
Development of 2-deoxystreptamine–nucleobase conjugates for the inhibition of oncogenic miRNA production†
Thi Phuong Anh
Tran‡
,
Sylvain
Poulet
,
Mélanie
Pernak
,
Anita
Rayar
,
Stéphane
Azoulay
 ,
Audrey
Di Giorgio
and
Maria
Duca
*
,
Audrey
Di Giorgio
and
Maria
Duca
*
Université Côte d'Azur, CNRS, Institut de Chimie de Nice (ICN), Nice, France. E-mail: maria.duca@univ-cotedazur.fr
First published on 16th December 2021
Abstract
The discovery of new original scaffolds for selective RNA targeting is one of the main challenges of current medicinal chemistry since therapeutically relevant RNAs represent potential targets for a number of pathologies. Recent efforts have been devoted to the search for RNA ligands targeting the biogenesis of oncogenic miRNAs whose overexpression has been directly linked to the development of various cancers. In this work, we developed a new series of RNA ligands for the targeting of oncogenic miRNA biogenesis based on the 2-deoxystreptamine scaffold. The latter is part of the aminoglycoside neomycin and is known to play an essential role in the RNA interaction of this class of RNA binders. 2-deoxystreptamine was thus conjugated to natural and artificial nucleobases to obtain new binders of the oncogenic miR-372 precursor (pre-miR-372). We identified some conjugates exhibiting a similar biological activity to previously synthesized neomycin analogs and studied their mode of binding with the target pre-miR-372.
Introduction
Targeting RNA using specific small molecules represents one of the great challenges of current medicinal chemistry and the first clinical successes of this paradigm are already on the market.1,2 First of all, a number of antibiotics targeting prokaryotic ribosomal RNA have been employed in therapy for many decades.3 Furthermore, a recent breakthrough has been made with the commercialization in 2020 of risdiplam as a modifier of RNA splicing in spinal muscular atrophy (SMA).4 This compound acts upon binding to the SMN2 mRNA, inducing the correction of splicing errors that cause the deadly pathology, and represents a major progress for treatment. Various therapeutically relevant RNAs are currently under study as innovative and original targets, such as viral RNAs and oncogenic RNAs.5 In the last class, microRNAs (miRNAs or miRs) are of particular interest as major regulators of gene expression. MicroRNAs are short non-coding RNAs that recognize mRNAs and control protein expression upon inducing their degradation.6 Each miRNA is responsible for the regulation of hundreds of proteins, thus being part of a massive regulation process inside cells. Despite being essential, miRNAs can be deregulated (overexpressed or underexpressed) and variations in the expression of miRNAs have been directly correlated with various pathologies such as cancer.7 For this reason, miRNAs have been studied as potential therapeutic targets.8Various strategies have been developed in particular to tackle the overexpression of oncogenic miRNAs. Oligonucleotides can directly inhibit the function of oncogenic miRNAs upon complementary hybridization.9 This specific approach is particularly successful, but still bears some limitations regarding its clinical application.10 Small molecules were also developed to inhibit the biogenesis of oncogenic miRNAs upon binding to oncogenic miRNA precursors such as primary miRNAs (pri-miRNAs) and pre-miRNAs.11 This approach is based on the search for RNA binders selective for a particular secondary and/or tertiary structure present in the precursors of the targeted miRNA which are called pre-miRNAs and pri-miRNAs and bear structures suitable for small-molecule binding. Among the most successful examples of this strategy, compounds from the group of Disney designed using the Inforna approach were demonstrated to be very specific inhibitors of miRNA biogenesis.12–15 Screening of compound libraries containing a collection of RNA binders also gave interesting results.16–20 Our group contributed to the field with the design of multimodal RNA ligands synthesized upon conjugation of various RNA binding domains bringing both affinity and selectivity for the target.21–24 More specifically, we built conjugates, whose general structure is illustrated in Fig. 1A, between aminoglycosides (such as neomycin) that are strong but non-selective RNA ligands and natural and artificial nucleobases that are heteroaromatic compounds able to selectively recognize RNA bases and RNA base pairs, respectively.25
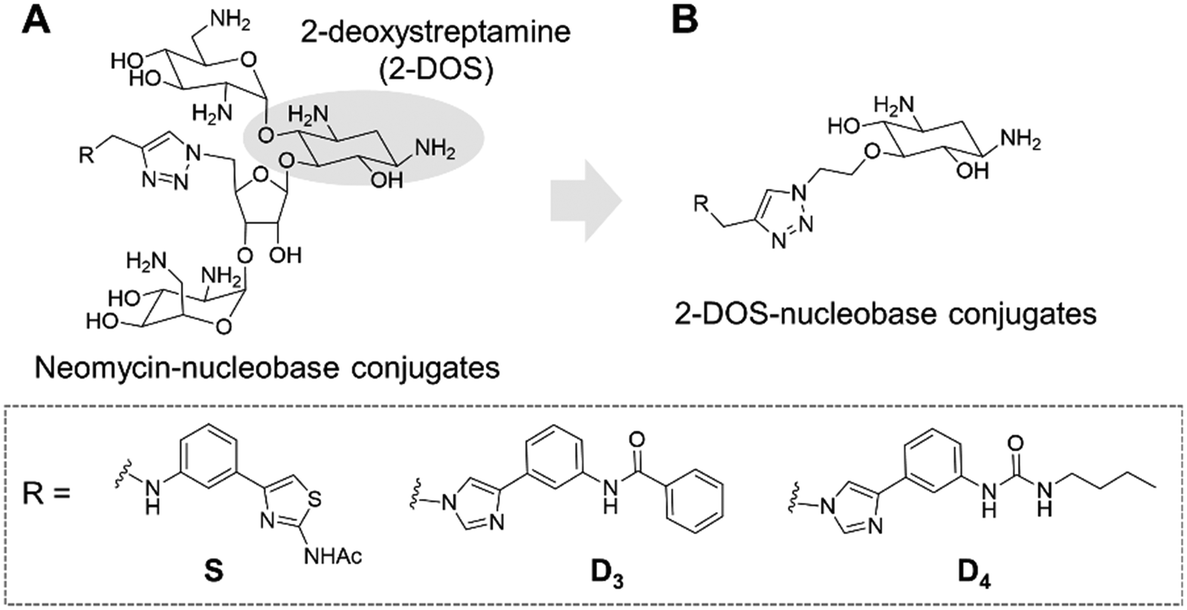 | ||
| Fig. 1 General structure of previously synthesized neomycin–nucleobase conjugates (A) and of the newly designed 2-DOS–nucleobase conjugates (B). The detailed structures for nucleobases S, D3 and D4 are illustrated in the dashed square. | ||
This led us to the identification of RNA ligands inducing the inhibition of the production of an oncogenic miRNA, miR-372, in vitro and in cancer cells overexpressing this miRNA. Some of the synthesized compounds were able to affect the biogenesis of a small set of miRNAs sharing common precursors or common binding sites with pre-miR-372 and, importantly, sharing the same protein targets. The main issue with these aminoglycoside conjugates is the size and the hydrophilicity of the obtained compounds. Both these features limit the perspectives for clinical applications because the physico-chemical properties are not drug-like and because they probably contribute to the lack of specificity. An analysis of aminoglycoside structures reveals that the vast majority contains the meso-1,3-diaminocyclitol 2-deoxystreptamine (2-DOS, Fig. 1A), glycosylated at the 4-position, as well as the 5- (neomycin class) or 6- (kanamycin–gentamicin class) position with a variety of aminosaccharides.26,27 In this work, we thus decided to replace the neomycin moiety with 2-deoxystreptamine, which is part of neomycin and is known to play an important role in the RNA binding ability of aminoglycosides. 2-DOS has been reported to bind weakly to two base units within a disrupted RNA helix. Solution studies carried out with 2-DOS have shown that it binds to 5′–3′ two-base steps (including GU, UG, and GG), albeit with a very low affinity (>1 mM).28 For this reason, various conjugates of 2-DOS have been previously reported not only as dimers for prokaryotic ribosomal RNA binding but also for the inhibition of miRNA maturation.29–31 Here, we report the synthesis of a series of 2-DOS conjugate (Fig. 1B) analogs of a previous neomycin-conjugated series in order to study their biological activities and structure–activity relationships in the context of the inhibition of miR-372 biogenesis. We found that some conjugates bear a very good affinity for the pre-miR-372 target and that, although the affinity is lower than that of their neomycin counterpart, the inhibition activity is maintained. After the evaluation of binding affinity, selectivity, inhibition activity and site of interaction, we identified one compound with very promising binding and inhibition properties compared to its neomycin counterpart and bearing much better physico-chemical properties for future development.
Methods
Chemistry
Reagents and solvents were purchased from Merck or Carlo Erba and used without further purification. All reactions that involved air- or moisture-sensitive reagents or intermediates were performed under an argon atmosphere. Flash column chromatography was carried out on silica gel (Merck; SDS 60 Å, 40–63 μM, VWR). Analytical TLC was conducted on pre-coated silica gel plates (60F254; Merck) and compounds were visualized by irradiation (λ = 254 nm) or by staining with the ninhydrin stain. 1H and 13C NMR spectra were recorded on Bruker AC 200, 400 and 500 MHz spectrometers. Chemical shifts are reported in parts per million (ppm, δ) referenced to the residual 1H resonance of the solvent (CDCl3δ 7.26; CD3OD δ 3.31; DMSO-d6δ 2.50; acetone-d6δ 2.05 ppm). Splitting patterns are labeled as follows: s (singlet), d (doublet), t (triplet), m (multiplet), and br (broad). Coupling constants (J) are listed in hertz (Hz). High-resolution mass spectrometry (HRMS) was carried out on an LTQ Orbitrap hybrid mass spectrometer with an electrospray ionization probe (Thermo Fisher Scientific, San Jose, CA) by direct infusion from a pump syringe, to confirm the correct molar mass and high purity of the compounds. HPLC analyses were performed using a Waters ARC UHPLC pump coupled to a Waters 2998 photodiode array detector and Waters Cortex® C18+ column (50 × 4.6 mm, 2.7 μm). Analyses were run at room temperature by using a gradient of CH3CN containing 0.1% TFA (eluent B) in water containing 0.1% TFA (eluent A) at a flow rate of 1.5 mL min−1. The employed gradient was 5 → 40% eluent B over 5 min and 40 → 100% eluent B over 2 min.Synthetic procedures
The preparation of Boc-protected 2-DOS derivatives 1, 2, 3 and 4a–h and the synthesis of modified nucleobases containing an alkyne chain 10a–h are described in the ESI.†2-Deoxystreptamine-4-O-[4-(adenyl-N-methyl)]ethyltriazole (5a). General procedure B was applied to a solution of 4a (100 mg, 0.146 mmol) in CH2Cl2 (3 mL) and water (1 mL) in the presence of TFA (557 μL, 7.32 mmol). Pure compound 5a was obtained after precipitation in CH2Cl2 and Chelex resin as a white solid in 60% yield (55.3 mg). Retention time 0.91 min; 1H NMR (500 MHz, CD3OD) δ ppm: 8.30–8.10 (m, 3H), 5.56 (br s, 2H), 4.64 (s, 2H), 4.50–4.45 (m, 1H), 4.15–4.10 (m, 1H), 3.40–3.32 (m, 3H), 3.30–3.25 (m, 1H), 3.15–3.10 (m, 1H), 2.40–2.30 (m, 1H), 1.55–1.45 (m, 1H); 13C NMR (100 MHz, CD3OD) δ ppm: 163.1, 162.8, 156.7, 143.1, 140.3, 129.9, 126.9, 82.5, 77.2, 74.5, 71.7, 55.8, 51.9, 51.4, 50.8, 29.8; MS (ESI) m/z 405.21063 [M + H]+ (C16H25O3N10 requires 405.21056).
2-Deoxystreptamine-4-O-[4-(uracil-N-methyl)]ethyltriazole (5b). General procedure B was applied to a solution of 4b (27 mg, 0.0408 mmol) in CH2Cl2 (0.6 mL) and water (0.2 mL) in the presence of TFA (149 μL, 2.04 mmol). Pure compound 5b was obtained after precipitation in CH2Cl2 and Chelex resin as a white solid in 98% yield (24.3 mg). Retention time 0.82 min; 1H NMR (500 MHz, CD3OD) δ ppm: 8.06 (s, 1H), 7.72 (d, J = 3.2 Hz, 1H), 5.68 (d, J = 3.2 Hz, 1H), 5.02 (s, 2H), 4.64 (t, J = 2.2 Hz, 2H), 4.50–4.45 (m, 1H), 4.15–4.10 (m, 1H), 3.40–3.35 (m, 4H), 3.20–3.10 (m, 1H), 2.40–2.30 (m, 1H), 1.80–1.70 (m, 1H); 13C NMR (125 MHz, CD3OD) δ ppm: 166.6, 152.7, 147.0, 126.1, 102.7, 82.6, 77.3, 74.6, 71.9, 51.7, 51.4, 50.8, 44.2, 29.9; MS (ESI) m/z 382.18375 [M + H]+ (C15H24O5N7 requires 382.18334).
2-Deoxystreptamine-4-O-[4-(cytosyl-N-methyl)]ethyltriazole (5c). General procedure B was applied to a solution of 4c (25.5 mg, 0.0385 mmol) in CH2Cl2 (0.6 mL) and water (0.3 mL) in the presence of TFA (149 μL, 1.93 mmol). Pure compound 5c was obtained after precipitation in CH2Cl2 and Chelex resin as a white solid in 70% yield (10.2 mg). Retention time 0.76 min; 1H NMR (500 MHz, CD3OD) δ ppm: 8.17 (s, 1H), 8.02 (d, J = 3.1 Hz, 1H), 6.11 (br s, 1H), 5.13 (s, 2H), 4.67 (t, J = 2.2 Hz, 2H), 4.50–4.40 (m, 1H), 4.20–4.10 (m, 1H), 3.50–3.35 (m, 4H), 3.20–3.10 (m, 1H), 2.45–2.40 (m, 1H), 1.90–1.80 (m, 1H); 13C NMR (125 MHz, CD3OD) δ ppm: 162.2, 150.9, 150.6, 150.3, 126.5, 95.1, 82.4, 77.3, 74.5, 71.8, 54.8, 52.2, 51.8, 51.5, 50.9, 45.4, 29.8; MS (ESI) m/z 381.19952 [M + H]+ (C15H25O4N8 requires 381.19933).
2-Deoxystreptamine-4-O-[4-((3-benzamidophenyl)imidazol-N-methyl)]ethyltriazole (5d). General procedure B was applied to a solution of 4d (60 mg, 0.0739 mmol) in CH2Cl2 (1.5 mL) and H2O (0.5 mL) in the presence of TFA (854 μL, 3.69 mmol). Pure compound 5d was obtained after precipitation in CH2Cl2 and Chelex resin as a white solid in 97% yield (55.0 mg). Retention time 2.78 min; 1H NMR (500 MHz, CD3OD) δ ppm: 8.26 (s, 1H), 8.00–7.95 (m, 3H), 7.70–7.50 (m, 8H), 5.60 (s, 2H), 4.70 (t, J = 2.0 Hz, 2H), 4.50–4.45 (m, 1H), 4.20–4.15 (m, 1H), 3.45–3.35 (m, 4H), 3.15–3.10 (m, 1H), 2.45–2.40 (m, 1H), 1.80–1.75 (m, 1H); 13C NMR (125 MHz, CD3OD) δ ppm: 169.1, 141.0, 136.0, 133.2, 131.0, 129.7, 128.7, 123.2, 82.6, 77.2, 74.5, 71.8, 51.8, 51.4, 50.8, 29.9; MS (ESI) m/z 533.26215 (C27H33O4N8 533.26193).
2-Deoxystreptamine-4-O-[4-(phenyl-(3-butylurea)-imidazol-N-methyl)]ethyltriazole (5e). General procedure B was applied to a solution of 4e (49 mg, 0.0692 mmol) in CH2Cl2 (0.5 mL) and H2O (0.2 mL) in the presence of TFA (265 μL, 3.46 mmol). Pure compound 5e was obtained after precipitation in CH2Cl2 and Chelex resin as a white solid in 97% yield (49.7 mg). Retention time 2.77 min; 1H NMR (400 MHz, CD3OD) δ ppm: 9.08 (s, 1H), 8.27 (s, 1H), 8.00–7.90 (m, 2H), 7.39 (t, J = 5.0 Hz, 1H), 7.60–7.45 (m, 2H), 5.61 (s, 2H), 4.75–4.70 (m, 2H), 4.50–4.40 (m, 1H), 4.30–4.20 (m, 1H), 3.40–3.30 (m, 3H), 3.22 (t, J = 9.8 Hz, 2H), 3.40–3.30 (m, 1H), 2.40–2.30 (m, 1H), 1.80–1.70 (m, 1H), 1.60–1.50 (m, 2H), 1.40–1.30 (m, 2H), 0.97 (t, J = 5.0 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ ppm: 158.1, 142.4, 140.2, 138.6, 137.8, 131.0, 121.1, 120.3, 117.0, 108.9, 98.5, 82.6, 77.2, 74.5, 71.8, 66.9, 51.8, 51.4, 50.8, 40.5, 33.3, 21.0, 14.1; MS (ESI) m/z 528.30426 (C25H38O4N9 528.30413).
2-Deoxystreptamine-4-O-[4-((3-(2-naphthamido)phenyl)-imidazol-N-methyl)]ethyltriazole (5f). General procedure B was applied to a solution of 4f (29 mg, 0.0336 mmol) in CH2Cl2 (0.5 mL) and H2O (0.2 mL) in the presence of TFA (129 μL, 1.68 mmol). Pure compound 5f was obtained after precipitation in CH2Cl2 and Chelex resin as a white solid in 87% yield (23.0 mg). Retention time 2.45 min; 1H NMR (400 MHz, CD3OD) δ ppm: 8.54 (s, 1H), 8.25 (s, 1H), 8.20–7.90 (m, 5H), 7.75–7.50 (m, 5H), 5.57 (s, 2H), 4.75–4.70 (m, 2H), 4.50–4.40 (m, 1H), 4.25–4.15 (m, 1H), 3.40–3.30 (m, 4H), 3.20–3.10 (m, 1H), 2.45–2.35 (m, 1H), 1.80–1.70 (m, 1H); 13C NMR (100 MHz, CD3OD) δ ppm: 141.0, 136.6, 134.1, 133.3, 131.0, 130.1, 129.5, 129.3, 128.9, 128.0, 125.1, 122.8, 82.6, 77.3, 74.6, 71.8, 54.8, 51.9, 49.8, 31.3, 17.2.
2-Deoxystreptamine-4-O-[4-((3-aminophenyl)-imidazol-N-methyl)]ethyltriazole (5g). General procedure B was applied to a solution of 4g (30 mg, 0.0372 mmol) in CH2Cl2 (0.5 mL) and H2O (0.2 mL) in the presence of TFA (143 μL, 1.86 mmol). Pure compound 5g was obtained after precipitation in CH2Cl2 and Chelex resin as a yellow solid in 87% yield (28.0 mg). Retention time 1.10 min; 1H NMR (500 MHz, CD3OD) δ ppm: 8.93 (s, 1H), 8.26 (s, 1H), 7.90 (s, 1H), 7.40–7.30 (m, 1H), 7.25–7.15 (m, 2H), 7.00–6.90 (m, 1H), 5.58 (s, 2H), 4.75–4.65 (m, 2H), 4.50–4.40 (m, 1H), 4.25–4.15 (m, 1H), 3.50–3.35 (m, 4H), 3.25–3.15 (m, 1H), 2.50–2.40 (m, 1H), 1.80–1.70 (m, 1H); 13C NMR (125 MHz, CD3OD) δ ppm: 137.3, 131.5, 126.6, 119.9, 119.4, 118.7, 115.4, 82.6, 77.2, 74.5, 71.8, 54.8, 51.8, 51.4, 50.8, 44.8, 29.8; MS (ESI) m/z 429.23608 (C20H29O3N8 429.23571).
2-Deoxystreptamine-4-O-[4-(2-oxo-2-((4-phenylthiazol-2-yl)amino)-N-ethylpropanamide)]ethyltriazole (5h). General procedure B was applied to a solution of 4h (16 mg, 0.033 mmol) in CH2Cl2 (0.5 mL) and water (0.2 mL) in the presence of TFA (129 μL, 1.65 mmol). Final compound 5h was obtained as a white solid in 98% yield (12 mg). Retention time 10.5 min (Phenomenex Synergi 4 μm Fusion-RP 80 Å column (250 × 4.6 mm); flow rate of 1 mL min−1, gradient 5 → 40% eluent B over 20 min); 1H NMR (400 MHz, MeOD) δ (ppm): 8.00–7.85 (m, 3H), 7.40–7.30 (m, 3H), 7.30–7.20 (m, 1H), 4.60–4.50 (m, 2H), 4.50–4.40 (m, 1H), 4.20–4.10 (m, 3H), 3.50–3.35 (m, 4H), 3.20–3.00 (m, 3H), 2.70–2.60 (m, 2H), 2.40–2.30 (m, 1H), 1.80–1.70 (m, 1H); 13C NMR (100 MHz, CD3OD) δ (ppm): 169.6, 159.2, 151.3, 135.8, 129.7, 129.0, 127.1, 108.7, 82.4, 77.3, 74.5, 72.0, 51.8, 51.4, 50.9, 43.4, 35.9, 29.8; MS (ESI) m/z 545.22974 (C24H33O5N8S 545.22891).
Results and discussion
Design of 2-DOS conjugates
The disubstituted 2-DOS-containing aminoglycosides represent a vast majority of this class of antibiotics and are strong RNA binders. The aim of this work is to assess if the 2-DOS moiety could replace neomycin in our previously synthesized pre-miR-372 ligands.23 The latter contained neomycin conjugated with natural and artificial nucleobases and some of them were efficient and selective inhibitors of oncogenic miR-372 maturation in vitro and in cells.23,25 Neomycin is known to bind to unpaired RNA bases at sites where the double helix is distorted by the presence of bulges or loops. Natural nucleobases should interact with unpaired bases via the formation of Watson–Crick–Franklin H-bonds and artificial nucleobases are meant to interact with paired bases via Hoogsteen H-bonds thus forming triplets.25 So, the conjugation of neomycin to natural nucleobases would direct the compounds on single-stranded regions of bulges and loops, and the conjugation of neomycin to artificial nucleobase conjugates should direct the ligands close to stem-bulge and stem-loop junctions. In our previous studies, we observed that artificial nucleobase conjugates were more efficient and selective binders of pre-miR-372 than natural base conjugates and some of them were able to inhibit its processing by the Dicer enzyme in vitro.23 Specific inhibition of proliferation of adenocarcinoma cancer cells (AGS) overexpressing miR-372 was also observed and this activity was linked to the inhibition of Dicer processing for miR-372. A study of the site of interaction of the best binders and inhibitors showed that they indeed were bound at stem-bulge junctions and close to the site of cleavage of the Dicer enzyme.21,24 Even with these promising results in hand, as well as with a complete structure–activity characterization of this series of conjugates,21,22,24 the size of the obtained compounds and their high hydrophilicity represent relevant drawbacks for the physico-chemical properties of this kind of conjugates. Furthermore, neomycin is known to be a toxic drug when administered and to lack specificity when used against a particular RNA. We thus decided to prepare new analogs of these conjugates by replacing the neomycin moiety by its substructure 2-DOS and to study the activity of these compounds against miR-372 maturation that we took as a model for comparison with our previously reported neomycin conjugates. 2-DOS represents a key structural feature of most aminoglycosides and occupies a central position in their structure as well as having an essential role for their interaction with prokaryotic ribosomal RNA. We thus wondered if the small 2-DOS moiety could be sufficient for binding and could lead to more favorable physico-chemical properties in the final conjugates. The most active compound in the neomycin series was the one containing nucleobase S.23 However, the 2-DOS analog of this conjugate was previously reported and did not show activity for miR-372 maturation inhibition.24 As illustrated in Scheme 1, we thus chose to conjugate 2-DOS with the natural nucleobases adenine (A), cytosine (C) and uracil (U) which were not studied previously.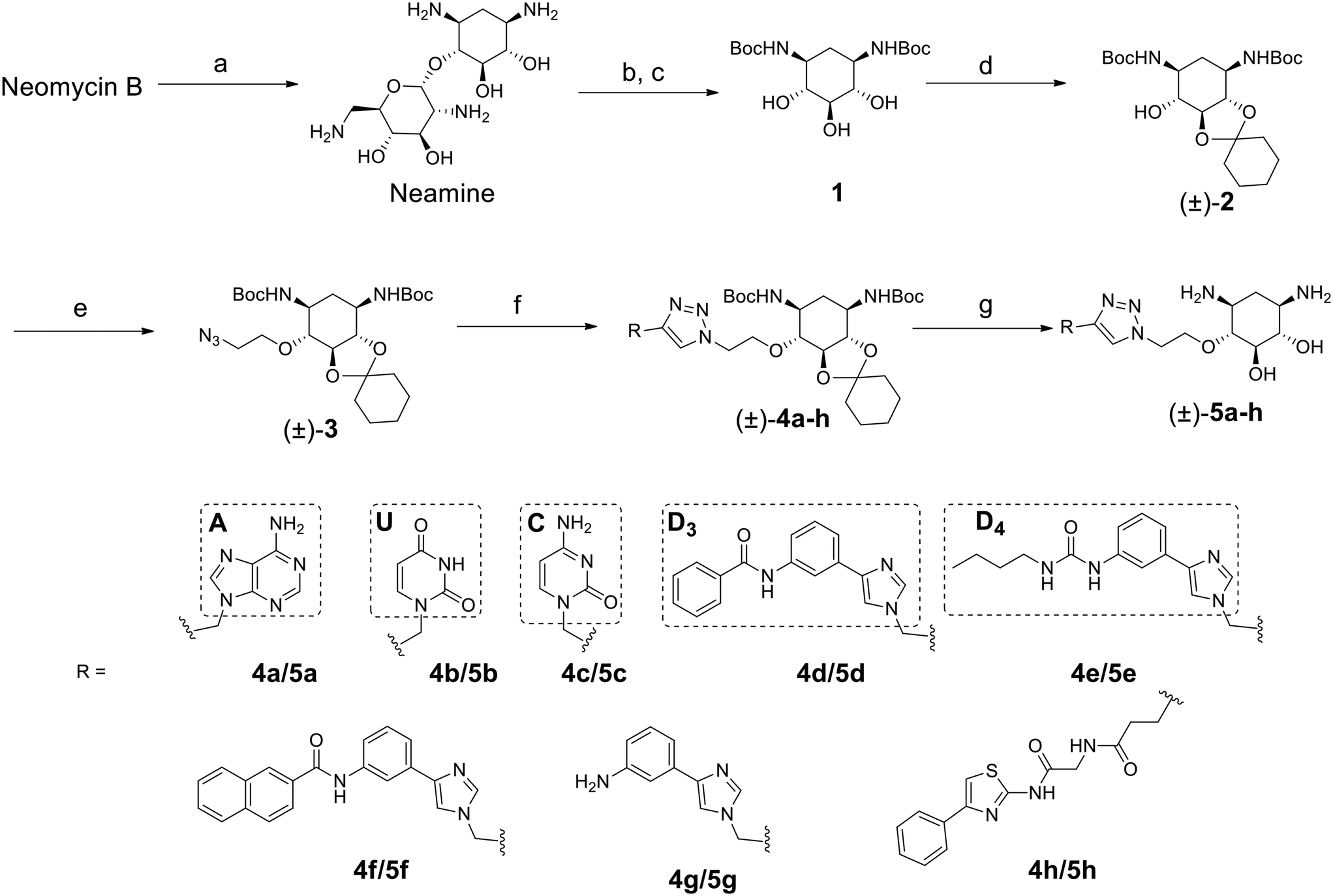 | ||
| Scheme 1 Synthesis of a new series of 2-DOS conjugates 5a–h. Reagents: a) HCl, reflux, overnight; b) 48% HBr aq., reflux, 2 days; c) Boc2O, NaOH, dioxane/H2O, overnight; d) 1,1-dimethoxycyclohexane, pTsOH, DMF, 50 °C, 55 mbar, 1 h; e) 2-azidoethyl-p-toluenesulfonate, NaH, THF, 30 °C, 3 h; f) 10a–h, CuI, DIPEA, CH3CN, from r. t. to 50 °C, from 1 h to overnight; g) TFA, CH2Cl2, H2O, r. t., overnight. | ||
We also chose 4-(3-benzamidophenyl)imidazole (nucleobase D3) which can form two hydrogen bonds with T·A and C·G pairs and 4-(3-butylamidophenyl)imidazole (nucleobase D4) which was found to recognize G·C base pairs with 3 hydrogen bonds (Fig. S1 in the ESI†), used for the previously prepared neomycin analogs. Furthermore, we decided to study two analogs of D3 and D4. To this aim, we used the unsubstituted 3-aminophenylimidazole and 4-(3-naphthylamidophenyl)imidazole in order to assess the influence of the substituent on the phenylimidazole moiety on biological activity. A new analog of the S-conjugate was also synthesized using a phenylthiazole moiety for comparison.24 For the synthesis of these conjugates, we employed 1,3-dipolar cycloaddition leading to the formation of a triazole linker suited for direct comparison with the previously reported compounds while also taking advantage of the straightforward synthesis of these new analogs (Fig. 1B).
Synthesis of 2-DOS conjugates
2-DOS is a meso compound that contains 5 contiguous stereogenic centers that pose a challenge for the preparation of enantiomerically pure conjugates. To date, the most practical method to obtain 2-deoxystreptamine is by the straightforward acidic degradation of neomycin.26 A more complex synthetic route was previously reported for the preparation of enantiomerically pure compounds.32 In this work, we first decided to employ the fastest synthetic pathway leading to the desired conjugates as racemic mixtures.As illustrated in Scheme 1, the 2-DOS moiety was prepared starting from commercially available neomycin upon acidic hydrolysis in the presence of a concentrated solution of HCl. This led, in quantitative yields, to neamine that was subjected to further acidic treatment with aqueous HBr. The protection of the amino groups with the Boc protecting group was then performed on the crude product in the presence of tert-butoxycarbonyl anhydride and NaOH in a H2O/dioxane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture leading to compound 1 in 50% yield over two steps. The next step was the protection of one vicinal diol in the presence of 1,1-dimethoxycyclohexane and pTsOH in DMF which led to the racemic compound (±)-2 in 98% yield. Following up with substitution of the remaining hydroxyl group with 2-azidoethyltosylate led to compound (±)-3 ready for 1,3-dipolar cycloaddition in 33% yield. The latter was performed using the appropriately modified heteroaromatic compounds 10a–h (Fig. S2 in the ESI†) containing the alkyne essential for the coupling reaction. The cycloaddition reaction was performed in CH3CN in the presence of CuI and DIPEA and led to the desired compounds (±)-4a–h in 46 to 87% yields. The final removal of the acetal and Boc protecting groups in CH2Cl2 in the presence of H2O and TFA led to the desired compounds (±)-5a–h in 60 to 97% yields as TFA salts. Complete removal of copper traces possibly remaining in the final product was performed upon treatment with Chelex resin. All synthesized compounds were also fully characterized by NMR, HRMS and HPLC for purity and the data are shown in the ESI.† The purity of the final products was found to be between 95% and 99%.
:1 mixture leading to compound 1 in 50% yield over two steps. The next step was the protection of one vicinal diol in the presence of 1,1-dimethoxycyclohexane and pTsOH in DMF which led to the racemic compound (±)-2 in 98% yield. Following up with substitution of the remaining hydroxyl group with 2-azidoethyltosylate led to compound (±)-3 ready for 1,3-dipolar cycloaddition in 33% yield. The latter was performed using the appropriately modified heteroaromatic compounds 10a–h (Fig. S2 in the ESI†) containing the alkyne essential for the coupling reaction. The cycloaddition reaction was performed in CH3CN in the presence of CuI and DIPEA and led to the desired compounds (±)-4a–h in 46 to 87% yields. The final removal of the acetal and Boc protecting groups in CH2Cl2 in the presence of H2O and TFA led to the desired compounds (±)-5a–h in 60 to 97% yields as TFA salts. Complete removal of copper traces possibly remaining in the final product was performed upon treatment with Chelex resin. All synthesized compounds were also fully characterized by NMR, HRMS and HPLC for purity and the data are shown in the ESI.† The purity of the final products was found to be between 95% and 99%.
Evaluation of the affinity, selectivity and inhibition activity
Once the new series of 2-DOS conjugates 5a–h were prepared, we evaluated these compounds for their ability to bind to pre-miR-372 and inhibit its processing by the Dicer enzyme in vitro and compared the results to the corresponding neomycin analogs when available. First of all, we studied the dissociation constants (KD) of all compounds for pre-miR-372 using a fluorescence-based assay where the target RNA is labeled with a fluorophore (FAM) at the 5′-end. Binding of an efficient ligand induces a change in the fluorophore environment which quenches fluorescence and plotting the variation of the fluorescence signal as a function of the concentration of the ligand allows for the measurement of KD.33 As illustrated in Table 1, all newly synthesized compounds 5a–h are good pre-miR-372 ligands with KD values in the low μM range (from 1.62 to 17.1 μM) except for 5c that bears a higher KD of 101 μM. It should be noted that 2-DOS and the nucleobases alone have no affinity for the target. A closer look at the obtained KD suggests that the D3 and D4 analogs (5d and 5e, respectively) are the strongest binders closely followed by the unsubstituted phenylimidazole analog 5g. Introduction of a naphthalene substituent as in 5f and of the phenylthiazole substituent as in 5h induces a slight loss of affinity while the natural nucleobase conjugates 5a–c are the weakest binders.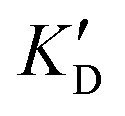 ) or DNA (100 eq.
) or DNA (100 eq. 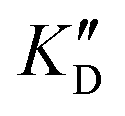 )
)
| ID | K D |
|
|
|
|
IC50 |
|
|---|---|---|---|---|---|---|---|
| 5a | 11.1 ± 2.3 | >1 mM | — | 47.1 | 4.2 | No inhibition | No inhibition |
| 5b | 17.1 ± 0.1 | >1 mM | — | >1 mM | — | No inhibition | No inhibition |
| 5c | 101 ± 18 | >1 mM | — | >1 mM | — | No inhibition | No inhibition |
| 5d | 2.52 ± 0.58 | 2.64 ± 0.52 | 1.0 | 2.96 ± 0.85 | 1.2 | 15.9 ± 1.8 | 29.5 ± 9.1 |
| 5e | 1.62 ± 0.49 | 3.65 ± 0.85 | 2.2 | 2.11 ± 0.81 | 1.3 | 72.4 ± 1.0 | No inhibition |
| 5f | 6.78 ± 0.46 | 8.51 ± 1.3 | 1.3 | 9.02 ± 1.2 | 1.3 | >100 μM | No inhibition |
| 5g | 3.53 ± 0.89 | 3.92 ± 0.52 | 1.1 | 4.27 ± 0.35 | 1.2 | 81.1 ± 2.3 | >500 μM |
| 5h | 5.13 ± 0.41 | 8.56 ± 2.0 | 1.7 | 11.0 ± 0.90 | 2.1 | 74.9 ± 12 | No inhibition |
| Neo-D 3 | 0.0189 ± 0.0017 | 0.0208 ± 0.0019 | 1.1 | 0.0244 ± 0.0018 | 1.3 | 8.30 ± 1.1 | 20.4 |
| Neo-D 4 | 0.0461 ± 0.0038 | 0.0164 ± 0.0012 | 1.1 | 0.0222 ± 0.0024 | 1.5 | 25.6 ± 1.1 | |
| 2-DOS | >1 mM | — | — | — | — | No inhibition | No inhibition |
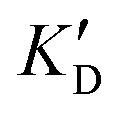
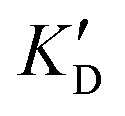
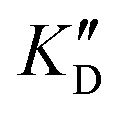
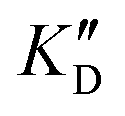
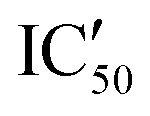
Selectivity is of course another important parameter to evaluate. To this aim, we chose to measure KD in the presence of two different competitors, tRNA and DNA, which are particularly abundant in cells. In both cases, 100 equivalents of the competitor were added to the mixture during KD measurement. An increase in KD (ratio 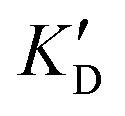 /KD for tRNA or
/KD for tRNA or 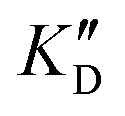 /KD for DNA higher than one) suggests a lack of selectivity since the compound can bind to other nucleic acid structures. The obtained results are illustrated in Table 1 and suggest that the natural nucleobase conjugates 5a–c were not selective in the presence of tRNA and DNA except for 5a that maintained some affinity in the presence of DNA (
/KD for DNA higher than one) suggests a lack of selectivity since the compound can bind to other nucleic acid structures. The obtained results are illustrated in Table 1 and suggest that the natural nucleobase conjugates 5a–c were not selective in the presence of tRNA and DNA except for 5a that maintained some affinity in the presence of DNA (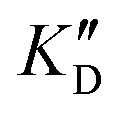 /KD = 4.2). Compounds 5d–5h maintained their affinity and seemed thus to be selective for a pre-miRNA structure such as pre-miR-372, compared to the other nucleic acid structures.
/KD = 4.2). Compounds 5d–5h maintained their affinity and seemed thus to be selective for a pre-miRNA structure such as pre-miR-372, compared to the other nucleic acid structures.
With these experiments, we assessed that all compounds were promising binders and that compounds 5d–5h were also selective for pre-miRNAs. We thus decided to verify if the compounds were also able to inhibit Dicer processing of pre-miR-372 by measuring the IC50 using a previously published cell-free assay where pre-miR-372 was double labeled with a fluorophore (FAM) and a quencher (DAB) at the 5′-end and at the 3′-end, respectively.23 When Dicer cleaves the pre-miRNA appropriately, the fluorescence signal increases, while when Dicer cleavage is inhibited, no fluorescence is observed. As shown by the IC50 values reported in Table 1, compounds 5a–5c were not able to induce inhibition and compounds 5e–5h had a weak inhibition activity starting at 72.4 μM for 5e to more than 100 μM for 5f. It is noteworthy that 5d was the most active compound with an IC50 of 15.9 μM. This value is in the same range as that of compound Neo-D3 with the latter bearing a much higher affinity for the target. This could be explained by a difference in the binding site that could be more favorable for inhibition in the case of compound 5d than that for compound Neo-D3 despite a loss in binding affinity.
Selectivity remains a major concern when designing new RNA ligands. We thus evaluated IC50 values in the presence of HEK293 cell lysates instead of using the recombinant enzyme. The conditions mimic the intracellular complexity and give hints about the potential selectivity in cells. With these experiments, we demonstrated that all compounds lost their ability to inhibit miR-372 maturation except for compound 5d whose 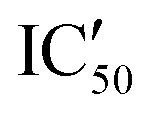 is 29.5 μM instead of 15.9 μM in the presence of pure enzymes.
is 29.5 μM instead of 15.9 μM in the presence of pure enzymes.
Despite the binding selectivity for all compounds against tRNA and DNA as well as the inhibition selectivity in the case of compound 5d, one could wonder what would be the affinity and the inhibition activity of these compounds against other pre-miRNAs. We thus measured KD and IC50 of the newly synthesized compounds for three supplementary pre-miRNAs: pre-miR-21, pre-miR-18a and pre-miR-148a. These three miRNAs are oncogenic and overexpressed in many types of cancers.34 Although their sequences and structures differ from those of pre-miR-372, the general shape of the pre-miRs remains similar, i.e. hairpin stem-loops with bulges. The obtained results (Table S1 in the ESI†) show that compounds 5a–c, the weakest binders of pre-miR-372, remain weak binders or lose affinity for the other miRNAs. Furthermore, 5a–c did not show any ability to inhibit Dicer cleavage. Compounds 5d–h instead maintain a similar affinity for all pre-miRNAs, but show major differences in inhibition activity. Indeed, compound 5d inhibits weakly pre-miR-21 maturation and is not active on pre-miR-18. Inhibition activity is maintained for pre-miR-148a but is lower than that for for pre-miR-372. The weak inhibitors of pre-miR-372 maturation 5e–h remain weak for the other tested miRNAs except for 5e that inhibits preferentially pre-miR-18 (IC50 20.3 μM) and pre-miR-148a (IC50 18.2 μM) cleavage. Compound 5d thus seems to be the most promising of the series for the inhibition of pre-miR-372 processing with an IC50 in the low μM range similar to one of its neomycin counterparts despite having a much higher KD value.
As mentioned above, compounds 5a–h form racemic mixtures. We thus wanted to verify if the enantiopure analog of the most promising compound of the series 5d showed any difference in activity, affinity and selectivity. The synthesis of this compound is similar to that for 5d but starts from the enantiopure compound 6 prepared following a previously reported procedure (Scheme 2).32
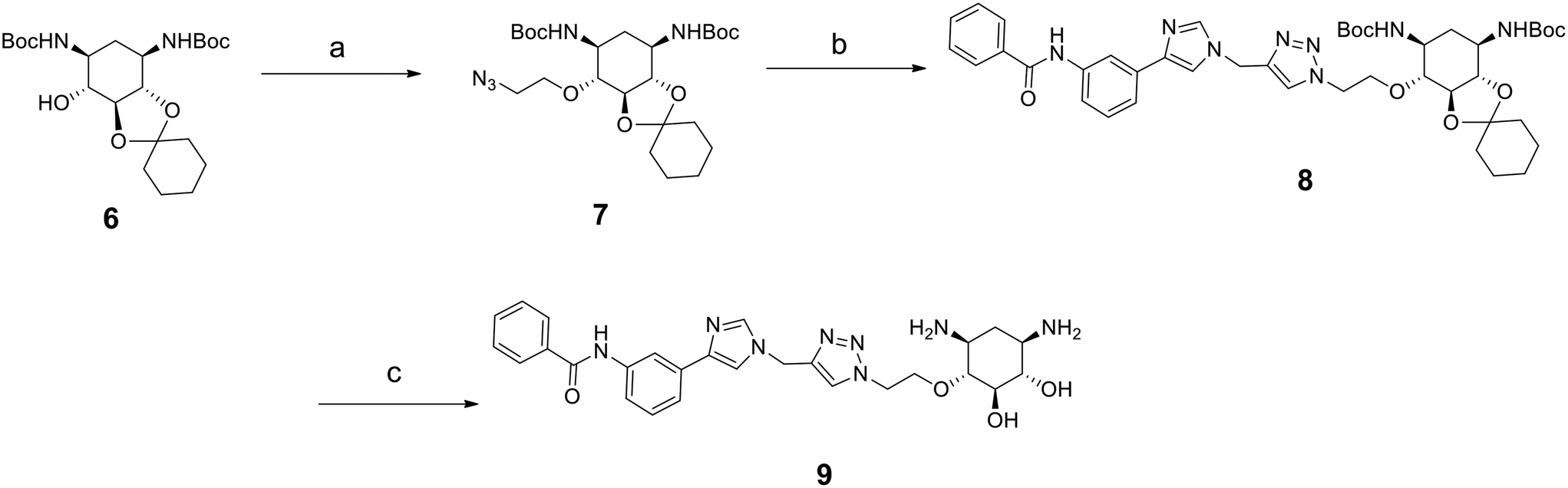 | ||
| Scheme 2 Synthesis of enantiopure compound 9. Reagents: a) 2-azidoethyl-p-toluenesulfonate, NaH, THF, 30 °C, 3 h; b) 10h, CuI, DIPEA, CH3CN, r. t., 5 h; c) TFA, CH2Cl2, H2O, r. t., overnight. | ||
The introduction of the 2-azidoethyl linker led to compound 7 in 20% yield and the 1,3-dipolar cycloaddition in the presence of 4-(3-benzamidophenyl)imidazole led to compound 8 in 56% yield. Final deprotection under acidic conditions led to the desired derivative 9 in 98% yield. The comparison of the affinity for pre-miR-372 between 9 and 5d revealed that compound 9 was a stronger binder with a KD of 0.627 μM. This improved affinity was then reflected in a better inhibition activity since 9 showed an IC50 of 12.6 μM. This enantiomer thus seems more favorable for binding and inhibition.
Altogether, these results show that there is a good correlation between KD and IC50 suggesting that RNA binding is the underlying mechanism of inhibition. Furthermore, the result obtained with compound 9 is particularly interesting since comparison of IC50 values between 9 and compound Neo-D3 shows that these are within the same range (8.3 μM vs. 12.6 μM, Fig. 2). Neomycin could thus be replaced by 2-DOS without losing activity.
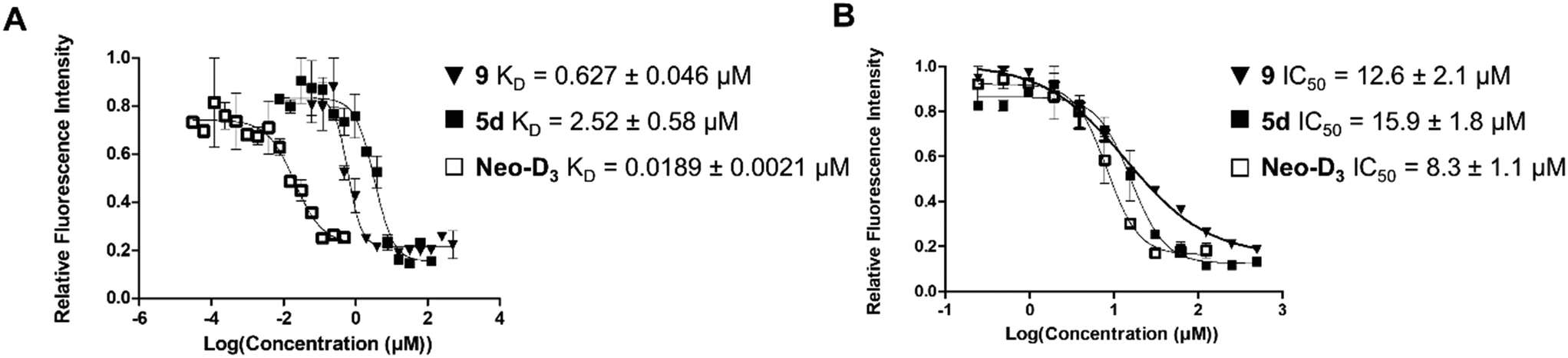 | ||
| Fig. 2 Comparison of the dissociation constants (KD, A) and of inhibition activities (IC50, B) of compounds 9, 5d and Neo-D3 against pre-miR-372. | ||
The explanation for the good inhibition activity of compound 9 despite having a higher KD value could come from a more favorable binding site for pre-miR-372 compared to the one for Neo-D3. This parameter could also explain the differences observed in the inhibition activities of other pre-miRNAs while keeping the affinity for them. Indeed, binding to a particular RNA is not sufficient to induce an inhibition activity since it is essential to bind to a functional site that in this case would be preferentially the cleavage site of the Dicer enzyme.
Molecular docking of compound 9 on pre-miR-372
To gain a better insight into the mode of binding of 9 and suggest an explanation for the good inhibition activity of this compound, similar to the one of the Neo-D3 analog despite having a much higher KD, we performed a molecular docking study of both compounds on the pre-miR-372 sequence. The sequence of the pre-miR-372 hairpin loop was generated from the miRBase database and then input to the MC-fold/MC-sym pipeline to construct a 3D model. For this model, we analyzed energy optimization using TINKER. Next, we conducted molecular docking for pre-miR-372 in the presence of compound 9 using the AutoDock program without defining any preferred binding site while performing the docking study on the entire sequence.35 As shown in Fig. 3, docking suggested that both compounds 9 and Neo-D3 bind to the cleavage site of Dicer even if differently. Compound Neo-D3 seems to interact with residues A45 and A46 corresponding to the cleavage site on one strand of the pre-miR-372 stem, while 9 has U31 and G32 corresponding to the cleavage site on the opposite strand. Regarding the other interactions, Neo-D3 interacts with other residues of the stem region (G20 to C25) while 9 interactswith residues of the loop region (G35-U36 and A40-G43). Both these binding sites involving the cleavage site of the enzyme, the compounds behave similarly in terms of inhibition activity.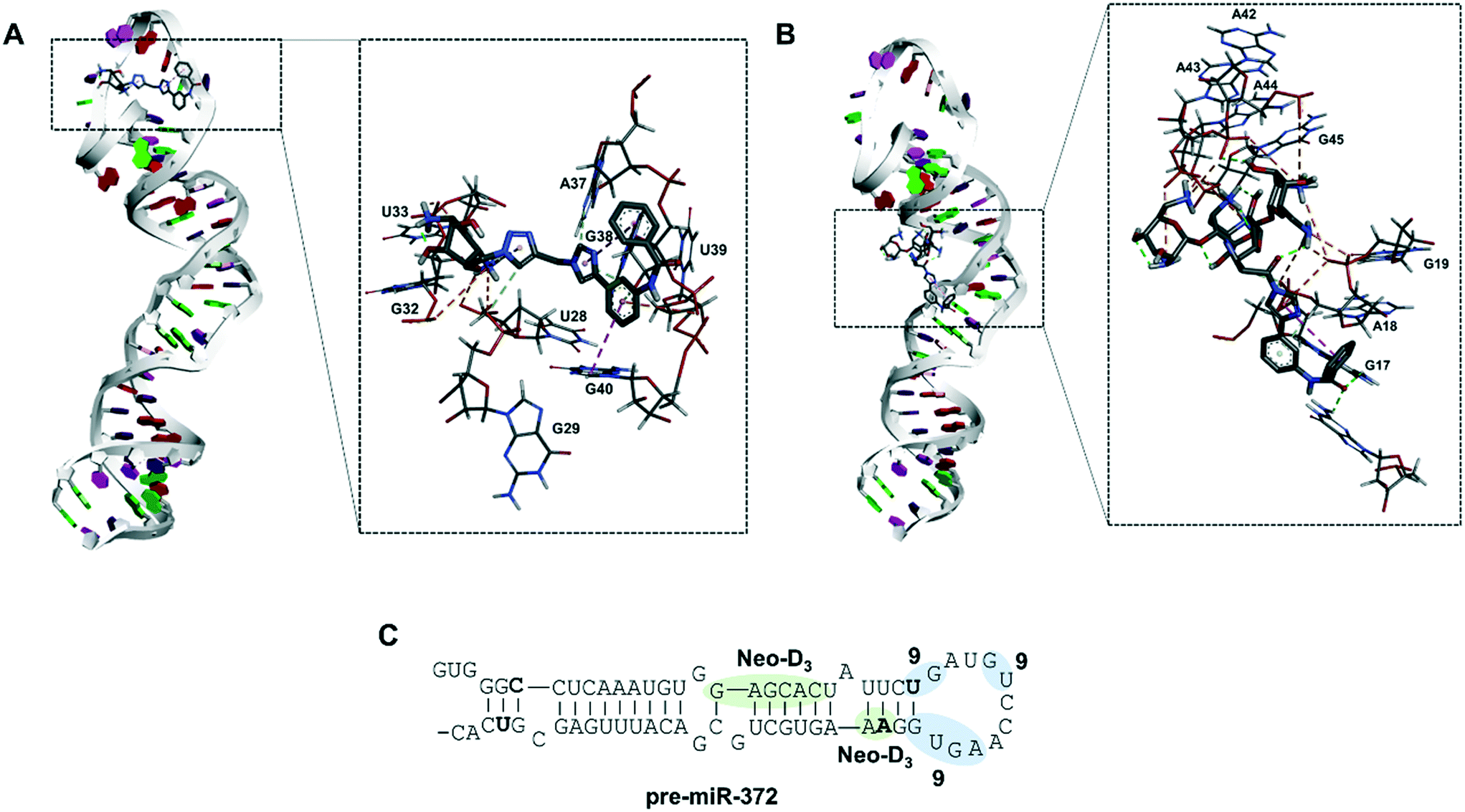 | ||
| Fig. 3 Molecular docking of compound DOS-D39 (A) and Neo-D3 (B) performed on a pre-miR-372 structure using Autodock 4 software. Primary and secondary structures of pre-miR-372 (C) and binding sites of compound 9 (light blue) and Neo-D3 (light green). | ||
In conclusion, we were able to synthesize a new series of 2-desoxystreptamine analogs conjugated to various heteroaromatic compounds such as natural and artificial nucleobases as new RNA binders. We performed a comparison of affinity, selectivity, inhibition activity and site of interaction for oncogenic pre-miR-372 with a previously synthesized series of ligands containing the neomycin aminoglycoside instead of 2-DOS. The aim of this work was to reduce the overall size and hydrophilicity of the RNA ligands and to verify if the biological properties could be maintained. We observed that all synthesized compounds are good RNA binders with selectivity when tested in competition with other nucleic acid structures. Some compounds were able to inhibit miR-372 maturation upon inhibiting Dicer cleavage of pre-miR-372 but only compound 5d showed an IC50 in the low micromolar range. Some selectivity was also observed once the compound tested against other pre-miRNAs. However, we already observed that compounds that are selective for a small set of pre-miRNAs can have a very specific biological effect and this could be an advantage for a comprehensive biological activity. The same evaluations performed on the enantiopure analog of 5d, compound 9, showed that this latter was an even better ligand and inhibitor concerning pre-miR-372. Finally, it was observed that both 5d and 9 are weaker binders of pre-miR-372 compared to their neomycin counterpart Neo-D3, but that they maintained a similar inhibition activity. Molecular docking suggested that this is due to the particular binding site that is located on the nucleotides being cleaved by the Dicer enzyme thus being functional for efficient inhibition. Further chemical optimization of these 2-DOS conjugates could thus lead to an even better inhibition activity and to study their intracellular activity.
Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We thank Dr. J. M. Guigonis (Plateforme Bernard Rossi, CEA TIRO, Nice, France) for HRMS analyses and Agence Nationale de la Recherche (ANR, France; ANR-11-JS07-011-01) for funding this work. A PhD fellowship to T. P. A. T. was supported by the Vietnam Ministry of Science and Technology (VIED grant 911), to S. P. by Université Côte d'Azur IDEX-JEDI, and to M. P. by ANRT and Sanofi. A post-doc fellowship to A. R. was supported by Université Côte d'Azur.References
- J. P. Falese, A. Donlic and A. E. Hargrove, Chem. Soc. Rev., 2021, 50, 2224–2243 RSC.
- K. D. Warner, C. E. Hajdin and K. M. Weeks, Nat. Rev. Drug Discovery, 2018, 17, 547–558 CrossRef CAS PubMed.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- H. Ratni, R. S. Scalco and A. H. Stephan, ACS Med. Chem. Lett., 2021, 12, 874–877 CrossRef CAS PubMed.
- A. Di Giorgio and M. Duca, MedChemComm, 2019, 10, 1242–1255 RSC.
- V. Ambros, Nat. Med., 2008, 14, 1036–1040 CrossRef CAS PubMed.
- M. Winkle, S. M. El-Daly, M. Fabbri and G. A. Calin, Nat. Rev. Drug Discovery, 2021, 20, 629–651 CrossRef CAS PubMed.
- F. Zamani and T. Suzuki, J. Med. Chem., 2021, 64, 7110–7155 CrossRef CAS PubMed.
- Z. Li and T. M. Rana, Nat. Rev. Drug Discovery, 2014, 13, 622–638 CrossRef CAS PubMed.
- S. T. Crooke, B. F. Baker, R. M. Crooke and X. H. Liang, Nat. Rev. Drug Discovery, 2021, 20, 427–453 CrossRef CAS PubMed.
- A. Di Giorgio, T. P. Tran and M. Duca, Future Med. Chem., 2016, 8, 803–816 CrossRef CAS PubMed.
- M. D. Disney and A. J. Angelbello, Acc. Chem. Res., 2016, 49, 2698–2704 CrossRef CAS PubMed.
- M. D. Disney, A. M. Winkelsas, S. P. Velagapudi, M. Southern, M. Fallahi and J. L. Childs-Disney, ACS Chem. Biol., 2016, 11, 1720–1728 CrossRef CAS PubMed.
- S. P. Velagapudi, M. D. Cameron, C. L. Haga, L. H. Rosenberg, M. Lafitte, D. R. Duckett, D. G. Phinney and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5898–5903 CrossRef CAS PubMed.
- S. P. Velagapudi, S. M. Gallo and M. D. Disney, Nat. Chem. Biol., 2014, 10, 291–297 CrossRef CAS PubMed.
- F. A. Abulwerdi, W. Xu, A. A. Ageeli, M. J. Yonkunas, G. Arun, H. Nam, J. S. Schneekloth, Jr., T. K. Dayie, D. Spector, N. Baird and S. F. J. Le Grice, ACS Chem. Biol., 2019, 14, 223–235 CrossRef CAS PubMed.
- K. Gumireddy, D. D. Young, X. Xiong, J. B. Hogenesch, Q. Huang and A. Deiters, Angew. Chem., Int. Ed., 2008, 47, 7482–7484 CrossRef CAS PubMed.
- Z. Shi, J. Zhang, X. Qian, L. Han, K. Zhang, L. Chen, J. Liu, Y. Ren, M. Yang, A. Zhang, P. Pu and C. Kang, Cancer Res., 2013, 73, 5519–5531 CrossRef CAS PubMed.
- C. Staedel, T. P. A. Tran, J. Giraud, F. Darfeuille, A. Di Giorgio, N. J. Tourasse, F. Salin, P. Uriac and M. Duca, Sci. Rep., 2018, 8, 1667 CrossRef PubMed.
- J. Zhang, S. Umemoto and K. Nakatani, J. Am. Chem. Soc., 2010, 132, 3660–3661 CrossRef CAS PubMed.
- C. Maucort, D. D. Vo, S. Aouad, C. Charrat, S. Azoulay, A. Di Giorgio and M. Duca, ACS Med. Chem. Lett., 2021, 12, 899–906 CrossRef CAS PubMed.
- D. D. Vo, C. Becquart, T. P. A. Tran, A. Di Giorgio, F. Darfeuille, C. Staedel and M. Duca, Org. Biomol. Chem., 2018, 16, 6262–6274 RSC.
- D. D. Vo, C. Staedel, L. Zehnacker, R. Benhida, F. Darfeuille and M. Duca, ACS Chem. Biol., 2014, 9, 711–721 CrossRef CAS PubMed.
- D. D. Vo, T. P. Tran, C. Staedel, R. Benhida, F. Darfeuille, A. Di Giorgio and M. Duca, Chemistry, 2016, 22, 5350–5362 CrossRef CAS PubMed.
- V. Malnuit, M. Duca and R. Benhida, Org. Biomol. Chem., 2011, 9, 326–336 RSC.
- S. Magnet and J. S. Blanchard, Chem. Rev., 2005, 105, 477–498 CrossRef CAS PubMed.
- J. L. Childs-Disney and M. D. Disney, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 123–140 CrossRef CAS PubMed.
- S. Yoshizawa, D. Fourmy, R. G. Eason and J. D. Puglisi, Biochemistry, 2002, 41, 6263–6270 CrossRef CAS PubMed.
- C. M. Klemm, A. Berthelmann, S. Neubacher and C. Arenz, Eur. J. Org. Chem., 2009, 2788–2794 CrossRef CAS.
- X. Liu, J. R. Thomas and P. J. Hergenrother, J. Am. Chem. Soc., 2004, 126, 9196–9197 CrossRef CAS PubMed.
- J. R. Thomas, X. Liu and P. J. Hergenrother, J. Am. Chem. Soc., 2005, 127, 12434–12435 CrossRef CAS PubMed.
- C. M. Klemm, A. Berthelmann, S. Neubacher and C. Arenz, Eur. J. Org. Chem., 2009, 2009, 2788–2794 CrossRef.
- B. Llano-Sotelo, E. F. Azucena, Jr., L. P. Kotra, S. Mobashery and C. S. Chow, Chem. Biol., 2002, 9, 455–463 CrossRef CAS PubMed.
- T. Dalmay and D. R. Edwards, Oncogene, 2006, 25, 6170–6175 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00345c |
| ‡ Present address: Department of Chemistry Engineering, Nha Trang University, 02 Nguyen Dinh Chieu, Nha Trang, Khanh Hoa, Vietnam. |
| This journal is © The Royal Society of Chemistry 2022 |