
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature triggered alternating copolymerization of epoxides and lactones via pre-sequenced spiroorthoester intermediates†
Hyuk-Joon
Jung
,
Chatura
Goonesinghe
and
Parisa
Mehrkhodavandi
 *
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: mehr@chem.ubc.ca
First published on 16th March 2022
Abstract
We report the alternating copolymerization of caprolactone and epoxide through the in situ formation of pre-sequenced spiroorthoester monomer. The reaction is catalyzed by the temperature triggered, bifunctional cationic indium complex (±)-[(NNiOtBu)In(CH2SiMe3)][B(C6F5)4] (1). 1 can catalyze the coupling of epoxide and lactone to form spiroorthoester at 60 °C and its double ring-opening polymerization at 110 °C to form poly(ether-alt-ester). The post-polymerization modification and degradation of the poly(ether-alt-ester) are further investigated.
Introduction
The precise control of monomer sequence in a macromolecule is an immediate and mostly unmet challenge in polymer synthesis.1 Perfectly alternating copolymers are the most basic sequence-controlled polymers where comonomer pairs (M1 and M2) are incorporated with a strictly alternating sequence into poly(M1-alt-M2).2 Precisely controlled alternating polymer sequences can affect the physical and mechanical properties of polymers and open the door to applications that are not achievable with corresponding homopolymers or gradient or block copolymers.3Despite well-studied comonomer pairs such as styrene and maleic anhydride,4 aziridine and CO,5 epoxides and CO2,6 epoxides and CO,7 epoxides and anhydride,8 alkenes and CO,9 and ethylene with other alkenes,10 perfectly alternating copolymerization is still a major challenge and has a limited scope of comonomer pairs. Alternating copolymerization of epoxides and lactones is rare. Endo and coworkers reported the anionic alternating copolymerization of bicyclic bis(γ-lactone) or 3,4-dihydrocoumarin with glycidyl ethers.11 These “non-homopolymerizable” lactones can be alternately co-polymerized with epoxides to form oligomers (Mn < 4 kDa). Coates and coworkers reported the alternating copolymerization of dihydrocoumarin with epoxides to provide alternating polyesters with improved molecular weights and dispersities in presence of cocatalyst, PPNCl (Mn 7–19 kDa).12
The strategy to control the alternating sequence without using comonomer pairs is the polymerization of pre-sequenced monomers, however, the synthesis of such monomers can require multiple steps, and may suffer from low yields and limited functional group tolerance in the resulting polymers.13 Namely, a wide range of spiroorthoesters (SOEs), a class of pre-sequenced monomer, can be synthesized through the coupling of epoxides and lactones (Scheme 1A).14 However, Lewis-acid-catalyzed formation and polymerization15 of spiroorthoesters to date have suffered from poor selectivity, resulting in the need for distillation, low monomer yields (<50%), low molecular weights, and high dispersities for the resulting polymers.16
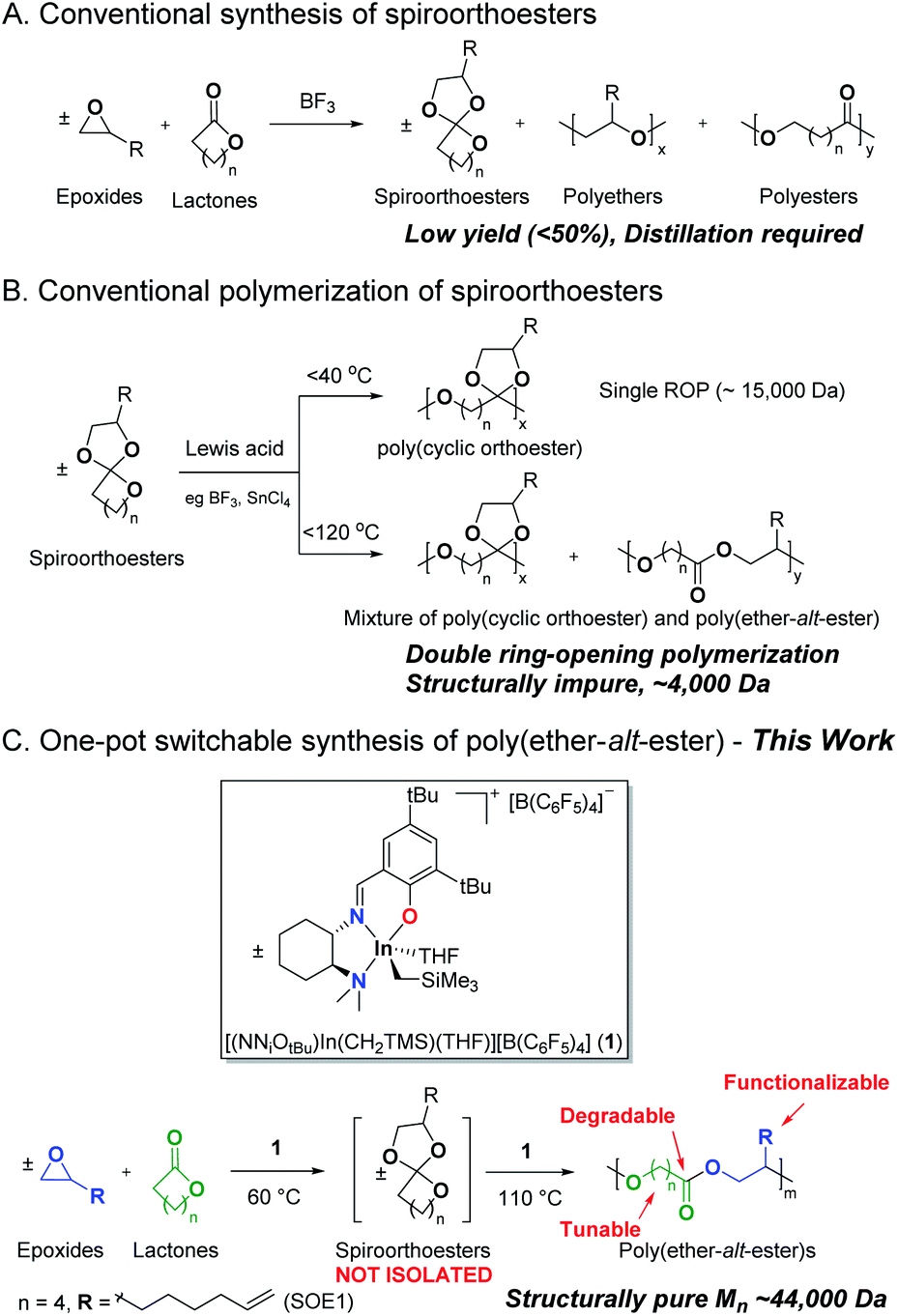 | ||
| Scheme 1 Synthesis and polymerization of spiroorthoesters. | ||
The cationic ring-opening polymerization of spiroorthoesters consists of a single ring-opening polymerization of the lactone cyclic unit to form poly(cyclic orthoester), followed by the opening of the second ring to form poly(ether-alt-ester) (Scheme 1B).17 The mechanism of spiroorthoester polymerization and the temperature-dependent polymer structure have been reported previously (Scheme S2†).18 To date, polymers from double ring-opening polymerization of spiroorthoesters are either low molecular weight oligomers19 or a mixture of low molecular weight single and double ring-opened polymers (Scheme 1B).15b,18b,c,20 To the best of our knowledge, this is the first example of structurally pure, high molecular weight poly(ether-alt-ester)s.
We have reported a series of neutral21 and cationic22 indium(III) complexes for the highly controlled ring-opening homo- or copolymerization of cyclic esters and ethers.23 In particular, we showed that cationic indium complex (±)-[(NNiOCm)In(CH2SiMe3)][B(3,5-(CF3)2C6H3)4] (Cm = Cumyl) catalyzes the coupling of an equimolar mixture of ε-caprolactone (ε-CL) and 1,2-epoxy-7-octene (EOE) to form 2-(hex-5-en-1-yl)-1,4,6-trioxaspiro[4.6]undecane (SOE1) in near quantitative isolated yield.24 We showed that 5, 6, and 7-membered lactones as well as epoxides with a range of functionality are converted to the respective spiroorthoesters quantitatively.24
Herein, we report a one pot formation of poly(ether-alt-ester) from ε-CL and EOE through the formation of pre-sequenced monomer SOE1 catalyzed by analogous cationic complex (±)-[(NNiOtBu)In(CH2SiMe3)][B(C6F5)4] (1). This temperature triggered copolymerization involves coupling of the epoxide and lactone at 60 °C and polymerizing the spiroorthoester intermediate at 110 °C, resulting in high molecular weight poly(ether-alt-ester) (Scheme 1C). In contrast to the state of the art,25 this is a simple and efficient strategy to synthesize functionalized aliphatic polyesters. We utilize the functionality in these polymers to prepare cross-linked materials. In addition, the ester linkages in these materials undergo hydrolytic degradation, offering a biodegradable alternative to conventional polymers.26
Results and discussion
Complexes (±)-[(NNiOtBu)In(CH2SiMe3)][B(C6F5)4] (1) and (±)-[(NNiOtBu)In(CH2SiMe3)2] (2) were synthesized according to a previous report.23d In a one pot reaction, the pre-sequenced monomer, SOE1, is synthesized via coupling of EOE and ε-CL with 1 in toluene at 60 °C for 24 h (Table 1, entry 1). Subsequently, the solvent is removed, resulting in a neat reaction mixture containing SOE1 and 1. This mixture is heated at 110 °C to form 100% double ring-opened product, the poly(ether-alt-ester) B, with molecular weights up to 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da (Fig. S8†).
000 Da (Fig. S8†).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Complex | [SOE]:[initiator] |
Temp. (°C) | Time (h) | Conv.b (%) | M n,GPC (g mol−1) | Đ | Yield (%) |
A:Bb |
|
| a All reactions were performed twice under a nitrogen atmosphere in neat. Initiator = 0.008 mmol. b Determined by analysis of the crude material by 13C{1H} NMR spectroscopy (151 MHz, C6D6, 25 °C). c Determined by GPC measurements with polystyrene as the standard in THF and corrected by Mark–Houwink's corrections. d One-pot reaction. Spiroorthoester synthesis: [1] = 3.75 mM, [EOE] = 0.25 M, [ε-CL] = 0.25 M; spiroorthoester polymerization: performed in neat. e Initiated by the neutral indium complex (2). f Oligomeric product formed. g Data from ref. 18b. h Polymerization reaction without 1. | |||||||||
| 1d | 1 | 67 | 110 | 24 | 100 | 17980 |
1.99 | 55 | 0100 |
| 2e | 2 | 200 | 110 | 24 | 0 | — | — | — | — |
| 3f | 1 | 200 | 80 | 24 | 34 | — | — | — | 5347 |
| 4 | 1 | 200 | 80 | 48 | 89 | 10760 |
1.28 | 24 | 694 |
| 5 | 1 | 200 | 110 | 24 | 100 | 23770 |
1.39 | 81 | 0100 |
| 6 | 1 | 400 | 110 | 24 | 100 | 44320 |
1.40 | 70 | 0100 |
| 7g | SnCl4 | 50 | 0 | 1 | — | 14400 |
4.69 | 84 | 1000 |
| 8g | SnCl4 | 50 | 80 | 1 | — | 2840 | 4.53 | 82 | 6238 |
| 9g | SnCl4 | 50 | 120 | 1 | — | 1900 | 2.95 | 79 | 2575 |
| 10h | — | 200 | 110 | 24 | 0 | — | — | — | — |
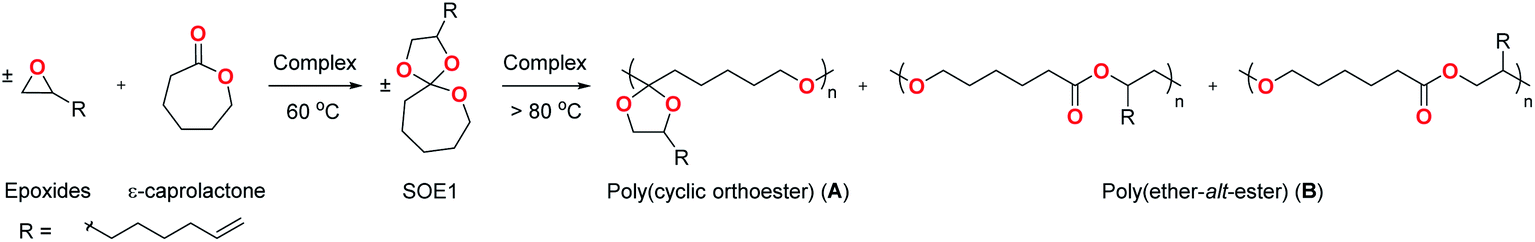
The exclusive formation of SOE1 is confirmed by the observation of the characteristic signals of the five-membered cyclic acetal (protons 7–8′) in the 1H NMR spectrum of the reaction mixture (Fig. 1a). After the double ring-opening polymerization, the 1H NMR spectrum of poly(ether-alt-ester) shows that protons 1 and 1′ in a seven-membered cyclic ether (3.56–3.60 ppm) are shifted to P1 (3.95 ppm) and 7–8′ (3.30–4.23 ppm) are shifted to P7–P8′ (3.21–3.39 and 5.20 ppm) (Fig. 1b). The ratio of regioisomers is almost 50:50 (peak area: P7:P7′–P8′ ≈ 1:5), meaning that intermediate poly(cyclic orthoester) undergoes the second ring-opening on either side of the cyclic acetal (Fig. S8 and S9†). A completely double ring-opened polymer structure is confirmed by the absence of the center carbon of SOE1 (C6 and C6′), the center carbon of poly(cyclic orthoester), and appearance of the ester carbonyl carbon of poly(ether-alt-ester) (PC6) in the 13C{1H} NMR spectrum of the product (Fig. 1c).18b
 | ||
| Fig. 1 One-pot synthesis of poly(ether-alt-ester) (Table 1, entry 1). 1H NMR spectra of (a) crude intermediate product of SOE1, (b) poly(ether-alt-ester), and (c) 13C{1H} NMR spectrum of poly(ether-alt-ester) (400 MHz, C6D6, 25 °C). | ||
FTIR spectroscopy confirms the formation of poly(ether-alt-ester) from double ring-opening polymerization. The most distinct difference between spiroorthoester/poly(cyclic orthoester) and poly(ether-alt-ester) is a strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching absorption at 1732 cm−1 that can be formed only by the double ring-opening polymerization. The νC–O frequencies of newly formed ether and ester linkages appear at 1094 and 1162 cm−1, respectively (Fig. S10†).
O stretching absorption at 1732 cm−1 that can be formed only by the double ring-opening polymerization. The νC–O frequencies of newly formed ether and ester linkages appear at 1094 and 1162 cm−1, respectively (Fig. S10†).
Since we have investigated the formation of various spiroorthoesters in a previous publication,24 we wanted to investigate the polymerization of isolated SOE1 with 1 to evaluate the impact of different reaction conditions on polymer characteristics. Importantly, as with SOE1 synthesis, neutral indium complex (2) was unreactive for the polymerization of SOE1 (Table 1, entry 2), and SOE1 was not polymerized in the absence of 1 (Table 1, entry 10).
Temperature impacts both the conversion and the selectivity of SOE1 polymerization. At 80 °C, polymerization of neat SOE1 reaches 34% conversion in 24 h. Importantly, the polymer products are a mixture of poly(cyclic orthoester) (A, 53%) and poly(ether-alt-ester) (B, 47%) (Table 1, entry 3). Increasing the reaction time to 48 h yields higher conversion of SOE1 (89%), with the resulting polymer mixture containing a significantly higher proportion of double ring-opened product (A:B = 6:94) (Table 1, entry 4). In contrast, polymerization of SOE1 at 110 °C yields solely double ring-opened poly(ether-alt-ester) in 24 h (Table 1, entry 5). Increasing the SOE1 to catalyst ratio forms polymers with Mn = 44000 g mol−1 (Table 1, entry 6). The molecular weight, dispersity, and degree of isomerization in this system are significantly superior to those reported by Endo and coworkers (Mn = 2840 g mol−1; Đ = 4.53; B = 38%) (Table 1, entry 8).18b
As these polymers are functionalizable, we investigated post-polymerization cross-linking via a thiolene reaction to transform the poly(ether-alt-ester) into a thermoset. The reaction of stoichiometric amounts of cross-linker 2,2′-(ethylenedioxy)diethanethiol (2SH) and the poly(ether-alt-ester) with Mn of 24 kDa (Table 1, entry 5) is initiated by 2,2′-azobis(2-methylpropionitrile) (AIBN). The transparent, flexible, and insoluble thiolene-cured polymer is generated in the shape of the mold in 5 minutes. FTIR spectroscopy shows that the cross-linked polymers lack the νS–H (2550–2600 cm−1) and νCC (910 and 1641 cm−1) frequencies (Fig. 2). Crosslinking improves polymer thermal stability significantly: the cross-linked polymer has a decomposition temperature of 240 °C while the non-cross-linked polymer decomposes at 177 °C. A change in glass transition temperature (Tg) is also observed after crosslinking: the cross-linked polymer has a higher Tg at −52 °C compared to Tg of non-cross-linked polymer at −69 °C. In addition, crystallization of the cross-linked polymer is observed (120 °C), whereas non-cross-linked polymer remains amorphous (Fig. S18–S23 and Table S1†).
 | ||
| Fig. 2 Cross-linking of poly(ether-alt-ester) via thiolene reaction (top). FTIR spectra of poly(ether-alt-ester) and cross-linked poly(ether-alt-ester) (bottom). | ||
Finally, we investigated the base-catalyzed degradation behavior of the polymers. Subjecting 44 kDa poly(ether-alt-ester) (Table 1, entry 6) to a 0.1 M solution of tetrabutylammonium hydroxide for 2 h results in a significant decrease in molecular weight (Fig. 3a). FTIR spectra show that the degradation products have a new νO–H frequency at 3700–3100 cm−1 which is consistent with the hydrolysis of the polymer (Fig. 3b). Importantly, there is a significant reduction in the νCO peak, indicating degradation at the ester linkage. The degradation of cross-linked poly(ether-alt-ester) was also attempted under the same degradation condition. A significant reduction in molecular weight and νCO peak were observed after 16 h (Fig. S27 and 28†).
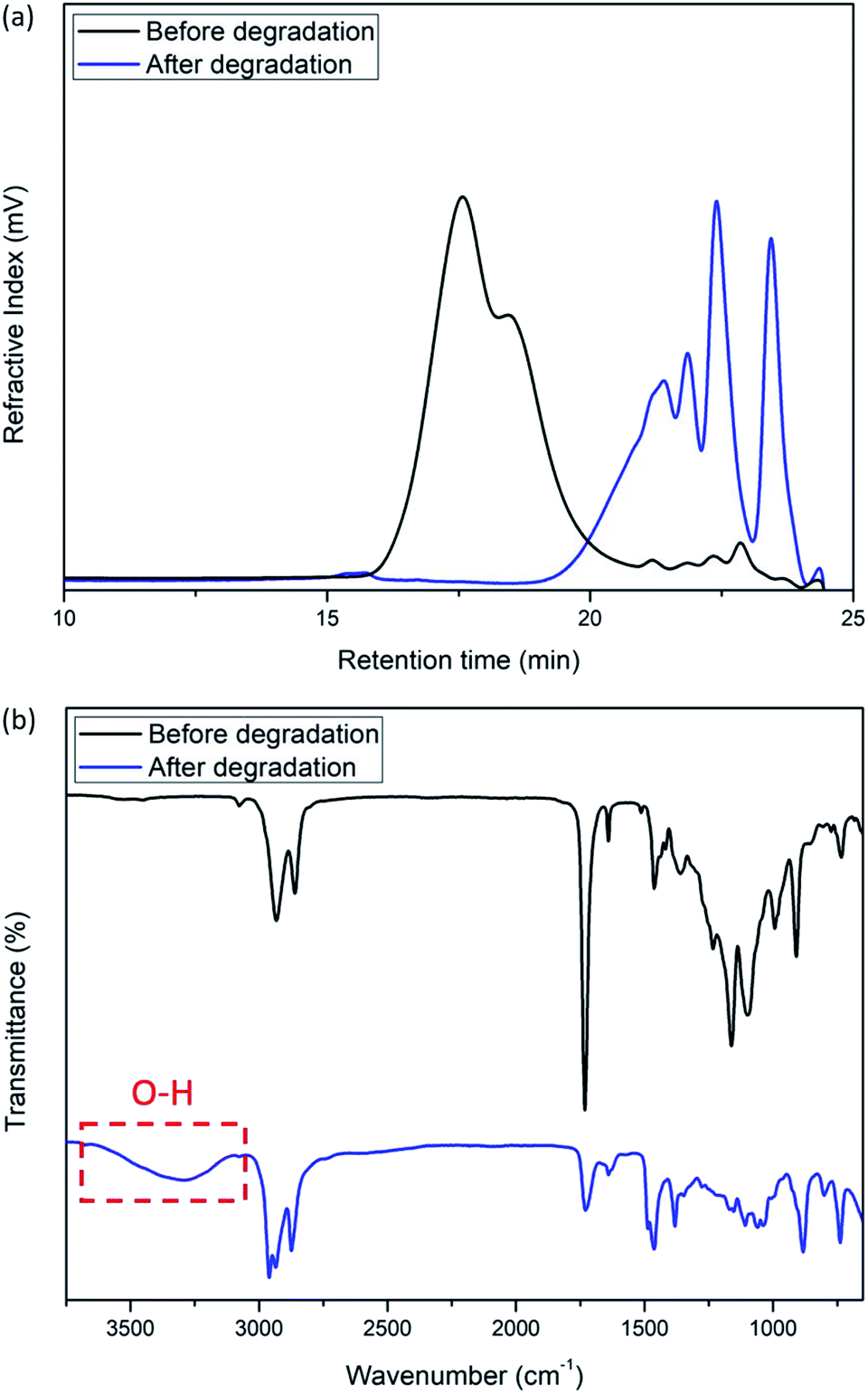 | ||
| Fig. 3 Degradation of poly(ether-alt-ester). GPC trace (a, top) and FTIR spectra (b, bottom) of poly(ether-alt-ester) and degradation products (condition: 0.1 M tetrabutylammonium hydroxide in MeOH/DCM, room temperature, 2 h). | ||
1 is clearly different than other simple Lewis acids reported for this reaction. Spiroorthoester polymerization with other Lewis acids can achieve higher molecular weights at low temperatures to form single ring-opened polymers; however, when the temperature is increased to generate the double ring-opened poly(ether-alt-ester)s, molecular weights are significantly curtailed (Table 1, entry 7).18b One reason for this difference may be the Lewis acidity of the respective catalysts.
The Lewis acidity of 1 was compared with other catalysts using a modified Gutmann–Beckett method with triethylphosphine oxide (TEPO) as a basic probe (Fig. 4).271 formed two species with TEPO due to the different solvations around indium metal centers.28 Interestingly, 1 is less Lewis acidic than BF3 and has similar Lewis acidity to SnCl4. Since spiroorthoester polymerization is not initiated by 2, supporting the cationic polymerization mechanism (Scheme S2†), we must also consider the effect of the counter anion, which plays an important role in chain growth during the cationic polymerization.29 With 1, the growing chain ends can be stabilized by the bulky, stable, and non-coordinating anion B(C6F5)4 so that chain transfer and termination reactions can be suppressed.30 Thus, 1 provides a combination of moderate Lewis acidity and a stabilizing counter anion that enable selectivity in spiroorthoester formation and control in spiroorthoester polymerization.
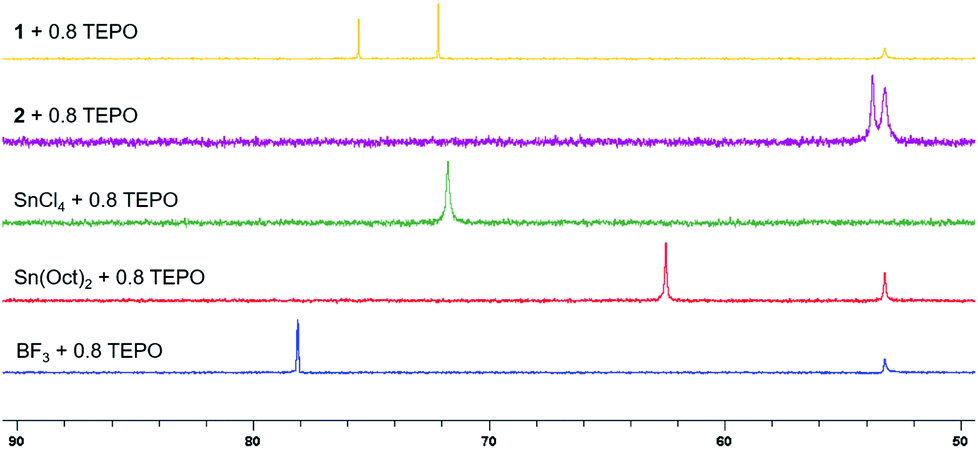 | ||
| Fig. 4 31P{1H} NMR spectra of TEPO showing the downfield shift with the increase in the Lewis acidity of the complexes (162 MHz, CDCl3, 25 °C). | ||
Conclusions
In summary, we report the first example of a sequence controlled alternating copolymerization of epoxides and lactones via a one pot generation of a pre-sequenced spiroorthoester monomer. This reaction is temperature triggered: Spiroorthoester formation is the sole reaction at 60 °C, while spiroorthoester polymerization is the sole reaction at 110 °C. The resulting poly(ether-alt-ester)s are crosslinkable and degradable. We will be expanding the substrate range in this reaction and investigating the properties and applications of the resulting polymers. We will also be exploring new systems to control the stereo- and regioselectivity in these reactions.Data availability
Additional data supporting this article were provided in ESI.†Author contributions
P. M. supervised the project. H.-J. J. carried out most of the experiments and wrote the initial manuscript draft. C. G. assisted with the synthesis of complexes and conducted Lewis acidity study. P. M. and H.-J. J. revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Council of Canada (NSERC) for supporting this work. H.-J. Jung acknowledges the UBC Chemistry department for the Award for Graduate Research Excellence. C.G. acknowledges the UBC Four Year Fellowship.Notes and references
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. V. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- (a) J. Li, R. M. Stayshich and T. Y. Meyer, J. Am. Chem. Soc., 2011, 133, 6910–6913 CrossRef CAS PubMed; (b) Z. W. Jia, J. X. Jiang, X. F. Zhang, Y. Q. Cui, Z. C. Chen, X. B. Pan and J. C. Wu, J. Am. Chem. Soc., 2021, 143, 4421–4432 CrossRef CAS PubMed.
- S. Bag, S. Ghosh, S. Paul, M. E. H. Khan and P. De, Macromol. Rapid Commun., 2021, 2100501 CrossRef CAS PubMed.
- (a) J. Li, E. Ding and W. R. Anderson, Chem. Commun., 2001, 1436–1437 Search PubMed; (b) L. Jia, H. L. Sun, J. T. Shay, A. M. Allgeier and S. D. Hanton, J. Am. Chem. Soc., 2002, 124, 7282–7283 CrossRef CAS PubMed.
- (a) A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed; (b) A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed; (c) G. W. Yang, C. K. Xu, R. Xie, Y. Y. Zhang, X. F. Zhu and G. P. Wu, J. Am. Chem. Soc., 2021, 143, 3455–3465 CrossRef CAS PubMed; (d) W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004–4011 RSC; (e) K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC; (f) G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- (a) M. Allmendinger, R. Eberhardt, G. Luinstra and B. Rieger, J. Am. Chem. Soc., 2002, 124, 5646–5647 CrossRef CAS PubMed; (b) J. T. Lee and H. Alper, Macromolecules, 2004, 37, 2417–2421 CrossRef CAS.
- (a) J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed; (b) M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed; (c) C. A. L. Lidston, B. A. Abel and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 20161–20169 CrossRef CAS PubMed; (d) W. T. Diment, G. L. Gregory, R. W. F. Kerr, A. Phanopoulos, A. Buchard and C. K. Williams, ACS Catal., 2021, 11, 12532–12542 CrossRef CAS; (e) M. J. Sanford, N. J. Van Zee and G. W. Coates, Chem. Sci., 2018, 9, 134–142 RSC; (f) N. Yi, T. T. D. Chen, J. Unruangsri, Y. Q. Zhu and C. K. Williams, Chem. Sci., 2019, 10, 9974–9980 RSC.
- (a) C. Bianchini and A. Meli, Coord. Chem. Rev., 2002, 225, 35–66 CrossRef CAS; (b) T. Fujita, K. Nakano, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2006, 128, 1968–1975 CrossRef CAS PubMed.
- (a) W. Fan, M. K. Leclerc and R. M. Waymouth, J. Am. Chem. Soc., 2001, 123, 9555–9563 CrossRef CAS PubMed; (b) X. F. Li, M. Nishiura, L. H. Hu, K. Mori and Z. M. Hou, J. Am. Chem. Soc., 2009, 131, 13870–13882 CrossRef CAS PubMed; (c) S. H. Li, D. T. Liu, Z. C. Wang and D. M. Cui, ACS Catal., 2018, 8, 6086–6093 CrossRef CAS.
- (a) K. Chung, T. Takata and T. Endo, Macromolecules, 1995, 28, 3048–3054 CrossRef CAS; (b) K. Uenishi, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4092–4102 CrossRef CAS; (c) K. Uenishi, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6750–6757 CrossRef CAS; (d) K. Uenishi, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3662–3668 CrossRef CAS; (e) K. Uenishi, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1661–1672 CrossRef CAS.
- N. J. Van Zee and G. W. Coates, Chem. Commun., 2014, 50, 6322–6325 RSC.
- (a) K. Nishimori and M. Ouchi, Chem. Commun., 2020, 56, 3473–3483 RSC; (b) C. K. Qu, Z. H. Li and J. P. He, Polym. Chem., 2018, 9, 3455–3460 RSC; (c) Y. Y. Lu, J. H. Swisher, T. Y. Meyer and G. W. Coates, J. Am. Chem. Soc., 2021, 143, 4119–4124 CrossRef CAS PubMed; (d) H. H. Liu, W. W. Liang, Y. Y. Lai, Y. C. Su, H. R. Yang, K. Y. Cheng, S. C. Huang and Y. J. Cheng, Chem. Sci., 2020, 11, 3836–3844 RSC.
- M. Fedtke, J. Haufe, E. Kahlert and G. Muller, Angew. Makromol. Chem., 1998, 255, 53–59 CrossRef CAS.
- (a) B. Trathnigg and G. Hippmann, Eur. Polym. J., 1981, 17, 1085–1087 CrossRef CAS; (b) Y. G. Hsu and Y. S. Wan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3680–3690 CrossRef CAS.
- (a) K. Bodenbenner, Justus Liebigs Ann. Chem, 1959, 623, 183–191 CrossRef CAS; (b) K. Yoshida, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2551–2558 CrossRef CAS; (c) N. Spegazzini, I. Ruisanchez, M. S. Larrechi, A. Serra and A. Mantecon, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3886–3899 CrossRef CAS.
- K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 1984, 22, 29–40 CAS.
- (a) T. Yokozawa, M. Sato and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1841–1846 CrossRef CAS; (b) S. Chikaoka, T. Takata and T. Endo, Macromolecules, 1992, 25, 625–628 CrossRef CAS; (c) S. Chikaoka, T. Takata and T. Endo, Macromolecules, 1991, 24, 6557–6562 CrossRef CAS; (d) S. Chikaoka, T. Takata and T. Endo, Macromolecules, 1991, 24, 331–332 CrossRef CAS.
- J. Canadell, A. Mantecon and V. Cadiz, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4211–4224 CrossRef CAS.
- (a) S. Chikaoka, T. Takata and T. Endo, Macromolecules, 1991, 24, 6563–6566 CrossRef CAS; (b) T. Endo and W. J. Bailey, J. Polym. Sci., Part C: Polym. Lett., 1980, 18, 25–27 CAS; (c) T. Endo, M. Okawara, N. Yamazaki and W. J. Bailey, J. Polym. Sci., Part A: Polym. Chem., 1981, 19, 1283–1286 CAS; (d) C. Y. Pan, Y. Wang and W. J. Bailey, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2737–2747 CrossRef CAS.
- (a) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS PubMed; (b) I. Yu, A. Acosta-Ramirez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS PubMed; (c) D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295–4297 RSC; (d) I. Yu, T. Ebrahimi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Dalton Trans., 2015, 44, 14248–14254 RSC; (e) T. Ebrahimi, D. C. Aluthge, B. O. Patrick, S. G. Hatzikiriakos and P. Mehrkhodavandi, ACS Catal., 2017, 7, 6413–6418 CrossRef CAS.
- (a) C. Diaz, T. Ebrahimi and P. Mehrkhodavandi, Chem. Commun., 2019, 55, 3347–3350 RSC; (b) C. Diaz, T. Tomkovic, C. Goonesinghe, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2020, 53, 8819–8828 CrossRef CAS; (c) C. Goonesinghe, H. Roshandel, C. Diaz, H. J. Jung, K. Nyamayaro, M. Ezhova and P. Mehrkhodavandi, Chem. Sci., 2020, 11, 6485–6491 RSC.
- (a) K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 CrossRef CAS PubMed; (b) H.-J. Jung, Y. Cho, D. Kim and P. Mehrkhodavandi, Catal. Sci. Technol., 2021, 11, 62–91 RSC; (c) A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS; (d) H. J. Jung, I. S. Yu, K. Nyamayaro and P. Mehrkhodavandi, ACS Catal., 2020, 10, 6488–6496 CrossRef CAS.
- H. J. Jung, C. Chang, I. Yu, D. C. Aluthge, T. Ebrahimi and P. Mehrkhodavandi, ChemCatChem, 2018, 10, 3219–3222 CrossRef CAS.
- (a) C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC; (b) D. Tian, P. Dubois, C. Grandfils and R. Jerome, Macromolecules, 1997, 30, 406–409 CrossRef CAS; (c) M. Trollsas, V. Y. Lee, D. Mecerreyes, P. Lowenhielm, M. Moller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619–4627 CrossRef; (d) X. D. Lou, C. Detrembleur and R. Jerome, Macromol. Rapid Commun., 2003, 24, 161–172 CrossRef CAS; (e) X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef CAS; (f) N. S. Teske, J. Voigt and V. P. Shastri, J. Am. Chem. Soc., 2014, 136, 10527–10533 CrossRef CAS PubMed.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- (a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS; (b) G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. R. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC.
- A. B. Kremer, R. J. Andrews, M. J. Milner, X. R. Zhang, T. Ebrahimi, B. O. Patrick, P. L. Diaconescu and P. Mehrkhodavandi, Inorg. Chem., 2017, 56, 1375–1385 CrossRef CAS PubMed.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: It includes the experimental section as well as the characterization of polymers. See DOI: 10.1039/d1sc06634j |
| This journal is © The Royal Society of Chemistry 2022 |