswas, H. K. Datta and P. Dastidar, Chem. Commun., 2022, 58, 969–972 RSC.
- P. Biswas, H. K. Datta and P. Dastidar, Biomater. Sci., 2022, 10, 6201–6216 RSC.
- C. P. Matos, Z. Adiguzel, Y. Yildizhan, B. Cevatemre, T. B. Onder, O. Cevik, P. Nunes, L. P. Ferreira, M. D. Carvalho, D. L. Campos, F. R. Pavan, J. C. Pessoa, M. H. Garcia, A. I. Tomaz, I. Correia and C. Acilan, Eur. J. Med. Chem., 2019, 176, 492–512 CrossRef CAS PubMed.
- M. Hussain, A. Javeed, M. Ashraf, N. Al-Zaubai, A. Stewart and M. M. Mukhtar, Pharmacol. Res., 2012, 66, 7–18 CrossRef CAS PubMed.
- C. Lang, C. Murgia, M. Leong, L.-W. Tan, G. Perozzi, D. Knight, R. Ruffin and P. Zalewski, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2007, 292, L577–L584 CrossRef CAS PubMed.
- B. Xing, C.-W. Yu, K.-H. Chow, P.-L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124(50), 14846–14847 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104(3), 1201–1218 CrossRef CAS PubMed.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39(8), 489–497 CrossRef CAS PubMed.
- J. Hou, A. F. Sapnik and T. D. Bennett, Chem. Sci., 2020, 11, 310–323 RSC.
- G. Cory, in Scratch-wound assay: Methods in Molecular Biology Cell Migration Developmental Methods and Protocols, ed. C. M. Wells and M. Parsons, Springer, London, 2011, pp. 25–30 Search PubMed.
- J. Deb, J. Majumder, S. Bhattacharyya and S. S. Jana, BMC Cancer, 2014, 14, 567 CrossRef PubMed.
- S. Toné, K. Sugimoto, K. Tanda, T. Suda, K. Uehira, H. Kanouchi, K. Samejima, Y. Minatogawa and W. C. Earnshaw, Exp. Cell Res., 2007, 313, 3635–3644 CrossRef PubMed.
- D. Ngabire, Y. Seong, M. Patil, I. Niyonizigiye, Y. Seo and G. Kim, Int. J. Oncol., 2018, 53, 2300–2308 CAS.
- B. Pucci, M. Kasten and A. Giordano, Neoplasia, 2000, 2, 291–299 CrossRef CAS PubMed.
- Q. Liu, Z. Peng, L. Shen and L. Shen, Front. Oncol., 2021, 11, 737760 CrossRef CAS PubMed.
- C. W. Scharenberg, M. A. Harkey and B. Torok-Storb, Blood, 2002, 99, 507–512 CrossRef CAS PubMed.
- A. Golebiewska, N. H. C. Brons, R. Bjerkvig and S. P. Niclou, Cell Stem Cell, 2011, 8, 136–147 CrossRef CAS PubMed.
- V. Aggarwal, H. S. Tuli, A. Varol, F. Thakral, M. B. Yerer, K. Sak, M. Varol, A. Jain, M. A. Khan and G. Sethi, Biomolecules, 2019, 9, 735 CrossRef CAS PubMed.
- R. Purpora, W. Massad, G. Ferrari, E. Reynoso, S. Criado, S. Miskoski, A. Pajares and N. A. García, Photochem. Photobiol., 2013, 89, 1463–1470 CrossRef CAS PubMed.
- S. S. Husain, I. L. Szabo and A. S. Tamawski, Am. J. Gastroenterol., 2002, 97, 542–553 CrossRef CAS PubMed.
- R. Kim, M. Emi, K. Tanabe and K. Arihiro, Cancer Res., 2006, 66, 5527 CrossRef CAS PubMed.
- D. G. DeNardo and L. M. Coussens, Breast Cancer Res., 2007, 9, 212 CrossRef PubMed.
- T. Marsh, K. Pietras and S. S. McAllister, Biochim. Biophys. Acta, 2013, 1832, 1070 CrossRef CAS PubMed.
- J. Liu, X. Geng, J. Hou and G. Wu, Cancer Cell Int., 2021, 21, 389 CrossRef CAS PubMed.
- B. Ruffell and L. M. Coussens, Cancer Cell, 2015, 27, 462–472 CrossRef CAS PubMed.
- R. Afik, E. Zigmond, M. Vugman, M. Klepfish, E. Shimshoni, M. Pasmanik-Chor, A. Shenoy, E. Bassat, Z. Halpern, T. Geiger, I. Sagi and C. Varol, J. Exp. Med., 2016, 213, 2315–2331 CrossRef CAS PubMed.
- S. Tiainen, R. Tumelius, K. Rilla, K. Hämäläinen, M. Tammi, R. Tammi, V.-M. Kosma, S. Oikari and P. Auvinen, Histopathology, 2015, 66, 873–883 CrossRef PubMed.
- S. Srinivas and D. Feldman, Anticancer Res., 2009, 29, 3605–3610 CAS.
- E. P. Chen and E. M. Smyth, Prostaglandins Other Lipid Mediators, 2011, 96, 14–20 CrossRef CAS PubMed.
- A. V. Timoshenko, G. Xu, S. Chakrabarti, P. K. Lala and C. Chakraborty, Exp. Cell Res., 2003, 289, 265–274 CrossRef CAS PubMed.
- C. Y. Wu, K. Wang, J. F. McDyer and R. A. Seder, J. Immunol., 1998, 161, 2723–2730 CrossRef CAS.
- B. Burkholder, R.-Y. Huang, R. Burgess, S. Luo, V. S. Jones, W. Zhang, Z.-Q. Lv, C.-Y. Gao, B.-L. Wang, Y.-M. Zhang and R.-P. Huang, Biochim. Biophys. Acta, 2014, 1845, 182–201 CAS.
- D. Skrombolas and J. G. Frelinger, Expert Rev. Clin. Immunol., 2014, 10, 207–217 CrossRef CAS PubMed.
- S. A. Rosenberg, J. Immunol., 2014, 192, 5451–5458 CrossRef CAS PubMed.
- T. Jiang, C. Zhou and S. Ren, OncoImmunology, 2016, 5, e1163462 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of the CCs (FT-IR spectra, SXRD, crystallographic table, ORTEP plot, PXRD plot); gel characterization (amplitude sweep, TEM, FT-IR); in vitro experiments (MTT assay, drug internalization, membrane permeability, nuclear condensation, cell migration, ROS, Hoechst efflux assay and cell cycle); in vivo experiments (tumor volume, images of the tumor bearing mice, component-treated tumor images, size, weight, body weight, drug internalization, annexin V-FITC, MMP, histopathology, ROS and Hoechst efflux assay); PGE2 assay; the in vivo immunomodulation study (PDF); Supplementary movies 1–5 (ZIP). CCDC 1918448–1918451 (CC-1–CC-4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3bm00758h |
| This journal is © The Royal Society of Chemistry 2023 |