
SnO2-supported single metal atoms: a bifunctional catalyst for the electrochemical synthesis of H2O2†
Santiago
Jimenez-Villegas
 a,
Sara R.
Kelly
b and
Samira
Siahrostami
*a
a,
Sara R.
Kelly
b and
Samira
Siahrostami
*a
aDepartment of Chemistry, University of Calgary, 2500, University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: samira.siahrostami@ucalgary.ca
bSUNCAT Center for Interface Science and Catalysis, Department of Chemical Engineering, Stanford University, Stanford, California 94305, USA
First published on 25th November 2021
Abstract
On-site hydrogen peroxide (H2O2) production via electrochemical methods, such as two-electron water oxidation reaction (2e-WOR) and two-electron oxygen reduction reaction (2e-ORR), offer an attractive alternative to the anthraquinone oxidation (AO) process. However, for 2e-WOR and 2e-ORR to hold any industrial relevance, inexpensive, stable, highly efficient, selective, and environmentally benign electrocatalysts must be developed. Designing a catalyst to meet such an extensive criterion remains a challenge. Single-atom catalysts (SACs), combining the benefits of heterogenous and homogenous catalysis, have drawn immense attention due to their distinguished catalytic performance. Ergo, we aim towards the exploration of a bifunctional SAC material capable of catalyzing the 2e-WOR and 2e-ORR to produce H2O2. Through density functional theory (DFT) calculations we investigate the catalytic activity, selectivity, and stability of SnO2-supported SACs. Considering 16 different single metal atoms, various promising candidates were identified. Particularly, Mn, Ti and Fe were found to be markedly active and selective 2e-WOR catalysts and W for the 2e-ORR. This work highlights the immense potential of bifunctional systems; a route towards increasing H2O2 yields while simultaneously minimizing manufacturing complexity and cost.
Introduction
With an array of industrial applications, hydrogen peroxide (H2O2) is a highly valued chemical oxidant. Notably, it has garnered significant attention in the fields of wastewater treatment and energy storage owning to its strong oxidative properties and environmentally benign by-products (H2O and O2). At present, over 5 million metric tons of H2O2 must be produced annually to meet demands.1 Unfortunately, H2O2 is primarily manufactured via the costly, and energetically intensive anthraquinone process.2 Additionally, the instability and susceptibility of H2O2 to degradation poses significant concerns on its safe distribution. Hence, to sustainably meet the global H2O2 demands, it is highly desirable that alternate onsite production methods be implemented.Electrochemical production methods have emerged as promising substitutes for the anthraquinone process. An alternate route with capabilities of in situ H2O2 generation is the two-electron water oxidation reaction (WOR).3 At the anode, the WOR evolves H2O2 from H2O, whereas H2 is produced at the cathode. Though the H2 evolved at the cathode is a valuable chemical fuel,4 the goal is to simultaneously minimize unwanted by-products and maximize H2O2 yields. A promising approach to accomplish this is through the replacement of the hydrogen evolution reaction with the oxygen reduction reaction (ORR).5Via the two-electron pathway of the ORR, H2O2 can also be produced at the cathode.
A tandem cell that couples the two-electron WOR at the anode with the ORR at the cathode, would theoretically give 200% faradaic efficiency (FE).6 Although tandem systems using bifunctional catalysts have been developed for other electrochemical reactions,7,8 their application in H2O2 production has only recently been reported.9,10 In 2017, Fuku et al. designed a photoelectrochemical system for H2O2 generation using WO3/BiVO4 and Au catalysts at the photoanode, and at the cathode, respectively.10 The cell design resulted in an FEanode(H2O2) ≈ 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) %, and a FEcathode(H2O2) ≈ 90%, achieving a total current efficiency FEtotal(H2O2) ≈ 140%. Following this, Shi et al. reported the two-sided H2O2 production using a photoelectrochemical cell while simultaneously generating electricity.9 Notably, their set-up involved low-cost, earth abundant materials (carbon at the cathode and BiVO4 at the anode) capable of producing H2O2 at a rate of 0.48 μmol min−1 cm−2. While these, and other reports,11 have demonstrated great advancements for paired H2O2 production, there is still a need for even more active and selective catalysts towards the two-electron ORR and WOR. Furthermore, these catalysts must be cost-effective, environmentally benign, and stable over long running periods.12
%, and a FEcathode(H2O2) ≈ 90%, achieving a total current efficiency FEtotal(H2O2) ≈ 140%. Following this, Shi et al. reported the two-sided H2O2 production using a photoelectrochemical cell while simultaneously generating electricity.9 Notably, their set-up involved low-cost, earth abundant materials (carbon at the cathode and BiVO4 at the anode) capable of producing H2O2 at a rate of 0.48 μmol min−1 cm−2. While these, and other reports,11 have demonstrated great advancements for paired H2O2 production, there is still a need for even more active and selective catalysts towards the two-electron ORR and WOR. Furthermore, these catalysts must be cost-effective, environmentally benign, and stable over long running periods.12
Particularly, catalyst stability has been a major issue for the 2e-WOR process.12 This is, in part, attributed to a highly oxidative environment that is required to promote H2O2 formation (i.e., >1.76 V vs. RHE). Given this limitation, metal oxides have been extensively studied as possible candidates for the 2e-WOR, owing to their ability to survive oxidative environments.3,12,13 These metal oxides were first introduced by Izgorodin et al. for H2O2 production in 2012 using a MnOx electrocatalyst.14 They found high catalytic activity using MnOx, with overpotentials around 150 mV at 1 mA cm−2. Following this, a myriad of metal oxides has been computationally and experimentally explored. Amongst many, these include ZnO,15 BiVO4,16 CaSnO3,17 SnO2 and TiO2 (ref. 16 and 18) where overpotentials as low as 40 mV and high selectivity (FE > 98%) have been unveiled.
Similar to the WOR, the production of H2O2via the 2e-ORR over metal oxides have been studied as green, cost-effective catalyst materials.19–26 Earlier reports by Abbott et al. showed the selective and active O2 conversion to H2O2 over RuO2 and Ru1−xMxO2 (M = Co, Ni, Zn) electrocatalysts, with calculated limiting potentials (UL) of ≈0.60 V.26 Following this, various strategies have been implemented in the design of metal oxide ORR catalysts. For instance, nano-structuring of metal oxides as in TiO2 nanotubes and nanoparticles or loading metal oxides such as Ni-based layered double hydroxide and CeO2 on a conductive, carbon support.22–25 These approaches have led to highly active (UL ≈ 0.40 V) and selective (FE > 95%) metal oxide-based electrocatalysts.
Unfortunately, metal oxides suffer from poor long-term stability in the 2e-ORR and WOR that partly originates from degradation by hydrogen peroxide. For example, the 2e-ORR over transition metal (TM) porphyrin catalysts have demonstrated to be both highly active, with overpotentials ≈0.4 V, and selective towards H2O2 production.27 However, under working cell conditions, H2O2 ceases to evolve past the ∼100 hour mark. This has been shown to originate from the degradation of the TM catalytic active site upon exposure to H2O2; ultimately resulting in an inactive catalyst system.28,29 To circumvent such stability issues, noble metal-based catalysts, resistant to H2O2 degradation, have been explored. To date, the state-of-the-art 2e-ORR electrocatalysts are composed of Pt- and Pd–Hg alloys,30,31 which are both costly materials. Thus, in search of a catalyst material that is well-suited for both the 2e-WOR and ORR, alternate approaches in catalyst development must be implemented.
Single atom catalysts (SACs) have attracted considerable interests as an alternate approach for improving catalyst activity, selectivity, and stability.32–34 When contrasted against their bulk counterparts, SACs benefit from low coordinated, isolated, catalytic active sites, unique electronic properties, and increased active surface area.35,36 Further, the efficiency of the single atom active site lowers the required metal loading, making them a financially viable material. While SACs have shown to be useful in the 2e-ORR due to their high activity,37–40 they have not been the focus of research as catalysts for the 2e-WOR.
Herein, we systematically analyse the catalytic properties of a series of metals anchored on a SnO2 support (M:SnO2) towards both the 2e-ORR and 2e-WOR. Our particular attention to SnO2 stems from the fact that it provides a low cost, corrosion resistant support.41,42 Additionally, SnO2 is stable across a wide range of pH and potential values making it a promising candidate for single atom catalysts.43 In this work, we aim at merging the high catalytic activity of SACs using an economical and stable SnO2 support, with the greater achievable H2O2 yields that can be attained via bifunctional systems.
Using density functional theory (DFT) calculations, we investigate the activity, selectivity, and stability of M:SnO2 catalysts towards H2O2 production. We show that Mn, Ti, and Fe embedded in the SnO2 support were both active and selective 2e-WOR catalysts, whereas W:SnO2 was found to be a promising SAC for the 2e-ORR.
Results and discussion
To model the proposed SnO2-based SACs, a surface Sn atom of a rutile SnO2 (110) structure was substituted with a single metal atom (Ru, V, Co, Pd, Pt, Ni, Mo, Ir, Cr, Mn, Fe, W, Ti, Sb, Nb, or Ta) (Fig. 1). The substitution resulted in a 25% surface metal loading and 6.25% bulk, relative to Sn atoms. We chose to study the 25% doping concentration because that is the lowest, we could afford in the simulation to balance the computational time and cost considering the large number of surfaces that we examined. This is also in line with previous reports44 and has been frequently used in the computational community to study the effect of single atoms doped in oxides. Higher concentrations to 50% or full mono layer would mimic an oxide over layer model on SnO2 which will likely show different trends in activity and selectivity but are not the scope of this study. After geometry optimization, minimal re-arrangement of the Mn:SnO2 surfaces, compared to that of bare SnO2, was observed. The ORR and WOR were carried out over two different surface-active sites, atop the single metal atom and on an adjacent Sn site.
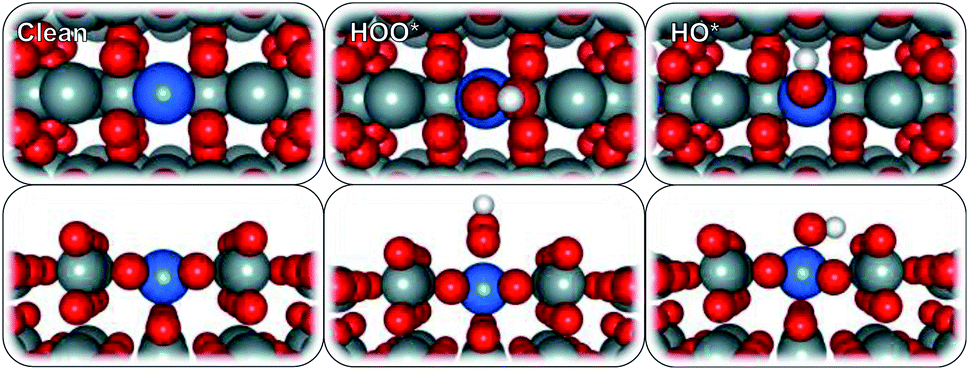 | ||
| Fig. 1 Top (top) and side (bottom) view of SnO2-supported single metal atom surfaces with adsorbed reaction intermediates (atoms in grey = Sn, red = oxygen, white = hydrogen, blue = Sn, Ru, V, Co, Pd, Pt, Ni, Mo, Ir, Cr, Mn, Fe, W, Ti, Sb, Nb, or Ta). | ||
Stability of SnO2-supported single metal atoms
We begin by investigating the stability of the single metal atoms embedded in the surface of SnO2. As discussed above, SnO2's tolerance to a wide range of pH values and applied potentials43 coupled with the material's low cost makes it a superb candidate for a bifunctional catalysts system. Additionally, the stability of the metal-support interaction plays a key role in the performance of SACs. A weak interaction may result in dissolution, diffusion, or agglomeration of the single metal atoms into a more thermodynamically preferred state, such as large nanoclusters.36 This would ultimately render an inactive catalyst. Formation energies of SAC systems and stability against dissolution under electrochemical ORR and WOR conditions were thus calculated (see ESI for details†). Fig. 2(A) displays the dissolution potential as a function of formation energy for the studied single metal atoms anchored in SnO2. The horizontal lines at U = 1.76 V and 0.70 V represent the standard potentials for the 2e-WOR and ORR, respectively. The vertical dashed line displays ΔEform = 0.0 eV, the upper limit of the stability of the M:SnO2 systems.
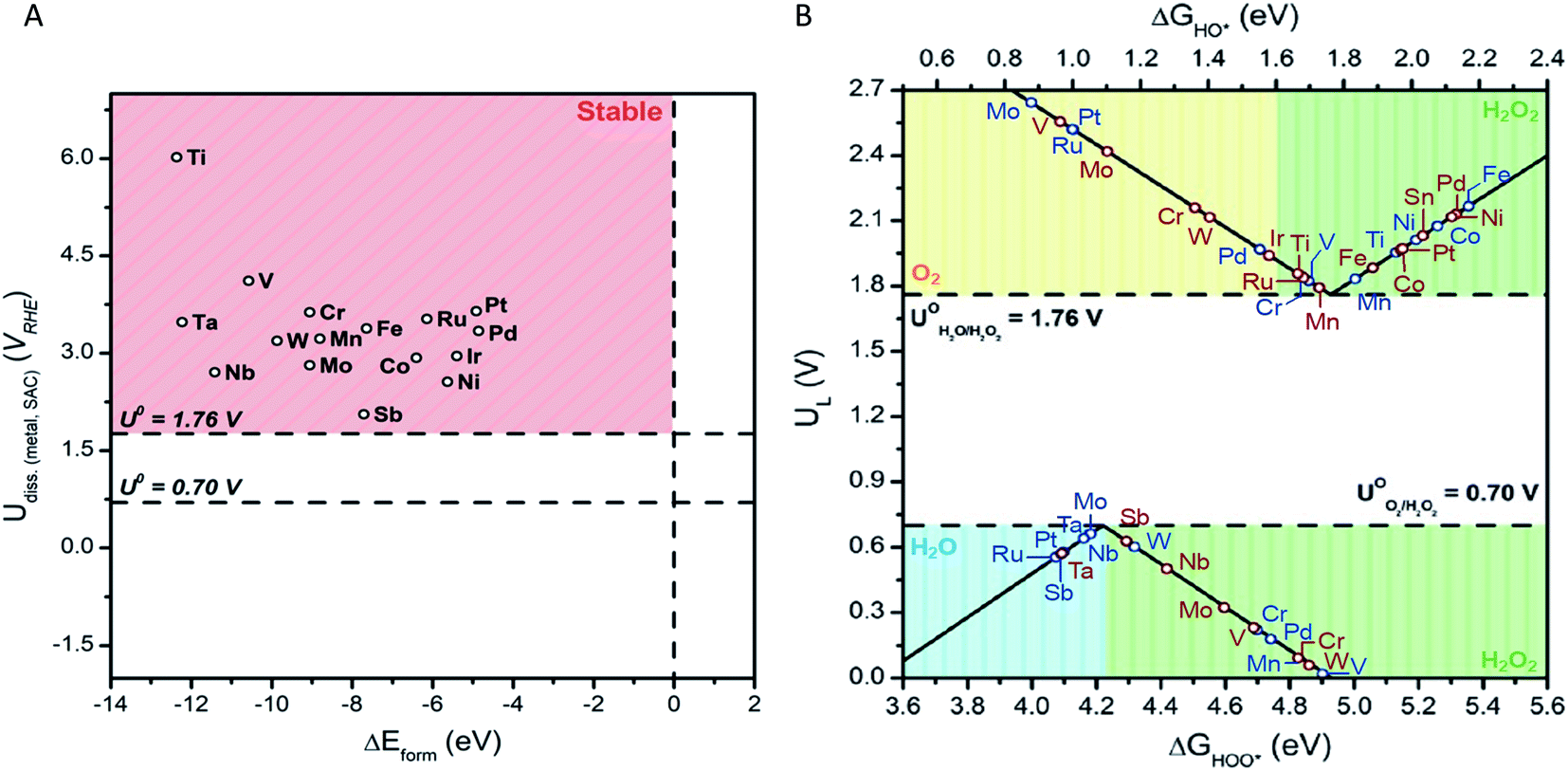 | ||
| Fig. 2 (A) Dissolution potentials vs. formation energies of single metal atom in SnO2. (B) Activity volcano plot for 2e-ORR (bottom) and 2e-WOR (top) for a variety of single metal atoms anchored on the surface of SnO2. Green, blue, and yellow shaded areas represent regions with high selectivity towards H2O2, H2O, and O2, respectively. Blue data points show activity of the catalyst when *OH or *OOH reaction intermediates are adsorbed on the surface of the single metal site, whereas red data points show activity of those adsorbed on the Sn site. | ||
The smaller the ΔEform value, the more energetically favoured the substitution of a surface Sn atom is. The SnO2-supported single metal systems chosen for this study all show negative ΔEform values. This is an indication that all of the studied metals form stable complexes when anchored on the surface of SnO2. Further, we find that the stability of the SACs system is influenced by the electronegativity difference between the single metal and the nearby Sn support atom (ESI – Fig. 1†). This correlation is in-line with previous reported SACs.45,46 Enhancement of catalyst stability can be obtained by increasing the electronegativity difference between Sn and single metal atom.
Dissolution potentials become relevant for the stability of the electrocatalyst systems under WOR and ORR operating conditions. The SnO2-supported SAC is considered stable if its dissolution potential is more positive than the corresponding 2e-ORR and WOR standard potentials. Dissolution potentials for the selected SACs were calculated to range between 2.62 to 6.05 V vs. RHE. This is well above 1.76 V and 0.7 V, demonstrating that the SnO2–metal systems are stable under working potentials.
Because the supported single metals may be prone to aggregation, we evaluated the cohesive energy of the metal bulk, Ecoh.47 The stability of the SAC against aggregation is determined by comparing Ecoh with ΔEform. If the calculated ΔEform is less than that of the Ecoh, the metal is stable against aggregation.48 We plot the difference between ΔEform and Ecoh for all the studied metals (ESI – Fig. 2†). The calculated values are all largely negative suggesting that the single metal has a stronger interaction with the SnO2 support than with neighbouring metal atoms. Further ruling out the possibility of aggregation of the single metal atoms after the M:SnO2 catalyst has been formed.
Overall, our stability calculations demonstrate that (1) all chosen M:SnO2 SACs form stable structures; (2) they are resistant to dissolution at minimum potentials required for both the 2e-WOR and ORR; and (3) stability is tuneable using SnO2 support.
We note that H2O2 is an oxidizing agent that may impact the long-term stability of the M:SnO2. Unfortunately, it is not trivial to estimate this type of stability using sole DFT calculations and the results of such calculations won't be conclusive. We think that H2O2 would increase the possibility of oxidizing the single metal atoms by splitting to OH radical and providing a local oxygen rich environment. This likely promotes oxidation of the SAC with strong tendency to bind oxygen, suppressing their activity. To verify this effect, an experimental study will be required which is out of the scope of this work.
Electrocatalytic activity and selectivity of SnO2-supported single metal atoms
Although stability requirements are met, electrocatalysis using metal oxides, such as SnO2, is often limited by poor electrical conductivity. To bypass this issue, strategies such as using dopant atoms or varying the lattice oxygen vacancies have been proposed. To this end, SnO2 has found applications in hydrogen production, fuel cells and ORR via metal doping and single metal atom systems.49–52The conversion of H2O and O2 to H2O2 is composed of a series of proton-coupled electron transfer reaction. The WOR involves the following:
| * + H2O → OH* + (H+ + e−) | (1) |
| OH* + H2O → H2O2 + (H+ + e−) + * | (2) |
| * + O2 + (H+ + e−) → OOH* | (3) |
| OOH* + (H+ + e−) → H2O2 + * | (4) |
We use the well-established volcano framework that follows Sabatier principle relating the binding free energy of intermediates to limiting potential, UL (i.e., the lowest (for 2e-WOR) and the highest (for 2e-ORR) potential at which all reaction steps are downhill in free energy). ΔGOOH or ΔGOH descriptors and UL were used to construct activity volcano maps for 2e-ORR and 2e-WOR over M:SnO2 SACS (Fig. 2(B)). The right and left leg of the 2e-WOR is determined by reaction free energies of the eqn (1) and (2), respectively. For the 2e-ORR volcano the right and left side of the volcano is related to the reaction free energies of the eqn (3) and (4), respectively. Based on the binding strength of key intermediates (OOH* and OH*) the volcano plot is divided into unique regions (shaded areas). These coloured partitions indicate selectivity towards H2O2 and other by-products from competing reactions.
Focusing on the 2e-ORR, an “ideal” catalyst would bind OOH* with an adsorption energy of ∼4.22 eV, with zero required overpotential to drive the reaction. However, deviations from ΔGOOH* = 4.22 eV (peak of the volcano) due to either stronger or weaker binding of OOH* will increase overpotential requirements, that is, lower electrocatalytic activity. Further, catalyst materials that weakly bind oxygen intermediates to the surface preserve the O–O bond during oxygen reduction. This ensures a high product selectivity towards H2O2 over H2O at the cost of low activity. For instance, the volcano analysis suggests that Mo, Nb, and Sb are highly active SACs for the ORR with UL of 0.66, 0.66, 0.57 V, respectively (Fig. 2(B)). However, OOH* binding energies of 4.18, 4.18, and 4.09 eV for Mo, Nb, and Sb, places these SACs in the blue region (ΔGOOH* < 4.22 eV) in which H2O is the preferred ORR product. On the other hand, SnO2-supported SACs that are well into the green region (inside of which H2O2 is produced) such as Mn (UL = 0.09 V), suffer from low catalytic activity. Turning our attention to the 2e-WOR, activity of the electrocatalyst is greatly influenced by the adsorption energy of OH* intermediate. Strong binding of the OH* species to the catalyst surface limits the oxidation of OH* to H2O2, while weak binding is associated with limited cleavage of HO–H bond (in H2O) to form OH* intermediate. Moreover, to ensure the suppression of competing WOR pathways, OH* adsorption energy must be greater (or equal) to 1.76 eV. Stronger OH* binding leads to further oxidation (OH* → O*) through the 4e-pathway, subsequently producing O2. This trend is illustrated in the WOR volcano plot (Fig. 2(B)). Without the addition of a single metal atom, SnO2 shows a UL of 2.03 V, in good agreement with previous reports.18 The introduction of single TM atoms such as Mn and Ti decrease limiting potentials (i.e., lower overpotential requirements) to 1.79 and 1.86 V, accordingly. To understand the reason behind the change in the activity, we plotted the density of states for pure SnO2 and example of Mn:SnO2 (ESI – Fig. 4†). This analysis indicates that the presence of Mn single atom increases the density of states at the Fermi level of SnO2 substrate originating from a high number of unoccupied d-electrons in Mn. These additional states increase the activity of inert SnO2 substrate and result in an optimum interaction with reaction intermediates.
With the above in mind, we plot the activity volcano using the lowest ΔG values for each M:SnO2 SACs with adsorbed oxygen intermediates (Fig. 3). From the series, W active site on the W:SnO2 SAC was found to be the most promising system for the 2e-ORR with a limiting potential of 0.6 V and ΔGOOH of 4.32 eV. Contrary to the ORR, the 2e-WOR was found to prefer the Sn site over the single metal site. Incorporating Mn, Ti, and Fe into the M:SnO2 SACs alters the catalytic activity of the adjacent Sn site. This resulted in a decrease in limiting potential, compared to that of SnO2 (2.03 V), of 1.79 V, 1.86 V and 1.88 V for Mn:SnO2, Ti:SnO2, and Fe:SnO2, respectively. Lastly, our results reveal the benefit of integrating these SnO2-supported single metal atom catalysts into a symmetrical cell. A perfect system producing H2O2via the 2e-ORR at the cathode and 2e-WOR at the anode requisites a 1.06 V standard cell potential. Incorporating a W:SnO2 SAC cathode and a Mn:SnO2 anode would require a minimum theoretical cell potential of 1.13 V, whereas an Fe:SnO2 anode yields 1.28 V, demonstrating only slight deviations from the ideal system.
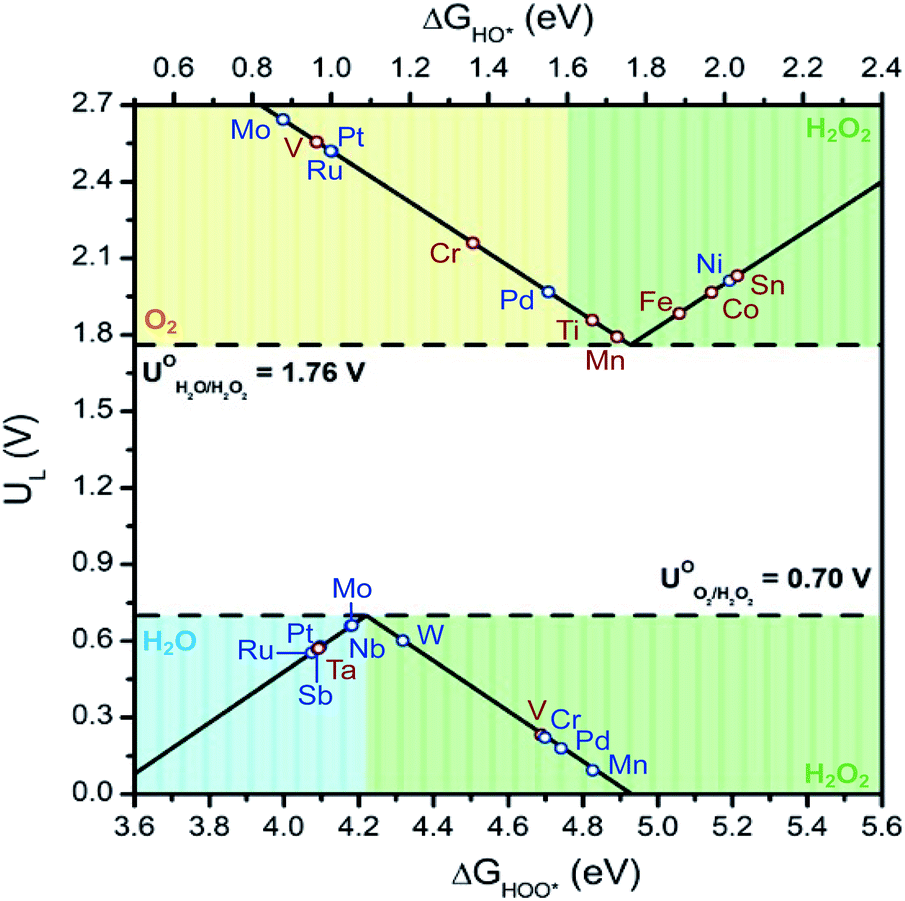 | ||
| Fig. 3 Activity volcano plot for 2e-ORR (bottom) and WOR (top) for the most stable M:SnO2 configurations. The M:SnO2 structure with the lowest ΔG between the adsorption of an oxygen intermediate on either the Sn site or on the single metal site, was selected as the most stable configuration. Green, blue, and yellow shaded areas represent regions with high selectivity towards H2O2, H2O, and O2, respectively. Blue data points show activity of the catalyst when *OH or *OOH reaction intermediates are adsorbed on the surface of the single metal site, whereas red data points show activity of those adsorbed on the Sn site. | ||
Conclusions
In closing, we explored the catalytic performance of SnO2-supported single metal atoms for the 2e-ORR and 2e-WOR. Through DFT calculations, we identified W:SnO2 to be a promising candidate for the 2e-ORR while Ti:SnO2, Fe:SnO2, and Mn:SnO2 for the 2e-WOR. Although the catalysts explored in this work do not outperform known, state-of-the-art, materials, we would like to emphasize the following: (1) SnO2 provides a stable cost-effective alternative to expensive materials such as Pt, and Ir; (2) the benefits of SACs can be accessed through a SnO2 support, without the stability issues often experienced with SACs; and (3) a common support material facilitates fabrication of bifunctional catalyst. Most importantly, the catalytic performance of the SnO2-supported SACs coupled with higher theoretical H2O2 yields obtained through the paired 2e-WOR and 2e-ORR brings us one step closer to completely shifting away from the costly, environmentally harmful AO process.
Methods
Computational details
All electronic structure calculations were performed using QUANTUM ESPRESSO program package. Atomic Simulation Environment (ASE) was used to handle the simulation. The electronic wavefunctions were expanded in plane waves up to a cutoff energy of 500 eV, while the electron density is represented on a grid with an energy cutoff of 5000 eV. Core electrons were approximated with ultrasoft pseudopotentials. The revised Perdew–Burke–Ernzerhof (RPBE) functional was used to describe the chemisorption properties. Rutile SnO2 and M:SnO2 surfaces were modelled using a 2 × 2 unit cell with a total of four layers for the (110) facet. The top two layers as well as adsorbates were allowed to relax while the bottom two layers were fixed at their bulk positions during the structure optimization. All calculations were done spin-polarized. A vacuum of about 18 Å was used to decouple periodic replicas along the z-direction. The Brillouin zone was sampled with (4 × 4 × 1) Monkhorst–Pack k-points. The ZPE and entropy changes were obtained from Man et al., and added to the calculated electronic energies using ΔG = ΔE + ΔZPE − TΔS.53,54 It is known that the DFT calculations for electrocatalysis need to accurately account for the effect of water on the adsorption energies of reaction intermediates. In a previous study, we explicitly studied the effect of water networks on rutile oxides.55 We showed that the interaction between water and oxide surface becomes weaker by adding more water and going to weak binding surfaces such as SnO2. We also found that the presence of the water layers has little impact on the predicted catalytic activities. Thus, we concluded that our DFT calculations are sufficiently accurate without inclusion of solvation corrections. The presented theoretical results in this work are based on thermodynamic analysis which have shown to be powerful in providing insights on the nature of active sites and guiding the design and optimization of various catalysts.56 We didn't consider the kinetic barriers in our analysis because a previous study by Tripković et al. shows that kinetic barriers for transferring proton to oxygen intermediates such as OOH*, and OH* is very small (∼0.2 eV).57 On the basis of this understanding the thermodynamic predicted activity volcano has been shown to be in close agreement with the predicted kinetic activity volcano.58 This analysis showed that there is a close connection between the kinetic and thermodynamic formulations for reactions involving oxygen intermediates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. J. V. and S. S. acknowledge the support from the University of Calgary's Canada First Research Excellence Fund Program, the Global Research Initiative in Sustainable Low Carbon Unconventional Resources. This research was enabled in part by support provided by computational resource at the University of Calgary (http://www.rcs.ucalgary.ca) and Compute Canada (http://www.computecanada.ca). The authors gratefully acknowledge Prof. Jens Nørskov for scientific advice and discussion.References
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- K. Wang, J. Huang, H. Chen, Y. Wang and S. Song, Chem. Commun., 2020, 56, 12109–12121 RSC.
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS.
- K. B. Tan, G. Zhan, D. Sun, J. Huang and Q. Li, J. Mater. Chem. A, 2021, 9, 5197–5231 RSC.
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1–21 Search PubMed.
- X. Shi, Y. Zhang, S. Siahrostami and X. Zheng, Adv. Energy Mater., 2018, 8, 1–9 Search PubMed.
- K. Fuku, Y. Miyase, Y. Miseki, T. Funaki, T. Gunji and K. Sayama, Chem.–Asian J., 2017, 12, 1111–1119 CrossRef CAS.
- C. Zhu, M. Zhu, Y. Sun, Y. Zhou, J. Gao, H. Huang, Y. Liu and Z. Kang, ACS Appl. Energy Mater., 2019, 2, 8737–8746 CrossRef CAS.
- S. Siahrostami, S. Jimenez Villegas, A. H. Bagherzadeh Mostaghimi, S. Back, A. B. Farimani, H. Wang, K. A. Persson and J. Montoya, ACS Catal., 2020, 10, 7495–7511 CrossRef CAS.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 17–19 Search PubMed.
- A. Izgorodin, E. Izgorodina and D. R. MacFarlane, Energy Environ. Sci., 2012, 5, 9496–9501 RSC.
- S. R. Kelly, X. Shi, S. Back, L. Vallez, S. Y. Park, S. Siahrostami, X. Zheng and J. K. Nørskov, ACS Catal., 2019, 9, 4593–4599 CrossRef CAS.
- X. Shi, S. Siahrostami, G. L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 1–12 CrossRef CAS.
- S. Y. Park, H. Abroshan, X. Shi, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2019, 4, 352–357 CrossRef CAS.
- V. Viswanathan, H. A. Hansen and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 4224–4228 CrossRef CAS PubMed.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 2100076 CrossRef CAS.
- J. F. Carneiro, R. S. Rocha, P. Hammer, R. Bertazzoli and M. R. V. Lanza, Appl. Catal., A, 2016, 517, 161–167 CrossRef CAS.
- E. C. Paz, L. R. Aveiro, V. S. Pinheiro, F. M. Souza, V. B. Lima, F. L. Silva, P. Hammer, M. R. V. Lanza and M. C. Santos, Appl. Catal., B, 2018, 232, 436–445 CrossRef CAS.
- V. S. Pinheiro, E. C. Paz, L. R. Aveiro, L. S. Parreira, F. M. Souza, P. H. C. Camargo and M. C. Santos, Electrochim. Acta, 2018, 259, 865–872 CrossRef CAS.
- J. Huang, J. Chen, C. Fu, P. Cai, Y. Li, L. Cao, W. Liu, P. Yu, S. Wei, Z. Wen and J. Li, ChemSusChem, 2020, 13, 1496–1503 CrossRef CAS PubMed.
- G. Cabello, R. A. Davoglio and E. C. Pereira, J. Electroanal. Chem., 2017, 794, 36–42 CrossRef CAS.
- M. A. Ghanem, A. M. Al-Mayouf, M. N. Shaddad and F. Marken, Electrochim. Acta, 2015, 174, 557–562 CrossRef CAS.
- D. F. Abbott, S. Mukerjee, V. Petrykin, Z. Bastl, N. B. Halck, J. Rossmeisl and P. Krtil, RSC Adv., 2015, 5, 1235–1243 RSC.
- P. Gouérec and M. Savy, Electrochim. Acta, 1999, 44, 2653–2661 CrossRef.
- H. Schulenburg, S. Stankov, V. Schünemann, J. Radnik, I. Dorbandt, S. Fiechter, P. Bogdanoff and H. Tributsch, J. Phys. Chem. B, 2003, 107, 9034–9041 CrossRef CAS.
- C. W. B. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- J. Kim, C. W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1–8 Search PubMed.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569–588 RSC.
- Y. Chen, Z. Huang, Z. Ma, J. Chen and X. Tang, Catal. Sci. Technol., 2017, 7, 4250–4258 RSC.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- S. K. Sahoo, Y. Ye, S. Lee, J. Park, H. Lee, J. Lee and J. W. Han, ACS Energy Lett., 2019, 4, 126–132 CrossRef CAS.
- J. Gao, H. bin Yang, X. Huang, S. F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef.
- K. Sasaki, F. Takasaki, Z. Noda, S. Hayashi, Y. Shiratori and K. Ito, ECS Trans., 2010, 33, 473–482 CrossRef CAS.
- D. S. Rivera Rocabado, T. Ishimoto and M. Koyama, SN Appl. Sci., 2019, 1, 1–15 CAS.
- D. R. Groot and J. A. N. Van Der Linde, J. South. Afr. Inst. Min. Metall., 2009, 109, 701–707 Search PubMed.
- M. García-Mota, A. Vojvodic, H. Metiu, I. C. Man, H. Y. Su, J. Rossmeisl and J. K. Nørskov, ChemCatChem, 2011, 3, 1607–1611 CrossRef.
- Y. Wang, E. Song, W. Qiu, X. Zhao, Y. Zhou, J. Liu and W. Zhang, Prog. Nat. Sci.: Mater. Int., 2019, 29, 256–264 CrossRef CAS.
- K. Yang, Y. Dai, B. Huang and M. H. Whangbo, Chem. Mater., 2008, 20, 6528–6534 CrossRef CAS.
- J. Hwang, S. H. Noh and B. Han, Appl. Surf. Sci., 2019, 471, 545–552 CrossRef CAS.
- Z. Yu, H. Xu and D. Cheng, Adv. Phys.: X, 2021, 6, 1905545 Search PubMed.
- Y. Senoo, K. Taniguchi, K. Kakinuma, M. Uchida, H. Uchida, S. Deki and M. Watanabe, Electrochem. Commun., 2015, 51, 37–40 CrossRef CAS.
- W. Dong, Y. Zhang, J. Xu, J. W. Yin, S. Nong, C. Dong, Z. Liu, B. Dong, L. M. Liu, R. Si, M. Chen, J. Luo and F. Huang, Cell Rep. Phys. Sci., 2020, 1, 100026 CrossRef.
- K. Stöwe and M. Weber, Zeitschrift für anorganische und allgemeine Chemie, 2020, 646, 1470–1480 CrossRef.
- R. Mohamed, T. Binninger, P. J. Kooyman, A. Hoell, E. Fabbri, A. Patru, A. Heinritz, T. J. Schmidt and P. Levecque, Catal. Sci. Technol., 2018, 8, 2672–2685 RSC.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. Siahrostami and A. Vojvodic, J. Phys. Chem. C, 2015, 119, 1032–1037 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef.
- V. Tripković, E. Skúlason, S. Siahrostami, J. K. Nørskov and J. Rossmeisl, Electrochim. Acta, 2010, 55, 7975–7981 CrossRef.
- H. A. Hansen, V. Viswanathan and J. K. NØrskov, J. Phys. Chem. C, 2014, 118, 6706–6718 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07562d |
| This journal is © The Royal Society of Chemistry 2022 |