
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurfactant-chaperoned donor–acceptor–donor NIR-II dye strategy efficiently circumvents intermolecular aggregation to afford enhanced bioimaging contrast†
Tianyang
Han
ab,
Yajun
Wang
ab,
Jiajun
Xu
ab,
Ningning
Zhu
ab,
Lang
Bai
ab,
Xiangping
Liu
ab,
Bin
Sun
ab,
Chenlong
Yu
 ab,
Qinglun
Meng
a,
Jiaqi
Wang
a,
Qi
Su
a,
Qing
Cai
c,
Kenneth S.
Hettie
d,
Yuewei
Zhang
e,
Shoujun
Zhu
*ab and
Bai
Yang
ab
ab,
Qinglun
Meng
a,
Jiaqi
Wang
a,
Qi
Su
a,
Qing
Cai
c,
Kenneth S.
Hettie
d,
Yuewei
Zhang
e,
Shoujun
Zhu
*ab and
Bai
Yang
ab
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, 130012, P. R. China. E-mail: sjzhu@jlu.edu.cn
bJoint Laboratory of Opto-Functional Theranostics in Medicine and Chemistry, First Hospital of Jilin University, Changchun, 130021, P. R. China
cHospital of Stomatology, Jilin University, Changchun, 130021, P. R. China
dMolecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University School of Medicine, Stanford, California 94305, USA
eSchool of Chemistry and Pharmaceutical Engineering, Jilin Institute of Chemical Technology, Jilin, 132022, P. R. China
First published on 2nd November 2022
Abstract
Fluorescence emission in the near-infrared-II (NIR-II) optical window affords reduced autofluorescence and light scattering, enabling deep-tissue visualization for both disease detection and surgical navigation. Small-molecule NIR-II dyes are preferable for clinical bioimaging applications, as the flexibility in their molecular synthesis allows for precise control of their optical and pharmacokinetic properties. Among the various types of dye, donor–acceptor–donor-based (D–A–D) dyes demonstrate exceptional photostability, whereas the frequently used PEGylation approach does not keep their intrinsic brightness enough in water environments due to their inherent effect of self-assembly. Here, we demonstrate that the commercially-available surfactants can serve as a dispersant to prevent molecular aggregation of PEGylated D–A–D dyes. Due to the favorable energetics for co-assembly between D–A–D dyes and surfactants, the formed surfactant-chaperoned dye strategy dramatically increases dye brightness. Accordingly, this effect provides remarkably improved performance for in vivo bioimaging applications. In parallel, we also investigate the D–A–D dye uptake and signal enhancement properties in the liver of murine models and demonstrate that the lumen-lining Kupffer cells can potentially disassemble PEGylated D–A–D aggregates such that their inherent brightness is restored. This phenomenon is similar to the surfactant-chaperoned dye strategy and our investigations provide a positive addition to better use of the current NIR-II fluorophores, especially for visualizing high-brightness required events.
Introduction
In the context of in vivo near-infrared fluorescence (NIR) bioimaging, imaging with near-infrared-II (NIR-II) small-molecule dyes is an attractive approach because they afford increased penetration depth, low autofluorescence, and relatively low biotoxicity, all of which allows for acquiring images with enhanced contrast levels when compared to traditional fluorophores that emit fluorescence in the visible/NIR-I spectral region.1–15 As NIR-II fluorescence-emitting fluorophores typically have extended hydrophobic π-conjugated groups,16–20 the dyes require modification with additional hydrophilic moieties to impart water solubility and biological biocompatibility. The commonly used hydrophilic groups include alkyl sulfonates, alkyl carboxylates, and polyethylene glycol (PEG)21–29 moieties and their numerous derivatives. Although this strategy can successfully overcome the water-solubility barrier that these dyes encounter, the quantum yield (Φ) of these fluorophores is considerably reduced compared to their unmodified counterparts.30–32 Fluorescence quenching upon transitioning from the oil phase to the water phase for current NIR-II fluorophores remains a major obstacle to their successful translation into the clinic. This loss in brightness has been attributed to the phenomenon often referred to as the aggregation-caused quenching (ACQ)33 effect. Thus, investigating the fluorescence quenching mechanism of NIR-II fluorophores is essential to improving the imaging capability of current NIR-II fluorophores.34Organic synthesis has allowed for the development of a diverse range of NIR-II small-molecule dyes, wherein the quintessential fluorophore types are cyanine35 and donor–acceptor–donor-based (D–A–D) dyes.36–38 NIR-II fluorescence D–A–D dyes exhibit exceptional photostability, and thereby eliminate any concerns of photobleaching upon implementation.39 To improve brightness, the use of PEG moieties is commonly pursued, as they can serve as shielding units (S) that are capable of modulating the quantum yield (Φ) and pharmacokinetic properties of NIR-II fluorescence S–D–A–D–S dyes. Presumably, extensive PEGylation could alter the solubility of S–D–A–D–S dyes such that they are transformed into an amphiphilic molecule, which consequently inherently tend to self-assemble. Unfortunately, this unavoidable aggregation effect is a common issue for all previously reported NIR-II D–A–D dyes.40 The self-assembly quenching behavior of these dyes could occur even at very low concentrations.
Much effort has been put forth in attempts of developing and employing effective strategies that are capable of overcoming the intermolecular aggregation of current PEGylated S–D–A–D–S dyes. Here, we synthesized two S–D–A–D–S small-molecule dyes that serve as model systems for investigating the intermolecular assembly and the consequent quenching of their NIR-II fluorescence emission. Rational donor substitution enabled the improvement of the NIR-II quantum yield of S–D–A–D–S fluorophores. Commercial surfactants were found to efficiently prevent the aggregation/self-assembly of our prepared PEGylated S–D–A–D–S dyes. As such, surfactant-chaperoned S–D–A–D–S dyes demonstrated a remarkable enhancement in their quantum yields in aqueous solution and upon performing in vivo imaging. We also found that the commonly-observed liver uptake and signal enhancement upon intravenous administration of S–D–A–D–S dyes may originate from dye disassembly by liver-lining Kupffer cells (KCs).26,41–44 These investigations both provided (i) insight into the effect of self-assembly of PEGylated S–D–A–D–S dyes and (ii) the development of a facile and efficacious surfactant-chaperoned strategy to overcome the inherent issue of intermolecular aggregation. This strategy will fundamentally promote the imaging efficiency that NIR-II fluorophores provide by significantly improving their brightness in aqueous media.
Results and discussion
Design and synthesis of D–A–D molecules by rational donor substitution
The literature has reported that the S–D–A–D–S structure underlying many NIR-II fluorophores allows for exceptional photostability, thereby providing the potential for long-term visualization of biological events for extended-duration in vivo imaging applications.26 Recently, the alkyl isomerization of donor groups has attracted much attention.45 This idea of donor-specific modification to overall electron delocalization of acceptor electrons and further enhancing emission wavelength has also recently attracted the interest of researchers.46 However, such an adjustment also reduces the molecular twist angle. This may lead to less charge transfer character which emphasizes the very strong acceptor effect, eventually leading stronger tendency to assembly quenching. To further confirm these conjectures, we designed and synthesized a pair of S–D–A–D–S structural isomers (Fig. 1a), IR-FH4 (n-hexyl substituent at the 4C-position) (Scheme S1†), and IR-FH3 (n-hexyl substituent at the 3C-position) (Scheme S2†). These modifications provide us the ability to investigate the effect of placing an alkyl side chain at the 3C, and 4C-position of the thiophenes. Polyethylene glycol-2000 (PEG2000)-alkyne (terminal functionalized) was used to impart these two molecules with solubility in aqueous media via using a click-reaction scheme (Scheme S3†).26,47 According to gel exclusion chromatography (SEC) data, the average molecular weight (Mw) of PEGylated product was 5500, indicating that PEGylation did not occur quantitatively. The incomplete occurrence of this click reaction may be due to the steric hindrance between the backbone structure and the PEG chain. This phenomenon has also been confirmed in similar dye structures.3 Gaussian calculation revealed that IR-FH4 with alkyl groups substituted on 4C-position had a smaller optical band gap and ground state dihedral angle (Fig. 1b and c).31 Results show that the 4C-dye has a smaller dihedral angle in the ground state, indicating that the electronic structure of this structure is more delocalized. The dihedral angle between BBTD and thiophene in the 4C-dye is only 0.8°, resulting in a longer absorption, emission wavelength (Fig. S1a, b, d, e, g, h, j and k†), and higher fluorescence quantum yield (Fig. S1c, f, i, l, S2a–d and Table S1†). However, fluorescence spectra characterization revealed that IR-FH4P showed a 20-fold decrease in aqueous solution versus that of DMSO (Fig. 1d). And the absorption spectrum also shows a strong blue shift in aqueous solution versus that of DMSO (Fig. 1e).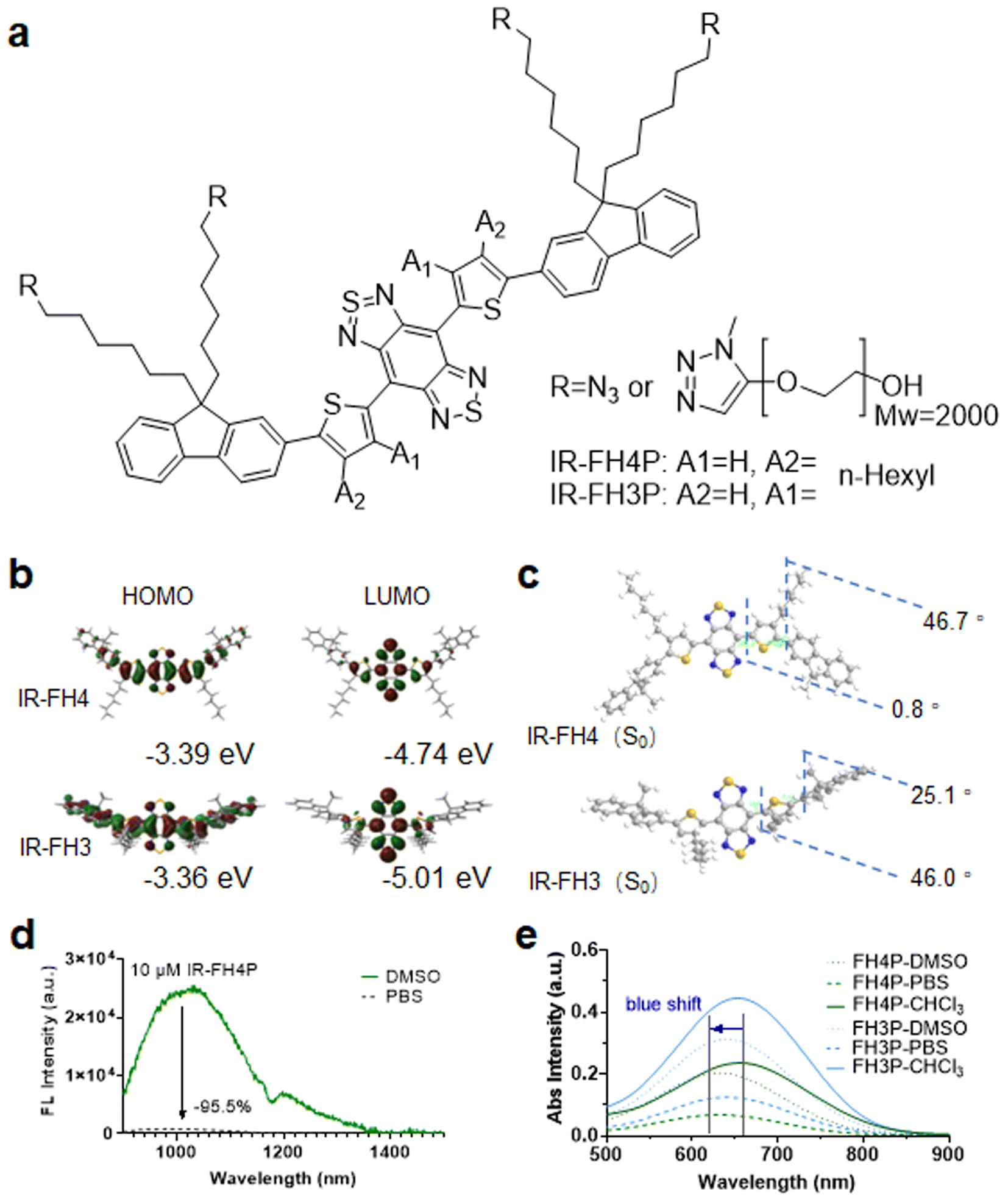 | ||
| Fig. 1 Chemical structure of S–D–A–D–S dyes and their photophysical properties. (a) Chemical structure of IR-FH4P and IR-FH3P. (b) Calculation of the optical bandgap for IR-FH4/IR-FH3 isomers using Gaussian (b31yp/6-31g(d)). (c) Calculation of dihedral angle for IR-FH4/IR-FH3 isomers through Gaussian (b31yp/6-31g(d)). PEG chains were replaced by a methyl group to simplify the calculation. (d) Fluorescence emission spectra of IR-FH4P (10 μM) in DMSO and PBS buffer. (e) Absorption spectra of IR-FH4P and IR-FH3P in DMSO, PBS, and CHCl3. | ||
Commercial surfactants efficiently conquered the inherent aggregation/assembly of PEGylated S–D–A–D–S dyes
Using transmission electron microscopy (TEM) characterization, we validated that IR-FH4P inherently formed relatively large nanoparticles (∼80 nm) (Fig. 2a) when dissolved in PBS buffer, consistent with the dynamic light scattering (DLS) data (Fig. S3a†). While the IR-FH3P shows a smaller size (∼55 nm) according to DLS data (Fig. S3b†). Conversely, it was well dispersed in organic solvents, such as the tested chloroform. Basically, IR-FH4P formed large spherical nanoparticles, resulting in fluorescence quenching in the aqueous environment. Even at very low concentrations, D–A–D fluorescent NIR dyes still form the aggregation/assembly of nanoparticles. However, the isomerization of the donor group of IR-FH4P amplifies this aggregation/assembly effect by reducing the structural twisting angle. This behavior was further confirmed by 1H NMR. The typical chemical shifts of IR-FH4P in a D2O and DMSO-D2O system substantially diminished with the increasing of the peak width, in contrast to the predicably accurate signal in CDCl3 (Fig. 2c). This phenomenon is in line with the previous self-assembly of macrocyclic molecules and aggregate-induced emission (AIE) molecules,48–52 indicating the aggregation feature of IR-FH4P. With a generalized understanding of the relationship between fluorescence quenching and molecular aggregation, it prompted us to resolve this pressing challenge via utilizing solubilizing technology such as a surfactant. Thus, we adopted a commonly used surfactant, Triton-X100 (Triton), to disassemble the IR-FH4P nanoparticles.53,54 DLS data verified that Triton-X100 chaperoned IR-FH4P underwent an intermediate state (large diameter in DLS test), and eventually changed into a disassembly state with a hydrodynamic diameter of ∼8 nm, similar to its fully dissolved state in the organic phase (Fig. 2e). Plotting the DLS size of seven cocktails of IR-FH4P and Triton-X100 at various concentration allowed us to identify the minimum Triton-X100 concentration (0.1% w/w) for completely disassembling the IR-FH4P in the aqueous solution (Fig. 2f). To further verify the feasibility of our strategy, concurrently, five common surfactants were employed to test for brightness enhancement. Consistent with our experimental design, we found that most of the tested surfactants, including the anionic, cationic, and non-ionic surfactants, can effectively enhance the NIR-II brightness of IR-FH4P, restoring the same brightness level as its organic-phase counterpart (Fig. 3a). Notably, the isomer IR-FH3P only exhibited 3-fold brightness enhancement when adding the same amount of Triton-X100, indicating the differences of solubility using surfactants for diverse S–D–A–D–S structures and that structure optimization is necessary (Fig. 3b). To investigate the disassembly mechanism of the surfactant and dye system, we tested the size evolution through a set of samples by IR-FH4P mixing with three typical surfactants (Fig. 3c). As expected, surfactant-chaperoned dyes preferentially formed co-assembled nanoparticles at lower surfactant concentration while when free of surfactant they essentially failed to self-assemble in the aqueous solution.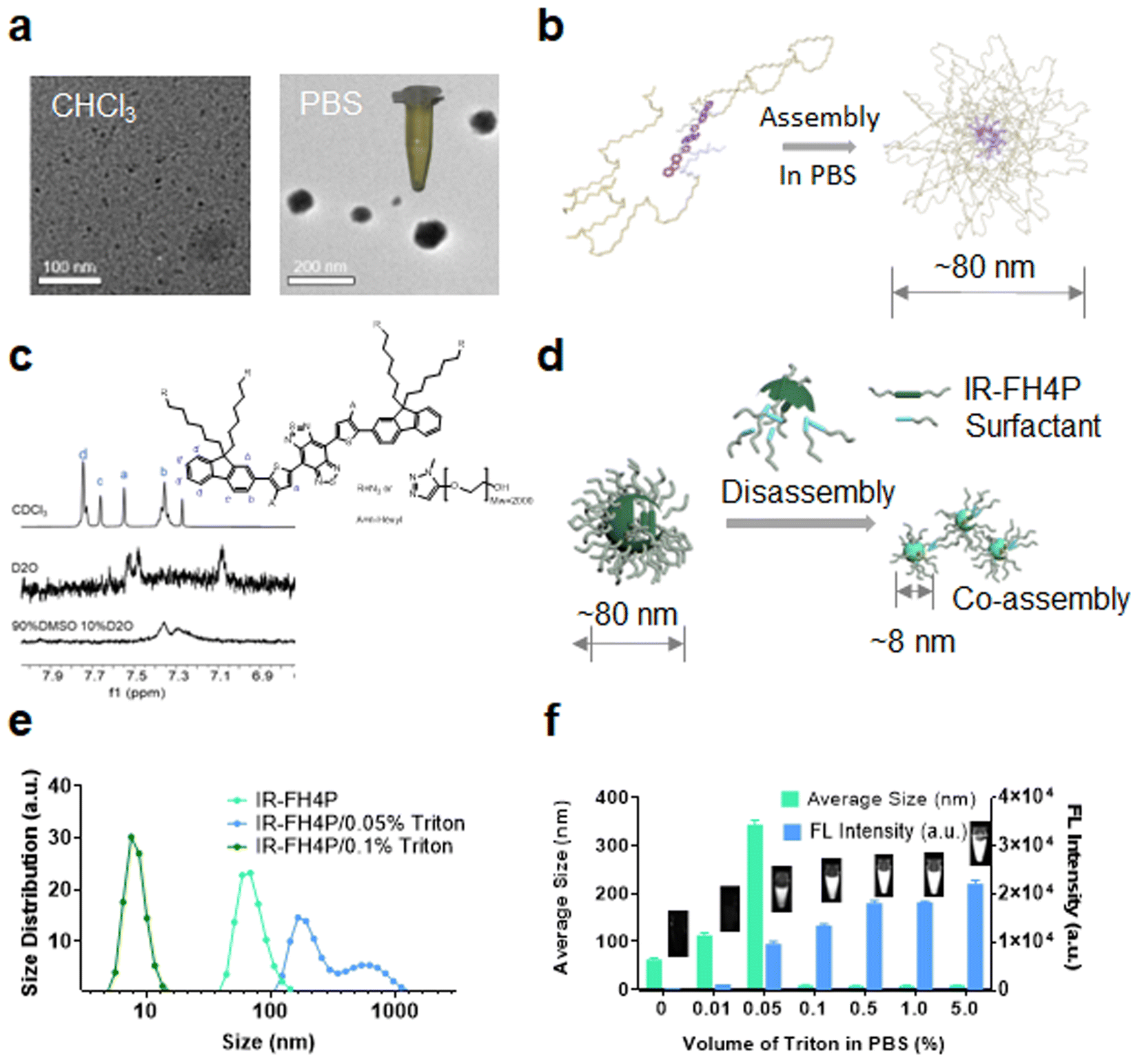 | ||
| Fig. 2 Self-assembly of IR-FH4P in PBS and disassembly of surfactant-chaperoned dye system. (a) TEM image of IR-FH4P (10 μM) in CHCl3 and PBS solution. (b) 3D model of IR-FH4P assembly in PBS. (c) 1H-NMR spectra of IR-FH4P (1 mg) in D2O, 90% DMSO-10% D2O, and CDCl3, respectively. (d) 3D model of IR-FH4P disassembly and subsequently co-assembly with surfactant molecules. (e) DLS size distribution of IR-FH4P (10 μM) with modified Triton-X100 concentration. (f) Dynamic light scattering (DLS) analysis and NIR-II brightness of IR-FH4P (10 μM) with changing Triton-X100 concentration. All data are expressed as mean ± SD. | ||
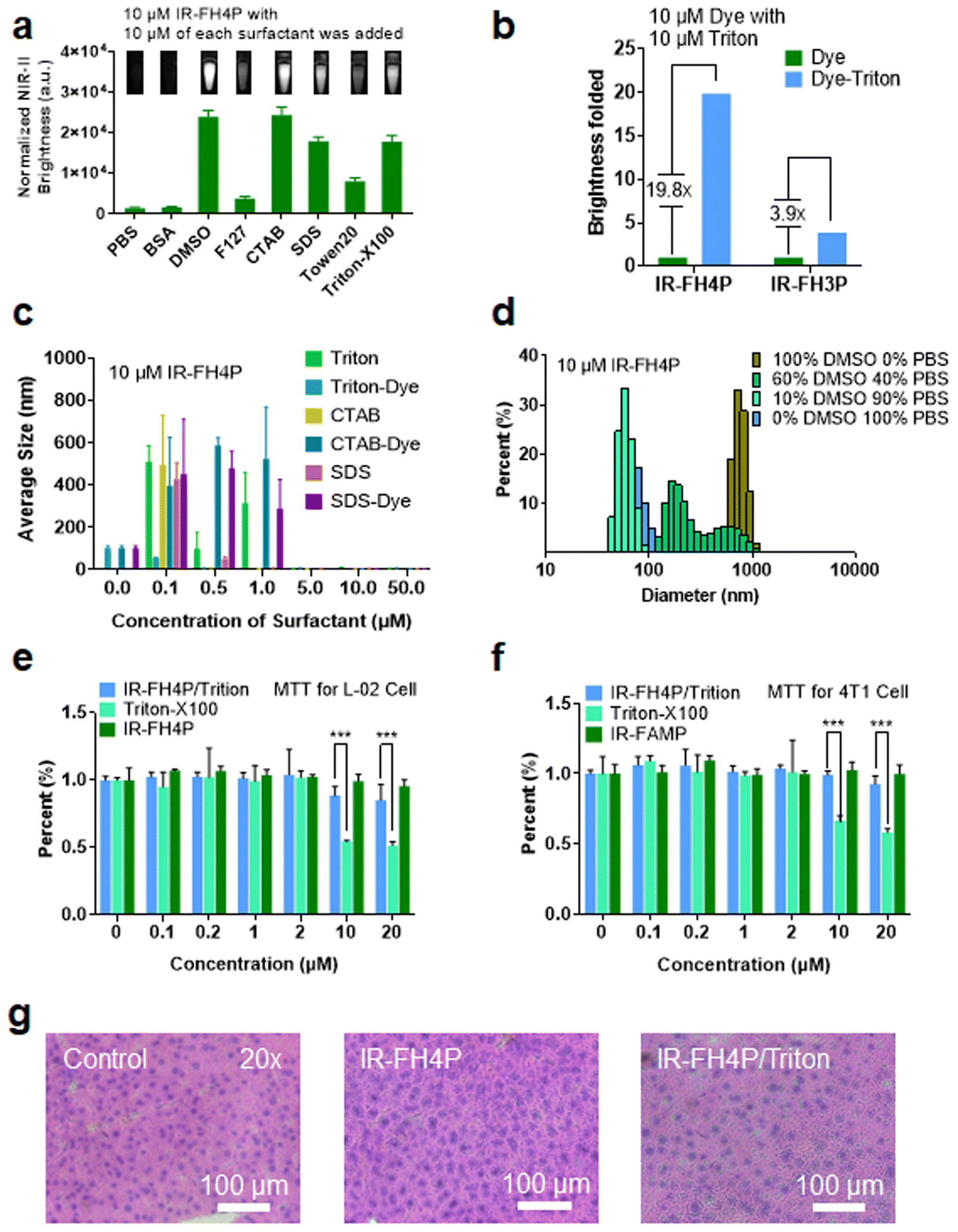 | ||
| Fig. 3 Commercial surfactant efficiently circumvented inherent aggregation/assembly effect(s) of IR-FH4P. (a) Fluorescence intensity of IR-FH4P (10 μM) by adding a set of five commercial surfactants in PBS, compared to the equivalent amount of IR-FH4P in PBS, BSA, and DMSO. (b) NIR-II brightness of IR-FH4P (10 μM) and IR-FH3P (10 μM) in pure PBS and pre-mixing Triton-X100 (10 μM) with PBS buffer. (c) DLS at different concentrations of Triton-X100, SDS, and CTAB with and without adding IR-FH4P (10 μM). (d) DLS of IR-FH4P (10 μM) in DMSO and DMSO/PBS mixture solutions. (e and f) Cell viability of IR-FH4P, Triton, and IR-FH4P/Triton-X100 co-assembly particles using L-02 and 4T1 cell lines. (g) Histological evaluation of the liver after intravenous administration of 24 hours of PBS, IR-FH4P, and IR-FH4P/Triton. All FL Intensity data are expressed as mean ± SD. The MTT data are expressed as mean ± SEM. | ||
Interestingly, a similar intermediate state of large-size distribution was observed in the mixed solution of different ratios of DMSO/PBS (from 10% DMSO to 100% DMSO, v/v). From the results of DLS (Fig. 3d) with the change of DMSO content in DMSO–PBS mixed solution, the particle size of the dye aggregates showed a similar trend in change to that of gradient-concentration surfactant systems. This result suggested that previous reports actually did not prevent the self-assembly and NIR-II signal quenching of S–D–A–D–S dyes by using DMSO to dissolve dyes as a stock solution and subsequently diluting them into PBS buffer even at low concentrations. For co-assembled small nanoparticles, toluene or THF rather than DMSO has been widely used in previous reports.55 We repeated the experiment with THF and found a similar trend of particle size variation in smaller ratios of THF/PBS (Fig. S3c†). As surfactants are generally toxic to living systems, we next conducted cell toxicity experiments using pure dye, pure surfactant, and a dye/surfactant mixture. To our surprise, the results showed that the toxicity of the dye/surfactant mixture was significantly lower than that of the single surfactant at the equivalent administered dosage (Fig. 3e and f). The dramatically reduced toxicity of the dye/surfactant probe further proved that surfactant and IR-FH4P co-assembled into a stable companion, thereby eliminating the toxicity concern for their in vivo application. To further confirm the low toxicity of this strategy, pathology analysis of liver from saline, IR-FH4P, and IR-FH4P/Triton administered cohorts of mice by hematoxylin and eosin (H & E) staining also verified that there was no obvious abnormal morphology (Fig. 3g).
Surfactant-chaperoned S–D–A–D–S dyes demonstrated remarkably-enhanced NIR-II brightness in vivo
After establishing the surfactant-chaperoned strategy for overcoming the issue of aggregation/self-assembly of S–D–A–D–S dyes, the next goal was to assess their ability to afford effective contrast levels in vivo. We first assessed whether bovine serum albumin (BSA) and mouse blood could disassemble the IR-FH4P, and we found that neither can enhance the NIR-II brightness significantly, indicating that there was no disassembly process. Next, 100 μM IR-FH4P/Triton-100X was diluted in PBS aqueous solution, BSA, and mouse whole blood from rat, respectively (10 μM), and no additional quenching phenomenon was observed before and after dilution (Fig. S3d†). Both IR-FH4P/Triton-100X and IR-FH3P/Triton-100X show a red shift in the absorption spectrum (Fig. S3e and f†). With the increase of dye concentration, IR-FH4P/Triton did not appear obvious aggregation quenching until 300 μM while the pure dyes were quenched at about 50 μM (Fig. S3g†). Both IR-FH4P and IR-FH4P/Triton-X100 show strong photostability under 808 nm laser excitation in PBS solution, BSA, and mouse blood (Fig. S4a and b†). To ensure the stability of the dye/surfactant complex, the as-prepared complex was stored for 14 days at room temperature. The changes in the absorption spectrum and fluorescence intensity were monitored by spectrophotometer spectra and InGaAs camera. No significant changes in optical properties were observed during 14 days of storage in a 4 °C environment. (Fig. S4c–f†).As our interest was to evaluate the enhanced brightness of surfactant-chaperoned dyes in vivo, we intravenously injected the surfactant-chaperoned IR-FH4P and surfactant-free IR-FH4P to compare their in vivo performance (Fig. 4a and b). The NIR-II signals of the IR-FH4P/Triton-X100-administered cohort were much higher than that of the surfactant-free IR-FH4P group within a short timeframe (approximately 12-fold enhancement at 60 s time point for the IR-FH4P/Triton-X100 model). The liver and skin/muscle signals of the surfactant-free IR-FH4P-injected cohort increased over time and reached an identical range to the IR-FH4P/Triton-X100 group at the 24 h time point (Fig. 4c–f). Long-term monitoring indicated that IR-FH4P could eventually be excreted out of the body within 7 days post-injection (Fig. S5†). Collectively, surfactant-chaperoned IR-FH4P provided a substantially enhanced brightness over a 24 h window, which may serve as a promising strategy in resolving the inherent intermolecular aggregation and fluorescence quenching issue for NIR-II fluorescence-emitting S–D–A–D–S dyes.
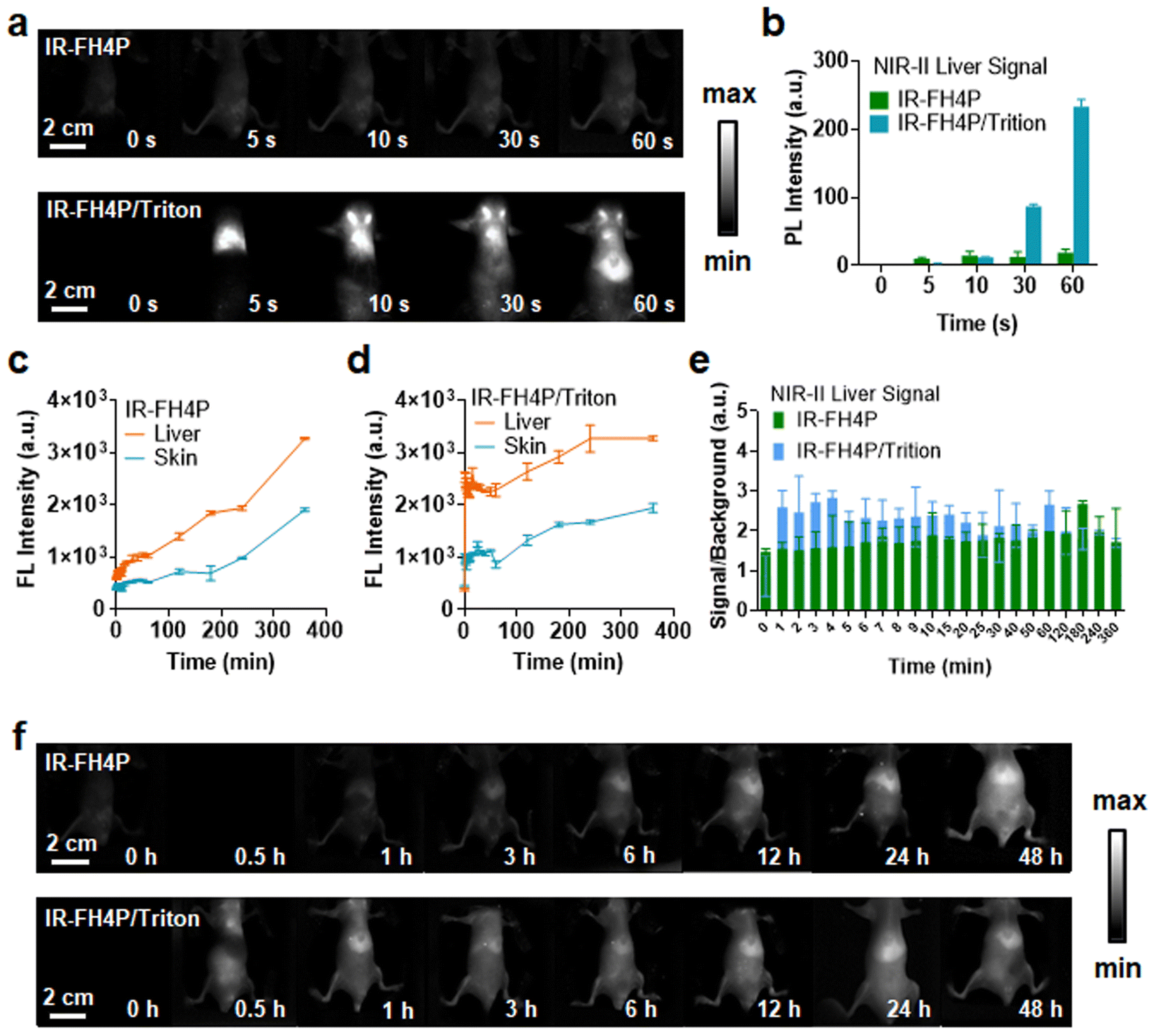 | ||
| Fig. 4 Surfactant-chaperoned IR-FH4P enhancement of NIR-II brightness of S–D–A–D–S dyes in vivo. (a) NIR-II fluorescence time points (imaging condition: 808 nm laser excitation with 88 mW cm−2 power density, 900 + 1100 nm long-pass filter) of mice in the supine position after intravenous administration of IR-FH4P and IR-FH4P/Triton-X100 taken within the short time window (injection dosage, 0.55 mg mL−1, 200 μL). (b) NIR-II fluorescence signal of the liver for IR-FH4P and IR-FH4P/Triton-X100 as a function of injected time points. (c) Selected time points from NIR-II imaging of mice in the supine position after intravenous administration of IR-FH4P. (d) Fluorescence intensity of both liver and skin for IR-FH4P/Triton-X100. (e) Comparison of the liver signal as a function of injection time points for surfactant-free IR-FH4P and IR-FH4P/Triton-X100 (10 μM Triton-100X). (f) Selected NIR-II imaging of IR-FH4P and IR-FH4P/Triton-X100-injected cohort of mice throughout long-term time points. All data are expressed as mean ± SD. | ||
Surfactant-chaperoned NIR-II fluorophore enabled high-performance tumor and lymph node imaging
After establishing the in vivo bioimaging ability of our surfactant-chaperoned dye strategy, we studied whether the IR-FH4P and IR-FH4P/Triton-X100 could provide high-quality lymph nodes (LNs) imaging in murine models.51 For this purpose, the shaved Balb/c mouse was set primely for popliteal and sacral LNs imaging. IR-FH4P/Triton-X100 mixture was intradermally injected (100 μM, 25 μL) on the right footpad while surfactant-free IR-FH4P of equivalent dye dosage was injected on the left footpad. As shown in Fig. 5a and b, very disparate NIR-II fluorescence intensities were apparently recorded, and roughly 3-fold brightness enhancement was quantitively plotted between using IR-FH4P/Triton-X100 and surfactant-free IR-FH4P in the first 30 minutes.56 We also studied whether the IR-FH4P/Triton-X100 dye maintained the D–A–D dyes' excellent photostability in LN imaging. Equivalent dosage of ICG (OD 808 nm = 0.5) in PBS and IR-FH4P/Triton-100X mixture were intradermally injected and compared through the same condition described above (Fig. 5c and d). The NIR-II signals of ICG injected left side quickly diminished in the first 30 minutes, while the LN signals of IR-FH4P/Triton-X100 injected on the right side remained consistent, providing potential for using IR-FH4P/Triton-X100 for long-term visualization of LN and image-guided surgical navigation.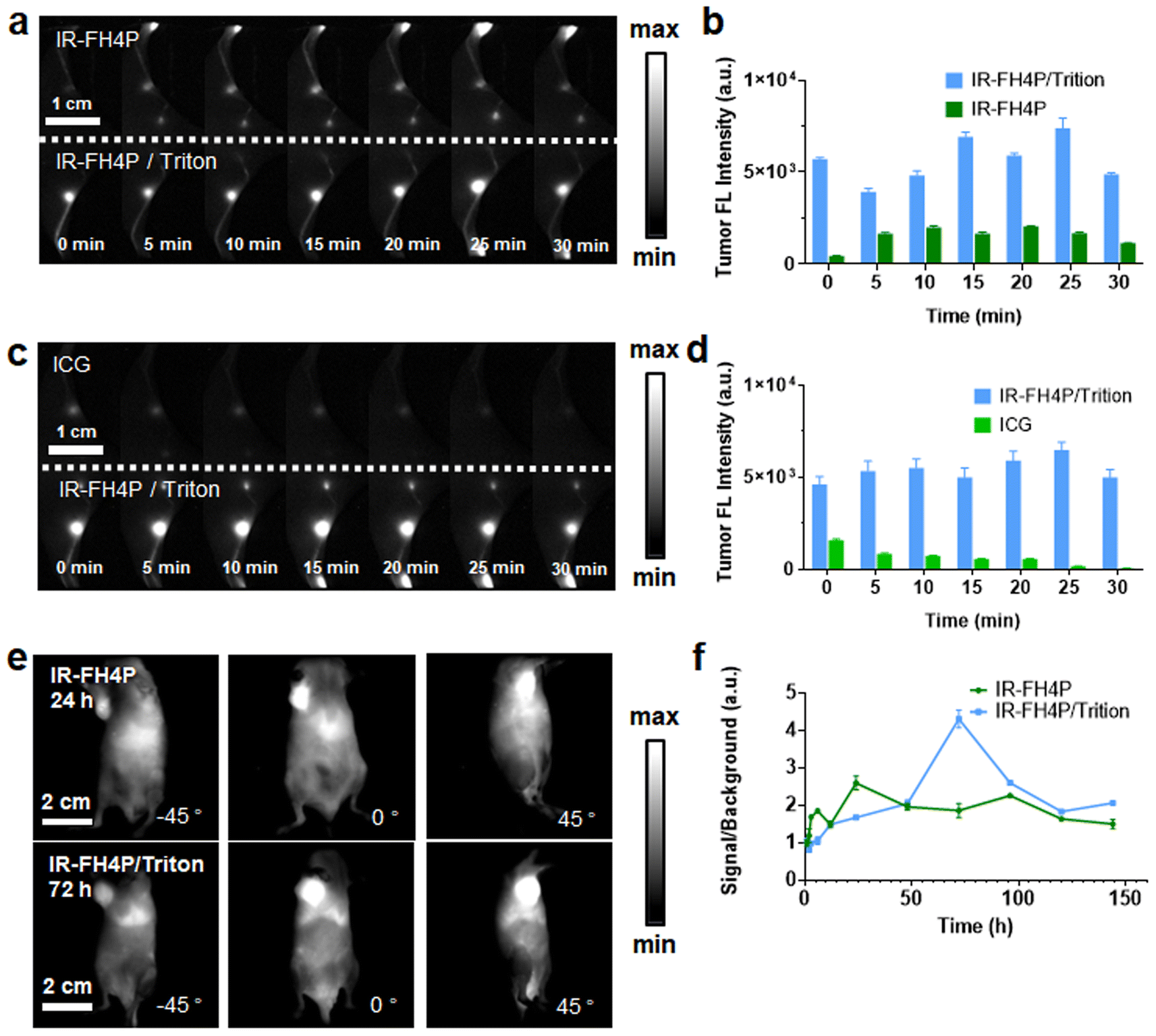 | ||
| Fig. 5 Surfactant-chaperoned NIR-II fluorophore enables bioimaging of tumor and lymph nodes with enhanced contrast levels. (a) NIR-II lymph node imaging of IR-FH4P/Triton-X100 injected into the right footpad and surfactant-free IR-FH4P into the left footpad at equivalent dye dosage (Imaging condition: 808 nm laser excitation with 88 mW cm−2 power density, 1100 nm long-pass filter). (b) Plotting lymph node signals as a function of post-injection time points for IR-FH4P/Triton-X100 and surfactant-free IR-FH4P at equivalent dye dosage. (c) NIR-II lymph node imaging of IR-FH4P/Triton-X100 injected in the right footpad and ICG in the left footpad at equivalent dye dosage. (d) Plotting lymph node signals as a function of post-injection time points for IR-FH4P/Triton-X100 and surfactant-free ICG at equivalent dye dosage. (e) Selected time points from NIR-II imaging of 4T1 tumor-bearing murine model after intravenous injection of IR-FH4P and IR-FH4P/Triton-X100. The maximum accumulation at the tumor site was recorded at ∼24 h for IR-FH4P and ∼72 h for IR-FH4P/Triton-X100. (f) Tumor-to-muscle ratios were recorded for two cohorts of mice throughout 144 h. All data are expressed as mean ± SD. | ||
Next, we imaged the tumors of two cohorts of 4T1 tumor-bearing mice with intravenously-administered IR-FH4P/Triton-X and surfactant-free IR-FH4P (Fig. S6a and b†). Due to the decrease in particle size from IR-FH4P to IR-FH4P/Triton-X100, it could be presumed that they would exhibit different tumor-enhanced permeability and retention (EPR) effects.57 Plotting tumor signal in two groups against post-injection time point revealed that the peak accumulation of IR-FH4P/Triton-X100 was much delayed compared to the surfactant-free IR-FH4P-injected group (72 h versus 24 h) (Fig. 5e). Notably, IR-FH4P/Triton-X100-injected cohort exhibited approximately 2-fold enhancement of signal-to-muscle ratio (Fig. 5f).56,58 The surfactant-chaperone strategy not only effectively enhances the fluorescence intensity of D–A–D dyes, but also provides better biocompatibility for D–A–D NIR-II fluorescence dyes by affecting their particle size, which further expands the imaging potential of a surfactant/dye cocktail.
Mechanism of NIR-II S–D–A–D–S fluorophore fluorescence enhancement in liver and Kupffer cells
We and previous reports repeatedly observed that the liver signal gradually increased after intravenous administration of a series of PEGylated S–D–A–D–S fluorophores. Previous reports exclusively attributed this phenomenon to their large size and long blood circulation. The above surfactant-chaperoned strategy prompted us to restrategize these attributions, and thus we considered that macrophages may also disassemble the S–D–AD–S fluorophores (which would lead to an enhancement in their NIR-II brightness). In this case, the signal enhancement in the liver could be roughly divided into two stages over time: a fluorophore accumulation stage and a fluorophore disassembly stage. Both stages can enhance the NIR-II fluorescence signal of accumulated fluorophores in the liver. In the first stage, the free dye accumulates in the liver (e.g., Kupffer cells) as it circulates, resulting in a certain brightness enhancement. The dye is further disassembled by Kupffer cells in the second stage, and the liver showed a further enhanced fluorescence signal (Fig. 6a). Based on this hypothesis, Kupffer cells, the main macrophages localized within the lumen of the liver, were used to examine their disassembly performance of IR-FH4P and IR-FH4P/Triton in vitro. By culturing KCs and incubating them with IR-FH4P for a set of typical periods (1, 3, 6, 12, 24, 48, 72 h), the trypsinized and collected KCs were subjected to NIR-II brightness testing (Fig. 6b). The quantified signal increased as a function of incubation time, positively correlating with liver signal after intravenous injection with IR-FH4P at the corresponding time points (Fig. 6c and d). Collectively, these results verified the accumulated IR-FH4P in the liver was potentially disassembled by KCs cells, which is consistent with our surfactant-chaperoned dye strategy. Interestingly, when we followed the same protocol using 4T1 cells, there was no further enhancement.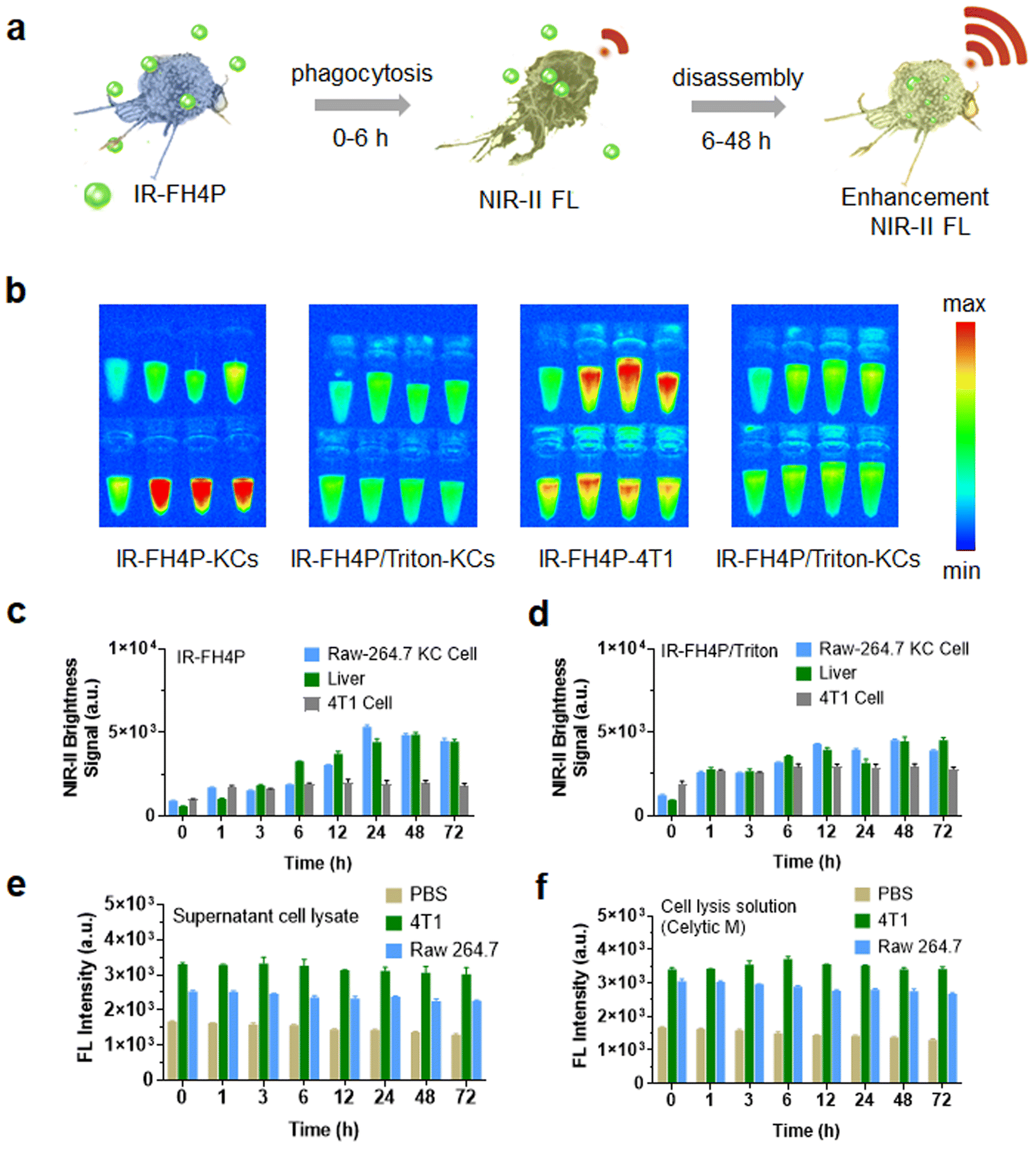 | ||
| Fig. 6 NIR-II brightness enhancement of IR-FH4P through KCs' phagocytosis and disassembly. (a) Suggested model of inherently aggregated NIR-II dye phagocytosed and subsequently disassembled by Raw-264.7 KC bioactivity. (b) NIR-II fluorescence of trypsinized Raw 264.7 KCs and 4T1 cells after incubation with IR-FH4P (10 μM) and IR-FH4P/Triton-X100 for different periods (1 × 106 cells were used for each time point). (c) and (d) Variation trend of NIR-II brightness of IR-FH4P and IR-FH4P/Triton-X100 in cultured Raw 264.7 and 4T1 cells was compared with liver signals after intravenous injection of IR-FH4P. (e) Fluorescence intensity of IR-FH4P (10 μM) incubated with cell lysate from 4T1 and Raw 264.7 (10 mg mL−1). (f) Fluorescence intensity of IR-FH4P (10 μM) incubated with cell lysis buffer (CeLytic M, the concentration of CeLytic M was slightly tuned to match their counterparts in 4T1/RAW lysate solutions). All data are expressed as mean ± SD. | ||
To further explore whether the disassemble mechanism exclusively depends on living cells, the cell lysate of KCs and 4T1 cells were incubated with equivalent IR-FH4P for predetermined periods (Fig. 6e). The NIR-II fluorescence intensity of dye-incubated cell lysates was similar with dye in pure lysate buffer (CeLytic M), indicating that the enhancement was solely caused by surfactant components in pure lysate buffer (Fig. 6f). These results verified that only living cells can enhance the NIR-II brightness of IR-FH4P, and we speculate that the endocytic process in KCs plays a role in solubilizing the dye similar to that of surfactants for disassembly of the IR-FH4P in aqueous solution.
Conclusion
We designed and synthesized a pair of NIR-II S–D–A–D–S dyes and investigated the influence of the alkyl chain substitutions at the C3- and C4-positions of the thiophene linker on their fluorescence properties. We found that alkyls substituted at the C4-position of the thiophene donor had better quantum efficiency compared with commonly used C3-position substitution. Our results will influence chemists and researchers in biomedical optics to reconsider the S–D–A–D–S design from the standpoint of substituting the donor. Previously synthesized D–A–D and S–D–A–D–S dyes were severely quenched when using PEGylation to impart water solubility.59 Consistent with previous results, the as-prepared IR-FH4P automatically formed homogeneous nanoparticles with a size of ∼80 nm in PBS buffer, inducing dramatic fluorescence quenching. To solve this issue, we developed a surfactant-chaperoned solubilization strategy, in which the surfactant can serve as a dispersant to disassemble and deliver the payload (aggregated dyes). Using this strategy, the S–D–A–D–S dyes were compelled to co-assemble with the surfactant to prevent intermolecular aggregation. A very small concentration of surfactant was sufficient to achieve ten-fold fluorescence enhancement of the PEGylated S–D–A–D–S fluorophore. Notably, the surfactant-chaperoned dye system displayed much lower cell toxicity compared to an equivalent amount of surfactant, thereby eliminating the toxicity concern for in vivo bioimaging.The NIR-II brightness enhancement from using surfactant-chaperoned S–D–A–D–S dye enabled remarkedly improved imaging contrast, especially within the short imaging window. In the short-term, surfactant-chaperoned IR-FH4P provided 3- to 10-fold fluorescence enhancement compared to that of the surfactant-free IR-FH4P group when used in in vivo applications. For lymph node mapping applications, our surfactant-chaperoned dye strategy provided increased imaging contrast (3-fold enhancement). Also, our strategy afforded a higher tumor-muscle ratio. The surfactant-chaperoned S–D–A–D–S dye was eventually excreted out of the body within 6 days post-injection, similar to that of the surfactant-free fluorophore. Our surfactant-chaperoned strategy is facile, low-cost (which may be universal to other various NIR-II dyes), and appears to circumvent the inherent fluorescence ACQ issue for in the lab, preclinical, and clinical usage.
Our investigations revealed that commonly observed NIR-II fluorescence enhancement in mice liver was partially attributed to the disassembly of NIR-II fluorophore by activity within liver Kupffer cells. Here, the inherently self-assembled NIR-II dyes first accumulated in the liver and were subsequently disassembled by Kupffer cell activity, thus further increasing the in vivo brightness. In parallel, this mechanism was further verified by the macrophage Raw cell experiment, in which the S–D–A–D–S dye incubated Raw cells showed a time-dependent brightness enhancement. Conversely, the cell lysate mixed and incubated with S–D–A–D–S dye did not increase the NIR-II brightness, indicating that the process of fluorescence enhancement exclusively depends on living cells and their corresponding biological processes. Our observations therefore explain the mechanism of liver enhancement by accumulating NIR-II fluorophores, guiding a direction of imaging liver and KCs using an inherent aggregated dye or even a hydrophobic dye structure. The inherent aggregated dye could function as a “turn-on” probe in the liver and KCs, which is much more favorable towards improving the bioimaging contrast levels.
Author contributions
T. H. and S. Z. conceived the project. S. Z. and B. Y. supervised the project. T. H. and Y. W. participated in the animal disease models. X. P. constructed theoretical models and ran operational simulations. T. H., N. Z., C. Y., Q. M., J. W., and Q. S. synthesized the dyes. T. H., J. X., Q. C., and B. S. conducted the animal experiments. T. H., J. X., and B. S. conducted the data analysis. T. H., Y. Z., K. H., and S. Z. wrote the manuscript. All authors participated in the manuscript editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFC2601900).Notes and references
- (a) Q. Fu, J. Ye, J. Wang, N. Liao, H. Feng, L. Su, X. Ge, H. Yang and J. Song, Small, 2021, 17, e2008061 CrossRef PubMed; (b) S. J. Woo, S. Park, J. E. Jeong, Y. Hong, M. Ku, B. Y. Kim, I. H. Jang, S. C. Heo, T. Wang, K. H. Kim, J. Yang, J. H. Kim and H. Y. Woo, ACS Appl. Mater. Interfaces, 2016, 8, 15937–15947 CrossRef CAS PubMed.
- R. R. Zhang, A. B. Schroeder, J. J. Grudzinski, E. L. Rosenthal, J. M. Warram, A. N. Pinchuk, K. W. Eliceiri, J. S. Kuo and J. P. Weichert, Nat. Rev. Clin. Oncol., 2017, 14, 347–364 CrossRef CAS PubMed.
- Y. T. Long and T. J. Meade, Chem. Sci., 2020, 11, 6940–6941 RSC.
- S. Zhu, S. Herraiz, J. Yue, M. Zhang, H. Wan, Q. Yang, Z. Ma, Y. Wang, J. He, A. L. Antaris, Y. Zhong, S. Diao, Y. Feng, Y. Zhou, K. Yu, G. Hong, Y. Liang, A. J. Hsueh and H. Dai, Adv. Mater., 2018, 30, e1705799 CrossRef PubMed.
- S. Zhu, B. C. Yung, S. Chandra, G. Niu, A. L. Antaris and X. Chen, Theranostics, 2018, 8, 4141–4151 CrossRef CAS PubMed.
- J. Zou, L. Li, J. Zhu, X. Li, Z. Yang, W. Huang and X. Chen, Adv. Mater., 2021, 33, e2103627 CrossRef PubMed.
- C. Sun, X. Sun, P. Pei, H. He, J. Ming, X. Liu, M. Liu, Y. Zhang, Y. Xia, D. Zhao, X. Li, Y. Xie and F. Zhang, Adv. Funct. Mater., 2021, 31 Search PubMed.
- E. D. Cosco, J. R. Caram, O. T. Bruns, D. Franke, R. A. Day, E. P. Farr, M. G. Bawendi and E. M. Sletten, Angew. Chem., Int. Ed., 2017, 56, 13126–13129 CrossRef CAS PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- C. Wang, H. Lin, X. Ge, J. Mu, L. Su, X. Zhang, M. Niu, H. Yang and J. Song, Adv. Funct. Mater., 2021, 31 Search PubMed.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan, L. Li, X. Mu, J. Wang, W. Mi, J. Luo, J. Xie and X. D. Zhang, Adv. Mater., 2019, 31, e1901015 CrossRef PubMed.
- S. I. Reja, M. Minoshima, Y. Hori and K. Kikuchi, Chem. Sci., 2020, 12, 3437–3447 RSC.
- F. Ding, Y. Zhan, X. Lu and Y. Sun, Chem. Sci., 2018, 9, 4370–4380 RSC.
- Y. Sun, M. Ding, X. Zeng, Y. Xiao, H. Wu, H. Zhou, B. Ding, C. Qu, W. Hou, A. Er-Bu, Y. Zhang, Z. Cheng and X. Hong, Chem. Sci., 2017, 8, 3489–3493 RSC.
- Q. Chen, C. Liang, C. Wang and Z. Liu, Adv. Mater., 2015, 27, 903–910 CrossRef CAS PubMed.
- B. Li, L. Lu, M. Zhao, Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7483–7487 CrossRef CAS PubMed.
- B. Guo, J. Chen, N. Chen, E. Middha, S. Xu, Y. Pan, M. Wu, K. Li, C. Liu and B. Liu, Adv. Mater., 2019, 31, e1808355 CrossRef PubMed.
- S. Li, Q. Deng, Y. Zhang, X. Li, G. Wen, X. Cui, Y. Wan, Y. Huang, J. Chen, Z. Liu, L. Wang and C. S. Lee, Adv. Mater., 2020, 32, e2001146 CrossRef PubMed.
- Y. Chen, B. Sun, X. Jiang, Z. Yuan, S. Chen, P. Sun, Q. Fan and W. Huang, J. Mater. Chem. B, 2021, 9, 1002–1008 RSC.
- Z. Ma, M. Zhang, J. Yue, C. Alcazar, Y. Zhong, T. C. Doyle, H. Dai and N. F. Huang, Adv. Funct. Mater., 2018, 28, 36 Search PubMed.
- T. Ishizawa, N. Fukushima, J. Shibahara, K. Masuda, S. Tamura, T. Aoki, K. Hasegawa, Y. Beck, M. Fukayama and N. Kokudo, Cancer, 2009, 115, 2491–2504 CrossRef PubMed.
- H. Shang, H. Fan, Q. Shi, S. Li, Y. Li and X. Zhan, Sol. Energy Mater. Sol. Cells, 2010, 94, 457–464 CrossRef CAS.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, H. Hyun, G. Park, Y. Xie, S. Bae, M. Henary and J. V. Frangioni, Nat. Biotechnol., 2013, 31, 148–153 CrossRef CAS PubMed.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed.
- X. Yi, F. Wang, W. Qin, X. Yang and J. Yuan, Int. J. Nanomed., 2014, 9, 1347–1365 CrossRef PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- H. Li, G. Parigi, C. Luchinat and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 6224–6233 CrossRef CAS PubMed.
- Y. Cao, R. C. Qian, D. W. Li and Y. T. Long, Chem. Commun., 2015, 51, 17584–17587 RSC.
- S. Zhu, Q. Yang, A. L. Antaris, J. Yue, Z. Ma, H. Wang, W. Huang, H. Wan, J. Wang, S. Diao, B. Zhang, X. Li, Y. Zhong, K. Yu, G. Hong, J. Luo, Y. Liang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 962–967 CrossRef CAS PubMed.
- Q. Yang, Z. Hu, S. Zhu, R. Ma, H. Ma, Z. Ma, H. Wan, T. Zhu, Z. Jiang, W. Liu, L. Jiao, H. Sun, Y. Liang and H. Dai, J. Am. Chem. Soc., 2018, 140, 1715–1724 CrossRef CAS PubMed.
- C. Qu, Y. Xiao, H. Zhou, B. Ding, A. Li, J. Lin, X. Zeng, H. Chen, K. Qian, X. Zhang, W. Fang, J. Wu, Z. Deng, Z. Cheng and X. Hong, Adv. Opt. Mater., 2019, 7, 15 Search PubMed.
- R. Tian, H. Ma, Q. Yang, H. Wan, S. Zhu, S. Chandra, H. Sun, D. O. Kiesewetter, G. Niu, Y. Liang and X. Chen, Chem. Sci., 2019, 10, 326–332 RSC.
- S. Gnaim, A. Scomparin, A. Eldar-Boock, C. R. Bauer, R. Satchi-Fainaro and D. Shabat, Chem. Sci., 2019, 10, 2945–2955 RSC.
- S. Thavornpradit, S. M. Usama, G. K. Park, J. P. Shrestha, S. Nomura, Y. Baek, H. S. Choi and K. Burgess, Theranostics, 2019, 9, 2856–2867 CrossRef CAS PubMed.
- G. Qian and Z. Y. Wang, Chem. - Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- G. Qian, J. P. Gao and Z. Y. Wang, Chem. Commun., 2012, 48, 6426–6428 RSC.
- J. Qian and B. Z. Tang, Chem, 2017, 3, 56–91 CAS.
- Q. Yang, Z. Ma, H. Wang, B. Zhou, S. Zhu, Y. Zhong, J. Wang, H. Wan, A. Antaris, R. Ma, X. Zhang, J. Yang, X. Zhang, H. Sun, W. Liu, Y. Liang and H. Dai, Adv. Mater., 2017, 29, 1605497 CrossRef PubMed.
- W. K. Tsai, C. I. Wang, C. H. Liao, C. N. Yao, T. J. Kuo, M. H. Liu, C. P. Hsu, S. Y. Lin, C. Y. Wu, J. R. Pyle, J. Chen and Y. H. Chan, Chem. Sci., 2019, 10, 198–207 RSC.
- P. Sun, Y. Chen, B. Sun, H. Zhang, K. Chen, H. Miao, Q. Fan and W. Huang, ACS Appl. Bio Mater., 2021, 4, 4542–4548 CrossRef CAS PubMed.
- H. Ma, C. Liu, Z. Hu, P. Yu, X. Zhu, R. Ma, Z. Sun, C.-H. Zhang, H. Sun, S. Zhu and Y. Liang, Chem. Mater., 2020, 32, 2061–2069 CrossRef CAS.
- Q. Fu, J. Ye, J. Wang, N. Liao, H. Feng, L. Su, X. Ge, H. Yang and J. Song, Small, 2021, 17, e2008061 CrossRef PubMed.
- X. Luo, D. Hu, D. Gao, Y. Wang, X. Chen, X. Liu, H. Zheng, M. Sun and Z. Sheng, ACS Nano, 2021, 15, 10010–10024 CrossRef PubMed.
- Y. Wang, T. Kadoya, L. Wang, T. Hayakawa, M. Tokita, T. Mori and T. Michinobu, J. Mater. Chem. C, 2015, 3, 1196–1207 RSC.
- P. Chowdhury and Y.-H. Chan, Mol. Syst. Des. Eng., 2022, 7, 702–719 RSC.
- C. Liu, H. Ma, Z. Hu, R. Tian, R. Ma, Y. Xu, X. Wang, X. Zhu, P. Yu, S. Zhu, H. Sun and Y. Liang, Front. Chem., 2021, 9, 739802 CrossRef CAS PubMed.
- B. Sun, Y. Kim, Y. Wang, H. Wang, J. Kim, X. Liu and M. Lee, Nat. Mater., 2018, 17, 599–604 CrossRef CAS PubMed.
- B. Sun, B. Shen, A. Urushima, X. Liu, X. Feng, E. Yashima and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 22690–22696 CrossRef CAS PubMed.
- Y. Zhang, S. Wang, X. Wang, Q. Zan, X. Yu, L. Fan and C. Dong, Anal. Bioanal. Chem., 2021, 413, 3823–3831 CrossRef CAS PubMed.
- X. Zhou, K. Zhang, C. Yang, Y. Pei, L. Zhao, X. Kang, Z. Li, F. Li, Y. Qin and L. Wu, Adv. Funct. Mater., 2021, 32 Search PubMed.
- J. Qi, C. Sun, A. Zebibula, H. Zhang, R. T. K. Kwok, X. Zhao, W. Xi, J. W. Y. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2018, 30, e1706856 CrossRef PubMed.
- Z. Sun, Y. Jiang and M. Stenzel, SmartMat, 2021, 2, 127–130 CrossRef CAS.
- J. Qi, Y. Fang, R. T. K. Kwok, X. Zhang, X. Hu, J. W. Y. Lam, D. Ding and B. Z. Tang, ACS Nano, 2017, 11, 7177–7188 CrossRef CAS PubMed.
- J. Yu, C. Wu, X. Zhang, F. Ye, M. E. Gallina, Y. Rong, I. C. Wu, W. Sun, Y. H. Chan and D. T. Chiu, Adv. Mater., 2012, 24, 3498–3504 CrossRef CAS PubMed.
- R. Tian, H. Ma, S. Zhu, J. Lau, R. Ma, Y. Liu, L. Lin, S. Chandra, S. Wang, X. Zhu, H. Deng, G. Niu, M. Zhang, A. L. Antaris, K. S. Hettie, B. Yang, Y. Liang and X. Chen, Adv. Mater., 2020, 32, e1907365 CrossRef PubMed.
- K. Shou, Y. Tang, H. Chen, S. Chen, L. Zhang, A. Zhang, Q. Fan, A. Yu and Z. Cheng, Chem. Sci., 2018, 9, 3105–3110 RSC.
- V. P. Baklaushev, A. Kilpelainen, S. Petkov, M. A. Abakumov, N. F. Grinenko, G. M. Yusubalieva, A. A. Latanova, I. L. Gubskiy, F. G. Zabozlaev, E. S. Starodubova, T. O. Abakumova, M. G. Isaguliants and V. P. Chekhonin, Sci. Rep., 2017, 7, 7715 CrossRef CAS PubMed.
- X. Zeng, Y. Xiao, J. Lin, S. Li, H. Zhou, J. Nong, G. Xu, H. Wang, F. Xu, J. Wu, Z. Deng and X. Hong, Adv. Healthcare Mater., 2018, 7, e1800589 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and structure characterization, quantum yield test, NIR-II tumor imaging, etc. See DOI: https://doi.org/10.1039/d2sc05651h |
| This journal is © The Royal Society of Chemistry 2022 |