
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferential copper-guided architectures of amyloid β peptidomimetics modulate oxidation states and catalysis†
Debasis
Ghosh
,
Mouli
Konar
,
Tanmay
Mondal
 and
Thimmaiah
Govindaraju
*
and
Thimmaiah
Govindaraju
*
Bioorganic Chemistry Laboratory, New Chemistry Unit and School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bengaluru 560064, Karnataka, India. E-mail: tgraju@jncasr.ac.in
First published on 31st March 2022
Abstract
Orchestration of differential architectures of designer peptidomimetics that modulate metal oxidation states to perform multiple chemical transformations remains a challenge. Cu-chelation and self-assembly properties of amyloid β (Aβ14-23) peptide were tuned by the incorporation of cyclic dipeptide (CDP) and pyrene (Py) as the assembly directing and reporting units, respectively. We explore the molecular architectonics of Aβ14-23 derived peptidomimetics (AkdNMCPy) to form differential architectures that stabilize distinct Cu oxidation states. The fibrillar self-assembly of AkdNMCPy is modulated to form nanosheets by the one-off addition of CuII. Notably, the serial addition of CuII resulted in the formation of micelle-like core–shell architectures. The micelle-like and nanosheet architectures were found to differentially stabilize CuII and CuI states and catalyze tandem oxidative-hydrolysis and alkyne–azide cycloaddition reactions, respectively.
Introduction
The controlled organization of designer molecular building blocks to generate well-defined material architectures with unique functional properties and applications is the guiding principle of molecular architectonics.1–3 Molecular recognition driven self-assembly and co-assembly through controlled noncovalent interactions are indispensable in the construction of diverse architectures. In the scheme of molecular architectonics, biomolecules play a key role as functional auxiliaries.4 The self-assembly of peptides and proteins has been shown to produce structurally and functionally diverse multifunctional architectures with potential applications in the fields of optoelectronics, sensing, bioimaging and biomaterials.5,6 Linear and cyclic peptides are attractive molecular building blocks to construct biomaterial scaffolds owing to their functional tolerability, ease of synthesis, biocompatibility, and enzyme mimetic ability.7,8 The self-assembly of amyloid-forming short and oligopeptides and peptidomimetics produces catalytic amyloids in the presence of metal ions with catalytic activity in various hydrolytic and redox chemical transformations.9–12 Ventura et al. recently demonstrated a pH dependent assembly of a short peptide with two divergent catalytic activities.13 Recently, CDPs are being used as versatile building blocks and auxiliaries to construct functional architectures.14,15 Gazit et al. reported a CDP-based nano-superstructure with efficient catalysis in the hydrolysis of para-nitrophenol acetate.16 The potential of CDP-driven assembly architectures as catalytic systems in multiple chemical reactions is largely unexplored.In the present work, we judiciously designed Aβ peptide-based CDP peptidomimetics and their metal-mediated assembly to produce differential architectures with distinct catalytic activities. We designed and synthesized peptidomimetics (AkdNMC and AkdNMCPy) of Aβ14-23 incorporated with unnatural CDP amino acid cyclo(L-Lys – L-Asp) (kd) and pyrene (Py) units at specific positions as shown in (Fig. 1 and S1†). Aβ14-23 (14HQKLVFFAE23D) has a self-recognition motif ‘KLVFF’ for amyloid assembly and metal-binding affinity.17
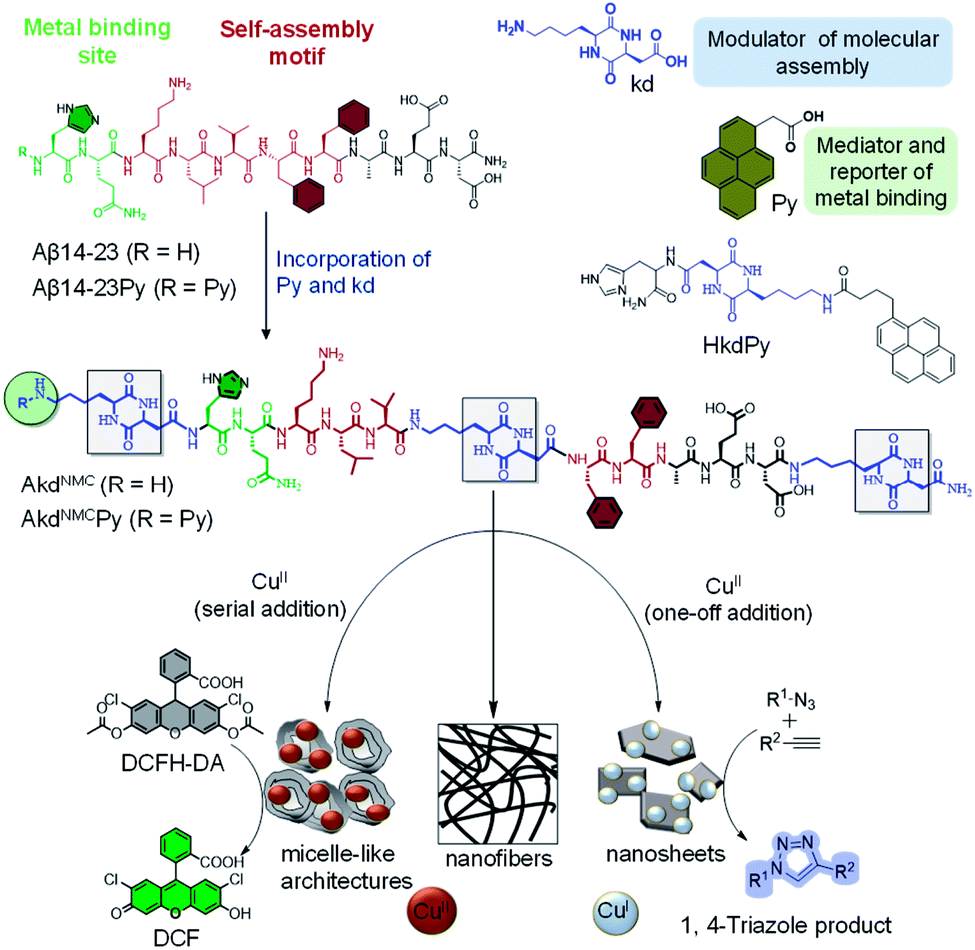 | ||
| Fig. 1 Design of Aβ14-23 peptidomimetics (AkdNMC and AkdNMCPy) incorporated with kd and Py units. HkdPy is a model peptidomimetic. Molecular assembly of AkdNMCPy in the absence and presence of one-off and serial additions of CuII to generate distinct architectures that stabilize CuII and CuI states with catalytic activity in tandem oxidative-hydrolysis of DCFDA and the alkyne–azide click reaction, respectively. | ||
Metal chelation of Aβ14-23 through N-terminal histidine (14H) prompted us to orchestrate its metal-directed assembly into differential functional architectures. To strengthen metal chelation and assembly, Aβ14-23 was incorporated with Py at the N-terminal and three kd units at the middle, and N and C-termini to obtain AkdNMCPy (Fig. 1).18 The kd with a rigid ring-core and multiple hydrogen bonding and metal coordinating sites (amide linkages) was anticipated to influence metal binding and self-assembly of AkdNMCPy. The fluorescent Py assists the monitoring of metal binding-mediated assembly of AkdNMCPy through its characteristic spectral properties. We investigated the molecular architectonics of AkdNMCPy upon binding to CuII in phosphate buffer saline (PBS, 10 mM, pH 7.4) (Fig. S2†). Cu is an important biocatalytic metal and capable of orchestrating distinct architectures with different oxidation states (CuII or CuI) and catalytic activity.19
We thus studied the assembly of AkdNMCPy (15 μM) by adding CuII in two distinct ways, (i) one-off addition: CuII (100 μM) was added in one shot, and (ii) serial addition: CuII (100 μM) was added as 20 aliquots of 5 μM each with an interval of 3.6 min (Fig. S3 and S4†). In the former case, AkdNMCPy assembly progressed upon immediate complexation with CuII to produce stable 2D sheet architectures,20,21 while the controlled complexation of AkdNMCPy to CuII in the latter case resulted in stepwise assembly to form sheets via metastable micelle-like core–shell architectures. Interestingly, CuII was reduced to CuI during sheet formation, while micelle-like architectures retained the CuII state. The distinct architectures prompted us to exploit their functional utility in CuII and CuI catalysed tandem oxidative-hydrolysis, and alkyne–azide click reactions, respectively. To the best of our knowledge, this is the first report of metal-induced differential architectures of Aβ-peptidomimetics that stabilize CuII and CuI states to accomplish distinctive catalytic activities.
Results and discussion
We probed the complexation of AkdNMCPy with CuII by UV-vis absorption, fluorescence, and circular dichroism (CD) in PBS (10 mM, 7.4 pH) (Fig. 2A–C). The AkdNMCPy emission intensity in the 350–400 nm region (pyrene) was gradually quenched as a function of the CuII concentration with the corresponding enhancement in the absorption intensity around 280–290 nm. This is attributed to a change in the microenvironment of Phe (F) residues because of CuII complexation induced assembly of AkdNMCPy. The absence of a Py excimer band ruled out possible Py–Py interactions. The decrease in the fluorescence lifetime (dynamic quenching) of Py (AkdNMCPy) from 140.8 to 113.9 ns in the presence of CuII supported Py–CuII interaction (Fig. 2D and Table S1†).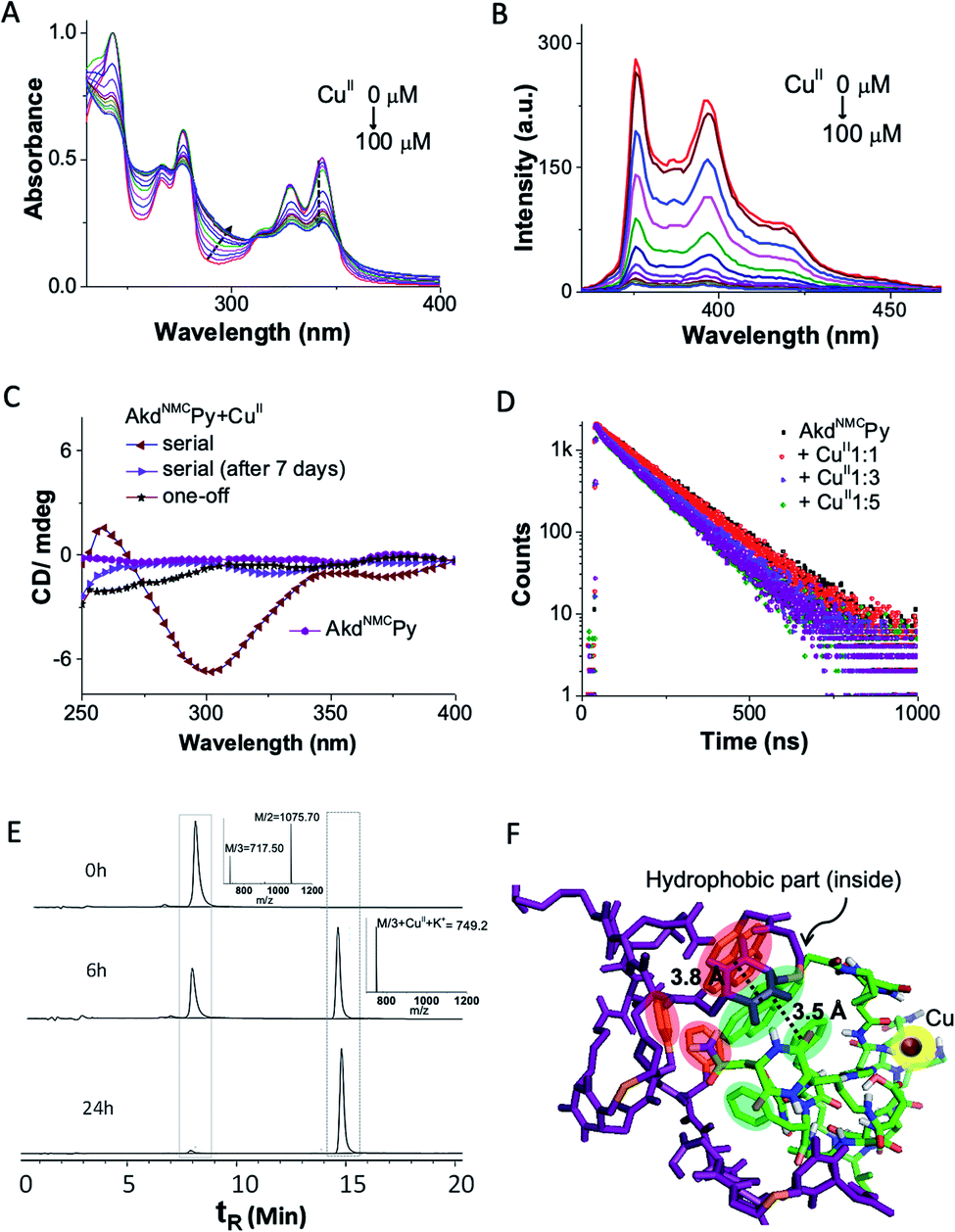 | ||
| Fig. 2 (A) Absorbance, and (B) fluorescence spectra of AkdNMCPy (15 μM) upon gradual addition of CuII (0 μM to 100 μM). (C) CD spectra of AkdNMCPy and upon addition of CuII (one-off and serial additions). (D) Fluorescence lifetime measurements of AkdNMCPy (λex 343 nm) with CuII. (E) Time-dependent LC of AkdNMCPy + CuII (inset MS data). (F) Molecular docking image of AkdNMCPy + CuII and AkdNMCPy with Py–Py and Py–Phe distances shown (dotted lines). | ||
This assumption was further supported by docking studies where AkdNMCPy was blindly docked with AkdNMCPy + CuII. The energy minimized docked pose showed a short F–Py distance (3.5 Å) as compared to the Py–Py distance (3.8 Å) (Fig. 2F, S5 and S6†).22,23 We speculate that AkdNMCPy adopted a spatial orientation to accommodate CuII that prevented Py–Py interaction while promoting F–Py interactions, as supported by the UV-vis absorption, fluorescence and docking studies.
The CD spectra revealed CuII-complexation-induced conformational change of AkdNMCPy. A distinct negative CD band observed around 298 nm indicated the generation of a chiral environment due to CuII complexation-induced assembly of AkdNMCPy. The spatial alignment of hydrophobic (F and Py) and hydrophilic moieties (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Asp, Glu and kd) of AkdNMCPy in the energy minimized docked structure of AkdNMCPy + CuII also suggested chirality induction upon complexation. Interestingly, the negative CD band at 298 nm disappeared over a period of 7 days suggesting two distinct assembly architectures for the AkdNMCPy + CuII complex with initial chirally active and final chirally inactive states (Fig. 2C).24 Fourier-transform infrared (FTIR) studies of AkdNMCPy and the NMR spectrum of a model peptidomimetic HkdPy with CuII confirmed the CuII–14H (AkdNMCPy) binding interactions (Fig. S7 and S8†).
O of Asp, Glu and kd) of AkdNMCPy in the energy minimized docked structure of AkdNMCPy + CuII also suggested chirality induction upon complexation. Interestingly, the negative CD band at 298 nm disappeared over a period of 7 days suggesting two distinct assembly architectures for the AkdNMCPy + CuII complex with initial chirally active and final chirally inactive states (Fig. 2C).24 Fourier-transform infrared (FTIR) studies of AkdNMCPy and the NMR spectrum of a model peptidomimetic HkdPy with CuII confirmed the CuII–14H (AkdNMCPy) binding interactions (Fig. S7 and S8†).
The morphological outcomes of AkdNMCPy + CuII assemblies were investigated by time-dependent transmission electron microscopy (TEM) imaging over a period of 1 to 14 days. AkdNMCPy (15 μm) self-assembled to form fibrillar aggregates in PBS (Fig. S9†). One-off addition of CuII (100 μm) into AkdNMCPy (15 μm) resulted in the formation of stable nanosheets. Atomic force microscopy (AFM) has confirmed the formation of nanosheets with ∼7 nm thickness and a 92.4 MPa DMT (Derjaguin–Muller–Toporov) modulus (Fig. 3A, S9 and S10†).25 The powder X-ray diffraction (PXRD) and selected area electron diffraction (SAED) patterns suggested the crystalline nature of the nanosheets (Fig. 3A and S11†). Serial addition of CuII (20 aliquots of 5 μM each) to AkdNMCPy formed micelle-like core–shell architectures. The docking results have shown that the core pocket constitutes a hydrophobic region (19F20F and Py), which is surrounded by the backbone and nucleophilic residues of AkdNMCPy to form CuII-induced micelle-like architectures. After 14 days of incubation, AkdNMCPy with serially added CuII (5 μM × 20 times) transformed from micelle-like architectures to 2D nanosheets (Fig. 3A). Field emission scanning electron microscopy (FESEM) images and the corresponding elemental mapping showed a higher Cu content in the micelle-like architecture compared to the nanosheets (Fig. S12†). We postulated that the micelle-like architectures formed initially were in a kinetically trapped metastable state, which restructured and transformed into thermodynamically stable 2D nanosheet architectures over time. Under similar conditions, AkdNMCPy + CuI (prepared by addition of CuSO4 + sodium ascorbate to AkdNMCPy), Aβ14-23, Aβ14-23Py, AkdNMC and HkdPy did not form sheets or micellar-like architectures (Fig. S13 and S18†).
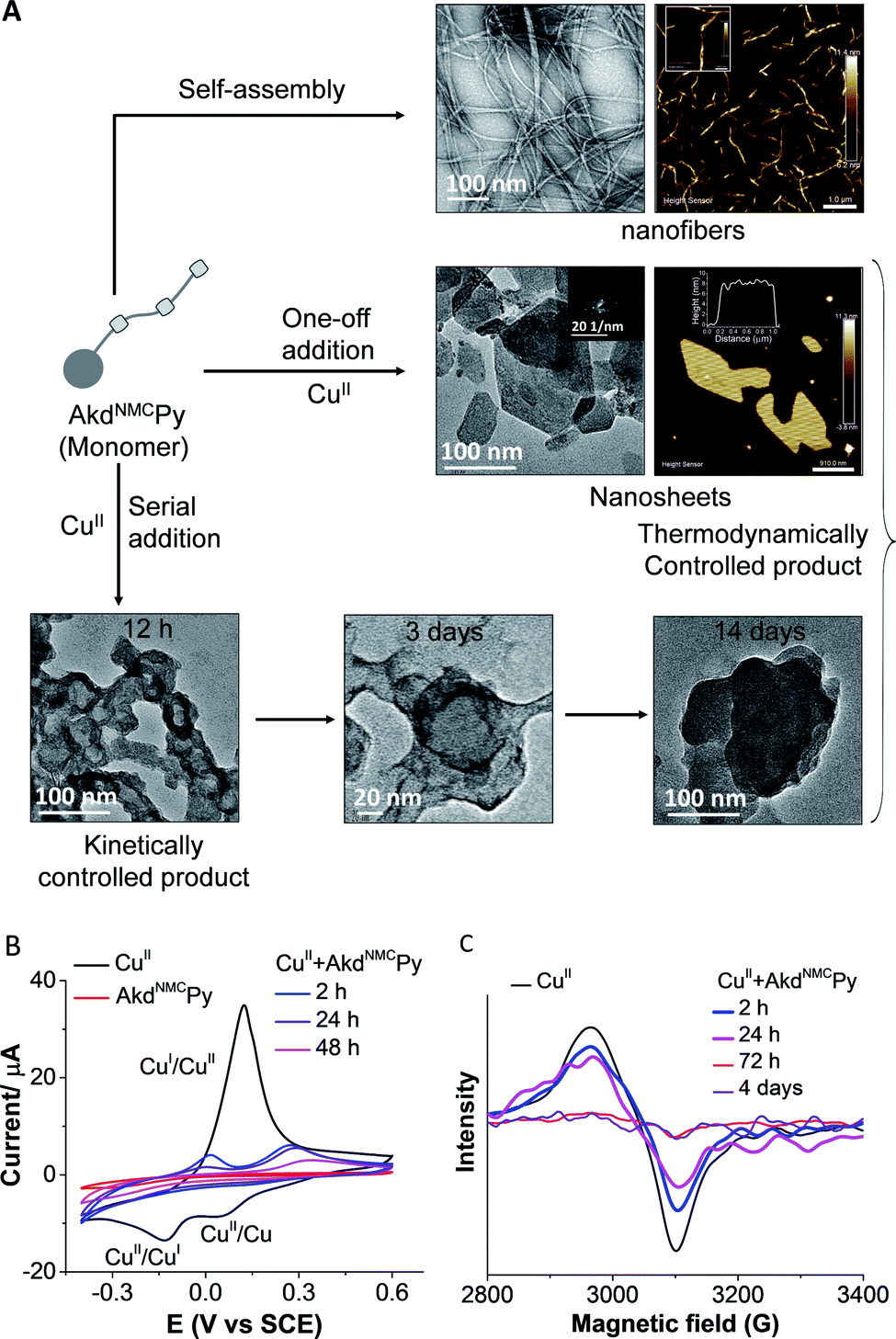 | ||
| Fig. 3 (A) Differential morphologies of AkdNMCPy in the absence and presence of CuII. AkdNMCPy self-assembles to form nanofibers. One-off addition of CuII produced 2D nanosheets (inset: SAED pattern of the sheets), while serial addition formed sheets through intermediate micelle-like core–shell architectures. (B and C) CV and EPR spectra of AkdNMCPy and CuII, and their complex at various time intervals. | ||
The collective experimental evidence confirmed the binding of AkdNMCPy with CuII followed by distinct assembly pathways to form nanosheets or micelle-like core–shell architectures which subsequently transformed into thermodynamically stable nanosheets. Next, the binding stoichiometry between AkdNMCPy and CuII, and the redox behavior of bound Cu ions were studied by liquid chromatography mass spectrometry (LCMS), cyclic voltammetry (CV) and electron paramagnetic resonance (EPR) experiments. Upon addition of CuII, LCMS spectra showed the gradual disappearance of the AkdNMCPy peak at retention time (tR) = 7.9 min and the concomitant appearance of a new peak at tR = 14.8 min (Fig. 2E and Table S2†), which confirmed the equimolar stoichiometric complexation between AkdNMCPy and CuII. The CV measurements of CuII solutions after mixing with AkdNMCPy showed a sharp decrease in cathodic (−15 μA at −0.18 V) and anodic (38 μA at 0.13 V) peak current intensities with time. At 48 h incubation with AkdNMCPy, the cathodic and anodic peaks of CuII completely disappeared inferring the complexation-induced decrease of free CuII in the presence of AkdNMCPy (Fig. 3B). We performed low-temperature EPR spectroscopy measurements to investigate the CuII coordination environment and oxidation state in the presence of AkdNMCPy.26 The EPR data showed diminishing of the CuII signal at 3100 gauss (G) in the presence of AkdNMCPy which is attributed to the reduction of CuII to diamagnetic CuI (Fig. 3C). Thus, we postulate that complexation of CuII by AkdNMCPy initially forms micelle-like architectures, which gradually reorganizes to nanosheets guided by the in situ reduction of CuII to CuI.
Next, we demonstrated the use of distinct architectures of AkdNMCPy as catalytic systems in performing chemical transformations specific to CuII and CuI. The catalytic utility of micelle-like architectures of AkdNMCPy + CuII was investigated in a tandem reaction involving oxidative-hydrolysis of 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Fig. 4A).9,27 The micelle-like architectures were prepared by the serial addition of CuII to AkdNMCPy, centrifugation, water washing, and finally dispersed in HEPES buffer (25 mM, pH 8) (Fig. S4†). The oxidative-hydrolysis of DCFH-DA by AkdNMCPy + CuII was monitored by recording the absorbance of 2′,7′-dichlorofluorescein (DCF) at λmax = 504 nm. The conversion of DCFH-DA into DCF was observed with an initial rate of 8.7 nM min−1 (Fig. 4B and S14†). The calculated KM and catalytic efficiency (kcat/KM) were 0.5 mM and 120 M−1 min−1, respectively, which are in agreement with reported peptide-based cascade reactions (Table S3†).9,27,28 The control reactions revealed that CuII, His, AkdNMCPy, AkdNMC, Aβ14-23, Aβ14-23Py and HkdPy were catalytically inefficient under similar assay conditions (Fig. 4B).
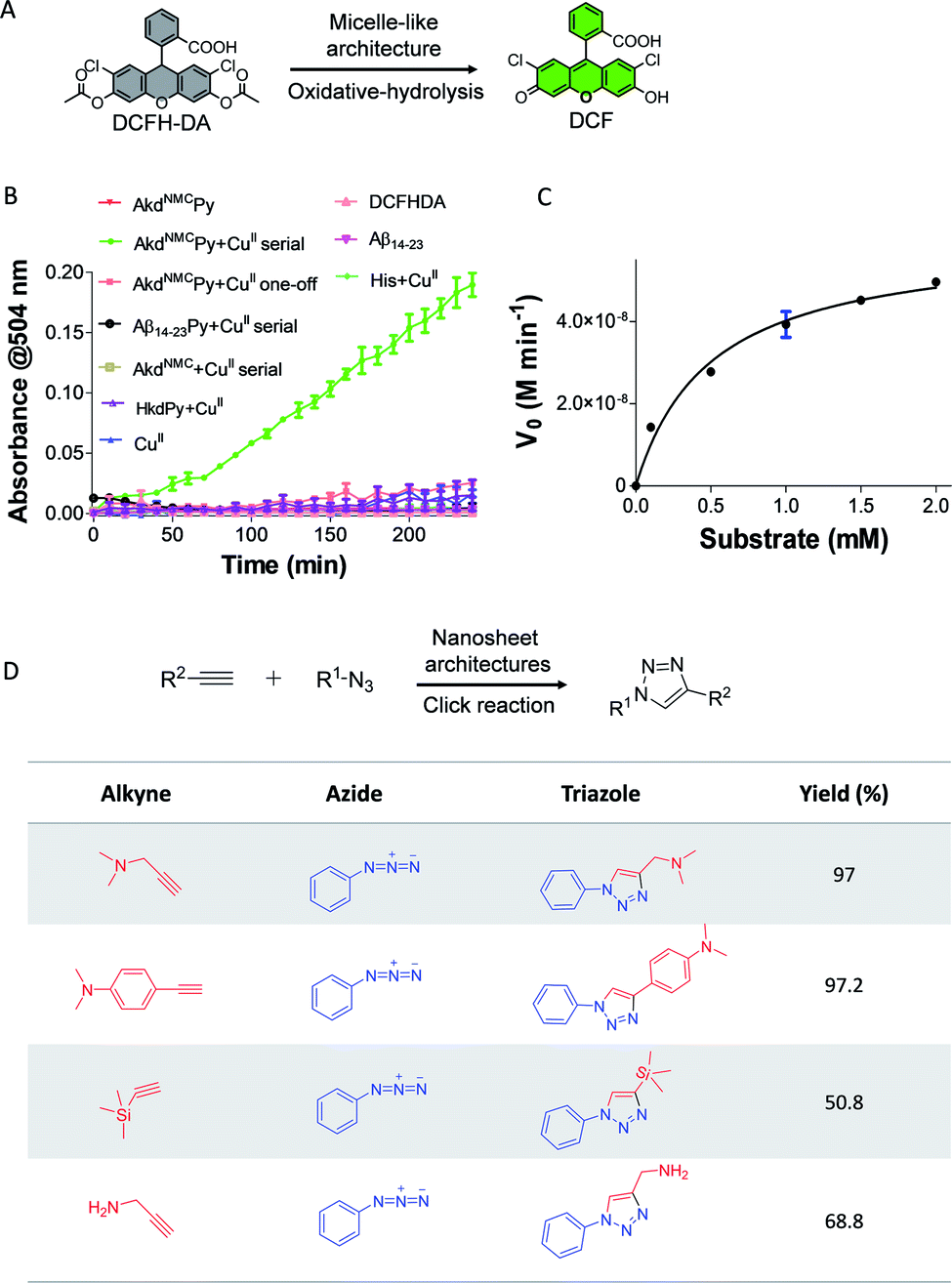 | ||
| Fig. 4 (A) Micelle-like architectures of catalyzed conversion of DCFH-DA to DCF in HEPES (25 mM, pH 8). (B) Absorbance vs. reaction time plots for oxidative-hydrolysis of DCFH-DA catalyzed by micelle-like architectures and other control systems. (C) The plot of initial rate (V0) vs. different substrate (DCFH-DA) concentrations. (D) Alkyne–azide click reaction catalyzed by nanosheets. Substrates with variable substituents and product yields calculated from LC-MS. | ||
Finally, we evaluated the activity of nanosheets of AkdNMCPy in the CuI-catalyzed alkyne–azide cycloaddition (CuAAC) reaction to form triazole products.29 The nanosheets were found to efficiently catalyze the reaction with varied combinations of azide and alkyne to yield 1,4-cycloaddition products as shown in Fig. 4D. The triazole adducts were quantitatively analyzed and characterized by LCMS (Table S4†). The product started forming within 10 min and reached completion by 60 min, as was monitored by NMR spectroscopy (Fig. 4D and S16†). Our study further revealed that nanosheet architectures of AkdNMCPy + CuI catalyze CuAAC reactions with a considerably faster rate and excellent yields compared to a standard CuSO4 + sodium ascorbate catalyst system (Fig. S15 and S17†). The detailed results from various spectroscopy techniques, oxidative-hydrolysis of DCFH-DA and CuAAC reactions confirmed the stabilization of the CuII state in micelle-like architectures and in situ reductions of CuII to CuI in nanosheets of AkdNMCPy.
We postulate that the complexation of CuII with AkdNMCPy initially forms micelle-like architectures, which gradually reorganize into nanosheets guided by the in situ reduction of the CuII to the CuI state. In one-off addition, the assembly of AkdNMCPy guided by the immediate complexation and reduction of CuII to CuI produces thermodynamically stable nanosheet architectures. On the other hand, serial addition led to the controlled complexation of AkdNMCPy to CuII resulting in step-by-step or cascade assembly processes, which forms meta-stable (kinetically controlled) micelle-like core–shell architectures. These meta-stable architectures finally transformed into thermodynamically stable nanosheets upon complete complexation and reduction of the CuII to the CuI state. The observed differential stabilization of copper oxidation states through the generation of distinct architectural forms allowed us to exploit their functional activity towards CuII and CuI catalysed oxidative-hydrolytic tandem, and alkyne–azide click reactions, respectively.
Conclusions
In summary, we demonstrated CDP and Cu-directed molecular architectonics of AkdNMCPy to form functional material architectures that differentially modulate Cu-oxidation states and catalytic activities. The fibrillar assembly of AkdNMCPy was engineered into thermodynamically stable nanosheets or kinetically controlled micelle-like architectures by one-off or serial additions of CuII to AkdNMCPy. The experimental studies showed that micelle-like architectures and nanosheets of AkdNMCPy differentially stabilized CuII and CuI states, which were efficient catalytic systems in tandem oxidation-hydrolysis and click reactions, respectively. This work is anticipated to inspire the design of artificial biocatalytic architectures capable of performing multiple chemical transformations. Further, the metal-complexation-guided distinct functional architectures that stabilize different oxidation states could help in understanding the complex redox activities of metalloproteins.Author contributions
D. G. and T. G. designed the project. D. G. synthesized the compounds and undertook the photophysical studies under the supervision of T. G. and D. G. M. K. and T. G. planned the experiments and analyzed the data. T. M. performed docking studies. D. G. and T. G. wrote the manuscript with input from all the authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Authors thank JNCASR, SwarnaJayanti Fellowship grant (DST/SJF/CSA-02/2015-2016), CEFIPRA grant (IFC/A/62T10-3/2020/963), DST, SERB grant (CRG/2020/004594), and DBT, India, for financial support, and Mohd Monis Ayyub for CV study, Bappaditya Roy for discussion.Notes and references
- M. Avinash and T. Govindaraju, Acc. Chem. Res., 2018, 51, 414–426 CrossRef CAS PubMed.
- H. Moorthy, L. P. Datta and T. Govindaraju, Chem.–Asian J., 2021, 16, 423–442 CrossRef CAS PubMed.
- K. Ariga, M. Nishikawa, T. Mori, J. Takeya, L. K. Shrestha and J. P. Hill, Sci. Technol. Adv. Mater., 2019, 20, 51–95 CrossRef CAS PubMed.
- B. Roy and T. Govindaraju, Bull. Chem. Soc. Jpn., 2019, 92, 1883–1901 CrossRef CAS.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. Bernardes, E. Gazit and T. P. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- K. Ariga and L. K. Shrestha, APL Mater., 2019, 7, 120903–120912 CrossRef.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- T. P. Knowles and M. J. Buehler, Nat. Nanotechnol., 2011, 6, 469–479 CrossRef CAS PubMed.
- Z. Lengyel, C. M. Rufo, Y. S. Moroz, O. V. Makhlynets and I. V. Korendovych, ACS Catal., 2018, 8, 59–62 CrossRef CAS PubMed.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- B. Roy, S. Pal and T. Govindaraju, ACS Appl. Mater. Interfaces, 2020, 12, 14057–14063 CrossRef CAS PubMed.
- C. Wang, J. Fei, K. Wang and J. Li, Angew. Chem., Int. Ed., 2020, 59, 18960–18963 CrossRef CAS PubMed.
- M. Díaz-Caballero, S. Navarro, M. Nuez-Martínez, F. Peccati, L. Rodríguez-Santiago, M. Sodupe, M. Teixidor and S. Ventura, ACS Catal., 2020, 11, 595–607 CrossRef.
- C. Balachandra, D. Padhi and T. Govindaraju, ChemMedChem, 2021, 16, 1–31 CrossRef PubMed.
- S. Manchineella and T. Govindaraju, ChemPlusChem, 2017, 82, 88–106 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, A. A. Orr, P. Makam, B. Redko, E. Haimov, Y. Wang, L. J. W. Shimon, S. R. Lazar, M. Ju, P. Tamamis, H. Dong and E. Gazit, et al. , Angew. Chem., Int. Ed., 2021, 60, 17164–17169 CrossRef CAS PubMed.
- P. Faller, C. Hureau and C. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- C. Madhu, C. Voshavar, K. Rajasekhar and T. Govindaraju, Org. Biomol. Chem., 2017, 15, 3170–3174 RSC.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- M. Avinash and T. Govindaraju, Nanoscale, 2012, 4, 6102–6117 RSC.
- I. Insua and J. Montenegro, J. Am. Chem. Soc., 2019, 142, 300–307 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- T. Mondal and B. Mandal, Chem. Commun., 2020, 56, 2348–2351 RSC.
- N. Giri and S. L. James, Chem. Commun., 2011, 47, 245–247 RSC.
- M. Pandeeswar, H. Khare, S. Ramakumar and T. Govindaraju, Chem. Commun., 2015, 51, 8315–8318 RSC.
- O. V. Makhlynets, P. M. Gosavi and I. V. Korendovych, Angew. Chem., Int. Ed., 2016, 55, 9017–9020 CrossRef CAS PubMed.
- M. Filice and J. M. Palomo, ACS Catal., 2014, 4, 1588–1598 CrossRef CAS.
- M. P. Friedmann, V. Torbeev, V. Zelenay, A. Sobol, J. Greenwald and R. Riek, PLoS One, 2015, 10, e0143948 CrossRef PubMed.
- Z. Du, D. Yu, X. Du, P. Scott, J. Ren and X. Qu, Chem. Sci., 2019, 10, 10343–10350 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2na00161f |
| This journal is © The Royal Society of Chemistry 2022 |