
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhthalocyanine reactivity and interaction on the 6H-SiC(0001)-(3 × 3) surface investigated by core-level experiments and simulations†
Anu
Baby
a,
Guillaume
Marcaud
b,
Yannick J.
Dappe
 c,
Marie
D’Angelo
b,
Jean-Louis
Cantin
b,
Mathieu G.
Silly
*d and
Guido
Fratesi
*e
c,
Marie
D’Angelo
b,
Jean-Louis
Cantin
b,
Mathieu G.
Silly
*d and
Guido
Fratesi
*e
aDipartimento di Scienza dei Materiali, Università di Milano-Bicocca, Via Roberto Cozzi 55, 20125 Milano, Italy
bInstitut des NanoSciences de Paris, Sorbonne Université and CNRS-UMR 7588, Paris 75005, France
cSPEC, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette Cedex, France
dSynchrotron SOLEIL, L'Orme des Merisiers, 91192 Saint-Aubin, France. E-mail: mathieu.silly@synchrotron-soleil.fr
eETSF and Dipartimento di Fisica “Aldo Pontremoli”, Università degli Studi di Milano, Via Celoria 16, 20133 Milano, Italy. E-mail: guido.fratesi@unimi.it
First published on 24th May 2022
Abstract
The adsorption of phthalocyanine (H2Pc) on the 6H-SiC(0001)-(3 × 3) surface is investigated using X-ray photoelectron spectroscopy (XPS), near edge X-ray absorption fine structure spectroscopy (NEXAFS), and density functional theory (DFT) calculations. Spectral features are tracked from the submonolayer to the multilayer growth regime, observing a significant modification of spectroscopic signals at low coverage with respect to the multilayer films, where molecules are weakly interacting. Molecules stay nearly flat on the surface at the mono and submonolayers. Previously proposed adsorption models, where the molecule binds by two N atoms to corresponding Si adatoms, do not reproduce the experimental spectra at the submonolayer coverage. We find instead that another adsorption model where the molecule replaces the two central H atoms by a Si adatom, effectively forming Si-phthalocyanine (SiPc), is both energetically more stable and yields in combination a better agreement between the experimental and simulated spectra. This suggests that the 6H-SiC(0001)-(3 × 3) surface may be a candidate substrate for the on-surface synthesis of SiPc molecules.
1 Introduction
SiC wide band gap semiconductors exist in more than 200 crystallographic structures or polytypes. SiC presents a wide band gap of 2.4–3.26 eV depending on the polytype, high thermal conductivity and high breakdown voltage.1 Due to these outstanding electronic properties compared to silicon, SiC is a promising material for application in a high-power and high-temperature device.2 As a wide band gap semiconductor, SiC is transparent to visible light and can be used as a transparent material for solar cell applications.3–5 Compared to other wide band gap semiconductors such as TiO2 and ZnO, SiC can be easily n or p doped.6 SiC can also play the role of active materials in p–n heterojunctions.SiC also undergoes various surface reconstructions depending on the surface stoichiometry,7–9 which can act as a template for molecular adsorption. SiC has also demonstrated its high potential for graphene growth with high quality on a large scale.10 The electronic properties of SiC reconstructed surfaces are very sensitive to their environment. Metallization of the 3C-SiC(100)-(3 × 2) semiconducting surface has been performed via atomic hydrogenation11,12 and negative differential resistance has been obtained by Ag adsorption on atomic silicon wires on 3C-SiC(100).13 Due to the high sensitivity of the electronic properties to the environment, SiC finds also application as a gas sensor14 and a biosensor.15
SiC also presents high potential for application in the synthesis of hybrid organic–inorganic heterostructures. SiC surface functionalization has been investigated finding its high potential for bio application as a biosensor.16,17 Molecule adsorption on the silicon terminated 6H-SiC(0001)-(3 × 3) surface has also been previously explored. It has been demonstrated experimentally by scanning tunneling microscopy (STM) and core level photoemission spectroscopy and theoretically that H2Pc18 and C6019 organic molecules deposited on 6H-SiC(0001)-(3 × 3) form a covalent bond with the Si adatom of the SiC surface. Singular insulating contact with the SiC surface has also been evidenced with a perylene molecule derivative.20
Phthalocyanine, chemically characterized for the first time in 1933 by Linstead and co-workers,21 presents peculiar electronic properties in terms of optical absorption, conductivity and long lived charge generation making it an important compound for various applications such as solar cell applications and medicine.22–24 The geometry of adsorption of the first layer of molecules on inorganic semiconductors plays a key role in the electronic properties of the interface and on the growth of an organic film.25,26 Organic–inorganic heterojunction interfaces remain insufficiently documented and need further investigation. For instance, only the first step of the growth of H2Pc molecules on 6H-SiC(0001)-(3 × 3) has been previously studied by STM.18,27 At the submonolayer coverage, corresponding to isolated molecules at the surface, it has been shown that H2Pc molecules adopt a bridge bond configuration involving Si–N covalent bonds between the H2Pc molecules and the Si adatom in the 6H-SiC(0001)-(3 × 3) surface reconstruction.18 Despite covalent bonds, H2Pc molecules adsorbed on the surface maintain the capability to rotate at the surface under the STM tip. H2Pc molecules also exhibit a peculiar sensitivity to the surface reconstruction underneath which results in a variation of electronic density localized on H2Pc legs.27 6H-SiC(0001)-(3 × 3) surface reconstruction is very sensitive to oxidation at the origin of dark sites observed by STM28,29 and occurs after depositing H2PcH2Pc.18,27 Further investigation involving electronic and chemical sensitive techniques is needed to fully determine the H2Pc/6H-SiC(0001)-(3 × 3) interactions.
In this work, we investigate the adsorption of H2Pc on the silicon terminated 6H-SiC(0001)-(3 × 3) surface by X-ray photoelectron spectroscopy (XPS), near edge X-ray absorption fine structure spectroscopy (NEXAFS), and density functional theory (DFT) calculations. Experiments highlight the interaction of the molecules with the surface taking place especially at the N atoms and strongly depending on H2Pc coverage. In addition to the adsorption sites previously identified,18 we propose that the molecules may be further stabilized by incorporating a Si adatom, thereby forming SiPc.
2 Materials and methods
2.1 Experiments
An on axis n-doped (0.07 Ω cm) 6H-SiC(0001) substrate (CREE Inc.) has been used to perform the experiments. All the experiments have been carried out at a base pressure of 5 × 10−10 mbars. The silicon rich 6H-SiC(0001)-(3 × 3) reconstructed surface is prepared using the standard protocol achieving a highly ordered surface.30 The substrate is first outgazed for 12 h at 600 °C under UHV conditions by direct current heating. The SiC substrate is flashed at temperature above 1150 °C to remove the native oxide. Silicon is then deposited on the SiC substrate at a temperature of 650 °C, followed by an annealing at 750 °C18,30 The quality of the surface reconstruction is checked by LEED. Metal free phthalocyanine, 29H, 31H-phthalocyanine (H2Pc), with a purity of 98% (Sigma-Aldrich) is evaporated in a UHV with a single filament effusion cell (Createc) optimized for organic material evaporation at a temperature of about 250 °C and a deposition rate of 2 Å min−1. The evaporation rate was calibrated in situ using a quartz crystal microbalance. We first deposit and measure one sixth of the monolayer, then molecules are evaporated further on the same sample achieving monolayer and multilayer coverages. High resolution core level photoemission spectroscopy (HRPES) and near edge X-ray absorption fine structure spectroscopy (NEXAFS) have been performed on the TEMPO beamline at the synchrotron SOLEIL.31 The beamline covers an energy range between 50 and 1500 eV with a resolving power better than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The HRPES measurement was done using a high energy resolution Scienta SES2002 photoelectron analyzer equipped with a delay line detector.32 NEXAFS is measured in partial electron yield. The partial electron yield is collected with a photoelectron analyzer measuring Auger electrons. The normalization of the absorption spectra is done with the photocurrent measured via a gold mesh. The core level photoemission spectra were deconvoluted according to the usual curve-fitting procedure. The secondary photoelectron background was removed using the Shirley background. The Voigt function and the Voigt doublet were used to fit, respectively, the C 1s and the Si 2p core levels with respect to a spin orbit splitting of 0.6 eV and a ratio of 0.5 between the Si 2p1/2 and Si 2p3/2 components. For the sake of clarity, the core level spectra are normalized to the maximum of the intensity of each spectrum.
000. The HRPES measurement was done using a high energy resolution Scienta SES2002 photoelectron analyzer equipped with a delay line detector.32 NEXAFS is measured in partial electron yield. The partial electron yield is collected with a photoelectron analyzer measuring Auger electrons. The normalization of the absorption spectra is done with the photocurrent measured via a gold mesh. The core level photoemission spectra were deconvoluted according to the usual curve-fitting procedure. The secondary photoelectron background was removed using the Shirley background. The Voigt function and the Voigt doublet were used to fit, respectively, the C 1s and the Si 2p core levels with respect to a spin orbit splitting of 0.6 eV and a ratio of 0.5 between the Si 2p1/2 and Si 2p3/2 components. For the sake of clarity, the core level spectra are normalized to the maximum of the intensity of each spectrum.
2.2 Theory
We have studied numerically the adsorption of H2Pc molecules on SiC(0001) by performing ab initio simulations based on density functional theory (DFT). For the exchange-correlation functional, we use the generalized gradient approximation in the form proposed by Perdew–Burke–Ernzerhof (GGA-PBE).33 Calculations are carried out using the QuantumESPRESSO package34,35 that implements pseudopotentials and plane waves. In analogy to previous works18 the surface is modeled by a periodically repeated slab containing, from top to bottom, the Si adatoms, trimers, adlayers for the reconstruction, a SiC bilayer, and a layer of H atoms saturating the dangling bonds on the back side. We study the geometry on the top side, in a 2 × 2 supercell of the 6H-SiC(0001)-(3 × 3) reconstructed surface (overall, the system contains 218 atoms). The coordinates are then optimized, apart from the SiC bilayer and saturating H atoms that are kept frozen. Brillouin zone integration is performed using a 3 × 3 k-point mesh. Wavefunctions are expanded over plane waves until a cutoff of 45 eV. To consider the influence of van der Waals interactions, GGA-PBE geometries are eventually further optimized including Grimme dispersion forces36 (PBE-D2).We compute the XPS core level shifts between inequivalent nitrogen atoms by performing self-consistent calculations with a N pseudopotential generated with a 1s full core hole (FCH)37 at the given atom site, on top of GGA-PBE geometries. Next, we evaluate NEXAFS within the half-core-hole approach (HCH)38,39 by using the xspectra code.40 As we adopt a pseudopotential approach, we obtain XPS and NEXAFS spectra up to an energy constant. Further details and the computational setup are given in our previous works.41,42
3 Results and discussion
3.1 Experiments
Survey photoemission spectra measured for the different sample preparation steps are presented in Fig. 1. The spectrum measured for the clean 6H-SiC(0001)-(3 × 3) surface exhibits three main peaks corresponding to C 1s, Si 2s, and Si 2p core levels. The presence of a slight amount of oxygen (O 1s) is also observed in the spectrum. The 6H-SiC(0001)-(3 × 3) surface is known to be very reactive to oxygen.28,29 Despite a low pressure (<5 × 10−10 bars) and low residual oxygen and water, the surface appears slightly contaminated. Fortunately, the oxidation process takes place in the second silicon layer in the surface reconstruction,43 which does not affect the Si adatom, a reactive site for molecular adsorption. After depositing one-sixth of a monolayer of H2Pc (submonolayer) on the surface, the presence of nitrogen is observed in the spectrum (Fig. 1). The amount of oxygen remains constant after the first deposit attesting the high purity of the deposited molecules. For the monolayer, nitrogen increases in agreement with the expected H2Pc coverage. The amount of oxygen has notably increased but remains contained. For the multilayer (≈20 layers), the carbon and silicon contributions coming from the substrate are clearly attenuated, and nitrogen and carbon contributions coming from H2Pc are now the main contributions to the spectrum. No oxygen contamination is observed.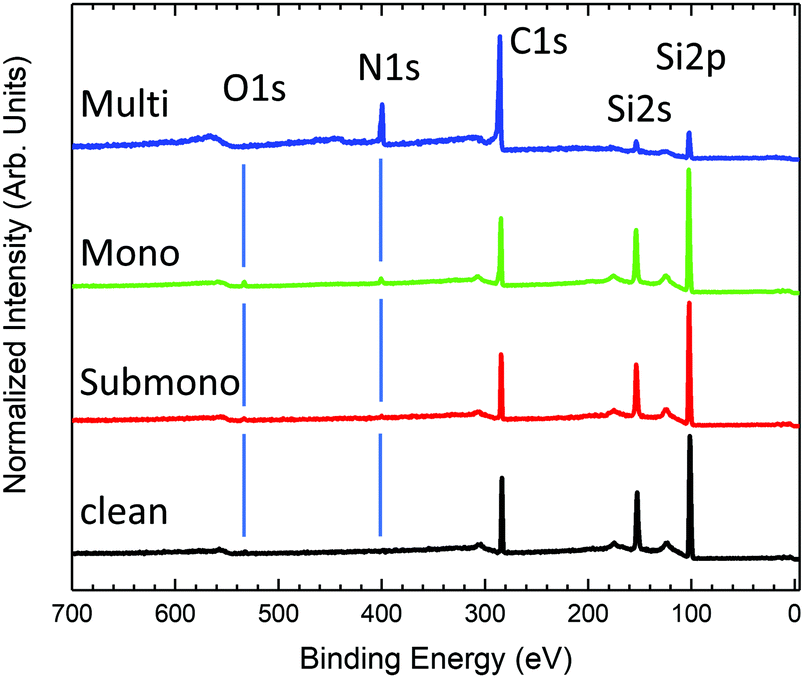 | ||
| Fig. 1 Photoemission survey measured at 825 eV photon energy for the clean 6H-SiC(0001)-(3 × 3) surface (black curve), one sixth of the H2Pc monolayer (submono, red curve), the H2Pc monolayer (green curve), and the multilayer (about 20 layers, blue curve). | ||
Fig. 2 presents the evolution of the C 1s core level spectra as a function of the H2Pc coverage. For the clean 6H-SiC(0001)-(3 × 3) surface, the C 1s core level exhibits only one contribution at 282 eV. As the surface is silicon terminated only one carbon corresponding to bulk contribution is visible. For the multilayer, the core level spectrum is measured at higher photon energy to probe more deeply into the bulk. At lower binding energy, the carbon bulk contribution is strongly attenuated but still visible and shifted to 282.46 eV. Three contributions are observed at higher binding energy corresponding to H2Pc molecules. The shape of the H2Pc C 1s core level spectrum is in accordance with the literature.44–46 The main contribution at 283.85 eV is attributed to the benzene carbon atoms and the second contribution at 285.25 eV is associated with the sum of pyrrole atoms and shake-up transition of benzene carbon atoms. The lowest component at 287.15 eV corresponds to shake-up contributions of pyrrole carbon atoms. For the monolayer, the bulk contribution is increased and lies at 282.22 eV. The feature corresponding to H2Pc molecules is now composed of two broader components compared to the multilayer attributed to benzene and pyrrole carbon atoms. The shake-up contributions are not discernable. For the submonolayer H2Pc feature is still composed of two contributions that are smaller than the bulk contribution located at 282 eV.
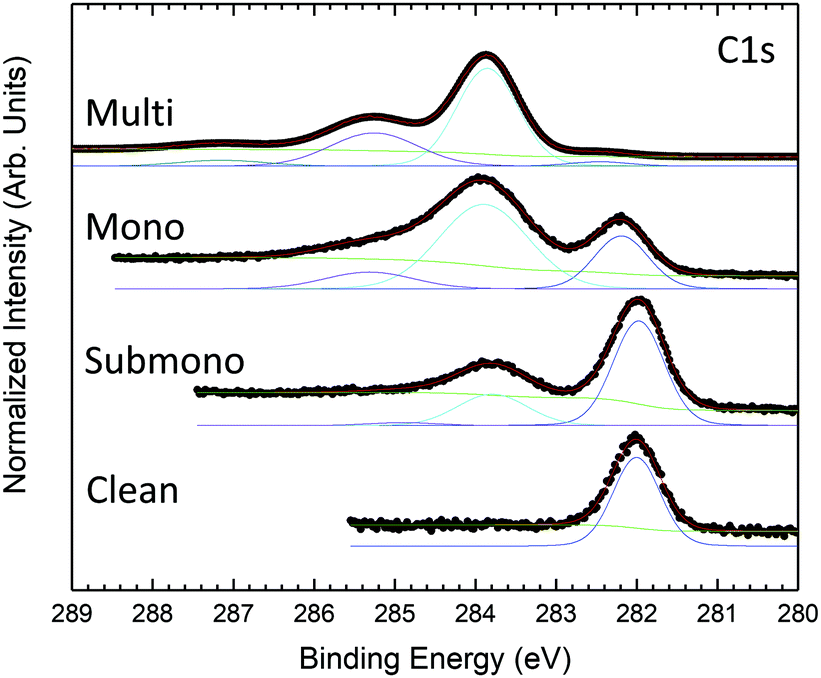 | ||
| Fig. 2 C 1s core level photoemission spectra measured at 335 eV photon energy for the clean 6H-SiC(0001)-(3 × 3) surface, and H2Pc coverages of one sixth of the monolayer and one monolayer and at 485 eV for the H2Pc multilayer. | ||
N 1s core level spectra are presented in Fig. 3. For the multilayer, the N 1S core level presents 3 contributions. The main component located at 397.77 eV is related to pyrrole and bridging nitrogen atoms. The second feature at 399.32 eV corresponds to center nitrogen atoms. The lowest component at higher binding energy corresponds to shake-up transition satellite. The N 1s core level is in good agreement with previous studies.47,48 For the monolayer, the N 1s core level is still deconvoluted into 3 contributions, the shake-up component at higher binding energy becomes marginal. Compared to the multilayer, the contributions appear broader but retain the same shape and relative intensities. For the sub-monolayer, the shake-up contribution is not yet visible. The N 1s core level spectrum appears strongly modified. The N 1s feature is still composed of two components but with inversed relative intensities compared to mono and multilayer H2Pc coverages. The modification in the shape indicates a stronger interaction between the nitrogen atoms of the H2Pc molecules and the substrate.
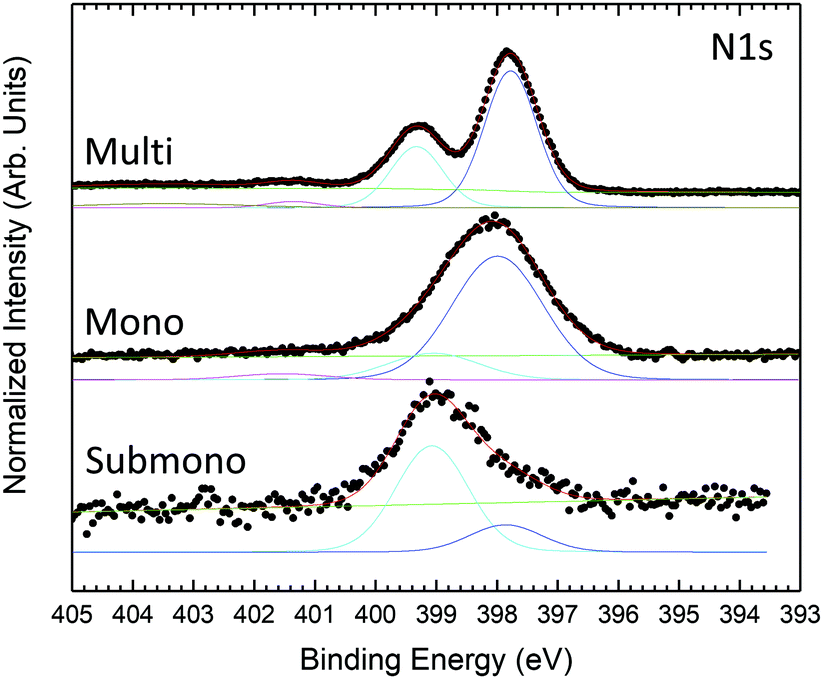 | ||
| Fig. 3 N 1s core level photoemission spectra measured at 600 eV photon energy for H2Pc coverages of one sixth of the monolayer, monolayer, and multilayer. | ||
To determine the interaction of the molecules with the surface, the Si 2p core level spectra are obtained and shown in Fig. 4. The Si 2p core level spectrum of the clean 6H-SiC(0001)-(3 × 3) surface, exhibiting the expected shape,19 is deconvoluted into five components. The bulk component is located at 100.66 eV and three surface states located at 100.13 eV, 99.34 eV and 98.64 eV correspond to the silicon atoms involved in the last Si–C bilayer (S3), silicon atoms composing the Si adlayer and the base of the tetramers (S2) and Si adatoms’ apex of the tetramers (S1), respectively. An additional component at a higher kinetic energy (101.54 eV) corresponds to the first oxidation state of silicon, in agreement with the presence of oxygen evidenced in the survey spectrum (Fig. 1). For the submonolayer and monolayer, the Si 2p, the surface states S1 to S3 are broadened and the S1 component corresponding to the adatom decreases in intensity. These results are in good agreement with the interaction of the Si adatom with the nitrogen atoms of H2Pc as proposed by Baffou et al.18 For the monolayer a second oxidation state appears at higher binding energy in agreement with the increasing amount of oxygen (Fig. 1).
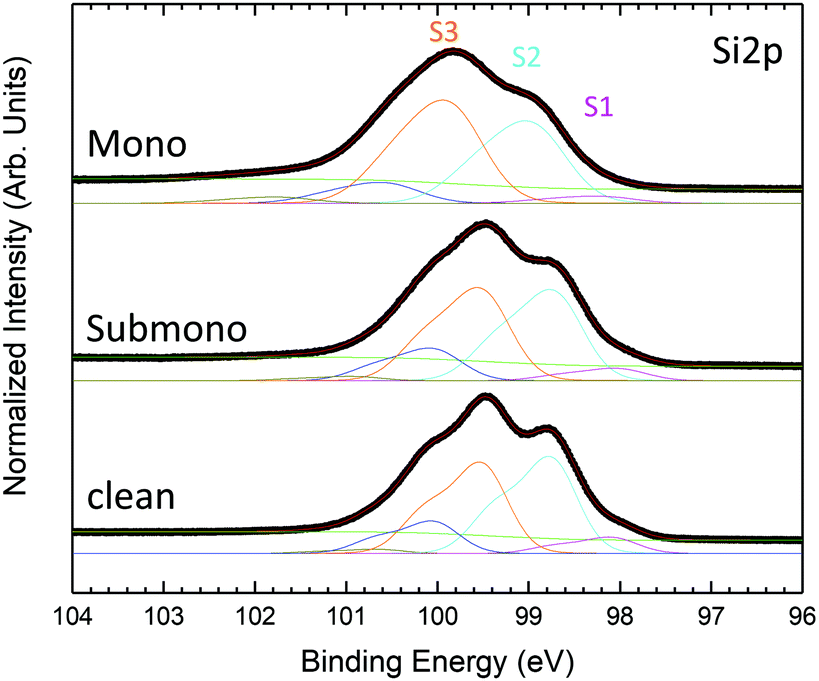 | ||
| Fig. 4 Si 2p core level photoemission spectra measured at 150 eV photon energy for H2Pc coverages of one sixth of the monolayer, the monolayer and the multilayer. | ||
The evolution of the N K-edge NEXAFS spectra measured at normal and grazing incidence as a function of H2Pc coverage is presented in Fig. 5. The NEXAFS spectrum of the multilayer exhibits a strong dichroic signal with enhanced sharp structures corresponding to 1s → π* transitions at grazing incidence.47,48 The dichroism observed indicates a preferential molecular orientation. Indeed, this dichroism is in good agreement with molecules lying perpendicular to the surface. For sub and monolayer coverages, the signal coming from 1s → π* transitions is strongly reduced and the dichroism is enhanced at normal emission in the opposite way to the signal measured for the multilayer. This result is in good agreement with H2Pc lying flat on the surface at the sub and monolayers according to STM studies of isolated H2Pc molecules lying flat on the surface.18 Nevertheless, the decrease in intensity results from the modification of the nitrogen bonds and especially double chemical bonds with Si adatoms of the surface. This result is in good agreement with the strong modification in the shape of the N 1s core level spectrum observed for the submonolayer (Fig. 3).
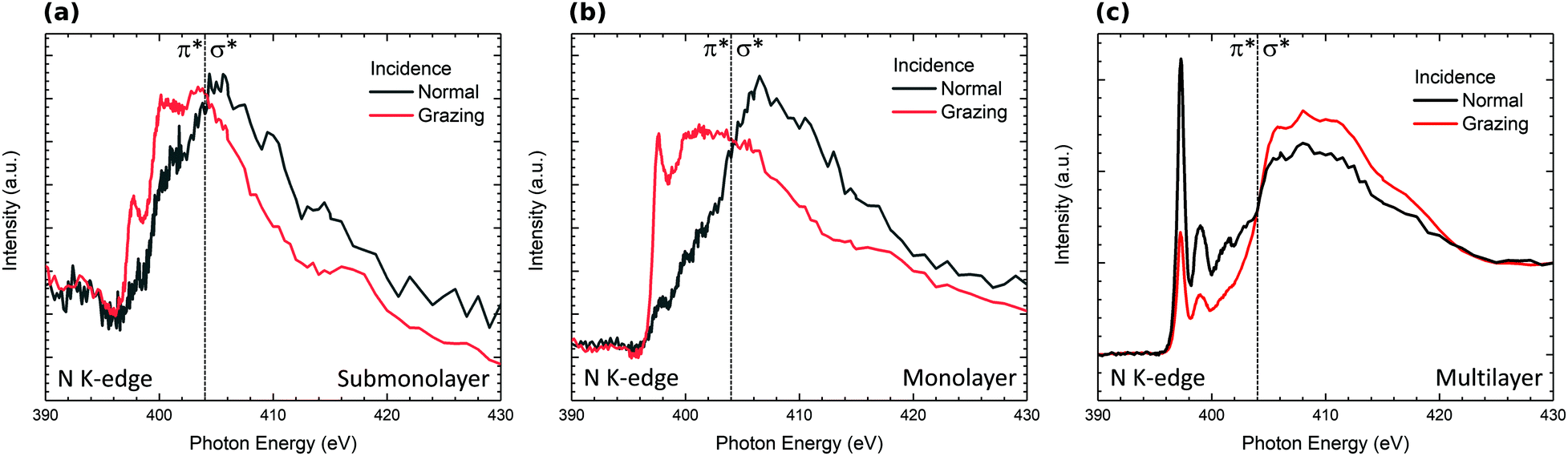 | ||
| Fig. 5 Nitrogen K-edge NEXAFS spectra measured at normal (E‖ substate) and grazing (E⊥ substrate) incidence for different H2Pc coverages: (a) subomolayer, (b) monolayer, and (c) multilayer coverages. Sharp features below 404 eV correspond to 1s → π* transitions and a broad feature above 404 eV corresponds to 1s → σ* transitions. | ||
To determine in more detail the adsorption geometry of the H2Pc molecules, the evolution of the NEXAFS intensity as a function of the angle with incoming light has been studied for sub and monolayer H2Pc coverages (Fig. 6(a and b)). The intensity of the 1s → π* transition has been integrated, normalized and plotted in Fig. 6(c). The experimental data are compared to theoretical intensity dependency as a function of molecular angle relative to the surface normal.49 The H2Pc molecules present angles of 40° and 30° relative to the normal of the surface for the submonolayer and monolayer, respectively. The H2Pc molecules appear not completely flat on the surface but present a small off axis angle which is reduced for the monolayer. The N 1s core level spectra have evidenced a strong interaction of the molecule with the substrate. Depending on the interaction strength with the substrate the adsorbed molecules can exhibit molecular distortions50 which can be the origin of the tilt of the molecule determined by NEXAFS.
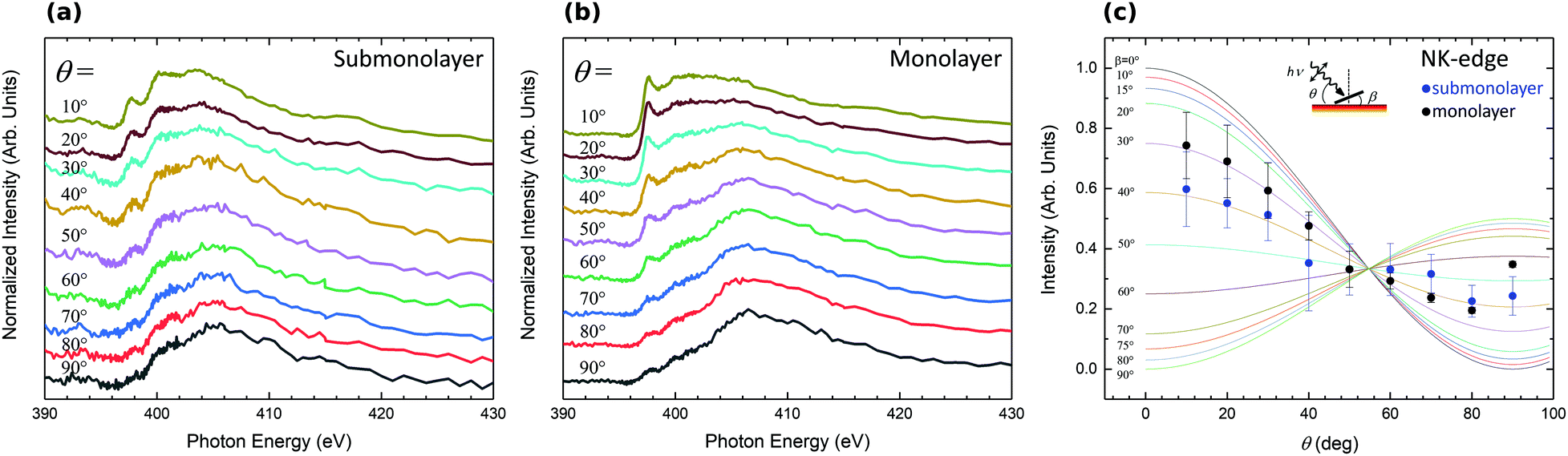 | ||
| Fig. 6 Angular-dependent N K-edge NEXAFS spectra for submonolayer (1/6th) (a) and monolayer (b) H2Pc molecules on the 6H-SiC(0001)-(3 × 3) surface surface. (c) Intensities of the main 1s → π* resonance against θ for submonolayer (blue circle) and monolayer (black circle) H2Pc coverages. The solid lines represent the theoretical angular variation of 1s → π* resonances calculated for various molecular tilt angles. The inset shows the schematic diagram of the measured geometry. | ||
3.2 Numerical simulations
To describe the interaction between the H2Pc molecule and the 6H-SiC(0001)-(3 × 3) surface we performed theoretical investigation of adsorbed molecules in a surface supercell whose lateral size is sufficiently large to consider individual H2Pc units. We consider initially the molecule at two adsorption sites as shown in Fig. 7(a and b). In the top position, Fig. 7(a), the molecule is placed with its center on top of a Si adatom and then relaxed. The molecule in this case is weakly bound and a marginal interaction to the surface is observed. In the bridge position, Fig. 7(b), two opposite N bridging N atoms are placed nearly above two of the Si adatoms, as proposed previously.18 The adsorption energy of H2Pc in the top and bridge configurations is reported in Table 1. We observe that the top site is less stable by nearly 1 eV and will not be discussed further. At the bridge site, the adsorption energy of the molecule to the 6H-SiC(0001)-(3 × 3) surface is −1.13 eV. We recall that this configuration has already been reported in great detail previously18,27 obtaining very similar results. The N atoms approach the Si adatoms thereby forming two N–Si bonds of length 1.90 Å. In the relaxed geometry the distance between the two Si adatoms results to be 8.85 Å (reducing by 0.43 Å from the initial value) whereas that between the nitrogen atoms attached to Si is 7.06 Å (increasing by 0.33 Å from the initial value). Including the dispersion forces enhances the adsorption strength by about 2 eV and reduces the distance between the outer parts of the molecule and the substrate (see Fig. S1 in the ESI†), but confirms the bridge as the most stable site and does not modify the the bond lengths given above.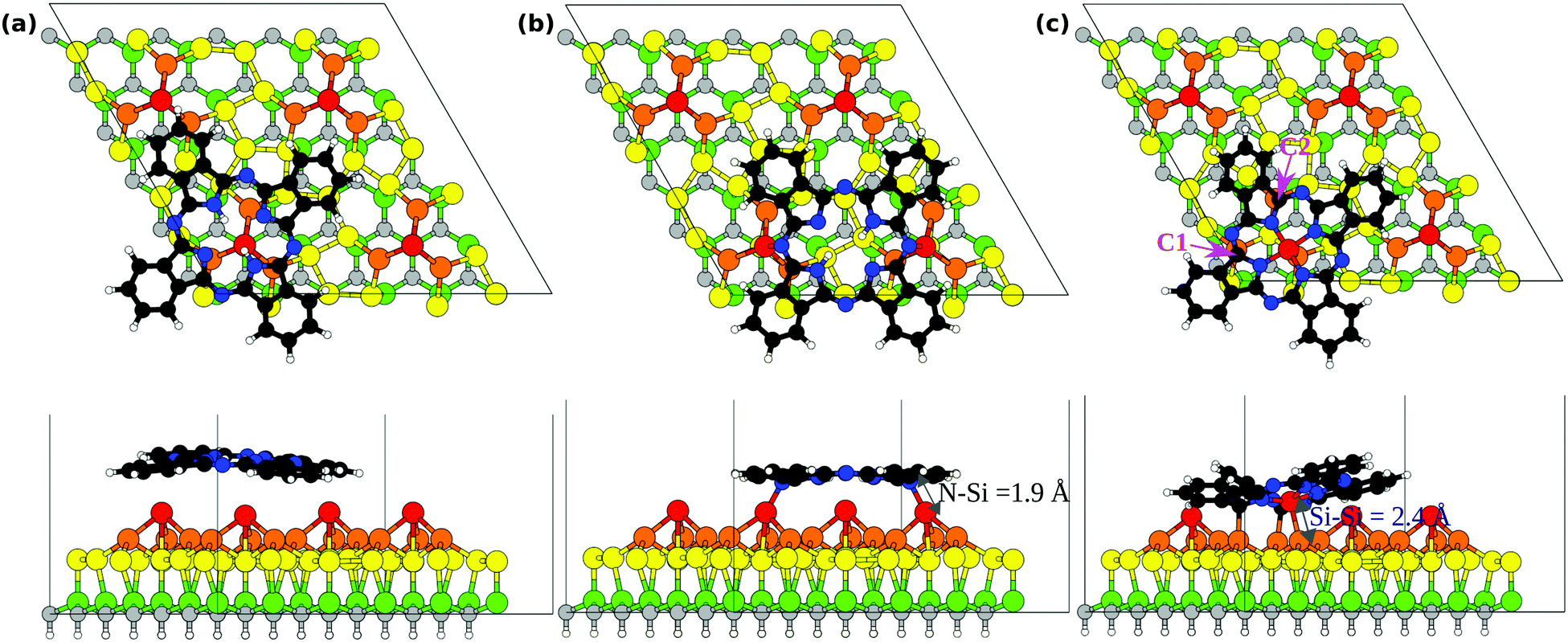 | ||
| Fig. 7 Optimized configurations obtained for (a) top and (b) bridge positions of H2Pc/6H-SiC(0001)-(3 × 3), (c) top positioned SiPc/6H-SiC(0001)-(3 × 3). The top panel shows the top view and the bottom panel shows the side view. The molecule is colored as follows: C-black, N-blue, H-white, and Si-red. The substrate is colored as follows: Si adatoms (first layer S1)-red, Si trimers (second layer S2)-orange, Si (third layer S3)-yellow, Si (fourth layer)-green, C-grey, and H-white. | ||
| Molecules | Ads. sites | Ads. energy PBE (eV) | Ads. energy PBE + D2 (eV) |
|---|---|---|---|
| H2Pc | Top | −0.23 | −2.18 |
| H2Pc | Bridge | −1.13 | −3.43 |
| SiPc | Top | −4.09 | −6.78 |
| SiPc | Bridge | −1.27 | −3.51 |
We now report the N 1s XPS for free and adsorbed H2Pc. In the gas phase, the molecule has three inequivalent N atoms as marked in the sketch above Fig. 8(a). N1 and N2 atoms constitute the cyclic tetrapyrrole nitrogen atoms where N1 attached to the hydrogen is the pyrrole-N, N2 the pyrrole aza-N and N3 the mesobridging-aza N. The spectrum is plotted in the bottom part of Fig. 8(a) where N2 has the lowest binding energy followed by N3 and N1, in full agreement with the experimental spectrum in the literature.47,48 Additionally this spectrum is also similar to the multilayer spectrum of the measured N 1s XPS shown in Fig. 3. This is an indication of the fact that the molecules of the higher layers essentially retain the pristine molecular electronic structure, so that the measured spectrum of the multilayer is very similar to that of the gas phase molecules.
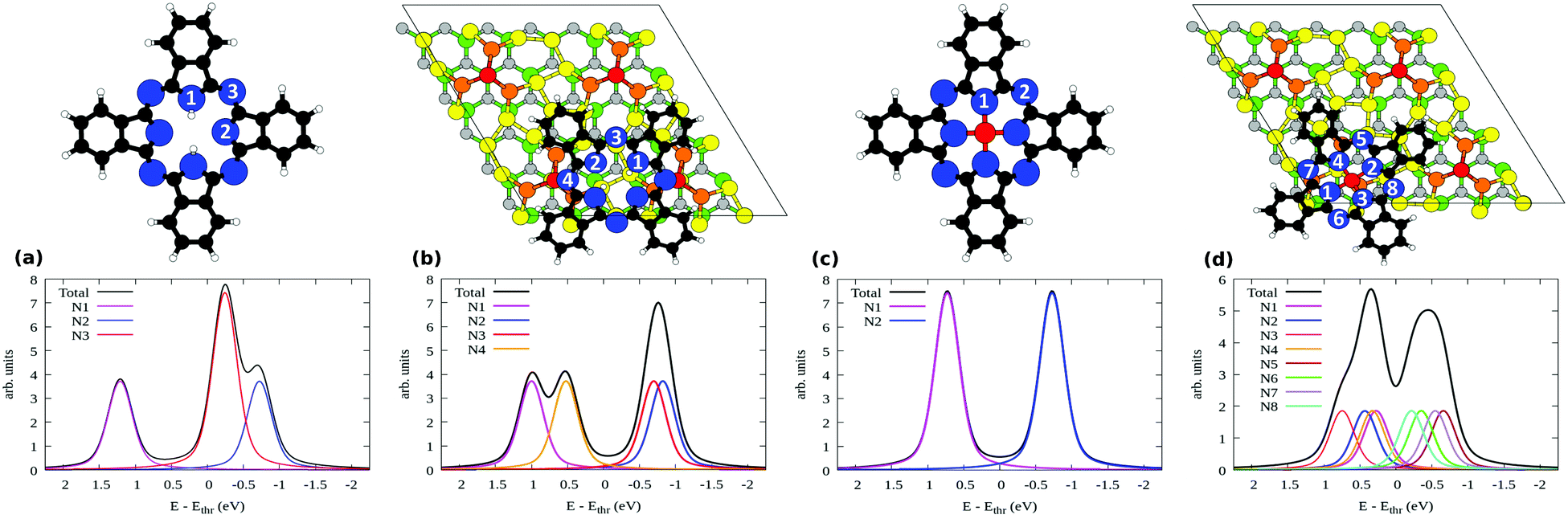 | ||
| Fig. 8 Simulated N 1s XPS of (a) H2Pc molecules, (b) bridge H2Pc/6H-SiC(0001)-(3 × 3), (c) SiPc, and (d) top SiPc/6H-SiC(0001)-(3 × 3). The inequivalent N atoms are numbered in the respective geometries shown above each plot. Refer to Fig. 7 for the color coding used here. | ||
Upon adsorption as in the bridge site, the N 1s XPS modifies as shown in Fig. 8(b). The spectrum is now composed of two well separated structures, produced by the four inequivalent N atoms: indeed, the bridging N atoms are no more equivalent, giving rise to contributions that we mark as N3 and N4 (see the top panel in Fig. 8(b)). The binding energy of the N4 atom is closer to that of N1, with which it groups, than N2 or N3. This points out that N4, bonded to the Si adatom, manifests a chemical environment similar to that of N1 bonded to the H atoms. Additionally, one can also notice that the N2 and N3 components constituting the second peak situated at lower binding energy have moved closer together than in the free molecule, reducing their separation from 0.48 to 0.12 eV. Overall, the agreement that we observed between the gas phase simulations and the multilayer measurements is lost, with the measured submonolayer as well as the monolayer spectra in Fig. 3 differing entirely from the result of Fig. 8(b). This suggests that the bridge configuration of H2Pc/SiPc(0001) is not the only case in the submonolayer and monolayer regimes in our experiments. Some other configurations may be present at low coverage, that we attempt to reveal by considering further models.
The capability of porphyrin and phthalocyanine deposited on a surface to substitute pretty easily the two central H atoms with a metal atom is well known.51,52 The surface reconstruction plays a key role in the molecule reactivity facilitating the metalation process.53 In the same way, it has been recently shown that silicon atoms can be substituted to H atoms to form nonmetal porphyrin.54 From the experimental point of view, our 6H-SiC(0001)-(3 × 3) surface presents silicon adatoms which can react with deposited H2Pc18 but also an excess of Si atoms, forming silicon clusters at the surface,30 suggesting that the substitution may occur by the Si atoms already present at the surface. Hence we simulated the geometry of the H2Pc molecule by removing the two central H atom and placing the molecule with its center above a Si adatom. The resulting structure obtained after optimization is shown in Fig. 7(c). The molecule initially with a void core embeds the Si adatom effectively forming an adsorbed silicon-phthalocyanine (SiPc) molecule. We name this the top SiPc/6H-SiC(0001)-(3 × 3) configuration. The adatom displaces from the pyramid center, keeping only one bond with a Si atom in the Si-triangle, with a Si–Si distance of 2.38 Å. As a consequence, the missing bonds of the other two Si atoms in the triangle are saturated by bonding to two carbon atoms, marked C1 and C2 in Fig. 7(c), with bond lengths of 2.02 and 2.04 Å. To achieve this configuration, the molecule distorts significantly, especially if we compare it with the nearly flat bridge configuration seen above.
Overall, three bonds are formed between the molecule and the substrate. This indicates a very strong interaction between the molecule and the substrate in this case. The adsorption energy results to be −4.09 eV taking into account also the adsorption energy on other Si adatom sites of the two H atoms removed from the center of the molecule. This is significantly more stable than the energy of H2Pc in the bridge position (−1.13 eV), see Table 1, despite the N–Si bonds are not formed for top SiPc. Displacing the so formed SiPc molecule from the top to the bridge site, thereby restoring the N–Si bonds but breaking the bonds with the Si triangle (0.8 eV larger than within bulk Si), yields to an energy cost of 2.82 eV (bridge SiPc, Eads= −1.27 eV). Removal of Si adatoms could be facilitated by surface oxidation43 that we observe by XPS. Also for SiPc configurations, including the dispersion forces enhances the adsorption energy (see Table 1) but keeps the energy ordering of the structures unchanged. Detailed configurations, with outer C atoms closer to the substrate, are reported in Fig. S1 in the ESI.† Both for its deformed structure and for a different substrate registry, SiPc at the top should be clearly distinguishable from H2Pc at the bridge as measured by STM. See also the simulated STM images presented in the ESI†, Fig. S2. Conversely, at the bridge site, SiPc and H2Pc would require good resolution to resolve four-fold and two-fold appearance around the center.
The above findings suggest that H2Pc in our experiments may react at Si adatom sites, thereby forming SiPc strongly bound to the surface. Although less common than their metal analogues, non-metal phthalocyanines are also synthesized. In particular, SiPc are promising molecules for applications in various fields including cancer phototherapy, NIR imaging, organic photovoltaics, organic electronics and photocatalysis55 also thanks to the uncommon hexacoordination of the Si atom that offers two axial ligands. In our case, the molecule is stabilized by the surface that acts as one of the two ligands, leaving ideally the second one free for further molecular functionalization. This is analogous to the recent on-surface synthesis of Si–Porphyrins on Ag surfaces upon evaporation of atomic Si.54
The incorporation of the central Si atom in place of the two H ones modifies the spectral characteristics of the molecule. The N 1s XPS for an hypothetical free SiPc molecule is given in Fig. 8(c). In this case, the pyrrole N atoms are equivalent as all bind to Si. As a consequence, all are found at higher binding energy with respect to the bridging N atoms, as was the case for the H-bonded pyrrole N1 atom in free H2Pc in Fig. 8(a).
When SiPc is adsorbed at the top site of 6H-SiC(0001)-(3 × 3) as shown in Fig. 7(c) no N atom is strictly equivalent by symmetry to another one. Its simulated N 1s XPS spectrum is shown in Fig. 8(d), where we have taken into account all of the eight inequivalent nitrogen atoms as indicated therein. Their spread in energy yields a smoother, double peaked structure. On the high-energy side we find the pyrrole N atoms bonded to Si (N1–N4), and on the low-energy side the bridging N atoms (N5–N8). Differences in the position of these atoms with respect to the substrate influence their binding energy, for example in the case of N5 and N7 that lie closer to the substrate than N6 and N8, and a lower binding energy is computed for the former pair than for the latter pair. The spectrum is overall less resolved and concentrated in a narrower energy range than that simulated both for free H2Pc (that was a good model for the multilayer case) and for bridge H2Pc/6H-SiC(0001)-(3 × 3). Considering the simultaneous presence of both molecular species, this results in a better agreement with the experimental spectrum of the monolayer regime that is composed of a wide and unresolved feature as seen in Fig. 3. Such findings support the possibility that top SiPc/6H-SiC(0001)-(3 × 3) molecules are also present in the monolayer regime. Differences in details of the adsorption configuration for various SiPc molecules may further smear the spectrum therefore improving agreement with experiments. We computed the XPS spectra also for PBE-D2 geometries obtaining relative core level energies that differ only marginally from the GGA-PBE ones (mean absolute error, 0.02 eV).
We further compare the two adsorption models, bridge H2Pc/6H-SiC(0001)-(3 × 3) and top SiPc/6H-SiC(0001)-(3 × 3), by means of their simulated N 1s NEXAFS spectra, shown in Fig. 9(a). There we distinguish between photon polarization along the surface normal (solid lines, transition to π* states) and in the surface plane (dashed lines, transition to σ* states). One can easily notice that the first peak present in the blue curve for the H2Pc bridge is missing from the red one for the SiPc top, similar to the missing π* peak in the NEXAFS measurements of the mono- and especially submonolayer in Fig. 5. An even stronger reduction of the first peak can be observed by comparing to free H2Pc whose NEXAFS spectrum is reported in Fig. 9(b). There, also the case of free SiPc is reported which presents a σ* resonance among the π* ones, much weaker for the surface-stabilized molecule. By detailing the contributions of individual N atoms to the spectrum as reported in the ESI†, Fig. S3, we can observe that for free H2Pc the first peak is mostly given by the four perypheral N3 atoms. For H2Pc bridge, the two perypheral N3 atoms that are not bonded to the Si adatoms still produce the first structure with π* symmetry. For top SiPc/6H-SiC(0001)-(3 × 3), there is no single identifiable peak due to the differences in the spectra among the 8 N species.
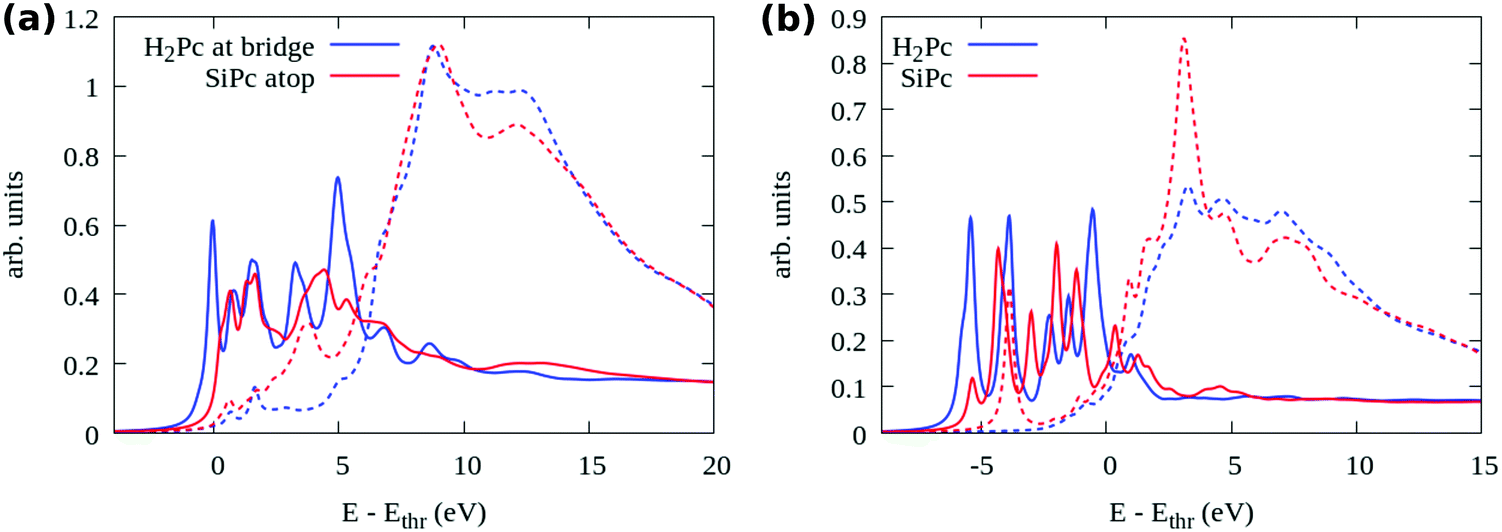 | ||
| Fig. 9 Comparison of the simulated N 1s NEXAFS between (a) bridge H2Pc/6H-SiC(0001)-(3 × 3) and top 6H-SiC(0001)-(3 × 3), (b) gas phase H2Pc and SiPc. The solid lines represent photon polarization along the surface normal and the dotted ones in the surface plane. | ||
The strong weakening of the π* contribution in the experimental spectrum of the submonolayer and monolayer regime is a further indication that the experimental observations may refer also to top SiPc/6H-SiC(0001)-(3 × 3) rather than only bridge H2Pc/6H-SiC(0001)-(3 × 3). Whereas in the experimental multilayer regime the π* resonances reappear in close similarity with the spectra simulated for the free molecules, stipulating the presence of H2Pc molecules that are weakly interacting. This is a new discovery regarding the reactivity of the 6H-SiC(0001)-(3 × 3) surface towards the H2Pc molecules converting them into SiPc at low coverage where the molecules are in direct contact with the surface, while remaining as H2Pc in the additional layers not in direct contact with the substrate.
From the experimental data, a remarkable surface reactivity with H2Pc has been evidenced. Especially for the submonolayer, the N 1s core level spectrum strongly differs and does not match with the calculation of bridge bonded H2Pc between N atoms and Si adatoms of the reconstructed surface as proposed in a previous STM study.18 Our calculations evidence a new adsorption model involving one silicon adatom substituted with hydrogen atoms. Differences between the monolayer and submonolayer are then induced by the molecular density. We speculate that for the monolayer, bridge bonding between N atoms of H2Pc and the Si adatoms of the SiC surface could still occur, while for the submonolayer, the spectroscopic features are in good agreement with a stronger interaction between H2Pc and the substrate leading to a Si insertion in H2Pc. The balance between intermolecular and molecule–substrate interactions could be one of the factor behind the spectroscopic signals observed between H2Pc monolayer and submonolayer coverages. Indeed, the competition between intermolecular and molecule–substrate interactions is known to play a key role in molecular organization even at a semiconductor surface.56–59 The surface preparation and molecule evaporation are particularly critical parameters in intermolecular and molecule–substrate interactions at the origin of changes in molecular packing and electronic properties of the molecular film. The evaporation rate used during molecule deposition plays a key role in molecule organization at the surface,60 it has been shown in previous work that a 2 Å min−1 evaporation rate is a critical value at which packing of molecules can change.61 Working at 250 °C corresponding to a 2 Å min−1 evaporation rate, a slight variation in evaporation can lead to different adsorption modes which could be the origin of the differences observed between the previous STM study for H2Pc/6H-SiC(0001)-(3 × 3)18 and the present work.
4 Conclusions
We have investigated the adsorption of metal-free phthalocyanine molecules evaporated on the 6H-SiC(0001)-(3 × 3) surface. Photoemission surveys guarantee the purity of the adsorbed layer, and although some O is present on the free surface, this remains buried by the molecules. X-Ray photoelectron spectra taken at the C 1s line allow identifying two molecular contributions from benzene and pyrrole atoms and the substrate one at lower binding energy. The N 1s spectrum highlights a strong interaction between the N atoms of H2Pc molecules and the substrate, with a single unresolved feature instead of the two peaks observed for thick layers. The interaction between N and Si atoms is also evidenced by the Si 2p spectrum. Near edge X-ray absorption fine structure spectra taken at the N 1s edge clarify adsorption geometry of molecules and preferential adsorption mode for the submonolayer and monolayer coverages. Molecules exhibit preferentially flat-down adsorption mode, with an observed moderate tilting which could include the effect of molecular distortion. The N interaction with the surface is further evidenced by the modification in the spectrum with the reduced intensity of the first π* resonances. By performing numerical simulations we find the bridge site as the stable adsorption site for H2Pc molecules as in the literature. However, the N 1s spectra computed for this case are not consistent with our measurements. At low coverage, another adsorption configuration is then identified where the molecule incorporates a Si adatom after loosing its two central H atoms. With a total of three bonds with the surface, it is energetically more stable by nearly 3 eV. Additionally, its simulated spectra yield a better agreement with the experimental ones. Effectively, a SiPc molecule is thereby formed, stabilized by the surface. This suggests 6H-SiC(0001)-(3 × 3) as a candidate substrate for the on-surface synthesis of non-metal SiPc molecules.Author contributions
A. B contributed in formal analysis, investigation, validation, visualization, writing – original draft, and writing – review & editing; G. M. contributed in investigation; Y. D. contributed in investigation; M. D. contributed in investigation; J. L. C. contributed in investigation; M. S. contributed in conceptualization, investigation, validation, visualization, writing – original draft, and writing – review & editing; G. F. contributed in conceptualization, formal analysis, investigation, validation, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge “NFFA” Nanoscience Foundries and Fine Analysis-Europe H2020-INFRAIA-2014–2015 (Grant Agreement No. 654360) having benefited from the access provided by the UMIL node, user-project ID 647. We acknowledge the CINECA award under the ISCRA initiative, for the availability of high-performance computing resources and support (grant HOMSI-HP10CEC0H6).Notes and references
- F. La Via, A. Severino, R. Anzalone, C. Bongiorno, G. Litrico, M. Mauceri, M. Schoeler, P. Schuh and P. Wellmann, Mater. Sci. Semicond. Process., 2018, 78, 57–68 CrossRef CAS.
- N. G. Wright, A. B. Horsfall and K. Vassilevski, Mater. Today, 2008, 11, 16–21 CrossRef CAS.
- M. Köhler, M. Pomaska, P. Procel, R. Santbergen, A. Zamchiy, B. Macco, A. Lambertz, W. Duan, P. Cao and B. Klingebiel, et al. , Nat. Energy, 2021, 6, 529–537 CrossRef.
- M. Syväjärvi, Q. Ma, V. Jokubavicius, A. Galeckas, J. Sun, X. Liu, M. Jansson, P. Wellmann, M. Linnarsson and P. Runde, et al. , Sol. Energy Mater. Sol. Cells, 2016, 145, 104–108 CrossRef.
- M. K. Sobayel, M. S. Chowdhury, T. Hossain, H. I. Alkhammash, S. Islam, M. Shahiduzzaman, M. Akhtaruzzaman, K. Techato and M. J. Rashid, Sol. Energy, 2021, 224, 271–278 CrossRef CAS.
- F. Roccaforte, P. Fiorenza, M. Vivona, G. Greco and F. Giannazzo, Materials, 2021, 14, 3923 CrossRef CAS PubMed.
- X. N. Xie, H. Q. Wang, A. T.-S. Wee and K. P. Loh, Surf. Sci., 2001, 478, 57–71 CrossRef CAS.
- L. Li, Y. Hasegawa, I. S.-T. Tsong and T. Sakurai, J. Phys. IV France, 1996, 06, C5–C172 Search PubMed.
- P. Soukiassian and F. Semond, J. Phys. IV France, 1997, 07, C6–C113 CrossRef.
- P. Avouris and C. Dimitrakopoulos, Mater. Today, 2012, 15, 86–97 CrossRef CAS.
- V. Derycke, P. G. Soukiassian, F. Amy, Y. J. Chabal, M. D. D'angelo, H. B. Enriquez and M. G. Silly, Nat. Mater., 2003, 2, 253–258 CrossRef CAS PubMed.
- M. G. Silly, C. Radtke, H. Enriquez, P. Soukiassian, S. Gardonio, P. Moras and P. Perfetti, Appl. Phys. Lett., 2004, 85, 4893–4895 CrossRef CAS.
- M. G. Silly, F. Charra and P. Soukiassian, Appl. Phys. Lett., 2007, 91, 223111 CrossRef.
- L. Sun, C. Han, N. Wu, B. Wang and Y. Wang, RSC Adv., 2018, 8, 13697–13707 RSC.
- A. Oliveros, A. Guiseppi-Elie and S. E. Saddow, Biomed. Microdevices, 2013, 15, 353–368 CrossRef PubMed.
- S. J. Schoell, M. Sachsenhauser, A. Oliveros, J. Howgate, M. Stutzmann, M. S. Brandt, C. L. Frewin, S. E. Saddow and I. D. Sharp, ACS Appl. Mater. Interfaces, 2013, 5, 1393–1399 CrossRef CAS PubMed.
- N. Yang, H. Zhuang, R. Hoffmann, W. Smirnov, J. Hees, X. Jiang and C. E. Nebel, Anal. Chem., 2011, 83, 5827–5830 CrossRef CAS PubMed.
- G. Baffou, A. J. Mayne, G. Comtet, G. Dujardin, P. Sonnet and L. Stauffer, Appl. Phys. Lett., 2007, 91, 073101 CrossRef.
- F. C. Bocquet, L. Giovanelli, Y. Ksari, T. Ovramenko, A. J. Mayne, G. Dujardin, F. Spillebout, P. Sonnet, F. Bondino and E. Magnano, et al. , J. Phys.: Condens. Matter, 2018, 30, 505002 CrossRef CAS PubMed.
- H. Yang, O. Boudrioua, A. J. Mayne, G. Comtet, G. Dujardin, Y. Kuk, P. Sonnet, L. Stauffer, S. Nagarajan and A. Gourdon, Phys. Chem. Chem. Phys., 2012, 14, 1700–1705 RSC.
- R. P. Linstead, Brit. Assoc. Advance. Sci. Rep., 1933, 465 Search PubMed.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed.
- A. M. Schmidt and M. J.-F. Calvete, Molecules, 2021, 26, 2823 CrossRef CAS PubMed.
- P.-C. Lo, M. Salomé Rodríguez-Morgade, R. K. Pandey, D. K.-P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- H. Lim, S. Yang, S.-H. Lee, J.-Y. Lee, Y. Lee, A. Bethavan Situmorang, Y.-H. Kim and J. Won Kim, J. Mater. Chem. C, 2021, 9, 2156–2164 RSC.
- M. H. Futscher, T. Schultz, J. Frisch, M. Ralaiarisoa, E. Metwalli, M. V. Nardi, P. Müller-Buschbaum and N. Koch, J. Phys.: Condens. Matter, 2018, 31, 064002 CrossRef PubMed.
- G. Baffou, A. J. Mayne, G. Comtet, G. Dujardin, L. Stauffer and P. Sonnet, J. Am. Chem. Soc., 2009, 131, 3210–3215 CrossRef CAS PubMed.
- F. Amy, P. Soukiassian, Y. K. Hwu and C. Brylinski, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165323 CrossRef.
- O. Kubo, T. Kobayashi, N. Yamaoka, S. Itou, A. Nishida, M. Katayama and K. Oura, Surf. Sci., 2003, 529, 107–113 CrossRef CAS.
- M. G. Silly, M. D'Angelo, A. Besson, Y. J. Dappe, S. Kubsky, G. Li, F. Nicolas, D. Pierucci and M. Thomasset, Carbon, 2014, 76, 27–39 CrossRef CAS.
- F. Polack, M. Silly, C. Chauvet, B. Lagarde, N. Bergeard, M. Izquierdo, O. Chubar, D. Krizmancic, M. Ribbens and J. Duval, et al. , AIP Conf. Proc., 2010, 1234, 185–188 CrossRef CAS.
- N. Bergeard, M. G. Silly, D. Krizmancic, C. Chauvet, M. Guzzo, J. P. Ricaud, M. Izquierdo, L. Stebel, P. Pittana and R. Sergo, et al. , J. Synchrotron Radiat., 2011, 18, 245–250 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli and M. Cococcioni, et al. , J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al. , J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- E. Pehlke and M. Scheffler, Phys. Rev. Lett., 1993, 71, 2338–2341 CrossRef CAS PubMed.
- M. Leetmaa, M. Ljungberg, A. Lyubartsev, A. Nilsson and L. Pettersson, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 135–157 CrossRef CAS.
- L. Triguero, L. G.-M. Pettersson and H. Ågren, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 8097 CrossRef CAS.
- C. Gougoussis, M. Calandra, A. Seitsonen and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075102 CrossRef.
- G. Fratesi, V. Lanzilotto, L. Floreano and G. P. Brivio, J. Phys. Chem. C, 2013, 117, 6632–6638 CrossRef CAS.
- A. Baby, G. Fratesi, S. R. Vaidya, L. L. Patera, C. Africh, L. Floreano and G. P. Brivio, J. Phys. Chem. C, 2015, 119, 3624–3633 CrossRef CAS.
- F. Amy, H. Enriquez, P. Soukiassian, P.-F. Storino, Y. J. Chabal, A. J. Mayne, G. Dujardin, Y. K. Hwu and C. Brylinski, Phys. Rev. Lett., 2001, 86, 4342–4345 CrossRef CAS PubMed.
- B. Brena, Y. Luo, M. Nyberg, S. Carniato, K. Nilson, Y. Alfredsson, J. Ålund, N. MÅrtensson, H. Siegbahn and C. Puglia, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 195214 CrossRef.
- J. Åhlund, K. Nilson, J. Schiessling, L. Kjeldgaard, S. Berner, N. MÅrtensson, C. Puglia, B. Brena, M. Nyberg and Y. Luo, J. Chem. Phys., 2006, 125, 034709 CrossRef PubMed.
- M. Vittorio Nardi, F. Detto, L. Aversa, R. Verucchi, G. Salviati, S. Iannotta and M. Casarin, Phys. Chem. Chem. Phys., 2013, 15, 12864–12881 RSC.
- Y. Alfredsson, B. Brena, K. Nilson, J. Åhlund, L. Kjeldgaard, M. Nyberg, Y. Luo, N. MÅrtensson, A. Sandell and C. Puglia, et al. , J. Chem. Phys., 2005, 122, 214723 CrossRef CAS PubMed.
- M.-N. Shariati, J. Lüder, I. Bidermane, S. Ahmadi, E. Göthelid, P. Palmgren, B. Sanyal, O. Eriksson, M. N. Piancastelli and B. Brena, et al. , J. Phys. Chem. C, 2013, 117, 7018–7025 CrossRef CAS.
- L. Cao, Y.-Z. Wang, T.-X. Chen, W.-H. Zhang, X.-J. Yu, K. Ibrahim, J.-O. Wang, H.-J. Qian, F.-Q. Xu and D.-C. Qi, et al. , J. Chem. Phys., 2011, 135, 174701 CrossRef PubMed.
- B. Amin, S. Nazir and U. Schwingenschlögl, Sci. Rep., 2013, 3, 1705 CrossRef.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS PubMed.
- D.-L. Bao, Y.-Y. Zhang, S. Du, S. T. Pantelides and H.-J. Gao, J. Phys. Chem. C, 2018, 122, 6678–6683 CrossRef CAS.
- J. Nowakowski, C. Wäckerlin, J. Girovsky, D. Siewert, T. A. Jung and N. Ballav, Chem. Commun., 2013, 49, 2347–2349 RSC.
- A. Baklanov, M. Garnica, A. Robert, M.-L. Bocquet, K. Seufert, J. T. Küchle, P. T.-P. Ryan, F. Haag, R. Kakavandi and F. Allegretti, et al. , J. Am. Chem. Soc., 2020, 142, 1871–1881 CrossRef CAS PubMed.
- K. Mitra and M. C.-T. Hartman, Org. Biomol. Chem., 2021, 19, 1168–1190 RSC.
- B. Baris, V. Luzet, E. Duverger, P. Sonnet, F. Palmino and F. Cherioux, Angew. Chem., Int. Ed., 2011, 50, 4094–4098 CrossRef CAS PubMed.
- Y. Makoudi, M. Beyer, J. Jeannoutot, F. Picaud, F. Palmino and F. Chérioux, Chem. Commun., 2014, 50, 5714–5716 RSC.
- Y. Makoudi, M. Beyer, S. Lamare, J. Jeannoutot, F. Palmino and F. Chérioux, Nanoscale, 2016, 8, 12347–12351 RSC.
- G. Copie, F. Cleri, Y. Makoudi, C. Krzeminski, M. Berthe, F. Cherioux, F. Palmino and B. Grandidier, Phys. Rev. Lett., 2015, 114, 066101 CrossRef CAS PubMed.
- M. E. Ardhaoui, P. Lang, F. Garnier and J. P. Roger, J. Chim. Phys., 1998, 95, 1367–1371 CrossRef.
- J.-H. Bae, S.-D. Lee and C.-J. Yu, Solid-State Electron., 2013, 79, 98–103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Geometries including dispersion forces; simulated STM images; and atomic contributions to the simulated N 1s NEXAFS spectra. See DOI: https://doi.org/10.1039/d2cp00750a |
| This journal is © the Owner Societies 2022 |