
Oleic acid/oleylamine ligand pair: a versatile combination in the synthesis of colloidal nanoparticles
Stefanos
Mourdikoudis
 *a,
Melita
Menelaou
*b,
Nadesh
Fiuza-Maneiro
c,
Guangchao
Zheng
d,
Shuangying
Wei
a,
Jorge
Pérez-Juste
ef,
Lakshminarayana
Polavarapu
*c and
Zdeněk
Sofer
*a
*a,
Melita
Menelaou
*b,
Nadesh
Fiuza-Maneiro
c,
Guangchao
Zheng
d,
Shuangying
Wei
a,
Jorge
Pérez-Juste
ef,
Lakshminarayana
Polavarapu
*c and
Zdeněk
Sofer
*a
aDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague, Technicka 5, 16628 – Prague 6, Czech Republic. E-mail: mourdikt@vscht.cz; soferz@vscht.cz
bDepartment of Chemical Engineering, Faculty of Geotechnical Sciences and Environmental Management, Cyprus University of Technology, 3036 Limassol, Cyprus. E-mail: melita.menelaou@cut.ac.cy
cCINBIO, Universidade de Vigo, Materials Chemistry and Physics, Department of Physical Chemistry, Campus Universitario Lagoas Marcosende, 36310 Vigo, Spain. E-mail: lakshmi@uvigo.es
dSchool of Physics and Microelectronics, Zhengzhou University, Zhengzhou, 450001, China
eCINBIO, Universidade de Vigo, Departamento de Química Física, Campus Universitario As Lagoas, Marcosende, 36310 Vigo, Spain
fGalicia Sur Health Research Institute (IIS Galicia Sur), 36310 Vigo, Spain
First published on 13th June 2022
Abstract
A variety of colloidal chemical approaches has been developed in the last few decades for the controlled synthesis of nanostructured materials in either water or organic solvents. Besides the precursors, the solvents, reducing agents, and the choice of surfactants are crucial for tuning the composition, morphology and other properties of the resulting nanoparticles. The ligands employed include thiols, amines, carboxylic acids, phosphines and phosphine oxides. Generally, adding a single ligand to the reaction mixture is not always adequate to yield the desired features. In this review, we discuss in detail the role of the oleic acid/oleylamine ligand pair in the chemical synthesis of nanoparticles. The combined use of these ligands belonging to two different categories of molecules aims to control the size and shape of nanoparticles and prevent their aggregation, not only during their synthesis but also after their dispersion in a carrier solvent. We show how the different binding strengths of these two molecules and their distinct binding modes on specific facets affect the reaction kinetics toward the production of nanostructures with tailored characteristics. Additional functions, such as the reducing function, are also noted, especially for oleylamine. Sometimes, the carboxylic acid will react with the alkylamine to form an acid–base complex, which may serve as a binary capping agent and reductant; however, its reducing capacity may range from lower to much lower than that of oleylamine. The types of nanoparticles synthesized in the simultaneous presence of oleic acid and oleylamine and discussed herein include metal oxides, metal chalcogenides, metals, bimetallic structures, perovskites, upconversion particles and rare earth-based materials. Diverse morphologies, ranging from spherical nanoparticles to anisotropic, core–shell and hetero-structured configurations are presented. Finally, the relation between tuning the resulting surface and volume nanoparticle properties and the relevant applications is highlighted.
 Stefanos Mourdikoudis | Stefanos Mourdikoudis is a chemical engineer who completed his PhD Degree in 2009 at the Aristotle University of Thessaloniki. Subsequently, he moved to the Laboratory of Coordination Chemistry at Toulouse as a Post-doc until 2011. A subsequent Research Fellowship allowed him to work at the University of Vigo. He worked for one year at Sorbonne University in Paris and between 2016–2020 he worked as a Researcher and Teaching Fellow at University College London. Currently, he is a researcher at the University of Chemistry and Technology Prague. His main research interests involve the chemical synthesis and characterisation of various types of nanoparticles destined for a range of applications including catalysis and biomedicine. |
 Melita Menelaou | Melita Menelaou received her Diploma in Chemical Engineering (2004) and PhD Degree (2009) from the Aristotle University of Thessaloniki (Greece). Dr Menelaou was a member of respected research groups in Asia (Japan – Advanced Institute of Materials Research) and Europe (Czechia – Central European Institute of Technology-Brno University of Technology; Spain – University of Barcelona; Greece – School of Chemical Engineering/School of Chemistry, Aristotle University of Thessaloniki). Currently, she is working in the Department of Chemical Engineering at Cyprus University of Technology. Her work focuses on the synthesis, characterization and application of a wide range of materials including nanomaterials with technological and biomedical applications. |
 Nadesh Fiuza-Maneiro | Nadesh Fiuza-Maneiro received her Master's Degree in Chemical Research and Industrial Chemistry from the University of Vigo in 2021. Currently, she is a PhD candidate in the Materials Chemistry and Physics group under the supervision of Dr Lakshminarayana Polavarapu. Her research interests are focused on the size and shape-controlled synthesis of chiral perovskites and the study of their self-assembly and optical properties. |
 Jorge Pérez-Juste | Jorge Pérez-Juste has a PhD from the University of Vigo (1999). He is currently an Associate Professor at the Biomedical Research Center at the University of Vigo. His research is focused on the synthesis and characterization of metal and semiconductor nanoparticles and the understanding of the mechanisms involved in the growth of nanoparticles, which determine their final size and shape. Besides, he is interested in the application of nanoparticles for photocatalysis. |
 Lakshminarayana Polavarapu | Lakshminarayana Polavarapu obtained his MSc in Chemistry from the University of Hyderabad (India) and PhD from the National University of Singapore. After Post-Doctoral research at CIC BiomaGUNE and the University of Vigo in Spain, he joined the Chair for Photonics and Optoelectronics at the LMU Munich (Germany) as an Alexander von Humboldt Fellow, and later continued as a Group Leader. In 2020, he became the Principal Investigator of the Materials Chemistry and Physics research group at the Centro de Investigaciones Biomedicas (CINBIO), University of Vigo. He is the co-author of 80 publications, which have received over 10000 citations thus far. |
 Zdeněk Sofer | Prof. Zdenek Sofer has been a Tenured Professor at the University of Chemistry and Technology Prague since 2019. He received his PhD at the University of Chemistry and Technology Prague, Czech Republic in 2008. During his PhD, he spent one year in Forschungszentrum Julich (Peter Grünberg Institute, Germany) and he did a Postdoctoral Stay at University Duisburg-Essen, Germany. Prof. Sofer's research interests are focused on 2D materials including graphene, layered chalcogenides and other 2D materials, their crystal growth, chemical modifications and derivatisation. His research covers various applications of 2D materials, which include energy storage and conversion, electronic, catalysis and sensing devices. He is an Associated Editor of FlatChem journal. He has published over 500 articles, which have received over 20 |
1. Introduction
A large range of different types of nanostructures with distinct compositions and shapes have attracted interest in the last few decades owing to their different properties compared to their bulk counterparts. These properties include plasmonic, magnetic and catalytic properties. The numerous types of nanomaterials have received attention due to their plethora of current and potential applications in various fields such as energy storage,1 chemical catalysis,2 electrocatalysis,3 biology,4 biomedicine,5 and environmental remediation.6 To obtain nanoparticles with the desired properties and potential for the above-mentioned applications, one of the most efficient routes for their production is the umbrella of colloidal bottom-up chemical synthetic approaches. When chemical synthesis is preferred, several parameters need to be adjusted thoroughly, such as the type and amount of precursors,7 solvents,8 surfactants9 and reducing agents.10 The surface ligands or surfactants are crucial for the controlled generation of nanomaterials with a well-tuned size, shape and surface and volume composition. Ligands are beneficial to provide a certain morphology to the particles during their growth, while also ensuring their dispersibility in selected solvents and their ability for post-synthetic functionalization. The different types of ligands include thiols, alkylamines, carboxylic acids, phosphines and phosphine oxides.9 Obviously, different ligands have distinct surface chemical behaviors, reactivity and molecular structures. For example, certain ligands bind strongly to the nanoparticle surface and others present rather weak bonding. Preferential binding to certain crystal facets is observed for some ligands, while others bind to all crystallographic domains in the same manner.10 Biocompatibility and ease of surface functionalization usually vary between different types of ligands. Therefore, due to the above-mentioned reasons, it is quite common to use more than one ligand when employing wet-chemical synthetic protocols. The combination of alkylamines with carboxylic acids and/or phosphines and thiols aims to endow numerous features, which can function in a synergistic way, producing nanoparticles with well-designed characteristics and functionalities.11–13Therefore, in this review, we decided to focus on the ligand pair of oleic acid (OAc) and oleylamine (OAm) for the controlled synthesis of diverse nanoparticle systems. OAc (cisCH3(CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)(CH2)7COOH) and OAm (cisCH3(CH2)7CHCH)(CH2)8NH2) are high-boiling point (≥350 °C) ligands, which are liquid at room temperature and cost-effective, and although they have approximately the same alkyl chain length (∼2 nm, see Scheme 1 and Fig. 6c for their linear molecular structures), they present different binding modes and strengths and distinct reducing capacities. Thermogravimetric analysis (TGA) is often used to determine the surface coverage of NPs with OAm and OAc capping ligands. The TGA measurements of OAm, OAc and OAm/OAc mixture are shown in Fig. 1.14 The TGA profiles of pure OAm and OAc demonstrate single degradation events occurring at around 240 °C and 280 °C, respectively. The OAm/OAc mixture exhibits two degradation events at approximately 225 °C and 335 °C, which are attributed to the degradation of OAm and OAc, respectively. The degradation temperatures of the adsorbed ligands compared to the pure ligand molecules can be elevated due to the fact that the surface-capped ligands are protected from degradation owing to their close proximity to the nanoparticles. FTIR measurements are also very useful to characterize the surface capping of OAm and OAc on different NP systems. For example, various peaks ascribed to OAm and/or OAc occur on surfactant-coated gold and silver NPs (i.e., methylene rocking mode at 723 cm−1, methylene stretching modes at 2800–3000 cm−1 and C–H stretching in CC–H at 3005 cm−1 (Fig. 2)).14 Certain modifications in the observed spectra of NP samples, e.g., the splitting of the free N–H bending peak at 1630 cm−1 in OAm into two peaks (at about 1570 and 1660 cm−1) in spectra of capped Au and Ag NPs denote the oxidation of the amine moiety to amide. The shifting of the carbonyl stretching peak appearing at 1711 cm−1 in OAc to higher wavenumbers in the spectra of capped gold and silver NPs indicates the chemical coordination of the carbonyl species on the nanoparticle surface, event after the post-synthetic washing stages (see also Fig. 10 for further FTIR peak assignments for OAm/OAc and Fig. 45). In the case of their 1H-NMR spectra, the 1H chemical shift for OAm is 2.56 ppm (CaH2 position), whereas that for OAc is 2.18 ppm.15 Tuning the OAm/OAc ratio enables nanoparticles with well-defined shapes to be obtained, which will have an effect also on their properties such as plasmonic and magnetic properties. OAm and OAc are classified as hard Lewis bases, and under certain conditions they can form complexes with the metal ions of the precursor compounds, which act as Lewis acids. The generated complexes can then be thermally decomposed/reduced, leading to the nucleation and growth of nanoparticles in the reaction pot. We will also present the role of the acid–base complex generated upon the use of both ligands under discussion together under certain conditions and display their distinct NMR spectra compared to the individual ligands. Upon completion of the colloidal synthesis and washing stages, the particles typically form stable dispersions in common organic solvents such as hexane, toluene and chloroform. However, ligand exchange protocols are often required for the transfer of the particles to water when hydrophilicity is needed.16 The sections of this review are categorized according to the different families of nanomaterials obtained in the presence of both OAc and OAm. These families include metal oxides, metal chalcogenides, metals, bimetallic structures, perovskites, upconversion particles and rare earth-based materials. Several morphologies, ranging from spherical nanoparticles to anisotropic, core–shell and hetero-structured shapes are demonstrated. We also discuss how tailoring the surface and volume properties of NPs by choosing suitable OAm and OAc ratios will affect their resulting performance in applications. The main insights derived are given in the concluding section herein, and also at the end of each section or sub-section for the different nanoparticle compositions.
CH)(CH2)7COOH) and OAm (cisCH3(CH2)7CHCH)(CH2)8NH2) are high-boiling point (≥350 °C) ligands, which are liquid at room temperature and cost-effective, and although they have approximately the same alkyl chain length (∼2 nm, see Scheme 1 and Fig. 6c for their linear molecular structures), they present different binding modes and strengths and distinct reducing capacities. Thermogravimetric analysis (TGA) is often used to determine the surface coverage of NPs with OAm and OAc capping ligands. The TGA measurements of OAm, OAc and OAm/OAc mixture are shown in Fig. 1.14 The TGA profiles of pure OAm and OAc demonstrate single degradation events occurring at around 240 °C and 280 °C, respectively. The OAm/OAc mixture exhibits two degradation events at approximately 225 °C and 335 °C, which are attributed to the degradation of OAm and OAc, respectively. The degradation temperatures of the adsorbed ligands compared to the pure ligand molecules can be elevated due to the fact that the surface-capped ligands are protected from degradation owing to their close proximity to the nanoparticles. FTIR measurements are also very useful to characterize the surface capping of OAm and OAc on different NP systems. For example, various peaks ascribed to OAm and/or OAc occur on surfactant-coated gold and silver NPs (i.e., methylene rocking mode at 723 cm−1, methylene stretching modes at 2800–3000 cm−1 and C–H stretching in CC–H at 3005 cm−1 (Fig. 2)).14 Certain modifications in the observed spectra of NP samples, e.g., the splitting of the free N–H bending peak at 1630 cm−1 in OAm into two peaks (at about 1570 and 1660 cm−1) in spectra of capped Au and Ag NPs denote the oxidation of the amine moiety to amide. The shifting of the carbonyl stretching peak appearing at 1711 cm−1 in OAc to higher wavenumbers in the spectra of capped gold and silver NPs indicates the chemical coordination of the carbonyl species on the nanoparticle surface, event after the post-synthetic washing stages (see also Fig. 10 for further FTIR peak assignments for OAm/OAc and Fig. 45). In the case of their 1H-NMR spectra, the 1H chemical shift for OAm is 2.56 ppm (CaH2 position), whereas that for OAc is 2.18 ppm.15 Tuning the OAm/OAc ratio enables nanoparticles with well-defined shapes to be obtained, which will have an effect also on their properties such as plasmonic and magnetic properties. OAm and OAc are classified as hard Lewis bases, and under certain conditions they can form complexes with the metal ions of the precursor compounds, which act as Lewis acids. The generated complexes can then be thermally decomposed/reduced, leading to the nucleation and growth of nanoparticles in the reaction pot. We will also present the role of the acid–base complex generated upon the use of both ligands under discussion together under certain conditions and display their distinct NMR spectra compared to the individual ligands. Upon completion of the colloidal synthesis and washing stages, the particles typically form stable dispersions in common organic solvents such as hexane, toluene and chloroform. However, ligand exchange protocols are often required for the transfer of the particles to water when hydrophilicity is needed.16 The sections of this review are categorized according to the different families of nanomaterials obtained in the presence of both OAc and OAm. These families include metal oxides, metal chalcogenides, metals, bimetallic structures, perovskites, upconversion particles and rare earth-based materials. Several morphologies, ranging from spherical nanoparticles to anisotropic, core–shell and hetero-structured shapes are demonstrated. We also discuss how tailoring the surface and volume properties of NPs by choosing suitable OAm and OAc ratios will affect their resulting performance in applications. The main insights derived are given in the concluding section herein, and also at the end of each section or sub-section for the different nanoparticle compositions.
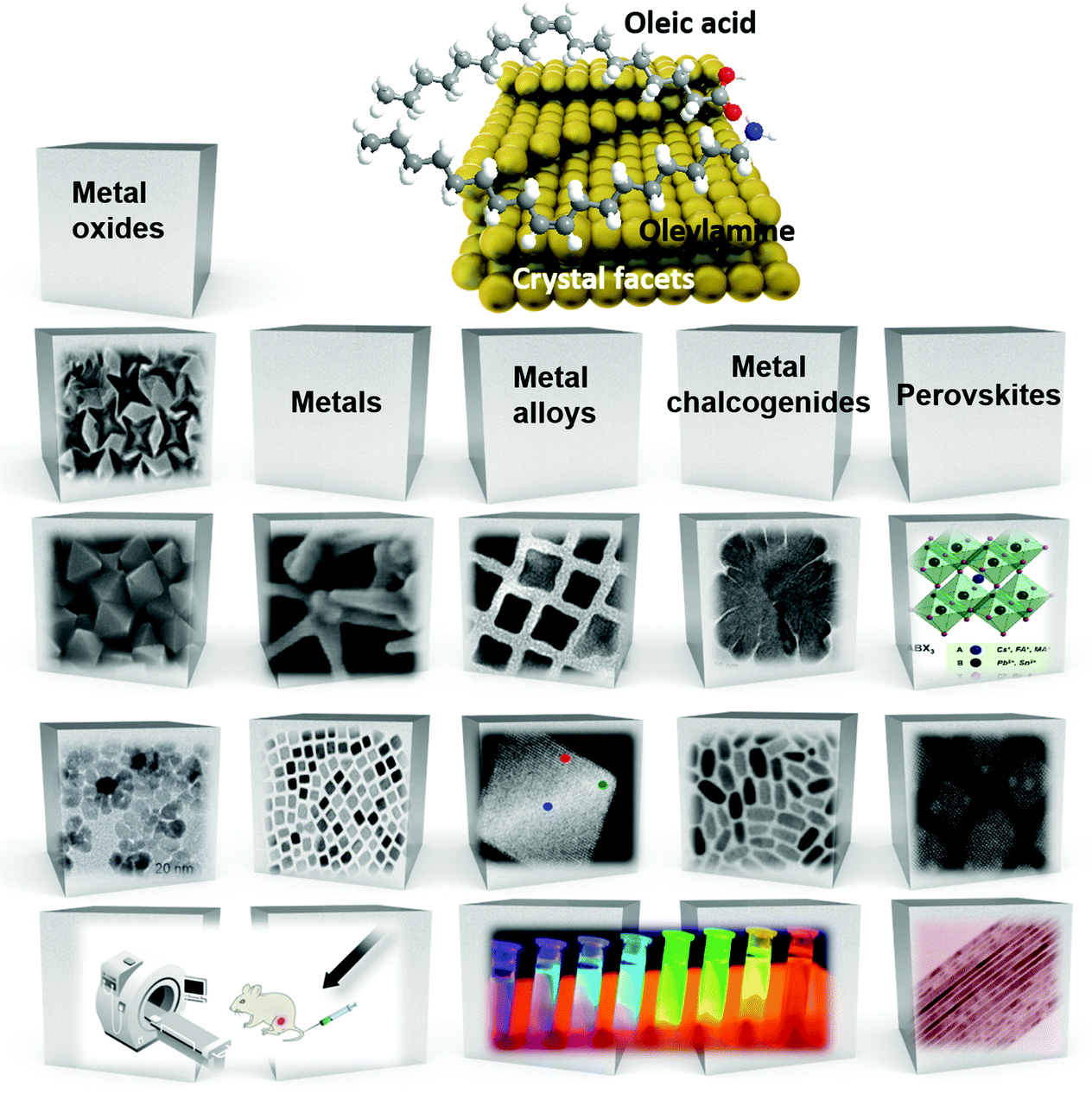 | ||
| Scheme 1 Overview of the different categories of nanomaterials prepared in the presence of OAm/OAc (at the stick models of the molecules, grey spheres correspond to C atoms, white spheres represent H, red spheres are O and blue colour denotes N atom). | ||
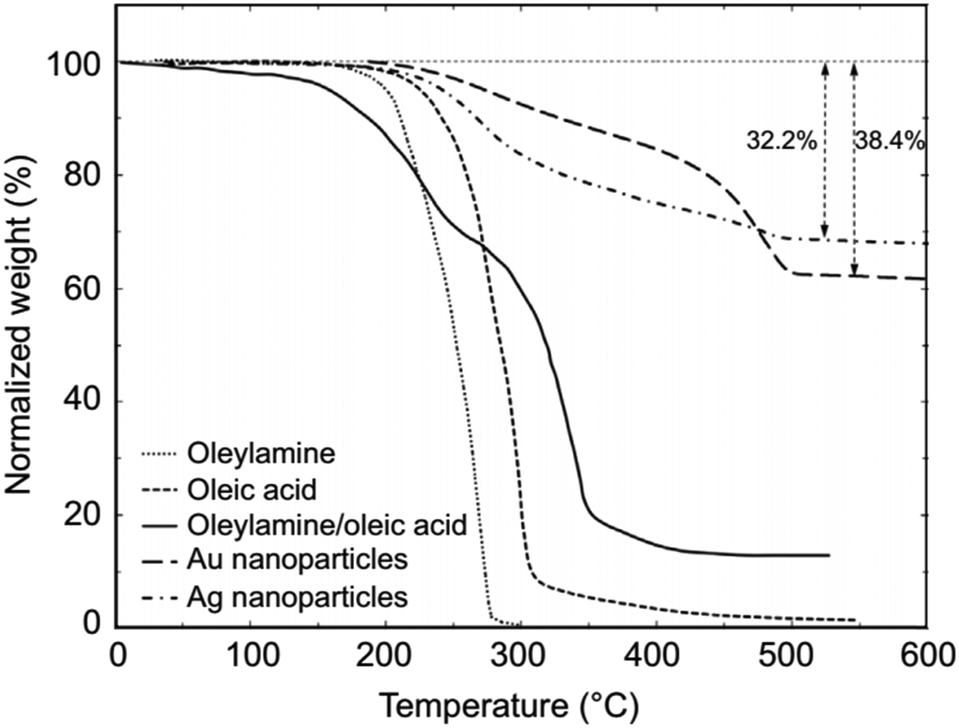 | ||
| Fig. 1 TGA plots of OAm, OAc, OAm/OAc mixture and OAm/OAc-modified gold and silver NPs.14 Originally published by and used with permission from ref. 14. Copyright 2017, Dove Medical Press Ltd. | ||
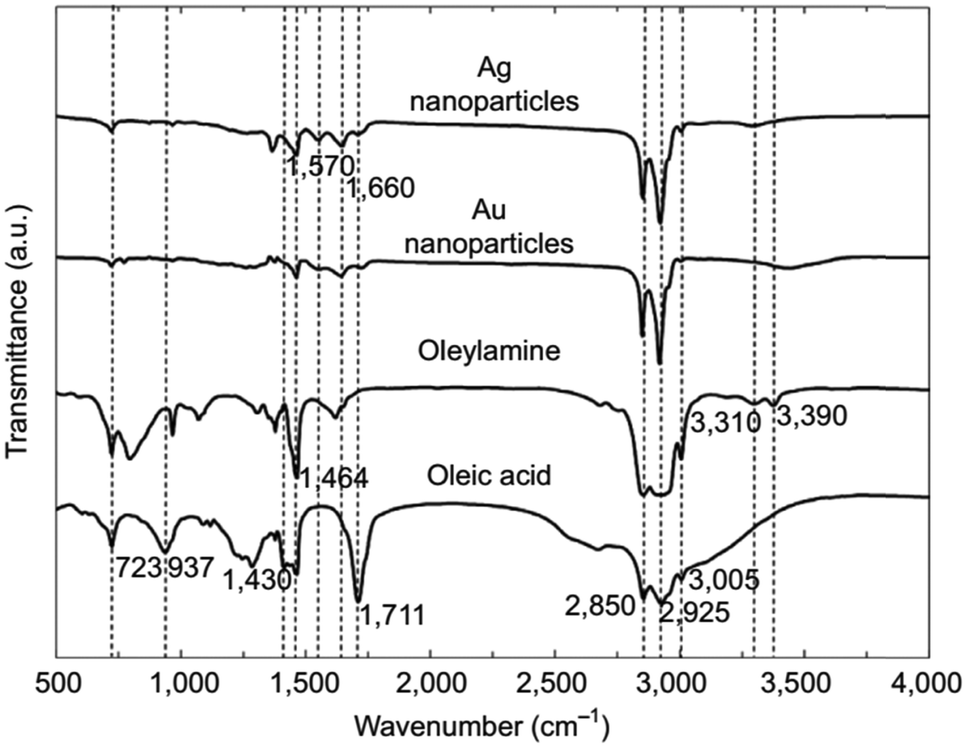 | ||
| Fig. 2 FTIR spectra of OAm, OAc and OAm/OAc-modified Au and Ag NPs.14 Originally published by and used with permission from ref. 14. Copyright 2017, Dove Medical Press Ltd. | ||
2. Iron oxide, metal ferrite and other oxide nanoparticles prepared with OAm/OAc
One of the main families of nanostructures prepared with the ligand pair under discussion refers to that composed of Fe-oxides and M-ferrites. The typical iron oxide compositions are hematite (α-Fe2O3), maghemite (γ-Fe2O3) and magnetite (Fe3O4). Common ferrites are CoFe2O4 an MnFe2O4. Besides spherical particles, other morphologies including anisotropic, core–shell shapes and heterostructures have also been explored to a significant extent, often by merging materials of different compositions. We start this section by describing magnetite nanostructures, before presenting the ones with a more oxidized form, that is, Fe2O3, followed by metal ferrite nanoparticles. The heterostructures and core–shell configurations will be discussed in a later section.2.1 Fe3O4 nanoparticles
Magnetite NPs are increasingly being used in fields related to biomedicine and environment, in addition to other fields. Mohapatra et al. prepared Fe3O4 NPs using FeCl2 as a precursor in a heated mixture of 1-octadecene (ODE), OAc and OAm under an inert atmosphere.17 The particle size was controlled between 2–12 nm by adjusting the amine to Fe mole ratio, whereas a bigger size (16 nm) was acquired by applying seed-mediated growth. The authors reported that the crystal field splitting energy (CFSE) of OAc is higher than that of OAm, and thus the spin orbit coupling was larger for OAm than OAc. This caused larger magnetocrystalline anisotropy for the surface layer of the OAm-coated NPs and the saturation magnetization Ms of these particles was higher compared to that of the OAc/OAm-capped particles.17 Another precursor, Fe(acac)3, was used to prepare Fe3O4 NPs in the presence of 1,2-dodecanediol, OAm, OAc and dibenzyl ether. The ligand pair provides a layer around each particle, which hinders their aggregation. The different functional groups between OAm and OAc result in distinct particle morphologies, depending on the amounts used. Homogeneous nanocrystals were generated only with a certain concentration of OAc and OAm. In that report, dodecanediol acted as the reducing agent.18Ivanco and co-workers also used iron acetylacetonate to produce spherical 6.5 nm iron oxide NPs with the ligand pair under discussion. Diphenyl ether was used as the reaction solvent under reflux conditions and 1,2-hexadecanediol as the reductant. The interparticle distance on a solid substrate could be tuned by the use of NPs with varying surfactant sizes.19 Wilson and Langell studied the surface composition of OAm/OAc-coated magnetite NPs via X-ray photoelectron spectroscopy (XPS), aiming to evaluate the nature of the ligand chemisorbed on the surface and determine the effects of the surfactant on the surface composition. The authors highlighted the capacity of OAm to serve as a mild reductant. The presence of a C–N bond from OAm and C–O and CO bonds from the monodentate carboxylate of OAc was confirmed in their spectra. Carboxylate adsorbates arising from exposure to acetic acid and other short-chain carboxylic acids have been reported to chemisorb strongly enough on transition metal oxide substrates. They act as good capping agent linkers, enabling the easier functionalization of the NP surface. The OAc/OAm pair can displace or react with most of the surface hydroxyl moieties generated under carboxylate adsorption. In that work, a slight excess of OAm resulted in the limited chemical reduction of Fe3O4 on the NP surface. OAm chemisorbed as an amine, whereas OAc was mainly in the form of monodentate carboxylate to link the long-chain alkyl tail to the magnetite surface. Even in its monodentate form, the carboxylate is more stable than the amine. Although OAm is decomposed at 525 K, desorbing nitrogen and depositing much of its aliphatic chain as graphitic C, the monodentate carboxylate decomposed slowly. Specifically, it lost most of its alkyl tail over a broad temperature range but kept its carboxylic linker even on the partially reduced oxide surface.20 Gao et al. prepared magnetite nanocubes using Fe(acac)3, OAm, OAc and trioctylphosphine oxide (TOPO) in ODE at 260 °C in an autoclave (Fig. 3). Adjusting the molar ratio of OAc to OAm was crucial to prepare monodisperse nanocubes. OAm apparently binds to the Fe centers on the NP surface in a rather weak manner, whereas OAc has a much stronger bond with the crystal surface due to its high oxophilicity. The nanocube morphology was no longer produced by further varying the OAm/OAc ratio, which led to the formation of triangular and other irregular-shaped nanostructures. It seems that OAc can modulate the growth rate along certain directions, which provides a range of NP morphologies.21
 | ||
Fig. 3 (a) Low- and (b) high-magnification TEM images of magnetite nanocubes prepared at 260 measured °C for 2 h using 0.1 mmol TOPO with a molar ratio 0.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for OAc to OAm. Reprinted with permission from ref. 21. Copyright 2010, the American Chemical Society. :1 for OAc to OAm. Reprinted with permission from ref. 21. Copyright 2010, the American Chemical Society. | ||
In another report, hydrophobic Fe3O4 NPs with sizes in the range of 7–12 nm were produced by using iron(III) isopropoxide as the precursor, water vapor as the hydrolysis agent and ethanol as the solvent. The OAm and OAc surfactants helped to control the nucleation and growth of the NPs in an autoclave reaction. It was found that OAm reacted with OAc to yield a dialkylamide, which acted as a new surfactant and the real capping agent of the NPs. 1H NMR indicated that the amide is bound at the NP surface through the amide proton (NH) probably via a hydrogen bond, while oxygen and nitrogen were not directly involved. The role of amine was fundamental in reducing Fe(III) to Fe(II), forming magnetite NPs. In fact, if no OAm was used, the above-mentioned reduction was inhibited and hematite (α-Fe2O3) was formed. In that case, OAc strongly interacted with specific hematite faces, promoting the growth of the non-interacting planes (Fig. 4).15
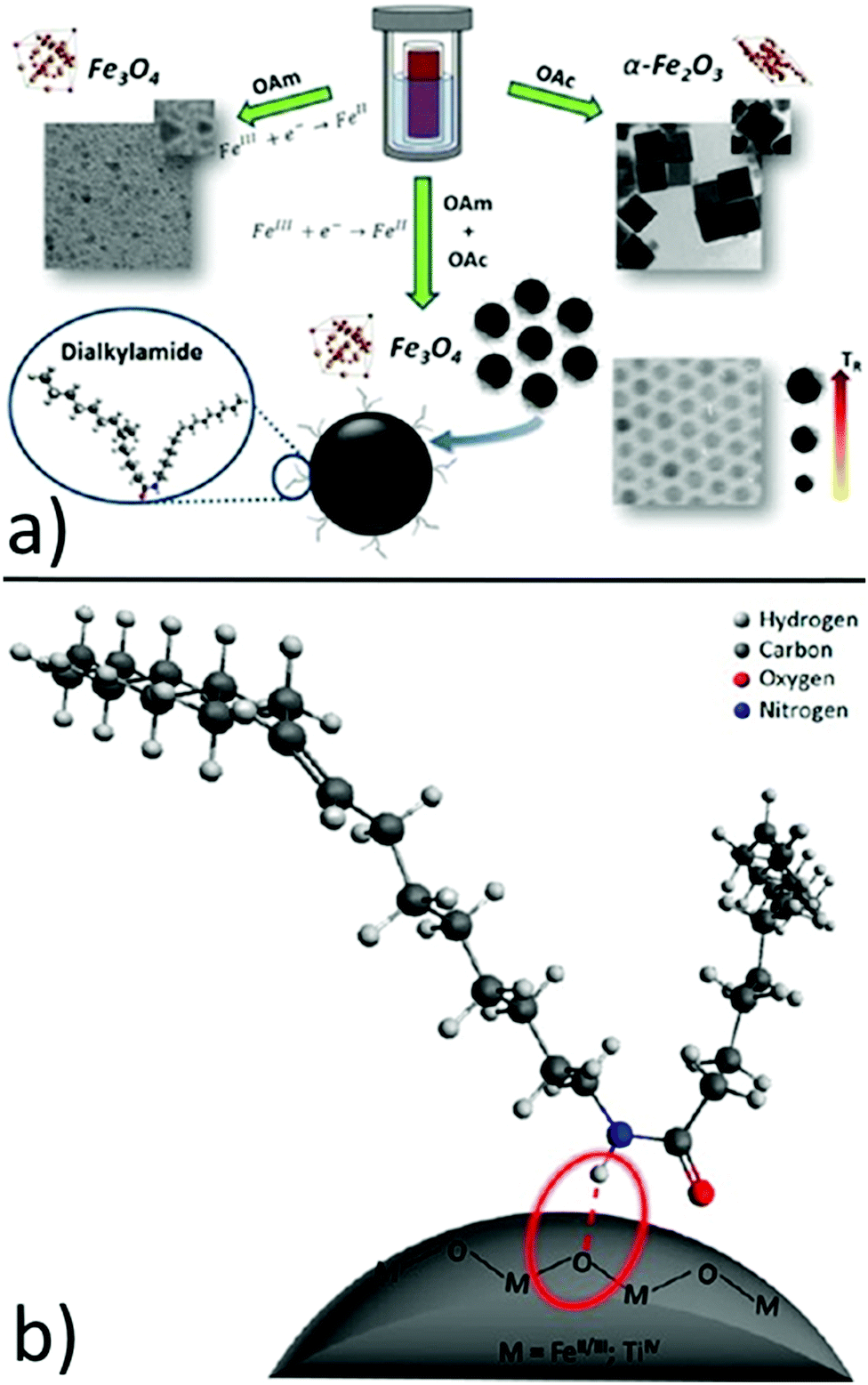 | ||
| Fig. 4 (a) Role of OAm, OAc and dialkylamine in the formation of iron oxide NPs and (b) schematic drawing of the dialkylamide-coated NPs (magnetite or anatase, bottom image). Reproduced with permission from ref. 15. Copyright 2015, the American Chemical Society. | ||
Liu and co-workers suggested that the long-chain decomposition products of ODE can also act in a supplementary way together with other surfactants to hinder particle aggregation. It has been reported that the OAm/OAc pair also helps to prevent, to some extent, further particle oxidation.22 If polypyrrole or poly(N-methylpyrrole), which is used as a reductant, is combined with OAm/OAc, ordered lamellar nanostructures would be impressively observed, probably via a template growth assisted by the OAc molecules.23 Sun's group reported in 2009 that the utilization of excess OAm is important to ensure a strongly reductive environment for the thermal decomposition of iron acetylacetonate.24 At that time, it was already shown that OAm can indeed serve as an alternative, inexpensive and even stronger reductant than 1,2-hexadecanediol, which was previously utilized to prepare magnetite NPs.25
The reductive conditions offered by the large amount of OAm used lowered the precursor decomposition temperature to 170 °C, at which small spinel-structured iron oxide NPs were synthesized.24 Actually, in that report, the sole use of OAm in benzyl ether was largely responsible for the lowering of the decomposition temperature, given that prior works showed that the combination of OAm, OAc and 1,2-hexadecanediol necessitated a high reaction temperature close to 300 °C to isolate the product, which was not magnetically responsive and contained FeO. The OAm-capped NPs showed good catalytic properties for the CVD growth of boron NWs.24 The above-mentioned work by Sun and Zeng was reported in 2002 on the synthesis of 4 nm magnetite NPs in the presence of iron acetylacetonate, 1,2-hexadecanediol, OAm/OAc and phenyl ether. Seed-mediated growth was employed to produce larger Fe3O4 NPs.25 Magnetization measurements on powder samples prepared with that protocol yielded a blocking temperature, Tb, at ∼20 K. Above this temperature, these 4 nm particles were superparamagnetic and the measured magnetization was 36.2 emu g−1.26
Salado et al. discussed that the –COO− group has higher affinity than the –NH− group of the deprotonated OAm to the iron oxide surface. They also commented that the existence of the ligand pair is required to retain kinetic control of the growth conditions to produce magnetite NPs with a well-defined size in the range of 3.5–7 nm.27 Horak and co-workers used Fe(II) oleate and mandelate complexes, which led to the formation of monodisperse particles in a controlled size range. In their report, the beneficial influence of the enhanced amount of carboxyl groups stabilizing the NPs at the growth stage was noticed. The authors mentioned that by using an equimolar ratio OAm/OAc, a mixture of carboxyl anions, protonated oleylamine and acid–base complex is generated. The carboxyl anions of OAc have high electron-donating capacity to the NP surface and offer good stabilization of the NPs during growth. A ligand exchange process was applied to transfer the magnetite NPs to an aqueous medium, forming stable dispersions with applicability in the biomedical domain. PEG derivatives containing phosphonic and hydroxamic groups were employed, given that they demonstrate good affinity to Fe.28
Guo et al. reported an easy solvothermal synthetic process involving Fe(acac)3, OAm and OAc and found that two intermediates, Fe(acac)x(OA)y and N-(cis-9-octadecenyl)-oleamide (OOA), played key roles in tuning the surface protection of the NPs and the reducibility of the reaction system. FTIR spectra illustrated that the dissolution of OAm in ODE is followed by the dissociation of amines due to the appearance of bands assigned to the N–H+ stretching. The OOA intermediate was produced from the condensation reaction of OAm and OAc. The free carboxyl groups of OAc were all converted to OOA or to an iron carboxylate complex upon heating to 200 °C. The presence of OAm contributed to a strong reductive environment, whereas when the OAc amount was low, the surface of the formed magnetic nanocrystals lacked sufficient protection. OAc preferred to bind with the (111) planes, and then slowed down the growth rate of these planes, enabling the formation of anisotropic triangular prisms. In fact, Fe(acac)x(OAc)y, which was produced in the early stages of the precursor preparation, was critical to tune the particle size and shape through the control of the amount of ‘free’ OAc in the reaction system. OOA also affected the reaction process by regulating its free energy, and also acted as a reductant, though weaker than OAm (Fig. 5).29
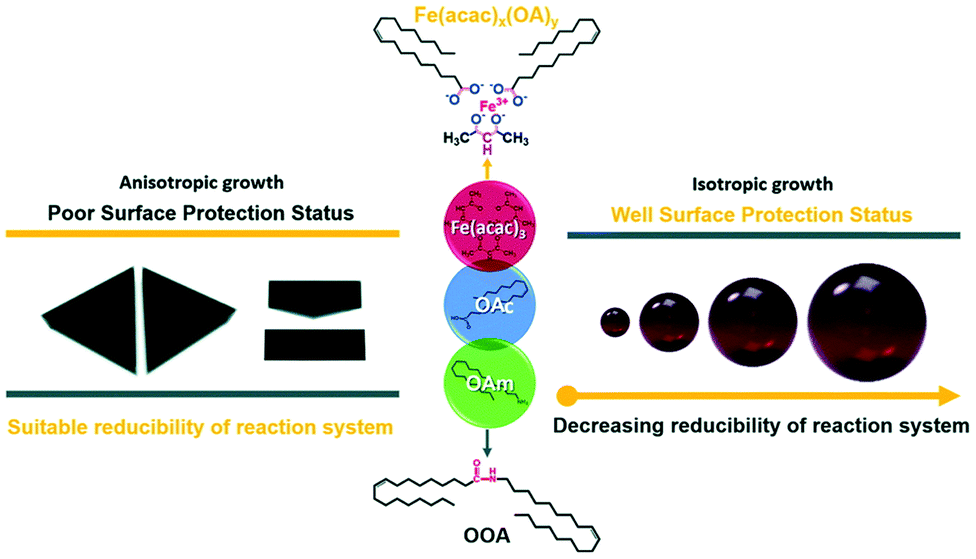 | ||
| Fig. 5 Scheme depicting the size and shape control regulated by Fe(acac)x(OAc)y and OOA.29 Copyright 2016, The Royal Society of Chemistry. | ||
Granata et al. reported that an interparticle distance of around 2 nm can depend on the thickness of the molecular capping, which is related to the chain length of the ligands used. The authors reported that the ligand coordination provides the CFSE generated from ‘d’ orbital splitting with a magnitude defined by the ligands having a certain coordination symmetry. When metal cations at the NP surface are coordinated with OAc, CFSE tends to be bigger and the spin–orbit coupling becomes lower. Magnetic anisotropy is formed by the spin–orbit coupling taking place at magnetic cations, which decreases with a decrease in the spin–orbit coupling, resulting in a decrease of the coercivity (see also ref. 17 above for a discussion on the CFSE and the magnetocrystalline anisotropy).30 Marchetti and co-workers reported that the O2− of the OAc molecules bonded covalently to magnetite NPs can reduce the surface spin disorder, and therefore the surface contribution to the anisotropy. Actually, Mössbauer spectroscopy revealed that sometimes these spinel oxides are composed of a mixture of magnetite and γ-Fe2O3 (maghemite). Under certain conditions, the OAc ligand enables NPs to exhibit magnetic properties very similar to that of the bulk. Even for small NP sizes, OAc does not perturb their magnetic properties, given that it ensures the absence of surface spin disorder.31
The synthesis of Fe3O4 and MnFe2O4 by the reduction of metallic salts in the presence of OAm/OAc in benzyl ether resulted in the formation of nanostructures with a size in the range of 10–80 nm and various morphologies (spheres, octahedral, and cubes). The OAm/OAc ratio significantly affected the growth rate of the nanocrystals. This effect was attributed to the varying binding ability of the two stabilizers on the crystal facets. The particle size was mostly influenced by modifying the quantity of the metal precursors, whereas the shape was tuned by adjusting the concentration of the stabilizers. Specifically, the size of the NPs was enhanced by reducing the relative concentration of OAm.32 In another report, monodisperse magnetite NPs were produced in big quantities by the pyrolysis of Fe(acac)3 using ODE as solvent and TOPO, OAm and OAc as surfactants at high temperature. In this case, OAc and OAm were injected at a specific molar ratio at 120 °C in the reaction mixture. The authors found that if all reagents were mixed at room temperature, followed by heating at 260 °C, the resulting product showed poor crystallization and wide size distribution, highlighting the beneficial role of their injection approach. TOPO affected the particle size only when using a certain molar ratio for OAm/OAc. The introduction of a given amount of CTAB in the reaction mixture, with a set OAc/OAm ratio of 1:16, enabled the tuning of the particle morphology from spherical NPs to triangles and rods.33
Wu et al. published a work in which propargyl focal point PAMAM-type dendrons with four carboxylic acid end groups displaced OAc and OAm on the surface of Fe3O4 NPs via the ‘grafted onto’ method.34 The PAMAM-type dendrons were chelated to the particle surface in a way that favored their stabilization. Actually, molecules that bear multiple carboxyl groups have stronger binding abilities and can be adsorbed more effectively on the surface of NPs. The resulting NPs in this work were very stable in water and doxorubicin (DOX) could be loaded in them, enabling thermosensitive drug release activity.34 Monodisperse hollow magnetite particles with sizes of 7 and 10 nm were produced through a high-temperature method involving ODE, a controlled OAm/OAc ratio, trimethylamine oxide and iron pentacarbonyl. Zwitterionic dopamine sulfonate (ZDS) was used to replace OAc/OAm and provide water solubility to the particles. This material demonstrated an efficient performance as a T1-weighted contrast agent for MRI, owing to its low saturation magnetization and spin-coupling shells.35
A solvent-free method was presented by Maity et al. for the preparation of Fe3O4 NPs with a defined size and shape. In fact, no ‘conventional’ solvent was used, but OAm and OAc were both used in excess, thus serving also as a solvent mixture due to their liquid nature at a broad temperature range, including room temperature. Fe(acac)3 was the iron source. The particle size was controlled by modifying the reaction temperature or time. The resulting NPs showed an increase in saturation magnetization with an increase in the reaction time and temperature. This was assigned to the enhancement in the crystallinity of the particles.36 Similarly, Nakaya et al. combined Fe(acac)3, 1,2-hexadecanediol and OAm/OAc to prepare magnetite NPs without the use of a ‘classic’ solvent. The excess ratio of ligands to metal ions could suppress nucleation through complexation with Fe ions and slowly induce NP growth.37 By adjusting the reaction time and temperature, monodisperse Fe3O4 NPs with a controlled size in the range of 5–20 nm were isolated. In another work, the combination of iron acetylacetonate with OAm/OAc was employed to provide magnetite NPs with a narrow size distribution in the range of 5–12 nm. The authors investigated the influence of the OAm/OAc ratio and they stated that carboxylic acid molecules are present as dimers in a non-polar solvent due to the hydrogen-bonding interaction, which results in a decrease in the electron-donating ability of the oxygen atoms in the molecules.38 The formation of an acid–base complex was highlighted also in that work, together with protonated oleylamine and carboxylate anions, depending on the OAc/OAm ratio used. When the OAc/OAm ratio was low (0.2), the particles appeared to be poorly coated by surfactants, with the formation of chain aggregates, poor dispersion and too small interparticle distances. The authors suggested that the concentration of carboxylic anion produced through the deprotonation reaction, which selectively binds to the particle surface competing with entities of weaker electron-donating ability, such as OAm, protonated OAm and acid–base complex, is low. Interestingly, the FTIR measurements in that report indicated that in the presence of the OAm/OAc ligand pair, the carboxylate groups would adsorb on the surface. The adsorption of OAm would be identified only when only OAm was used. However, in the case of mixed ligands, deprotonated carboxylic acid would preferentially bind to the NP surface. The acid–base complex formation between OAm and OAc can be represented as follows:39
| RCOOH + RCNH2 ↔ RCOO−:RCNH3+ | (1) |
| RCOOH ↔ RCOO− + H+ and RCNH2 + H+ ↔ RCNH3+ | (2) |
Gao et al. prepared monodisperse Fe3O4 NPs with a size in the range of 5–12 nm via a facile solvothermal approach using the OAm/OAc pair, TOPO, diethylene glycol, iron acetylacetonate and a temperature of up to 260 °C. The beneficial role of the amine/acid ligand pair was demonstrated, where if no OAc was used, the product did not show good dispersibility, as observed by TEM imaging. When excess OAc was added, the viscosity of the reaction medium significantly increased, hindering the diffusion of the reaction species, resulting in a significant degree of aggregation. Otherwise, a ‘reasonable’ amount of OAc would make the surface Fe atoms of the NPs coordinate with the COOH groups of OAc, creating a steric-stabilizing layer, which prevents aggregation and promotes the growth of monodisperse NPs. Still, the DEG–OAm–OAc reaction medium is quite viscous, but if it allows the diffusion of growth species, even at a slow rate, it will hinder particle aggregation.40
Fe(acac)3, hydrazine, EG, β-cyclodextrin, OAm and OAc were combined to synthesize 3D-assemblies of Fe3O4 nanocrystals in a one-pot process. The generation of spherical magnetite aggregates was largely dependent on the use of cyclodextrin, whereas OAm and OAc ameliorated the shape of the constituent magnetic NPs in the assembled spheres.41 Nano-Fe3O4-loaded tubular carbon nanofibers (nano-Fe3O4-TCNF) were produced by a suitable process involving the addition of TCNF to organic-phase reactions of Fe(acac)3 with 1,2-hexadecanediol, using also the OAm/OAc pair. Magnetite NPs were dispersed and loaded on the C surface. After annealing, the resulting nanocomposite was employed as a negative electrode for Fe/air batteries. The downsizing of the conductive magnetite particles was deemed to have contributed to the good electrochemical characteristics of the material. It seems that the thermal treatment at 500 °C did not deteriorate the quality of the surfactant-capped NPs.42 In another report, EG, iron acetylacetonate, OAm and OAc were combined in a solvothermal heating approach to prepare hydrophobic magnetite NPs. It was shown that although OAc could promote particle growth, it also increased the thickness of the organic coating, thus decreasing the magnetic properties of the particles. By tuning the OAc amount, cuboid shapes could be isolated. Stearic acid was also beneficial for the growth of the NPs. The ratio between the OAm and EG solvents affected the product composition, shape and magnetic properties.43
In 2009, Harada and Hatton reported that twofold symmetric rectangular NP superlattices could be formed by the slow evaporation of the carrier solvent from colloidal dispersions of OAm/OAc-coated magnetite NPs (Fig. 6). The two ligands were expected to align in parallel and alternating directions and lead to the formation of ladder-like molecular networks, as depicted in Fig. 6a. The two molecules interact strongly with each other via van der Waals forces between the neighboring aliphatic chains. As mentioned above, OAm and OAc have very similar chain lengths and the schematic representation of the NP assembly is consistent with the ‘real’ NP assembly structure derived by TEM. In fact, the van der Waals interaction forces between NPs are present over a large length scale, often inducing a close-packed arrangement for the NPs. In that work, the size of the superlattices reached microscale values with no evident disorders or defects, and it could be tuned by adjusting the amount of OAc in the evaporating colloid.44 Harris et al. presented a comprehensive qualitative model revealing the crucial role of the acid–base complex formation (Fig. 8) between the ligand pair under discussion. The authors reported that OAm binds only in a single motif on the particle surface, whereas OAc has three different binding motifs, i.e., monodentate, bridged and chelating. Ignoring the effect of electron affinity, it was suggested that OAc had a statistically greater probability to bind to surface iron atoms than OAm if they are competing for the same Fe(II) or Fe(III) species on the NP surface. A surfactant double layer could be created with OAc molecules near the NP core and OAm entities, providing an outer layer not bonded to the NP surface atoms. The reason given for the unlikely existence of OAm on the surface layer was that it cannot be understood how it would bind to Fe3O4 and that OAc cannot form a carboxylate with OAm and with the magnetite surface simultaneously. Due to the generation of the acid–base complex with OAm, plenty of OAc molecules have a dissociated hydroxyl group compared to only OAc, and this promotes adsorption. In addition, when the electrostatic pressure and steric hindrance increase, some OAc molecules are forced closer to the NP surface Fe atoms, and thus more bonds can form between the iron atoms and OAc species. Still, the authors stated that an OAc-only system does not possess a lower binding energy in comparison to OAc/OAm systems. The addition of OAm resulted in more stable NPs. The use of OAc alone led to the formation of a viscous red-brown product, which was difficult to purify and characterize, while the exclusive use of OAm produced NPs in a quite low yield. The authors discussed the phenomena taking place with the use of different OAm/OAc ratios, and they concluded that an optimum ratio permits a good trade-off between NP surface charge, free proton concentration in the dispersion medium and ζ-potential.39
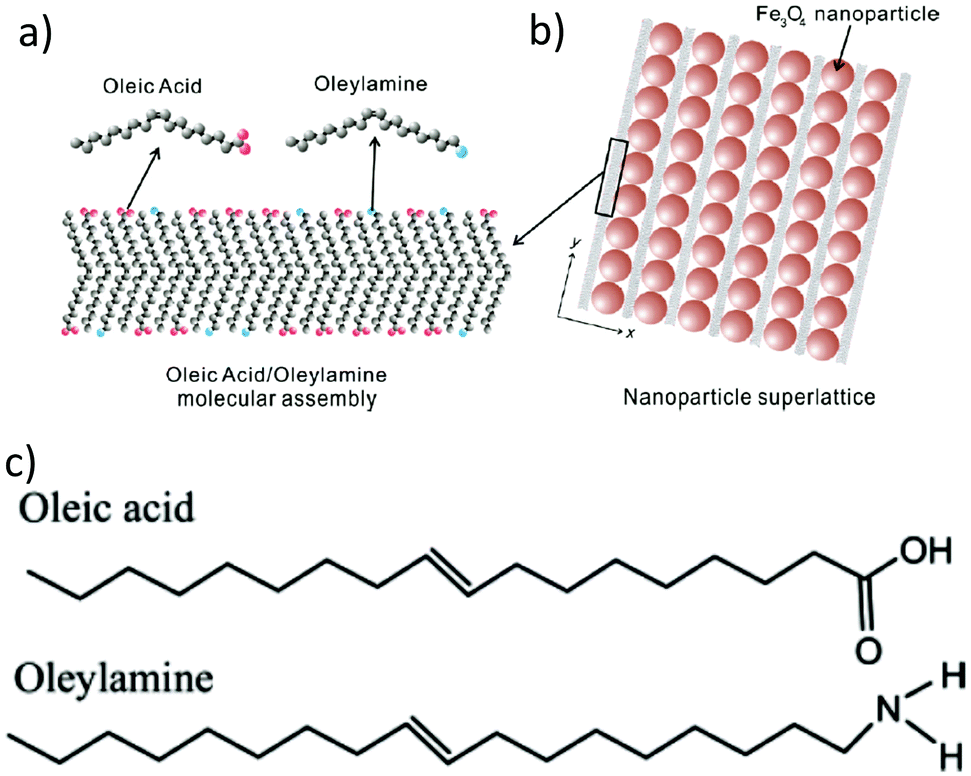 | ||
| Fig. 6 (a) Schematic illustration of a molecular assembly of OAc and OAm and (b) NP superlattice constructed by the cooperative assembly of NPs and fatty molecules. Reproduced with permission from ref. 44. Copyright 2009, the American Chemical Society. (c) Molecular structure of OAc and OAm. Reprinted with permission from ref. 41. Copyright 2005, the American Chemical Society. | ||
Asadi and co-workers found that an optimum OAc/OAm ratio of 3 permitted the tuning of the particle size with nanometer accuracy by just modifying the reaction heating rate. Fig. 7 demonstrates the characteristic TEM images of their samples, which were produced in the presence of iron(III) acetylacetonate, 1,2-hexadecanediol and benzyl ether.45 The authors discussed that the free protons (H+) are neutralized in the reaction medium by OAm through the formation of RCNH3+ or by the desorption of a bound OAc moiety (RCOO−) from the NP surface. Thus, it was clear that besides the type and concentration of the ligand in the reaction mixture, the ratio between the two ligands exhibited a critical effect on the reaction evolution, and consequently on the size, shape and distribution of the NPs. Using the above-mentioned optimum surfactant ratio, the binding energy of oleate on the NP surface was maximized, yielding the most stable growth species, with decelerated but homogeneous growth kinetics. Excess OAc provoked an increased concentration of free protons in the reaction pot. Then, due to the limited amount of amine species, the free protons would not be fully neutralized. Consequently, the electrostatic pressure and chemical potential between the medium and the NP surface increased, causing the desorption of the bonded oleate from the surface. In this way, the NP surface would become unprotected, leading to anisotropic colossal growth and large-faceted NPs.45
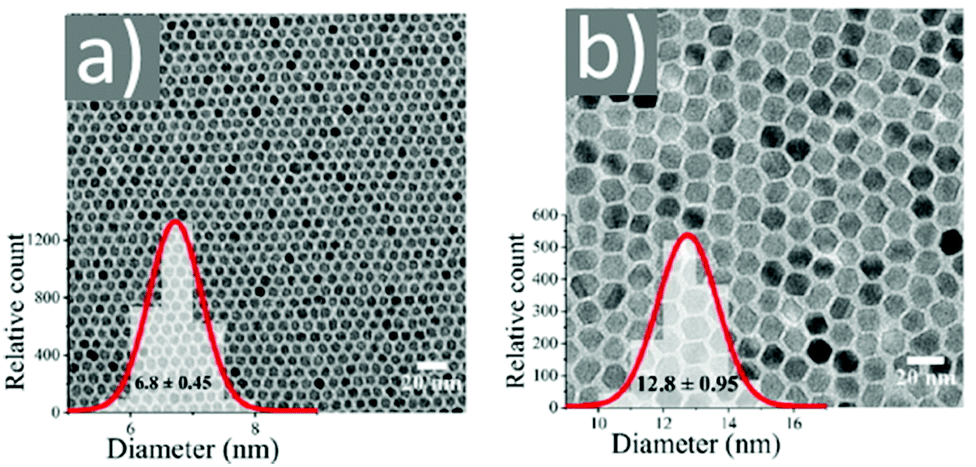 | ||
| Fig. 7 TEM images of samples obtained at OAc/OAm ratio of 3 at different heating rates. Reprinted with permission from ref. 45. Copyright 2018, the American Chemical Society. | ||
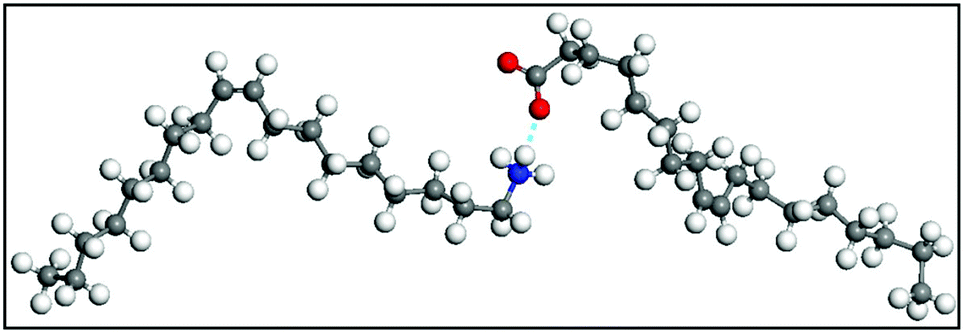 | ||
| Fig. 8 Acid–base complex formation between two OAc molecules. The blue dotted line denotes the hydrogen bond between the deprotonated H+ and oxygen molecule. The blue atom is nitrogen. Red atoms are oxygen. Grey atoms are carbon and white atoms are hydrogens. Reprinted with permission from ref. 39, Copyright 2015, the American Chemical Society. | ||
In another report, magnetite NPs covered by OAm, OAc and 1-stearoyl-2-hydroxy-3-glicerno-sn-phosphocholine (18LPC) could interact with the abundant human plasma proteins HSA (human serum albumin) and HTF (human transferrin), where the type and strength of the interaction was dependent on the nature of the protein. Significant binding was confirmed for HSA-iron oxide NPs, as revealed by fluorescence quenching experiments. The amphiphile-coated particles were envisaged for potential biomedical applications as biocompatible nanodevices.46 Sondari and colleagues discussed that the bonding pattern of the carboxylic acids on the Fe3O4 NP surface was a combination of molecules bonded symmetrically and molecules bonded at an angle to the surface. According to their results, OAc was chemisorbed on the particles as a carboxylate and their NPs could be used as wastewater treatment agents.47 Another work reported the synthesis of spherical 16 nm and cubic 20 nm NPs in the presence of the ligand pair under discussion. It was mentioned that OAc is known to coordinate to the [111] facet of the growing crystal, which favors the generation of cubes and octahedral nanostructures. Also these researchers mentioned that OAm coordinates in no preferential mode to all the crystal facets, resulting in the production of nanospheres. When bigger sizes were pursued, the use of less OAm was proposed, providing low polydispersity and the generation of a minor portion of octahedral particles and nanocubes. In that case, the concentration of OAc affected the NP size to a lower extent compared to OAm.48
A continuous flow approach for the synthesis of OAm/OAc-coated magnetite nanocrystals was also published, where the authors mentioned that when the concentration of OAm/OAc reached 1.2 M, the viscosity of the reaction medium increased, given that the diffusion coefficient of the solute is inversely correlated with the solvent viscosity. When the viscosity of the solvent increased, the nanocrystal growth rate decreases and NPs of smaller sizes were produced. However, in a lower concentration range (0.3 to 0.6 M for OAm/OAc), the reactivity of the monomer decreased when the concentration increased to 0.6 M. This occurs because of the stronger binding effect. Consequently, the supersaturation degree of the reaction medium decreased, favouring the growth of bigger particles due to the decreased number of nuclei created during the nucleation step.49 Rossi and co-workers presented an economically appealing route to produce Fe3O4via the thermal decomposition of Fe(acac)3. The role of iron(III)-oleate as the actual precursor to obtain the NPs was analysed when OAc was combined with iron acetylacetonate. Combining OAc and OAm led to an increase in the precursor decomposition temperature to T > 200 °C, preventing the generation of Fe-oleate, which would decompose at lower temperatures. Still, the presence of both OAm and OAc was crucial to achieve good size control. Adding mono or di-alcohols did not impair the OAm–OAc interaction or the decomposition temperature, ameliorating the size control, and relatively cheap molecules could function quite well such as 1,2-octanediol and cyclohexanol.50
Sweigart and co-workers reported that HBF4 can generate ammonium salts with OAm and possibly result in NP solubility in DMSO by forming a bilayer with the aliphatic chains of OAc on the NP surface in DMSO. The ligand exchange process applied endowed solubility to the particles in polar solvents such as DMSO and acetone, whereas the NPs were not soluble in non-polar solvents such as THF, dichloromethane and hexane. Dispersibility in polar aprotic organic solvents such as DMSO and dimethylformamide (DMF) facilitates the processing of the obtained NPs for applications in catalysis, gas storage and electronic materials.51 Mohapatra et al. published a seedless thermolysis route to prepare monodisperse Fe3O4 NPs in the presence of OAm and OAc. The particles had a controlled size in the range of 3–32 nm and their heating efficiency was measured. The authors suggested that excess OAm can increase the deprotonation of OAc and result in the formation of acid–amine complex. With an OAm/OAc ratio of 1.2, the increased deprotonation of OAc molecules and their highest electron-donating ability resulted in preferential binding with cations on specific Fe3O4 facets and led to anisotropic shapes. The acid–amine complex pair maintained the pH value of the reaction medium at 8–9, promoting the formation of crystalline magnetite. A higher OAc concentration could shift the pH to acidic conditions, increasing the NP size. Alternatively, excess OAm offered a sufficient reductive environment, allowing the decomposition of Fe(acac)3 even at the moderate temperature of 200 °C.52 7.1 nm magnetite NPs were prepared using the same precursor together with the ligand pair discussed in this review. The replacement of the OAc/OAm pair by polymeric ligands (Fig. 9) resulted in an important increase in Dh. The interparticle distances in Langmuir–Schaefer monolayers could be tuned in this way, as studied by atomic force microscopy (AFM) and grazing-incidence small-angle X-ray scattering (GISAXS) technique.53
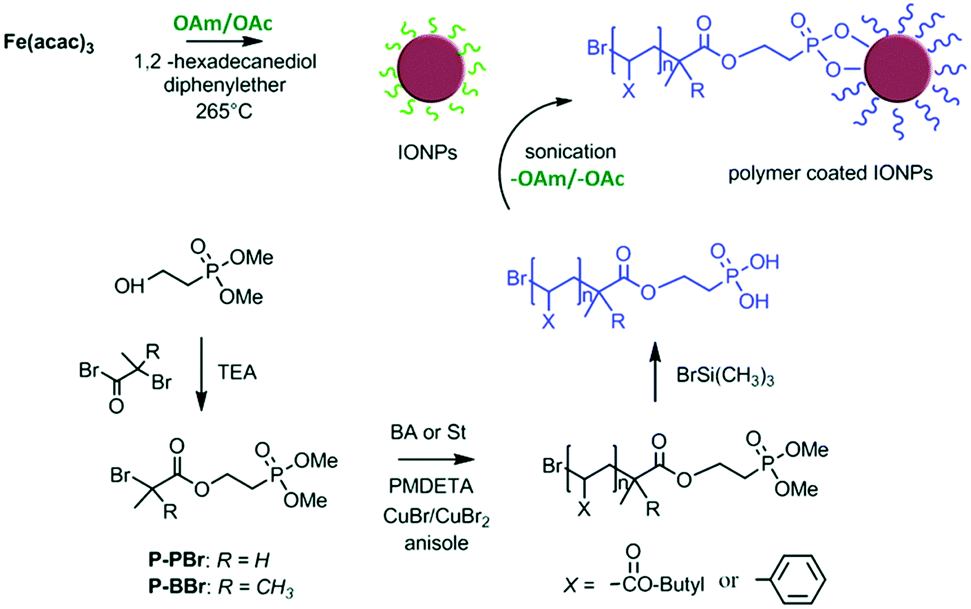 | ||
| Fig. 9 Schematic illustration of iron oxide NPs coated by poly(n-butyl acrylate) or polystyrene ligands.53 Copyright 2019, The Royal Society of Chemistry. | ||
Eom et al. used the ligands under discussion to prepare magnetite and cobalt ferrite NPs using benzyl ether as a solvent. A spherical shape was observed for CoFe2O4 NPs when both OAc and OAm were employed, whereas the use of pure OAc resulted in highly crystalline nanocubes. In the case of Fe3O4, the sole use of OAc resulted in either nanocubes or hexagonal shape, depending on the synthesis temperature. Higher temperatures induced the formation of nanohexagons.54 Klokkenburg et al. reported the FTIR characterization of magnetite NPs dispersed in cyclohexane, which were prepared using OAm/OAc pair. The most relevant peak assignments for both in situ and ex situ spectra are depicted in Fig. 10.55
 | ||
| Fig. 10 Assignment of the most relevant bands observed in the FTIR spectra of OAm/OAc-coated Fe3O4 NPs. Reprinted with permission from ref. 55. Copyright 2007, Elsevier. | ||
Therefore, for magnetite nanostructures, one can summarize that OAm/OAc can control the morphology of the produced particles, usually by tuning the ratio between the two ligands as they normally bind in different ways on the facets, thus favouring the growth in distinct modes. The interparticle distances and protection against aggregation are also controlled, and an acid–base complex may often form as a result of the reaction between the two ligands. OAm provides more reductive conditions in the medium, affecting the precursor decomposition temperature. The particles may also be transferred in water by applying ligand exchange processes. Table 1 presents the main information related to the synthesis of the various magnetite nanostructures produced using OAm/OAc and discussed in this review.
| Size/morphology | OAm/OAc ratio | Fe precursor/temperature | Ref. |
|---|---|---|---|
| 2–16 nm/nearly spherical | 2/3 | FeCl2/240 °C | 17 |
| 10 nm/mainly hexagonal | 1/1 | Fe(acac)3/300 °C | 18 |
| 6.5 nm/spherical | 1/1 | Fe(acac)3/265 °C | 19 |
| 5 nm/not specified | 1.5/1 | Fe(acac)3/300 °C | 20 |
| 12 nm/cubic | 2.5/1 | Fe(acac)3/260 °C | 21 |
| 7–12 nm/rhombic, triagonal, hexagonal | 1.5/1 and varying | Fe(III) isopropoxide/140–220 °C | 14 |
| 5–20 nm/nearly spherical and cubic-like | 2.5/1 | Fe(acac)3/200 °C | 22 |
| 3–10 nm/faceted and cubic | 1/1 | Fe(acac)3/180 °C | 23 |
| 3–20 nm/slightly faceted | 1/1 | Fe(acac)3/265 °C | 25 |
| 8–27 nm/cubic-line to slightly faceted | From 4/1 to 1/4 | Fe(III) oleate or Fe(III) mandelate/320 °C | 28 |
| 7–37 nm/triangular prism to quasi-spherical | Varying in a range | Fe(acac)3/240 °C | 29 |
| 4 nm/spherical | 1/1 | Fe(acac)3/245 °C | 31 |
| 10–50 nm/spherical, octahedral, cubic | From 0/1 to 2/1 | Fe(acac)3/Reflux T | 32 |
| ∼10 nm/spheres, triangles, rods | Varying | Fe(acac)3/260 or 315 °C | 33 |
| 7 and 10 nm/hollow | Varying | Fe(acac)3/240–250 °C | 35 |
| 5–20 nm/spherical | 1/1 | Fe(acac)3/200–300 °C | 37 |
| 5–12 nm/spherical to faceted and cubic | Varying | Fe(acac)3/250–320 °C | 38 |
| 5–12 nm/nearly spherical | 2.5/1 | Fe(acac)3/260 °C | 40 |
| Varying sizes/mostly spherical to agglomerated-irregular | From 5/1 to 1/5 | Fe(acac)3/180 °C | 39 |
| 7–13 nm/mostly spherical to faceted | From 1/3 to 1/1 | Fe(acac)3/300 °C | 45 |
| 4–8 nm/nearly spherical to faceted | 1/1 | Fe(acac)3/250 °C (flow synthesis) | 49 |
| 3–32 nm/nearly spherical | From 6/1 to 3/1 | Fe(acac)3/240 °C | 52 |
2.2 Fe2O3 nanostructures
Fe(III) oxide can occur in the form of hematite (α-Fe2O3), which crystallizes in the rhombohedral lattice system and has weak magnetic properties. It can also be found in the form of maghemite (γ-Fe2O3), which is a polymorph of hematite but it possesses a spinel structure, like magnetite, and has strong magnetic properties.Wu et al. published the synthesis of α-Fe2O3 oblique and truncated nanocubes in the presence of acetylacetonate (Hacac), OAm and OAc using a solvothermal protocol (Fig. 11). In the absence of OAc, truncated nanocubes with certain exposed facets were produced. In fact, OAc has a strong chelating ability to Fe atoms and its non-polar tail group provides steric hindering, thus endowing an excellent stabilizing function. The crystal phase of the iron oxides produced were accurately regulated by adjusting the volume ratio of OAm/Hacac, as shown by XRD and Raman monitoring measurements. In particular, for high OAm/Hacac ratios, pure Fe3O4 would be isolated because the excess amount of OAm offers quite strong reductive conditions for the reduction of Fe3+ to Fe2+.56
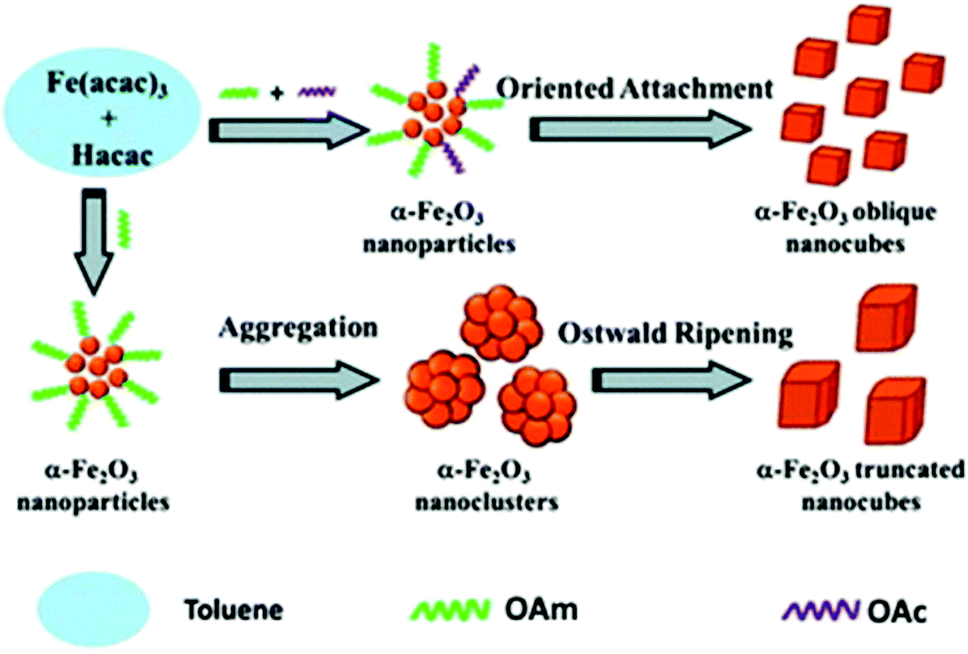 | ||
| Fig. 11 Scheme depicting the process for the generation of oblique and truncated hematite nanocubes.56 Copyright 2013, The Royal Society of Chemistry. | ||
Petridis and colleagues confirmed through FTIR measurements that OAc is bound to the NP surface through carboxylate anions, in a chemisorption manner. Its coordination mode is reported to be chelating bidentate. Alternatively, OAm appeared to be bound on the particle surface via the unpaired electron group of the amine moiety. Regarding the magnetic properties, the type of capping ligand (either OAm or OAc) did not modify the particle coercivity, while the saturation magnetization did not seem to be affected by the type of ligand, but rather by the particle size.57 In another report, the use of OAc and OAm helped to incorporate maghemite NPs in a host template composed of polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) diblock copolymer. Also, in that work, FTIR showed that the particle surface was dominantly coated by OAc, which coordinates to the surface in a stronger way compared to OAm. The high affinity between the PS block and organic ligands facilitated the good dispersibility of the NPs in the block copolymer.58
Actually, the OAm/OAc coating of γ-Fe2O3 NPs can also affect the maghemite-hematite phase transition behavior. For example, laser irradiation of 15 mW could induce this transition for the uncoated 4 nm NPs, but 120 mW would be needed for coated NPs of a similar size to make this transition happen.59 Meledandri et al. prepared maghemite NPs via the thermal decomposition of Fe(acac)3 in the presence of OAm/OAc. By varying the surfactant/precursor ratio, they managed to obtain particles in the range of 3–6 nm. Their saturation magnetization was in the expected range of values for γ-Fe2O3 NPs of this size.60 Also, when applying the microwave-assisted synthesis of maghemite NPs, the addition of OAc helped to limit the extent of NP aggregation, in comparison to the use of only OAm. In that work, an increase in the amount of OAc led to a shift from superparamagnetic to paramagnetic properties.61 Zhang et al. used an MW method to produce maghemite NPs in the presence of iron acetylacetonate, OAm, OAc and biochar (Fig. 12). The resulting product was a nanocomposite consisting of 6–17 nm particles and biochar. They suggested that OAm acts as a reductant and weak capping agent, while OAc plays the role of main capping agent. The single use of OAm led to larger NPs due to its weak capping function. In that paper, the presence of oxygen in air perturbed the reducing action of OAm, resulting in the most oxidized form of iron.62 Under similar conditions, the use of inert Ar gas led to the formation of a wustite (FeO) composition, as shown by the group of Shouheng Sun in 2007.63 Wustite NPs could be stable for one month in an organic solvent in air. The amino group of OAm was deemed to hinder rapid oxidation.64 The distinct capping abilities between OAm and OAc would cause selective facet passivation during growth, leading to varying NP morphologies.62
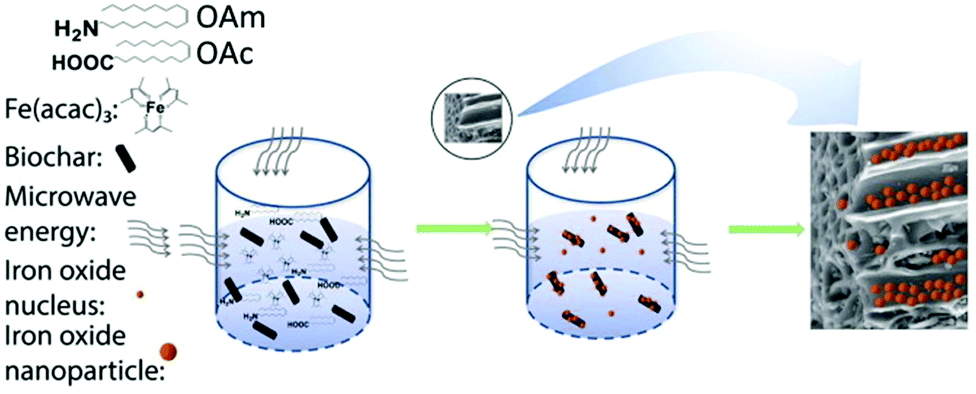 | ||
| Fig. 12 Growth mechanism for iron oxide NP-biochar nanocomposites. Reproduced with permission from ref. 62. Copyright 2019, Elsevier. | ||
Regarding the bonding mode of the surfactants, Marchetti and co-workers mentioned that Fe3+ surface ions can be complexed with OAc via carboxylate heads with monodentate and bidentate coordination, whereas their complexation with alkylamine ligands takes place through –NH2 groups. In their work, FTIR spectra denote the prominent presence of OAc as a capping ligand (with OAm being possibly a minor surface component). The minimum distance between the surfaces of two adjacent NPs was around 3 nm (Fig. 13). The presence of interparticle interactions was confirmed by magnetic measurements.65
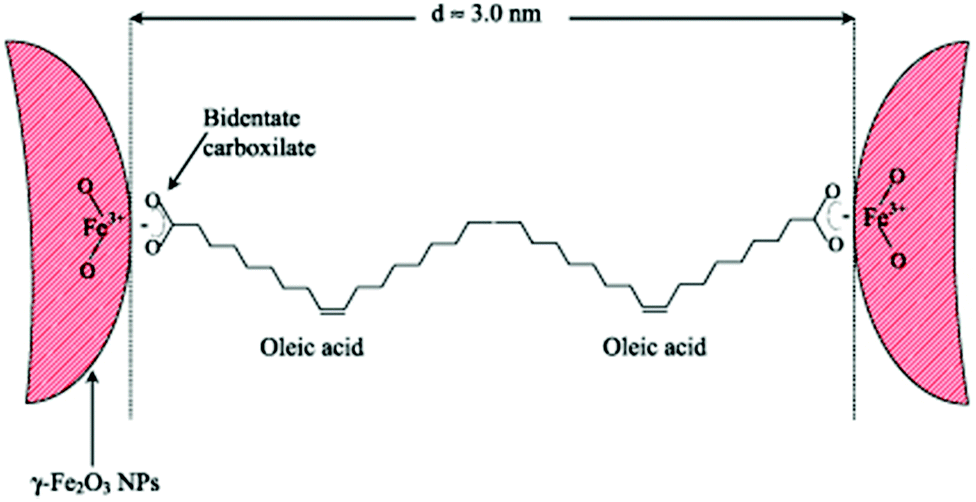 | ||
| Fig. 13 Scheme depicting maghemite NPs coated by OAc, in an arrangement at the minimum distance between them. Reprinted with permission from ref. 65. Copyright 2013, Institute of Physics. | ||
The use of oxidizing agents such as trimethylamine-N-oxide (TMAO) can also help to isolate NPs of iron(III) oxide composition, as shown by Mathur and co-workers. In their work, iron(II) and iron(III) oxalates were the Fe sources. The role of OAc and how its oleate groups can enable ligand exchange with TMAO to occur was discussed. The role of ligands and how they can radically affect the precursor decomposition temperature, and thus the reaction pathway and the resulting NP size and crystalline phases was analyzed.66 Superparamagnetic iron oxide NPs coated with OAm and OAc could also be formulated in vitamin E (D-α-tocopheryl-co-poly(ethylene glycol) 1000 succinate) micelles through a facile solvent-exchange method. The resulting composites showed potential for nanothermotherapy and MRI applications, exhibiting improved thermal, magnetic, cellular uptake, cytotoxicity, therapeutic and imaging properties than commercial and other materials previously reported in the literature.67 In a synthesis involving imidazolium ionic material as the reaction medium, OAm and 1,2-hexadecanediol were crucial to achieve the formation of maghemite nanorods. In that work, different shapes were also produced, such as nanocubes and nanospheres, and the presence of OAc played a significant role in the control of morphology. For instance, the formation of cubes was attributed to the preferred stabilization of iron oxide {100} surfaces by OAc.68 In the case of iron-iron oxide core–shell NPs produced using Fe(CO)5 as an iron precursor and ODE as the solvent with OAm/OAc surfactants, the presence of a crystalline Fe3O4 shell offers better stability toward deep core oxidation for prolonged time.69
Another iron salt, that is, Fe(II) acetate, was used as a precursor to prepare iron oxide NPs with a size in the range of 5–43 nm. 1,2-Dodecanediol was used as the reducing agent and combinations of OAm, OAc, TOP and polyvinylpyrrolidone (PVP) were used as dispersants. The minimum temperature to obtain crystalline NPs was 240 °C. Increasing the amounts of OAm and TOP was associated with an increase in particle size. It has been proposed that OAm and TOP may form stable complexes with the individual metal atoms of the precursor prior to nucleation. These complexes can increase the decomposition temperature, thus suppressing nucleation and leading to larger NPs.70 Farber et al. commented that a drawback of OAc-coated NPs is the absence of functional groups for the chemical attachment of proteins and ligands to their surface. However, for example, Pluronic F127 coating can associate with the hydrocarbon tails of OAc via its hydrophobic polypropylene oxide chains. In their work, they managed to coat iron oxide NPs with hydrophobized aldehyde-rich dextrans. These particles were initially treated with OAc and had a spherical shape with a size in the range of 15–45 nm. The magnetic properties of the particles were mostly retained after their functionalization with dextran. Therefore, the combination of attractive magnetic properties, capacity for further surface binding of biological entities and particle stability in water render these nanomaterials particularly interesting for magnetically-assisted drug delivery.71
Zanchet and colleagues decomposed Fe(acac)3 in the presence of 1,2-hexadecanediol, OAm, OAc and benzyl ether for the production of 5–10 nm magnetite NPs. The authors used 2,3-dimercaptosuccinic acid (DMSA) to transfer the NPs to water. The particles were superparamagnetic at room temperature and the preliminary tests to incorporate HeLa cells and MRI measurements were promising.72 In fact, OAm is known to form deprotonated OAc given that the carboxylate moiety has greater reactivity than the carboxylic acid group for the surface of Fe-oxide NPs. When both diol and OAc are added in the reaction mixture, it can be suggested that Fe binds to both the diol and oleate simultaneously, and these two entities can control the formation of NPs. As mentioned above, FTIR spectroscopy has demonstrated that the carboxylate group of oleate molecules is chelated to Fe in a bidentate mode. When dispersed in a hydrophobic solvent such as heptane, the long carbon chains of oleic acid in OAc-capped NPs are extended in the solvent. At high temperatures, it is proposed that the diffusion of oleate complexes is enhanced and that a big population of complexes deposits on the surface of small NPs, followed by the creation of an oleate bilayer.73 Sun and co-workers proposed that the generation of NPs advances via the replacement of the acetylacetonate of the iron salt by 3 equiv. of OAc/OAm, which are needed to secure monodisperse particle sizes. The authors mentioned that iron oxide particles are produced by the assembly of their respective atomic species. These species are produced through the partial reduction and decomposition of iron oleate in the presence of OAm. The addition of a biotinylated-phospholipid, DSPE-PEG(2000)biotin, to the initial hydrophobic iron oxide NPs provided a robust double-layer structure on the surface of the particles, where its inner layer is the starting oleate/OAm coating and the outer layer is the phospholipid. This surface treatment can help make these NPs suitable for the attachment of biomolecules and act as a label for highly sensitive biosensing performance, separation and imaging applications.74
As a summary for α-Fe2O3, it can be noted that also in this case, the different binding modes between OAm and OAc lead to varying passivation degrees in different facets, affecting the particle morphology. FTIR is an important tool to study the binding modes of the ligands. The role of the complexes formed between ligands and metals prior to nucleation has also been discussed. These nanomaterials can be further functionalized with suitable biomolecules, thus becoming water-dispersible and biocompatible.
2.3 Metal ferrite nanostructures
Maenosono and co-workers argued that the coordination ability of TOPO to the surface of iron oxide and metal ferrite NPs is much more reliable than that of OAm or OAc. Their MFe2O4 NPs (M = Mn, Fe, Co, Ni and Zn) had sizes in the range of 3–4 nm and their synthesis was low-cost, MW-mediated and employed 1-dodecanol. The authors envisaged that the fine tuning of the structure of these ferrite NPs and their corresponding magnetic properties will render them promising materials for magnetic data storage, catalysis, sensing, and ferrofluid-related applications.75 In another report, NiFe2O4 NPs were prepared by decomposing nickel and iron acetylacetonates in the presence of OAm and OAc, which functioned simultaneously as solvents, stabilizers and reductants. Tuning the OAc/OAm ratio helped to control the size and size distribution of the 10 nm particles. The upgrading of a heavy crude oil reaction with low hydrogen pressure was the catalytic property investigated for these NPs.76 Cannas et al. demonstrated that 1H NMR is a technique that can be employed to identify signals denoting the presence of hydrogen bonding, arising from the existence of the acid–base complex. As mentioned above, the reaction of an OAc molecule with an OAm molecule can lead to the generation of the aforementioned complex. Interestingly, FTIR spectra showed that the OAc group can undergo modification to an elaidic group upon bonding to the NP core. The authors showed that their CoFe2O4 NPs are capped by a shell of residual surfactant (arising from the OAc and OAm mixture) and this layer also remained unmodified after a silica-coating step. The cytotoxicity of these NPs was studied on human cells using trypan blue and MTT assays.77 Cobalt ferrite and iron manganese oxide NPs, capped by OAc and OAm, were stable in toluene for several months at room temperature, as reported by Arenholz and co-workers. These researchers used single-source precursors to synthesize their particles and the effect of the decomposition temperature, reaction time and precursor amount were investigated. It was found that the stoichiometry of the cobalt ferrite was a function of precursor concentration, whereas the stoichiometry of iron manganese oxide was not dependent on it.78 The group of Begin-Colin demonstrated that oleate entities coming from OAc and precursors offer a reducing atmosphere, resulting in core–shell NPs, with a CoxFe1−xO@CoyFe3−yO4 composition. The use of OAm ligand promoted the decomposition of the precursor and less reducing medium, with a higher oxidation of ferrous cations, yielding NPs with a homogeneous composition, without a core–shell structure. This is not in full agreement with above-mentioned reports, which focused on the reductive conditions offered by the presence of OAm. The authors preferred the use of commercial iron stearate rather than Fe-oleate, given that the latter has been reported to be less stable because of its high sensitivity to the synthesis, washing and storage conditions. Amine species promoted the decomposition of stearates, and therefore increased the monomer concentration, resulting in the generation of a big population of nuclei with a small size, according to the La Mer theory. Ways to control the particle shape from spherical to cubic were also presented in this work.79Liang et al. commented that in OAm-mediated decomposition, the amine can initiate nucleophilic attack on the carboxylic groups of the acac ligand upon gaining energy, thus resulting in crystal nucleation and growth. Actually the broad use of metal acetylacetonates as precursors for iron oxide and ferrite NPs is owing to their environmentally benign nature and low sensitivity to moisture. In that work, liquid cell TEM was employed for particle characterization, which is a technique that enables NP growth to be monitored in situ, which can help to reveal the growth mechanisms taking place.80 In another report, MnxFe1−xO concave nanocubes (Fig. 14) were prepared through a thermolysis process. The size and shape of the resulting particles were tailored by modifying the OAm/OAc ratio, precursor concentration, reaction temperature and reaction time. Highly branched cubic mesocrystals with sizes larger than 100 nm were produced by mixing 1-ODE as a co-solvent with OAm/OAc. The addition of octadecene made the nanocubes less protected by OAc, and their high surface energy initiated aggregation, and thus the formation of highly branched cubic aggregates. It was reported that a 1:1 volume ratio of OAc and OAm provides a sufficient amount of OAc to regulate the competitive growth in the 〈111〉 direction over the 〈100〉 direction, causing the generation of a truncated octahedron shape. Actually, the carboxylate groups of OAc can create stable complexes with the Fe2+ and Mn2+ ions on the MnxFe1−xO nuclei. Thus, the ratio of OAc/OAm should not be too high, otherwise the formation of nanocrystals would be prevented. Alternatively, OAm, as a weaker ligand, permits the fast decomposition of metal oleate complexes, causing a homogeneous nucleation of MnxFe1−xO through the quick decomposition of a big fraction of the precursors. A large amount of OAm would lead to growth of all crystal planes in the same manner, causing the formation of a spherical shape. The particle morphology can be also varied by modifying the ratio between manganese and iron cations, given that these cations possess distinct binding strengths with OAc.81 MnFe2O4 NPs with sizes in the range of 5–12 nm were capped by OAm and OAc during their synthesis, and subsequently these ligands were replaced by meso-2,3-dimercaptosuccinic acid, enabling the dispersion of the NPs in aqueous medium. This replacement left carboxyl groups to extend from the NP surfaces, offering electrostatic repulsion suitable for dispersion in water. Phenyl ether and 1,2-hexadecanediol were also used as reagents in that work, besides Mn- and Fe-acetylacetonates.82 Kawabe and co-workers prepared nickel zinc ferrite NPs via co-precipitation and the particles were simultaneously coated on carbon microcoils (CMC). SEM imaging illustrated that the use of OAm and OAc facilitated better coverage of the NPs on the CMC. An optimum ratio between the particles and CMC was found to achieve the highest coverage of the CMC by the NPs. In that case, Ni-, Zn- and Fe–nitrate salts were used as precursors.83
 | ||
| Fig. 14 TEM images of Mn0.15Fe0.85O concave nanocubes with different shapes and sizes by modifying the total concentration of precursors at a constant heating rate.81 Copyright 2014, The Royal Society of Chemistry. | ||
In 2004, Sun and co-workers published the synthesis of magnetite (Fig. 15a) and MFe2O4 (M = Fe, Co, Mn) NPs in the presence of metal acetylacetonate salts, 1,2-hexadecanediol, OAm and OAc in phenyl or benzyl ether solvents at high temperature. A seed-mediated growth approach was employed to increase the particle size from 3 up to 20 nm. The use of the OAm/OAc ligand pair was crucial for the generation of well-defined NPs. The exclusive use of OAc led to a viscous red-brown product, which was hard to isolate and characterize. Conversely, the single use of OAm resulted in Fe-oxide particles at a quite lower yield compared to the reaction in the presence of both OAm and OAc.39,84 Subsequently, the same group followed the above-mentioned synthetic route and published another paper focusing on the shape-controlled preparation and shape-induced texture of MnFe2O4 NPs. In that work, the particle size was mainly tailored by modifying the precursor concentration, whereas the shape was tuned by adjusting the quantity of the stabilizing agents. The careful evaporation of the carrier solvent (hexane) resulted in the formation of nice NP superlattices.85 Iwamoto et al. prepared spinel ferrite NPs with Nd and B elements doped in their crystal structure. The polyol process was used, involving tetraethylene glycol (TEG), OAm and OAc, together with the precursors of each element (Fe, Nd, and B). It was found that prolonging the refluxing time led to an increase in the particle diameter. As expected, at a fixed amount of OAm and OAc, the ligand content per NP surface area decreased as the mean particle size increased. Regarding the magnetic properties, the magnetic anisotropy was enhanced by inserting Nd and B elements in the spinel, and the ferrite NPs possessed higher coercivity in comparison to the magnetite NPs.86 The production of Fe(II), Co(II) and Ni(II) ferrite NPs (Fig. 15b) starting from (multi)metallic single-source precursors was achieved in the presence of OAm/OAc, benzyl ether and 1,2-dodecanediol. The particle size was in the range of 9–25 nm. The complexation of metal ions by oleate or OAm renders the M-ion soluble in non-polar solvents. Variations in the magnetic properties among the different samples were attributed to their morphological changes. In fact, the substitution of Fe(II) in the spinel structure by Co(II) led to a radical increase in coercivity at low temperatures, whereas substituting Ni(II) reduced the coercivity values.87
 | ||
| Fig. 15 (a) 12 nm Fe3O4 NPs. Reprinted with permission from ref. 84. Copyright 2004, the American Chemical Society. (b) Fe3O4 NPs. Reproduced with permission from ref. 87. Copyright 2011, Elsevier. (c) CoFe2O4 NPs.88 Copyright 2015, The Royal Society of Chemistry. | ||
Crouse and Barron studied the effect of tuning the total ligand concentration and the ratio OAm/OAc, aiming to achieve improved control of the size distribution of Co–Fe–O NPs. 1,2-Hexadecanediol was also used, together with iron- and cobalt-acetylacetonates, and different synthetic conditions were explored. It was suggested that metal alkoxides could have been formed in the presence of diol. The authors indicated that under certain conditions, the diol may compete with OAm/OAc, possibly resulting in a bigger size distribution for the produced particles. Larger NPs with a tendency to form faceted shapes were obtained in OAc-rich conditions than in OAm-rich conditions. The role of OAm was critical when aiming to maintain a spherical shape. Adjusting the Co:Fe ratio also contributed to the growth mode to some extent. Specifically, the Co-rich compositions affected the particle size and shape.89
Another type of ferrites, that is, CuxFe3−xO4 NPs with sizes in the range of 5–7 nm were produced using Cu(HCOO)2 and Fe(CO)5 in the presence of OAm/OAc. Surface functionalization was performed using a hydrophilic multifunctional polymeric ligand containing PEG(800) moieties and a fluorophore. The final water-soluble, superparamagnetic and fluorescent particles showed potential as MRI contrast agents for specific cell targeting.90 It was suggested that even repeated washing cannot remove easily the covalently attached monolayer of OAm/OAc on ferrite particles due to the chemical bonding of the surfactants on the NP surface. In certain cases, a decrease in saturation magnetization was reported, which can be due to the surface spin disorder, often described as the so-called magnetically dead layer, and the increase in surface-to-volume ratio. The melting temperatures of pure OAc and OAm are 13–14 °C and 21 °C respectively. This implies that the Brownian relaxation of Zn-substituted iron oxide NPs contributes, at a higher T, to the magnetic moment recorded during ZFC–FC scans, which will affect the mode of the temperature dependence, in addition to significantly increasing the magnitude of susceptibility. Pyrolysis of the surfactants at 500 °C can generate a carbon coating around the NPs, yielding a promising sorbent for steroid contaminants and dyes through a magnetic solid-phase extraction process.91
Cobalt ferrite NPs with a monodisperse size have been prepared using the synthetic protocol under discussion in benzyl ether. The treatment with tetraethyl ortho-silicate (TEOS) resulted in the formation of CoFe2O4-SiO2 nanocomposites. The dilution in the silica matrix aimed to weaken the interparticle dipolar interactions. The effects of these interactions were evaluated after annealing the as-prepared and silica-diluted CoFe2O4 NPs at various temperatures.92
A more unusual composition, that is, Fe2TiO5 NPs, was produced using Fe(NO3)3·9H2O and Ti(OC3H7)4 through a sol–gel approach. Ethanol was the solvent and oxalic acid acted as the chelating agent. Surfactants as OAm/OAc, sodium dodecyl sulfate and CTAB were tested for their effect on particle size. A smaller NP size was obtained in the presence of SDS. The resulting particles were evaluated as photocatalysts for the decolorization of RhB and MB dyes under UV light irradiation.93 The self-organization of the colloids consisting of NPs larger than 4 nm could be explained in terms of the interplay between isotropic steric repulsive forces due to surfactant layers, isotropic attractive van der Waals and anisotropic attractive dipolar interactions. Besides the chain length of the OAc molecule, which is around 2 nm, as mentioned above, its width has been reported to be of 0.5 nm. To estimate the number of OAc molecules attached to the particles surface, one can assume that every adsorbed molecule covers an area of 0.5 nm2. The tail of an OAc molecule can take any orientation due to its thermal motion. It has been reported that oleic, dodecanoic and citric acid may decrease the surface spin disorder, leading to quite high magnetization values (at least normalized ones to non-organic content). Of course, the OAc adsorption efficiency can depend on the solvent used as well as on other synthetic parameters.94 Cobalt ferrite NPs with a size of 11 nm were prepared using the OAm/OAc ligand pair and were supported on Vulcan XC-72. The resulting nanocomposite had a good accommodation/dispersion of the NPs over the Vulcan support, owing to the existence of OAm/OAc and the use of a suitable liquid self-assembly method. The composite showed very good activity as an electrode material for lithium-air and lithium-ion batteries.95 Ligand stripping of the OAm/OAc-capped MgFe2O4 NPs with Et3OBF4 enabled the removal of the surface ligands, e.g., OAm/OAc or carboxylate residues from the particle surface. Aiming to isolate agglomeration-free dispersions of NPs in aqueous medium targeted for photocatalytic reactions, OAm/OAc surfactants could be substituted by citric acid. The surface stabilization positively affected the adsorption of compounds and boosted the degradation of contaminants.96 The formation of an acid–base complex was supported in another work by Verma and Pravarthana via FTIR spectroscopy, where OAm was found to deprotonate OAc. The complex functioned as a binary capping agent and the free surfactants were removed by applying washing steps.97
Thanh and co-workers reported the synthesis of CoFe2O4 NPs (Fig. 15c) by thermally decomposing the corresponding metal acetylacetonate salts in organic solvents, in the presence of OAm/OAc and 1,2-hexadecanediol or octadecanol.88 These authors highlighted the different strengths and selective binding energies of OAm and OAc with the surfaces of the NPs to achieve good shape control. Using high ligand quantities, the OAc species was suggested to rapidly attach to the lowest-energy {100} facets and prevented the growth of the particles in the [100] direction by creating a dense ligand layer, causing the generation of cubic NPs. When the metal salts complexed with OAc to generate metal oleates, it was proposed that the decomposition temperature of these complexes increased from 190 °C to 300 °C, which affected the growth evolution and the final particle monodispersity. A scheme illustrating the shape evolution of the nanostructures according to the solvents, reducing agents, ligand concentrations and reaction time is shown at Fig. 16.88 PMAO was used as an encapsulating agent for the water transfer of OAm/OAc-capped cobalt ferrite NPs. The reaction process for this encapsulation is depicted in Fig. 17 and the surface modification was confirmed by techniques such as FTIR and TGA. The water-dispersible NPs exhibited a very good performance in magnetic hyperthermia and showed minimal cell cytotoxicity.98
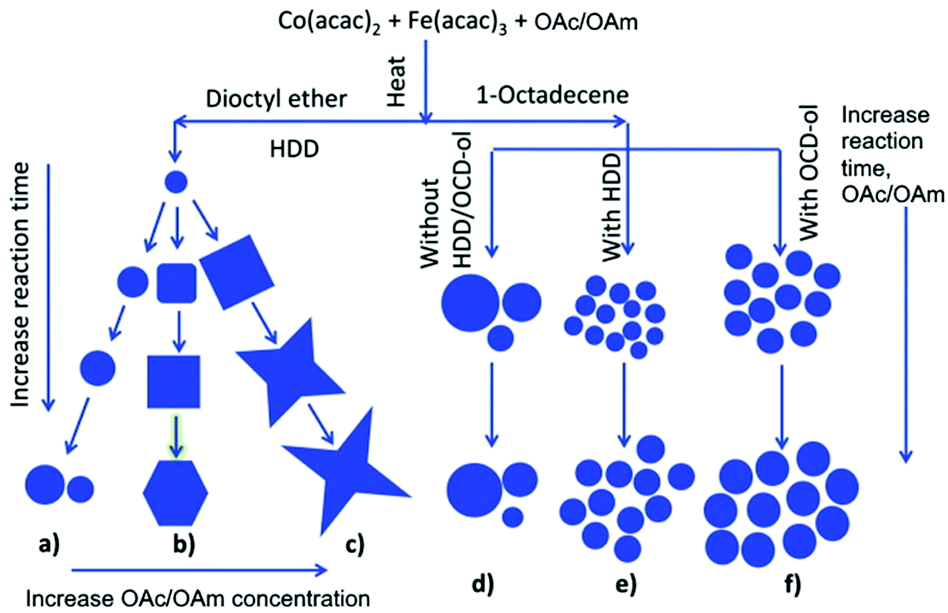 | ||
| Fig. 16 Schematic representation of the shape evolution of CoFe2O4 NPs under different synthetic conditions: (a–c) dioctyl ether with hexadecanediol at a range of reaction times and low (a), medium (b) and high concentrations of OAc and OAm (c) and (d–f) in ODE at varying reaction times and concentrations of OAc and OAm without diol/ODE-ol (d), with diol (e) and with ODE-ol (f).88 Copyright 2015, The Royal Society of Chemistry. | ||
 | ||
| Fig. 17 Scheme illustrating the encapsulation of OAm/OAc-coated NPs with PMAO through hydrophobic interactions.98 Copyright 2018, The Royal Society of Chemistry. | ||
Saez-Puche and colleagues stated that OAc can protect NPs in the presence of OAm from van der Waals attraction, minimize interparticle interactions and limit growth because the organic layer constitutes a steric barrier to the transfer of mass. It has been reported that OAc can lead to high coercivity values due to its cumulative effect on surface spin disorder, large strain and surface anisotropy. Similarly, an increase in magnetization due to anisotropy effects induced by the ligand coating was also reported for OAc-capped magnetite NPs.99 In the case of MnFe2O4 NPs, Hu et al. demonstrated that the elevated affinity of OAc to metal ions on the surface of NPs results in a smaller particle size compared to the use of ligands with weaker affinity, such as OAm and 1,2-hexadecanediol. In their work, when these reagents were used as single surfactants, the particle shape varied from asteroidal to cubic and nearly spherical. When mixed surfactants were employed, the particle shape was modified, for instance, asteroidal particles turned to nearly spherical ones when hexadecanediol was mixed with OAc. The magnetic properties of the different types of particles were a function of the size and surface state of the NPs.100
Thus, in the case of mixed metal ferrite nanomaterials, besides the binding modes and strengths, which affect the particle growth and final shape, the role of the ligand pair under discussion has also been discussed on how it influences the magnetic properties of different materials. Ligand exchange with suitable surfactants may improve the applicability of NPs in both the ‘bio’ and catalytic fields.
2.4 Miscellaneous oxide nanostructures
 | ||
| Fig. 18 Scheme demonstrating the possible growth steps for ceria nanoflowers. Reprinted with permission from ref. 101. Copyright 2008, Wiley-VCH. | ||
 | ||
| Fig. 19 TEM images of CeO2@hexaniobate nanocomposites. Reproduced with permission from ref. 102. Copyright 2019, Wiley-VCH. | ||
Colvin and co-workers managed to transfer cerium oxide nanocrystals to water by using a combination of poly(acrylic acid)-octyl amine (PAAOA) and OAc as phase transfer agents. Regarding the reactivity of nanocrystalline ceria with oxidants such as hydrogen peroxide, the tightly packed layer of OAm ligands on the particle surface could act as a strong barrier for electron transfer. In this study, the authors observed a transition from Ce(III) to Ce(IV) by injecting H2O2. An increase in the amount of OAm led to the formation of larger and more elliptical nanocrystals.103 Imagawa et al. reported that OAc plays an essential role in the formation of rods in the CeO2 system. By modifying the amount of OAm under ligand pair conditions, the aspect ratio of the NRs could be tuned. In the case of spherical particles, a very small amount of OAm resulted in irregular morphologies and a broad size distribution.104
 | ||
| Fig. 20 SEM images of (a) nano-octahedra composed of Co3O4 and (b) CoO.106 Copyright 2012, The Royal Society of Chemistry. | ||
When 3D self-assembled CoO nanoflowers were obtained in the presence of OAm/OAc, it was suggested that the ratio of the ligand pair may play a key role in preventing crystal growth along a certain crystal plane direction, thus generating a flower-like morphology. In particular, it was noticed that a decrease in OAm/OAc ratio led to an increase in the mean NFs size.107 Dai and Teng demonstrated that when cobalt acetate tetrahydrate is decomposed at 300 °C under inert gas in the presence of OAm/OAc, the amount of OAm can define the composition of the resulting NPs. Specifically, CoO or Co would be obtained by changing the quantity of OAm due to its reducing action. Alternatively, OAc could accelerate the growth rate and influence the final structure and morphology of the particles. An octahedral shape could be derived due to surfactant control over the nanocrystal facets in the course of growth owing to the strong coordination of OAc on the O2-rich facet of the nanocrystal or coordination of OAc through a cobalt-rich facet.108
In fact the binding capabilities of the passivating OAm/OAc ligands can be controlled to a significant degree by adjusting the reaction temperature, yielding different sizes, structures and surface chemistry of CoO NPs upon the thermal decomposition of cobalt acetate. The reaction atmosphere (O2 or N2) can also affect the binding capabilities of the surfactants, being stronger in the case of oxygen, providing a smaller NP size. A larger size of around 300 nm was obtained when nitrogen gas was added to the reaction mixture. The magnetic behaviour of the resulting products was discussed by analysing the role of uncompensated surface spins and defects.109 Interestingly, Yan and co-workers showed that when synthesizing CoO and Mn3O4 starting from simple formate precursors, the addition of OAc provided poorly crystalline products, and thus the sole use of OAm was preferred as solvent. The injection method was not chosen when using the aforementioned precursors for Co and Mn, and it was observed that the precursor concentration rather than the reaction time affected importantly the NP crystal size. However, when aiming to produce CuOx, it was found that the injection method could remarkably improve the size uniformity of the produced NPs. For that system, a suitable quantity of OAc facilitated the control of the size distribution of the NPs in a narrow range. The size polydispersity was worse when OAc or OAm were used alone.110 Zhu et al. produced In2O3 NPs via the thermal decomposition of In(dipy)3Cl3·2H2O in OAm/OAc under an inert gas atmosphere. It was observed that the single use of OAc did not yield any NPs, while a large amount of OAm without OAc resulted in the quick growth and aggregation of the produced particles. The higher oxophilicity of OAc was responsible for its stronger binding to the surface atoms of the NPs compared to OAm. Therefore, an equiatomic ratio of OAm/OAc was chosen and the particle size could be tuned from 10 to 50 nm, depending on the molar ratio of precursor to combined OAm/OAc solvent.111 In2O3 nanocubes (Fig. 21a) were produced by Lee and co-workers in the presence of OAm/OAc, H2O and trioctylamine. For the solubility of the prepared particles, trioctylamine alone cannot act as an efficient ligand, where the presence of OAm or OAc is required for their solubility. The size of the nanocubes could be tuned from 8 to 12 nm by changing the OAc/OAm ratio from 1/7 to 1/3.112
 | ||
| Fig. 21 TEM images of (a) In2O3 nanocubes. Reprinted with permission from ref. 112. Copyright 2006, the American Chemical Society. (b) MnO nanocubes.113 Copyright 2009, The Royal Society of Chemistry. (c) Star-like MnO nanocrystals.114 Copyright 2014, The Royal Society of Chemistry. | ||
Mn3O4 or MnO nanostructures with sizes in the range of 5–25 nm could be produced through the decomposition of Mn(acac)2 by simply modifying the nucleation temperature. The reaction medium also contained paraffin oil, OAc, OAm and dodecanol. Paraffin oil is a non-polar solvent, which does not generally participate in the reaction, and thus it can be simply used as a suitable injection solvent for a fast decrease in the reaction temperature. Dodecanol plays a crucial role in the control of the NP size and the high yield of monodisperse NPs. OAm offered alkaline conditions, facilitating the formation of self-assembled superlattices for the as-prepared NPs. The use of an excess of all three ligands (OAm, OAc, and dodecanol) was beneficial for the particle growth in all crystal planes on the surface of the manganese oxide nuclei. Interestingly, FTIR measurements showed that the Mn3O4 NPs were coated by dodecanol, whereas the MnO NPs (Fig. 21b) were capped by OAm, showing that these ligands played a key role in each one of the oxide compositions, correspondingly. However, the role of all these surfactants was important for the formation of monodisperse, non-aggregated NPs.113 Gao and colleagues pyrolyzed Mn(II) acetate in 1-ODE using the OAm/OAc ligand pair and produced both MnO and Mn3O4 nanocrystals. It was found that the Mn2+–OAm coordination was kinetically driven and promoted the generation of a Mn3O4 phase owing to the relatively low electronegativity of the N in OAm. Alternatively, the Mn2+-oleate coordination was thermodynamically driven and could hinder the central metal ion (Mn2+) from being oxidized due to the somewhat high electronegativity of O from the oleate ligand. Considering the hard and soft acid and base theory, Mn2+ is a hard acid, whereas deprotonated OAc and OAm are hard bases. However, the deprotonated OAc shows stronger hardness than OAm. Thus, the OAm/OAc ligand pair provides a molecular switch for modifying the chemical environment of the Mn-precursor, leading to either OAm-dominant or OAc-dominant coordinating conditions. These conditions can largely determine the oxidation state of Mn in the resulting NPs. In fact, the thermal decomposition of OAc may also assist the formation of Mn3O4 by supplying O. An OAc-dominant environment will give rise to branched MnO NPs (Fig. 21c); however, the prolonged heating treatment of these nanocrystals will degrade the oleate ligands and make the system OAm-dominant. This will induce the generation of a Mn3O4 phase, and also convert the branched MnO nanocrystals to spherical Mn3O4 ones.114
:1, the aforementioned amide can be formed, whereas if, for example, OAc is in slight excess compared to OAm, then an equilibrium between the amide and excess OAc occurs. This will also affect the growth mode and the final shape of the resulting titania NPs. The preferential growth on certain facets is also affected by the reaction time. Jardiel and co-workers employed titanium(IV) tetrabutoxide and ethanol together with OAm/OAc in their work. Additional surfactants such as trifluoroacetic acid, arginine and 11-aminoundecanoic acid were tested, aiming to acquire TiO2 nanostructures with a tunable morphology.119,120
Dinh et al. published a versatile approach based on the solvothermal method to prepare various shapes of titania nanocrystals including rhombic, dumbbell, spherical and truncated, among others. Water vapor as the hydrolysis agent accelerated the reaction rate, while OAm and OAc were used as surfactants. The carboxylic acid functional groups bind tightly on the TiO2 surface, promoting the generation of a nanorod shape. For the material under discussion, OAc binds strongly to the anatase {001} faces, while OAm prefers to adhere on the {101} faces. The OAm/OAc acid–base pair catalyst can also contribute to a higher condensation rate without affecting the hydrolysis rate. OAc can react with titanium n-butoxide or hydroxyalkoxide to generate carboxyalkoxide moieties, which slow down the hydrolytic condensation process. OAm can favor the nonhydrolytic condensation process by aminolysis reaction with titanium carboxyalkoxide. In the absence of OAc, the exclusive formation of rhombic shape was noticed. When OAm was not used, a mixture of spherical NPs and nanodots was obtained. A scheme presenting the morphology evolution of the TiO2 NCs according to the OAc/OAm ratio is shown in Fig. 22.121 The anatase TiO2 NPs prepared in the presence of OAm/OAc can also occur in 16 nm elongated bipyramid or 8 nm uniform bipyramid morphologies, as shown by Xing and co-workers. It is worth mentioning that the OAm and OAc layers can be removed by treating the NPs with NaBH4 in chloroform, with no grain growth taking place.122
 | ||
| Fig. 22 Scheme demonstrating the morphological evolution of titania NCs depending on the OAc/OAm ratio added. Reprinted with permission from ref. 121. Copyright 2009, the American Chemical Society. | ||
Another Ti source, titanium sulfate, was used by Luo et al. to produce anatase TiO2 NPs with several shapes in the presence of OAm/OAc and combinations of other reagents such as H2SO4. Upon increasing the reaction time and temperature, functional groups or even C chains of OAm and OAc could be destroyed by sulfuric acid. While the adsorption and protection action of OAm and OAc deteriorated, the unstable nano-subunits of TiO2 could aggregate rapidly, thus decreasing the surface activation energy and becoming a relatively stable phase with low energy after nucleation. By changing the reaction parameters, the crystal growth evolution was modified and NPs with either porous mesocrystals or uniform hollow morphology were produced.123
The combination of supercritical methanol, OAm/OAc, PEGME/citric acid and titanium(IV) tetraisopropoxide led to the formation of hierarchically porous anatase titania microparticles, where primary 5–9 nm TiO2 NPs were generated firstly, which subsequently loosely aggregated to form secondary 0.2–1.5 μm particles in the presence of PEGME/CA. Interestingly, OAc–TiO2 had a larger crystallite size than the bare TiO2 particles produced in supercritical methanol, possibly because the OAc molecules reacted with methanol in its supercritical state to generate methyl oleate. FTIR measurements showed a relatively low degree of surface modification by OAc, indicating that the use of OAc did not result in effective suppression of the crystal growth. The electrochemical performance of the C-coated TiO2 particles, obtained by calcination in the presence of surface modifiers was evaluated. The primary particles prepared using PEGME/CA were smaller than in the case of the use of OAm/OAc ligands. An excellent performance for the final materials was observed when they were used as anodes in sodium- and lithium-ion batteries. However, the electrochemical activity for the carbon-coated titania particles obtained starting with OAm/OAc-capped NPs did not increase, given that this ligand pair did not lead to effective suppression of the crystallite size.124 Li et al. bought commercial needle-shaped rutile TiO2 NPs, the surface of which they treated with HCl before testing their chemical reactivity with dispersants. For this purpose, the adsorption amounts of OAc, OAm, oleyl phosphate and oleyl alcohol were measured and compared. OAc can adsorb chemically on TiO2 by unidentate bonding between the carboxylic acid group and Ti4+. Actually, oleyl phosphate and OAm adsorb also on titania through chemical interaction, whereas oleyl alcohol does not. The dispersion activity of the dispersants was related to the type of functional groups, given that these moieties determine the interaction and the quantity of dispersant absorbed on the powder. OAc, phosphate and OAm demonstrated very good efficiency for dispersing the HCl-treated titania. The alkaline nature of OAm helped toward a better efficiency, and its suitability can be explained by the fact that the treated powder is acidic. Hydrogen bonding between the –OH groups of the phosphate-based dispersants can offer an even better dispersion performance for TiO2. In addition, OAm was most efficient in introducing electrical double layer repulsions, which dispersed TiO2. Oleyl phosphate achieved a very effective steric repulsion.125,126 Another finding worth noting is that OAm was adsorbed predominantly on 3:1 anatase:rutile hydrophobic TiO2, whereas OAc showed preferential adsorption on rutile hydrophilic TiO2.127
Morazzoni and co-workers produced anatase titania nanocrystals via the solvothermal decomposition of titanium butoxide in the presence of OAm and OAc as morphology-adjusting agents. OAc demonstrated selective binding on the anatase {001} face, whereas OAm binds mostly to the anatase {101} face, which limits the growth in that respective direction. Carefully modifying particle morphology and the exposed crystal faces will help to tailor the photocatalytic performance of TiO2 nanocrystals. In that study, the {001} surfaces played a crucial role in the photocatalytic process of phenol mineralization with O2 as an oxidant by providing oxidation sites, while the {101} surfaces offered reductive sites and were only indirectly involved in photocatalysis.128 Hydrophobic OAc-capped titania NRs and OAm/OAc-capped magnetite NPs became hydrophilic after their surface modification with short-chain carboxylic acids (acrylic, acetic and methacrylic acids). The acrylic acid-exchanged particles were well-dispersed for several months in polar solvents, unlike the case of the acetic- or methacrylic-acid exchanged solutions, which did not show stability. This was attributed to the presence of OAc left on the TiO2 surface.129 Indium-tin oxide NPs with an average size of 6 nm were produced via a hot-injection route in the presence of OAm/OAc surfactants. Indium- and tin-acetates were the precursors and 1-ODE was the reaction solvent. 3-Aminopropyldimethylethoxysilane was used to replace the initial ligands and the aminated NPs became electroactive through functionalization with ferrocenoyl chloride. These electroactive nanomaterials are deemed candidate materials for electrochemical charge storage systems.130 Liu and co-workers produced anatase TiO2 NPs with a tailored size and aspect ratio using a simple solvothermal route with titanium butoxide as the precursor, OAc/OAm as the ligands and ethanol as the solvent. Ethanol is an environmentally friendly solvent and OAc (component of vegetable oil) is relatively environmentally benign. The effect of the OAc/OAm ratio on the size, aspect ratio and shape of TiO2 NPs was investigated. The morphology of the resulting particles is affected by the hydrolysis and condensation rates. The hydrolysis reaction rate using OAc/OAm is much slower than that in aqueous reaction medium, which influences the shape evolution of the particles.131 Gonzalo-Juan et al. described how the production of coordination complexes at intermediate reaction stages can be adjusted to grow different shapes of TiO2 such as NWs, NRs and NPs. The distinct binding strengths of OAm/OAc in their solvothermal approach affected the shape of the final TiO2 nanomaterials. In their work, in the absence of OAm, OAc induced the anisotropic growth along the [001] plane of the nanostructures with a low aspect ratio. Higher OAm concentrations promoted the onset of anisotropic growth along the [101] plane, favouring the generation of NCs with a lower aspect ratio such as nanorods. Alternatively, an OAc/OAm ratio of 75:25 resulted in an isotropic growth mode and the produced particles had an aspect ratio of around 1. Fig. 24 schematically depicts the preferential growth patterns for TiO2 NCs along the different planes.132
In fact, the OAm/OAc pair can help TiO2 NPs to achieve a better dispersion in the poly(ethylene terephthalate) matrix compared to commercial TiO2 (P25). Accordingly, PET/TiO2 nanocomposite mats (Fig. 23) with remarkable thermal stability and mechanical properties were produced by electrospinning.133
 | ||
| Fig. 23 SEM images of (a) PET/TiO2 nanofibers with 5 wt% TiO2 and (b) 10 wt% TiO2. Reprinted with permission from ref. 133. Copyright 2020, Wiley-VCH. | ||
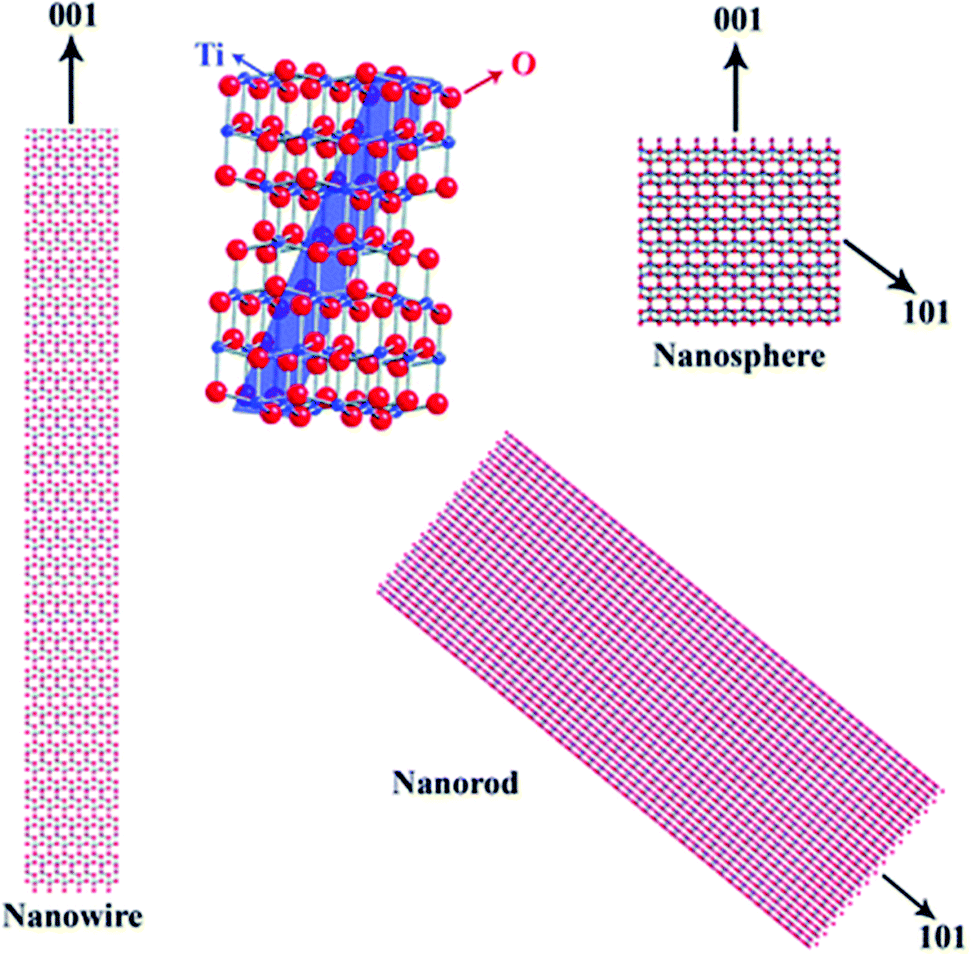 | ||
| Fig. 24 Scheme presenting the distinct preferential growth modes along the [001] and [101] planes of titania nanostructures.132 Copyright 2010, The Royal Society of Chemistry. | ||
Tang and co-workers produced Mn-doped ZnO nanocrystals with a hexagonal structure via the thermal decomposition of Zn-acetate and Mn-acetate using OAm/OAc at a range of temperatures, ligand ratios and Mn doping amounts. Accordingly, the particle size could be tuned in the range of 47–375 nm, and the resulting shape could be varied from hexagonal nanopyramids (Fig. 25a) to hexagonal nanodisks and irregular nanospheres. The reported protocol was quite easy to reproduce and was deemed as scalable for large production. When OAc was added in excess (4:1 to OAm), no ZnO:Mn was formed, probably because of the formation of a Zn–OAc chelate complex which prevented the decomposition of the Zn(II) acetate with a low amount of OAm. An increased ratio of OAm led to a decrease in size and the particle shape was modified from hexagonal to irregular spheres.137 Zeng and colleagues published the one-pot thermolysis of Zn(acac)2 and Mg(acac)2 in the presence of OAm/OAc and managed to introduce Mg in the ZnO lattice with no phase separation for concentrations up to 20% (Fig. 25b and c). The ZnO phase did not show any significant selectivity (that is, ‘preferential binding affinity’) for one of the two ligands, whereas MgO demonstrated a strong selectivity for OAc. Varying the amount of OAm used could lead to either a nanoflower (with low OAm quantity) or nanoparticle configuration (with high OAm quantity).138
 | ||
| Fig. 25 (a) SEM image of Mn-doped ZnO hexagonal bipyramids. Reproduced with permission from ref. 137. Copyright 2018, Elsevier. (b and c) TEM images of MgZnO nanostructures with varying Mg doping. Reproduced with permission from ref. 138. Copyright 2015, Wiley-VCH. | ||
Other oxides with several different compositions have also been synthesized using the OAm/OAc ligand pair. In particular, BiVO4 with multiple morphologies and/or porous structures were produced with bismuth nitrate and ammonium metavanadate precursors, sodium hydroxide as pH-controlling agent, ethanol and EG as solvent and/or dodecylamine, OAm/OAc as ligands. The choice of the surfactants was critical for the formation of high-surface-area porous BiVO4 NPs. The pH of the precursor solution affected the surface area of the product to a significant extent. Excellent photocatalytic activity was observed for porous olive-like BiVO4 particles for the catalysis of phenol degradation using a small amount of hydrogen peroxide under visible light irradiation.139 CaWO4 nanocrystals were prepared by Lee et al. using surfactants such as citric acid, CTAB and a mixture of OAm/OAc. Regardless of the initial capping layer, the original surfactant could be subsequently replaced by bioinert block copolymer micelle enclosure using a solvent-exchange method. 1H NMR and TGA measurements indicated that the OAm/OAc coating was not fully replaced with the new enclosure formed by the PEG/PnBA/PEG-PLA block copolymers. Thus, the possible effect of residual OAm/OAc on the toxicity of the particles should be assessed prior to their use in biomedical applications.140
OAc/OAm-coated Gd2O3 nanoplates were encapsulated in an N-dodecyl-PEI–PEG polymer to produce hydrophilic clusters for T1-weighted MRI (Fig. 26). The resulting material had a surface chemistry of lower toxicity compared to the initial nanoplates.141 Watt and co-workers prepared PbO nanocrystals via a one-pot method by introducing O2 gas in an air- and moisture-free complex of Pb with OAc and OAm in 1-ODE at 190 °C. Thin films of the nanocrystals were fabricated using the 1,2-ethanedithiol-based dip-coating process.142 The synergistic effect of OAc and OAm was also suitable for the preparation of MoO3−x nanocrystals in a top-down approach starting from bulk a-MoO3, where in this work, OAc offered H+ ions, while OAm provided free electrons. The amine group from OAm coordinated with Mo6+ to release H2O molecules. The crystal lattice of the bulk precursor then broke down to yield nanocrystals, while oxygen deficiencies could also be formed. Strong localised surface plasmon (LSPR) absorption in the Vis and NIR region between 400 and 1100 nm was observed for the resulting particles. When only OAc or OAm was used as a single solvent, the absorption in the visible region was very weak, implying that no oxygen defects were formed under that conditions.143 Nanocrystals with the CsxWO3 composition were prepared in the presence of OAm/OAc, and in that work the modification of the ligand pair ratio led to the formation of three different shapes, including hexagonal prisms, truncated cubes and pseudospheres. Increasing the quantity of OAm induced the generation of more faceted shapes. For certain ligand ratios, the product consisted of mixed crystalline phases, for example, WO2.72 rods and CsxWO3 nanostructures.144 3D symmetric dendrites of BaWO4 were produced with a solvothermal approach in water-hexane bilayer solutions in the presence of barium-oleate complex in the hexane layer and sodium tungstate in the water layer. A phase transfer reaction was carried out, where aqueous Ba2+ passed in hexane which was mediated by the use of Na-oleate and OAm/OAc. Increasing the concentration of OAm or the temperature resulted in the formation of sharper and longer branches and higher-order hierarchical structures. Protonated OAm is formed due to its basicity in the presence of water and can react with Ba-oleate to form adducts composed of protonated OAm cations and oleate anions. The release rate of free Ba2+ ions from the Ba-oleate will be faster when using OAm. Then the released free Ba(II) ions will react with WO42− ions to produce BaWO4 particles.145 Zou and co-workers published a facile and reproducible one-pot method to produce a series of metal oxides (CdO, PbO, ZnO, SnO and Ga2O3) with their respective bulk materials as precursors. The OAm/OAc ligand pair was used for each oxide. Although the whole concept for this synthetic route resembles a top-down approach, the nanoscale oxide materials were formed from molecular complexes via a bottom-up strategy. FTIR measurements showed that OAc was converted to M-OAc and all these metal complexes had an ionized structure. An increase in the reaction temperature led to the dissolution of bulk metal oxides, and after exceeding a critical value, homogeneous nucleation occurred spontaneously, followed by the growth stage. In the case of CdO, the molar ratio of OAm/OAc was studied regarding its effect on the particle size and shape.146 Rodriguez and colleagues published a paper on the synthesis and structural characterization of modified hafnium tert-butoxide materials for use as sources to produce HfO2 NPs. A solvothermal process using OAm/OAc as the solvent was chosen based on the tendency of this system to form a nanowire shape. The resulting products could generate hafnia dots, plates and rods, depending on the type of precursor molecule used.147 Uranium dioxide NPs were prepared via the thermal decomposition of uranyl acetylacetonate in the presence of OAm/OAc in octadecene solvent. The minimum number of stable nuclei was produced at an OAm/OAc ratio of 1:1 and increasing the amount of either OAm or OAc resulted in an increase in the population of stable nuclei, accompanied with a decrease in the final NP size (Fig. 27). FTIR spectra demonstrated that OAm reacted with OAc in a condensation way to produce N-(cis-9-octadecenyl)oleamide (OOA). However, OOA was not found on the NP surface, while the particles were passivated by OAc via chelating bidentate interaction.148 Barron and co-workers prepared binary and ternary metal oxide NPs via the thermal decomposition of metal acetylacetonate salts, which were the sources of Fe, Mn, Pd, Cu, Al and Gd, respectively. Benzyl ether was the solvent, 1,2-hexadecanediol the accelerating agent and OAm/OAc were the surface ligands. Besides the capping agents, OAm/OAc seemed to also play a role in the decomposition of the precursors. For example, Cu(acac)2 readily forms complexes with diol or OAm (and possibly benzyl ether), and these coordination compounds should be deemed as the actual copper sources, leading to the formation of Cu-O fragments for particle growth.149
 | ||
| Fig. 26 Preparation of Gd2O3@N-dodecyl-PEI–PEG clusters for use as excellent T1-weighted MRI agents. Reprinted with permission from ref. 141. Copyright 2020, Elsevier. | ||
 | ||
| Fig. 27 (a–c) TEM images of UO2 NPs with OAc/OAm ratios of 1:3, 1:1 and 3:1, respectively. (d) TEM image of NPs produced using uranyl acetylacetonate (0.4 mmol), 1-ODE (1 g), OAc (10 g) and OAm (10 g). Scale bar: 10 nm. Reprinted with permission from ref. 148. Copyright 2006, the American Chemical Society. | ||
The influence of OAm/OAc ratio on the morphology of Y2O3 NPs was reported in another work, where shapes such as hexagons, rods, irregular morphologies and plates could be isolated. In the initial growth stages, the OAm bonded to the particle surface poisoned the {111} plane, providing a platelet shape and stabilizing the {101} planes. OAc preferentially absorbed on the {111} planes and these competing conditions affected the particle morphology.150
GeO2 particles were prepared via a solvothermal route involving OAm/OAc, which showed potential for use in anodes of lithium-ion batteries. The authors discussed the growth evolution of the OAm/OAc-capped oxide particles based on the formation of complexes of Ge with OAm and OAc. For example, a (Ge–OAm)4+ complex may form in the beginning, which can be partially substituted later by oleate ions due to their stronger binding, resulting in Ge-oleate. Afterwards, the latter complex will decompose to give GeO2. The OAm/OAc ligand (and solvent) pair mixture assists the generation of small nanoplates, which finally assemble to generate an ellipsoidal shape with a porous structure. However, the hydrophobic ligand pair deteriorates the battery performance. Thus, removing the ligands with glacial acetic acid can improve the electrochemical activity, and this performance can be improved by wrapping the particles with rGO/PANI composites.151 Li4Ti5O12/carbon nanocomposites were produced by first preparing LTO NPs in supercritical methanol, followed by calcination under a mild reducing atmosphere. The influence of surface modifiers with distinct functional groups and chain lengths (OAm, OAc, and hexylamine) was investigated regarding their effect on the particle size, shape, crystallinity and electrochemical behavior. Interestingly, the OAm-capped LTO particles had the smallest crystallite size, probably because the surface modification with OAm radically inhibited the crystal growth. Apparently, the reaction of the amino group of OAm with the hydroxyl groups of LTO was more robust compared to their reaction with the carboxylic groups of OAc. A higher degree of surface coverage is a critical factor to more efficiently delay the interparticle aggregation during the calcination step. If OAm is compared with hexylamine, a worthy observation is that the use of a longer chain length would result in a larger organic content, even though the modifier with a shorter chain length would provide a higher degree of surface coverage. The optimum rate performance of the LTO/C nanocomposite prepared with OAm was attributed to the combination of smaller crystallite size, larger C content, uniform carbon layer coverage, larger Ti3+ content and bigger population of defects. These characteristics would lead to the quick diffusion of electrons and Li+ inwards/outwards of the individual LTO NPs.152 Three-dimensional hierarchical superstructures of BaCrO4 were produced via the hydrothermal reaction of Ba(NO3)2 and Na2CrO4 in an aqueous-hexane bilayer system. The OAm/OAc pair was used to generate hydrophobic Ba-oleate micelles. The acid–base chemical equilibrium is responsible for the presence of deprotonated OAc and protonated OAm in the medium. The deprotonated OAc would interact with Ba(II) to yield Ba-oleate. Stabilization of the latter with OAm would induce the formation of micelles with hydrophobic outer parts, which are stable in solvents such as toluene. Subsequently, Ba(II) would react with CrO42− at the interface of the hexane and water phases, resulting in the nucleation of BaCrO4.153
MgO nano/microparticles with numerous shapes and porous structures were produced using a surfactant-mediated solvo- or hydrothermal process in dodecylamine or OAc solvent. The ligands tested included PVP, PEG, CTAB, OAm and triblock copolymer P123 or F127. Depending on the solvent used, different degrees of crystallinity were observed. Calcination below 550 °C in an oxidative atmosphere could remove F127, CTAB and OAm molecules. The obtained MgO particles showed a propensity to possess a hexagonal shape when surfactants such as F127, P123 and OAm were added to the reaction system.154 Sub-2 nm uniform oxide NCs (TiO2, ZnO and Nb2O5, among others) were produced by the thermal decomposition of suitable precursors in the presence of 1-ODE and OAm/OAc. The interaction between the ligands and inorganic moieties resulted in well-organized layered mesostructures.155
In the case of thorium dioxide NPs, OAc exhibited selective adsorption on the (110) surface, resulting in the transformation of octahedrons to NRs. OAm and TOPO did not modify the equilibrium shape of the ThO2 NPs. The (111) surface was the most stable facet at a low OAc coverage, whereas the (110) surface became the most stable facet when the OAc coverage was greater than 2 ligand/nm2. Modifying the relative stability of the surface had a significant effect on the equilibrium shape of ThO2, as illustrated in Fig. 28.156 A high-temperature, quick solvothermal route was employed to produce (NH4)xWO3 with OAm/OAc as the solvent and WCl6 as tungsten precursor. When only OAc was used as the solvent, an unknown phase was isolated, not possessing the typical composition of ammonium tungsten bronze. The addition of a small amount of OAm in the reaction pot resulted in the formation of a mixture of small nanocubes and bundles of nanofibers, given that not all the active particle sites were surfactant-coated. A large amount of OAm led to homogeneous oblong blocks possessing a glossy appearance with a relatively low reaction yield, owing to the increase in pH value associated with excess OAm.157 WCl6 was used as the tungsten source for the synthesis of 3 nm WO3 quantum dots in the presence of OAm/OAc and ethanol. A CuCl2 ligand exchange treatment was applied to replace the long chains of the OAc and OAm capping agents. This treatment was required because it is beneficial for improving the performance of the tungsten oxide material toward the sensing of H2S.158
 | ||
| Fig. 28 Surface energies of ThO2 as a function of the coverage of ligands for (a) OAc, (b) acac, (c) OAm, and (d) OP(Oct)3 and (e) evolution of the most stable shape of the nanocrystal with increasing OAc coverage.156 Copyright 2018, The Royal Society of Chemistry. | ||
It was reported that both OAc and OAm can serve as ligands at 305 °C, but the acid is a more active ligand at 330 °C. The amine is more labile at 330 °C, which actually assists the decomposition of Sm-oleate but cannot actively participate as a capping agent. Consequently, at that temperature, Sm2O3 particles (Fig. 29a) were mainly coated by OAc, with the regular growth mode resulting in a square platelet shape. At a lower temperature of 305 °C, the mobility of the amines decreased to some extent, and thus both acid and amine could function as surface ligands. However, the labile and weak binding mode of amines led to more rapid and perhaps irregular particle growth. An increase in the amount of OAm caused faster decomposition of the Sm-oleate precursor. The formation of square platelets, which were mostly capped by OAc, was favored at 330 °C, while a flower shape was produced at 305 °C, and in the latter case the particles were coated by both OAm and OAc.159
 | ||
| Fig. 29 TEM images of: (a) Sm2O3 NPs. Reprinted with permission from ref. 159. Copyright Nature Publishing Group. (b) Ag2S NPs. Reprinted with permission from ref. 160. Copyright 2019, Elsevier. (c) CdIn2S4 NPs.161 Copyright 2015, The Royal Society of Chemistry. (d) ZnxCd1−xS NRs. Reproduced with permission from ref. 162. Copyright 2010, Elsevier. | ||
To conclude this sub-section on metal oxides with miscellaneous compositions, one of the main topics discussed in different reports is again the control of the crystal growth in certain directions depending on the preferential binding modes and binding strengths of the different ligands on specific facets. The formation of coordination complexes at intermediate reaction stages can also affect the final particle size and shape.
3. Metal chalcogenides
Ag-Based
Metal chalcogenide nanostructures have received significant interest due to their morphology- and composition-dependent semiconductor properties. They can be used as solar absorber materials and have found applications in photovoltaics and photocatalysis. In a report presenting the solvothermal synthesis of AgAbS2 NPs, OAm complexed with Ag+ and OAc complexed with Sb3+. The amino groups of OAm and the carboxylic groups of OAc combined with the metal cation to form a metal complex. In fact, the excellent coordination ability of OAm with metal cations led to the generation of Sb(OA)3 solvate, starting with SbCl3 as the antimony source. Sulfur anions could be easily dissolved in OAm to form an anionic ligand solution, which aided the size and shape control of the NPs. Thus, OAm, an excellent surfactant and weak chelating agent, would dissolve silver cations and sulfur anions, while OAc would dissolve antimony cations. Actually, S–OAm solutions functioning as sulfur precursors occur mostly as alkylammonium polysulfides at low temperatures. When heating the reaction mixture to higher temperatures, the polysulfides would react with the excess OAm to generate H2S. This compound can integrate with the metal precursor to produce AgSbS2 NPs under suitable conditions.163In the case of the Ag2S system, the sole use of 1-dodecanethiol as the ligand resulted in cluster formation. Adding OAc helped to significantly improve the dispersity of the NPs but it caused an important decrease in the emission intensity of the quantum dots. The presence of a small amount of OAm could hinder the formation of undesirable Ag NPs and assisted the improvement in fluorescent intensity at high temperature. A strong emission in the second NIR window was noticed. Probably, the affinity of OAm for metals and sulfur was the main factor that ensured the absence of metallic Ag in the final product (Fig. 29b). After ligand exchange, the hydrophilic Ag2S QDs were used for whole-body blood vessel imaging and tumor imaging in living mice.160
Jiang and Chen noticed that increasing the amount of long-chain OAc or decreasing the amount of short-chain 1-octanethiol could slow down the growth of Ag2S QDs due to steric factors and render their synthesis more controllable. Ag(I) ions could be reduced by OAm to undesirable Ag NPs. Actually, OAm would activate the monomers and accelerate the nucleation and growth of the Ag2S particles in the beginning of the reaction. Aiming to prevent the reduction of Ag(I) ions during the synthesis, 1-octanethiol was added to the reaction mixture. This thiol is a soft Lewis base ligand and Ag(I) is a very soft Lewis acid, while OAm and OAc are classified as hard Lewis base ligands. The bond between Ag(I) and thiol should be stronger than that between Ag(I) and OAm (or OAc). Therefore, the standard redox potential of Ag(I) is lower when Ag(I) ions are coordinated by thiols, and thus these ions will be harder to reduce. However, OAc is expected to bond to Ag stronger compared to OAm and the strong coordination bond between the monomers and ligands will decrease the reactivity of the monomers. Based on this, the authors evaluated the photoluminescence properties of their QDs in a range of different OAc concentrations. Higher OAc concentrations led to the preparation of smaller Ag2S QDs. On the contrary, the presence of OAm was not convenient to produce small-size QDs because of its above-mentioned accelerating effect on the growth.164
Cd-Based
A triple ligand system (TOPO–OAm–OAc) was very efficient for the synthesis of high-quality CdSe NPs within a short reaction time using a capillary microreactor with a serpentine microchannel in the heating section (Fig. 30). To achieve small-sized particles, a high molar ratio of Se to Cd was required, and the concentration of OAc was kept at low levels. Accordingly, massive nucleation could occur and the slow growth of TOPO-capped CdSe NPs could be ensured by diluting the precursor solution with non-coordinating 1-ODE. In the case of OAm, it has been reported to act as a favorable ligand for the fabrication of CdSe NPs via micro-reaction, owing to its low melting point, strong packing density and low reactivity in air. Improved monomer reactivity was observed for an amine-Cd complex. The addition of OAm induced a blue shift in the absorption peak and high particle concentration. In fact, CdSe NCs with a high photoluminescence quantum yield were obtained when all the above-mentioned ligands were simultaneously present.165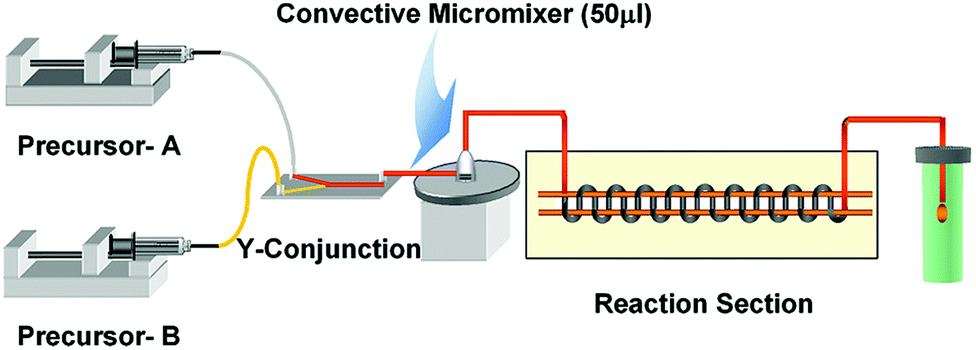 | ||
| Fig. 30 Schematic illustration of the capillary microreactor with a serpentine channel in the heated part used to produce high-quality CdSe nanocrystals. Reprinted with permission from ref. 165. Copyright 2009, the American Chemical Society. | ||
Al-Salim et al. published a work in 2007 on the synthesis of CdSexSy in eight solvents with distinct coordinating properties such as amines, phosphines, 1-ODE and TOPO. NMR and IR characterization showed that the particles prepared in non-coordinating solvents were stabilized in solution mostly by OAc, whereas the particles produced in coordinating solvents were stabilized by the solvent, phosphine, phosphine sulfide ligands and some OAc. Some of these surfactants can potentially act as metal-site-coordinating ligands such as TOPO and OAc (via oxygen), amines (via nitrogen) and TOP-sulfide (via the S atom). In addition, certain ligands are potential chalcogen-site-coordinating molecules such as TOP and triphenylphosphine (via the P atom) and primary and secondary amines (via weak hydrogen bonding). OAc was demonstrated to increase the growth rate of CdSe QDs given that the number of nuclei generated after injecting TOPSe into cadmium oleate in 1-ODE was reduced by the increase of OAc amount but remained stable after the initial nucleation step. In the 1H NMR spectra, OAc displays a multiplet signal centered at 5.35 ppm because of its alkene hydrogen atoms. The amide, which can be formed by the reaction of OAc with OAm, exhibited a signal at 5.95 ppm because of the amide proton of the dialkylamide. Coordinating solvents favored the formation of CdSe NPs with a hexagonal crystal structure, while weakly- or non-coordinating solvents gave rise to cubic crystal structures.166
CdSe QDs coated with TOPO–OAm–OAc in toluene were reported to display bright and uniform luminescence-displaying green color upon exposure to 365 nm UV lamp. However, the QDs dispersed in pyridine/chloroform exhibited a blue-green color of lower intensity. In fact, FTIR measurements showed that ligand exchange with pyridine was not effective to fully remove OAm and OAc from the particles surface. The complexation of OAm or OAc with monomers and Cd precursor hampered the nucleation and resulted in early ripening, which is associated with larger rather than smaller NP sizes. OAm, being labile, is less effective in decelerating the crystal growth in comparison to OAc and TOP. Both OAc and OAm passivated the majority of vacancies and trap sites on the CdSe surface, and consequently enhanced the quantum yield and photostability. A sufficient amount of OAc and OAm residual ligands on the NP surface after pyridine treatment created a high potential barrier for charge transfer to take place between the polymer and CdSe QDs, rendering the quenching mechanism due to charge transfer being less likely, where energy transfer would then be initiated, which was prominent in the case of MEH-PPV:CdSe–OAm nanocomposites.167
Abazovic and co-workers employed a hot-injection route to produce CdIn2S4 and ZnIn2S4 NPs. When the OAm/OAc ligand pair was used, 20–30 nm mesocrystals were self-organized in marigold-like structures. OAc could act as a complexing and capping agent, and the ratio between In ions and OAc was a crucial parameter to control the synthesis of the NPs. To speed up the degradation of the metal-OAc complex, the addition of OAm played a key role. The basicity of OAm facilitated the more rapid decomposition of the above-mentioned complex. Taking into account that Cd(II) has a weaker binding energy with OAc than Zn(II), the same principle for OAm utilization was employed to produce ZnIn2S4. The OAm/OAc pair offered a suitable platform for the generation of complex hierarchical morphologies. XRD measurements illustrated that the decomposition of metal-oleates and subsequent nucleation, even in the presence of OAm, is the dominant process controlling the overall kinetics of the CdIn2S4 NPs (Fig. 29c).161 ZnxCd1−xS semiconductor NPs with a tunable composition and morphology were prepared in the presence of OAc/OAm and paraffin oil via a non-injection, scalable and reproducible approach. When OAc was omitted, the particle shape was modified from spherical to nanorods (Fig. 29d). All the resulting particles had a hexagonal wurtzite crystal structure and the shape transition was not composition-induced.162 CdS nanopencils were produced in the presence of CdS seed NPs, OAm/OAc and dimethyldistearylammonium chloride (DDAC) at 260 °C. Although the reaction could proceed in the absence of OAc, the resulting particles were small with a broad size distribution. The presence of OAc was required to achieve uniform CdS nanopencils. Subsequently, Au NPs could be deposited on CdS to form CdS–Au nanopencils (Fig. 31a).168
 | ||
| Fig. 31 TEM images of (a) Cd–Au nanopencils. Reprinted with permission from ref. 168. Copyright 2010, the American Chemical Society. (b) CuFeSe2 NCs. Reprinted with permission from ref. 169. Copyright 2015, the American Chemical Society. (c and d) CuInS2 nanostructures. Reproduced with permission from ref. 170. Copyright 2014, the American Chemical Society. | ||
The OAm exchange of oleate-capped zinc blende CdSe NPs was reported to offer optical properties similar to that of OAm-capped wurtzite CdSe NPs. Before OAm exchange, the ZB-CdSe NPs displayed characteristic FTIR spectral features corresponding to OAc ligands being deprotonated and bonded on their surface in the form of negatively charged oleate. The OAm exchange narrowed the energy gap between the first and second excitonic absorption bands, and this narrowing effect was size-dependent. Quenching, subsequent recovery and enhancement of the photoluminescence emission were observed due to OAm exchange. The chelating bidentate interaction between the carboxylate group and surface Cd cation was weakened in the presence of OAm. This effect can be ascribed to the formation of hydrogen bonding between oleate and OAm on the surface of CdSe NCs. OAm can bond on the particle surface through hydrogen bonding with oleate groups. An alternative bonding mechanism also proposed is that OAm bonds to the surface oleate via hydrogen bonding and a coordinating bond to a surface Cd cation. The lone-pair of electrons of the N atom of OAm can be donated to the empty orbital of the surface Cd cation. High-temperature annealing helped to replace the oleate ligands from the NP surface by OAm.171 CdSe nanoplatelets were produced in a mixture of 1-ODE and OAm for the wurtzite lattice or in 1-ODE/OAc for the ZB lattice. Especially in the latter case, OAc helped to hinder platelet aggregation. In the case of the wurtzite structure, no aggregation was observed in chloroform in the presence of OAm and tributylphosphine, even if OAc was absent.172
A capillary microreactor was employed to continuously produce NIR-emitting CdSexTe1−x nanocrystals. TOPO or TOP was used as the solvent for the anion precursor, while OAc and OAm were the solvents for the cation precursor. Then, an easy single-step coating process was applied, taking advantage of the dissolution of cadmium oxide and free elemental S in OAc. Consequently, a very thin CdS shell was grown on the as-prepared CdSexTe1−x nanocrystals in an epitaxial manner. Also, in this case, OAc was the potential metal site-coordinating ligand (via oxygen) and TOP served as the potential chalcogen site-coordinating ligand (via the P atom).173 Kolny-Olesiak and co-workers prepared different shapes of CdTe NPs by modifying the composition of the solvent (1-ODE and OAm mixture). At a later stage of the reaction with a high OAm content, also OAc depletion played an important role, resulting in the destabilization of the particles and promoting nanowire formation. The in situ-formed CdO NPs (by the reaction of Cd-oleate with OAm) acted as an additional source of Cd monomer during the synthesis and affected the growth of the CdTe NPs.174 Hassinen et al. produced CdTe QDs in the presence of tetradecylphosphonic acid and OAm. Adding OAc did not lead to the release of any amine or phosphonate anhydride ligands from the particle surface. Nevertheless, if phosphonic acid was added to the partially OAc-coated particles in a high-temperature ligand exchange process, OAc moieties would be released from the surface. Forced ligand exchange after ligand stripping with bis(trimethylsilyl) selenium would lead to the presence of OAc species on the ligand shell and cause hydrolization of the TDPAanh ligands to TDPA ligands. Also, the bound OAm was removed from the particle surface in this way.175
The single source precursor approach was used for the synthesis of CdSe QDs through the decomposition of 2-(N,N-dimethylamino)ethylselenolate of cadmium. The OAc ligand induced the formation of a direct band gap, whereas OAm showed only a broadband emission. PMMA was used as a dispersant polymer and the annealed CdSe/PMMA film was studied regarding its photoluminescent properties as a function of precursor/ligand ratio. The carboxylate groups of the OAc molecules bind strongly to the Cd surface atoms through electrostatic interactions, while the amino terminations of the OAm moieties would display a lower binding affinity to the II–VI QD surfaces. Considering that the head groups of the ligands adsorb readily at the NP surface, the ligands are expected to play a significant role in particle formation in the polymer matrix as well as in the control of the particle size and the resulting optical properties of the nanocomposite films. The PL spectra denoted that the OAc entities form a shell surrounding the QD surface, interacting with the Cd dangling bonds. The low degree of grafting for the OAm species would be associated with a bigger number of surface defects.176
In the case of CdS nanostructures, different shapes such as irregular, spheres and nanorods were produced by adjusting the solvent ratio of OAm/OAc. Ill-defined and agglomerated particles were obtained when an insufficient concentration of OAm and OAc was employed, which is associated with their poor capping ability. Increasing the concentration of OAm offered a more controllable morphology owing to its excellent capping nature compared to OAc. A higher monomer concentration of OAm assisted the anisotropic growth of NRs (c-axis) in the preferential (002) orientation, as shown by XRD monitoring.177
The reaction between OAm and OAc was shown to be essential for the formation of CdTe NWs in the presence of CdO. It assisted the activation of the Cd precursor and contributed to the creation of favorable reaction conditions for NW formation by oriented attachment. Especially, the use of OAm as a solvent was crucial to achieve nanowires. The formation of amide, caused by the reaction between OAm and OAc, could not occur in the absence of CdO. Excess OAc hindered the generation of metallic Cd in the course of the reaction. Increasing the amount of TOP hampered the formation of nanowires, while the OAc did not show any detrimental effect in obtaining NWs. Alternatively, it helped to develop a dense ligand shell around the NPs and limit aggregation.178 Recently, Zamkov and co-workers demonstrated a thermodynamically-driven aggregative growth for the shape control of CdS (and CdSe) NCs. Several insights were presented by the authors. For instance, in an OAm/CdCl2 environment, a low dispersion of particle sizes persisted for the duration of the synthesis, whereas the aggregative growth in the presence of OAc featured the formation of two NC populations at intermediate stages. The use of OAm/OAc mixtures led to the generation of very thick (>4 nm diameter) rods. The main strategy presented by the authors was the replacement of the atomic precursors with small-size NCs as building blocks, which provided a more facile way to safely predict the shape evolution of the resulting particles.179 An organometallic hot-injection route was used by Adegoke and Park to epitaxially passivate the surface of CdSe QDs with a non-toxic In2S3 shell layer, thus avoiding the use of the highly toxic CdS (Fig. 32). Hexadecylamine and OAm were used as both capping agents and reductants, while TOP, OAc, lauric acid and 1-dodecanethiol were the surface-capping agents. The lattice mismatch between CdSe and In2S3 was ∼14% and the final particles were transferred to water and showed excellent cell viability, as revealed by the MTT cytotoxicity assay.180
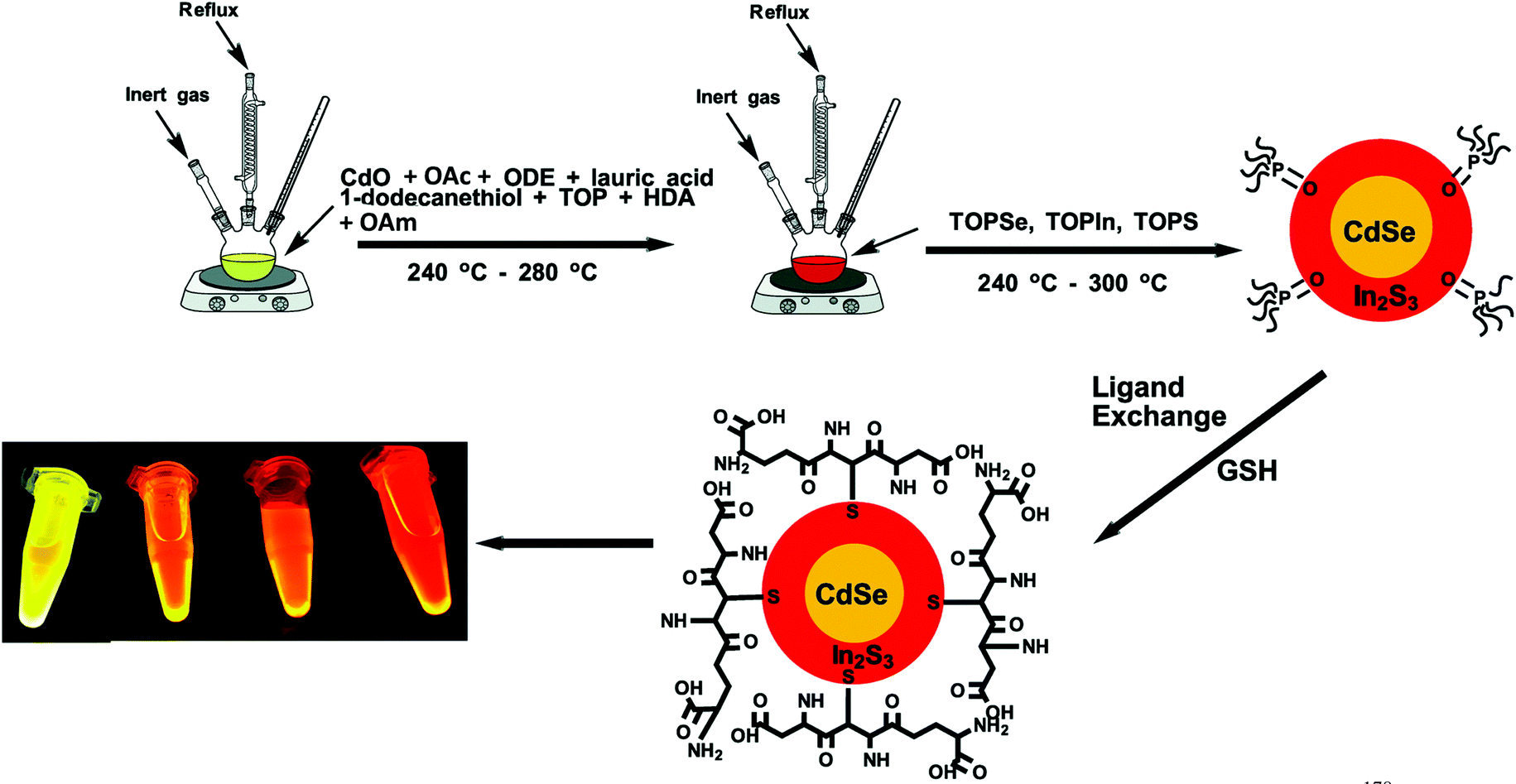 | ||
| Fig. 32 Scheme depicting the organometallic synthesis and ligand exchange reaction of GSH-coated CdSe/In2S3 QDs.180 Copyright 2017, The Royal Society of Chemistry. | ||
Cu-Based
Copper-based chalcogenide nanostructures comprise a highly attractive category of materials, which occur in a broad range of elemental compositions and crystal phases. Elongated CuInS2 nanomaterials such as nanorods are promising for applications in solar energy conversion, given that the interconnected nanocrystals offer extended pathways for the enhancement of electrical conductivity, unlike their single spherical-shaped counterparts. The group of Kolny-Olesiak observed that modifying the amount of OAc in a system containing OAm, OAc, copper and indium precursors affected the reactivity of the monomers, as described in their paper on CuInS2 nanostructures (Fig. 31c and d). Consequently, the size of the copper sulfide seeds formed in situ after the injection of tert-dodecanethiol was affected. These seeds will then function as a sulfur source. The seed formation implies the higher reactivity of Cu compared to In in the reaction solution. It seems that indium ions are mostly coordinated by OAc and OAm in the initial reaction stages. The use of a small amount of OAc led to the formation of relatively larger seeds (8 nm), which resulted in the formation of hybrid copper sulfide-copper indium disulfide nanocrystals as intermediates during the growth process of NRs.170Using large amounts of OAc resulted in smaller seeds (4 nm), which gradually turned to copper indium sulfide NRs. The reaction between OAm and OAc at high temperatures appeared to be the critical factor triggering the attachment between NRs to form multipods and networks. The 1H NMR spectra suggested that quick exchange dynamics occurred in the metal-oleylamine (or OAc) complexes. OAc can protonate acetate ions, and the acetic acid formed in this reaction will be removed from the solution upon heating. OAm seemed to be bound to the ends, rather than to the sides of the nanorods. The formation of networks of end-to-end NRs is attributed to the decrease in stabilizing effect of OAm, which was removed in a certain rate from the solution through its metal cation-catalyzed reaction with OAc.170
Wurtzite CuInS2 NPs (Fig. 33a) were prepared by using copper- and indium-nitrate precursors with dodecanethiol. The size and shape of the NPs were controlled by adjusting the Cu/In ratio together with the addition of more ligands such as OAm and OAc. For example, when OAc substituted a half of dodecanethiol by volume, uniform NRs were produced. Combining 3 mL of OAc, 3 mL of OAm and 4 mL of thiol led to the formation of quasi-spherical NPs with a mean size of 8 nm. The hard bases OAm and OAc could function cooperatively with the thiol to provide a suitable coordination. The hard bases could coordinate with In(III) on the lateral surface of the nanocrystals, and then guide the wurtzite copper indium disulfide phase to grow along the c axis. OAc could prevent the massive aggregation of NRs, while also inducing a type of attraction between the different nanorods.181
 | ||
| Fig. 33 TEM images of (a) CuInS2 nanorods.181 Copyright 2011, The Royal Society of Chemistry. (b) Cu2−xSe nanocrystals.182 Copyright 2020, Elsevier, reprinted with permission. (c) Cu2−xSe nanodisks. Copyright 2012, The Royal Society of Chemistry.183 | ||
Chang and Waclawik produced zinc blende CuInS2 nanostructures in the presence of copper iodide, indium acetate and 1-dodecanethiol using 1-ODE or OAc at 220 °C. The thermodynamically metastable wurtzite crystal structure was favored when the solvent was replaced by OAm or TOPO. Metal ions are Lewis acids, while OAm and TOPO are Lewis bases, and thus metal complexes are often generated during the reaction. In that report, OAc was deemed to be less likely to form complexes with metal ions because of its acidic property. The chalcopyrite phase (obtained also in the case of 1-ODE solvent) is similar to the zinc blende phase due to the fact that it can be regarded as resembling a double zinc blende structure along the c-axis.184 Li et al. produced alloyed CuInS2–ZnS NRs via a heating method in mixed OAm/OAc as the solvent. Cu-, In- and Zn-acetates were used as the precursors, while the source of sulfur was tert-dodecanethiol. It was found that indium oleate could react at high temperatures with OAm to form indium hydroxide. The synthesis started with the generation of copper sulfide particles, which later transformed to CuInS2–ZnS via the incorporation of In- and Zn-ions.185 For the preparation of Fe- and Zn-substituted CuInS2 NPs, Tremel and co-workers employed OAc (a hard Lewis base), OAm (a borderline Lewis base compared to OAc) and thiourea (a soft Lewis base) to form suitable complexes. Considering that Fe(III) is a strong Lewis acid similar to Ga(III), OAc can be used to form an appropriate complex but Zn(II) is not a strong Lewis acid, and therefore OAm was chosen also as a borderline coordinating solvent. The authors investigated the complex formation, structure and optical properties for the resulting particles using several characterization techniques. The prepared CuIn1−xMxS2 (M = Fe, Zn, and Ga) colloidal dispersions were stable for many weeks and these materials were considered potential agents for low-cost ink jet-printed solar cells.186 Regarding the formation of the chalcopyrite structure, the role of OAc and the reaction temperature was highlighted by Vahidshad et al., where combining OAc with OAm or using a mixture of OAm with 1-ODE favored the production of a chalcopyrite structure as a dominant product. Concerning the particle morphology, the trigonal shape was prevalent when a mixture of OAc/OAm/1-ODE solvents was used, unlike the case of only OAm solvent (mixture of semi-trigonal particles and full triangles). OAm could react with In(III), a hard Lewis acid, while thiourea could react with Cu(I), a soft Lewis acid, besides also serving as a source of sulfide ions. Increasing the thiourea to OAm ratio led to an enhancement in chalcopyrite production with respect to wurtzite.187 In the case of the CuInS2 system, it was reported that OAc could reduce the precursor reactivity, leading to a decrease in the population of nuclei formed during the nucleation step. Consequently, this would maintain the presence of a considerable quantity of precursors during the growth step, causing an increase in the NP size. 1-ODE and OAc could help to carefully regulate the kinetic growth of the NPs and obtain well-defined nanocrystals. A mixture of OAm and 1-ODE (1:1 in volume) led to the formation of rectangle-like NRs and quasi-round bodies. Thinner nanorods were obtained upon replacing half the amount of OAm with OAc.188 Lu et al. also corroborated through their findings that the obtained crystal phase can be tuned to be either chalcopyrite or wurtzite by modifying the ratio between TOP/OAc/OAm. TOP, being a soft base, shows a tendency to bind with Cu+ ions, which is a soft acid, thus regulating the coordination functions of the solvent with the copper cations. This suppresses the reactivity and slows down the NP growth rate. Then, the copper and indium cations would have increased possibility to be distributed in an ordered manner, promoting the formation of a chalcopyrite phase as the TOP ratio increased.189 For the synthesis of the system under discussion, the strong reduction property of OAm is a crucial feature. OAm can expedite the precursor decomposition. It can reduce elemental S to S2− and it can also reduce Cu(II) to Cu(I). The latter can react with OAm to form [Cu(OAm)n]+. These complexes will hinder the formation of binary copper chalcogenides such as CuS or Cu2S. The possible mechanism for the formation of CuInS2 is presented in Fig. 34.190 OAm and OAc were used as structure-directing agents for the assembly of QDs into 1D quantum chain superstructures through a stepwise heterogeneous reaction approach. High-quality lead sulfide (PbS) and copper indium sulfide (CuInS2) QDs could be readily crosslinked into microscale QCs via the interdigitation of vulcanized solid sulfur precursors. The chain-like structures can be used for Li-ion batteries applications.191
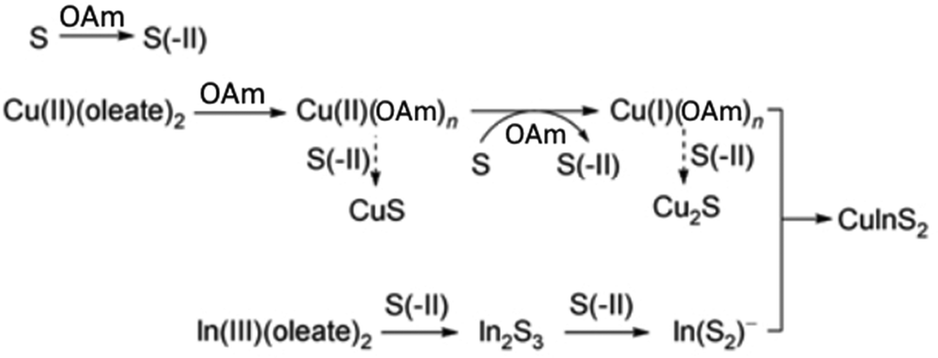 | ||
| Fig. 34 Scheme illustrating the proposed synthetic mechanism for CIS NCs. Reprinted with permission from ref. 190, Copyright 2012, Springer Nature. | ||
Regarding the CuFeSe2 system (Fig. 31b), the effect of OAc on its shape evolution was investigated by varying the amount of ligand in the reaction process. A high-temperature route was employed in the presence of OAm solvent, while the precursors were Cu(II) acetylacetonate, Fe(III) acetylacetonate and Ph2Se2.169 Gabka et al. prepared Cu–Fe–S NCs with varying compositions (from copper-rich to iron-rich) and sizes using OAm/OAc, CuCl, FeCl3 and thiourea. The crystal structure could be either chalcopyrite or high bornite. It was found that ODE could be formed in situ from OAm through the elimination-hydrogenation reaction taking place between OAm and the NP surface.192 Vahidshad et al. published a report on the synthesis of CuGaS2 NPs using copper chloride, Ga-acetylacetonate and thiourea in a solvent medium composed of either single OAm or a mixture of OAm/OAc and 1-ODE. The morphologies of the produced NPs were either elongated, polygonal or a combination of both, as a function of the particle crystal structure. This phase could be mostly wurtzite, chalcopyrite, or a balanced mixture of both crystal structures. The addition of OAc favored the formation of an increased percentage of chalcopyrite in the product. The temperature and amount of OAm also affected the resulting crystal structure to a significant extent. For example low temperatures and increased or exclusive use of OAm promoted the generation of wurtzite.193
The OAm/OAc ratio was studied regarding its effect on the LSPR properties of Cu2−xSe NPs (Fig. 33b) produced via a hot-injection route. In that work, OAm/OAc was used as both the solvent and ligand pair, while CuCl and elemental selenium powder were the Cu and Se sources. In general, OAc often coordinates to the NP surface through carboxylate groups, which are negatively charged and can act as electron donors, resulting in a decrease in the free carrier density. Alternatively, OAm will coordinate to the surface through its amino group, which will not influence the free carrier density of the NPs.182 A phosphine-free hot-injection protocol was employed to synthesize pure-phase monodisperse berzelianite Cu2−xSe nanoplates using elemental Se, OAm and 1-dodecanethiol. Besides being dissolved in OAm/thiol, Se was also tested as a precursor combined with 1-ODE, sole OAm or sole OAc, respectively. Obvious LSPR behavior was observed in the case of the nanoplates prepared at 180 °C.194 Shen et al. presented a facile non-injection approach to produce Cu2−xSe NPs with several shapes (hexagonal self-assembled nanodisks (Fig. 33c), elongated hexagonal bipyramids and trigonal bipyramids). To achieve this, the ratio of Cu-stearate, OAc, OAm and Se-ODE was adjusted. Interestingly, FTIR measurements implied that the surface of the prepared NPs was only coated by OAm, even though both OAm and OAc were used as stabilizers. When a high concentration of OAc was added, the growth of the (200) facets appeared to be too fast. Triangular Cu2−xSe NPs showed a propensity to assemble in three-dimensional superlattices when the reaction conditions were OAm-rich.183 Small Cu2−xSe NPs with a size of <5 nm were synthesized using Cu(II) stearate with NaHSe in the presence of OAm/OAc at 95 °C at the toluene/water interface. The potential photovoltaic properties of the nanocrystals were evaluated by fabricating NC-sensitized TiO2 solar cells, which demonstrated a high fill factor, implying their possible application in photovoltaic devices.195 Combining OAm with OAc (and 1-ODE) was also proven to be beneficial for the controllable synthesis of the Cu2−xS system. Hierarchical nanostructures, truncated octahedral morphologies, nanorods and spherical Cu2−xS were reported. CuCl was the copper source, while elemental S was the sulfur precursor. When single OAc was used as the solvent, no plasmonic peaks appeared in the NIR region, indicating the presence of a stoichiometric Cu2S phase. Depending on the amount of 1-ODE and OAm, Cu-rich or Cu-deficient phases with different shapes could be isolated.196
In the case of the CuInSe2 system, a nanoplate-like morphology was obtained with the sole use of OAm, while its partial replacement with OAc and 1-ODE resulted in a bunch of hierarchical structures. Using pure OAc or a mixture of OAc with 1-ODE led to a nanocube-like morphology with sizes in the range of 25–50 nm.197 A relatively easy, non-injection process was employed for the synthesis of CuInSe2 and CuGaSe2 hexagonal microplates. OAc was critical to generate the microplate morphology, taking into account that this shape was not observed in the absence of OAc.198 A facile, relatively green and cost-effective approach was reported for the synthesis of high-quality Cu2ZnSnS4 NPs with a single source precursor. OAm/OAc were used and the reaction temperature was lower than 220 °C. It was found that OAc plays a key role in the generation of these quaternary nanocrystals. OAc was suggested to bind to the surface of the particles and it may also form stable complexes with them, since the aforementioned composition is not the most thermodynamically stable one formed during the course of the synthetic process. If no OAm was used, byproducts appeared in the work under discussion even after applying a reaction temperature of 300 °C.199
It is generally accepted that deprotonated OAc (carboxylate anions, C17H33COO−) can interact selectively with certain crystal facets due to their high electron-donating capacity. In the case of CuSbS2 nanostructures prepared at high temperature, it must be considered that Cu+ and Sb3+ are soft and hard acids, respectively, and therefore the coordinating power between the metal ions and the hard Lewis bases OAm and OAc followed the order of Sb3+ > Cu+. Thus, Cu+ could form a complex with Tu (a sulfur source) and Sb could form a complex with OAc and OAm. Given that the Cu-Tu complex is more reactive than the Sb-complex, heating the mixture causes the decomposition of the former to generate Cu2−xS. Then, the Sb–OAm–OAc complex will decompose and be incorporated in the Cu2−xS lattices by ion exchange to yield CuSbS2 and 3Cu+. 1-ODE could not significantly influence the size and shape of the resulting particles, considering that nanoplatelets were obtained when OAm was used as the solvent, with or without dilution by 1-ODE. In contrast, OAc modified the shape to nanobricks. The use of a low amount of OAc resulted in the formation of smaller particles but would reduce the shape homogeneity for the resulting Su(sulfur powder)–OAm–OAc-ODE sample. Combining OAm and ODE with Su resulted in the formation of nanoplatelets with the best shape homogeneity (Fig. 35a).200 Cu2ZnSnS4 nanocrystals were prepared from copper, zinc and tin dithiocarbamate complexes in the presence of OAm/OAc. These particles could be readily dispersed in non-polar solvents such as toluene. Subsequently, an extraction process was employed to transfer the NPs to a K2S solution in formamide. Accordingly, the nanocrystals were electrostatically stabilized with S2− in formamide and HS− in the aqueous medium. Interestingly, the FTIR spectra showed that residual alkyl ligands were still left on the NP surface, which are probably arranged in rather compact configurations, and in this manner, they do not extend into the polar solvent (e.g., water or formamide). A carefully performed extraction process could maintain the original size and structure of the CZTS NPs.201 Babu and co-workers observed that the sole use of OAm resulted in the formation of nearly spherical Cu2ZnSnS4 NP, while single OAc or an OAm/OAc mixture promoted the generation of hierarchical microspheres and nanodisc-like structures. The formation of disc-like structures could be attributed to the structural direction of the aliphatic carboxy anion from OAc while interacting with the NP surface. Nanodiscs and nanoflower-like shapes may arise from the formation of hexagonal-shaped Cu2−xS NPs after the generation of the corresponding nuclei. A lower amount of OAm was associated with sheet-like morphologies. Regarding the resulting crystal structure, the combination of elemental sulfur with OAm promoted the formation of zinc blende NPs, whereas 1-dodecanethiol in OAm favored the formation of the wurtzite phase.202 It was reported that the phase of CZTS nanocrystals could be tuned from kesterite to wurtzite using dodecanethiol-S solution and OAc instead of OAm in the presence of metal precursors during the synthetic procedure. Combining OAc with the thiol assisted the tuning of the size of the spherical particles in the range of 8–20 nm. Kesterite is the most thermodynamically stable phase of CZTS and the differences in the reactivity and coordination strength of the different reagents will determine the final crystal structure (kesterite or wurtzite).203
 | ||
| Fig. 35 SEM image of (a) CuSbS2 nanostructures200 and (b) TEM image of Ni3S4 nanoprisms.204 Copyright 2018 and 2014, The Royal Society of Chemistry, respectively. | ||
In the case of the Cu2ZnSnSe4 system, the nanoplates obtained using OAm/OAc were slightly different than those produced with 1-ODE/OAc. Morphological similarities were noticed to some extent in the hierarchical configuration obtained using OAm/OAc/1-ODE. The tetragonal phase of CZTSe was obtained for all the above-mentioned samples.205 Copper tin selenide NPs were produced via a hot-injection process under N2 gas in the presence of OAm/OAc and 1-ODE at 230 °C. CuCl2·2H2O, SnCl4·2H2O and elemental Se powder were the precursors. The presence of OAm and OAc helped to obtain highly crystalline nanostructures with a tailored morphology. In the absence of OAc, agglomerated particles were produced. Regarding the formation mechanism, it seems that small Cu2−xSe NPs are generated initially upon injecting the Se precursor in the metal-oleate mixture. Subsequently, the diffusion of Sn4+ ions resulted in the formation of Cu2SnSe3 NPs.206
Fe-Based
Schaak and co-workers published a paper on the hot-injection synthesis of Fe–Ge nanostructures in the presence of OAm/OAc, GeI4, Fe(CO)5 and hexamethyldisilazane (HMDS). The reaction temperature was a crucial factor for the control of the resulting particle shape and composition. At 260 °C, nanospheres of Ni2In-type Fe3Ge2 were produced. Applying a temperature of 300 °C converted the spherical nanocrystals to CoGe-type FeGe NWs.207 Zhang et al. prepared iron sulfide nanostructures with several morphologies (nanoparticles, nanoribbons and nanoplates) through a single-source precursor approach in mixed solvents composed of OAm/OAc/1-ODE. The cubic Fe3S4 phase was obtained using Fe(Ddtc)3 precursor. This was attributed to the weak binding of OAm or OAc on the Fe ion. When Fe(Ddtc)2(Phen) was used as the iron source, it was suggested that the Phen moiety from the decomposed precursor acted as a chelating ligand, strongly coordinating to the Fe phases. Consequently, a five-ring structure with iron ions was created, leading to a nanoplate shape with Fe7S8 composition. Especially the presence of OAm was indispensable for the production of iron sulfide nanostructures, since a mixture of OAc/ODE solvents did not result in any product. The formation of FeSx nanoribbons was suggested to arise from the template-direction of micellar structures formed via the self-assembly of the capping ligands.208 FeSex nanoflakes with a tetragonal crystal structure and Se-deficiency were produced using ferrous chloride and selenium trioctylphosphine via a solution-based route in OAm/OAc. Adding OAc at a certain stage of the heating process was crucial to acquire the tetragonal PbO-type phase. The sequence of the addition of OAm and OAc in the reaction pot was reported to affect the nucleation and growth evolution of the nanocrystals. OAm can form a complex with Fe, and it was observed that when no OAc was used, nanoflakes with an inhomogeneous morphology were produced. Nevertheless, the much earlier presence of OAc in the reaction mixture could affect the nucleation and delay the growth of FeSex, providing a poorly crystalline product. It has to be noted that the complexing ability between the N atom of OAm to Fe atom and the O atom in OAc to Fe atom is different. Fe(II) can be present in a more stable manner in amines than in organic acids. The early presence of OAc and the absence of OAm will not properly preserve the state of Fe, resulting in the formation of different products (e.g., monoclinic Fe3Se4).209 In the case of FeCoS2 nanocrystals obtained in the presence of OAm/OAc with metal acetylacetonate precursors and elemental sulfur, initially large and uniform 2D microsheets were formed. Further heating at higher temperatures in the presence of excess OAc transformed the sheets to monodisperse spherical NPs via a digestive ripening process. OAc was bound to the NPs via the oxygen atoms of the carboxylic group in three different modes, i.e., monodentate, bridged and chelating. OAm binds to the NPs surface through the N atom of its amino group. OAc facilitated the formation of the final spherical shape, but OAm was largely responsible for the formation of the initial uniform sheets. Unlike OAm and OAc, in that work it was found that 1-ODE was detrimental to the formation of monodisperse particle sizes and homogeneous shapes. It neither assisted the formation of sheets, nor was it found to act as a digestive ripening agent.210Ni-Based
Ethylenediamine-based compounds were used as single-source precursors to produce NiS (and MnS) nanocrystals. Irregular shapes were obtained when OAc was combined with dodecanethiol, whereas the combination of thiol with OAm led to faceted cubic-shaped rhombohedral-phase NiS particles. Combining OAm with thiol provoked the formation of OLAH+ and dodecane thiolate anion. The anion could bind to the surfaces of the NiS crystals better than the neutral thiol, contributing to isotropic particle growth. With an increase in the reaction time, shape evolution to faceted nanocubes occurred. Regarding the MnS composition, particles with a rock-salt structure and hexagonal shape were isolated in a mixture of OAm-ODE by adjusting the reaction temperature. With a suitable OAm/ODE ratio, monodisperse MnS nanocubes can be formed. In the case of NiS particles, the exclusive use of OAm resulted in uncontrollable nucleation and formation of polydisperse NPs.211 A temperature-controlled injection method was employed for the synthesis of rGO-functionalized nickel sulfide NPs. Single (OAm) or mixed (OAm, OAc, ODE) solvent systems were tested. Consequently, nanocrystals with a controlled phase (NiS, NiS2, and Ni3S4) were deposited on rGO (reduced graphene oxide) sheets. When OAc was used, FTIR measurements illustrated its complete chemisorption on the NPs surface. OAm could assist the reduction of GO NPs, given that rGO has lower reactivity with S than GO. However, when OAm was used as a single solvent, the interaction between OAm and GO was stronger than that between the nickel and sulfur ions, causing the generation of uncontrolled nickel sulfide phases on rGO. Thus, to overcome this issue, the mixed solvent approach was used (Fig. 36), yielding phase-controlled NPs with a homogeneous size. The injection method enabled the sulfur ions to react slowly with the well-dispersed nickel ions, ensuring the generation of specific single phases.212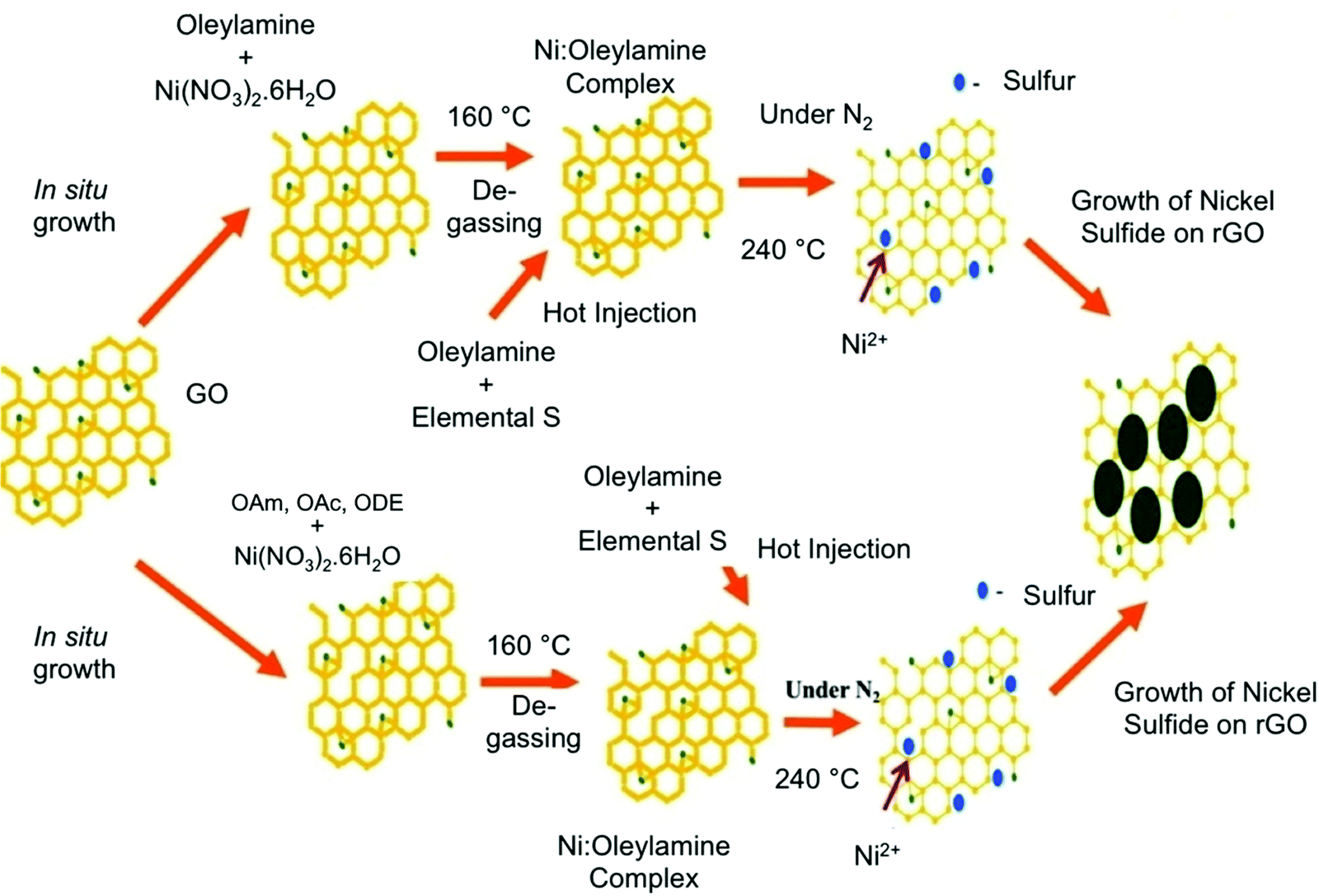 | ||
| Fig. 36 Scheme representing the growth of rGO/nickel sulfide structures in different solvent mixtures. Reproduced with permission from ref. 212. Copyright 2017, Wiley-VCH. | ||
Ni3S4 NPs were produced via the decomposition of two different nickel precursors in the presence of 1-dodecanethiol and OAm/OAc. The choice of precursor led to a change in the triangular nanoprism shape to tetrahedra (nanopyramids). By keeping the concentration of OAm stable, an increase in the amount of OAc resulted in an increase in the size of the nanoprisms, but a further enhancement of its concentration led to particle aggregation and a broad size distribution. A variation in the OAc and OAm concentrations had no impact on the particle shape when nickel chloride was used as the precursor (Fig. 35b). However, it influenced the shape in the case of the Ni(acac)2 precursor (tetrahedral pyramids, NWs, NRs and butterflies were formed).213
Ni-Se nanostructures of different stoichiometries were produced via colloidal synthesis in the presence of OAm or OAm/OAc mixture. OAm served as a reductant and/or stabilizer, whereas OAc acted as a co-surfactant to produce different stoichiometries. The sole use of OAm was mainly associated with thermodynamic-type growth in all directions, leading to the formation of spherical particles.214
Pb-Based
In the case of the PbS system, FTIR measurements indicated the formation of oleyl ammonium oleate salt through the reaction of OAc with OAm. This ammonium salt is the medium where the chemical reaction for the generation of PbS takes place. The spectral features of PbS QDs as a function of temperature, reaction time and Pb/S ratio were studied by Razumov and co-workers.215 Zhao et al. produced PbS QDs in the presence of ligands such as OAm, OAc and TOP. It was found that the amphiphilic polymer approach, using PMAO-PEG, could transfer the particles to water without losing their initial structure and photostability when OAc or OAc/TOP were the starting ligands. Nevertheless, OAm caused severe ligand etching, inducing Ostwald ripening and resulting in a double size distribution and total photoluminescence loss within a short time after transfer to the aqueous medium. Etching and Ostwald ripening resulted in several unpassivated surface atoms, which deteriorated the PL properties to a significant extent.216 Shape-controlled PbS NPs were prepared using Pb-acetate and dodecanethiol, while additional reagents such as OAm, OAc and 1-ODE were also tested. By using suitable mixtures of surfactants, shapes such as octahedral, starlike (Fig. 37a), cubic, truncated octahedral and truncated cubic were produced. Increasing the reaction temperature could cause the binding of OAm to the NP surface to be weaker, thus inducing the acceleration of the nanocrystal growth rate. However, a bigger aggregation risk is also associated with the weaker binding of OAm.217 Warner and Cao showed that the suitable combination of OAm, OAc and TOP could tune the morphology of the PbS nanostructures from cubic and stars to rods (Fig. 37b) and branched NWs. Although some previous reports indicated that OAm has the tendency to coordinate with the {111} PbS facets and prevent the formation of nanocubes, this was not confirmed by the work under discussion.218 | ||
| Fig. 37 SEM image of (a) star-like PbS nanostructures. Reprinted with permission from ref. 217. Copyright 2012, the American Chemical Society. (b) TEM image of PbS nanorods. Reproduced with permission from ref. 218. Copyright 2008, Institute of Physics. | ||
A Pb-stearate complex and OAm-selenium were employed as precursors to produce monodisperse PbSe nanocrystals with a tailored morphology. Five different shapes (quasi-spherical, cubic, octahedral, cubooctahedral and star-shaped) were generated, while the particle size could be tuned from 18 to 50 nm by adjusting the amount of OAc, keeping the amount of OAm unchanged. OAm was effective and required during the hot-injection process, but if used in excess it led to the etching of the NP surface. A low concentration of OAc favored the passivation of {100} facets, producing quasi-spherical and cubic NPs (Fig. 38a), while a high OAc amount promoted the generation of octahedral and star-shaped NPs with {111} facets. More OAc also suppressed the activity of the monomers, resulting in a lower population of nuclei in the reaction pot, and therefore PbSe NPs with a larger size were synthesized.219 Shao et al. investigated the use of OAc, OAm and dibenzylamine in the shape-controlled synthesis of PbSe nanocrystals (Fig. 38b). OAc seemed to possess a more intense interaction with PbSe than that of DBA, as it was shown that the use of OAc can modify the nanostructure shape from cubes to octahedrons. However, FTIR indicated that DBA had a stronger interaction with the NPs than OAm. The synergistic effect between OAm, OAc and DBA assisted the growth of hexagonal pod-like nanostars with well-defined morphologies. If no OAc was used, cubic PbSe NPs were obtained.220 Zhang et al. reported the synthesis of small and surface-passivated PbSe QDs by adding SnCl2 as a nucleation promoting agent and PbCl2 as an in situ halide-passivating agent (Fig. 38c). Surface passivation is very important for these types of NPs. For example, oxidation will lead to the loss of OAc ligands and surface Pb atoms, forming dissolvable Pb-oleate. In the work under discussion, XPS spectra showed that halide adlayers were created on the top of PbSe QDs, preventing their oxidation and offering stable photoluminescent behavior.221
 | ||
| Fig. 38 (a) TEM image of PbSe nanocubes.219 Copyright 2013, The Royal Society of Chemistry. (b) SEM image of star-shaped PbSe NCs. Reprinted with permission from ref. 220. Copyright 2014, the American Chemical Society. (c) TEM image of 7 nm PbSe QDs. Reproduced with permission from ref. 221. Copyright 2015, the American Chemical Society. | ||
Sn-Based
SnS QDs coated by OAm, OAc and TOP ligands were produced via a commonly employed solvothermal route. The particles had a mean size of 8 nm and were applied as the hole-transporting material in n–i–p-structure perovskite solar cells, achieving a power conversion efficiency of 13.7%. Very good air stability was demonstrated, and the device performance remained stable after 1000 h of storage under ambient conditions.222 Greenidge and co-workers used a variety of high boiling point solvent combinations to produce tin disulfide thin nanosheets. The use of single OAm resulted in mixed phases, while solvent combinations such as OAm/OAc/ODE and OAm/OAc led to single-phase 2H-SnS2, without signs of phase impurities. OAm could induce relatively fast nucleation and particle growth. Upon mixing with OAc, the slower nucleation and growth steps affected the particle shape, leading to flat and well-defined edges.223 In fact, it has been reported that the ratio of OAm cis to trans isomers and the amount of OAc can also influence the size and thickness of tin sulfide nanosheets, as well as their crystal structure (α-SnS or π-SnS). For example OAc was reported to form a highly ordered monolayer on the (100) surfaces of PbS, and thus suppressing an increase in the nanosheet thickness. 98% cis OAm promoted the formation of densely packed ligands, lowering the surface energy, and then additionally hindering the sheet growth in the [010] direction. A tin(II) oleate complex was formed after dissolving tin(II) chloride in a mixture of OAm and OAc. Oleylammonium chloride was formed simultaneously, separating the Sn from the Cl ions. It is worth noting that hexamethyldisilazane aids the formation of nanosheets. In the absence of OAc, the crystal edges would be unshaped. This ligand improved the hexagonal shape of nanosheets and it preferentially attached to the (101) and (100) crystal facets.224 Thin films of SnS were produced with nanocrystals as their constituting units using low-cost solution phase methods. The exchange of the starting OAm/OAc/TOP surface ligands helped to enhance the electronic coupling and charge carrier mobilities by decreasing the interparticle distance, ensuring the immobilization of the NCs.225 Liang et al. investigated the role of reaction temperature and the volume of OAc in the synthesis of SnS NPs using Sn(OAc)x and S–OAm precursors. In that work, increasing the volume of OAc resulted in crystal aggregation. Possibly, this occurred due to the coalescence of the destabilized NPs.226Zn-Based
The phase transfer behavior of CdSe/ZnS, InP/ZnS and CuInS/ZnS QDs coated with OAc, OAm and octadecylamine ligands from organic medium to water was studied by Marsan et al. Besides the case of CuInS-based NPs, particles capped with amine-group surfactants seemed to transfer more slowly to water than those capped with OAc.227 The Hyeon group, in 2005, published a report on the synthesis of ZnS nanostructures of different shapes in organic medium. When OAc was replaced by OAm, 10 nm quasi-spherical nanocrystals were formed. Under certain reaction conditions, which allowed kinetic control and oriented attachment mechanism to take place, ZnS nanorods could also be formed.204 An OAm/OAc mixture was successfully employed to produce ZnIn2S4 nanostructures with a hot-injection process. OAc was expected to form quite stable complexes with Zn(II), meaning that a high temperature and prolonged reaction time would be needed to achieve nanoparticle synthesis. OAm accelerated the decomposition of the complexes not only owing to its reducing role, but its amine entity is also a nucleophile and can attack the carbonyl group in the cation-acid complex, followed by cation release from the complex.228 Wurtzite and zinc blende ZnSe NPs were synthesized via the thermal decomposition of Zn(ac)2 in the presence of OAm/OAc. TOP-Se was the selenium precursor. It was found that the OAc complex started to form ZnSe NPs at a higher temperature (274 °C) than in the case of OAm (220 °C). A stronger coordination of OAc with Zn monomers than that with OAm was reported. A higher supersaturation (higher concentration of monomers) during the nucleation stage would result in thermodynamically stable ZB phase. Alternatively, the wurtzite phase is favored when low supersaturation occurs.229 In the case of the Zn0.5Cd0.5Se system, the mean particle size increased from 4 to 9 nm with an increase in OAm quantity from 4 to 10 mL. The formation of the [Zn(OAc)2-]OAm complex was followed by its decomposition to form ZnO, which grew on the Zn0.5Cd0.5Se surface. Especially high OAm contents induced the formation of ZnO more easily.230Miscellaneous metal chalcogenides
Bi2Se3 nanoflowers consisting of numerous thin nanosheets were produced via a hot-solution injection method in the presence of OAm/OAc. Adjusting the ligand ratio allowed the NF size to be tuned from 100 nm to 1 μm. Characterization techniques indicated that Bi-oleate was reduced initially by OAm to Bi-rich NPs, which subsequently reacted with the Se precursor to yield Bi2Se3 nanosheets. Ostwald ripening helped to grow and assemble the nanosheets into hierarchical nanoflowers.231 Also in the case of bismuth, OAm and hexadecylamine have nitrogen as the ligating atom, and therefore the interaction of these ligands with Bi cations is weaker than that of the OAc. For the Bi2S3 system prepared in the presence of OAm/OAc/hexadecylamine, respectively, XRD showed a bias of orientations towards the (211) crystal planes when using OAm. The highest crystallinity degree was observed in the OAm-capped sample. Bismuth dithiocarbamate was the Bi source in that work.232The adjustment of the OAm/OAc ratio was shown to be important for the formation of ultrathin MoSe2 nanosheets. Porous network-like or discrete flower-like nanostructures (Fig. 39a) with a large specific surface area and abundant exposed edge sites could be selectively formed. Under certain conditions, OAc could accelerate the reaction of Se-ODE with the Mo precursor, causing the production of numerous nanosheets with a small size. The passivation effect of OAm/OAc on the curled nanosheets facilitated their stability.233
 | ||
| Fig. 39 (a) TEM image of MoSe2 nanoflower. Reprinted with permission from ref. 233. Copyright 2017, Elsevier. (b) TEM image of GeSe nanobelts. Reproduced with permission from ref. 234. Copyright 2012, the American Chemical Society. (c) SEM image of Sb2Se3 NRs.235 Copyright 2020,The Royal Society of Chemistry. | ||
GeSe nanobelts (Fig. 39b) were produced in a one-pot approach using a mixture of GeI4, TOP-Se, OAm/OAc and HMDS heated at 320 °C. Diffuse reflectance measurements implied that the particles possessed NIR absorption with an indirect band gap of 1.1 eV. Interesting electrical properties were recorded, with a resistivity of approximately 360 Ω cm.234 Tuning the OAc/OAm volume ratio helped to control the oxidation extent of Sb2Se3 NRs (Fig. 39c), which spanned 1.20–51.5% by increasing the OAc ratio, as shown by XPS. Pure OAc yielded a mixture of nanotube and nanorod morphologies. An OAc/OAm volume ratio of 5:4 provided a highly crystalline product with orthorhombic phase and high purity. The nanorods produced in OAc/OAm displayed a higher photoresponsivity, stable photo-switching, lower dark current and higher photocurrent/dark current ratio than that produced in single OAm or single OAc. Varying the amount of OAc did not lead to an important modification of the optical absorption and bandgap energies of the Sb2Se3 NRs. The modest degree of surface oxidation (20–30%) was associated with the superior photoresponsivity of the nanorods.235 Han et al. presented a phase transfer-based route to produce shape-controlled HgS nanocrystals using different solvents or temperatures. Hg(II) ions were transferred from water to toluene in the presence of OAc or OAm, followed by sulfidation at a given temperature. The use of only OAc led to a spherical NP morphology with coarse surfaces. Dendritic morphologies were obtained when an OAm/OAc mixture was used at different ratios (Fig. 40a). Beyond a certain range of ratios, cubic, fan-like or large spherical NPs were isolated. Possibly, OAm self-organized into reverse micelles, where HgS was generated. The amount of OAm influenced the resultant NP shape by changing the structure of the OAm reverse micelles. The hydrocarbon amide formation, produced by the condensation reaction between OAm and OAc, also affected the reverse micelles and the final NP morphology.236 The single-source precursor approach was employed to produce Pd4S with different shapes at 195 °C. OAm medium resulted in flower-like structures, while a mixture of OAc and ODE led to a spherical shape. The latter non-reducing and weakly ligating mixture caused the Pd4S structures to self-assemble rapidly after their formation, yielding nanospheres. The strong coordinating and reducing functions of OAm were responsible for the nanoflower shape in this medium, where the slow self-assembly of quickly formed OAm-capped Pd4S clusters induced the formation of the flower-like shape.237
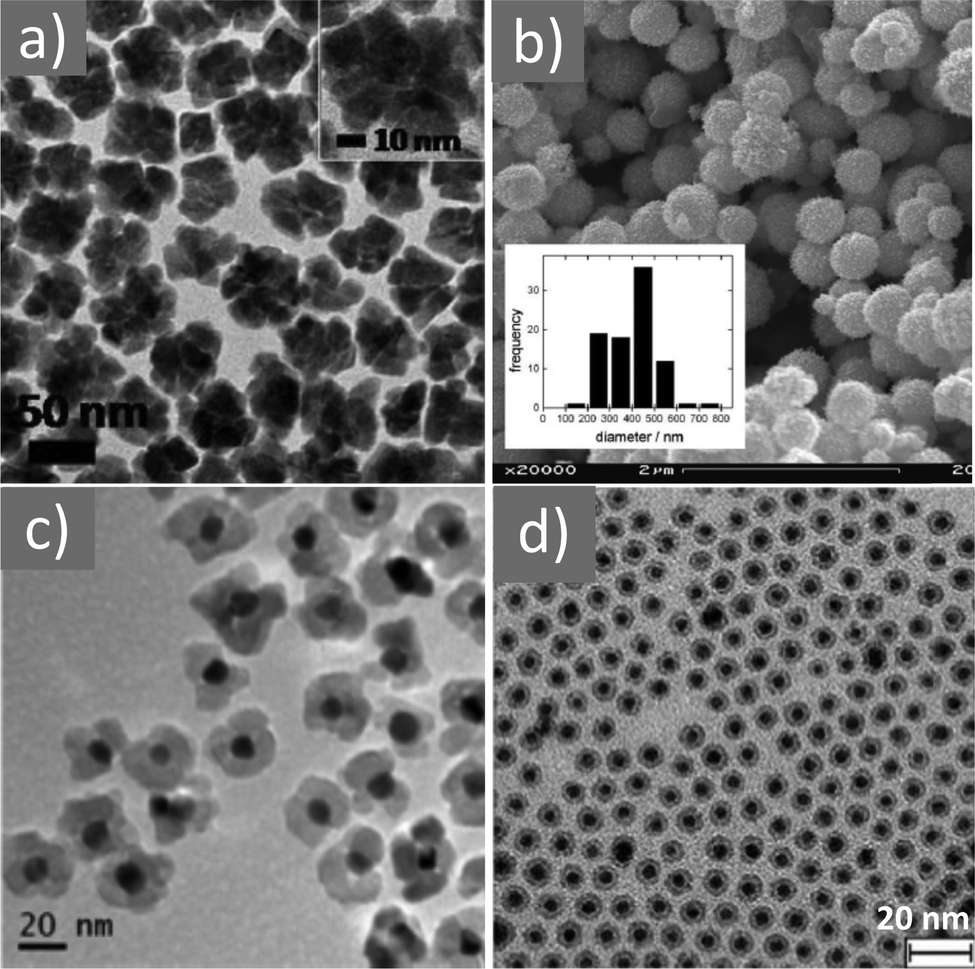 | ||
| Fig. 40 (a) TEM image of HgS nanocrystals.236 Copyright 2014, The Royal Society of Chemistry. (b) SEM image of Fe3O4@Au nanocomposites. Reprinted with permission from ref. 238. Copyright 2013, Elsevier. (c) TEM image of FePt/MgO NPs. Reproduced with permission from ref. 239. Copyright 2008, the American Chemical Society. (d) TEM image of Pd-Fe3O4 hybrid NPs.240 Copyright 2014, The Royal Society of Chemistry. | ||
Kawai and colleagues reported the production of tetrapod-shaped EuSe NPs via the thermal reduction of europium chloride and an organic selenide complex in a medium composed of hexadecylamine (HDA) and OAm/OAc. A small amount of OAc induced anisotropic crystal growth. The coordination ability of the carboxyl group in OAc may be stronger than that of the amine group in HDA. The carboxyl groups are considered to play a significant role in stabilizing the Eu(III) precursor complexes. The OAm would also support the stabilization of the europium-based complexes in HDA. All these phenomena affected the crystal growth mode of the EuSe nanostructures. Applications in domains such as magneto-optics are envisaged for these lanthanide nanocrystals.241 OAm, OAc and dodecanethiol were used for the synthesis of AuSe nanostructures. The different reducing powers and binding energies of the reagents resulted in distinct growth modes and resulting shapes. OAm and OAc capping resulted in belt-like structures, while the thiol led to short rod-like morphologies. The lowest amount of impurities was observed in the case of OAm-capped NPs.242 Dandelion-like Co9S8 and sphere-like Co3S4 nanostructures were produced using Co(acac)2 as the cobalt source and 1-dodecanethiol as the S source in a mixture of heated OAm/OAc. The molar ratio of Co:S precursor and the solvent composition affected the crystal phase and shape control of the resulting cobalt sulfide particles. The sphere-like particles displayed a better electrocatalytic performance for the hydrogen evolution reaction compared to the dandelion-like particles. The large surface area and high electrical conductivity of the sphere-like Co3S4 explain their higher catalytic activity.243
In summary, regarding metal chalcogenides nanostructures, one can highlight that also in this case, the role of OAm/OAc is crucial to tune the particle morphology, and also the crystal structure (e.g., wurtzite or chalcopyrite). OAc acts as a hard Lewis base and OAm is a borderline Lewis base compared to OAc. These properties affect their ability to form coordination complexes with metals in the reaction pot. Finally, the way that OAm/OAc may influence the optical (photoluminescent, fluorescent) properties of metal chalcogenide NPs was discussed. For instance, excess OAm can cause severe ligand etching and loss of photoluminescence.
4. Heterostructured and core–shell nanostructures
Multifunctional nanomaterials with complex configurations such as core–shell and heterostructured (island-type, onion-type, dimers, core-satellite, etc.) morphologies combine the different properties of all their components, aiming for improved suitability in a range of fields. Mohammad and Arfin produced Fe3O4@Au core–shell NPs by growing gold onto pre-synthesized magnetite NPs. OAm/OAc formed a capping monolayer surrounding the NPs, protecting them from interparticle interactions. Diphenyl ether and 1,2-hexadecanediol were also used, while the metal sources were iron acetylacetonate and gold acetate.244 Of course, sometimes the gold shell was not homogeneously distributed around the spherical magnetic core. Phase transfer to water using tetramethylammonium hydroxide (TMAOH) provided negatively charged hydroxyl groups on the NP surface, which replaced the initial hydrophobic OAc/OAm coating.245 In fact, initially, the iron oxide NPs were strongly stabilized by OAc, and in the case of the chloroauric acid precursor, AuCl4− competed and finally displaced OAc from the surface of the Fe3O4 core NPs by a ligand exchange mechanism. Varying the OAm/OAc ratio, the particle size could be tuned, for example, in the range of 100–500 nm, as shown by Lloret et al.238 It has been suggested that preferential absorption on the {111} facets occurs when the size of the head group of the additive ligands is comparable to the spacing of the Au atoms on the facet. XRD measurements can show if OAm absorbs preferentially on the {111} facets, which would result in very slow growth along the {111} orientations and prominence of the {111} facets. In that work, the deposition of Au on the iron oxide NPs led to the formation of Fe3O4@Au particles, which agglomerated to form larger aggregates. Subsequently, OAm-controlled anisotropic deposition of gold continued to occur on these aggregates, promoting the development of branched nanostructures (Fig. 40b).238 In another report, iron oxide–Au core–shell NPs were produced via the thermal decomposition of Fe(CO)5 in the presence of OAm/OAc in octyl ether. Oxidation of the Fe cores took place from the surface to the inner region and the oxidation was incomplete in the interior part. Oxidation could occur probably during the washing and redispersion stages, before the gold coating. A subsequent coating step helped to grow Au on the initial NPs by the reduction of gold acetate using 1,2-hexadecanediol. Size monodispersity and capping with OAm/OAc mediated the formation of 2D-arrays.246Salado et al. presented the synthesis of water-soluble magnetite@Au NPs functionalized with biocompatible and fluorescent molecules and they studied their interaction with cell cultures. The initial synthesis of magnetite involved OAm/OAc as hydrophobic ligands, followed by the thermolytic reduction of gold(III) acetate to grow Au on the Fe3O4 seeds. To confer biocompatibility, initial treatment of the product with PMAO, and then further conjugation with either PEG or glucose helped in performing the cytotoxicity and cell studies. The gold shell was also helpful for the aimed biocompatibility and it protected the magnetite core from further oxidation. In addition, it enabled the possibility to attach more ligands, and it also enhanced the superparamagnetism of the NPs by softening the interactions between them.247 The pyrolysis of Fe–nitrate with OAm/OAc led to the synthesis of FexOy@C NPs, which were then added to a heated mixture of OAm and silver nitrate in different concentrations. The final product was a mixture of iron oxide crystalline NPs (surrounded by an amorphous C shell) and Ag crystalline NPs. Different shapes such as ellipses, quadrangles and hexagons with rounded corners having sizes in the range 20–40 nm could be isolated.248 The group of Shouheng Sun, in 2008, published the synthesis of fcc-FePt/Fe3O4 NPs with platinum acetylacetonate and iron pentacarbonyl precursors in the presence of OAm/OAc. Coating of the FePt NPs with MgO through the thermal decomposition of Mg(acac)2 helped to hamper particle sintering upon high temperature reductive annealing (Fig. 40c). In this way, the fcc-FePt core could be converted to the tetragonal fct-FePt phase with more attractive magnetic properties without undesirable side effects. The final hard magnetic NPs could be stabilized by hexadecanethiol/OAc and dispersed in hexane.239 Normally, particles capped by OAm/OAc are only soluble in non-polar or weakly polar solvents. TMAOH was also shown to be efficient for the water transfer of initially OAm/OAc-capped Fe/Fe-oxide core–shell NPs. In fact, TMAOH can bind to the empty spaces available at the surface of NPs between OAm and OAc, offering hydrophilic behavior.249
Heterostructured Au–Fe3O4 dimer nanostructures coated with OAm/OAc were prepared by a method combining seed-mediated growth and high temperature decomposition. The surfactant coating was replaced by DMSA by applying a ligand exchange process. In fact, the charge transfer of Au NPs may partially reduce magnetite at the Au–Fe3O4 interface to the antiferromagnetic FeO phase. Thus, high magnetic anisotropy can be observed for the particles, which is attributed to the exchange bias between FeO and Fe3O4. Improved heating efficiency, as reflected by the elevated specific absorption rate (SAR) values, was achieved for the DMSA-coated dimer particles.250 Pariti et al. produced superparamagnetic Au–Fe3O4 bifunctional NPs via a single step hot-injection precipitation method. OAm helped to reduce Au(III) in chloroauric acid precursor to Au(0) seeds, which simultaneously oxidized Fe(0) from iron pentacarbonyl to form the bifunctional particles. The size of the NPs was affected by the injection time of the gold precursor, the amount of OAm (OAc was also present), the reflux time and the ratio between the two precursors. Subsequently, L-cysteine was attached to the NPs to enable the evaluation of their cytotoxicity.251 In a somewhat different approach, 8–20 nm Au–Fe3O4 NPs were synthesized by decomposing Fe(CO)5 on the surface of gold NPs in the presence of OAc and OAm. Subsequent coating with PEG enabled cytotoxicity measurements to be carried out. Actually, PEG helped to align the oil phase of the NPs to the water phase, dopamine reacted with OAc on the NP surface and the amino entity of dopamine reacted with the carboxyl group of PEG. This ensured that PEG was only connected to the Fe3O4 NPs.252
Schaak's group showed that Pt–Fe3O4 heterodimers can react with Pb and Sn acetylacetonates at 180–200 °C in a solution of benzyl ether, OAm/OAc and tert-butylamine borane (TBAB) to generate PtPb–Fe3O4 and Pt3Sn–Fe3O4 heterodimers, respectively. Actually, the PtPb–Fe3O4 dimers could spontaneously aggregate to form colloidally stable (PtPb–Fe3O4)n flower-like nanostructures through a process analogous to a molecular condensation reaction.253 Bao and co-workers investigated the attachment of Pt NPs on surfactant-capped Fe-oxide NPs. When the latter particles were coated with OAc, the Pt attachment proved to be challenging because of the strong binding between the carboxylic groups and iron oxide surface. In the case of OAc/TOPO or OAc/OAm-coated NPs, an important increase in Pt attachment was noticed. Single OAc was associated with a uniform spherical shape for the iron oxide NPs, while particles with OAc/TOPO capping were less spherical but still homogeneous. Irregular morphologies were observed for the OAm/OAc-capped particles. However, these ill-defined shapes contained numerous corners and edges, which could act as preferred heterogeneous nucleation sites, thus facilitating the attachment of platinum.254 X-ray absorption near-edge spectroscopy (XANES) measurements were employed to study the thermal evolution of OAm/OAc-capped Pt-rich FePt/Fe3O4 heterodimers. An increase in temperature led to a gradual reduction of magnetite to FeO assisted by the thermolysis of the ligands, whereas at temperatures higher than 550 K, the Fe3Pt composition started to form. Interestingly, it was noted that ‘secondary’ reducing agents such as C, CO and H2 could be produced via the thermolysis of OAc and OAm at high temperatures, causing possible modifications in the oxidation state of the heterodimers.255
Ag–Fe3O4 nanohybrids were fabricated through a simple one-step reaction of Ag-acetate with Fe-acetylacetonate in the presence of 1,2-dodecanediol, OAm and OAc. These ligands were necessary for successful particle formation. OAm and diol acted as reductants for the generation and stabilization of silver particles. OAc and diol were crucial especially for the magnetite component. No heterodimers or even core–shell NPs could be produced in the absence of one of the above-mentioned ligands. The final Ag–Fe3O4 dimers displayed higher sensitivity for the detection of 2-naphthalenethiol compared to Ag or core–shell Ag@Fe3O4 NPs.256 Socolovsky and co-workers prepared Ag–Fe3O4 nanoheterostructures in two steps, where Ag seeds were initially produced with a tailored size according to the chosen OAm/OAc ratio, and then magnetite NPs were grown onto those seeds in a second stage. The interaction of the seeds with the metal surface was mediated by the electron-dense carboxylic group of OAc. The denser presence of these groups over the surface would generate the charge compensation required to permit nucleation on multiple facets. The use of a large amount of OAc and Ag NPs with a size of around 32 nm induced high charge compensation conditions and resulted in the formation of flower-like structures. Alternatively, low OAc quantities and 23 nm Ag seeds led to a decrease in the charge compensation, hindering the magnetite nucleation in numerous facets and promoting a dumbbell-like morphology.257 Kim and Song produced Pd-Fe3O4 hybrid NPs by initially synthesizing spherical 4 nm palladium NPs, which were used as seeds. Subsequently, iron pentacarbonyl was thermally decomposed on the surface of Pd in the presence of OAm/OAc with a variable ratio. Further growth and air oxidation followed. When OAm was the only surfactant added, highly homogeneous concentric Pd@Fe3O4 yolk-shell particles were obtained (Fig. 40d). In the case of OAc as a single ligand, the product was large metal clumps without the formation of crystals. OAm acted as a typical stabilizer; however, OAc was strongly bound to Fe, forming an Fe-oleate complex with a high nucleation barrier. Increasing the OAc concentration decreased the number of Fe atomic seeds and slowed down the growth rate, resulting in a gradual modification of the particle symmetry. Adjusting the OAc/OAm ratio was crucial to tune the structure and the crystal domain size of the resulting hybrid particles.240 Yadong Li and co-workers grew magnetite NPs on pre-synthesized metal seeds (M = Au, Ag, Pt, and Pd) to produce M-Fe3O4 heterostructures. The authors used both OAm and OAc as capping agents to complex ferric ions, and also to reduce the required reaction temperature. The particle size of the magnetite NPs increased with a decrease in the concentration of the capping agents.258 Hyeon's group presented the gram-scale synthesis of Pd-Fe3O4 heterodimer NCs via the thermolysis of Fe-acetylacetonate and Pd-acetylacetonate using OAm and OAc. The heterodimer NPs were composed of a 6 nm-sized Pd nanosphere and 30 nm-sized faceted Fe3O4 nanocrystal, exhibiting soft ferrimagnetic behavior, where high saturation magnetization and low coercivity values were recorded.259
MoS2–MoO2 composites were synthesized via a hot-injection route involving MoO3 and S powder as precursors with OAm/OAc solvents. Upon increasing the reaction temperature and prolonging the reaction time, OAc reorganized the octahedral units of MoO3 and a low valence of Mo gradually became prevalent. OAc can react with MoO3, modifying its bonding environment rather than forming a new composite. The OAm can first reduce the Mo-precursor to MoO2 as the core, followed by the replacement of the O atoms by S atoms to create an MoS2 shell, leading to the formation of an MoS2–MoO2 core–shell structure. The detailed reaction mechanism considered responsible for this synthesis is suggested in Fig. 41.260 Kanehara and co-workers reported that the stability of Co9S8/PdSx nanoacorns can be improved by adding OAm/OAc stabilizers. Especially, the affinity of this ligand pair to surface cobalt ions helps in this direction. For example, nanoacorns treated with TOP or dodecanethiol did not appear to form stable solutions for a prolonged time.261
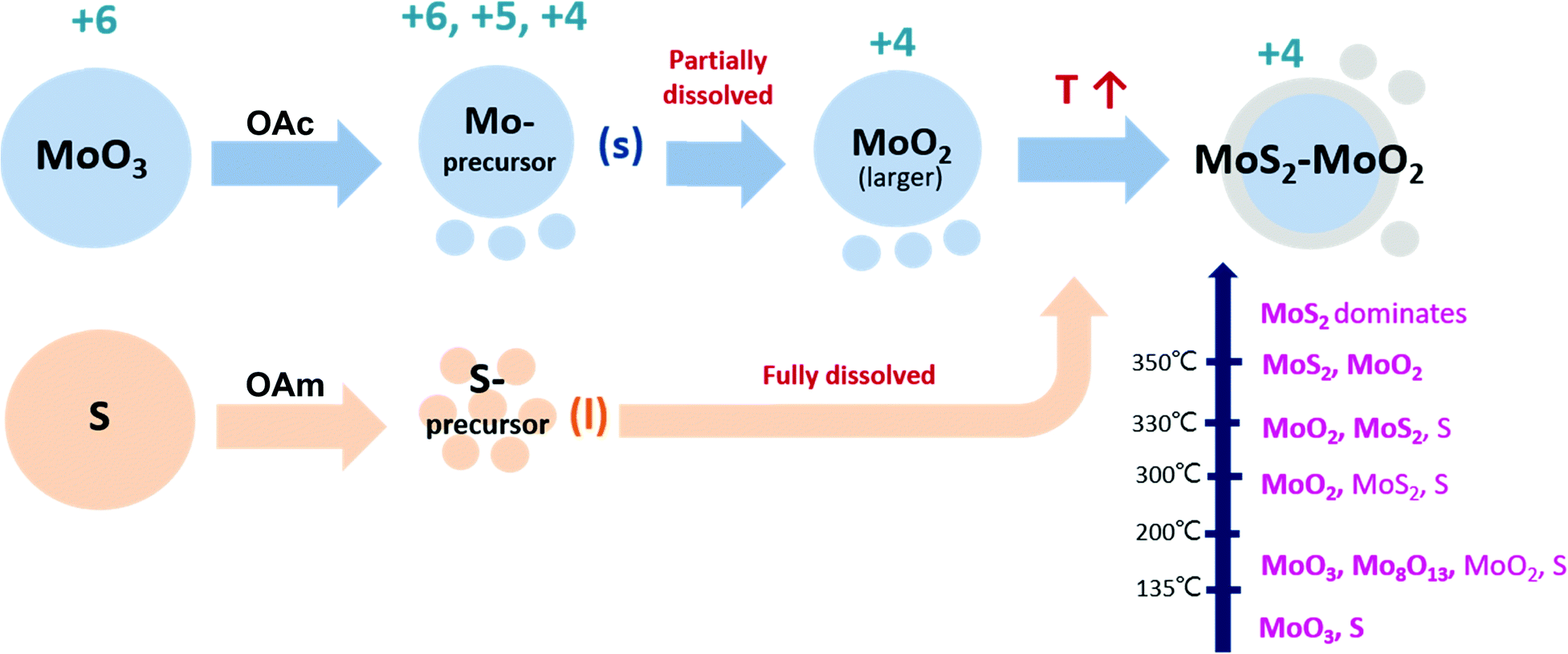 | ||
| Fig. 41 Schematic representation of the reaction mechanism leading to the formation of MoS2–MoO2 composite structures in the presence of OAm/OAc. Reproduced with permission from ref. 260. Copyright 2017, Elsevier. | ||
Lee and co-workers reported the synthesis of PbSe NRs and PbSe/CdSe heterojunction NRs through direct Cd-to-Pb cation exchange in CdSe NRs. The use of suitable ligand-cation combinations permitted cation exchange, while leaving the nanoparticle shape unchanged. For instance, solvation of the Cd cations using OAm enables the cation exchange process, which would not be feasible using OAc instead of OAm. Given that Cd(II) and OAm are harder acid–base combinations than Pb(II) and OAc, Cd–OAm or Pb-oleate is more stable than Pb–OAm or Cd-oleate complexes. Pb-oleate on its own is not capable to drive enough Cd-to-Pb cation exchange given that OAc is less capable of solvating Cd2+, which diffused out of the NRs during the conversion. OAm exhibits strong hard–hard interaction with Cd(II), and additional OAm in the mixture of CdSe NCs with Pb-oleate can offer a driving force for the cation exchange. In fact, the authors of the work under discussion presented in detail the phenomena taking place to achieve the heterojunctions and the reader is directed to that paper to access these insights, and also see Fig. 42.262
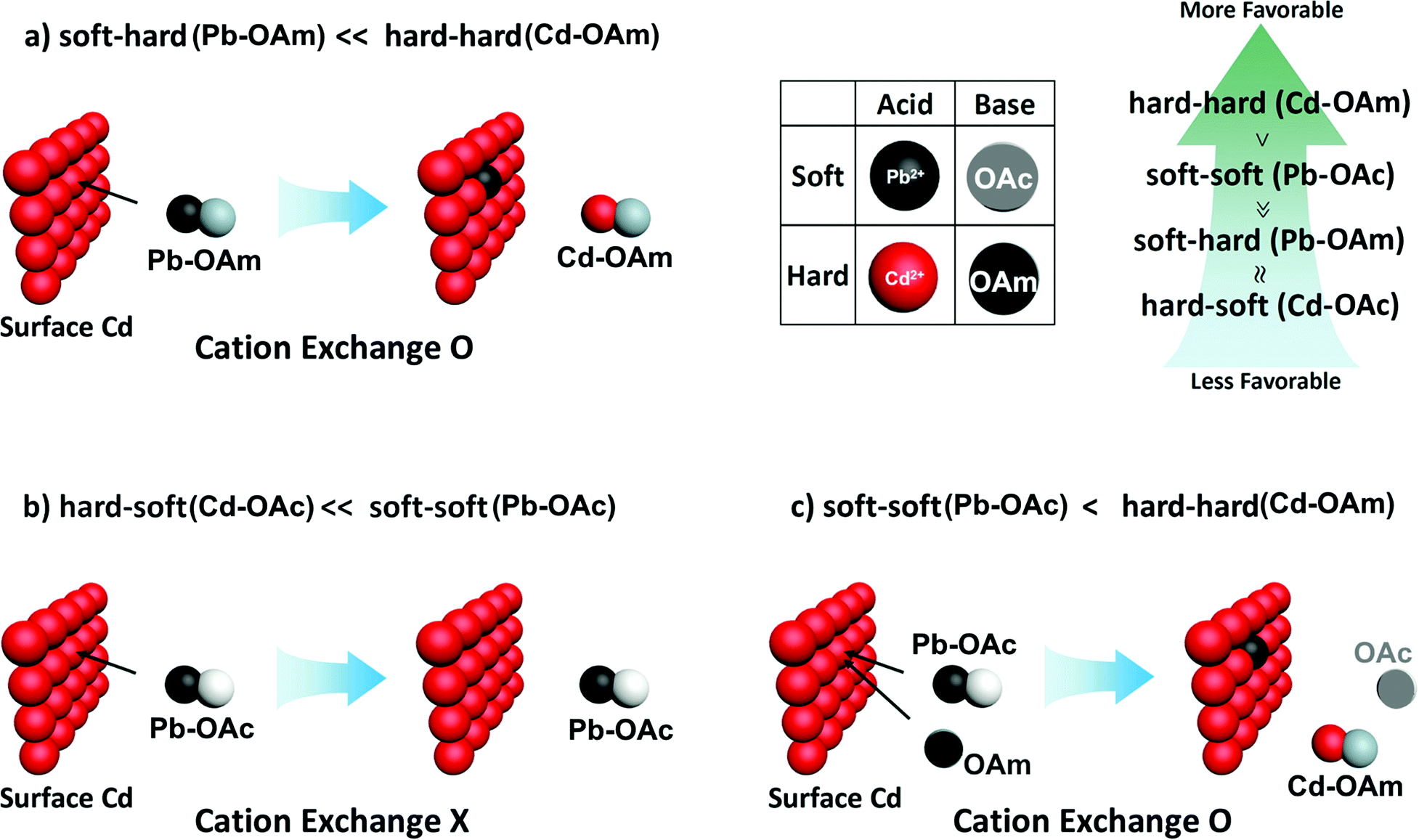 | ||
| Fig. 42 Cd-to-Pb cation exchange when (a) PbCl2–OAm, (b) Pb-oleate, and (c) Pb-oleate–OAm is used as a source of Pb. Reproduced with permission from ref. 262. Copyright 2015, the American Chemical Society. | ||
High-quality Ag2S–CdS hetero-nanostructures were produced through an easy one-pot method based on the thermal decomposition of two single source precursors in OAm/OAc. OAc was shown to be conducive to the generation of spherical Ag2S–CdS structures and OAm promoted the formation of matchstick-like Ag2S–CdS morphologies. High yields of these configurations could be obtained by careful control of the growth kinetics. It was suggested that OAc favored mostly the formation of an isotropic shape, while OAm induced, in principle, the growth of 1D NCs by preferentially binding CdS to a certain crystal facet.263 In another report, the quick decomposition of Zn-acetate by microwave irradiation in the presence of OAm/OAc led to the formation of hexagonal ZnO nanopyramids. When gold ions were present, the formed Au NCs initially served as heterogeneous nuclei for the nucleation and growth of ZnO nanopyramids, where the decomposition of zinc acetate requires higher temperatures than the reduction of Au ions. When only OAc was used, the growth of ZnO pyramids seemed to be rapid but it did not lead to the generation of well-defined nanopyramid shapes. Pure OAm was related to the slow formation of ZnO nanopyramids and most of the NPs formed did not contain an Au core. Uniform hybrid Au–ZnO nanopyramids were produced when both OAc and OAm were present within an optimum molar ratio range (50–75% OAm). Further insights into the contribution of OAm/OAc to produce high-quality gold-zinc oxide nanopyramids can be found in the corresponding ref. 264. Porous ceria nanocubes produced using the hot injection technique could form a stable monolayer over H2O and chloroauric acid subphase in a Langmuir–Blodgett trough due to the presence of surface-capped OAc and OAm. CeO2–Au nanocomposites were produced by exploiting the capacity of OAm to reduce AuCl4− ions to Au(0) via the LB method. Gold seeds were only formed site-selectively on the ceria nanocubes.265 The same group published a method to couple porous CeO2 nanocubes with Ag NPs, where OAm and OAc-capped 3D porous ceria cubes were first produced by a hot-injection method. Subsequently, 2,4-dimethylphenol was employed to modify the particle surface and acted as a reducing and capping agent to generate Ag NPs on the porous CeO2 support at room temperature. The final CeO2–Ag nanocomposite displayed a higher catalytic performance in click reactions in comparison to the unsupported Ag NPs and porous ceria nanocubes. The nanocomposites were also found to exhibit good catalytic activity in the carboxylation of terminal alkynes.266
Sun's group reported a general route to produce NiAu alloy NPs via the co-reduction of nickel acetylacetonate and HAuCl4·2H2O at 220 °C using OAm/OAc. After electro-anodization, the NiAu alloy NPs were converted to core/shell NiAu/Au NPs, which exhibited increased activity for the HER. OAm acted as the solvent, surfactant and reductant, while both OAm and OAc were stabilizers. First-principles calculations proposed that the Au sites with low coordination numbers around the shell were responsible for the improved electrocatalytic performance.267 Cannas et al. studied the molecular interactions in the incorporation mechanism of colloidal hydrophobic NPs in the hydrophilic core of reverse microemulsions. OAm/OAc were the chosen hydrophobic capping molecules, while Igepal-CO520 was the surfactant. A mechanism was hypothesized to take place based on the tail-to-tail intercalation of OAc in the Bi2S3 NP capping layer with Igepal CO520, followed by the interaction of the polar head species of Igepal with water to create an external water shell. The detachment of the OAc monomers from the NP surface was attributed to their high affinity for the Igepal micelles, rather than direct exchange with the Igepal molecules. Readers can find more insights and suggestions on the role of OAm/OAc in this process in the respective ref. 268. OAm/OAc-capped FePt NPs were modified through a ligand exchange protocol with (3-mercaptopropyl)-trimethoxysilane. FTIR spectra demonstrated that OAc was initially chemisorbed on the FePt NPs as a carboxylate, while modification with 3-MPTS resulted in the appearance of Si–OCH3 bands. The final FePt@Ti–SiO2 composite particles were utilized for the heterogeneous catalytic epoxidation reaction using H2O2.269
Ag@CuInS2 core–shell structures were prepared by initially producing Ag core NPs, which were afterwards coated with a CuInS2 shell through the thermal decomposition of a metal thiolate complex. The OAm/OAc mixture was employed for the synthesis of the composites, and their size increased up to 11 nm by increasing the concentration of OAc. The ligand mixture reacted with the metal cation to create metal-OAc complexes. These complexes were deemed to be solvated by several OAm molecules. Dodecanethiol was also used as a reagent. The authors described in detail the suggested mechanism for the final particle formation based on the ‘hard and soft (Lewis) acid and base’ (HSAB) theory, the masking effect of OAc for In(III) and the partial exchange of Cu(I) by In(III) on the initial Cu2S shell to form CuInS2.270 OAm and OAc can be used, besides ZnS, to passivate QDs and study their effects on the incident-photon-to-current efficiency (IPCE) performance of solar cells. Dai et al. reported that OAm passivation may decrease the device performance, unlike OAc passivation, which can improve the IPCE. OAc coating caused a redshift in the IPCE onset wavelength due to the delocalization of excitons in the CdS QDs. The etching effect and surface restructuring provoked by the presence of OAm were considered to be responsible for the decrease in performance in that case.271 Tu and co-workers reported the synthesis of green luminescent CdSe/ZnS NCs through a microfluidic reaction. A triple ligand system (TOPO/OAc/OAm) was employed to produce small CdSe NPs initially. Subsequently, zinc diethyl dithiocarbamate was added as a precursor to form a ZnS shell around the CdSe NCs. The role of OAm was critical for the assisted decomposition of the zinc precursor at relatively low temperatures and within shorter reaction times compared to previous reports.272
Magnetic fluorescent FePt@CdSe core–shell NPs were produced via the sequential addition of precursors, with tetraethylene glycol as the solvent and reductant. The OAm/OAc pair was also used and their ratio affected the size and composition of the FePt core. The amount of precursors was also a factor influencing the size of both the core and shell components.273 Zhang et al. produced Li3V2(PO4)3 NPs through a high-temperature (320 °C) approach in organic medium. The OAm/OAc ligand pair helped to prevent the uncontrolled growth and aggregation of the particles. FTIR measurements indicated that residual organic compounds from the OAm/OAc solvent were present on the surface of the intermediate components, suppressing particle overgrowth. The surface electronic conductivity of the as-prepared NPs was increased through a following carbon-coating treatment, producing LVP@C NCs. To achieve this, in situ polymerization of dopamine and carbonization were performed. Improved lithium storage activity was achieved for the final carbon-coated composites.274
To conclude for this sub-section, it is interesting to highlight that ‘secondary’ reducing agents such as C, CO and dihydrogen can be generated through the thermolysis of OAc and OAm at high temperatures, affecting the oxidation state of heterodimer nanostructures. The reactions of OAm or OAc with metal-containing precursors affect the evolution of the synthetic process, yielding heterostructures, depending also on the other synthetic conditions applied.
5. Upconversion and rare earth-based nanostructures
Eu-Based
Lanthanide oxysulfides are known as non-toxic materials with a sufficient degree of chemical stability. They are candidate materials for applications in optoelectronic and magnetic devices for biological cell imaging and drug delivery. Nevertheless, synthesizing Ln2O2S can be complicated, given that the hard acid Ln(3+) may interact simultaneously with a hard (O2−) and soft (S2−) base. Eu2O2S NPs were produced via the solvothermal decomposition of an Eu-complex containing acda−, which is the anion of 2-aminocyclopentene-1-dithiocarboxylic acid. Individual OAm or combined with TOP or OAc at 280 °C enabled the production of several morphologies such as thin nanoplates, nanospheres and short rods. Ultrathin 3D hexagonal nanoplates with a surface diameter of 8–12 nm and thickness of 0.3 nm were obtained when OAc and ODE were added to the reaction pot together with OAm. Also in this system, the different binding capacity between OAm and OAc was considered crucial for the acquisition of different shapes. The replacement of OAc and ODE by TOP led to the cancelation of the preferential growth in specific directions by reducing the surface energy. In this case, spherical particles were formed.275 Cai et al. used OAm/OAc as combined surfactants in a thermal decomposition route to produce Gd2O2S:Eu NPs. The product consisted of nanocubes with a mean size of 25 nm. A well-established phospholipid surface modification strategy was introduced to transfer the hydrophobic OAm/OAc-coated particles to an aqueous medium.276 Cho and Huh prepared SrMoO4:Eu3+,Tb3+ nanophosphors with a diameter of 5 nm through solvothermal reactions in a hexane-water bilayer system composed of Sr-oleate, Eu-oleate and Tb-oleate complexes in hexane and an aqueous (NH4)6Mo7O24 solution. A simple phase-transfer method was utilized to produce the above-mentioned oleate complexes in hexane from aqueous Sr(NO3)2, Eu(NO3)3 and Tb(NO3)3 solutions with the assistance of Na-oleate, OAc and OAm. The emission colour of the transparent suspensions of SrMoO4:Eu3+,Tb3+ nanophosphors could be finely tuned over the green-red spectral region.277 YVO4:Bi3+, Eu3+ NPs were produced in water using a citrate precursor, and then they were surface-modified with either OAm or octylamine to transfer them to pentane. The surface of the citrate-coated NPs was modified through electrostatic interaction by alkylammonium cations, which were formed in water. The dried NPs became hydrophobic and could be redispersed in non-polar solvents containing OAc. Interestingly, their hydrodynamic sizes are smaller than that of the hydrophilic NPs dispersed in aqueous solution before hydrophobization. The bidentate coordination of OAc on the NP surface was stronger than the monodentate adsorption of alkylammoniums on the NP surface. The downsizing was attributed to the release of the aggregated NPs.278 Du et al. published the fluorophilic-assisted synthesis of highly luminescent self-organized ultrathin EuOF NWs with a uniform diameter of around 1.8 nm. Eu(CF3COO)3 was the europium precursor, while OAm/OAc were used as the solvents for heating the mixture to 310 °C. Increasing the amount of OAc resulted in the generation of ∼4 nm EuOF spherical NCs, which was attributed to the retarded fluorination due the presence of a high population of free OAc ligands. Alternatively, when excess OAm was used, Eu2O3 NCs rather than EuOF NWs were produced because of the high fluorophilicity of the free OAm ligands through F–N binding. An OAm/OAc ratio of 1:1 was favorable for the formation of EuOF NWs. Under these conditions, the reactivity of the F− ions produced by the cleavage of the Eu(CF3COO)3−x(OA)x intermediate was assumed to be maximized. Hence, the highest reactivity of the F− ions drove the preferential 1D-growth of the thin EuOF NWS along the 〈111〉 direction via the bridging interactions of the F− anions among the crystalline seeds.279
NdFeB
NdFeBO polycrystalline NPs were produced via the coprecipitation of Nd(ac)3 and Fe(acac)3 in the presence of OAc and OAm as complexing agents and CTAB as the surfactant at 300 °C. The OAm/OAc pair helped to stabilize the metallic precursors by controlling their deposition rate. The above-mentioned reagents together with CTAB were crucial to hinder irregular growth patterns and particle aggregation. The oxide NPs were embedded in the CaO matrix, followed by the reduction of calcium toward Nd2Fe14B NPs. The complexation between Nd(ac)3 with OAm decreased the reduction potential of Nd.280 In fact, the OAm/OAc ligand pair was employed in colloidal chemical bottom-up approaches, and also in top-down methods, such as high-energy ball-milling. The ball-milling of 40 μm Nd2Fe14B powder under an inert atmosphere in the presence of heptane and OAm/OAc led to 15 nm isolated NPs after 20 h. These particles exhibited improved magnetocrystalline anisotropy compared to the starting powder.281Rare-earth fluorides
Lithium-based upconverting NPs were produced with a size of 5 nm by controlling the relative amount of OAm and OAc. The carboxylic group of OAc can be deprotonated, therefore yielding metal-oleates upon interaction with the rare earth (RE)3+ ions. This interaction is both electrostatic and through the lone electron pairs featured by the O atoms in the carboxylic group. In the case of OAm, the ligand species can interact with Re(III) only via the lone electron pair on the N atom of the primary amine group. More insights into the role of OAm and OAc on the mechanism of the formation of LiYF4:Tm3+,Yb3+ upconverting NPs were reported by Vetrone and co-workers.282 The size of the particles could be tuned in the range of 5–90 nm by adjusting the surfactant ratio. A high concentration of OAm led to the production of sub-5 nm cores, which were not thermodynamically stable. However, injecting OAc, which has stronger coordinating ability than OAm, achieved size-focusing of the primary cores into slightly larger but thermodynamically stable NPs.282 Liu and co-workers produced LiYF4:Yb0.2Er0.02 NPs through the thermal decomposition method. When the chosen solvent system was OAc/1-ODE, the samples had a smaller size and stronger emission in comparison to the case of the OAc/OAm/1-ODE medium.283To summarize for this sub-section, we choose to highlight that OAm/OAc can be also used as an efficient ligand pair in top-down synthesis methods such as ball-milling. The choice of these long-chain surfactants can be beneficial to obtain remarkable magnetic properties. Tuning the OAm/OAc ratio is crucial to obtain size-controlled upconversion particles with competitive emission properties.
6. Monometallic nanoparticles
6.1 Magnetic metal NPs
Magnetic metal nanostructures show large potential in numerous applications, including magnetic data storage, biomedicine, environmental science and catalysis. Research and detailed studies on MNPs have become one of the most active and exciting fields in materials science. An important challenge is to produce MNPs that exhibit a high degree of monodispersity, low level of aggregation and high magnetic saturation. Thus, the MNP size, crystallinity and magnetization saturation must be all tuned for these materials to be employed in the aforementioned applications.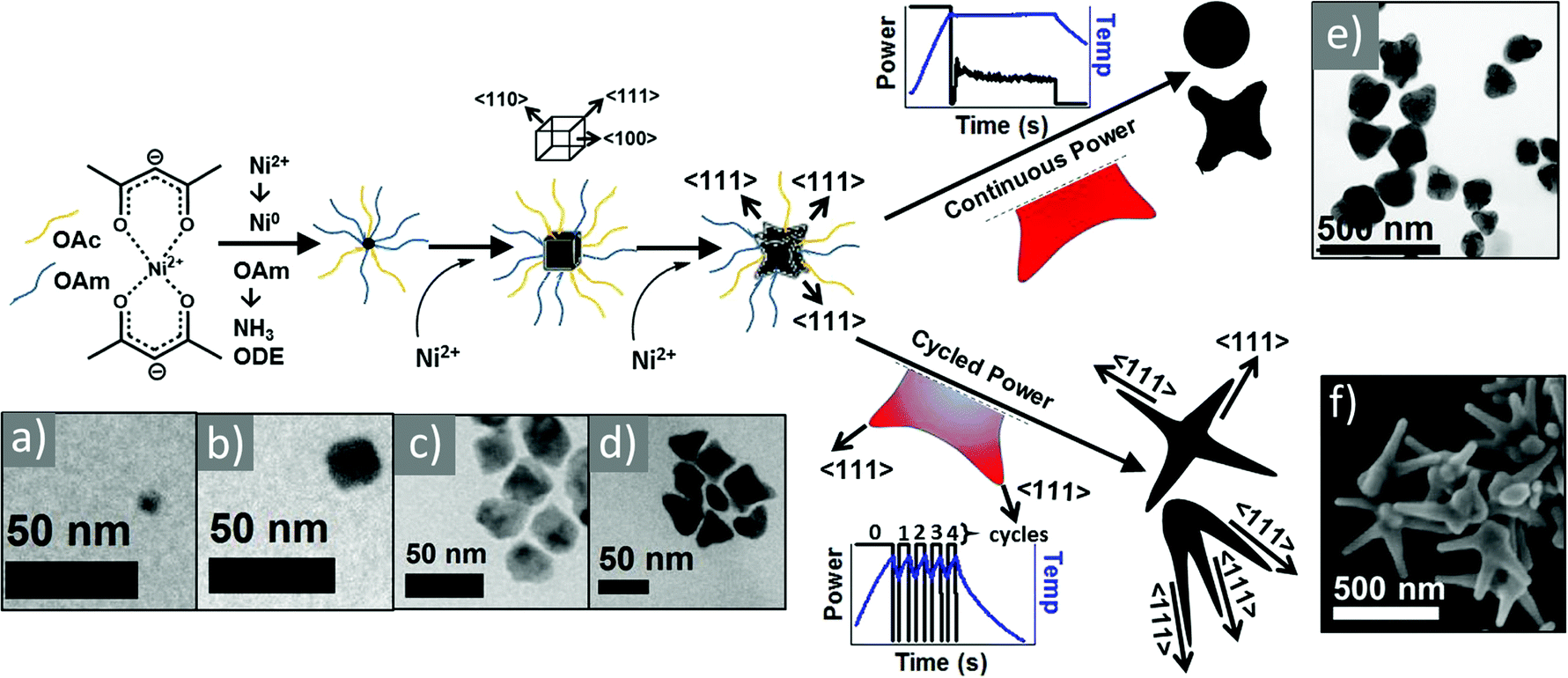 | ||
| Fig. 43 Proposed mechanism of nickel multipod growth during cycled microwave heating. Initial nucleation and growth leads to cubic shape (a–c), evolving toward (111) overgrowth (d), and kept under continuous power (e). High aspect ratio multipod structures will form under cycled conditions, as depicted in (f). Reprinted with permission from ref. 291. Copyright 2018, the American Chemical Society. | ||
6.2 Noble metal (non-plasmonic) nanostructures
Noble metal nanostructures with controlled morphologies play important roles in many fields, such as catalysis, optics and sensors. Previously reported works have shown that the special properties of noble metal nanostructures are often size- and shape-dependent. Their final shape and size are usually determined by a combination of factors such as the effect of reagents including precursors, ligands, reductants and solvents. Thermodynamic and kinetic influences on the particle morphology may also arise through a variation in the reaction temperature and time.Single-crystalline Pd nanowire arrays in porous anionic aluminium oxide template were prepared by Gao et al. using a simple high-temperature liquid-phase reduction method. In this synthetic process, OAc was used to modify the inner wall of the pores and Pd2+ complex with OAm was filled in the channel of the anodic aluminium oxide (AAO) template. Subsequently, the complex was reduced to provide OAm-capped Pd nanowires. This article suggested that OAc could improve the environment of the inner wall of the pores due to the negative charge of its hydrophilic group (COO−), which can be combined with the positively charged wall by electrostatic attraction, leading to the formation of uniform Pd nanowires.296 Dish-shaped palladium nanostructures were obtained from Pd(acac)2 in the presence of OAm and OAc. This process was correlated with the kinetic-controlled evolution of two-dimensional (2D) Pd nanosheets, which served as intermediate growth species. The authors described that the morphology of the Pd-(111)-plane-based nanodish is closely related with the growth of secondary crystals along the rims of the Pd-(111) planes.297 Pod-like Pd nanostructures were synthesized by Tilley and co-workers at room temperature in a reaction vessel with a controlled reaction pressure. OAm/OAc were present in a 1:1 ratio. The carboxylic-acid functionalities of OAc were shown to induce structural changes. In this system, kinetically controlled growth was achieved with the use of OAc, while the single use of OAm exclusively resulted in thermodynamic growth conditions.298 To grow branched Pd nanostructures, Tilley's group employed the addition of Pd seeds to a solution containing bis(acetonitrile) palladium(II) dichloride, OAm/OAc in a stoichiometric ratio and toluene. The palladium salt was reduced under a hydrogen atmosphere in a pressurized reaction vessel at room temperature. Controlling the particle size was achieved by a simple manipulation of the seed concentration. The selective use of OAm and OAc was found to be critical for the synthesis of these structures, while Pd icosahedra enclosed by low-index {111} facets were produced when OAm/OAc was replaced with hexadecylamine.299
Pt nanocrystals with octahedral and cubic shapes were synthesized by Choi via the reduction of Pt(acac)2 in the presence of OAm, OAc and benzyl alcohol with no metal additives. By controlling the volume ratio of OAm/OAc, the reduction kinetics was manipulated, and thus the shape of the nanocrystals was modified. OAm plays the role of the coordination ligand, namely, coordination of its amine group with Pt ions occurs, resulting in the formation of Pt complexes, which caused the slow reduction of the Pt precursor. Therefore, the reduction rate of the Pt precursor can be modulated by controlling the volume of OAm in the reaction mixture, achieving the kinetically controlled synthesis of Pt nanocrystals. Octahedral and cubic shapes of Pt nanocrystals were obtained using OAm/OAc ratios of 1 and 0.25, respectively.300 Vulcan carbon-supported cube-shape Pt NPs with various metal loadings were synthesized in the presence of OAm/OAc, Pt(acac)2 and W(CO)6. The nanoparticle suspension was mixed with Vulcan carbon to obtain Pt/C catalysts. The TEM micrographs of the Pt nanocubes together with carbon-supported platinum particles with various loading amounts are depicted in Fig. 44.301 The catalytic activity of a range of materials is often affected by the presence of surfactants; thus, removing surfactants without altering the particle structure is a prerequisite for examining facet-dependent particle catalytic activity in a highly reliable way. A simple procedure for removing PVP and OAm/OAc from shape-controlled Pt nanocubes using electrochemical potential cycling in 0.5 M NaOH was described. The Pt nanocubes were synthesized using OAm/OAc as surfactants in a hexane/ethanol mixture through one-pot polyol synthesis.302
 | ||
| Fig. 44 TEM micrographs of (a) unsupported Pt nanocubes and carbon-supported cubic Pt nanoparticles: (b) 10, (c) 20, (d) 30, (e) 40 and (f) 50 wt% Pt/C catalysts. Reprinted with permission from ref. 301. Copyright 2017, Elsevier. | ||
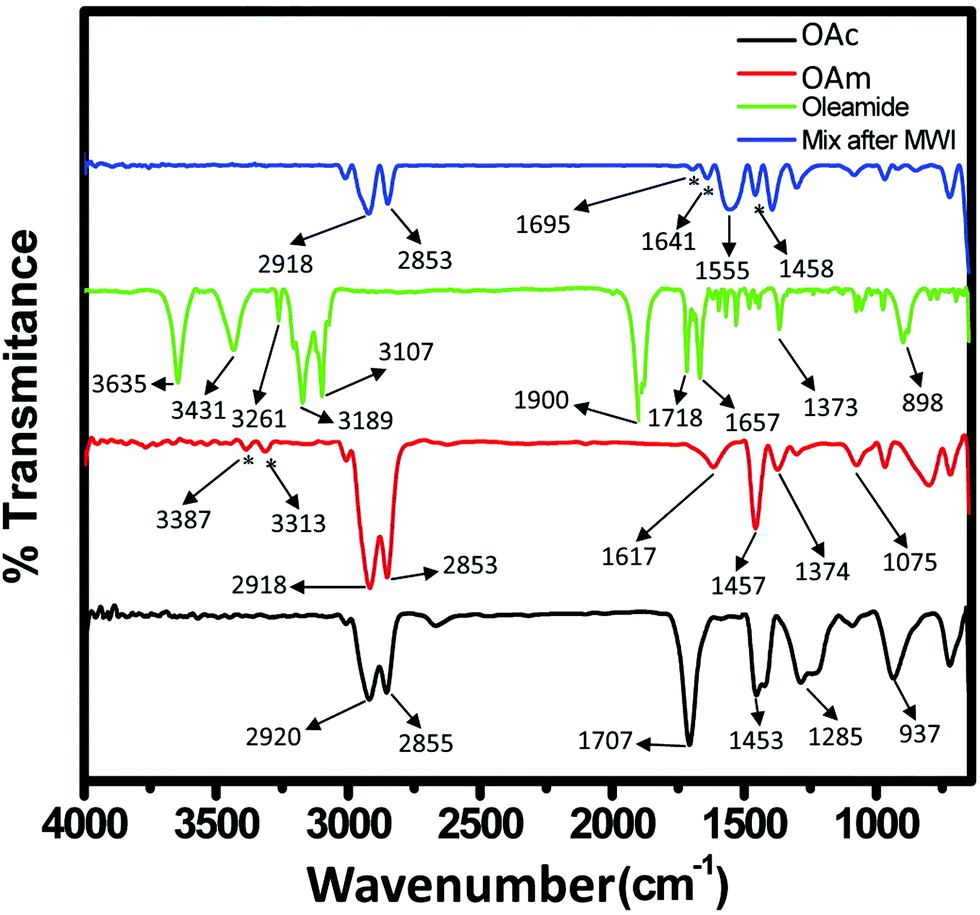 | ||
| Fig. 45 FTIR spectra of OAc, OAm, oleamide, and the reaction mixture consisting of OAc, OAm and HAuCl4 after microwave irradiation. Reprinted with permission from ref. 303. Copyright 2010, the American Chemical Society. | ||
The reduction of (methylcyclopentadienyl)(1,5-cyclooctadiene)Ir with hexadecanediol in the presence of four different capping ligand combinations between OAc/OAm, TOP, tetraoctylammonium bromide (TOAB) and tetraoctylphosphonium bromide (TOPB) led to the isolation of Ir nanocrystals. The OAc/OAm-coated nanocrystals were of the highest quality, with the narrowest size and shape distribution among the prepared Ir nanocrystals. These OAm/OAc-capped fcc Ir NPs were tested for their ability to catalyze the hydrogenation of 1-decene, and in this case, no catalytic activity was observed.304
For the sub-section of noble metal-non plasmonic nanostructures, we choose to highlight that the removal of OAm/OAc from shape-controlled NPs is crucial to reliably test their face-selective catalytic activity.
6.3 Plasmonic nanoparticles
O group shifting to 1695 cm−1, as well as the disappearance of the NH2 stretching at 3380 and 3313 cm−1 indicate the formation of dioleamide.303 The impact of mixing OAm and OAc as a liquid matrix for sputtering of Au NPs on the particle size, uniformity and their colloidal and compositional stability was investigated by Yonezawa and co-workers. Equal volumes of OAc and OAm resulted in the smallest Au NPs with very good colloidal stability. This finding was attributed to the improved viscosity and synergistic protecting capability of the mixed liquids compared with single-component liquid media.305 Sun et al. used a one-pot synthesis to develop a series of hydrophobic Au NPs ranging from short nanobars and long nanorods to ultrathin and ultralong nanowires by adjusting the ratio of chloroauric acid to OAm, OAc and triphenylsilane. The silk-like ultralong and ultrathin nanowires were found to have a single crystalline structure. Gold spherical NPs with large sizes were obtained from gold nanodots via a seed-mediated growth approach. These NPs and ultralong nanowires showed excellent surface-enhanced Raman scattering (SERS) activity in organic solvent.306 In another report, Au NWs were produced by the reduction of HAuCl4 in OAc and OAm. OAm served as both the reducing agent and stabilizer. By varying the volume ratio of OAm and OAc, the authors could prepare NWs with controlled diameters. When linked between two gold electrodes, the 9 nm-diameter Au NWs showed good electron conductivity.307 Zhang and co-workers studied the phase transfer of hydrophobic monodisperse OAm/OAc-capped Au nanocrystals to the aqueous phase using many different ligands, such as oleate and CTAB, and discovered that the original particle properties, including plasmonic, magnetic, catalytic and fluorescent properties, were well retained after transfer to water.308
 | ||
| Fig. 46 (a and b) SEM images of Cu NWs at different magnifications. (c) Optical image of a Cu NW suspension. (d) XRD patterns of the as-prepared Cu NWs with CuCl2 and CuCl2·2H2O acting as the copper source, correspondingly. Reprinted with permission from ref. 312. Copyright 2012, The Chemical Society of Japan. | ||
Ultra-long Cu NWs were also synthesized on a large scale via a hydrothermal reaction when OAm and OAc were used as dual ligands. In that work, silicon composites were combined with Cu NWs to investigate the influence of the latter material on the thermal conductivity. It was found that ultra-long copper nanowires enhanced the thermal conductivity of the Si composites, where a substantial increase was noticed with an increase in the volume fraction of the nanowire fillers. The big length of the NWs together with their large aspect ratio facilitated the formation of effective thermal conductivity networks.314 The simple and cost-effective bio-based synthesis of Cu NWs was reported by Pivot and co-workers. These researchers used an aqueous plant extract (Eucalyptus globulus) as the reducing/stabilizing agent and OAm/OAc as the surfactants. Phenolic compounds were found to play a key role in this bioreduction process, allowing almost exclusively a nanowire shape to be obtained. The use of OAc/OAm was essential to manipulate the size and aspect ratio of the NWs. These bio-based Cu NWs were resistant to oxidation when stored in ethanol and even after exposure to air.315 Furthermore, Rossi et al. explored the synthesis of Cu NPs via the thermal decomposition of copper(II) acetate in diphenyl ether with OAc/OAm in the presence or absence of 1,2-octanediol. Consequently, air-stable Cu(0) NPs were formed. The authors found that the presence of these capping ligands significantly reduced the bactericidal activity exhibited by the Cu NPs against the Gram-negative bacteria Escherichia coli.316 Cu/Cu2O NPs were produced via thermal decomposition using OAc and OAm. In this work, the OAm/OAc combination led to a single nucleation step and controlled the growth of NPs, yielding a high shape homogeneity, while the particles were also protected against oxidation. The Cu NPs were further dispersed on acid-functionalized multiwalled carbon nanotubes decorated on glassy carbon (GC) electrodes. The electrochemical activity of the produced composite material on the simultaneous determination of paracetamol and dopamine was studied.317 Courty et al. reported the synthesis of Cu NPs based on the reduction of an organometallic precursor (CuCl(PPh3)3) by TBAB in the presence of OAm (or dodecylamine (DDA)). The addition of OAc stabilized the Cu NPs for several weeks under a nitrogen atmosphere. Considering that OAm exhibits a longer hydrocarbon chain length than DDA, the authors discussed that OAm tends to remain in the solvent, which favors the NP growth and allows the formation of spherical NPs of larger sizes.318
To summarize the family of plasmonic monometallic NPs, the OAm/OAc pair can affect their size, shape, homogeneity and colloidal and compositional stability, together with their resulting optical properties. Extended superlattices may be formed through the controlled self-assembly of very monodisperse NP populations.
7. Bimetallic and trimetallic nanostructures
Co-Based NPs
Various synthetic techniques have been published in the literature for the fabrication of transition metal alloys with a controlled size and shape, including thermal decomposition, chemical reduction, chemical vapor deposition and the polyol process. A controlled morphology can be achieved by taking advantage of the flexibility of using different concentrations of metal ions in organic solvents. In this context, the chemical affinity of the capping agents with the produced NPs is of primary importance for a successful synthetic reaction. Amongst the above-mentioned methods, the chemical reduction process seems very convenient to prepare nanometer-sized alloy particles. FexCo1−x (x = 0.2, 0.4, 0.6, and 0.8) alloy NPs were synthesized via a superhydride reduction route using OAc and OAm as stabilizing agents. The FTIR study indicated the presence of an organic coating on the particle surface, which helped in stabilizing the FeCo alloy NPs in air at room temperature. A variation in Ms was observed based on varying alloy compositions, reduction in particle size, altered crystal anisotropies and spin canting at the surface of the NPs.319 Panda and co-workers synthesized bcc Fe50Co50 alloy NPs via the simultaneous chemical reduction of FeCl3 and CoCl2 precursors in diphenyl ether using superhydride as the reducing agent and OAc/OAm as capping agents. The magnetic hysteresis studies showed the ferromagnetic behavior of the NPs at room temperature, which was explained by the authors based on the fine particle magnetism, which indicates the effects of surface and size on the magnetization.320 Moreover, the formation mechanism of monodisperse Co50Ni50 and Co80Ni20 particles was studied by Sharma et al., where these particles were prepared via the simultaneous thermolytic reduction of Ni(acac)2 and Co(acac)2 in the presence of OAc, OAm and TOP as surfactants. The authors proposed that the metal complexes firstly decompose at 200 °C to form complexes with TOP. These metal-phosphine complexes decompose at 240 °C in the presence of 1,2-hexadecanediol reductant, thus signalling the onset of the nucleation stage. Afterwards, refluxing the reaction solution at the boiling point temperature of the solvent will lead to the formation of monodisperse CoNi alloy NPs. The proposed reaction mechanism and a TEM image of the Co50Ni50 nanoparticles are shown in Fig. 47. The Co50Ni50 NPs are superparamagnetic at room temperature, whereas the Co80Ni20 NPs are ferromagnetic.321 Adopting a modified polyol process, Saravanan et al. synthesized AgCo NPs in the form of spherical aggregates through the simultaneous reduction of cobalt acetate and AgNO3 in a coordinating solvent containing OAc/OAm under an Ar atmosphere. Both XRD and UV-Vis studies demonstrated the presence of individual particles of Ag and Co in the AgCo colloids. Typical ferromagnetic characteristics for the Ag/Co NPs were observed at room temperature by VSM studies.322 | ||
| Fig. 47 (a) Proposed reaction mechanism of the simultaneous reductive thermal decomposition of Ni(acac)2 and Co(acac)2 and (b) bright field TEM image of Co50Ni50 nanoparticles. Reprinted with permission from ref. 321. Copyright 2010, Elsevier. | ||
Mn-Based NPs
Nanostructured permanent magnetic materials have attracted considerable attention due to their numerous scientific and technological applications in magnetic recording devices, micro-electro-mechanical-systems (MEMS), and the so-called next generation hard/soft phase nanocomposite permanent magnets. Several techniques have been proposed to synthesize these materials, where mechanical milling is one of the most popular. This is because it is relatively simple, inexpensive and efficient in producing nanocrystalline powders in bulk quantities. Over the last few years, the use of surfactants during high-energy ball milling (SA-HEBM) has been increasingly reported. For instance, SA-HEBM was utilized to process the Mn54Al46 alloy. In that work, OAm/OAc was added together with n-heptane solvent during milling. Two different products were isolated. i.e., a sediment powder consisting of sub-micron sized Mn–Al flakes and a colloidal suspension in the milling medium containing Mn–Al NPs. The colloid was composed of Mn–Al nanocrystalline particles with a lamellar morphology. Magnetic measurements of these colloidal NPs demonstrated almost non-magnetic behavior at 300 K.323 Low-temperature phase (LTP) Mn–Bi nanosheets/nanoflakes were also prepared via SA-HEBM with OAm/OAc surfactants. The authors investigated the effect of ball milling time on the coercivity of LTP Mn–Bi nanosheets. The results showed that a milling time from tens of minutes to several hours causes an increase in coercivity for the Mn–Bi powders. The strong magnetic properties of these materials indicated their great potential for applications as high performance nanocomposite permanent magnets without the addition of any rare earth elements.324Pd-Based NPs
Among the Pd-magnetic metal NPs, Pd–Ni NPs have been investigated by many research groups due to their prominent electrocatalytic activity. The development of these bimetallic (or trimetallic) NPs has been achieved through various synthetic approaches, including electrodeposition and other physical and chemical methods. A one-pot synthesis of monodisperse Pd–Ni alloy nanoparticles was implemented through the reduction of Pd(acac)2 and Ni(acac)2 by TBAB in the presence of OAc and OAm stabilisers. The authors mentioned that the key to the formation of homogeneous NPs was the sequential nucleation-interdiffusion mechanism and the simultaneous reduction of both metal precursors during the one-pot synthetic scheme.325 Zhu and co-workers used a different solution-based one-pot method to synthesize a series of Pd–Ni bimetallic NPs, namely, through the reduction of PdCl2 and Ni(acac)2 in the presence of OAm, OAc and benzyl alcohol. The as-prepared Pd–Ni bimetallic NPs had core–shell structures with a Pd-rich core and Ni-rich shell. A TEM image of these NPs and a schematic illustration of their formation are depicted in Fig. 48. These particles were highly active in air and could be used as catalysts for the Miyaura–Suzuki coupling reaction.326 Cruz-Martínez et al. reported the synthesis and characterization of trimetallic nanocatalyst NiPdPt (60:20:20 wt%) NPs supported on Vulcan carbon. The metal precursors Pd(acac)2, Ni(acac)2 and Pt(acac)2 were dissolved in OAm/OAc to provide NiPdPt NPs. The electrochemical evaluation of the synthesized NPs revealed that the NiPdPt/C nanocatalyst has an enhanced mass activity and specific activity in the oxygen reduction reaction (ORR) in acidic medium compared to commercial Pt/C.327 Furthermore, thin films of nanoporous palladium (np-Pd) were produced from binary Pd–Ni precursors with the following compositions: 6, 20, 22, 24 and 30 at% Pd. These Pd–Ni precursors were subjected to dealloying in sulfuric acid to study the effect of their initial composition on the morphology of dealloyed films. To achieve finer porosity, OAm and OAc were added as surfactants during the dealloying process in a 1:1 ratio.328
 | ||
| Fig. 48 (a) TEM image of as-prepared Pd–Ni bimetallic NPs and (b) schematic illustration of the formation of Pd–Ni bimetallic NPs. Reprinted with permission from ref. 326. Copyright 2014, Springer Nature. | ||
Monodisperse AgPd alloy NPs were obtained using a high-temperature organic-phase surfactant-assisted protocol, involving the co-reduction of CH3COOAg and Pd(acac)2. OAm/OAc and 1-ODE were present in the reaction pot. This strategy resulted in the formation of four different compositions of AgPd alloy NPs. These particles were assembled on mesoporous graphitic carbon nitride (mpg-C3N4), rGO and Ketjenblack using a liquid-phase self-assembly method. Among the three supports tested, the mpg-C3N4@AgPd catalysts provided the best catalytic activity for the hydrolytic dehydrogenation of ammonia borane.329 Moreover, Bao and co-workers synthesized monodisperse PdAu NPs with three different compositions to study their electrocatalytic activity towards the methanol oxidation reaction (MOR). The NPs were prepared via an emulsion-assisted synthetic strategy using ternary metal precursors with various Pd to Au precursor molar ratios in the presence of OAm/OAc. The particles showed superior electrocatalytic activities in the MOR compared with the pure Pd NPs prepared via the same method.330 Besides the aforementioned bimetallic alloy NPs, the core–shell configuration of bimetallic NPs has received increasing attention in heterogeneous catalysis owing to their enhanced properties especially compared to their monometallic counterparts. Monodisperse AgPd core–shell NPs with a controllable Pd shell thickness were synthesized via the thermal decomposition of PdBr2 over as-prepared Ag NPs in an OAm/OAc mixture. The core–shell particles were assembled on rGO and the resulting composite was employed to catalyze the transfer hydrogenation of nitroarenes to anilines using ammonia borane. The authors demonstrated that the thickness of the Pd shell had a significant effect on the catalytic activity of the rGO@AgPd catalysts.331
It has to be noted that bimetallic and trimetallic NPs can be resistant to oxidation, at least partially, through the help of an organic coating on their surface. Depending on their composition, these NPs can be used in applications ranging from permanent magnets to chemical catalysis and electrocatalysis (e.g., ORR and MOR).
Pt-Based alloy nanostructures
Yamashita et al. studied the catalytic activity, magnetic properties, and water-compatibility of FePt NPs synthesized via the thermal decomposition of Fe(CO)5, followed by the reduction of Pt(acac)2 in the presence of OAc and OAm. Hydrophobic FePt NPs with an Fe-rich core and Pt-rich shell were developed, and subsequently treated with g-cyclodextrin (g-CD), which rendered them water-dispersible. FePt-g-CD exhibited superparamagnetic behavior at 300 K and acted as an efficient nanocatalyst for aqueous hydrogenation reactions.339 To extend the ordered domains of self-assembled nanoscale FePt, NPs of this alloy were synthesized via the thermal decomposition of Fe(CO)5 and chemical reduction of Pt(acac)2 dispersed in a mixture of hexane and octane in the presence of OAm/OAc. The self-assembly of FePt NPs was patterned on a silicon wafer, aiming to increase the ordered domains. The Si wafer used as a template had patterned holes of a photoresist film and helped to direct the stacking direction of the FePt NPs.340 A different chemical synthesis of FePt nanocubes and their self-assembly into FePt nanocrystal superlattice arrays was published by Sun and co-workers in 2006. A controlled texture in the self-assembled superlattices and magnetic alignment were observed for these materials. The Fe50Pt50 nanocubes were synthesized by mixing OAc and Fe(CO)5 with a benzyl ether/octadecene solution containing Pt(acac)2 and OAm. The authors mentioned that the key to the success of the nanocube synthesis is the reaction temperature (205 °C) rather than 300 °C, which is the temperature normally used for the synthesis of FePt. They also demonstrated that the shape is controlled by adding OAc first during the reaction. Self-assembly of these FePt nanocubes created a (100) textured array. In addition, thermal annealing induced an internal particle phase change from fcc to fct and changed the nanocube assembly from superparamagnetic to ferromagnetic.341 In another work, size-controlled FePt3 concave nanocubes (CNCs) were prepared using a high-temperature organic medium containing OAm and OAc. The TEM images of the as-prepared FePt3 NCs indicate the selective growth of highly uniform cubic nanostructures with curved surfaces, as shown in Fig. 49a–f. The particle size and concavity could be controlled by adjusting several parameters such as the ratio of OAm and OAc, the physicochemical properties of the metal carbonyl, the metal valence in the precursor and the ratio of metal precursors. The TEM images of the Pt–Fe NCs grown under various synthetic conditions are depicted in Fig. 49g–l. The FePt3 CNCs exhibited a high performance in both the hydrogenation of styrene and reduction of 4-nitrophenol.342 To study the structural and magnetic properties of monodisperse FexPt1−x NPs, Salgueirino and co-workers synthesized Fe60Pt40 NPs stabilized by different ligands (OAc/OAm or TMAOH). The particles were self-assembled in 3D dispersions and several parameters were analysed in detail, such as the competition of the surface, finite-size effects and magnetic interparticle interactions, which altogether controlled the overall macroscopic magnetic behavior. The results demonstrated that for the FePt NPs with an identical size distribution but different surface chemistry, substantial differences in the effective magnetic anisotropy were determined.343
 | ||
| Fig. 49 (a) TEM image of as-prepared Pt3Fe CNCbs; (b) 3D perspective illustration model of a concave cube; (c) HRTEM of a single Pt3Fe CNCb; (d) fast Fourier transform pattern of the CNCb shown in (c); (e) a stepped surface highlighted from the HRTEM image; and (f) scheme of the stepped surface composed of a mixture of high-index facets with {100} terraces and {110} steps. TEM images of Pt–Fe NCs grown under various synthetic conditions: (g) volume ratio of OAm/OAc 10:1); (h) volume ratio of OAm/OAc 4:1; (i) W(CO)6 was replaced with Cr(CO)6; (j) Fe(acac)3 was replaced with FeCl2·4H2O; (k) amount of Pt(acac)2 was increased; and (l) Fe precursors were absent (second column). Reproduced with permission from ref. 342. Copyright 2015, Wiley-VCH. | ||
The effect of the total dispersant concentrations of OAm and OAc in a FePt-nanoparticle/toluene coating solution on a self-assembled nanoparticle array was examined, where the self-assembly led to long-range periodicity on a flat SiO2/Si substrate. The metal precursors were Fe(CO)5 and Pt(acac)2. At a surfactant concentration greater than 0.8 vol%, FePt NPs with an average size of 4.4 nm were assembled in a two-dimensional and hexagonal-close-packed ordered array with 8.3 nm pitches on the flat surface. Self-assembled NPs were organized on the bottom surface of the patterned substrate.344 Lukehart et al. used single-source molecular precursors for the synthesis of FePt NPs exhibiting high room-temperature coercivity, namely, to a solution of toluene, OAc and OAm, a known polyheteronuclear cluster complex, Pt3Fe3(CO)15, was added. Metal ordering was accomplished by annealing surface-capped fcc FePt powder under getter gas (9:1 N2/H2).345 The environmental concerns due to the high toxicity of carbonyl precursors caused many groups to focus their efforts on isolating and studying FePt NPs in the presence of alternative metal sources. Sirisathitkul et al. produced FePt3 NPs with Fe(acac)3 or iron(III) hexafluoroacetylacetone (Fe(hfac)3) precursors, together with Pt(acac)2 and OAm/OAc. FePt3 NPs were formed by either precursor, but particles with a more uniform size and shape were obtained using Fe(hfac)3. Alternatively, the agglomeration of NPs in the case of Fe(acac)3 severely affected their long-range self-assembly.346 Monodisperse FePt NPs were synthesized via the one-pot chemical reduction of Fe(acac)3 and Pt(acac)2 using 1,2-hexadecanediol as a reducing agent or OAm/OAc. Annealing of the as-prepared particles resulted in a significantly improved coercivity, especially when the diol was added in the synthetic protocol.347 A similar reaction between Fe(acac)3 and Pt(acac)2 in a modified polyol process using two different solvents, namely, dioctyl ether and benzyl ether as well as OAm/OAc led to the isolation of FePt NPs. OAm was suggested to control the size and number of initial Pt-rich cores, whereas OAc affected the formation of the Fe shell, which eventually resulted in the formation of FePt NPs. An increase in the amount of surfactants reduced the Fe:Pt ratio and increased the size of the NPs synthesized in dioctyl ether.348 In another report, the mechanism for the formation of FePt NPs via a pyrolysis approach was investigated. In that case, an alternative iron source was used, namely, iron(III) ethoxide (Fe(OEt)3), in the presence of Pt(acac)2, OAc and OAm. This study investigated the influence of various factors such as the reaction temperature, heating rate and total amount as well as the input molar ratios of precursors and capping agents. The authors determined the following important aspects of the reaction mechanism: (i) the FePt NPs are rapidly formed at 250–297 °C by the allelocatalytic decomposition of the precursors; (ii) the formation of FePt NPs is dominated by the nucleation of Pt followed by a slow growth process of Fe (and Pt) atoms; and (iii) the Fe and Pt atoms are stabilized by OAc and OAm.349 Thanh and colleagues also used Fe(OEt)3 to synthesize multicore FePt NPs with high magnetic saturation. They performed a systematic study on the NP morphology, composition and magnetic properties by tuning the overall amounts and molar ratios of OAc to OAm, while the ratio of Fe(OEt)3 to Pt(acac)2 was kept at 2:1. The volume of solvent, (dioctyl ether or dibenzyl ether), was also stable. The results indicated that the coordination of the Fe and Pt intermediates with OAc and OAm, respectively, hinders deposition of each respective metal toward the growth of discrete and multicore NPs.350 In addition, FePt NPs were synthesized via the decomposition of another iron source, i.e. FeCl2, and the reduction of Pt(acac)2 in phenyl ether in the presence of 1,2-hexadecanediol. OAc and OAm surfactants were added to the solvent as a protective ligand pair. The FePt NPs were finally formed by adding LiBEt3H superhydride to the reaction mixture, which facilitated the release of Fe atoms from FeCl2 and their alloying with Pt.351
As mentioned above, an effective strategy to assist the aqueous dispersibility of NPs is to use ligand exchange. Spherical, ferromagnetic Fe–Pt NPs were prepared via the simultaneous polyol reduction of Fe(acac)3 and Pt(acac)2 in the presence of 1,2-hexadecanediol and OAm/OAc, with phenyl ether as the solvent. The as-prepared FePt particles were coated by OAc, which allowed dispersion of the particles in hydrocarbon solvents to cast thin films and perform solution-phase chemistry. The OAc ligands were replaced with 11-mercaptoundecanoic acid, offering dispersibility in water for the particles, while their physical properties were retained.352
To summarize the FePt-based NPs, annealing at high temperatures can convert their as-prepared disordered fcc phase to an ordered fct (tetragonal) phase with improved magnetic features. This post-synthetic thermal treatment can lead to the transformation of the surface capping ligands to pure C, being in the form of a graphitic carbon layer, encapsulating the magnetic NPs. Ligand-exchange protocols can be also applied for the FePt system, allowing the aqueous dispersion of the NPs with no important loss in their magnetic and structural features.
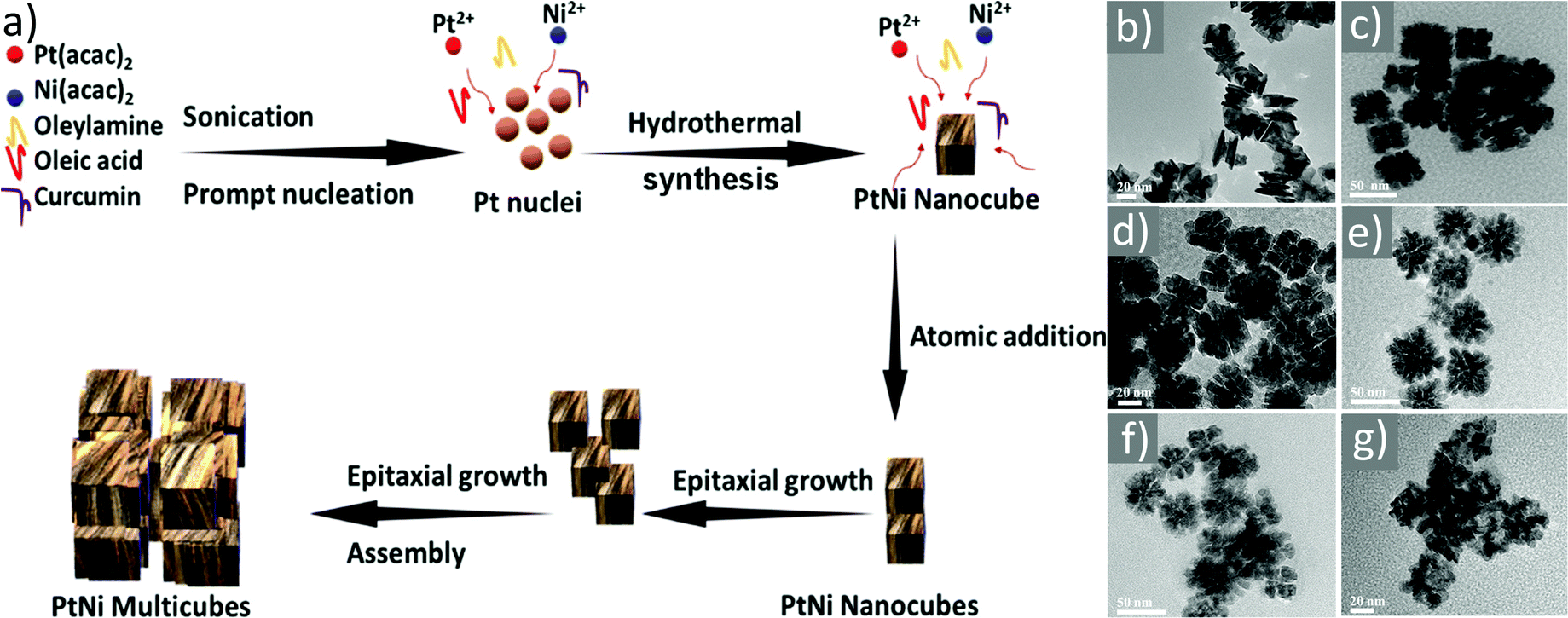 | ||
| Fig. 50 (a) Proposed synthetic mechanism of PtNi multi-cubes and TEM image of Pt–Ni prepared by fixing all the reaction conditions except (b) without oleic acid and (c) using 0.5 mL of oleic acid, (d) 0.5 mL of HCHO, (e) without HCHO, (f) 60 mg of curcumin, and (g) without curcumin. Reprinted with permission from ref. 353. Copyright 2020, the American Chemical Society. | ||
Highly composition-segregated Pt–Ni nanocrystals with a controllable shape and high yield were prepared via the selective use of dodecyltrimethylammonium chloride (DTAC) and by controlling the OAm/OAc ratio; in particular, well-defined tetrahexahedral Pt–Ni nanocrystals (THH Pt–Ni NCs) and rhombic dodecahedral Pt–Ni nanocrystals (RDH Pt–Ni NCs) were produced in the presence of Pt(acac)2 and Ni(acac)2 precursors.354 The formation of NixPt1–x NPs via the reduction of Pt(acac)2 and Ni(acetate)2 in the presence of OAc/OAm and 1,2-hexadecanediol was also studied, where the authors could tune the composition of the NixPt1–x nanocrystals by varying the Ni:Pt initial ratio; in that work, the particle size depended on the concentration of OAc. OAm mainly affected the final particle shape. The results led to the assumption of a single nucleation event taking place, followed by a growth process in the absence of Ostwald ripening. This type of growth can be extended by the continuous injection of the precursors, which allowed the possibility of particle size control.355
NixPt1−x NPs were also synthesized via the reduction of Ni(acetate)2 and Pt(acac)2 in the presence of OAc, OAm, and 1,2-hexadecanediol. Several other stabilizing agents such as TOPO were tested to prepare stable colloids. In another set of experiments, the concentration effect of the stabilizing agents was investigated. The authors found that an increase in the concentration of OAc led to a decrease in particle size when keeping the amount of OAm constant. However, increasing the concentration of OAm resulted in the formation of polycrystalline and irregular-shaped particles. Thus, the colloidal stability was mainly determined by OAc, whereas the primary influence of OAc had to do with the shape of the particles.356 Pt–Ni nanourchins with an average size of 50 nm were successfully prepared in the presence of Pt(acac)2 and Ni(acac)2. Benzyl ether together with the OAm/OAc pair constituted the reaction medium. W(CO)6 was also added in some of the synthetic reactions as it was proposed that W(0) formed during its decomposition can reduce Pt(II) and accelerate the formation of Pt nuclei. Interestingly, the addition sequence of the above-mentioned reagents was found to affect the morphology of the resulting Pt–Ni NPs. When OAm was added last, Pt–Ni nanourchins were formed, while a mixture of Pt–Ni octahedra and branch-like nanostructures were obtained when OAc was added at the end. If OAc was the last added solvent, well-controlled Pt–Ni octahedra with a very high yield were produced when W(CO)6 was also introduced in the reaction pot.357 Uniform Pt–Ni octahedra with the use of OAm/OAc surfactants and W(CO)6 as a source of CO were produced by Xia and co-workers, where they aimed to promote the formation of {111} facets in the presence of Ni. Fig. 51a shows a TEM image of the as-obtained Pt–Ni octahedra with an average edge length of 9 nm. The high-resolution TEM and scanning TEM-high angle annular dark field (STEM-HAADF) images of a single Pt–Ni octahedron are presented in Fig. 51b and c, respectively. The atomic composition profile at different sites of a Pt–Ni octahedron was determined by EDS spot analysis (Fig. 51d). Through the introduction of benzyl ether as the solvent, the coverage of both surfactants on the surface of the resultant Pt–Ni octahedra significantly decreased, while the octahedral shape was retained. By further removing the surfactants through acetic acid treatment, the authors observed specific activity in the ORR higher than that of the state-of-the-art Pt/C catalyst.358
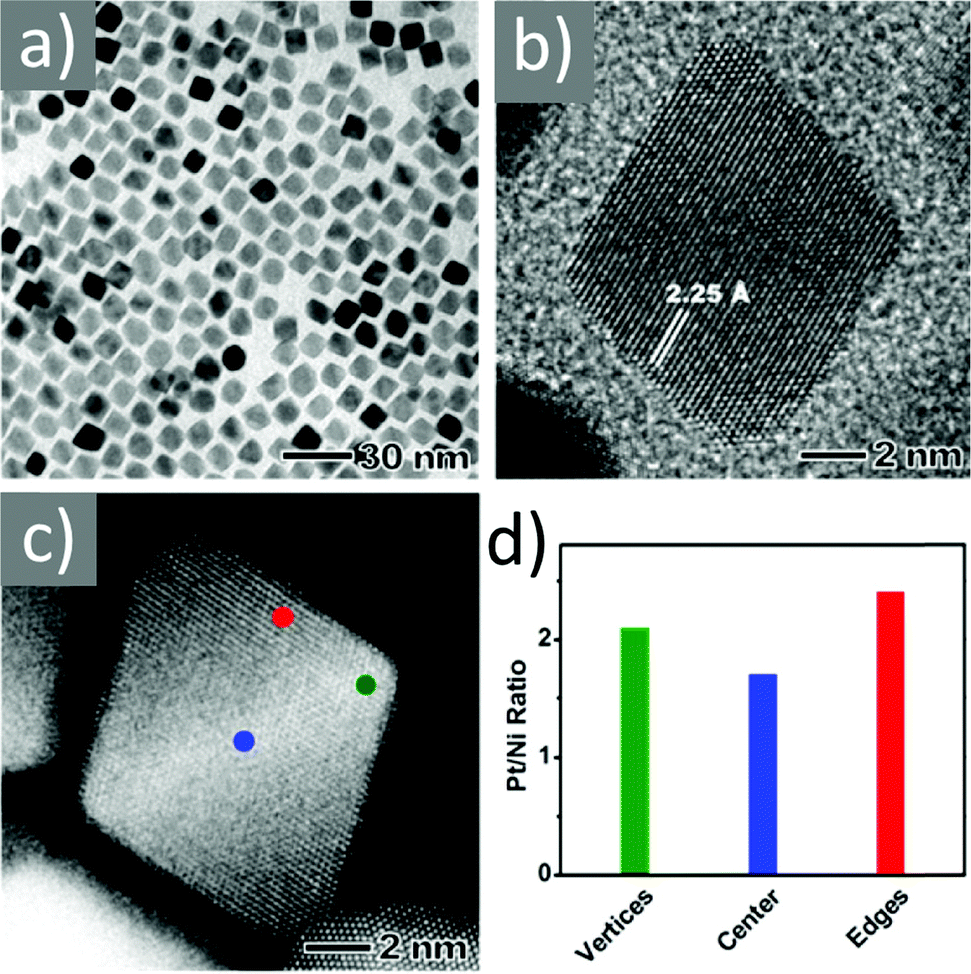 | ||
| Fig. 51 (a) TEM and (b) HRTEM images of as-obtained 9 nm Pt–Ni octahedra, (c) STEM-HAADF image and (d) EDS spot analysis of an as-obtained Pt–Ni octahedron. Reprinted with permission from ref. 358. Copyright 2013, the American Chemical Society. | ||
Several synthetic reactions were explored by Reyes-Rodríguez et al. to study the effect of the hot-injection temperature on the size distribution of Ni–Pt NPs. Here, Ni–Pt polyhedral NPs were synthesized through a thermochemical route using the OAm/OAc pair as solvents, reductants and stabilizing agents simultaneously. It was found that the injection of precursors in OAm/OAc at 180 °C resulted in the formation of paramagnetic NPs, and these particles possessed homogeneous polyhedral structures and showed greater Pt accumulation on their edges and corners. Ni–Pt polyhedral NPs with larger sizes and high polydispersity were obtained when the injection temperature was increased closer to the reduction temperature (around 270 °C).359 Pt–Ni octahedra were also synthesized by Xia and co-workers with controllable sizes and compositions in continuous-flow droplet reactors given that these reactors enable a linear increase in the production throughput. For the production of Pt–Ni octahedra, Pt(acac)2, Ni(acac)2 and W(CO)6 were dissolved in a mixture of OAm/OAc and benzyl ether, followed by pumping into a polytetrafluoroethylene tube. The size and composition of the resulting particles were tuned by modifying the composition of the solvent and/or adjusting the amount of Ni(acac)2.360
Fcc NiPt NPs were synthesized via the reduction of Ni(acac)2 and Pt(acac)2 in the presence of OAc/OAm. These precursors were chosen by the authors because they were supposed to have a similar reaction rate (presumably because they are both metal-acetylacetonate salts). In addition to being less toxic than carbonyl precursors, they are also less volatile and do not produce much vapour during the reaction. Their non-volatile nature is also beneficial to achieve good reproducibility of the reaction. By employing the colloidal coating deposition method, the authors could further encapsulate Ni–Pt NPs within silica shells (NiPt@SiO2). By controlling the molar ratio between the NiPt particles and the silica precursor, shells with a tunable thickness (8 to 16 nm) were formed.361 Da Silva and co-workers studied the efficiency of colloidal chemistry for the synthesis of alloyed Ni–Pt NPs, without phase segregation or with a core–shell structure, and these particles had a tailored elemental composition from pure Ni to pure Pt. Consequently, fcc NixPt1−x NPs (Ni3Pt, NiPt and NiPt3) were synthesized via the co-reduction of Ni(acac)2 and Pt(acac)2 in the presence of 1,2-hexadecanediol, benzyl ether and OAm/OAc. The principle of this procedure is based on the correlation between the oxidation-reduction potential of the metal cations present in the precursor molecules and the required synthesis temperature to nucleate particles without phase segregation.362
In the case of NiPt-based systems, we choose to highlight in summary that the addition sequence of OAm and OAc to the reaction pot can influence the morphology of the resulting particles.
Among the 3d-transition metal-Pt alloys, nanometer-sized Pt–Cu is one of the most interesting materials in this category due to its high stability against catalyst poisoning. In addition, the abundance and relatively low cost of copper make this material appealing for wide research and use. Several routes to obtain Pt–Cu nanocrystals with various morphologies have been reported. These shapes include nanocubes, nanospheres and nanorods, among others. Fang and co-workers synthesized Pt–Cu nano-octahedra through the simultaneous reduction of Pt(acac)2 and copper(I)/(II) chloride in the presence of W(CO)6 and OAm/OAc at high temperature. Comparative studies of the electrocatalytic activity of Pt–Cu NCs, {111}-terminated Pt–Cu nano-octahedra and {100}-bounded Pt–Cu nanocubes were carried out. Their performance in formic acid oxidation was used as an example to demonstrate the importance of controlling the shape of these nanocatalysts.367 The same group published the synthesis of Pt3Cu2 nano-octahedra using a similar synthetic approach. Using both the TEM tomography and GISAXS techniques, the authors identified that these nano-octahedra can assemble into an open structure and be arranged tip-to-tip to form a bcc superlattice with a low packing efficiency.368 Liu et al. used a colloidal chemical pathway to synthesize size- and shape-controlled CuPt NPs and NRs with tunable lengths and aspect ratios. To achieve this, Pt(acac)2, Cu(acac)2, 1,2-hexadecanediol and OAm/OAc were mixed with 1-octadecene. OAm was required to form CuPt nanorods, presumably to stabilize the (100) faces during growth. By varying the OAm/OAc ratio and the reduction rate, solvent and temperature, the morphology and nanorod length were tuned. The TEM images of the CuPt nanorods are shown in Fig. 52. The NPs were found to be highly active for CO oxidation compared with a commercial supported Pt nanoparticle catalyst of comparable particle size.369
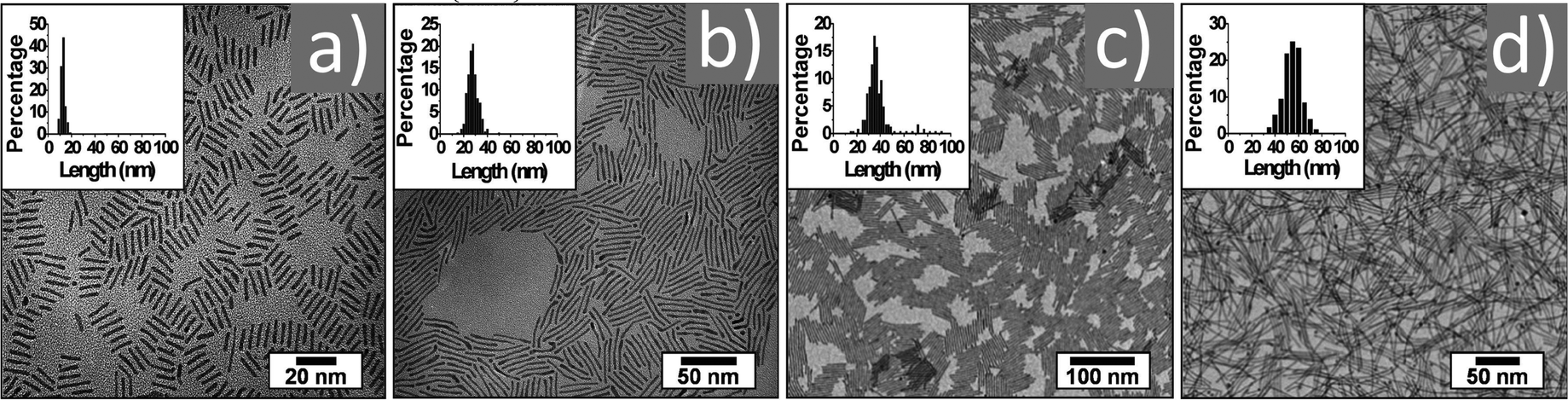 | ||
| Fig. 52 (a–d) TEM images of CuPt nanorods with various average lengths (histograms of lengths are shown in the corresponding insets). Reprinted with permission from ref. 369. Copyright 2009, the American Chemical Society. | ||
Pt–Cu concave cuboctahedral nanoalloys enclosed by high-index facets were prepared with the assistance of 1,2-dodecanediol and OAm/OAc through a one-pot co-reduction method in the presence of different metal precursors than that mentioned above, namely, H2PtCl6 and Cu(acac)2. The synergistic capping effect of OAc/OAm stabilized the {100} and {111} facets, which eventually led to the formation of concave cuboctahedral nanostructures. The morphology and microstructure of the as-prepared Pt–Cu concave cuboctahedrons and a schematic representation of their formation mechanism are shown in Fig. 53. The electrocatalytic performance of the product in the MOR was significantly improved in comparison to that exhibited by spherical Pt–Cu NPs and pure Pt nanocatalysts.370
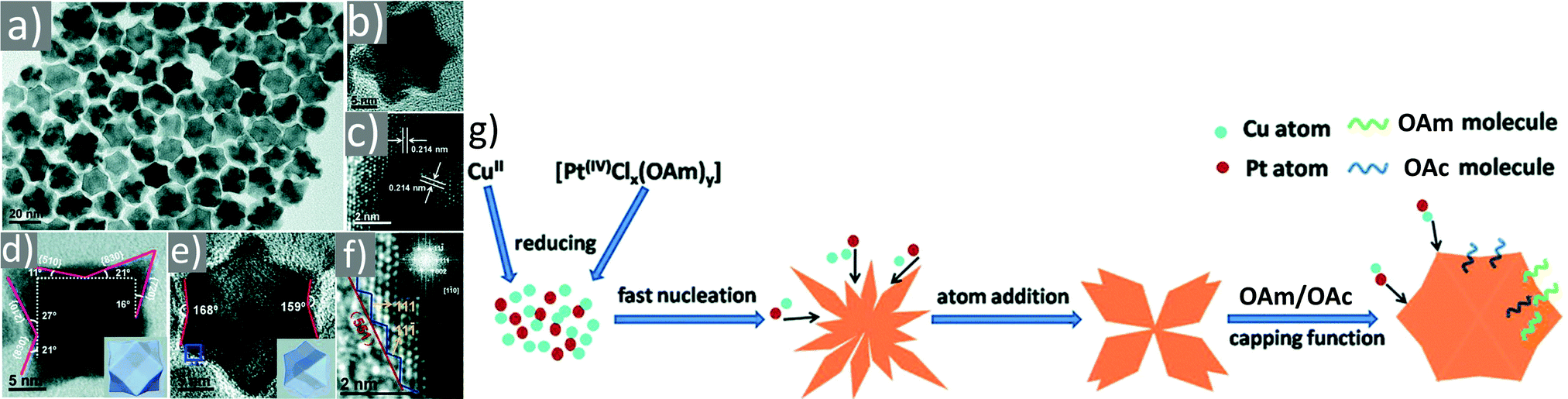 | ||
| Fig. 53 (a–f) Morphology and microstructure of as-prepared Pt–Cu concave cuboctahedrons and (g) scheme suggesting the formation mechanism of a Pt–Cu concave cuboctahedron.370 Copyright 2016, The Royal Society of Chemistry. | ||
Monodisperse Pt–Ru NPs were synthesized via the co-reduction of Pt(acac)2 and Ru(acac)3 in a reaction mixture composed of 1,2-hexadecanediol, OAm/OAc and octyl ether. The authors found that the carbon-supported Pt–Ru colloidal nanoalloy could be activated by a simple acid treatment under mild conditions and the resulting catalyst displayed remarkably ameliorated catalytic activity in the MOR compared to that of a commercially available Pt–Ru catalyst.371 Although surface capping agents can greatly influence the growth mode of nanocrystals in solution, the generation of a certain morphology can hardly be understood without a good comprehension of the main concepts and principles that define shape formation. The synthesis of Pt–Ag alloy nanowires in the presence of OAm/OAc was reported by Yang and co-workers. The TEM study revealed the formation of sphere-like NPs using a Pt(acac)2/Ag-stearate ratio of 1:4 (Fig. 54a) and 1:2 (Fig. 54b), whereas worm-like particles were obtained at a stoichiometric precursor ratio (Fig. 54c). The length of the NWs decreased for higher Pt/Ag precursor ratios (2:1 Pt to Ag, Fig. 54e), while NWs were hardly spotted when the molar ratio was modified to 4:1 (Fig. 54f). The oriented attachment growth mode was suggested to be responsible for the coalescence of the metal NPs, which were formed in solution to yield larger Pt–Ag NWs. Both Pt- and Ag-rich alloy nanostructures formed sphere-like or faceted NPs under the same reaction conditions.372
 | ||
| Fig. 54 TEM images of Pt–Ag nanostructures made at different metal precursor molar ratios. Details are mentioned in the text. Reprinted with permission from ref. 372. Copyright 2010, the American Chemical Society. | ||
The effect of temperature on the formation of Pt–Pd NP superlattices assisted by OAm/OAc was examined through TEM monitoring and fast Fourier transform (FFT) analysis. The spherical Pt–Pd NPs superlattices were prepared at room temperature via drying-mediated self-assembly, and they were capped by the OAm/OAc ligand pair. Their restructuring was investigated at elevated temperatures. The critical temperature that induced a phase transition in the Pt–Pd superlattices from a crystalline state to an amorphous state was in the range of 100–110 °C. The temperature-induced restructuring study showed that bigger 3D Pt–Pd superlattices could be produced through the sintering of smaller superlattices.373
Alloying a non-noble metal with Pt is becoming a popular approach to obtain Pt-based catalysts with much reduced Pt usage and enhanced Pt activity. Monodisperse Pt3Sn alloy NPs were synthesized via the controlled co-reduction of Pt(acac)2 and Sn(acac)2 in a mixture of 1-ODE and OAm/OAc. In that work, OAm also functioned as a reductant. The key factor for the successful synthesis of Pt3Sn NPs was the co-injection of Pt(acac)2 and Sn(acac)2 at high temperatures, which also enabled the tuning of the size of the resulting particles. These Pt3Sn NPs were highly active for CO and methanol oxidation, while their activity and stability could be further improved by a post-synthetic heating treatment.374 Nikles and co-workers also reported the synthesis of cubic Pt3Sn alloy NPs via the co-reduction of Pt(acac)2 and Sn(acac)2 in a high-boiling solvent, trioctylamine, in the presence of OAm/OAc. The as-prepared particles were catalytically inactive. To activate the NPs for catalysis, the initial ligands were removed by treatment with TMAOH solution, which was also confirmed by FTIR measurements. The activated Pt3Sn particles exhibited high activity in the MOR and CO oxidation.375 Villullas and colleagues applied a modified polyol method using 1,2-hexadecanediol, metal acetylacetonates and OAm/OAc to produce Pt–Fe/C catalysts with nominal Pt:Fe compositions of 70:30 and 50:50. Surface segregation, i.e., substitution of Fe with Pt atoms in the first few atomic layers resulted in Pt-enriched surfaces. Thus, portions of the as-prepared Pt–Fe/C materials were heat-treated under an H2 atmosphere to induce Pt segregation. The resulting nanocatalysts demonstrated better catalytic activity in the ORR than that of Pt/C.376 In another work, aiming to synthesize Mn–Pt nanocubes, Pt(acac)2 was dissolved in benzyl (or phenyl) ether in the presence of OAc/OAm, followed by the injection of a certain amount of an Mn2(CO)10 solution at 160 °C. The OAm/OAc combination was essential to obtain particles with a uniform size and shape. The Mn–Pt NCs showed higher ORR activity than commercial catalysts.377
In the present sub-section, the shape formation, oriented attachment growth mechanism and superlattice generation were discussed. Also in this case, OAm/OAc often needed to be removed from the particle surface by treatment with different chemical reagents to render the produced NPs catalytically active.
8. Halide perovskite nanocrystals
The successful use of the OAm/OAc ligand-pair has also been extended to the synthesis of colloidal halide perovskite NCs with excellent shape and size control.378–380 Over the past 7 years, halide perovskites, in the form of both thin films and NCs, have emerged as a new class of semiconductor materials with many interesting optical and electronic properties.378,381–383 Metal halide perovskites (MHPs) exhibit the general chemical formula of ABX3, where A and B are monovalent and divalent cations, respectively, while X is a monovalent halide anion (Cl, Br, and I). As shown in the typical cubic crystal structure of bulk MHPs, the B-cation has six nearest-neighbor halide (X) anions, whereas the A-cation sits in a cavity formed by eight corner-sharing BX6 octahedra (Fig. 55a).378 These are classified into two types, as follows: (1) organic–inorganic hybrid perovskites if the A-site cation is organic, e.g., methylammonium (MA+) and formamidinium (FA+) and (2) inorganic perovskites if the A-site cation is inorganic, e.g., Cs+. The optical band gap, and thus the emission color of perovskites (thin films and nanocrystals) can be easily tuned across the visible spectrum of light by varying the halide composition (Fig. 55b and c).378,384 The halide composition is easily controllable either by tuning the precursor ratio or through post-synthetic halide ion exchange.380,384,385 Although perovskites were known since the 1800s, they came into the limelight in 2009 after the demonstration of their use as photosensitizers in solar cells by Miyasaka and co-workers.386 Since then, there has been great progress in perovskite photovoltaics with their power conversion efficiency surpassing 25%.387 This is due to the combination of many interesting properties such as large charge carrier diffusion lengths, defect-tolerant electronic structure (Br and I-based perovskites), and high mobilities of charge carriers (compared to organic semiconductors).378,383,388 Alternatively, highly luminescent colloidal perovskites were first realized in the case of hybrid perovskites by Schmidt et al. in 2014.389,390 However, the morphology of the prepared colloids in their work is still under debate.378 The first colloidal synthesis of well-defined inorganic (CsPbX3) perovskite NCs with excellent control over their particle size and size monodispersity using the OAm/OAc ligand pair was reported by Protesescu et al. in 2015.380 Their synthesis is based on the typical hot-injection method, which was previously applied to the classical semiconductor colloidal NCs and other NC systems.378 Later, the use of the OAm/OAc ligand pair was also extended to the room temperature synthesis of CsPbX3 NCs by Li et al. in 2016.391 The CsPbX3 NCs, especially the Br and I-based NCs, exhibited near-unity photoluminescent quantum yields and superior thermodynamic stability compared to hybrid perovskite NCs.378,380,388 Following these early reports, research on colloidal perovskite NCs has virtually exploded.378 A wide range of synthetic methods has been reported to prepare monodisperse halide perovskite NCs, particularly for the CsPbX3 system.378 For example, CsPbX3 NCs obtained through the ultrasonication approach using the OAm/OAc ligand pair generally exhibit over 90% monodispersity and high crystallinity (Fig. 55d and e).384 The OAm/OAc pair has been widely used in various synthesis methods to achieve shape-, size-, and composition-control of colloidal perovskite NCs.378 A wide range of morphologies including nanocubes, nanowires, nanorods, nanoplatelets, and hexapods has been synthesized in this way.378,379 We direct readers to previous reviews for more details on the shape- and composition-controlled synthesis of metal halide perovskite NCs (inorganic and organic–inorganic hybrid) and their properties together with potential applications.378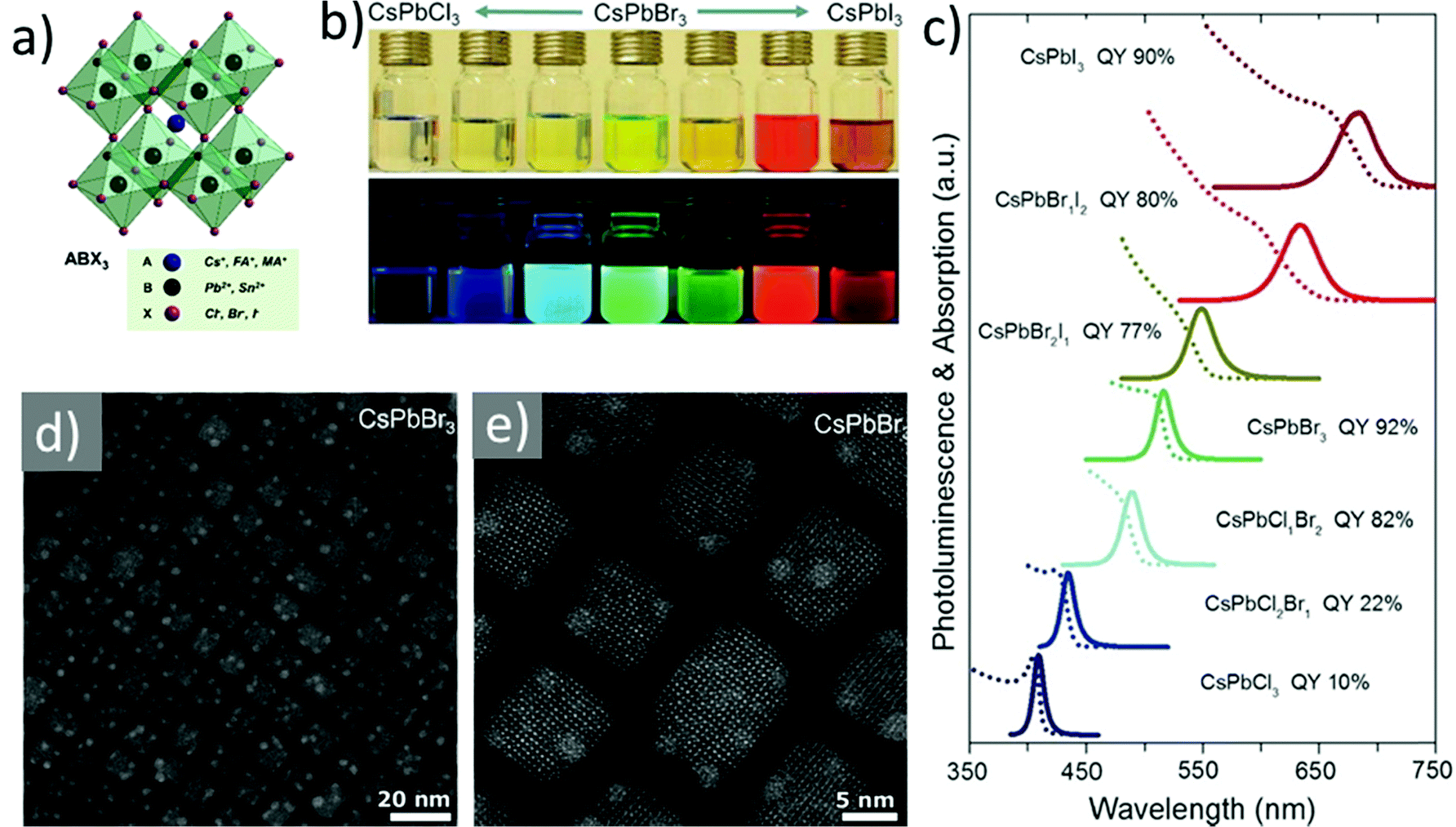 | ||
| Fig. 55 (a) Schematic illustration of cubic crystal structure of ABbX3 perovskites, where A = Cs+, methylammonium (MA+), and formamidinium (FA+); B = Pb2+ and Sn2+; X = Cl−, Br− and I−. (b) Photograph of the CsPbX3 NCs of different halide compositions under room light (top) and UV light (bottom) illumination. The samples were synthesized via a ligand-assisted ultrasonication approach using the OAm/OAc ligand pair. (c) Corresponding absorption and PL spectra of the CsPbX3 NCs of different halide compositions. The photoluminescence quantum yield of each sample is also provided in the graph. (d and e) HAADF-STEM image of CsPbBr3 NCs under two different magnifications. Panels ‘b’ and ‘c’ are adapted with permission from ref. 384. Copyright 2016, Wiley-VCH. | ||
In this section, firstly, we provide an overview of the surface chemistry of colloidal perovskite NCs synthesized using the OAm/OAc ligand pair. In this regard, we discuss how the OAm and OAc ligands can bind to the surface of perovskite NCs based on the existing literature. Secondly, we summarize the shape-controlled synthesis of perovskite NCs using this ligand pair. Finally, we discuss the outstanding questions of OAm/OAc ligand pair-protected perovskite NCs. As discussed in earlier sections, the ligands on the surface of NCs are crucial for their stabilization and play an important role in their optical and electronic properties.378,388 Especially, surface ligands control the photoluminescence efficiency of colloidal semiconductor nanocrystals.388 The detachment of ligands from a semiconductor NC surface leads to the formation of surface defects, which trap photogenerated electrons or holes before they recombine, leading to a decrease in photoluminescence quantum yield (PLQY).388 Therefore, it is fundamental to understand the surface chemistry of colloidal semiconductor nanocrystals to improve their optical properties, and thus corresponding potential applications.378,388 The surface chemistry of colloidal perovskite NCs is more complex due to the weak ionic interactions between their surface and ligands.392 Generally, long-chain alkyl amines in combination with alkyl acids have been used as ligands in the synthesis of colloidal perovskite NCs. Among the various acid–base pairs, the OAm/OAc pair has been the most extensively used ligand pair for the stabilization of colloidal perovskite NCs.378 The presence of the OAm/OAc acid–base pair in the reaction medium leads to the formation of the oleate ion and oleylammonium cation due to proton transfer from acid to amine.393 Over the years, significant efforts have been devoted to understanding the binding of ligands to perovskite NCs using nuclear magnetic resonance spectroscopy (NMR) and are still being intensively investigated.392,394–396
Based on a survey of the available literature reports, there are several different ways that the oleate ion and oleylammonium cation bind to the surface of perovskite NCs, as illustrated in Fig. 56. In this regard, using 1H solution-NMR spectroscopy, De Roo et al.392 firstly performed a detailed study on the surface chemistry of CsPbBr3 perovskite NCs synthesized with the OAm/OAc ligand pair. They identified the presence of protonated oleylamine (oleylammonium cation) by analyzing the difference in the concentrations of free oleylamine and oleic acid in the 1H NMR spectra of CsPbBr3 NCs. However, the positive cross-peaks (arising due to the lack of interaction with NC surface) in the nuclear Overhauser effect spectroscopy (NOESY) NMR spectrum of the NCs revealed that octadecene solvent and OAc were not bound to the NC surface. Based on the 1H NMR and NOESY-NMR results, it was proposed that the oleylammonium bromide molecules formed in the reaction medium act as ligand. The oleylammonium bromide ligands bind to the bromine atoms on the NC surface either through cation-anion electrostatic interactions or through the H⋯Br hydrogen bridge, while the Br− ions bind to Cs+ or Pb+ on the surface of the CsPbBr3 NCs (Fig. 56a).392 Understandably, the highly ionic nature of perovskites prefers ionic ligands in comparison with ligands that have a covalently binding nature. Because of its ionic nature, the ligand binding to perovskite NCs is highly dynamic, and therefore the ligands can be easily detached from the NC surface during their purification by washing, aging, and dilution.392,397 These processes lead to the creation of surface traps, and thus to a reduction in PL efficiency. However, by adding an excess amount of OAm/OAc ligand pair to the perovskite NC solution during its purification, the PL efficiency and colloidal stability could be retained. Interestingly, the addition of an excess amount of OAm to the NC solution also led to an improvement in PL efficiency due to the formation of oleylammonium oleate, which can tightly bind to the perovskite NC surface.392
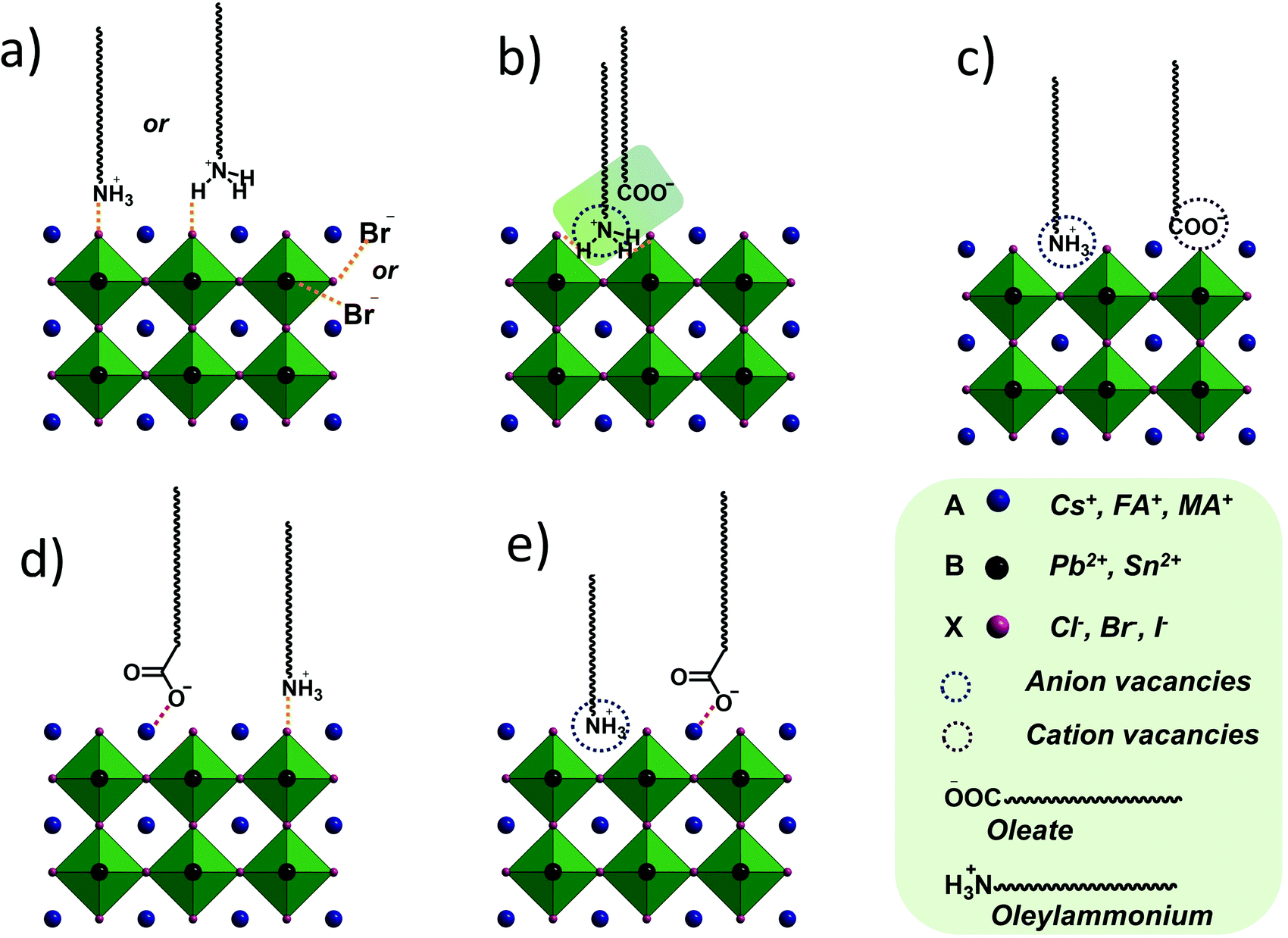 | ||
| Fig. 56 Schematic illustration showing different ways by which the OAm/OAc pair (i.e., oleate ion and oleylammonium cation) can bind to the CsPbBr3 perovskite NC surface: (a) oleylammonium cations interact with bromide atoms, and the bromide atoms interact with Cs+ or Pb+ cations. (b) Oleylammonium cations occupy the Cs+ positions on the surface and form hydrogen bonds with the neighboring Br− ions, while the oleate molecules neutralize the surface charge and do not interact with the NC surface. (c) Oleylammonium cations occupy some of the Cs+ positions, while the oleate molecules occupy the halide vacancies on the surface. (d) Oleate and oleylammonium cations form electrostatic interactions with the Cs+ and Br− ions of the NC surface, respectively. (d) Oleylammonium cations occupy Cs+ vacancies, while oleate molecules bind to the rest of the Cs+ cations. | ||
According to the above discussion, it is clear that a small amount of OAm needs to be added during the purification (washing with an antisolvent) stage of perovskite NCs to retain their high PL efficiency. A slightly different ligand binding mechanism was reported for CsPbBr3 NCs by Ravi et al.,398 where they proposed (based on XPS and 1H NMR spectroscopy) that the oleylammonium cations act as capping ligands by replacing some of the Cs+ ions and forming hydrogen bonds with surrounding Br− ions on the NC surface (Fig. 56b). Interestingly, it was also shown that the oleate molecules do not interact with the NC surface but help to maintain charge neutrality by forming an equilibrium with oleylammonium capping ligands (Fig. 56). Furthermore, the theoretical studies by Brinck and Infante399 suggest that oleylammonium cations act as capping ligands by replacing 50% of the “A” cations of the NC surface. Nevertheless, in contrast to early works, they proposed that the oleate molecules fill the halide vacancies on the NC surface, as illustrated in Fig. 56c. Another ligand-binding mechanism was reported by Brutchey and co-workers,395 where the authors proposed that both oleate and oleylammonium ions interact dynamically with the particle surface and likely bind to the Cs+ and Br− ions of the CsPbBr3 NC surface, respectively (Fig. 56d). However, in their recent study, 1H and 133Cs NMR spectra suggested that the alkylammonium ligands substitute some of the Cs+ ions on the NC surface, while the oleate molecules bind to the rest of the Cs+ ions (Fig. 56e).394 Therefore, different groups proposed slightly different ligand binding mechanisms for the CsPbBr3 perovskite NCs synthesized with the OAm/OAc ligand pair. Despite the ambiguity of ligand binding chemistry on the perovskite NC surface, tremendous progress has been made in the shape-controlled synthesis of colloidal perovskite NCs using the OAm/OAc ligand pair.378,379
According to the above discussion, it can be concluded that OAm/OAc are also efficient for the shape-controlled synthesis of perovskite nanocrystals with remarkable optical and electronic properties. The way by which entities such as oleylammonium cations, for instance, act as capping ligands has been studied via different characterization techniques and described in detail in several reports.
Shape and composition-controlled synthesis of MHP NCs using the OAm/OAc ligand pair
°C. In general, perovskite precursor-ligand complexes crystallize into nanoplatelets at lower reaction temperatures.407,408,410,411 However, it has been challenging to achieve precise control over the size of perovskite nanocubes, given that only limited success has been reported in this direction. For instance, Dong et al.412 demonstrated the preparation of size-tunable quantum-confined CsPbBr3 nanocubes based on the halide ion equilibrium in the reaction medium together with temperature control. The perovskite nanocubes with strong 3D-quantum confinement can be called quantum dots. The size of the obtained CsPbBr3 nanocubes strongly depends on the Br equilibrium between a nanocube and the reaction medium as the Br atoms diffuse inwards and outwards of the nanocubes. Thus, an increase in the Br content in the reaction medium leads to a decrease in the nanocube size. The concentration of Br was controlled by adding ZnBr2 together with the PbBr2 precursor in the reaction pot. The precise control over the size of quantum-confined nanocubes enabled a better understanding of their size-dependent optical properties.
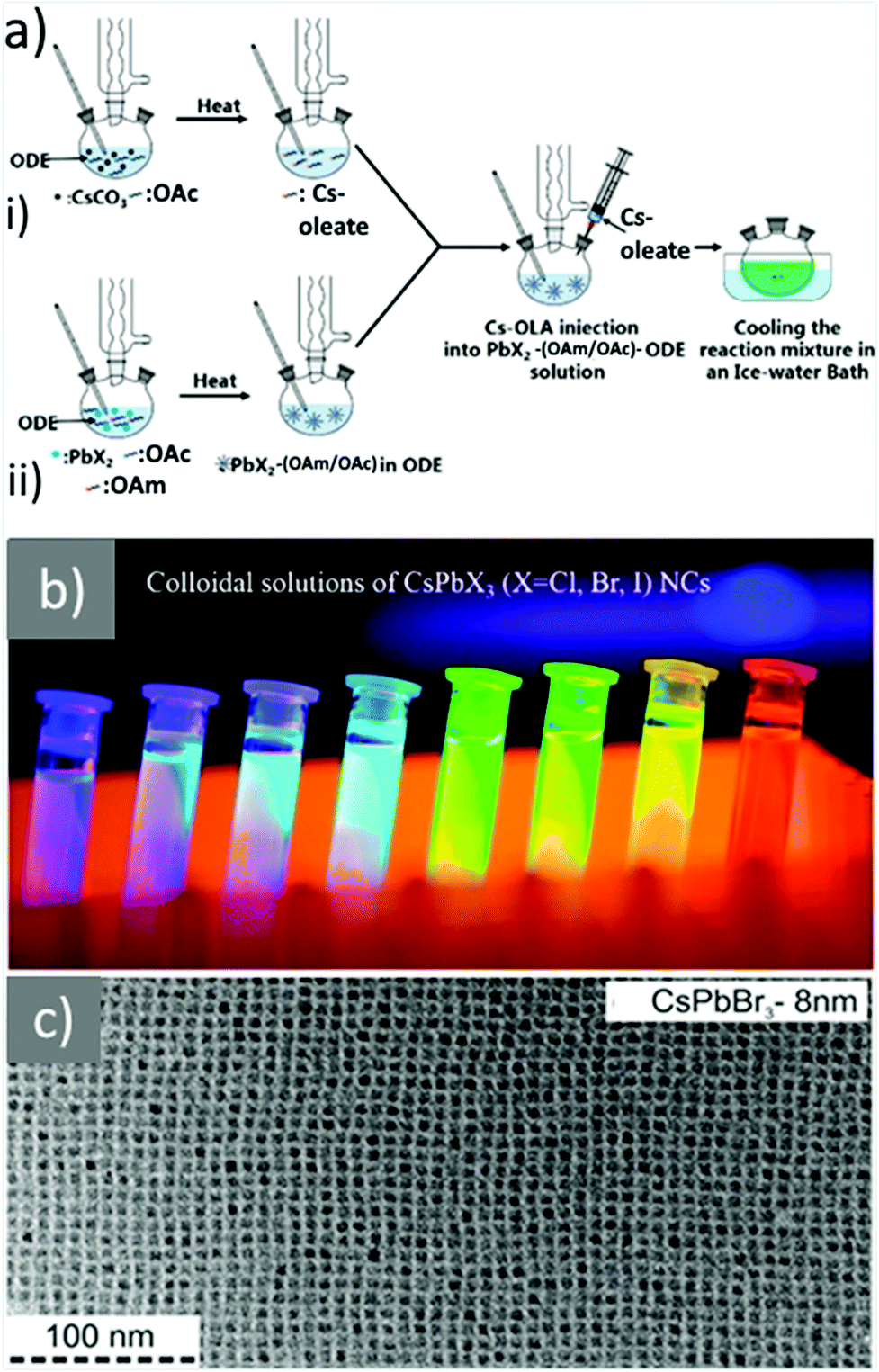 | ||
| Fig. 57 (a) Schematic illustration of the two-step hot-injection synthesis of colloidal CsPbX3 NCs. Step (i) Cs-oleate stock solution is prepared by dissolving Cs2CO3 in OAc at high temperature. Step (ii) pre-synthesized Cs-oleate is injected in the reaction medium (PbX2 dissolved in 1-ODE using OAm/OAc ligands) at a high temperature, and then the resulting solution is cooled in an ice bath. (b) Photograph of the colloidal solutions of CsPbX3 NCs prepared by the hot-injection synthesis method. (c) Transmission electron microscopy image of the corresponding CsPbBr3 NCs. Panel (a and b) are reprinted with permission from ref. 378. Copyright 2021, the American Chemical Society. Panel (c) is reproduced with permission from ref. 380. Copyright 2015, the American Chemical Society. | ||
The dimensions of perovskite NCs are not only controllable by the reaction temperature, but also through varying the ligand concentration during their synthesis.393,413 For instance, Dutta et al.413 demonstrated the size-controlled synthesis of CsPbBr3 nanocubes by modifying the amount of OAm. They could reduce the size of the CsPbBr3 nanocubes down to ∼3.5 nm by increasing the amount of OAm-HBr without altering the reaction temperature. The role of the OAm/OAc ligand pair in the size-controlled synthesis of CsPbX3 NCs was systematically studied by Manna and co-workers by varying the acid–base equilibrium (by adjusting the ratio of OAm to OAc) in the reaction mixture.393 It was found that the size of the CsPbBr3 nanocubes decreased gradually from 16.4 nm to 4 nm with a narrow distribution by changing the OAm/OAc ratio together with reaction temperature in the hot-injection synthesis (Fig. 58). As shown in Fig. 58a, the CsPbBr3 NCs obtained with the minimum concentration of OAm/OAc pair required for the solubilization of PbBr2 exhibited a narrow size distribution. The size of the nanocubes was enhanced from 4.0 to 7.6 nm by increasing the reaction temperature from 120 °C to 150 °C. At reaction temperatures higher than 200 °C (which require a higher ligand concentration to dissolve PbBr2), larger nanocubes with a broad size distribution were obtained. Interestingly, all the nanocube samples exhibited a narrow emission because the broad size distribution of large nanocubes has less influence on their PL peak position considering that their size is much larger than the exciton Bohr diameter (Fig. 58c). Furthermore, it was found that the CsPbBr3 NCs crystallize in the orthorhombic crystal phase regardless of their dimensions (Fig. 56d). However, at higher concentrations of the OAm/OAc pair during the synthesis, the reaction yielded a non-fluorescent Cs4PbBr6 phase depending on the reaction temperature.393 More interestingly, it was found that the reaction yielded CsPbBr3 nanoplatelets (NPls) at high concentration of oleylammonium species; otherwise, it led to the formation of nanocubes (Fig. 58). The thickness of the CsPbBr3 NPls decreased with an increase in the concentration of the oleylammonium cation with respect to the Cs+ ion (Fig. 58h–j), and the reaction yielded one monolayer (1 ML) of nanosheets with a size in the microscale, without the introduction of Cs+ ions (Fig. 56k). These results suggest that the hot-injection synthesis can produce perovskite NPls at higher temperatures using elevated concentrations of oleylammonium cation molecules.
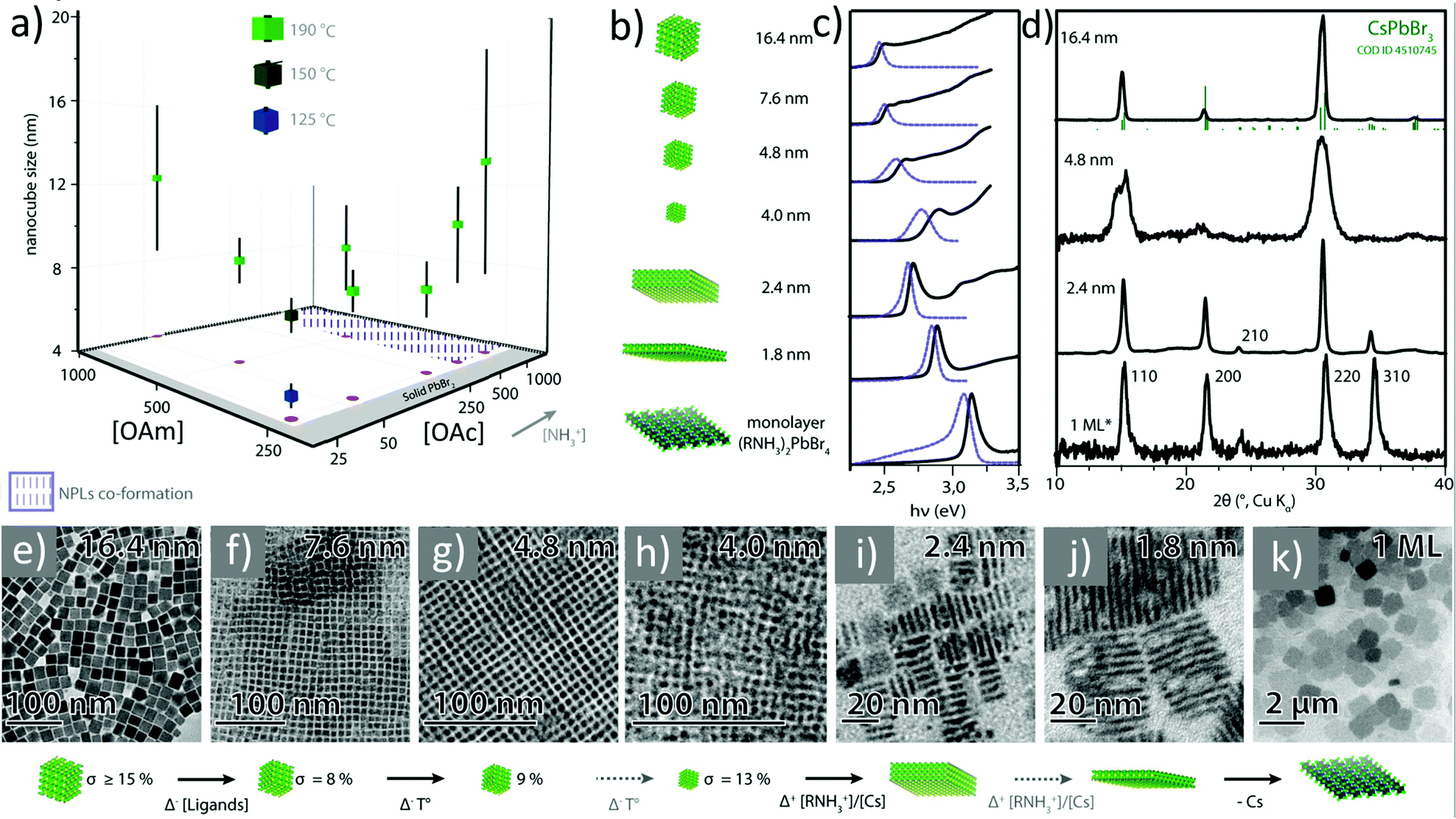 | ||
| Fig. 58 (a) Different sizes of CsPbBr3 nanocubes obtained in hot-injection synthesis at different concentrations of OAm and OAc and different reaction temperatures (size distributions are designated with vertical bars). (b) Schematic illustration showing the range of CsPbBr3 nanocubes, nanoplatelets and nanosheets obtained using only OAm/OAc as ligands. (c) UV-Vis absorbance (black solid line) and photoluminescence (blue dashed line) spectra of the corresponding NCs. XRD patterns (d) and TEM images (e–k) of the illustrated (in Fig. 58b) samples. Figure is adapted with permission from ref. 393. Copyright 2018, the American Chemical Society. | ||
Over the years, the hot-injection method using the OAm/OAc pair has been extended to produce perovskite NCs of various compositions including organic–inorganic hybrid (MAPbX3 and FAPbX3) NCs,414 doped ABX3 NCs,415 Cs4PbBr6 0D NCs378 and Pb-free NCs.416,417 The underlying chemistry is very similar in the synthesis of various types of perovskite NCs using the OAm/OAc pair.378 The ligands first help to dissolve the precursors, and then bind to the prepared NCs. However, organic–inorganic hybrid nanocubes using the OAm/OAc pair have been rarely reported compared with inorganic perovskite NCs. For instance, Vybornyi et al.418 applied the hot-injection method for the synthesis of MAPbBr3 nanocubes using OAm/OAc. They could tune the morphology from nanocubes to nanoplatelets (NPls) and NWs by adjusting the reaction conditions with an increased concentration of OAc with respect to OAm. Later, this method was further extended to the synthesis of MAPbI3 nanocubes by Korgel and co-workers.419
The main challenge associated with the hot-injection synthesis of hybrid perovskite NCs is that small molecules such as MA+ can easily degrade at high reaction temperatures. However, a hot-injection route was successfully implemented for the synthesis of FAPbX3 NCs.408,420 Protesescu et al.420 demonstrated the hot-injection synthesis of green emissive FAPbBr3 nanocubes in the presence of OAm/OAc. In their synthesis, FA+ and Pb-acetate precursors were initially dissolved in octadecene using OAc, and then pre-synthesized oleylammonium bromide (OAm-Br) was injected at 130 °C. Very recently, Otero-Martínez et al.408 reported that the injection of FA-oleate into PbBr2–OAm–OAc dissolved in octadecene could produce high-quality FAPbX3 nanocubes with a controllable size by tuning the reaction temperature.
The OAm/OAc pair has been further applied in the hot-injection synthesis of lead-free double perovskite (A2B+B3+X6) NCs.416,417,421–424 In 2018, Creutz et al.417 adapted the hot-injection method to synthesize lead-free Cs2AgBiX6 (X = Cl, Br) double perovskite NCs using OAm/OAc. Their synthesis was based on the injection of trimethylsilyl halide (TMS-X) precursors into an octadecene solution containing Cs, Ag and Bi precursors, which were dissolved by the assistance of OAm/OAc, to obtain Cs2AgBiX6 NCs. In that study it was possible to realize the necessity of the presence of OAm due to the stabilization of the nanocrystal surface, taking into account that when OAm was absent, the presence of an insoluble material was observed. Adding small amounts of OAc helped to avoid the presence of impurities in the final particles. The authors also demonstrated that the halide composition of the double perovskite NCs is tunable by applying post-synthetic halide exchange reaction. Similarly, Bekenstein et al.416 reported the synthesis of Cs2AgBiX6 (X = Cl, Br) NCs via the hot-injection of a halide source ((TMS-X) or HX) into a metal acetate solution containing the OAm/OAc pair. Later, Zhou et al.421 extended the classical hot-injection synthesis of CsPbX3 NCs to Cs2AgBiBr6 nanocrystals by swiftly injecting Cs-oleate into a precursor solution consisting of BiBr3, AgNO3, ODE, HBr and OAm/OAc at high temperature. The synergistic behavior of the OAm/OAc pair was demonstrated after observing the bulk formation in the absence of OAc and the inability to form BiBr3 without OAm due to the fact that OAm acts as a complexing agent for Bi+3 ions. Therefore, the relevance of the OAm/OAc pair in the crystallization of the Cs2AgBiBr6 NCs becomes clear. Furthermore, a similar approach, involving the injection of Cs-oleate into a precursor solution in the presence of OAm/OAc, was employed for the synthesis of Cs2AgSb1−yBiyX6 (X: Br, Cl; 0 ≤ y ≤1) and Cs2AgInCl6 NCs.425,426 For more details on the synthesis of double perovskite NCs, we direct the reader to previously published reviews.378,422
Inspired by the hot-injection synthesis of CsPbX3 NCs, a wide range of relatively simple methods such as hot-plate approach,408 ligand-assisted ultrasonication,384,402,427 solvothermal process,428 mechanical grinding,429 and microwave-assisted430 synthesis have been developed to obtain high-quality halide perovskite NCs in the presence of OAm/OAc. We suggest that readers check the available comprehensive reviews for more detailed insights into the different synthesis methods reported for metal halide perovskite NCs.378,379 For instance, Tong et al.384 published a method for the one-pot single-step synthesis of CsPbX3 NCs via ultrasonication of the precursor salts in ODE in the presence of OAm/OAc, yielding high-quality NCs with extremely high PLQYs (Fig. 55). Similarly, Chen et al.428 employed a solvothermal route, in which a mixture of perovskite precursors together with OAm/OAc was heated in an autoclave to obtain highly emissive CsPbX3 perovskite nanocubes. The authors showed that this approach could be also adapted to produce CsPbX3 NWs. It is important to note that most of the single-step synthesis methods are based on applying an external stimulus (such as heat, ultrasonication, MW irradiation and mechanical grinding) to a mixture of precursors and OAm/OAc pair (with additional ligands in some cases).384,428–430
Alternatively, the OAm/OAc pair has also been used in the ligand-assisted reprecipitation (LARP) of perovskite NCs at room temperature.389,391,431 In 2016, Li et al.391 demonstrated the RT synthesis of highly luminescent CsPbX3 NCs via the LARP approach, in which a DMF solution containing the corresponding salt precursors together with OAm/OAc was injected into toluene to provide crystallized perovskite NCs, as illustrated in Fig. 59a and b. It was suggested that the ligand pair mediated the size control of the NCs and favored their solubility in non-polar solvents. Later, Levchuk et al.414 applied the LARP approach for the synthesis of highly luminescent FAPbX3 nanocubes in the presence of OAm/OAc. Their approach was based on the rapid injection of a DMF solution containing PbX2 and FAX precursors together with OAm/OAc in chloroform. Importantly, it was found that the morphology of the NCs was tunable from nanocubes to NPls of different thicknesses by varying the ratio of OAm/OAc. A slightly different LARP approach was used by Minh et al. to obtain FAPbX3 nanocubes.432 In their approach, a DMSO solution containing the precursors (FAX and PbX2) and OAm was injected into OAc-containing toluene, resulting in the formation of stable FaPBX3 NCs. The authors could tune the size of the nanocubes by varying the amount of OAm used in the reprecipitation reaction. It should be noted that the use of DMF in LARP could affect the stability of the crystallized perovskite NCs. In contrast to classical LARP, Huang et al.406 demonstrated the polar-solvent-free synthesis of ABX3 (A: MA, FA and Cs) NCs through the spontaneous crystallization of precursor-ligand complexes in toluene (Fig. 59c). Interestingly, the shape of perovskite NCs was controllable from nanocubes to NPls by varying the ratio of monovalent cation (A+) to Pb2+ (Fig. 59c).
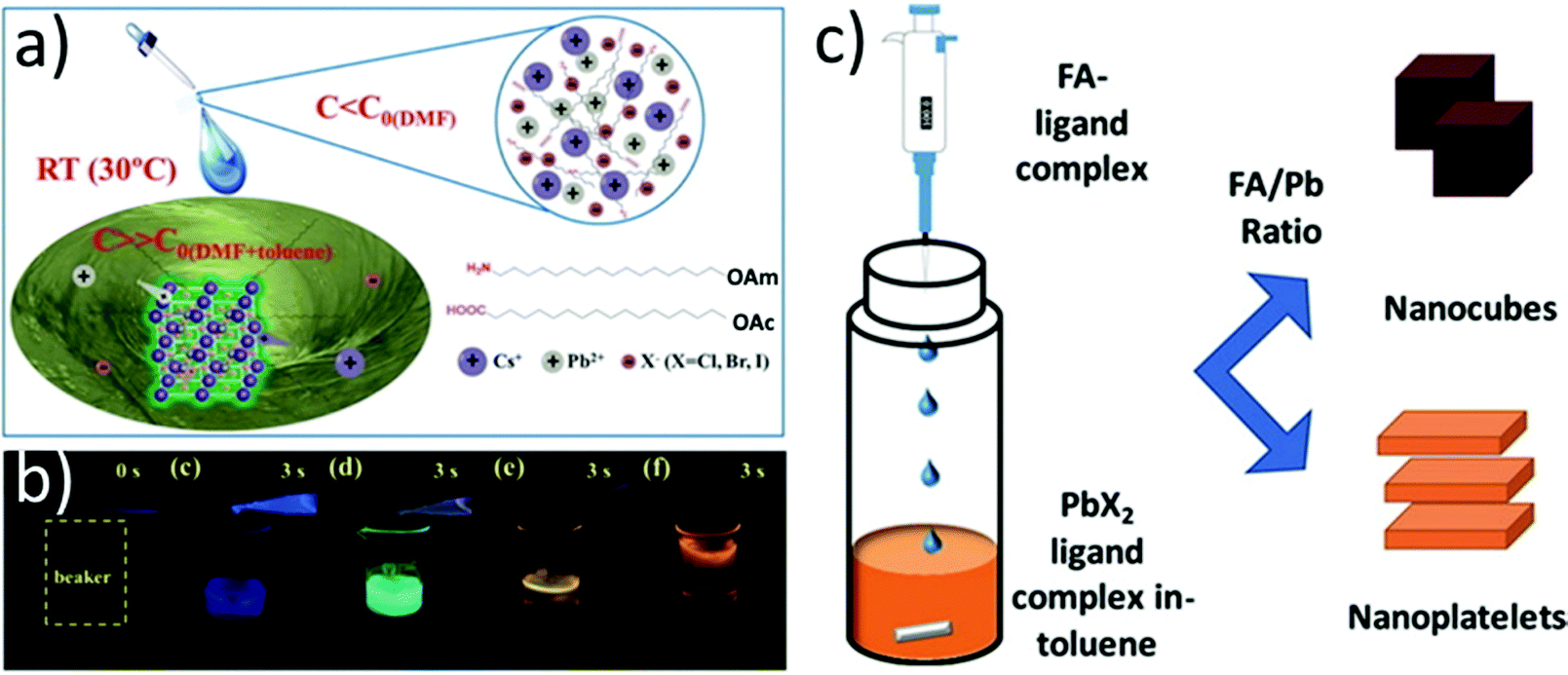 | ||
| Fig. 59 (a) Schematic illustration of the synthesis of CsPbX3 (X = Cl, Br, and I) nanocubes via supersaturated recrystallization at RT. The precursors (Cs+, Pb2+, and X− ions) dissolved in good solvent (DMF) are introduced into a bad solvent (toluene) to crystallize perovskite nanocubes. (b) Photographs of pure toluene (0 s) and colloidal solutions of the corresponding CsPbX3 nanocubes under UV illumination in the dark. Images (a and b) are adapted with permission from ref. 391. Copyright 2016, Wiley-VCH. (c) Spontaneous crystallization of perovskite NCs in organic medium upon introducing A-oleate (e.g., FA-oleate) and PbX2-(OAm/OAc) pre-prepared precursor solutions in it. The shape of NCs can be controlled from nanocubes to NPls by varying the ratio of A-cation to Pb2+ ratio. Image (c) is adapted with permission from ref. 433. Copyright 2019, Wiley-VCH. | ||
In this sub-section, it was highlighted that the OAm/OAc pair helps to dissolve the perovskite precursors in the reaction pot, and afterwards it binds to the NC surface. This is a crucial function to achieve high-quality nanomaterials with a controllable size.
Self-assembly of nanocubes
In general, perovskite nanocubes, especially inorganic ones, exhibit a high degree of monodispersity (with 5–10% variation), regardless of the synthesis method,378–380,384,391,393,408,413,428,433–439 such as OAm-capped Au NPs reported a couple of decades ago.440–442 It is well known that OAm/OAc-capped (in general, long chain ligand-capped) Au and classical semiconductor NCs tend to form perfectly ordered superlattices.443–447 The self-assembly of CsPbBr3 perovskite NCs into highly luminescent colloidal supercrystals is driven by the van der Waals interactions between the hydrophobic ligands in the perovskite NCs under high concentration conditions.400 This self-assembly process has been observed for gold NCs.448–450 The influence of the OAm/OAc ligands in the self-assembly could be also extended to NWs, which is discussed in the next section. Inspired by the previous knowledge acquired on the self-assembly techniques used for classical colloidal NCs, researchers reported different methods to obtain long-range ordered perovskite superlattices.378,400,434,451 In this regard, Tong et al. found that OAm/OAc-coated monodisperse CsPbBr3 nanocubes spontaneously self-assemble into superlattices in a concentrated colloidal solution, and the superlattices tend to settle down at the bottom of the glass vial due to their larger mass (Fig. 60a).400 However, these superlattices adapt irregular shapes without any control over their dimensions. Alternatively, the superlattices obtained on a substrate via the solvent evaporation technique exhibit a cubic shape with better uniformity in their dimensions.434,447 The long-range ordering and the size of the superlattices can be enhanced by slow solvent evaporation in a glovebox, as demonstrated by Manna and co-workers.447 Furthermore, it has been illustrated that 2D superlattices composed of supercrystal building blocks can be formed through the templated approach.452 The 2D superlattices exhibit enhanced linear and non-linear optical properties through light amplification caused by the diffraction of light at specific excitation wavelengths.453 We refer to the self-assembly section of the published comprehensive review below for more insights into the self-assembly of perovskite NCs and the optical properties of the resulting superlattices.378 | ||
| Fig. 60 (a) Schematic illustration of the synthesis of CsPbBr3 nanocube superlattices via the ultrasonication of concentrated precursor dispersions. (b and c) HAADF-STEM images of CsPbBr3 nanocube superlattices with different magnifications obtained via the spontaneous self-assembly of OAm/OAc-capped CsPbBr3 nanocubes in solution. The insets of figure ‘c’ are a photograph of the colloidal solution and schematic view of a supercrystal. (d) Self-assembly of CsPbBr3 nanocubes into superlattices by solvent evaporation and (e) corresponding HAADF-STEM image (inset: magnified view of individual NCs. Images in ‘(d and e)’ are reprinted with permission from ref. 434. Copyright 2018, Nature Publishing Group. | ||
CsPbX3 nanowires
As discussed previously in the surface chemistry section, the ligands on perovskite NCs are characterized by a highly dynamic binding mode due to their weak ionic interactions with the NC surface.388,392–397 Therefore, the ligands detach easily from the NC surface, which is also facet selective. Consequently, CsPbX3 nanocubes often transform into nanowires under specific reaction conditions.402,453–456 For instance, Zhang et al. first observed the formation of single crystalline CsPbX3 NWs under a prolonged reaction (90 min), which were initially developed for CsPbX3 nanocubes (Fig. 61a).457 After performing a control experiment by changing the solvent from ODE to OAm, a deceleration of the kinetics could be reasonably assumed together with the preferential formation of NWs. This type of growth was considered to follow a 1D surfactant-driven mode. Later, Tong et al. found that the CsPbBr3 nanocubes obtained via ultrasonication of the precursors together with the OAm/OAc pair gradually converted to NWs through oriented attachment.402 It was possible to determine the stabilizing effect of OAm through the creation of bilayers on the surfaces of the NWs when excess OAm was added. The OAm-assisted bilayers acted in a protecting manner against the degradation of the final product. Several research groups demonstrated the transformation of perovskite nanocubes into NWs through the selective removal of the ligands from specific facets in different ways.402,453,455,458 Bakr et al.453 described the desorption of OAm/OAc by selective photoinduced induction, which resulted in a decrease in the steric repulsion between adjacent NCs. The carboxylate-rich environment induced the formation of NWs. Moreover, the use of small amounts of didodecyldimethylammonium sulfide (DDA+S−) together with OAc was essential for a self-assembly process to occur, favoring longitudinal growth and producing the NW shape.453 Manna and co-workers reported the synthesis of colloidal CsPbBr3 NWs with width that is tunable down to few-unit-cell thick, where this level of thickness allows strong quantum confinement to be observed using a mixture of alkyl amines and a short alkyl carboxylic acid in the synthesis initially developed for nanocubes.456 These researchers obtained NWs with 10–20 nm width by controlling the ratio of octylamine to OAm and by increasing the reaction time to 50 min.456 Interestingly, replacing OAc with a short-chain carboxylic acid (octanoic or hexanoic acid) resulted in thinner NWs. In particular, the width of the NWs was tunable from 10 nm (bulk region) to 3.5 nm (strong quantum confinement region) by increasing the ratio of short-chain acid to amine ligands used in the hot-injection synthesis.456 It is important to note that the yield of thin NWs was higher at lower reaction temperatures (70 °C). In contrast, Zhang et al.459,460 noticed that the addition of dodecylamine to the OAm/OAc mixture in a typical hot-injection synthesis of CsPbBr3 NCs resulted in the formation of strongly quantum-confined single crystalline ultrathin NWs (Fig. 61b).459 These NWs resemble the ultrathin gold NWs obtained by reducing polymeric strands of OAm–AuCl complexes.461,462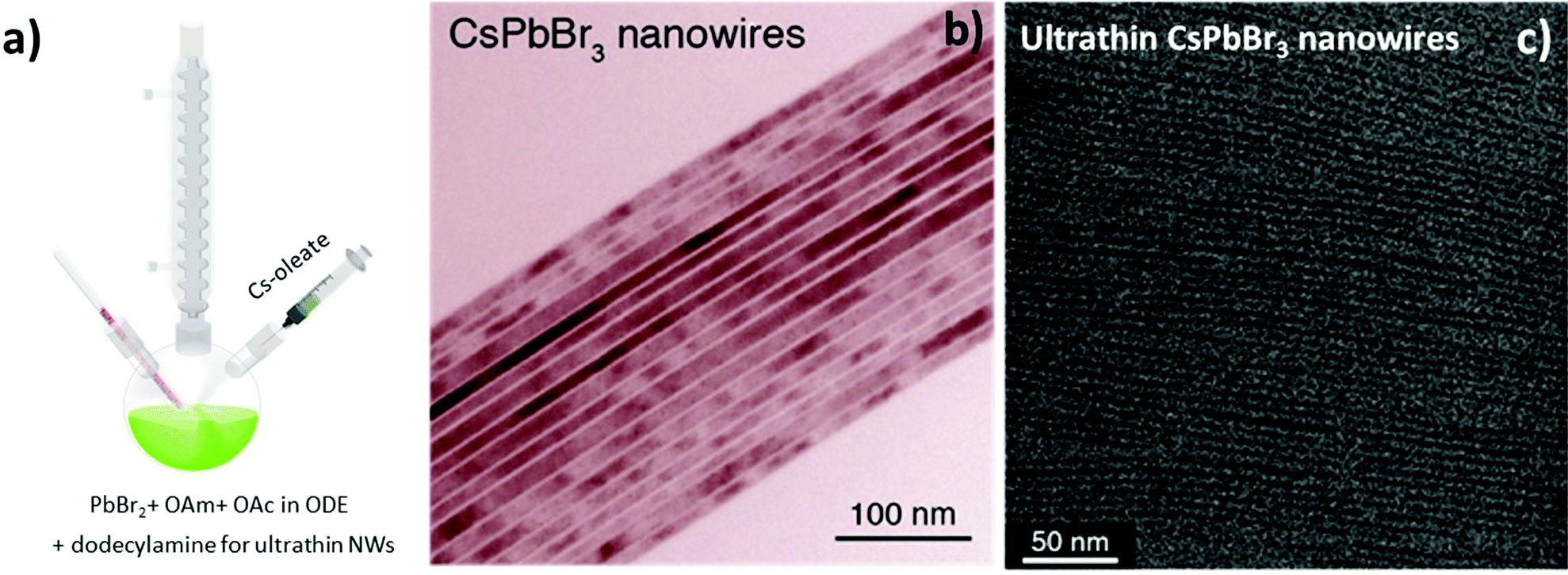 | ||
| Fig. 61 (a) Schematic illustration of the synthesis of CsPbBr3 NWs. TEM images of bulk-like (b) and ultrathin (c) CsPbBr3 nanowires. The bulk-like NWs were prepared by prolonging the reaction time (90 min) for the synthesis of the nanocube (see Fig. 57) applying the hot-injection method at 150 °C. The ultrathin NWs were prepared at 160 °C for 20 min using dodecylamine together with OAm/OAc ligand pair. Panel ‘a’ is adapted with permission from ref. 457. Copyright 2015, the American Chemical Society. Panel ‘b’ is adapted with permission from ref. 459. Copyright 2016, the American Chemical Society. | ||
A focus on ABX3 nanoplatelets
Perovskite nanoplatelets (NPls) have received increasing attention due to their strong quantum confinement effects, which enable a tunable emission across the visible spectrum of light. This can be achieved by controlling the thickness of the Br- and I-based NPls rather than having a mixed halide composition.378,407,409–411,414,463–471 The OAm/OAc pair has been successfully applied in the synthesis of halide perovskite NPls.407,410,411,433,468 It was found that the ratio of OAm/OAc together with the reaction temperature plays a critical role in the formation of NPl.393,414,472 Colloidal perovskite NPls were first reported in 2015 via the LARP of precursors in an organic solvent using octylammonium bromide ligands.469,470 The prepared NPls showed thickness-dependent optical properties due to their strong quantum confinement effects.469,470 Sichert et al.470 demonstrated that the thickness of NPls decreases with an increase in the ratio of octylammonium bromide to methylammonium bromide. However, the lateral size of the NPls appeared to be rather polydisperse. Later, it was found that inorganic CsPbX3 NPls obtained with OAm/OAc exhibit better monodispersity and tend to form face-to-face stacks.407,410,433,468In this regard, Bekenstein et al.410 first reported that the hot-injection synthesis initially developed for CsPbX3 nanocubes, yielded NPls when the reaction temperature is decreased to ≤130 °C, and the thickness of the NPls decreases with a decrease in the temperature. The authors isolated NPls of different thicknesses by applying several purification steps and drew a relation between thickness vs optical band gap of CsPbBr3 NPls. Later, Akkerman et al.468 employed a reprecipitation approach, using the Cs-oleate precursor together with OAm/OAc in DMF solvent. The precipitation of NPls was induced by acetone and the authors could tune the thickness of NPls down to the monolayer-level by controlling the amount of HBr added to the reaction medium. It was proposed that the addition of HBr led to the formation of oleylammonium cations, which replaced the Cs+ ions on the particle surface. Consequently, this prevented the growth in certain directions, leading to an anisotropic growth pattern. Therefore, the concentration of oleylammonium cations plays a critical role in the thickness control of NPls. Thus, it is possible to tune the thickness even at a relatively high temperature by varying the amount of the above-mentioned cations through control over OAm/OAc equilibrium, as described in a systematic study on the hot-injection synthesis of CsPbBr3 NCs by Manna and co-workers.393 This approach was also further extended to hybrid systems to obtain monodisperse MAPbBr3 and FAPbBr3 NPls with thickness adjustment by varying the OAm/OAc ratio.414,472 Alternatively, Bohn et al.407 reported the acetone-induced reprecipitation of Cs-oleate and PbBr2–OAm–OAc complex to NPls in toluene solution without using the polar DMF (Fig. 62). The authors demonstrated that the thickness could be precisely tuned from 2–6 monolayers by varying the ratio of Cs to Pb cations in the reaction medium (Fig. 62c). The NPls tend to form face-to-face stacks, enabling a straightforward thickness measurement (Fig. 62d–f).407 Furthermore, it has been shown that the perovskite precursors (A-oleate and PbX2–OAm–OAc complexes) spontaneously crystallize into nanocubes or NPls of different thicknesses in organic medium without the addition of acetone (Fig. 59c).433 In principle, all these synthetic methods using the OAm/OAc ligand pair are also applicable to doped perovskite NPls by adding dopant precursors in the reaction medium.473–476 Recently, the ligand pair under discussion was also used in the hot-injection synthesis of lead-free Cs2AgBiX6 (X = Cl, Br, I) double perovskite NPls by Chen and co-workers.477 In their synthesis, Cs-oleate was injected into an ODE solution containing Ag, Bi and halide precursors together with OAm/OAc.477,478 The thickness of the produced NPls was controlled from monolayer to few layer-thick by adjusting the reaction temperature.478 For more details, we recommend the reader to refer to our previous review for further description of the synthesis, optical properties and applications of halide perovskite NPls.411
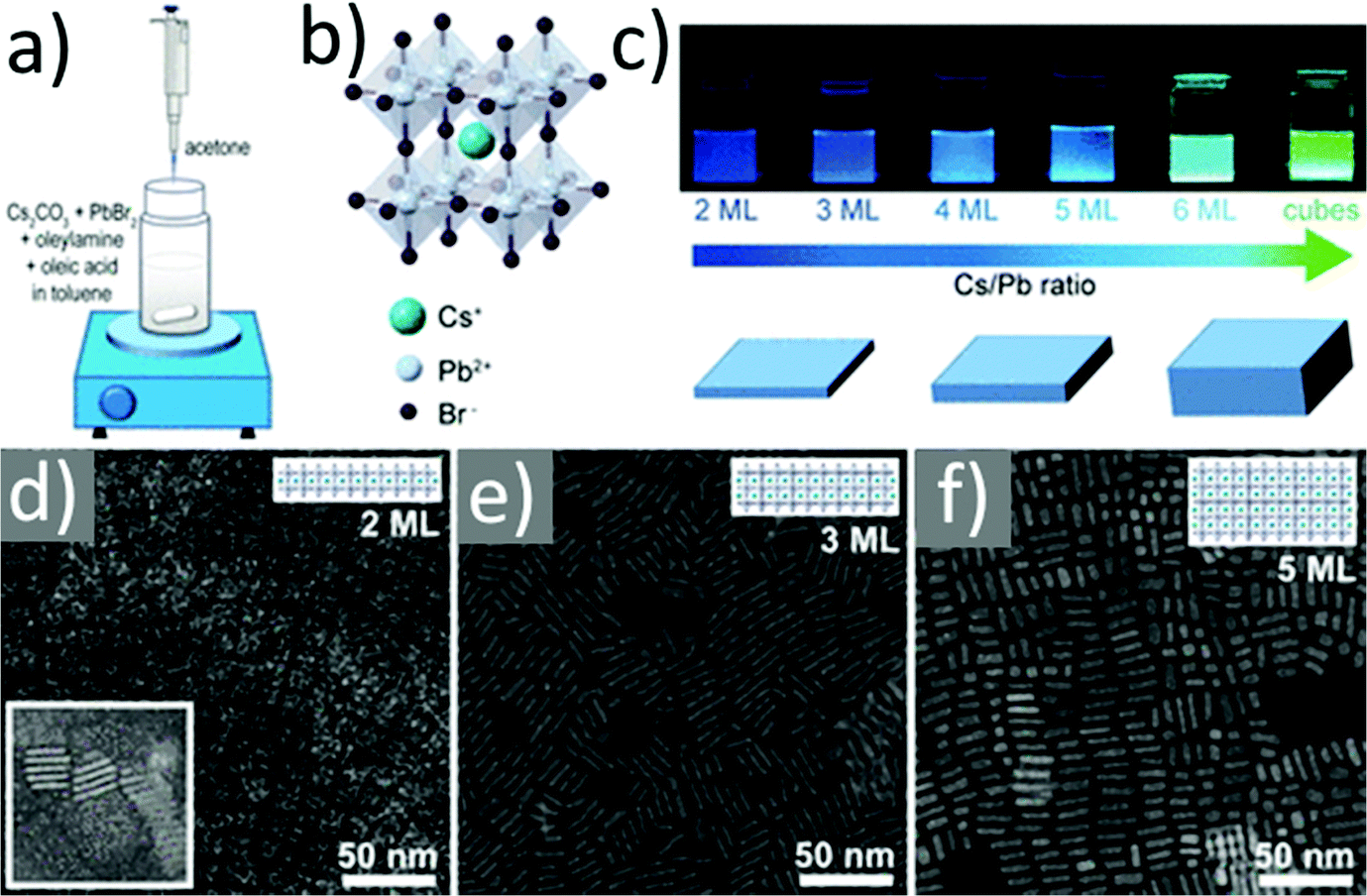 | ||
| Fig. 62 (a) Schematic illustration of the synthesis of CsPbBr3 NPls by reprecipitation of precursors using acetone as an antisolvent. NPls crystallize upon the addition of acetone in a toluene solution containing a mixture of Cs-oleate and PbBr2–OAm–OAc. (b) Scheme of the typical cubic crystal structure of CsPbBr3 perovskites. (c) Photographs of the colloidal NPl dispersions (with increasing Cs/Pb ratio) under UV light illumination. The emission color gradually changes from blue to green with increasing Cs/Pb ratio. (d–f) HAADF-STEM images of 2, 3, and 5 ML NPls. Inset in (d): HAADF-STEM image showing stacked 2 ML NPls standing on their edges. Figure is adapted with permission from ref. 407. Copyright 2018, the American Chemical Society. | ||
Table 2 summarizes the main information related to some examples of different nanomaterials synthesized in the presence of OAm/OAc and depicted/described in this review.
| Type/Fig. | Synthesis method | Applications | Ref. |
|---|---|---|---|
| Magnetite nanocubes/3 | Autoclave heating in 1-ODE at 260 °C | Not reported | 21 |
| 7–12 nm Fe3O4 NPs/4 | Decomposition of Fe(III) isopropoxide in ethanol | Not reported | 15 |
| Hematite nanocubes/11 | Solvothermal process in the presence of Hacac | Not reported therein | 56 |
| Mn0.15Fe0.85O nanocubes/14 | Thermolysis method | Not reported therein | 81 |
| CeO2@hexaniobate nanocomposites/19 | Two-stage solvothermal process | Catalysis-photocatalysis, SPR-based sensing, battery components | 102 |
| CoO nano-octahedra/20 | Thermal decomposition of Co(acac)2 | Magnetic particle imaging (MPI) | 106 |
| Mn-Doped ZnO hexagonal bipyramids/25a | Thermal decomposition | Not reported | 137 |
| Uranium dioxide NPs/27 | Thermal decomposition | Not reported | 148 |
| CdS–Au nanopencils/31a | Deposition of Au NPs on pre-synthesized CdS | Not reported | 168 |
| Wurtzite CuInS2 NRs/33a | Decomposition of nitrate precursors in dodecanethiol | Not reported | 181 |
| Star-like PbS nanostructures/37a | Decomposition of Pb-acetate in dodecanethiol | Biological applications, MRI | 217 |
| GeSe nanobelts/39b | Heating GeI4 and TOP-Se in HMDS at 320 °C | Good electrical properties | 234 |
| Pt nanocubes/44 | Thermolytic reduction of Pt(acac)2 | Not reported therein | 301 |
| FePt3 concave nanocubes/49 | High-temperature synthesis in organic medium | Hydrogenation of styrene and reduction of 4-nitrophenol | 342 |
| PtNi nano-octahedra/51 | Synthesis in benzyl ether with W(CO)6 | Good activity in ORR | 358 |
| CsPbBr3 nanocubes/58 | Hot-injection process | Photoluminescence properties | 393 |
| CsPbBr3 NWs/61a | Prolonged reaction for 90 min | Not reported | 457 |
9. Summary and outlook
This review extensively analyzed all the different aspects of the utility of oleylamine and oleic acid as a versatile chemical reagent pair for the colloidal synthesis of a wide variety of nanomaterials. We previously showed that OAm can be applied as a solvent, ligand and reductant, while it can also form complexes with metals, which can be decomposed to provide nanoparticles.479 Petit and co-workers recently revisited the role of OAm, demonstrating that it induces the disproportionation of the starting metal complexes in the case of Co (and Ni) NPs, and this process does not invoke a reducing role in OAm.480 Baranov et al. presented purification protocols to remove impurities from commercial OAm and discussed how these processes affect/benefit the synthesis of nanomaterials.481 In fact, Claridge and co-workers have recently investigated the effect of trans and saturated alkyl impurities in technical-grade OAm on the nanocrystal growth of Au NWs.482 In the current review, we showed that the combination of OAm with OAc constitutes a polyvalent reagent pair with roles not strictly limited to that of ligands. The morphology, composition and crystal structure of several different nanostructure types can be tuned by varying the ratio between OAm and OAc. In certain cases, even presumably subtle modifications such as changing the addition sequence of these two reagents in the reaction pot may affect the final particle morphology. The preferential binding patterns of these two molecules in different facets result in the blocking of growth in certain directions, while allowing/favoring growth in different directions. Consequently, the final size and shape of the produced structures can be controlled. The capping ligands protect the particles from aggregation, allowing their dispersion in different media, and in some cases, they also offer partial protection against oxidation. If post-synthetic annealing at very high temperatures is applied, the organic ligands will be decomposed and a graphitic carbon layer may form at the outer surface of the particles, encapsulating them.OAm and OAc are categorized as hard Lewis bases, which can form complexes with the metal ions of the precursors (which are Lewis acids). These complexes will then decompose to generate nanoparticles. Under certain reaction conditions, an acid–base complex may be formed as a result of the reaction occurring between OAm and OAc, and this complex can also play its own role during synthesis. In particular, this complex can act as a reductant and capping agent and it can be detected by characterization techniques such as 1H NMR and FTIR. The surface capping of the synthesized nanomaterials by OAm/OAc, which can be normally stored in several organic solvents, affects a range of properties, such as magnetic, optical, and catalytic, not always in a beneficial manner. Thus, on certain occasions, ligand stripping techniques are used for the removal of ligands from the NP surface, aiming for a better catalytic performance. In other cases, ligand exchange protocols are preferred for the transfer of the NPs in aqueous medium. Biocompatible, water-soluble new ligands will facilitate the use of NPs in biological/biomedical applications, sometimes with the assistance of further surface functionalization steps. When OAm/OAc are employed to synthesize monodisperse spherical particles, the interparticle distances are controlled and extended superlattices may be formed after a careful self-assembly process and solvent evaporation. Finally, even though OAm/OAc are normally used for colloidal chemical ‘bottom up’ synthesis routes, their use in top-down approaches such as ball milling has been shown to also be efficient. Besides categories such as metals, metal alloys, metal oxides, metal chalcogenides and other materials for which the use of OAm/OAc is well established, recently this ligand pair was also proven to be beneficial for the synthesis of perovskite nanostructures. Also, for the latter family of materials, size and shape control were achieved to a remarkable extent, which enabled the optical properties of these materials to be tuned, improving their device performance. Multiple theories have been proposed to explain the binding modes of OAm/OAc and the growth mechanisms responsible for the acquisition of 1D-structures, for instance. Nevertheless, despite the emergence of several interesting theories, outstanding questions are still under debate to obtain a full comprehension of the function of OAm and OAc during the growth of nanomaterials. For instance, the conditions under which OAm and OAc can form complexes with metal precursors or react with each other are understood to some extent, but still they cannot be fully predicted in advance, given that several parameters in the reaction pot may also affect the evolution of a given reaction. For example, it is not that clear how often metal–ligand and acid–amine complexes can be present simultaneously in the reaction pot. Further studies are required to investigate whether metal–ligand and acid–amine complex formation reactions may continuously compete with each other or if they can often proceed in parallel. The chemical behaviors of OAm and OAc alone are quite well known, but the degree of impact of the OAm/OAc pair on the nanoparticle growth will also depend on other parameters such as solvent, precursors, co-surfactants, and reaction temperature. It seems that further experiments in the lab are still necessary to get more detailed and precise insights on the nanoparticle growth evolution. This is because the data accumulated thus far may not be always sufficient to just insert in a software that would let us reliably know the final outcome of a certain reaction. Thus, although the OAm/OAc pair has been broadly used for a wide range of nanomaterials, we strongly believe that it will continue to attract interest due to the low cost, ease, and multi-functionality use of these reagents in the nanotechnology industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC-CZ program (project LL2003) from the Ministry of Education Youth and Sports (MEYS). L. P. acknowledges the support from the Spanish Ministerio de Ciencia e Innovacion through Ramon y Cajal grant (RYC2018-026103-I), the Spanish State Research Agency (Grant No. PID2020-11737RA-I00) and the grant from the Xunta de Galicia (ED-431F2021/05).References
- X. Song, F. Xiao, X. Li and Z. Li, Nanotechnology, 2019, 31, 094001 CrossRef PubMed.
- S. Mourdikoudis, V. Montes-Garcia, S. Rodal-Cedeira, N. Winckelmans, I. Perez-Juste, H. Wu, S. Bals, J. Perez-Juste and I. Pastoriza-Santos, Dalton Trans., 2019, 48, 3758 RSC.
- D. Chen, C. Li, H. Liu, F. Ye and J. Yang, Sci. Rep., 2015, 5, 11949 CrossRef PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904 CrossRef CAS PubMed.
- S. K. Sharma, N. Shrivastava, F. Rossi, L. D. Tung and N. T. K. Thanh, Nano Today, 2019, 29, 100795 CrossRef CAS.
- G. Papadopoulos, T. Asimakidou, D. Karfaridis, I. Kellartzis, G. Vourlias, M. Mitrakas and K. Simeonidis, Chem. Rev., 2019, 119, 4819 CrossRef.
- H. Chang, B. H. Kim, S. G. Lim, H. Baek, J. Park and T. Hyeon, Inorg. Chem., 2021, 60, 4261 CrossRef CAS PubMed.
- F. Douglas, D. A. MacLaren and M. Murrie, RSC Adv., 2012, 2, 8027 RSC.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819 CrossRef CAS.
- Reducing agents in colloidal nanoparticle synthesis. ed. S. Mourdikoudis, Royal Society of Chemistry, 2021 10.1039/9781839163623.
- O. Margeat, D. Ciuculescu, P. Lecante, M. Respaud, C. Amiens and B. Chaudret, Small, 2007, 3, 451 CrossRef CAS PubMed.
- O. Adegoke, K. Takemura and E. Y. Park, ACS Omega, 2018, 3, 1357 CrossRef CAS.
- S. Ananthakumar, J. R. Kumar and S. M. Babu, Optik, 2017, 130, 99 CrossRef.
- M. Seyhan, W. Kucharczyk, U. E. Yarar, K. Rickard, D. Rende, N. Baysal, S. Bucak and R. Ozisik, Nanotechnol., Sci. Appl., 2017, 10, 69 CrossRef CAS PubMed.
- C. Cara, A. Musinu, V. Mameli, A. Ardu, D. Niznansky, J. Bursik, M. A. Scorciapino, G. Manzo and C. Cannas, Cryst. Growth Des., 2015, 15, 2364 CrossRef CAS.
- M. Branca, M. Ibrahim, D. Ciuculescu-Pradines, K. Philippot and C. Amiens, Water Transfer of Hydrophobic Nanoparticles: Principles and Methods, in Handbook of Nanoparticles, ed. M. Aliofkhazraei, Springer International Publishing, 2016 DOI:10.1007/978-3-319-15338-4_29.
- J. Mohapatra, A. Mitra, D. Bahadur and M. Aslam, AIP Conf. Proc., 2013, 1512, 318 CrossRef CAS.
- F. Li and X. Han, AIP Conf. Proc. Chem., 2018, 1944, 020053 CrossRef.
- M. Benkovicova, A. Holos, J. Kollar, J. Mosnacek, Y. Halahovets, M. Kotlar, M. Jergel and J. Ivanco, AIP Conf. Proc., 2018, 1996, 020003 CrossRef.
- D. Wilson and M. A. Langell, Appl. Surf. Sci., 2014, 303, 6 CrossRef CAS.
- G. Gao, X. Liu, R. Shi, K. Zhou, Y. Shi, R. Ma, E. Takayama-Muromachi and G. Qiu, Cryst. Growth Des., 2010, 10, 2888 CrossRef CAS.
- R. Shim, G. Gao, R. Yi, K. Zhou, G. Qiu and X. Liu, Chin. J. Chem., 2009, 27, 739 CrossRef.
- Y. Tan, Z. Zhuang, Q. Peng and Y. Li, Chem. Mater., 2008, 20, 5029 CrossRef CAS.
- Z. Xu, C. Shen, Y. Hou, H. Gao and S. Sun, Chem. Mater., 2009, 21, 1778 CrossRef CAS.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS PubMed.
- K. Parvin, J. Ma, J. Ly, X. C. Sun, D. E. Nikles, K. Sun and L. M. Wang, J. Appl. Phys., 2004, 95, 7121 CrossRef CAS.
- J. Salado, M. Insausti, L. Lezama, I. Gil de Muro, E. Goikolea and T. Rojo, Chem. Mater., 2011, 23, 2879 CrossRef CAS.
- V. Patsula, E. Petrovsky, J. Kovarova, R. Konefal and D. Horak, Colloid Polym. Sci., 2014, 292, 2097 CrossRef CAS.
- X. Guo, W. Wang, Y. Yang and Q. Tian, CrystEngComm, 2016, 18, 9033 RSC.
- C. Granata, R. Russo, E. Esposito, A. Vettoliere, M. Russo, A. Musinu, D. Peddis and D. Fiorani, Eur. Phys. J. B, 2013, 86, 272 CrossRef.
- L. A. Cano, M. V. Cagnoli, S. J. Stewart, E. D. Cabanillas, E. L. Romero and S. G. Marchetti, Hyperfine Interact., 2010, 195, 275 CrossRef CAS.
- D. Dadarlat, I. Craciunescu, R. Turcu and C. Tripon, Int. J. Thermophys., 2017, 38, 86 CrossRef.
- R. Shi, X. Liu, G. Gao, R. Yi and G. Qiu, J. Alloys Compd., 2009, 485, 548 CrossRef CAS.
- X. Wu, X. He, L. Zhong, S. Lin, D. Wang, X. Zhu and D. Yan, J. Mater. Chem. C, 2011, 21, 13611 RSC.
- W. Liu, G. Deng, D. Wang, M. Chen, Z. Zhou, H. Yang and S. Yang, J. Mater. Chem. B, 2020, 8, 3087 RSC.
- D. Maity, S.-G. Choo, J. Yi, J. Ding and J. M. Xue, J. Magn. Magn. Mater., 2009, 321, 1256 CrossRef CAS.
- M. Nakaya, R. Nishida and A. Muramatsu, Molecules, 2014, 19, 11395 CrossRef PubMed.
- T. Kikuchi, R. Kasuya, S. Endo, A. Nakamura, T. Takai, N. Metzler-Nolte, K. Tohji and J. Balachandran, J. Magn. Magn. Mater., 2011, 323, 1216 CrossRef CAS.
- R. A. Harris, P. M. Shumbula and H. van der Walt, Langmuir, 2015, 31, 3934 CrossRef CAS.
- G. Gao, R. Shi, W. Qin, Y. Shi, G. Xu, G. Qiu and X. Liu, J. Mater. Sci., 2010, 45, 3483 CrossRef CAS.
- Y. Hou, H. Kondoh, M. Shimojo, E. O. Sako, N. Ozaki, T. Kogure and T. Ohta, J. Phys. Chem. B, 2005, 109, 4845 CrossRef CAS.
- A. Ito, L. Zhao, S. Okada and J.-I. Yamaki, J. Power Sources, 2011, 196, 8154 CrossRef CAS.
- X. Sun, S. Wang, Y. Wang and K. Sun, J. Supercond. Novel Magn., 2019, 32, 2903 CrossRef CAS.
- T. Harada and T. A. Hatton, Langmuir, 2009, 25, 6407 CrossRef CAS PubMed.
- H. S. Dehsari, R. A. Harris, A. H. Ribeiro, W. Tremel and K. Asadi, Langmuir, 2018, 34, 6582 CrossRef.
- I. R. Krauss, A. Picariello, G. Vitiello, A. De Santis, A. Koutsioubas, J. E. Houston., G. Fragneto and L. Paduano, Langmuir, 2020, 36, 8777 CrossRef.
- A. Haryono, S. B. Harmani and D. Sondari, Mater. Sci. Forum, 2013, 737, 153 Search PubMed.
- N. V. Pulkova, S. A. Tonevitskaya, V. M. Gerasimov, P. G. Rudakovskaya, A. G. Mazhuga and D. A. Sakharov, Nanotechnol. Russ., 2015, 10, 570 CrossRef CAS.
- M. Jiao, L. Jing, X. Wei, C. Liu, X. Luo and M. Gao, Nanoscale, 2017, 9, 18609 RSC.
- F. B. Effenberger, R. A. Couto, P. K. Kiyohara, G. Machado, S. H. Masunaga, R. F. Jardim and L. M. Rossi, Nanotechnology, 2017, 28, 115603 CrossRef PubMed.
- S. B. Kim, C. Cai, J. Kim, S. Sun and D. A. Sweigart, Organometallics, 2009, 28, 5341 CrossRef CAS.
- J. Mohapatra, F. Zeng, K. Elkins, M. Xing, M. Ghimire, S. Yoon, S. R. Mishra and J. P. Liu, Phys. Chem. Chem. Phys., 2018, 20, 12879 RSC.
- M. Benkovicova, A. Holos, P. Nadazdy, Y. Halahovets, M. Kotlar, J. Kollar, P. Siffalovic, M. Jergel, E. Majkova, J. Mosnacek and J. Ivanco, Phys. Chem. Chem. Phys., 2019, 21, 9553 RSC.
- Y. Eom, M. Abbas, H. Y. Noh and C. G. Kim, RSC Adv., 2016, 6, 15861 RSC.
- M. Klokkenburg, J. Hilhorst and B. H. Erne, Vib. Spectrosc., 2007, 43, 243 CrossRef CAS.
- W. Wu, R. Hao, F. Liu, X. Su and Y. Hou, J. Mater. Chem. A, 2013, 1, 6888 RSC.
- P. Dallas, A. B. Bourlinos, D. Niarchos and D. Petridis, J. Mater. Sci., 2007, 42, 4996 CrossRef CAS.
- L. Cano, A. E. Di Mauro, F. Petronella, E. Fanizza, M. Striccoli, M. L. Curri and A. Tercjak, J. Phys. Chem. C, 2015, 119, 6435 CrossRef CAS.
- Y. El Mendili, F. Gasset, N. Randrianantoandro, N. Nerambourg, J.-M. Greneche and J.-F. Bardeau, J. Phys. Chem. C, 2015, 119, 10662 CrossRef CAS.
- C. Meledandri, J. K. Stolarczyk, S. Ghosh and D. F. Brougham, Langmuir, 2008, 24, 14159 CrossRef CAS PubMed.
- T. A. Lastovina, A. P. Budnyk, M. A. Soldatov, Y. V. Rusalev, A. A. Guda, A. S. Bogdan and A. V. Soldatov, Mendeleev Commun., 2017, 27, 487 CrossRef CAS.
- Y. Zhang, M. Z. Joel., Y. He, D. Weathersby, F. Han, G. Rimal, J. Tang and Q. Dai, Mater. Lett.: X, 2019, 3, 100020 CAS.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329 CrossRef CAS PubMed.
- M. Nakaya, R. Nishida and A. Muramatsu, Chem. Lett., 2013, 42, 863 CrossRef CAS.
- I. O. Perez de Berti, M. V. Cagnoli, G. Pecchi, J. L. Alessandrini, S. J. Stewart, J. F. Bengoa and S. G. Marchetti, Nanotechnology, 2013, 24, 175601 CrossRef CAS PubMed.
- C. Cavelius, K. Moh and S. Mathur, Cryst. Growth Des., 2012, 12, 5948 CrossRef CAS.
- P. Chandrasekharan, D. Maity, C. X. Yong, K.-H. Chuang, J. Ding and S.-S. Feng, Biomaterials, 2011, 32, 5663 CrossRef CAS PubMed.
- Y. Wang and H. Yang, Chem. Eng. J., 2009, 147, 71 CrossRef CAS.
- H. Khursid, V. Tzitzios, W. Li and C. G. Hadjipanayis, J. Appl. Phys., 2010, 107, 09A333 CrossRef.
- Y. J. Suh, D. S. Kil, K. S. Chung, H. S. Lee and H. Shao, J. Magn., 2008, 13, 106 CrossRef.
- S. Farber, D. E. Ickowicz, K. Melnik, I. Yudovin-Farber, D. Recko, A. Rampersaud and A. J. Domb, J. Nanopart. Res., 2014, 16, 2425 CrossRef.
- P. S. Haddad, T. M. Martins, L. D’Souza-Li, L. M. Li, K. Metze, R. L. Adam, M. Knobel and D. Zanchet, Mater. Sci. Eng., C, 2008, 28, 489 CrossRef CAS.
- S. Belaid, S. Laurent, M. Vermeersch, L. Vander Elst, D. Perez-Morga and R. N. Muller, Nanotechnology, 2013, 24, 055705 CrossRef PubMed.
- J. Xie, S. Peng, N. Brower, N. Pourmand, S. X. Wang and S. Sun, Pure Appl. Chem., 2006, 78, 1003 CrossRef CAS.
- M. Harada, M. Kuwa, R. Sato, T. Teranishi, M. Takahasi and S. Maenosono, ACS Appl. Nano Mater., 2020, 3, 8389 CrossRef CAS.
- O. Morelos-Santos, A. I. Reyes de la Torre, P. Schacht-Hernandez, B. Portales-Martinez, I. Soto-Escalante, A. M. Mendoza-Martinez, R. Mendoza-Cruz, J. J. Velazquez-Salazar and M. Jose-Yacaman, Catal. Today, 2021, 360, 20 CrossRef CAS.
- C. Cannas, A. Musinu, A. Ardu, F. Orru, D. Peddis, M. Casu, R. Sanna, F. Angius, G. Diaz and G. Piccaluga, Chem. Mater., 2010, 22, 3353 CrossRef CAS.
- K. O. Abdulwahab, M. A. Malik, P. O’Brien, G. A. Timco, F. Tuna, C. A. Muryn, R. E. P. Winpenny, R. A. D. Pattrick, V. S. Coker and E. Arenholz, Chem. Mater., 2014, 26, 999 CrossRef CAS.
- W. Baaziz, B. P. Pichon, Y. Liu, J.-M. Greneche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halte, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063 CrossRef CAS.
- W.-I. Liang, X. Zhang, K. Bustillo, C.-H. Chiu, W.-W. Wu, J. Xu, Y.-H. Chu and H. Zheng, Chem. Mater., 2015, 27, 8146 CrossRef CAS.
- Z. Li, Y. Ma and L. Qi, CrystEngComm, 2014, 16, 600 RSC.
- D.-H. Kim, Y. T. Thai, D. E. Nikles and C. S. Brazel, IEEE Trans. Magn., 2009, 45, 64 CAS.
- M. Shima, K. Oguri, Y. Ohya, M. Gomi, Y. H. Ikuhara, Y. Sasaki, Y. Hishikawa and K. Kawabe, IEEE Trans. Magn., 2013, 49, 4824 CAS.
- S. Sun, H. Zeng, B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273 CrossRef CAS.
- H. Zeng, P. M. Rice, S. X. Wang and S. Sun, J. Am. Chem. Soc., 2004, 126, 11458 CrossRef CAS PubMed.
- T. Iwamoto, Y. Komorida, M. Mito and A. Takahara, J. Colloid Interface Sci., 2010, 345, 143 CrossRef CAS PubMed.
- K. P. Naidek, F. Bianconi, T. C. R. da Rocha, D. Zanchet, J. A. Bonacin, M. A. Novak, M. das Gracas, F. Vaz and H. Winnischofer, J. Colloid Interface Sci., 2011, 358, 39 CrossRef CAS PubMed.
- L. T. Lu, N. T. Dung, L. D. Tung, C. T. Thanh, O. K. Quy, N. V. Chuc, S. Maenosono and N. T. K. Thanh, Nanoscale, 2015, 7, 19596 RSC.
- C. A. Crouse and A. R. Barron, J. Mater. Chem., 2008, 18, 4146 RSC.
- K. O. Abdulwahab, M. A. Malik, P. O’Brien, G. A. Timco, F. Tuna, R. E. P. Winpenny, R. A. D. Pattrick, V. S. Coker and E. Arenholz, J. Mater. Chem. C, 2014, 2, 6781 RSC.
- L. Kubickova, J. Koktan, T. Korinkova, M. Klementova, T. Kmjec, J. Kohout, A. Weidenkaff and O. Kaman, J. Magn. Magn. Mater., 2020, 498, 166083 CrossRef CAS.
- Y. Xu, Y. Ma, S. Xu, G. Zheng and Z. Dai, J. Mater. Sci., 2015, 50, 4486 CrossRef CAS.
- M. Ramezani, A. Davoodi and A. Malekizad, J. Mater. Sci.: Mater. Electron., 2015, 26, 3957 CrossRef CAS.
- M. B. Fernandez van Raap, P. Mendoza Zelis, D. F. Coral, T. E. Torres, C. Marquina, G. F. Goya and F. H. Sanchez, J. Nanopart. Res., 2012, 14, 1072 CrossRef.
- T. Sener, E. Kayhan, M. Sevim and O. Metin, J. Power Sources, 2015, 288, 36 CrossRef CAS.
- K. Kirchberg, A. Becker, A. Bloesser, T. Weller, J. Timm, C. Suchomski and R. Marschall, J. Phys. Chem. C, 2017, 121, 27126 CrossRef CAS.
- S. Verma and D. Pravarthana, Langmuir, 2011, 27, 13189 CrossRef CAS PubMed.
- P. H. Nam, L. T. Lu, P. H. Linh, D. H. Manh, L. T. Thanh Tam, N. X. Phuc, P. T. Phong and I.-J. Lee, New J. Chem., 2018, 42, 14530 RSC.
- M. Virumbrales-del Olmo, A. Delgado-Cabello, A. Andrada-Chacon, J. Sanchez-Benitez, E. Urones-Garrote, V. Blanco-Gutierrez, M. J. Torralvo and R. Saez-Puche, Phys. Chem. Chem. Phys., 2017, 19, 8363 RSC.
- H. Hu, Z.-q Tian, J. Liang, H. Yang, A.-T. Dai, L. An, H.-X. Wu and S.-P. Yang, Nanotechnology, 2011, 22, 085707 CrossRef PubMed.
- H.-P. Zhou, Y.-W. Zhang, H.-X. Mai, X. Sun, Q. Liu, W.-G. Song and C.-H. Yan, Chem. – Eur. J., 2008, 14, 3380 CrossRef CAS PubMed.
- T. Rostamzadeh, S. Adireddy, C. Davis-Wheeler Chin, M. H. Abdallah and J. B. Wiley, ChemNanoMat, 2019, 5, 1373 CrossRef CAS.
- S. S. Lee, H. Zhu, E. Q. Contreras, A. Prakash, H. L. Puppala and V. L. Colvin, Chem. Mater., 2012, 24, 424 CrossRef CAS.
- H. Imagawa, A. Suda, K. Yamamura and S. Sun, J. Phys. Chem. C, 2011, 115, 1740 CrossRef CAS.
- M. Kuwa, M. Harada, R. Sato and T. Teranishi, ACS Appl. Nano Mater., 2020, 3, 2745 CrossRef CAS.
- R. Shi, G. Chen, W. Ma, D. Zhang, G. Qiu and X. Liu, Dalton Trans., 2012, 41, 5981 RSC.
- X. Li, J. Zhang, Z. Liu, M. Liu, H. Lin and R. Che, J. Mater. Chem. C, 2014, 2, 5216 RSC.
- Q. Dai and J. Tang, Nanoscale, 2013, 5, 7512 RSC.
- Q. Dai and J. Tang, RSC Adv., 2013, 3, 9228 RSC.
- X. Sun, Y.-W. Zhang, R. Si and C.-H. Yan, Small, 2005, 1, 1081 CrossRef CAS.
- P. Zhu, W. Wu, J. Zhou and W. Zhang, Appl. Organometal. Chem., 2007, 21, 909 CrossRef CAS.
- C. H. Lee, M. Kim, T. Kim, A. Kim, J. Paek, J. W. Lee, S.-Y. Choi, K. Kim, J.-B. Park and K. Lee, J. Am. Chem. Soc., 2006, 128, 9326 CrossRef CAS PubMed.
- H. Si, H. Wang, H. Shen, C. Zhou, S. Li, S. Lou, W. Xu, Z. Du and L. S. Li, CrystEngComm, 2009, 11, 1128 RSC.
- H. Zhang, L. Jing, J. Zeng, Y. Hou, Z. Li and M. Gao, Nanoscale, 2014, 6, 5918 RSC.
- T. Kida, T. Doi and K. Shimanoe, Chem. Mater., 2010, 22, 2662 CrossRef CAS.
- R. O. da Silva, T. G. Conti, A. F. de Moura, D. G. Stroppa, L. C. G. Freitas, C. Ribeiro, E. R. Camargo, E. Longo and E. R. Leite, ChemPhysChem, 2009, 10, 841 CrossRef CAS PubMed.
- L. Zhu, M. Wang, T. K. Lam, C. Zhang, H. Du, B. Li and Y. Yao, Sens. Actuators, B, 2016, 236, 646 CrossRef CAS.
- G. Wang, W. Lu, J. Li, J. Choi, Y. Jeong, S.-Y. Choi, J.-B. Park, M. K. Ryu and K. Lee, Small, 2006, 2, 1436 CrossRef CAS PubMed.
- D. G. Calatayud, M. Rodriguez and T. Jardiel, Bol. Soc. Esp. Ceram. Vidrio, 2015, 54, 159 CrossRef CAS.
- D. G. Calatayud, T. Jardiel, M. Rodriguez, M. Peiteado, D. Fernandez-Hevia and A. C. Caballero, Ceram. Int., 2013, 39, 1195 CrossRef CAS.
- C.-T. Dinh, T.-D. Nguyen, F. Kleitz and T.-O. Do, ACS Nano, 2009, 3, 3737 CrossRef CAS PubMed.
- H. Zhu, Q. Li, Y. Ren, L. Fan, J. Chen, J. Deng and X. Xing, Adv. Mater., 2016, 28, 6894 CrossRef CAS PubMed.
- L. Luo, J. Hui, Q. Yu, Z. Zhang, D. Jing, P. Wang, Y. Yang and X. Wang, CrystEngComm, 2012, 14, 7648 RSC.
- W. Devina, D. Nam, J. Hwang, C. Chandra, W. Chang and J. Kim, Electrochim. Acta, 2019, 321, 134639 CrossRef CAS.
- C.-C. Li, S.-J. Chang and M.-Y. Tai, J. Am. Ceram. Soc., 2010, 93, 4008 CrossRef CAS.
- M.-S. Kuo, S.-J. Chang, P.-H. Hsieh, Y.-C. Huang and C.-C. Li, J. Am. Ceram. Soc., 2016, 99, 445 CrossRef CAS.
- T.-Y. Yang, S.-J. Chang, C.-C. Li and P.-H. Huang, J. Am. Ceram. Soc., 2017, 100, 56 CrossRef CAS.
- M. D’Arienzo, J. Carbajo, A. Bahamonde, M. Crippa, S. Polizzi, R. Scotti, L. Wahba and F. Morazzoni, J. Am. Chem. Soc., 2011, 133, 17652 CrossRef PubMed.
- D. Q. Vo, E.-J. Kim and S. Kim, J. Colloid Interface Sci., 2009, 337, 75 CrossRef CAS PubMed.
- J. J. P. Roberts, K. T. Vuong and R. W. Murray, Langmuir, 2013, 29, 474 CrossRef CAS PubMed.
- Z. Xu, Y. Kan and C. Liu, Mater. Res. Bull., 2018, 107, 80 CrossRef CAS.
- I. Gonzalo-Juan, J. R. McBride and J. H. Dickerson, Nanoscale, 2011, 3, 3799 RSC.
- H. Gallah, F. Mighri, A. Ajji and J. Bandyopadhyay, Polym. Adv. Technol., 2020, 31, 1612 CrossRef CAS.
- R. Turgeman and A. Gedanken, Cryst. Growth Des., 2006, 6, 2260 CrossRef CAS.
- S. Suehiro, K. Horita, A. D. Pramata, M. Yuasa, Y. Ishiwata, K. Shimanoe and T. Kida, J. Asian Ceram. Soc., 2016, 4, 319 CrossRef.
- S.-H. Choi, E.-G. Kim, J. Park, K. An and N. Lee, J. Phys. Chem. B, 2005, 109, 14792 CrossRef CAS PubMed.
- Y. Zhang, F. Han, Q. Dai and J. Tang, J. Colloid Interface Sci., 2018, 517, 194 CrossRef CAS PubMed.
- X. Li, Y. Liu, J. Song, J. Xu and H. Zeng, Small, 2015, 11, 5097 CrossRef CAS PubMed.
- H. Jiang, H. Dai, X. Meng, K. Ji, L. Zhang and J. Deng, Appl. Catal., B, 2011, 105, 326 CrossRef CAS.
- J. Lee, N. J. Rancilio, J. M. Poulson and Y.-Y. Won, ACS Appl. Mater. Interfaces, 2016, 8, 8608 CrossRef CAS PubMed.
- W. Cai, Y. Zhang, J. Wang, Z. Wang, Y. Tian, H. Liu, H. Pan, L. Fu, W. Chen, C. Wu, X. Wang and G. Liu, Chem. Eng. J., 2020, 380, 122473 CrossRef CAS.
- C. A. Cattley, A. Stavrinadis, R. Beal, J. Moghal, A. G. Cook, P. S. Grant, J. M. Smith, H. Assender and A. A. R. Watt, Chem. Commun., 2010, 46, 2802 RSC.
- S. Qi, G. Liu, L. Tan, J. Chen, Y. Lou and Y. Zhao, Chem. Commun., 2020, 56, 4816 RSC.
- T. M. Mattox, A. Bergerud, A. Agrawal and D. J. Milliron, Chem. Mater., 2014, 26, 1779 CrossRef CAS.
- H.-Y. Jung and Y.-D. Huh, Cryst. Res. Technol., 2015, 50, 284 CrossRef CAS.
- P. Zhang, Y. Wang, Y. Sui, C. Wang, B. Liu, G. Zou and B. Zou, CrystEngComm, 2012, 14, 5937 RSC.
- T. J. Boyle, L. A. M. Steele, P. D. Burton, S. M. Hoppe, C. Lockhart and M. A. Rodriguez, Inorg. Chem., 2012, 51, 12075 CrossRef CAS PubMed.
- H. Wu, Y. Yang and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 16522 CrossRef CAS PubMed.
- A. W. Orbaek, L. Morrow, S. J. Maguire-Boyle and A. R. Barron, J. Exp. Nanosci., 2015, 10, 324 CrossRef CAS.
- L. J. Treadwell, T. J. Boyle, N. S. Bell, M. A. Rodriguez, B. R. Muntifering and K. Hattar, J. Mater. Sci., 2017, 52, 8268 CrossRef CAS.
- S. Sarkar, R. Borah, A. L. Santhosha, R. Dhanya, C. Narayana, A. J. Bhattacharyya and S. C. Peter, J. Power Sources, 2016, 306, 791 CrossRef CAS.
- A. Nugroho, D. Yoon, K. Y. Chung and J. Kim, J. Supercrit. Fluids, 2015, 101, 72 CrossRef CAS.
- Y.-S. Cho and Y.-D. Huh, Mater. Lett., 2011, 65, 3618 CrossRef CAS.
- Z. Zhao, H. Dai, Y. Du, J. Deng, L. Zhang and F. Shi, Mater. Chem. Phys., 2011, 128, 348 CrossRef CAS.
- Z. Huo, C.-K. Tsung, W. Huang, M. Frady, R. Yan, X. Zhang, Y. Li and P. Yang, Nano Lett., 2009, 9, 1260 CrossRef CAS PubMed.
- G. Wang, E. R. Batista and P. Yang, Phys. Chem. Chem. Phys., 2018, 20, 17563 RSC.
- M. Yan, H. Gu, Z. Liu, C. Guo and S. Liu, RSC Adv., 2015, 5, 967 RSC.
- H. Yu, Z. Song, Q. Liu, X. Ji, J. Liu, S. Xu, H. Kan, B. Zhang, J. Liu, J. Jiang, L. Miao and H. Liu, Sens. Actuators, B, 2017, 248, 1029 CrossRef CAS.
- A. K. Guria, K. Dey, S. Sarkar, B. K. Patra, S. Giri and N. Pradhan, Sci. Rep., 2014, 4, 6514 CrossRef CAS PubMed.
- F. Lu, Y. Gong, W. Ju, F. Cheng, K. Zhang, Q. Wang, W. Wang, J. Zhong, Q. Fan and W. Huang, Inorg. Chem. Commun., 2019, 106, 233 CrossRef CAS.
- M. V. Carevic, M. I. Comor, M. N. Mitric, T. S. Barudzija, S. P. Ahrenkiel and N. D. Abazovic, CrystEngComm, 2015, 17, 8492 RSC.
- Z. Chen, T. Tian, Y. Song, J. Yang and J. Hu, J. Alloys Compd., 2010, 506, 804 CrossRef CAS.
- L. Yu, J. Jia, G. Yi and W. Wang, Chem. Lett., 2017, 46, 88 CrossRef CAS.
- P. Jiang and Z. Chen, New J. Chem., 2017, 41, 5707 RSC.
- H. Yang, W. Luan, S.-T. Tu and Z. M. Wang, Cryst. Growth Des., 2009, 9, 1569 CrossRef CAS.
- N. Al-Salim, A. G. Young, R. D. Tilley, A. J. McQuillan and J. Xia, Chem. Mater., 2007, 19, 5185 CrossRef CAS.
- A. Mehta, S. N. Sharma, P. Chawla and S. Chand, Colloid Polym. Sci., 2014, 292, 301 CrossRef CAS.
- M. Saruyama, M. Kanehara and T. Teranishi, J. Am. Chem. Soc., 2010, 132, 3280 CrossRef CAS PubMed.
- W. Wang, J. Jiang, T. Ding, C. Wang, J. Zuo and Q. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 2235 CrossRef CAS PubMed.
- J. Li, M. Bloemen, J. Parisi and J. Kolny-Olesiak, ACS Appl. Mater. Interfaces, 2014, 6, 20535 CrossRef CAS PubMed.
- O. Chen, Y. Yang, T. Wang, H. Wu, C. Niu, J. Yang and Y. C. Cao, J. Am. Chem. Soc., 2011, 133, 17504 CrossRef CAS PubMed.
- S. J. Lim, W. Kim and S. K. Shin, J. Am. Chem. Soc., 2012, 134, 7576 CrossRef CAS PubMed.
- Z. Wan, W. Luan and S.-T. Tu, J. Colloid Interface Sci., 2011, 356, 78 CrossRef CAS PubMed.
- X. Jin, J. Li, J. Parisi and J. Kolny-Olesiak, J. Nanopart. Res., 2011, 13, 6963 CrossRef CAS.
- A. Hassinen, R. Gomes, K. de Nolf, Q. Zhao, A. Vantomme, J. C. Martins and Z. Hens, J. Phys. Chem. C, 2013, 117, 13936 CrossRef CAS.
- F. Limosani, R. Carcione and F. Antolini, J. Vac. Sci. Technol., 2020, 38, 012802 CrossRef CAS.
- R. Aruna-Devi, L. Marasamy, J. Cruz-Gomez, S. A. Mayen-Hernandez, F. de Moure-Flores and J. Santos-Cruz, Mater. Lett., 2021, 282, 128856 CrossRef CAS.
- X. Jin, M. Kruszynska, J. Parisi and J. Kolny-Olesiak, Nano Res., 2011, 4, 824 CrossRef CAS.
- J. Cassidy, D. Harankahage, J. Ojile, D. Porotnikov, L. Walker, M. Montemurri, B. S. L. Narvaez, D. Khon, M. D. E. Forbes and M. Zamkov, Chem. Mater., 2022, 34, 2484–2494 CrossRef CAS.
- O. Adegoke and E. Y. Park, New J. Chem., 2017, 41, 1303 RSC.
- X. Lu, Z. Zhuang, Q. Peng and Y. Li, CrystEngComm, 2011, 13, 4039 RSC.
- D. Zhu, L. Wang, Z. Liu and A. Tang, Appl. Surf. Sci., 2020, 509, 145327 CrossRef CAS.
- H. Shen, H. Wang, H. Yuan, L. Ma and L. S. LiW, CrystEngComm, 2012, 14, 555 RSC.
- J. Chang and E. R. Waclawik, CrystEngComm, 2013, 15, 5612 RSC.
- J. Li, B. Kempken, V. Dzhagan, D. R. T. Zahn, J. Grzelak, S. Mackowski, J. Parisi and J. Kolny-Olesiak, CrystEngComm, 2015, 17, 5634 RSC.
- Y. Vahidshad, M. N. Tahir, A. I. zad, S. M. Mirkazemi, R. Ghazemzadeh and W. Tremel, J. Mater. Chem. C, 2015, 3, 889 RSC.
- Y. Vahidshad, M. N. Tahir, S. M. Mirkazemi, A. I. Zad, R. Ghazemzadeh and W. Tremel, J. Mater. Sci.: Mater. Electron., 2015, 26, 8960 CrossRef CAS.
- S. Asgary, K. Mirabbaszadeh, P. Nayebi and H. Emadi, Mater. Res. Bull., 2014, 51, 411 CrossRef CAS.
- S. H. Lu, T. F. Green, A. J. Wang, D. Zheng, Y. L. Li and Y. S. Wang, Mater. Sci. Eng. B, 2016, 203, 19 CrossRef CAS.
- S. J. Peng, Y. L. Liang, F. Y. Cheng and J. Liang, Sci. China: Chem., 2012, 55, 1236 CrossRef CAS.
- B. Hou, M. Sohn, Y.-W. Lee, J. Zhang, J. I. Sohn, H. Kim, S. N. Cha and J. M. Kim, Nano Energy, 2019, 62, 764 CrossRef CAS.
- G. Gabka, P. Bujak, J. Zukrowski, D. Zabost, K. Kotwica, K. Malinowska, A. Ostrowski, I. Wielgus, W. Lisowski, J. W. Sobczak, M. Przybylski and A. Pron, Phys. Chem. Chem. Phys., 2016, 18, 15091 RSC.
- Y. Vahidshad, S. M. Mirkazemi, M. N. Tahir, A. I. Zad, R. Ghasemzadeh and W. Tremel, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 187 CrossRef.
- W. Wu, J. Zhang, X. Zheng, H. Liu, Y. Tian, Q. Hao and C. Liu, Chem. Lett., 2018, 47, 657 CrossRef CAS.
- X.-J. She, Q. Zhang, C.-F. Wang and S. W. Chen, RSC Adv., 2015, 5, 90705 RSC.
- M. Senthilkumar, C. I. Mary, G. Manobalaji and S. M. W. Babu, Mater. Sci. Semicond. Process., 2019, 104, 104685 CrossRef CAS.
- J. R. Kumar, S. Ananthakumar and S. M. BabuW, J. Mater. Sci.: Mater. Electron., 2016, 27, 12418 CrossRef.
- Y. Luo, Colloid J., 2009, 71, 375 CrossRef CAS.
- C. Zou, L. Zhang, D. Lin, Y. Yang, Q. Li, X. Xu, X. Chen and S. Huang, CrystEngComm, 2011, 13, 3310 RSC.
- S. Moosakhani, A. A. S. Alvani, R. Mohammadpour, J. Sainio, Y. Ge and S.-P. Hannula, CrystEngComm, 2018, 20, 1527 RSC.
- B. S. Tosun, B. D. Chernomordik, A. A. Gunawan, B. Williams, K. A. Mkhoyan, L. F. Francis and E. S. Aydil, Chem. Commun., 2013, 49, 3549 RSC.
- S. Ananthakumar, J. R. Kumar and S. M. Babu, Optik, 2017, 130, 99 CrossRef.
- T. Ajjammouri, S. Aazou, O. Mahboub, Z. Laghfour, M. Bouzbib, M. Abd-Lefdil, A. Ulyashin, A. Slaoui and Z. Sekkat, 2016 International Renewable and Sustainable Energy Conference (IRSEC), 2016, pp. 797–801 DOI:10.1109/IRSEC.2016.7983993.
- J. H. Yu, J. Joo, H. M. Park, S.-I. Baik, S. W. Kim, S. C. Kim and T. Hyeon, J. Am. Chem. Soc., 2005, 127, 5662 CrossRef CAS PubMed.
- S. Ananthakumar, J. R. Kumar and S. M. Babu, Optik, 2016, 127, 10360 CrossRef CAS.
- S. Ananthakumar and S. M. Babu, J. Inorg. Organomet. Polym. Mater., 2019, 29, 477 CrossRef CAS.
- D. D. Vaughn, D. Sun, J. A. Moyer, A. J. Biacchi, R. Misra, P. Schiffer and R. E. Schaak, Chem. Mater., 2013, 25, 4396 CrossRef.
- Y. Zhang, Y. Du, H. Xu and Q. Wang, CrystEngComm, 2010, 12, 3658 RSC.
- L. Chen, H. Zhan, X. Yang, Z. Sun, J. Zhang, D. Xu, C. Liang, M. Wu and J. Fang, CrystEngComm, 2010, 12, 4386 RSC.
- A. Shetty, A. Saha, M. Makkar and R. Viswanatha, Phys. Chem. Chem. Phys., 2016, 18, 25887 RSC.
- L. Tian, L. Y. Yep, T. T. Ong, J. Yi, J. Ding and J. J. Vittal, Cryst. Growth Des., 2009, 9, 352 CrossRef CAS.
- R. Karthikeyan, D. Thangaraju, N. Prakash, M. Arivandhan and Y. Hayakawa, ChemistrySelect, 2017, 2, 2187 CrossRef CAS.
- Q. Liu, A. Diaz, A. Prosvirin, Z. Luo and J. D. Batteas, Nanoscale, 2014, 6, 8935 RSC.
- M. A. Airo, F. Otieno, L. Mxakaza, A. Ipadeola, R. S. Kadzutu-Sithole, L. F. E. Machogo-Phao, C. Billing, M. Moloto and N. Moloto, RSC Adv., 2020, 10, 39509 RSC.
- I. A. Shuklov, V. F. Toknova, D. V. Demkin, G. I. Lapushkin, L. M. Nikolenko, A. A. Lizunova, S. B. Brichkin, V. N. Vasilets and V. F. Razumov, High Energy Chem., 2020, 54, 183 CrossRef CAS.
- H. Zhao, D. Wang, M. Chaker and D. Ma, J. Phys. Chem. C, 2011, 115, 1620 CrossRef CAS.
- Y. Wang, A. Tang, K. Li, C. Yang, M. Wang, H. Ye, Y. Hou and F. Teng, Langmuir, 2012, 28, 16436 CrossRef CAS PubMed.
- J. H. Warner and H. Cao, Nanotechnology, 2008, 19, 305605 CrossRef PubMed.
- C. Li, T. Bai, F. Li, L. Wang, X. Wu, L. Yuan, Z. Shi and S. Feng, CrystEngComm, 2013, 15, 597 RSC.
- G. Shao, G. Chen, W. Yang, T. Ding, J. Zuo and Q. Yang, Langmuir, 2014, 30, 2863 CrossRef CAS PubMed.
- Z. Zhang, C. Liu and X. Zhao, J. Phys. Chem. C, 2015, 119, 5626 CrossRef CAS.
- Y. Li, Z. Wang, D. Ren, Y. Liu, A. Zheng, S. M. Zakeeruddin, X. Dong, A. Hagfeldt, M. Gratzel and P. Wang, ACS Appl. Energy Mater., 2019, 2, 3822 CrossRef CAS.
- D. Thangaraju, R. Marnadu, V. Santhana, A. Durairajan, P. Kathirvel, J. Chandrasekaran, S. Jayakumar, M. A. Valente and D. C. Greenidge, CrystEngComm, 2020, 22, 525 RSC.
- M. M. Kobylinski, C. Ruhmlieb, A. Kornowski and A. Mews, J. Phys. Chem. C, 2018, 122, 5784 CrossRef CAS.
- A. de Kergommeaux, J. Faure-Vincent, A. Pron, R. de Bettignies and P. Reiss, Thin Solid Films, 2013, 535, 376 CrossRef CAS.
- B. Y. J. Liang, Y.-M. Shen, S.-C. Wang and J.-L. Huang, Thin Solid Films, 2013, 549, 159 CrossRef CAS.
- D. Marsan, H. Sengul and A. M. A. Ozdil, Environ. Sci.: Nano, 2019, 6, 879 RSC.
- M. V. Carevic, T. D. Savic, N. D. Abazovic, M. N. Mitric, Z. A. Stojanovic, S. P. Ahrenkiel and M. I. Comor, Mater. Res. Bull., 2017, 87, 140 CrossRef CAS.
- T. Omata, H. Uesugi and M. Kita, J. Cryst. Growth, 2014, 394, 81 CrossRef CAS.
- Y.-A. Chen, K.-H. Chou, Y.-Y. Kuo, C.-Y. Wu, P.-W. Hsiao, P.-W. Chen, S.-H. Yuan and D.-S. Wuu, Nanomaterials, 2019, 9, 999 CrossRef CAS PubMed.
- D. Li, M. Fang, C. Jiang, H. Lin, C. Luo, R. Qi, R. Huang and H. Peng, J. Nanopart. Res., 2018, 20, 228 CrossRef.
- D. C. Onwudiwe and V. M. Nkwe, Heliyon, 2020, 6, e04505 CrossRef PubMed.
- W. Guo, Y. Chen, L. Wang, J. Xu, D. Zeng and D.-L. Peng, Electrochim. Acta, 2017, 231, 69 CrossRef CAS.
- D. D. Vaughn II, D. Sun, S. M. Levin, A. J. Biacchi, T. S. Mayer and R. E. Schaak, Chem. Mater., 2012, 24, 3643 CrossRef.
- F. Guan, L. Ji, Y. Cui, T. Wang, S. Li, L. Li, J. Zhang and J. Wang, CrystEngComm, 2020, 22, 6189 RSC.
- L. Han, P. Hou, Y. Feng, H. Liu, J. Li, Z. Peng and J. Yang, Dalton Trans., 2014, 43, 11981 RSC.
- V. V. Singh, U. Kumar, S. N. Tripathi and A. K. Singh, Dalton Trans., 2014, 43, 12555 RSC.
- P. Lloret, G. Longinotti, G. Ybarra, L. Socolovsky and C. Moina, Mater. Res. Bull., 2013, 48, 3671 CrossRef CAS.
- J. Kim, C. Rong, Y. Lee, J. P. Liu and S. Sun, Chem. Mater., 2008, 20, 7242 CrossRef CAS.
- M. Kim and H. Song, J. Mater. Chem. C, 2014, 2, 4997 RSC.
- A. Tanaka, T.-a Adachi, Y. Hasegawa and T. Kawai, J. Alloys Compd., 2009, 488, 538 CrossRef CAS.
- L. F. E. Machogo, R. K. Sithole, N. Phao, T. Kolokoto, S. S. Gqoba, M. Mlambo, M. J. Moloto, P. M. Shumbula, P. S. Mdluli and N. Moloto, Mater. Sci. Eng., B, 2021, 263, 114878 CrossRef CAS.
- Y. Pan, Y. Liu and C. Liu, Appl. Surf. Sci., 2015, 357, 1133 CrossRef CAS.
- F. Mohammad and T. Arfin, Adv. Mater. Lett., 2014, 5, 315 CrossRef.
- C. Iacovita, C. Stiufiuc, A. Florea, R. Stiufiuc and C. M. Lucaciu, Dig. J. Nanomater. Biostructures, 2015, 10, 1209 Search PubMed.
- I.-C. Chiang and D.-H. Chen, Nanotechnology, 2009, 20, 015602 CrossRef PubMed.
- J. Salado, M. Insausti, L. Lezama, I. Gil de Muro, M. Moros, B. Pelaz, V. Grazu, J. M. de la Fuente and T. Rojo, Nanotechnology, 2012, 23, 315102 CrossRef CAS PubMed.
- D. A. Petrov, C.-R. Lin, R. D. Ivantsov, S. G. Ovchinnikov, S. M. Zharkov, G. Y. Yurkin, D. A. Velikanov, Y. V. Knyazev, M. S. Molokeev, Y.-T. Tseng, E.-S. Lin, I. S. Edelman, A. O. Baskakov, S. S. Starchikov and I. S. Luybutin, Nanotechnology, 2020, 31, 395703 CrossRef CAS PubMed.
- H. Khurshid, S. Balakrishnan, L. Colak, M. J. Bonder and G. C. Hadjipanayis, IEEE Trans. Magn., 2009, 45, 4877 CAS.
- Y. Yamamoto, J. Ogasawara, H. Himukai and T. Itoh, Appl. Phys. Lett., 2016, 109, 142406 CrossRef.
- A. Pariti, P. Desai, S. K. Y. Maddirala, N. Ercal, K. V. Katti, X. Liang and M. Nath, Mater. Res. Express, 2014, 1, 035023 CrossRef CAS.
- D. Liu, X. Li, C. Chen, C. Li, C. Zhou, W. Zhang, J. Zhao, J. Fan, K. Cheng and L. Chen, Oncol. Lett., 2018, 15, 8079 Search PubMed.
- M. J. Bradley, A. J. Biacchi and R. E. Schaak, Chem. Mater., 2013, 25, 1886 CrossRef CAS.
- S. Palchoudhury, Y. Xu, W. An, C. H. Turner and Y. Bao, J. Appl. Phys., 2010, 107, 09B311 CrossRef.
- S. J. A. Figueroa, S. J. Stewart, T. Rueda, A. Hernando and P. de la Presa, J. Phys. Chem. C, 2011, 115, 5500 CrossRef CAS.
- J. Huang, Y. Sun, S. Huang, K. Yu, Q. Zhao, F. Peng, H. Yu, H. Wang and J. Yang, J. Mater. Chem., 2011, 21, 17930 RSC.
- O. Moscoso-Londono, D. Muraca, P. Tancredi, C. Cosio-Castaneda, K. R. Pirota and L. M. Socolovsky, J. Phys. Chem. C, 2014, 118, 13168 CrossRef CAS.
- F.-h Lin, W. Chen, Y.-H. Liao, R.-a Doong and Y. Li, Nano Res., 2011, 4, 1223 CrossRef CAS.
- Y. Jang, J. Chung, S. Kim, S. W. Jun, B. H. Kim, D. W. Lee, B. M. Kim and T. Hyeon, Phys. Chem. Chem. Phys., 2011, 13, 2512 RSC.
- C.-L. Wu, P.-C. Huang, S. Brahma, J.-L. Huang and S.-C. Wang, Ceram. Int., 2017, 43, S621 CrossRef CAS.
- T. Teranishi, Y. Inoue, M. Saruyama, M. Nakaya and M. Kanehara, Chem. Lett., 2007, 36, 490 CrossRef CAS.
- D. Lee, W. D. Kim, S. Lee, W. K. Bae, S. Lee and D. C. Lee, Chem. Mater., 2015, 27, 5295 CrossRef CAS.
- Y. Zhang, S. Shen and Q. Wang, CrystEngComm, 2014, 16, 9501 RSC.
- N. P. Herring, K. A. Zeid, M. B. Mohamed, J. Pinsk and M. S. El-Shall, Langmuir, 2011, 27, 15146 CrossRef CAS PubMed.
- S. Das, G. Bhattacharjee, B. Satpati, M. Kumar, S. Deka, M. K. Ghosalya, C. S. Gopinath and T. Bala, New J. Chem., 2018, 42, 1379 RSC.
- S. Das, P. Mondal, S. Ghosh, B. Satpati, S. Deka, S. M. Islam and T. Bala, New J. Chem., 2018, 42, 7314 RSC.
- H. Lv, Z. Xi, Z. Chen, S. Guo, Y. Yu, W. Zhu, Q. Li, X. Zhang., M. Pan, G. Lu, S. Mu and S. Sun, J. Am. Chem. Soc., 2015, 137, 5859 CrossRef CAS PubMed.
- M. A. Scorciapino, R. Sanna, A. Ardu, F. Orru, M. Casu, A. Musinu and C. Cannas, J. Colloid Interface Sci., 2013, 407, 67 CrossRef CAS PubMed.
- K. Mori, K. Sugihara, Y. Kondo, T. Takeuchi, S. Morimoto and H. Yamashita, J. Phys. Chem. C, 2008, 112, 16478 CrossRef CAS.
- T. Kuzuya, T. Kuwada, H. Hamanaka and S. Hirai, Mater. Trans., 2017, 58, 65 CrossRef CAS.
- Q. Dai, S. Maloney, W. Chen, U. Poudyal and W. Yang, Nanotechnology, 2016, 27, 225401 CrossRef PubMed.
- W. Lian, H. Yang, N. Fan and S.-T. Tu, Nanoscale Res. Lett., 2008, 3, 134 CrossRef.
- T. T. Trinh, D. Mott, N. T. K. Thanh and S. Maenosono, RSC Adv., 2011, 1, 100 RSC.
- C. Zhang, Y. Liu, J. Li, K. Zhu, Z. Chen, S. Liao and X. Zhang, RSC Adv., 2018, 8, 19335 RSC.
- A. B. Ghosh, N. Saha, A. Sarkar, A. K. Dutta, P. Biswas, K. Nag and B. Adhikary, New J. Chem., 2016, 40, 1595 RSC.
- Y. Zhan, F. Ai, F. Chen, H. F. Valdovinos, H. Orbay, H. Sun, J. Liang, T. E. Barnhart, J. Tian and W. Cai, Small, 2016, 12, 2872 CrossRef CAS PubMed.
- Y.-S. Cho and Y.-D. Huh, Bull. Korean Chem. Soc., 2015, 36, 282 CrossRef CAS.
- Y. Iso, S. Takeshita and T. Isobe, Chem. Lett., 2012, 41, 1681 CrossRef CAS.
- Y.-P. Du, Y.-W. Zhang, Z.-G. Yan, L.-D. Sun and C.-H. Yan, J. Am. Chem. Soc., 2009, 131, 16364 CrossRef CAS PubMed.
- H. Wei, F. Wang, J. Du, W. Xia, J. Zhang, J. P. Liu and A. Yan, IEEE Trans. Magn., 2015, 51, 2301204 Search PubMed.
- K. Simeonidis, C. Sarafidis, E. Papastergiadis, M. Angelakeris, I. Tsiaoussis and O. Kalogirou, Intermetallics, 2011, 19, 589 CrossRef CAS.
- T. Cheng, R. Marin, A. Skripka and F. Vetrone, J. Am. Chem. Soc., 2018, 140, 12890 CrossRef CAS PubMed.
- D. Zhang, G. De, L. Zi, Y. Xu and S. Liu, Mater. Res. Express, 2016, 3, 075005 CrossRef.
- T. Lin, H. Shao, Z. Guo, J. Luo and J. Hao, Rare Met., 2009, 28, 241–244 CrossRef CAS.
- H. Shao, Y. Huang, H. S. Lee, Y. J. Suh and C. O. Kim, J. Appl. Phys., 2006, 99, 08N702 CrossRef.
- S. B. Dalavi and R. N. Panda, J. Magn. Magn. Mater., 2015, 374, 411–416 CrossRef CAS.
- S. I. Cha, C. B. Mo, K. T. Kim and S. H. Hong, J. Mater. Res., 2005, 20, 2148–2153 CrossRef CAS.
- J. P. Mehta, B. R. Knappett, G. Divitini, E. Ringe, P. A. Midgley, D. Fairen-Jimenez and A. E. H. Wheatley, Part. Part. Syst. Charact., 2018, 35, 1800120 CrossRef.
- H. T. Yang, T. Ogawa, D. Hasegawa and M. Takahashi, Phys. Status Solidi A, 2007, 204, 4013–4016 CrossRef CAS.
- P. Li, W. Jiang and F. Li, J. Sol-Gel Sci. Technol., 2013, 65, 359–366 CrossRef CAS.
- P. N. Vakil, D. A. Hardy and G. F. Strouse, ACS Nano, 2018, 12, 6784–6793 CrossRef CAS PubMed.
- Ö. Metin, S. Özkar and S. Sun, Nano Res., 2010, 3, 676–684 CrossRef.
- Ö. Metin, V. Mazumder, S. Özkar and S. Sun, J. Am. Chem. Soc., 2010, 132, 1468–1469 CrossRef PubMed.
- Y. Chen, Y. Zhang, S. Zhang, L. Yu, P. Zhang and Z. Zhang, Tribol. Lett., 2013, 51, 73–83 CrossRef CAS.
- X. Gao, H. Liu, K. Hidajat and S. Kawi, ChemCatChem, 2015, 7, 4188–4196 CrossRef CAS.
- S. Cheng-Min, Y. Tian-Zhong, X. Cong-Wen, Z. Huai-Ruo, T. Ji-Fa, B. Li-Hong, L. Chen, L. Jian-Qi and G. Hong-Jun, Chin. Phys. B, 2008, 17, 2191–2196 CrossRef.
- X. Yin, M. Shi, K. S. Kwok, H. Zhao, D. L. Gray, J. A. Bertke and H. Yang, Nano Res., 2018, 11, 3442–3452 CrossRef CAS.
- J. Watt, N. Young, S. Haigh, A. Kirkland and R. D. Tilley, Adv. Mater., 2009, 21, 2288–2293 CrossRef CAS.
- L. Graham, G. Collins, J. D. Holmes and R. D. Tilley, Nanoscale, 2016, 8, 2867–2874 RSC.
- S.-I. Choi, Adv. Powder Technol., 2016, 27, 1862–1867 CrossRef CAS.
- K. Jukk, N. Kongi, K. Tammeveski, R. M. Arán-Ais, J. Solla-Gullón and J. M. Feliu, Electrochim. Acta, 2017, 251, 155–166 CrossRef CAS.
- H. Yang, Y. Tang and S. Zou, Electrochem. Commun., 2014, 38, 134–137 CrossRef CAS.
- M. B. Mohamed, K. M. AbouZeid, V. Abdelsayed, A. A. Aljarash and M. S. El-Shall, ACS Nano, 2010, 4, 2766–2772 CrossRef CAS PubMed.
- C. A. Stowell and B. A. Korgel, Nano Lett., 2005, 5, 1203–1207 CrossRef CAS PubMed.
- M. T. Nguyen, K. Wongrujipairoj, H. Tsukamoto, S. Kheawhom, S. Mei, V. Aupama and T. Yonezawa, ACS Sustainable Chem. Eng., 2020, 8, 18167–18176 CrossRef CAS.
- J. Neng, C. Xiang, K. Jia, X. Nie and P. Sun, Appl. Sci., 2019, 9, 935 CrossRef.
- C. Wang, Y. Hu, C. M. Lieber and S. Sun, J. Am. Chem. Soc., 2008, 130, 8902–8903 CrossRef CAS PubMed.
- L. Wu, J. Yu, L. Chen, D. Yang, S. Zhang, L. Han, M. Ban, L. He, Y. Xu and Q. Zhang, J. Mater. Chem. C, 2017, 5, 3065–3071 RSC.
- M. Veith, K. Moh and C. Cavelius, Adv. Eng. Mater., 2012, 14, 825–829 CrossRef CAS.
- P. Jeevanandam, C. K. Srikanth and S. Dixit, Mater. Chem. Phys., 2010, 122, 402–407 CrossRef CAS.
- L. Li, F. Hu, D. Xu, S. Shen and Q. Wang, Chem. Commun., 2012, 48, 4728–4730 RSC.
- W. Huang, J. Li, F. Han, G. Zhang, R. Suna and C.-P. Wong, J. Chin. Chem. Soc., 2017, 64, 1354–1359 CrossRef CAS.
- M. Tan and M. D. Balela, MATEC Web Conf., 2015, 27, 03003 CrossRef.
- L. Zhang, J. Yin, W. Yu, M. Wang and H. Xie, Nanoscale Res. Lett., 2017, 12, 462 CrossRef PubMed.
- R. J. B. Pinto, J. M. F. Lucas, F. M. Silva, A. V. Girão, F. J. Oliveira, P. A. A. P. Marques and C. S. R. Freire, J. Cleaner Prod., 2019, 221, 122–131 CrossRef CAS.
- F. B. Effenberger, M. A. Sulca, M. T. Machini, R. A. Couto, P. K. Kiyohara, G. Machado and L. M. Rossi, J. Nanopart. Res., 2014, 16, 2588 CrossRef.
- M. Devaraj, R. Saravanan, R. K. Deivasigamani, V. Kumar Gupta, F. Gracia and S. Jayadevan, J. Mol. Liq., 2016, 221, 930–941 CrossRef CAS.
- M. A. B. Aissa, B. Tremblay, A. Andrieux-Ledier, E. Maisonhaute, N. Raouafia and A. Courty, Nanoscale, 2017, 7, 3189–3195 RSC.
- S. B. Dalavi, M. Manivel Raja and R. N. Panda, New J. Chem., 2015, 39, 9641–9649 RSC.
- S. B. Dalavi, M. M. Raja and R. N. Panda, AIP Conf. Proc., 2014, 1591, 241–243 CrossRef CAS.
- S. Sharma, N. S. Gajbhiye and R. S. Ningthoujam, J. Colloid Interface Sci., 2010, 351, 323–329 CrossRef CAS PubMed.
- P. Saravanan, K. Srinivasa Rao, M. Premkumar and A. K. Singh, J. Alloys Compd., 2011, 509, 3880–3885 CrossRef CAS.
- P. Saravanan, D. Deepika, J.-H. Hsu, V. T. P. Vinod, M. Černík and S. V. Kamat, RSC Adv., 2015, 5, 92406–92417 RSC.
- R. Liu, M. Zhang, E. Niu, Z. Li, X. Zheng, R. Wu, W. Zuo, B. Shen, F. Hu and J. Sun, J. Appl. Phys., 2014, 115, 17A742 CrossRef.
- K. Lee, S. W. Kang, S.-U. Lee, K.-H. Park, Y. W. Lee and S. W. Han, ACS Appl. Mater. Interfaces, 2012, 4, 4208–4214 CrossRef CAS PubMed.
- J. Xiang, P. Li, H. Chong, L. Feng, F. Fu, Z. Wang, S. Zhang and M. Zhu, Nano Res., 2014, 7, 1337–1343 CrossRef CAS.
- H. Cruz-Martínez, M. M. Tellez-Cruz, H. Rojas-Chávez, C. A. Ramírez-Herrera, P. Calaminici and O. Solorza-Feria, Int. J. Hydrogen Energy, 2019, 44, 12463–12469 CrossRef.
- J. Schoop and T. J. Balk, Metall. Mater. Trans. A, 2014, 45A, 2309–2314 CrossRef.
- H. Kahri, M. Sevim and Ö. Metin, Nano Res., 2017, 10, 1627–1640 CrossRef CAS.
- Z. Yin, M. Chi, Q. Zhu, D. Ma, J. Sune and X. Bao, J. Mater. Chem. A, 2013, 1, 9157–9163 RSC.
- Ö. Metin, H. Can, K. Şendil and M. S. Gültekin, J. Colloid Interface Sci., 2017, 498, 378–386 CrossRef PubMed.
- R. Medwal, N. Sehdev, Govind and S. Annapoorni, Appl. Phys. A: Mater. Sci. Process., 2012, 109, 403–408 CrossRef CAS.
- I. Zafiropoulou, E. Devlin, N. Boukos, D. Niarchos, D. Petridis and V. Tzitzios, Chem. Mater., 2007, 19, 1898–1900 CrossRef CAS.
- R. Medwal, S. Gautam, S. Gupta, K. H. Chae, K. Asokan, G. R. Deen, R. S. Rawat, R. S. Katiyar and S. Annapoorni, IEEE Magn. Lett., 2018, 9, 5504105 CAS.
- N. Shukla, C. Liu, P. M. Jones and D. Weller, J. Magn. Magn. Mater., 2003, 266, 178–184 CrossRef CAS.
- G. Gupta, M. N. Patel, D. Ferrer, A. T. Heitsch, B. A. Korgel, M. Jose-Yacaman and K. P. Johnston, Chem. Mater., 2008, 20, 5005–5015 CrossRef CAS.
- S. B. Dalavi and R. N. Panda, J. Magn. Magn. Mater., 2017, 428, 306–312 CrossRef CAS.
- P. de la Presa, T. Rueda, A. Hernando, J. M. Ramallo-López, L. J. Giovanetti and F. G. Requejo, J. Appl. Phys., 2008, 13, 103909 CrossRef.
- K. Mori, N. Yoshioka, Y. Kondo, T. Takeuchi and H. Yamashita, Green Chem., 2009, 11, 1337–1342 RSC.
- M. Chen, D. E. Nikles, H. Yin, S. Wang, J. W. Harrell and S. A. Majetich, J. Magn. Magn. Mater., 2003, 266, 8–11 CrossRef CAS.
- M. Chen, J. Kim, J. P. Liu, H. Fan and S. Sun, J. Am. Chem. Soc., 2006, 128, 7132–7133 CrossRef CAS PubMed.
- C. Wang, C. Lin, B. Zhao, L. Zhang, A. Kumbhar, G. Fan, K. Sun, J. Zhang, S. Chen and J. Fang, ChemNanoMat, 2015, 1, 331–337 CrossRef CAS.
- D. Serantes, M. Spasova, D. Baldomir, M. Farle and V. Salgueirino, Chem. Mater., 2010, 22, 4103–4110 CrossRef CAS.
- Y. Fujihira, T. Hachisu, S. Shitanda, K. Aikawa, A. Sugiyama, J. Mizuno, S. Shoji, T. Asahi and T. Osaka, J. Electrochem. Soc., 2016, 163, D171–D174 CrossRef CAS.
- R. D. Rutledge, W. H. Morris, M. S. Wellons, Z. Gai, J. Shen, J. Bentley, J. E. Wittig and C. M. Lukehart, J. Am. Chem. Soc., 2006, 128, 14210–14211 CrossRef CAS PubMed.
- P. Sarmphim, S. Soontaranon, C. Sirisathitkul, P. Harding, S. Kijamnajsuk, B. Chayasombat, S. Pinitsoontorn and J. Chingunpitak, Bull. Pol. Acad.: Tech., 2017, 65, 79–84 CAS.
- K. E. Elkins, G. S. Chaubey, V. Nandwana and J. P. Liu, J. Nano Res., 2008, 1, 23–30 CAS.
- K. Chokprasombat, K. Koyvanich, C. Sirisathitkul, P. Harding and S. Rugmai, Trans. Indian Inst. Met., 2016, 69, 733–740 CrossRef CAS.
- S. Saita and S. Maenosono, Chem. Mater., 2005, 17, 6624–6634 CrossRef CAS.
- L. A. W. Green, T. T. Thuy, D. M. Mott, S. Maenosono and N. T. K. Thanh, RSC Adv., 2014, 4, 1039–1044 RSC.
- H. Shao, Y. Huang, H. S. Lee, Y. Jae Suh and C. O. Kim, J. Magn. Magn. Mater., 2006, 304, e28–e30 CrossRef CAS.
- X. Sun, C. J. Thode, J. K. Mabry, J. W. Harrell, D. E. Nikles, K. Sun and L. M. Wang, J. Appl. Phys., 2005, 97, 10Q901 CrossRef.
- F. Wu, K. Eid, A. M. Abdullah, W. N. C. Wang, Y. Lan, A. A. Elzatahry and G. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 31309–31318 CrossRef CAS PubMed.
- J. Ding, L. Bu, S. Guo, Z. Zhao, E. Zhu, Y. Huang and X. Huang, Nano Lett., 2016, 16, 2762–2767 CrossRef CAS PubMed.
- K. Ahrenstorf, H. Heller, A. Kornowski, J. A. C. Broekaert and H. Weller, Adv. Funct. Mater., 2008, 18, 3850–3856 CrossRef CAS.
- K. Ahrenstorf, O. Albrecht, H. Heller, A. Kornowski, D. Gçrlitz and H. Weller, Small, 2007, 3, 271–274 CrossRef CAS PubMed.
- Q. Chang, Y. Xu, S. Zhu, F. Xiao and M. Shao, Front. Energy, 2017, 11, 254–259 CrossRef.
- S.-I. Choi, S. Xie, M. Shao, J. H. Odell, N. Lu, H.-C. Peng, L. Protsailo, S. Guerrero, J. Park, X. Xia, J. Wang, M. J. Kim and Y. Xia, Nano Lett., 2013, 13, 3420–3425 CrossRef CAS PubMed.
- J. L. Reyes-Rodríguez, A. Velázquez-Osorio, O. Solorza-Feria, D. Bahena-Uribe and J. Roque, MRS Commun., 2017, 7, 947–952 CrossRef.
- G. Niu, M. Zhou, X. Yang, J. Park, N. Lu, J. Wang, M. J. Kim, L. Wang and Y. Xia, Nano Lett., 2016, 16, 3850–3857 CrossRef CAS PubMed.
- Y. Li, X. L. Zhang, R. Qiu, R. Qiao and Y. S. Kang, J. Phys. Chem. C, 2007, 111, 10747–10750 CrossRef CAS.
- C. M. Da Silva, A. Girard, M. Dufond, F. Fossard, A. Andrieux-Ledier, V. Huc and A. Loiseau, Nanoscale Adv., 2020, 2, 3882–3889 RSC.
- X. Zhu, L. Huang, M. Wei, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2021, 281, 119460 CrossRef CAS.
- H. Miao, S. Hu, K. Ma, L. Sun, F. Wu, H. Wang and H. Li, Catal. Commun., 2018, 109, 33–37 CrossRef CAS.
- E. I. Santiago, L. C. Varanda and H. M. Villullas, J. Phys. Chem. C, 2007, 111, 3146–3151 CrossRef CAS.
- W. Li, Z. Chen, L. Xu and Y. Yan, J. Power Sources, 2010, 195, 2534–2540 CrossRef CAS.
- J. Zhang, H. Yang, B. Martens, Z. Luo, D. Xu, Y. Wang, S. Zou and J. Fang, Chem. Sci., 2012, 3, 3302–3306 RSC.
- J. Zhang, Z. Luo, B. Martens, Z. Quan, A. Kumbhar, N. Porter, Y. Wang, D.-M. Smilgies and J. Fang, J. Am. Chem. Soc., 2012, 134, 14043–14049 CrossRef CAS PubMed.
- Q. Liu, Z. Yan, N. L. Henderson, J. C. Bauer, D. W. Goodman, J. D. Batteas and R. E. Schaak, J. Am. Chem. Soc., 2009, 131, 5720–5721 CrossRef CAS PubMed.
- X.-X. Wan, D.-F. Zhang and L. Guo, CrystEngComm, 2016, 18, 3216–3222 RSC.
- Y. H. Lee, G. Lee, J. H. Shim, S. Hwang, J. Kwak, K. Lee, H. Song and J. T. Park, Chem. Mater., 2006, 18, 4209–4211 CrossRef CAS.
- Z. Peng, H. You and H. Yang, ACS Nano, 2010, 4, 1501–1510 CrossRef CAS PubMed.
- G. Ren and Y. Xing, Nanotechnology, 2009, 20, 465604 CrossRef PubMed.
- Y. Liu, D. Li, V. R. Stamenkovic, S. Soled, J. D. Henao and S. Sun, ACS Catal., 2011, 1, 1719–1723 CrossRef CAS.
- Z. Liu, D. Reed, G. Kwon, M. Shamsuzzoha and D. E. Nikles, J. Phys. Chem. C, 2007, 111, 14223–14229 CrossRef CAS.
- A. R. Malheiro, J. Perez and H. M. Villullas, J. Power Sources, 2010, 195, 3111–3118 CrossRef CAS.
- Y. Kang and C. B. Murray, J. Am. Chem. Soc., 2010, 132, 7568–7569 CrossRef CAS PubMed.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X.-G. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C.-J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- H. Huang, L. Polavarapu, J. A. Sichert, A. S. Susha, A. S. Urban and A. L. Rogach, NPG Asia Mater., 2016, 8, e328 CrossRef CAS.
- Q. Zhang and Y. Yin, ACS Cent. Sci., 2018, 4, 668–679 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- Y. Tong, E. Bladt, M. F. Aygüler, A. Manzi, K. Z. Milowska, V. A. Hintermayr, P. Docampo, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2016, 55, 13887–13892 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D’Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- J. Ye, M. M. Byranvand, C. O. Martínez, R. L. Z. Hoye, M. Saliba and L. Polavarapu, Angew. Chem., Int. Ed., 2021, 60, 21636–21660 CrossRef CAS PubMed.
- L. C. Schmidt, A. Pertegás, S. González-Carrero, O. Malinkiewicz, S. Agouram, G. Mínguez Espallargas, H. J. Bolink, R. E. Galian and J. Pérez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef CAS PubMed.
- S. Gonzalez-Carrero, R. E. Galian and J. Pérez-Prieto, J. Mater. Chem. A, 2015, 3, 9187–9193 RSC.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS PubMed.
- S. R. Smock, Y. Chen, A. J. Rossini and R. L. Brutchey, Acc. Chem. Res., 2021, 54, 707–718 CrossRef CAS PubMed.
- S. R. Smock, T. J. Williams and R. L. Brutchey, Angew. Chem., Int. Ed., 2018, 57, 11711–11715 CrossRef CAS PubMed.
- Y. Chen, S. R. Smock, A. H. Flintgruber, F. A. Perras, R. L. Brutchey and A. J. Rossini, J. Am. Chem. Soc., 2020, 142, 6117–6127 CrossRef CAS PubMed.
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32, 5410–5423 CrossRef CAS.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS PubMed.
- S. ten Brinck and I. Infante, ACS Energy Lett., 2016, 1, 1266–1272 CrossRef CAS.
- Y. Tong, E.-P. Yao, A. Manzi, E. Bladt, K. Wang, M. Döblinger, S. Bals, P. Müller-Buschbaum, A. S. Urban, L. Polavarapu and J. Feldmann, Adv. Mater., 2018, 30, 1801117 CrossRef PubMed.
- M. Imran, P. Ijaz, D. Baranov, L. Goldoni, U. Petralanda, Q. Akkerman, A. L. Abdelhady, M. Prato, P. Bianchini, I. Infante and L. Manna, Nano Lett., 2018, 18, 7822–7831 CrossRef CAS PubMed.
- Y. Tong, B. J. Bohn, E. Bladt, K. Wang, P. Müller-Buschbaum, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2017, 56, 13887–13892 CrossRef CAS PubMed.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed.
- R. K. Behera, S. Das Adhikari, S. K. Dutta, A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2018, 9, 6884–6891 CrossRef CAS PubMed.
- N. Mondal, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 32–39 CrossRef CAS.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- B. J. Bohn, Y. Tong, M. Gramlich, M. L. Lai, M. Döblinger, K. Wang, R. L. Z. Hoye, P. Müller-Buschbaum, S. D. Stranks, A. S. Urban, L. Polavarapu and J. Feldmann, Nano Lett., 2018, 18, 5231–5238 CrossRef CAS PubMed.
- C. Otero-Martínez, D. García-Lojo, I. Pastoriza-Santos, J. Pérez-Juste and L. Polavarapu, Angew. Chem., Int. Ed., 2021, 60, 26677–26684 CrossRef PubMed.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- C. Otero-Martínez, J. Ye, J. Sung, I. Pastoriza-Santos, J. Pérez-Juste, Z. Xia, A. Rao, R. L. Z. Hoye and L. Polavarapu, Adv. Mater., 2022, 2107105, DOI:10.1002/adma.202107105.
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS PubMed.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2018, 3, 329–334 CrossRef CAS.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS PubMed.
- D. Rossi, D. Parobek, Y. Dong and D. H. Son, J. Phys. Chem. C, 2017, 121, 17143–17149 CrossRef CAS.
- Y. Bekenstein, J. C. Dahl, J. Huang, W. T. Osowiecki, J. K. Swabeck, E. M. Chan, P. Yang and A. P. Alivisatos, Nano Lett., 2018, 18, 3502–3508 CrossRef CAS PubMed.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118–1123 CrossRef CAS PubMed.
- O. Vybornyi, S. Yakunin and M. V. Kovalenko, Nanoscale, 2016, 8, 6278–6283 RSC.
- Y. Zhang, C. J. Thomas, A. Guillaussier, D.-M. Smilgies and B. A. Korgel, J. Phys. Chem. C, 2019, 123, 17555–17565 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Bertolotti, N. Masciocchi, A. Guagliardi and M. V. Kovalenko, J. Am. Chem. Soc., 2016, 138, 14202–14205 CrossRef CAS PubMed.
- L. Zhou, Y.-F. Xu, B.-X. Chen, D.-B. Kuang and C.-Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- H. Tang, Y. Xu, X. Hu, Q. Hu, T. Chen, W. Jiang, L. Wang and W. Jiang, Adv. Sci., 2021, 8, 2004118 CrossRef CAS PubMed.
- R. S. Lamba, P. Basera, S. Bhattacharya and S. Sapra, J. Phys. Chem. Lett., 2019, 10, 5173–5181 CrossRef CAS PubMed.
- C. Wang, Y. Liu, Y. Guo, L. Ma, Y. Liu, C. Zhou, X. Yu and G. Zhao, Chem. Eng. J., 2020, 397, 125367 CrossRef CAS.
- B. Yang, F. Hong, J. Chen, Y. Tang, L. Yang, Y. Sang, X. Xia, J. Guo, H. He, S. Yang, W. Deng and K. Han, Angew. Chem., Int. Ed., 2019, 58, 2278–2283 CrossRef CAS PubMed.
- Y. Liu, Y. Jing, J. Zhao, Q. Liu and Z. Xia, Chem. Mater., 2019, 31, 3333–3339 CrossRef CAS.
- S. Paul, E. Bladt, A. F. Richter, M. Döblinger, Y. Tong, H. Huang, A. Dey, S. Bals, T. Debnath, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2020, 59, 6794–6799 CrossRef CAS PubMed.
- M. Chen, Y. Zou, L. Wu, Q. Pan, D. Yang, H. Hu, Y. Tan, Q. Zhong, Y. Xu, H. Liu, B. Sun and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1701121 CrossRef.
- L. Protesescu, S. Yakunin, O. Nazarenko, D. N. Dirin and M. V. Kovalenko, ACS Appl. Nano Mater., 2018, 1, 1300–1308 CrossRef CAS PubMed.
- Q. Pan, H. Hu, Y. Zou, M. Chen, L. Wu, D. Yang, X. Yuan, J. Fan, B. Sun and Q. Zhang, J. Mater. Chem. C, 2017, 5, 10947–10954 RSC.
- F. Zhang, H. Zhong, C. Chen, X.-G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- D. N. Minh, J. Kim, J. Hyon, J. H. Sim, H. H. Sowlih, C. Seo, J. Nam, S. Eom, S. Suk, S. Lee, E. Kim and Y. Kang, Chem. Mater., 2017, 29, 5713–5719 CrossRef CAS.
- H. Huang, Y. Li, Y. Tong, E.-P. Yao, M. W. Feil, A. F. Richter, M. Döblinger, A. L. Rogach, J. Feldmann and L. Polavarapu, Angew. Chem., Int. Ed., 2019, 58, 16558–16562 CrossRef CAS PubMed.
- G. Rainò, M. A. Becker, M. I. Bodnarchuk, R. F. Mahrt, M. V. Kovalenko and T. Stöferle, Nature, 2018, 563, 671–675 CrossRef PubMed.
- S. K. Mehetor, H. Ghosh and N. Pradhan, ACS Energy Lett., 2019, 4, 1437–1442 CrossRef CAS.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- Y. Tong, M. Fu, E. Bladt, H. Huang, A. F. Richter, K. Wang, P. Müller-Buschbaum, S. Bals, P. Tamarat, B. Lounis, J. Feldmann and L. Polavarapu, Angew. Chem., Int. Ed., 2018, 57, 16094–16098 CrossRef CAS PubMed.
- B. K. Patra, H. Agrawal, J.-Y. Zheng, X. Zha, A. Travesset and E. C. Garnett, ACS Appl. Mater. Interfaces, 2020, 12, 31764–31769 CrossRef CAS PubMed.
- C. Shen, C. Hui, T. Yang, C. Xiao, J. Tian, L. Bao, S. Chen, H. Ding and H. Gao, Chem. Mater., 2008, 20, 6939–6944 CrossRef CAS.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509–2511 CrossRef CAS.
- L. Polavarapu and Q.-H. Xu, Nanotechnology, 2009, 20, 185606 CrossRef PubMed.
- C. Y. Lau, H. Duan, F. Wang, C. B. He, H. Y. Low and J. K. W. Yang, Langmuir, 2011, 27, 3355–3360 CrossRef CAS PubMed.
- M. Asbahi, K. T. P. Lim, F. Wang, H. Duan, N. Thiyagarajah, V. Ng and J. K. W. Yang, Langmuir, 2012, 28, 16782–16787 CrossRef CAS PubMed.
- Z. Chen, J. Moore, G. Radtke, H. Sirringhaus and S. O'Brien, J. Am. Chem. Soc., 2007, 129, 15702–15709 CrossRef CAS PubMed.
- J. S. van der Burgt, J. J. Geuchies, B. van der Meer, H. Vanrompay, D. Zanaga, Y. Zhang, W. Albrecht, A. V. Petukhov, L. Filion, S. Bals, I. Swart and D. Vanmaekelbergh, J. Phys. Chem. C, 2018, 122, 15706–15712 CrossRef CAS PubMed.
- S. Toso, D. Baranov, C. Giannini, S. Marras and L. Manna, ACS Mater. Lett., 2019, 1, 272–276 CrossRef CAS PubMed.
- E. N. Lang, A. G. Porter, T. Ouyang, A. Shi, T. R. Hayes, T. C. Davis and S. A. Claridge, ACS Nano, 2021, 15, 10275–10285 CrossRef PubMed.
- C.-W. Yang, C.-Y. Chiu and M. H. Huang, Chem. Mater., 2014, 26, 4882–4888 CrossRef CAS.
- J.-e Park, D. R. Hickey, S. Jun, S. Kang, X. Hu, X.-J. Chen and S.-J. Park, Adv. Funct. Mater., 2016, 26, 7791–7798 CrossRef CAS.
- J. M. Carreño, N. Passarelli, C. Otero-Martínez, L. Polavarapu, L. A. Pérez, J. S. Reparaz, M. I. Alonso and A. Mihi, Adv. Opt. Mater., 2022, 2101324 CrossRef.
- D. Vila-Liarte, M. W. Feil, A. Manzi, J. L. Garcia-Pomar, H. Huang, M. Döblinger, L. M. Liz-Marzán, J. Feldmann, L. Polavarapu and A. Mihi, Angew. Chem., Int. Ed., 2020, 59, 17750–17756 CrossRef CAS PubMed.
- J. Pan, X. Li, X. Gong, J. Yin, D. Zhou, L. Sinatra, R. Huang, J. Liu, J. Chen, I. Dursun, A. M. El-Zohry, M. I. Saidaminov, H.-T. Sun, O. F. Mohammed, C. Ye, E. H. Sargent and O. M. Bakr, Angew. Chem., Int. Ed., 2019, 58, 16077–16081 CrossRef CAS PubMed.
- Y. Liu, M. Guo, S. Dong, X. Jiao, T. Wang and D. Chen, J. Mater. Chem. C, 2018, 6, 7797–7802 RSC.
- B. Pradhan, A. Mushtaq, D. Roy, S. Sain, B. Das, U. K. Ghorai, S. K. Pal and S. Acharya, J. Phys. Chem. Lett., 2019, 10, 1805–1812 CrossRef CAS PubMed.
- M. Imran, F. Di Stasio, Z. Dang, C. Canale, A. H. Khan, J. Shamsi, R. Brescia, M. Prato and L. Manna, Chem. Mater., 2016, 28, 6450–6454 CrossRef CAS PubMed.
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230–9233 CrossRef CAS PubMed.
- J.-K. Sun, S. Huang, X.-Z. Liu, Q. Xu, Q.-H. Zhang, W.-J. Jiang, D.-J. Xue, J.-C. Xu, J.-Y. Ma, J. Ding, Q.-Q. Ge, L. Gu, X.-H. Fang, H.-Z. Zhong, J.-S. Hu and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 11705–11715 CrossRef CAS PubMed.
- D. D. Zhang, Y. Yu, Y. Bekenstein, A. B. Wong, A. P. Alivisatos and P. D. Yang, J. Am. Chem. Soc., 2016, 138, 13155–13158 CrossRef CAS PubMed.
- D. D. Zhang, Y. M. Yang, Y. Bekenstein, Y. Yu, N. A. Gibson, A. B. Wong, S. W. Eaton, N. Kornienko, Q. Kong, M. L. Lai, A. P. Alivisatos, S. R. Leone and P. D. Yang, J. Am. Chem. Soc., 2016, 138, 7236–7239 CrossRef CAS PubMed.
- Z. Huo, C.-k Tsung, W. Huang, X. Zhang and P. Yang, Nano Lett., 2008, 8, 2041–2044 CrossRef CAS PubMed.
- X. Lu, M. S. Yavuz, H.-Y. Tuan, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2008, 130, 8900–8901 CrossRef CAS PubMed.
- W. Zhai, J. Lin, Q. Li, K. Zheng, Y. Huang, Y. Yao, X. He, L. Li, C. Yu, C. Liu, Y. Fang, Z. Liu and C. Tang, Chem. Mater., 2018, 30, 3714–3721 CrossRef CAS.
- V. A. Hintermayr, A. F. Richter, F. Ehrat, M. Döblinger, W. Vanderlinden, J. A. Sichert, Y. Tong, L. Polavarapu, J. Feldmann and A. S. Urban, Adv. Mater., 2016, 28, 9478–9485 CrossRef CAS PubMed.
- M. C. Weidman, A. J. Goodman and W. A. Tisdale, Chem. Mater., 2017, 29, 5019–5030 CrossRef CAS.
- M. C. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef CAS PubMed.
- C. M. Guvenc and S. Balci, ChemNanoMat, 2021, 7, 1249–1257 CrossRef CAS.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D’Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- P. Tyagi, S. M. Arveson and W. A. Tisdale, J. Phys. Chem. Lett., 2015, 6, 1911–1916 CrossRef CAS PubMed.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- D. Yang, Y. Zou, P. Li, Q. Liu, L. Wu, H. Hu, Y. Xu, B. Sun, Q. Zhang and S.-T. Lee, Nano Energy, 2018, 47, 235–242 CrossRef CAS.
- I. Levchuk, P. Herre, M. Brandl, A. Osvet, R. Hock, W. Peukert, P. Schweizer, E. Spiecker, M. Batentschuk and C. J. Brabec, Chem. Commun., 2017, 53, 244–247 RSC.
- W. J. Mir, M. Jagadeeswararao, S. Das and A. Nag, ACS Energy Lett., 2017, 2, 537–543 CrossRef CAS.
- Z.-J. Li, E. Hofman, A. H. Davis, A. Khammang, J. T. Wright, B. Dzikovski, R. W. Meulenberg and W. Zheng, Chem. Mater., 2018, 30, 6400–6409 CrossRef CAS.
- Z. Chen, L. Dong, C. Zhou, B. Zhou, Z. Zheng, R. Chen and J. Zang, CrystEngComm, 2021, 23, 793–803 RSC.
- Q. Cao, A. Ilyas, S. Zhang, Z. Ju, F. Sun, T. Liu, Y. Yang, Y. Lu, X. Liu and R. Deng, Nanoscale, 2021, 13, 11552–11560 RSC.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- J. Huang, S. Zou, J. Lin, Z. Liu and M. Qi, Nano Res., 2021, 14, 4079–4086 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzan, Chem. Mater., 2013, 25, 1465 CrossRef CAS.
- A. Vivien, M. Guillaumont, L. Meziane, C. Salzemann, C. Aubert, S. Halbert, H. Gerard, M. Petit and C. Petit, Chem. Mater., 2019, 31, 960 CrossRef CAS.
- D. Baranov, M. J. Lynch, A. C. Curtis, A. R. Carollo, C. R. Douglass, A. M. Mateo-Tejada and D. M. Jonas, Chem. Mater., 2013, 31, 1223 CrossRef.
- E. N. Lang, C. J. Pintro and S. A. Claridge, Chem. Mater., 2022, 34, 5273–5282 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |