
Optimization of phospholipid chemistry for improved lipid nanoparticle (LNP) delivery of messenger RNA (mRNA)†
Ester
Álvarez-Benedicto
,
Lukas
Farbiak
,
Martha
Márquez Ramírez
,
Xu
Wang
,
Lindsay T.
Johnson
 ,
Osamah
Mian
,
Erick D.
Guerrero
and
Daniel J.
Siegwart
*
,
Osamah
Mian
,
Erick D.
Guerrero
and
Daniel J.
Siegwart
*
Department of Biochemistry, Simmons Comprehensive Cancer Center, The University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA. E-mail: Daniel.Siegwart@UTSouthwestern.edu
First published on 30th November 2021
Abstract
Lipid nanoparticles (LNPs) have been established as an essential platform for nucleic acid delivery. Efforts have led to the development of vaccines that protect against SARS-CoV-2 infection using LNPs to deliver messenger RNA (mRNA) coding for the viral spike protein. Out of the four essential components that comprise LNPs, phospholipids represent an underappreciated opportunity for fundamental and translational study. We investigated this avenue by systematically modulating the identity of the phospholipid in LNPs with the goal of identifying specific moieties that directly enhance or hinder delivery efficacy. Results indicate that phospholipid chemistry can enhance mRNA delivery by increasing membrane fusion and enhancing endosomal escape. Phospholipids containing phosphoethanolamine (PE) head groups likely increase endosomal escape due to their fusogenic properties. Additionally, it was found that zwitterionic phospholipids mainly aided liver delivery, whereas negatively charged phospholipids changed the tropism of the LNPs from liver to spleen. These results demonstrate that the choice of phospholipid plays a role intracellularly by enhancing endosomal escape, while also driving organ tropism in vivo. These findings were then applied to Selective Organ Targeting (SORT) LNPs to manipulate and control spleen-specific delivery. Overall, selection of the phospholipid in LNPs provides an important handle to design and optimize LNPs for improved mRNA delivery and more effective therapeutics.
Introduction
Lipid nanoparticles (LNPs) are an established concept for nucleic acid delivery.1–9 The first siRNA therapeutic approved by the U.S. FDA in 2018, called Onpattro, was a LNP formulation that delivers siRNA into liver hepatocytes.10 More recently, LNP delivery of mRNA has gained timely importance, including utility in multiple COVID-19 vaccines against SARS-CoV-2.11 LNPs are typically composed of four key components: ionizable cationic lipids, zwitterionic phospholipids (PL), cholesterol, and poly(ethylene glycol) (PEG) lipids (Fig. 1A). Fundamentally, the ionizable cationic lipid plays an important role since it can be positively charged below its pKa to mediate loading of negatively charged RNA during self-assembly into LNPs, uncharged at neutral pH to minimize toxicity, and become positively charged again following cellular uptake to facilitate endosomal escape as the endosomal pH decreases.12,13 Because of its active role in nucleic acid delivery, ionizable lipids have been the primary focus of LNP study and development.4,7,14–20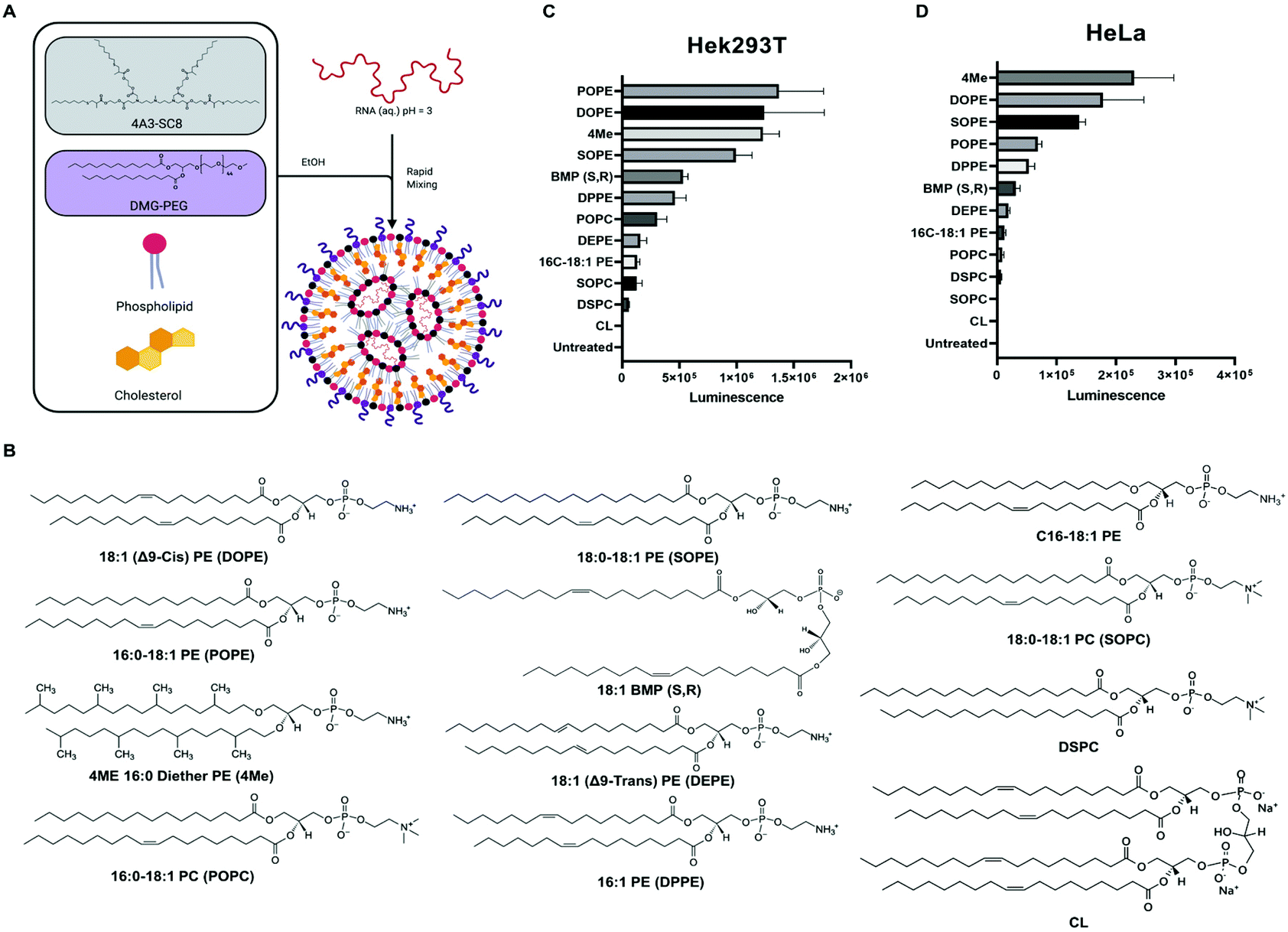 | ||
| Fig. 1 Phospholipids with PE headgroup increase LNP-mediated mRNA delivery. (A) Model illustrating LNP components and formulation process. (B) Structure of phospholipids used for in vitro mRNA delivery screen. Results for luciferase protein activity following delivery of firefly luciferase mRNA to HEK293T cells (C) and HeLa cells (D). | ||
Recently, however, it has become clearer that all four components of LNPs play essential roles at various stages of the delivery process. For example, variants of cholesterol21,22 and PEG lipids23–28 have been shown to modulate LNP physical properties, nanostructure, and ultimate efficacy. Zwitterionic phospholipids are commonly referred to as “helper lipids”, implying that they are less critical for efficacy. While PLs have been used and studied in small molecule drug and siRNA containing liposomes for decades,29–32 there is more to learn in understanding their location and function within mRNA containing LNPs and in cells. Both Onpattro and the mRNA vaccines use 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) as the “helper lipid” in their LNP formulation following its traditional use in liposome-mediated drug delivery.33–41 Computational and experimental studies have suggested that phospholipids may aid in the solubilization of RNAs inside of aqueous pockets within LNPs.42,43 We, and others, have further studied how the solubilizing forces may improve LNP construction.43–45 We showed, for example, that increasing molar proportions of phospholipid into LNPs improved delivery efficacy of sgRNA and mRNA, but did not affect siRNA delivery efficacy (see ESI in ref. 43). Zwitterionic PLs are thought to have an assistive role during LNP assembly by stabilizing electrostatic interactions between ionizable cationic lipids, RNA, and water molecules.25,46 However, less is known about their potential biological or physical intracellular behaviour. Thus, more work remains to be done to better understand the impact of phospholipids on LNP delivery of mRNAs.
Natural and naturally-derived PLs have been used for decades in liposomes for various drug delivery applications with typically high biocompatibilities.47 It remains intriguing to consider how cells use a variety of natural lipids in various cellular membranes, suggesting a broad space for lipid discovery and application to LNPs. Eukaryotic membranes are rich in glycerophospholipids such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS).48 Depending on the location within cells, the PL composition varies significantly. The plasma membrane is rich in sphingolipids and sterols, whereas endosomal membranes are rich in bis(monoacylglycerol)phosphate (BMP).48 Lipids with PC and PS groups provide bilayer membrane stability, whereas PE-lipids introduce membrane curvatures and increase tension, which in turn facilitates membrane fusion.30,49,50 These observations concerning the compositions of natural membranes within cells, coupled to the essential endosomal escape requirement facilitated by LNPs, prompted us to consider how inclusion of alternative phospholipids in LNPs might serve as active lipids to improve cellular delivery of mRNAs.
RNA therapies used in the clinic, such as Onpattro and the first-in-class SARS-CoV-2 vaccines, use DSPC as the PL in their four-component LNP formulation.10,11 Despite DSPC's wide utility and proven efficacy in LNP formulations for siRNA delivery and COVID-19 vaccines, substituting DSPC with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) greatly increased luciferase mRNA delivery in vivo up to four-fold.44,51,52 Thus, phospholipid identity can significantly affect mRNA delivery suggesting PLs play a functional role in nucleic acid delivery. Phospholipids with PC headgroups can adopt a cylinder phase that forms stable lipid bilayer.47 In contrast, a lipid with PE headgroups can adopt a cone shape that transitions to hexagonal conformation (HII) associated with membrane fusion properties.47 An enhanced fusion of polyplexes with the plasma membrane which resulted in greater transfection efficiency was observed when substituting DSPC for DOPE.53 Based on these precedents, one can hypothesize that the differences in phospholipid structure may increase RNA delivery potency by physical and/or biological mechanisms.
To study the role of phospholipids in LNPs, we decided to screen several structurally diverse phospholipids with PE and PC headgroups, varying chain length, degree of saturation, and methyl substitution (Fig. 1B). Additionally, we included lipids that are unique to organelle membranes, such as BMP and cardiolipin (CL), exclusive to endosome and mitochondrial membranes, respectively. Both lipids have unique chemical structures that could provide benefits for endosomal escape due to similarities with the endosomal membrane or disrupt the integrity of the endosomal membrane due to striking structural differences. Additionally, BMP and CL have molecular shapes of inverted cone and cone, respectively, which produce similar effects to PE lipids.47
Overall, the results described here demonstrate that phospholipids have a functional role in nucleic acid delivery. Phospholipids with PE head groups were consistently more efficient for in vitro and in vivo mRNA delivery. Moreover, DOPE-containing LNPs enabled significantly more endosomal escape compared to DSPC-containing LNPs, which appeared to be more sequestered in lysosomes after endocytosis. Additionally, LNPs with negatively charged phospholipids had an increased spleen targeting effect in vivo compared to neutral phospholipids that were most effective in liver delivery of luciferase mRNA. Therefore, we conclude that all components of the LNP play an essential role and should be systematically studied for mRNA delivery. Further knowledge on the role PLs play on LNP-mediated nucleic acid delivery could lead to improved delivery of mRNA therapeutics including current SARS-CoV-2 and future vaccines,54 as well as to set the foundation for the development of novel vaccines and therapeutics against a variety of diseases.55
Materials and methods
Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC), N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-phosphatidylethanolamine (NBD-PE), N-(lisamine Rhodamine B sulfonyl)-phosphatidylethanolamine (Rh-PE), cholesterol (Chol), sphingomyelin (SM) from porcine brain, phosphatidylinositol (PI) from soybean, 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC), sn-(3-(9Z-octadecenoyl)-2-hydroxy)-glycerol-1-phospho-sn-3′-(1′-(9Z-octadecenoyl)-2′-hydroxy)-glycerol (BMP-S,R), and sn-(3-oleoyl-2-hydroxy)-glycerol-1-phospho-sn-1′-(3′-oleoyl-2′-hydroxy)-glycerol (BMP-S,S) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol was purchased from Sigma Aldrich. DMG-PEG2000 (Sunbright GM-020) was purchased from NOF America Corporation. The dendrimer ionizable amino lipid 4A3-SC8 was synthesized in our lab according to a previously reported protocol.17
:1), 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC), N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-phosphatidylethanolamine (NBD-PE), N-(lisamine Rhodamine B sulfonyl)-phosphatidylethanolamine (Rh-PE), cholesterol (Chol), sphingomyelin (SM) from porcine brain, phosphatidylinositol (PI) from soybean, 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC), sn-(3-(9Z-octadecenoyl)-2-hydroxy)-glycerol-1-phospho-sn-3′-(1′-(9Z-octadecenoyl)-2′-hydroxy)-glycerol (BMP-S,R), and sn-(3-oleoyl-2-hydroxy)-glycerol-1-phospho-sn-1′-(3′-oleoyl-2′-hydroxy)-glycerol (BMP-S,S) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol was purchased from Sigma Aldrich. DMG-PEG2000 (Sunbright GM-020) was purchased from NOF America Corporation. The dendrimer ionizable amino lipid 4A3-SC8 was synthesized in our lab according to a previously reported protocol.17
Methods
:30:30:1.5. The mRNA was dissolved in the 100 mM citrate buffer pH 3.0. The aqueous solution and ethanol solution were rapidly mixed for 30 seconds at a 3:1 volume ratio and were incubated at room temperature for 15 min to allow LNP assembly. For in vitro experiments, PBS solution was added to reach a 10 mM citrate concentration. For in vivo experiments, LNPs were purified by dialysis in sterile PBS with 3.5 kDa cut-off for 2 hours. The mol ratio of 4A3-SC8 to mRNA was 10000:1 and the weight ratio was 23:1.
:15:30:30:3:x, where x was varied to change the molar percentage of the SORT phospholipid from 5% to 40% (x = 3.315, 7, 15.75, 27, 42). SORT LNPs were purified by dialysis in sterile PBS with 3.5 kDa cut-off for 2 hours. The mol ratio of 4A3-SC8 to mRNA was 10000:1 and the weight ratio was 23:1.
:38.5:1.35:10. The mol ratio of C12-200 to mRNA used was 10000:1 which is equivalent to a weight ratio of 25:1.
:3000 dilution) and Hoechst 33342 (0.1 mg ml−1) for 15 min at 37 °C. Finally, cells were imaged by confocal microscopy (LSM 700, Zeiss). Pearson's coefficient was calculated using PSC Colocalization plugin for Image J following the protocol previously described.56
:18:1:1:20:30) in chloroform followed by 3 h of dry vacuum to give a thin layer. The thin layer was resuspended in PBS buffer pH 7.4 for a 1 mM final concentration of total lipid and sonicated for 20 min (10 s sonication, 20 s rest). Endosome mimicking liposomes were prepared mixing BMP, DOPC, DOPE, NBD-PE, N-Rh-PE, PI (molar ratio 10:50:18:1:1:10) in chloroform. Liposomes were prepared following the same procedure described before. LNPs were prepared as described previously. In a black bottom 96-well plate, 100 μL of PBS were added per well with pH 7.4 or pH 5.5 for plasma membrane and endosome mimicking liposomes, respectively. Then, 1 μL of 1 mM mimicking liposomes was added to each well and LNP with different formulations (n = 3, per formulation) were added for a ratio of 1:10 (liposome:LNP by concentration). The control for minimal fluorescence (Fmin) was wells untreated with LNP; and the control for maximum fluorescence (Fmax) was treated with 8 μL of Triton X-100. Plate was incubated at 37 °C for 5, 15, 30, 45, 60, and 90 min. Finally, fluorescence for each time point was measured in a plate reader (Tecan), λexcitation = 465 nm, and λemission = 520 nm. Lipid fusion was calculated as (Fsample − Fmin)/(Fmax − Fmin) × 100.
Results and discussion
Selection of PLs and LNP formulation
Our lab and others have studied how the composition of LNPs should be engineered to productively deliver diverse nucleic acids including siRNA,17 miRNA,17 mRNA,52,57,58 sgRNA,43,59 and ribonucleoproteins.60 The LNPs used here are composed of four core components: dendrimer-based ionizable amino lipids, cholesterol, zwitterionic phospholipids, and PEG lipids. Dendrimer-based ionizable amino lipids have been systematically tested to determine the structure–activity relationship (SAR) between the amine core (4A3), the alkyl peripheries (SC8), and the nucleic acid delivery potential. 4A3-SC8 has been consistently active for the delivery of short (siRNA)17 to long (mRNA, sgRNA, ssDNA) nucleic acids.61,62A systematic series of LNPs were prepared by introducing a variable phospholipid in each formulation while maintaining the ionizable cationic lipid (4A3-SC8), cholesterol, and PEG-lipid (PEG-DMG) constant. In all LNPs, the molar ratio of the components was fixed at 4A3-SC8/cholesterol/PEG-DMG/variable phospholipid = 38.5/30/1.5/30 (mol mol−1). This strategy allowed us to systematically test the mRNA delivery efficiency of each phospholipid as a single variable in the context of LNPs. Each formulated LNP was characterized with respect to diameter, polydispersity index (PDI), zeta potential, and mRNA encapsulation (Fig. S1†). All formulated LNPs had RNA encapsulation above 80%, with the exception of CL-LNP which exhibited 50% encapsulation efficiency (Fig. S1A†). Zeta potential values for all LNPs were between −2 mV and −4 mV, (Fig. S1B†). LNPs were similar in size, approximately 100 nm (Fig. S1C†), and PDI values were below 0.2, indicating uniform size across all LNPs (Fig. S1D†). LNPs were stable at room temperature and at 4 °C for at least 72 hours regardless of the phospholipid used in the formulation (Fig. S2†). We concluded that the identity of the phospholipids studied in this manuscript did not significantly affect LNP stability. A gel retardation assay examining the LNPs and free mRNA shows that all LNPs successfully encapsulated mRNA and inhibited mRNA migration compared to free mRNA (Fig. S3†). The results agree with results determined by the Ribogreen assay (Fig. S1A†). Lastly, we used Transmission Electron Microscopy (TEM) to image the various LNPs as a function of the incorporated phospholipid (Fig. S4†). These results showed that there are no appreciable differences in morphology observed when different phospholipids are used to formulate the LNPs. All LNPs were spherical in shape. No obvious trends were observed in the physical characterization that explained the differences observed in the in vitro mRNA delivery (Fig. 1).
SAR determination of zwitterionic PLs for in vitro mRNA delivery
We initially evaluated each LNP for its ability to deliver luciferase (Luc) mRNA to two representative cell lines as a first readout. Waterfall plots revealed that PE-containing lipids (DOPE, POPE, SOPE, 4ME) were more efficient at in vitro mRNA delivery in HEK293T and HeLa cell lines than PC-containing lipids (Fig. 1C and D, respectively). Acknowledging that in vitro delivery potential does not always translate to in vivo delivery potential,28 top-performing phospholipids in LNPs contained hydrophobic chains with unsaturated bonds. Whether one chain or both chains contained an unsaturation seemed to have no difference in mRNA delivery efficiency. Additionally, the bulkier methyl-substituted chains present in 4ME facilitated mRNA delivery to the same degree of unsaturated/saturated chains. BMP (S, R) enhanced delivery compared with standardly used DSPC but less so when compared to top PE-containing lipids. The fusogenic nature of PE lipids may enhance in vitro LNP delivery of mRNA. LNPs can be formulated with different ionizable amino lipids, such as the benchmark C12-200, that might affect how PLs impact mRNA delivery. We found that C12-200 LNPs formulated with DOPE outperformed DSPC formulated C12-200 LNPs (Fig. S5†). These results suggest phospholipids could enhance LNP uptake resulting in enhanced delivery of mRNA into the cytosol. Alternatively, it could be a result of enhanced LNP fusion with endosomal membranes causing their destabilization and increasing endosomal escape of mRNA. To further investigate these questions, we quantified cellular uptake and endosomal escape.Uptake and transfection of LNPs for in vitro mRNA delivery
After identification of the top- and worst-performing LNPs, we set out to determine whether the limiting step for effective delivery could be uptake of LNPs or endosomal escape. LNPs enter cells through endocytosis pathways. For effective transfection, mRNA must escape the endosomes before they fuse with lysosomes and the mRNA is degraded. To quantify uptake, LNPs were formulated with Cy5-labeled mRNA to measure the percentage of cells that were positive for Cy5 fluorescence. Mean fluorescence intensity (MFI) was used to measure the amount of LNPs that were endocytosed by the cells. These results were coupled to parallel experiments involving delivery of mCherry mRNA. mCherry positive cells allowed us to quantify the effective transfection of mRNA, which reflects the amount of mRNA that escaped the endosome and was translated into protein. The distribution of Cy5 fluorescence showed there was no difference between the uptake of LNPs formulated with different phospholipids (Fig. 2A). This was confirmed by the number of Cy5 positive cells (Fig. 2B) and the Cy5 MFI observed for each treatment group (Fig. 2C). With respect to functional delivery, mCherry fluorescence emission matched the results of the in vitro luminescence screen (Fig. 2D–F). The same experiments were performed using HEK293T cells (Fig. S6†) confirmed the results observed in HeLa cells. Overall, we found that LNP uptake was not dependent on PL identity and did not explain the differences observed in effective mCherry mRNA delivery. These results suggest that phospholipids may play a role in facilitating the endosomal escape of mRNA. We therefore hypothesized that the increased endosomal escape was due to enhanced fusion with the endosomal membrane.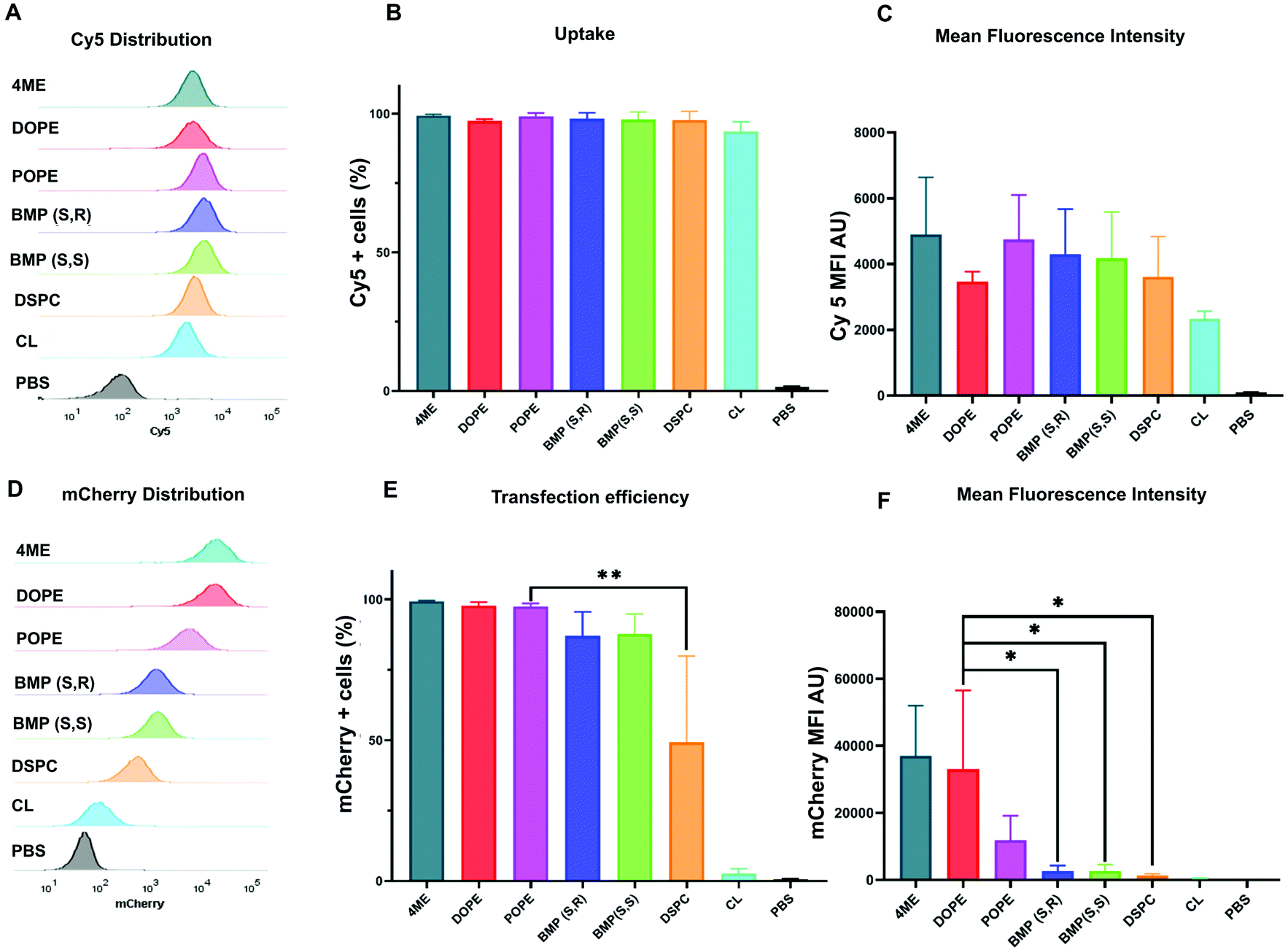 | ||
| Fig. 2 Uptake of Cy5-LNPs is not affected by phospholipid identity. Flow cytometry was used to determine (A) distribution of Cy5 fluorescence, (B) percentage of Cy5 positive cells, and (C) mean fluorescence intensity. Distribution of mCherry fluorescence (D), percentage of mCherry positive cells, (E) and mean fluorescence intensity of each LNP (F) obtained by flow cytometry after 48 h incubation of HeLa cells with Cy5-labeled mRNA-LNP (250 ng N = 4 ± stdev, one-way ANOVA **** p < 0.0001, Tukey's test ** p < 0.001, * p < 0.01. | ||
Endosomal escape of DOPE- and DSPC-LNPs
Flow cytometry experiments of Cy5-labeled LNPs ruled out uptake as the limiting step for increased mRNA delivery observed with the different formulations. This led us to consider that PLs may enhance endosomal escape of mRNA. If endosomal escape is enhanced by PLs, colocalization of Cy5-labeled LNPs and lysosomes would likely be affected (Fig. 3A). Confocal microscopy was used to quantify the colocalization between lysosomes and DOPE- and DSPC-LNPs, the top- and worst-performing LNPs, respectively. Fig. 3B includes representative confocal images of cells labelled with LysoTracker green and treated with Cy5 labelled DSPC- or DOPE-LNPs. The Pearson correlation coefficient was used to measure the colocalization of lysosomes and LNPs. The Pearson coefficient can have values from −1 to 1, values close to −1 indicate the two signals oppose themselves; 0, no colocalization; and 1, perfect colocalization.63 Larger sized images and additional images used to calculate the Pearson's Coefficient are included in Fig. S7 and S8.† The Pearson's coefficient values are listed in Fig. S9.† DSPC-LNPs had a higher coefficient than DOPE-LNPs (Fig. 3C), indicating that DSPC-LNPs remain more trapped in the lysosomes failing to deliver mRNA into the cytosol adequately.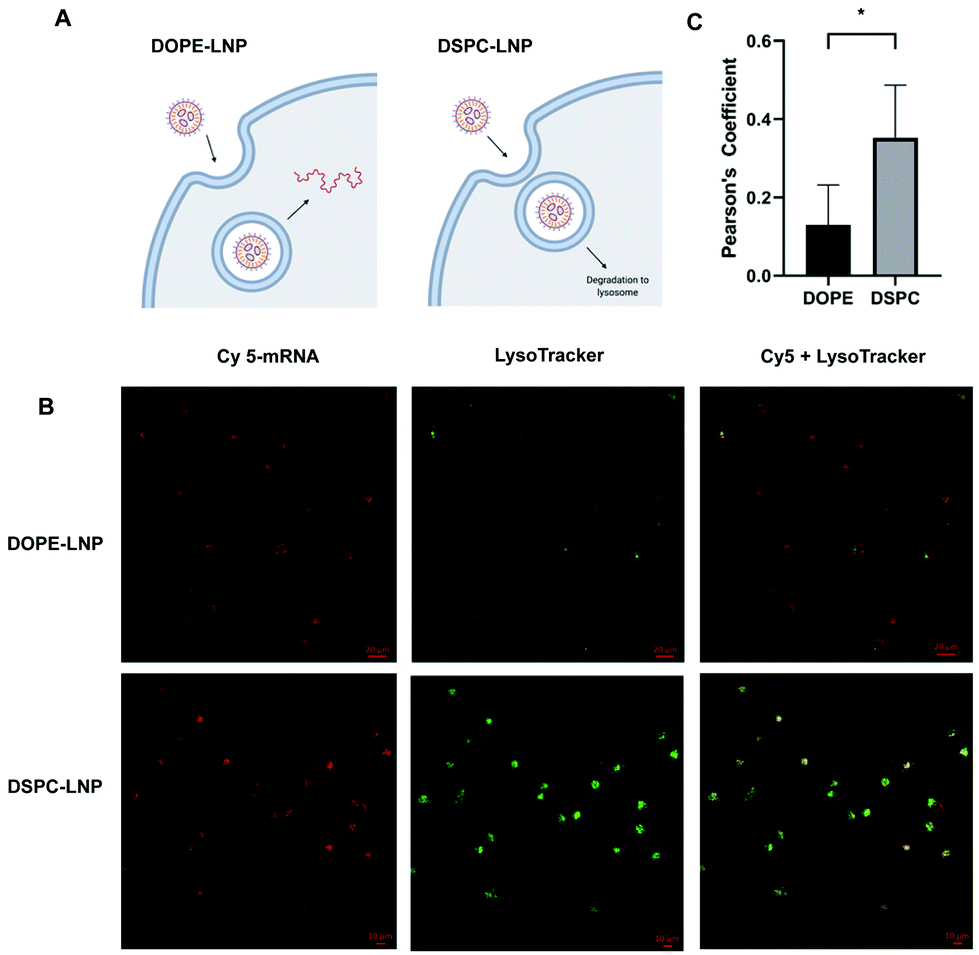 | ||
| Fig. 3 DOPE enhances endosomal escape. (A) Proposed model of endosomal escape for DOPE-LNP and DSPC-LNP. (B) Representative images of HeLa cells treated with Cy5-labeled mRNA (280 ng) LNPs (red), stained with LysoTracker (green) imaged 24 h later using confocal microscopy. (C) Pearson coefficient of colocalization between LNPs (red) and lysosomes (green) obtained using the PSC Colocalization plugin from ImageJ. (C) N = 4 ± stdev, Unpaired t test * p < 0.05. | ||
LNP fusion with membrane mimicking liposomes
Uptake and transfection experiments led us to hypothesize that PLs in LNPs could be enhancing endosomal escape. This was confirmed by higher colocalization of DSPC-LNPs with lysosomes, which have consistently achieved the lowest levels of mRNA transfection. In contrast, DOPE-LNPs had lower colocalization with lysosomes. PE lipids have been highlighted for their ability to change their conformation from cone shape to HII at endosomal pH, which creates membrane instability and promotes fusion.50 We set out to determine if the fusogenic nature some PLs would enhance LNP mixing with membrane mimicking liposomes. This would allow us to further understand PL interactions with biological membranes in the context of LNPs and determine whether phospholipids can enhance endosomal escape in an in vitro model system.The lipid content (molar percentage) of the plasma membrane (PM) and the late endosome membrane (LEM) were mimicked, such that model plasma membrane liposomes had a higher concentration of cholesterol and phosphatidylserine, whereas late endosome model membranes were rich in BMP.64 Analogs of DOPE N-Rh-PE and NBD-PE—fluorophore and quencher, respectively—were included to label the resulting liposomes and enable membrane fusion detection via FRET. The dequenching of N-Rh-PE measured lipid fusion as a result of LNP-induced mixing with liposomes (Fig. 4A).
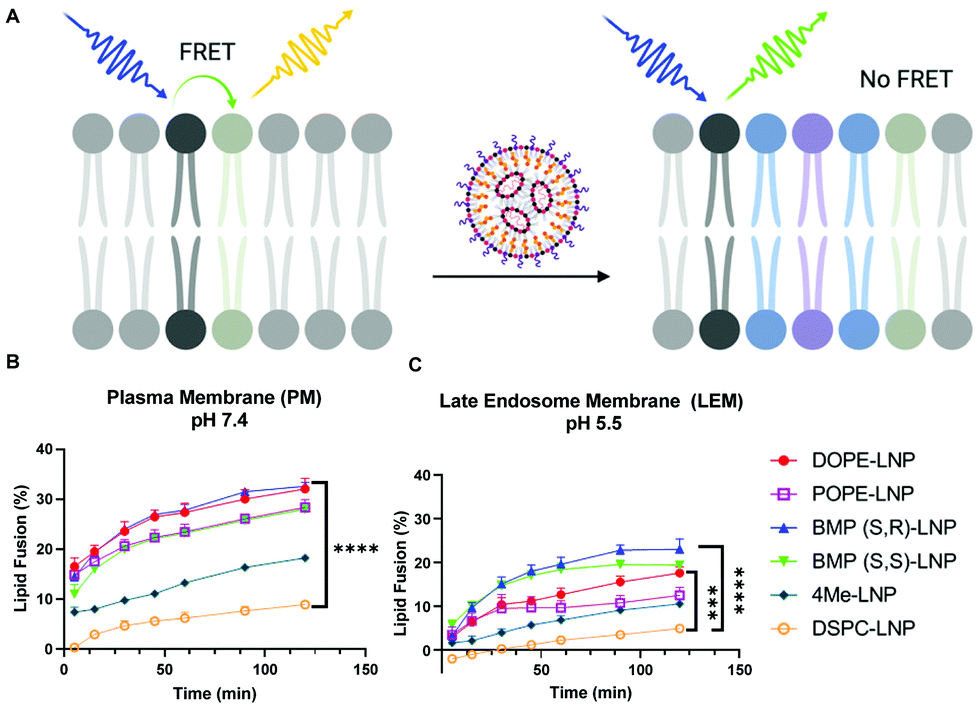 | ||
| Fig. 4 DOPE- and BMP-LNP enhance lipid fusion with liposome-mimicking biological membranes. (A) Lipid fusion model of biological membrane mimic in which a liposome containing a quencher lipid (black) and fluorescent lipid (light green) is mixed with unlabeled LNP(blue). Separation of the FRET pair increases 520 nm emission measured in a plate reader. Results of lipid fusion experiments at different time points after incubation of LNPs with plasma membrane (B) and endosomal membrane (C) mimicking liposomes. N = 3 ± stdev, one-way ANOVA **** p < 0.0001, Tukey's test ** p < 0.001, * p < 0.01. | ||
PE-containing LNPs, as well as BMP-LNPs, had higher lipid mixing with both PM (Fig. 4B) and LEM (Fig. 4C) liposome model membranes. DSPC-LNPs consistently displayed the weakest ability to fuse with membranes. These results agree with previous reports that highlight DOPE's fusogenic nature.47,53 In contrast, DSPC has a molecular shape that resembles a cylinder that provides stability to bilayer membranes.30 The Pearson's coefficient values obtained in Fig. 3B indicate that DSPC-LNPs colocalize with lysosomes more than DOPE-LNP. The lipid mixing results—taken together with the lysosomal colocalization coefficients—further suggest that PE-formulated LNPs enhance endosomal escape. Conversely, due to DSPC-LNP's lack of lipid mixing, these LNPs are less able to escape the endosomes and less mRNA is delivered effectively to the cytosol.
Optimized LNPs delivered mRNA in vivo
After characterizing LNP-mediated mRNA delivery efficacy in vitro, we next studied whether the results obtained translated in vivo. Organ luminescence was quantified 6 hours after IV injection of low dose 0.03 mg kg−1 Luc mRNA in LNPs with variable PLs. The 6-hour time point was used for all in vivo experiments because it has been previously determined to be the time point with highest protein expression after mRNA delivery.52 DOPE-, POPE-, and 4ME-LNPs achieved the highest luminescence overall, which targeted mainly the liver (Fig. 5A and C). BMP-LNPs did not have luminescence as high as PE-containing LNPs (Fig. 5A and C). Interestingly, BMP-LNPs predominantly delivered to the spleen (Fig. 5B). Overall, PE-containing LNPs outperformed BMP- and PC-containing LNPs both in vitro and in vivo. BMP-based LNPs could provide an advantage for immune cell delivery as they also outperformed PC-LNPs and offered spleen tropism.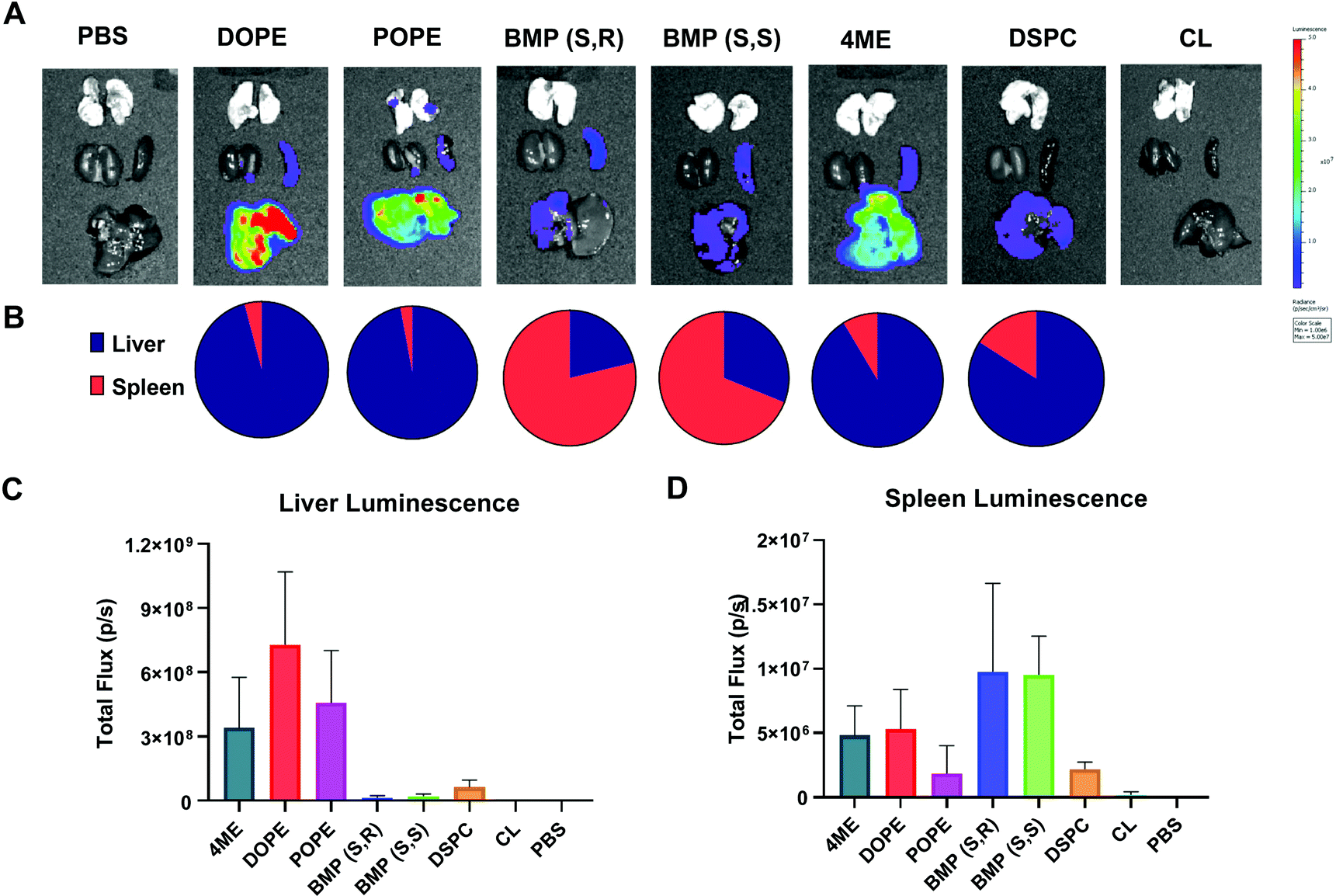 | ||
| Fig. 5 DOPE-, POPE-, and 4ME-LNPs are liver predominant whereas BMP-LNPs are spleen predominant. Female C57BL/6 were injected 0.03 mg kg−1 Fluc mRNA IV and imaged 6 h later after IP injection of luciferin. (A) IVIS Lumina imaging of dissected organs after IV injection of Fluc mRNA (0.03 mg kg−1). (B) Liver to spleen ratio of luminescence. Quantification of total and average luminescence in liver (C) and spleen (D). N = 3 ± stdev, one-way ANOVA **** p < 0.0001, Tukey's test ** p < 0.001, * p < 0.01. | ||
Application of PLs to the formation of selective ORgan targeting (SORT) LNPs
Our lab recently developed a strategy called Selective ORgan Targeting (SORT), wherein the addition of a SORT molecule to LNPs creates SORT LNPs that can target specific organs including the liver, lungs, and spleen (Fig. 6A).57–60 We discovered that incorporation of a SORT lipid (e.g. permanently cationic lipids, anionic lipids, zwitterionic lipids, and ionizable amino lipids) enables tissue-specific gene delivery and gene editing as a function of the chemistry and amount of the added supplemental molecule. Notably, inclusion of anionic lipids promoted exclusive protein expression in the spleen. From our results in Fig. 5A and B, the four-component formulation that contained anionic BMP exhibited preference for spleen delivery over liver delivery and could be considered a SORT LNP. BMPs are negatively charged lipids and aid spleen delivery, which is consistent with our previous results.59 Therefore, we decided to further explore the role of BMP as a SORT molecule in five-component LNPs to increase efficacy.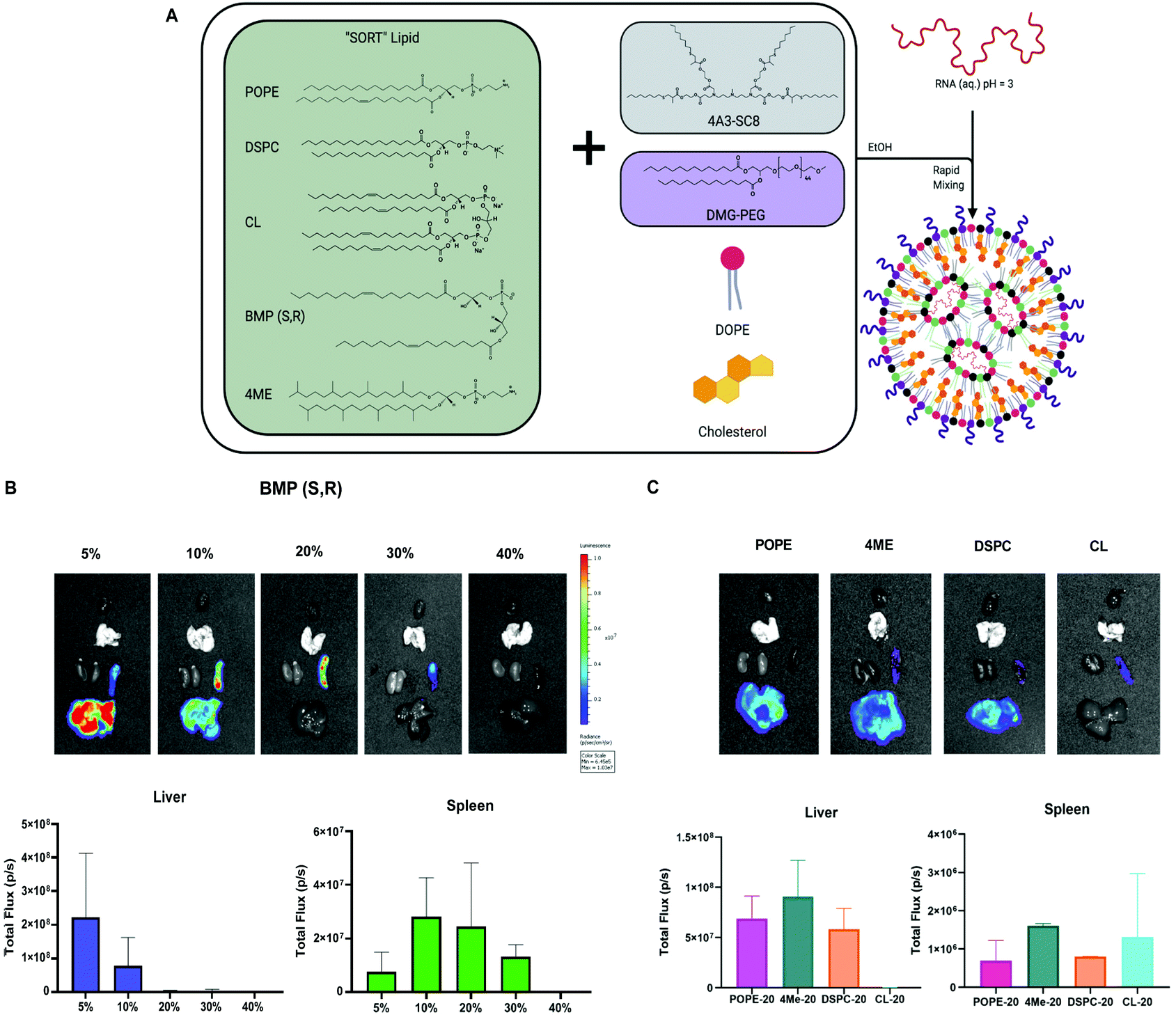 | ||
| Fig. 6 Phospholipids function in SORT LNPs to drive spleen and liver organ targeting. (A) Model of SORT LNP composition and formulation process. (B) Evaluation of anionic BMP phospholipid as a SORT molecule for spleen and liver mRNA delivery. (C) Evaluation of zwitterionic phospholipids as SORT molecules. | ||
We formulated LNPs using 4A3-SC8:DOPE:cholesterol:PEG following the molar ratios of the basic SORT LNP, 15:15:30:3. Next, we titrated in BMP to achieve 5, 10, 20, 30 and 40% of the total lipid composition. A table summarizing the molar ratios is present in Fig. S10.† Each SORT LNP was characterized with respect to diameter, polydispersity index (PDI), zeta potential, and mRNA encapsulation (Fig. S11†) and no differences were observed that explained the results of the in vivo mRNA delivery. After IV injection, we were able to obtain spleen specific delivery at 20 and 30%.
Since 20% incorporation enabled the highest luminescence, we decided to focus on a series of formulations at 20% to study the remaining PLs as SORT molecules. The LNPs containing 4ME, POPE and DSPC achieved similar delivery efficiency to the liver when incorporated into the SORT formulation at 20%. Interestingly, CL-LNP exhibited spleen-specific delivery although lower in efficacy (Fig. 6C). These results indicate that choice of PLs in a four component LNP increase overall delivery efficiency. Moreover, in the context of SORT LNPs, PLs can change organ tropism from liver to spleen and further increase spleen delivery efficacy.
Conclusions
The field of mRNA therapeutics has quickly gained importance due to the worldwide impact of mRNA LNP vaccines. As the mRNA field moves forward, it is important to understand the fundamental role(s) of all lipid components that comprise LNPs. Although efforts have mainly focused on the ionizable cationic lipid, understanding the role of the other molecules within LNPs are important for safety and efficacy. Here, we demonstrated that the chemistry of the phospholipid plays an important role in endosomal escape and can enhance the delivery of mRNA both in vitro and in vivo when incorporated as part of a 4-component LNP. Furthermore, we demonstrated that negatively charged lipids drive spleen tropism in vivo in the context of 4- and 5-component SORT LNPs. Together these results break the paradigm of the phospholipid being considered as a “helper lipid”. It has also been observed that PLs can alter the activity of LNPs designed for siRNA and DNA.46,65,66 Studying the roles of PLs in additional LNP types and for different cargoes is an important area of ongoing and future research. Moreover, we demonstrated how alternative phospholipids in LNP formulations can enhance delivery and offer organ targeting. Enhanced in vivo delivery of mRNA LNPs via inclusion of PLs with higher endosomal escape potential is expected to lead to improved mRNA vaccines against SARS-CoV-2 and future viruses, as well as improve development of novel therapeutics against a variety of diseases.Author contributions
Ester Álvarez-Benedicto: conceptualization, methodology, investigation, formal analysis, writing – original draft, writing – review & editing, visualization, funding acquisition. Lukas Farbiak: conceptualization, methodology, investigation, writing – review & editing, visualization. Martha Márquez Ramírez: investigation, writing – review & editing. Lindsay T. Johnson: investigation, writing – review & editing. Xu Wang: investigation. Osamah Mian: investigation, writing – review & editing. Erick D. Guerrero: investigation. Daniel J. Siegwart: conceptualization, methodology, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. J. S. acknowledges financial support from the National Institutes of Health (NIH) National Institute of Biomedical Imaging and Bioengineering (NIBIB) (R01 EB025192-01A1), the Cystic Fibrosis Foundation (CFF) (SIEGWA18XX0), the Cancer Prevention and Research Institute of Texas (CPRIT) (RP190251), the American Cancer Society (ACS) (RSG-17-012-01), and the Welch Foundation (I-1855). We acknowledge the UTSW Tissue Resource, supported in part by the National Cancer Institute (5P30CA142543), and the Moody Foundation Flow Cytometry Facility. E. A. B acknowledges financial support from the National Science Foundation Graduate Research Fellowship Program (NSF GRFP) (2018270395). D. J. S. and E. A. B. acknowledge support from the Howard Hughes Medical Institute (GT15136).Notes and references
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed.
- K. Whitehead, R. Langer and D. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- K. J. Kauffman, M. J. Webber and D. G. Anderson, J. Controlled Release, 2016, 240, 227–234 CrossRef CAS PubMed.
- H. X. Wang, M. Li, C. M. Lee, S. Chakraborty, H. W. Kim, G. Bao and K. W. Leong, Chem. Rev., 2017, 117, 9874–9906 CrossRef CAS PubMed.
- K. A. Hajj and K. A. Whitehead, Nat. Rev. Mater., 2017, 2, 17056 CrossRef CAS.
- J. B. Miller and D. J. Siegwart, Nano Res., 2018, 11, 5310–5337 CrossRef CAS.
- T. Wei, Q. Cheng, L. Farbiak, D. G. Anderson, R. Langer and D. J. Siegwart, ACS Nano, 2020, 14, 9243–9262 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- L. A. Jackson, E. J. Anderson, N. G. Rouphael, P. C. Roberts, M. Makhene, R. N. Coler, M. P. McCullough, J. D. Chappell, M. R. Denison, L. J. Stevens, A. J. Pruijssers, A. McDermott, B. Flach, N. A. Doria-Rose, K. S. Corbett, K. M. Morabito, S. O’Dell, S. D. Schmidt, P. A. Swanson, II, M. Padilla, J. R. Mascola, K. M. Neuzil, H. Bennett, W. Sun, E. Peters, M. Makowski, J. Albert, K. Cross, W. Buchanan, R. Pikaart-Tautges, J. E. Ledgerwood, B. S. Graham and J. H. Beigel, N. Engl. J. Med., 2020, 383, 1920–1931 CrossRef CAS PubMed.
- A. Wittrup, A. Ai, X. Liu, P. Hamar, R. Trifonova, K. Charisse, M. Manoharan, T. Kirchhausen and J. Lieberman, Nat. Biotechnol., 2015, 33, 870–876 CrossRef CAS PubMed.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS PubMed.
- I. M. Hafez, N. Maurer and P. R. Cullis, Gene Ther., 2001, 8, 1188–1196 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. X. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. M. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- D. F. Zhi, S. B. Zhang, S. H. Cui, Y. A. Zhao, Y. H. Wang and D. F. Zhao, Bioconjugate Chem., 2013, 24, 487–519 CrossRef CAS PubMed.
- K. Zhou, L. H. Nguyen, J. B. Miller, Y. Yan, P. Kos, H. Xiong, L. Li, J. Hao, J. T. Minnig, H. Zhu and D. J. Siegwart, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 520–525 CrossRef CAS PubMed.
- O. Zelphati and F. C. Szoka, Jr., Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11493–11498 CrossRef CAS PubMed.
- N. Pardi, S. Tuyishime, H. Muramatsu, K. Kariko, B. L. Mui, Y. K. Tam, T. D. Madden, M. J. Hope and D. Weissman, J. Controlled Release, 2015, 217, 345–351 CrossRef CAS PubMed.
- S. Ramishetti, I. Hazan-Halevy, R. Palakuri, S. Chatterjee, S. Naidu Gonna, N. Dammes, I. Freilich, L. Kolik Shmuel, D. Danino and D. Peer, Adv. Mater., 2020, 32, e1906128 CrossRef PubMed.
- K. Paunovska, A. J. D. Sanchez, C. D. Sago, Z. B. Gan, M. P. Lokugamage, F. Z. Islam, S. Kalathoor, B. R. Krupczak and J. E. Dahlman, Adv. Mater., 2019, 31, 1807748 CrossRef PubMed.
- S. Patel, N. Ashwanikumar, E. Robinson, Y. Xia, C. Mihai, J. P. Griffith, S. Hou, A. A. Esposito, T. Ketova, K. Welsher, J. L. Joyal, O. Almarsson and G. Sahay, Nat. Commun., 2020, 11, 983 CrossRef CAS PubMed.
- Y. J. Bao, Y. Jin, P. Chivukula, J. Zhang, Y. Liu, J. Liu, J. P. Clamme, R. I. Mahato, D. Ng, W. B. Ying, Y. T. Wang and L. Yu, Pharm. Res., 2013, 30, 342–351 CrossRef CAS PubMed.
- Q. G. Xu, L. M. Ensign, N. J. Boylan, A. Schon, X. Q. Gong, J. C. Yang, N. W. Lamb, S. T. Cai, T. Yu, E. Freire and J. Hanes, ACS Nano, 2015, 9, 9217–9227 CrossRef CAS PubMed.
- X. W. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Y. Du, Y. Y. C. Tam, P. J. C. Lin, S. Chen, J. K. Narayanannair, K. G. Rajeev, M. Manoharan, A. Akinc, M. A. Maier, P. Cullis, T. D. Madden and M. J. Hope, Mol. Ther.–Nucleic Acids, 2013, 2, e139 CrossRef CAS PubMed.
- S. C. Wilson, J. L. Baryza, A. J. Reynolds, K. Bowman, M. E. Keegan, S. M. Standley, N. P. Gardner, P. Parmar, V. O. Agir, S. Yadav, A. Zunic, C. Vargeese, C. C. Lee and S. Rajan, Mol. Pharm., 2015, 12, 386–392 CrossRef CAS PubMed.
- K. A. Whitehead, J. Matthews, P. H. Chang, F. Niroui, J. R. Dorkin, M. Severgnini and D. G. Anderson, ACS Nano, 2012, 6, 6922–6929 CrossRef CAS PubMed.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biochem., 1965, 13, 238–IN227 CAS.
- P. R. Cullis, M. J. Hope and C. P. S. Tilcock, Chem. Phys. Lipids, 1986, 40, 127–144 CrossRef CAS PubMed.
- R. Yang, X. Zhang, F. Li, L. Ding, B. Li, H. Sun and Y. Gan, Colloids Surf., A, 2013, 436, 434–442 CrossRef CAS.
- R. Kolašinac, C. Kleusch, T. Braun, R. Merkel and A. Csiszár, Int. J. Mol. Sci., 2018, 19, 346 CrossRef PubMed.
- V. K. Sharma, K. K. Sarwa and B. Mazumder, J. Liposome Res., 2014, 24, 83–89 CrossRef CAS PubMed.
- C. Rupp, H. Steckel and B. W. Müller, Int. J. Pharm., 2010, 395, 272–280 CrossRef CAS PubMed.
- M. Anderson and A. Omri, Drug Delivery, 2004, 11, 33–39 CrossRef CAS PubMed.
- G. Gregoriadis, P. D. Leathwood and B. E. Ryman, FEBS Lett., 1971, 14, 95–99 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- M. H. Gaber, K. Hong, S. K. Huang and D. Papahadjopoulos, Pharm. Res., 1995, 12, 1407–1416 CrossRef CAS PubMed.
- F. Passero, D. Grapsa, K. Syrigos and W. Saif, Expert Rev. Anticancer Ther., 2016, 16, 697–703 CrossRef CAS PubMed.
- E. A. Forssen and M. E. Ross, J. Liposome Res., 1994, 4, 481–512 CrossRef.
- R. Zhang, R. El-Mayta, T. J. Murdoch, C. C. Warzecha, M. M. Billingsley, S. J. Shepherd, N. Gong, L. Wang, J. M. Wilson, D. Lee and M. J. Mitchell, Biomater. Sci., 2021, 9, 1449–1463 RSC.
- A. K. K. Leung, I. M. Hafez, S. Baoukina, N. M. Belliveau, I. V. Zhigaltsev, E. Afshinmanesh, D. P. Tieleman, C. L. Hansen, M. J. Hope and P. R. Cullis, J. Phys. Chem. C, 2012, 116, 22104–22104 CrossRef CAS.
- J. B. Miller, S. Zhang, P. Kos, H. Xiong, K. Zhou, S. S. Perelman, H. Zhu and D. J. Siegwart, Angew. Chem., Int. Ed., 2017, 56, 1059–1063 CrossRef CAS PubMed.
- K. J. Kauffman, J. R. Dorkin, J. H. Yang, M. W. Heartlein, F. DeRosa, F. F. Mir, O. S. Fenton and D. G. Anderson, Nano Lett., 2015, 15, 7300–7306 CrossRef CAS PubMed.
- Y. Dong, J. R. Dorkin, W. Wang, P. H. Chang, M. J. Webber, B. C. Tang, J. Yang, I. Abutbul-Ionita, D. Danino, F. DeRosa, M. Heartlein, R. Langer and D. G. Anderson, Nano Lett., 2016, 16, 842–848 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, J. Leung, Y. Y. C. Tam and P. R. Cullis, Nanoscale, 2019, 11, 21733–21739 RSC.
- J. Li, X. Wang, T. Zhang, C. Wang, Z. Huang, X. Luo and Y. Deng, Asian J. Pharm. Sci., 2015, 10, 81–98 CrossRef.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- S. Simões, V. Slepushkin, N. Düzgünes and M. C. Pedroso De Lima, Biochim. Biophys. Acta, Biomembr., 2001, 1515, 23–37 CrossRef.
- H. Ellens, J. Bentz and F. C. Szoka, Biochemistry, 1984, 23, 1532–1538 CrossRef CAS PubMed.
- B. Li, X. Luo, B. Deng, J. Wang, D. W. McComb, Y. Shi, K. M. Gaensler, X. Tan, A. L. Dunn, B. A. Kerlin and Y. Dong, Nano Lett., 2015, 15, 8099–8107 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, Y. Jia, L. Farbiak, K. Zhou, S. Zhang, Y. Wei, H. Zhu and D. J. Siegwart, Adv. Mater., 2018, 30, e1805308 CrossRef PubMed.
- Z. Du, M. M. Munye, A. D. Tagalakis, M. D. I. Manunta and S. L. Hart, Sci. Rep., 2014, 4, 7107 CrossRef PubMed.
- K. K. L. Phua, H. F. Staats, K. W. Leong and S. K. Nair, Sci. Rep., 2015, 4, 5128 CrossRef PubMed.
- N. Veiga, M. Goldsmith, Y. Granot, D. Rosenblum, N. Dammes, R. Kedmi, S. Ramishetti and D. Peer, Nat. Commun., 2018, 9, 4493 CrossRef PubMed.
- Y. Yan, L. Liu, H. Xiong, J. B. Miller, K. Zhou, P. Kos, K. E. Huffman, S. Elkassih, J. W. Norman, R. Carstens, J. Kim, J. D. Minna and D. J. Siegwart, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5702–E5710 CrossRef CAS PubMed.
- S. M. Lee, Q. Cheng, X. Yu, S. Liu, L. T. Johnson and D. J. Siegwart, Angew. Chem., Int. Ed., 2021, 60, 5848–5853 CrossRef CAS PubMed.
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson, L. Farbiak and D. J. Siegwart, Nat. Mater., 2021, 20, 701–710 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- T. Wei, Q. Cheng, Y.-L. Min, E. N. Olson and D. J. Siegwart, Nat. Commun., 2020, 11, 3232 CrossRef CAS PubMed.
- H. Xiong, S. Liu, T. Wei, Q. Cheng and D. J. Siegwart, J. Controlled Release, 2020, 325, 198–205 CrossRef CAS PubMed.
- L. Farbiak, Q. Cheng, T. Wei, E. Alvarez-Benedicto, L. T. Johnson, S. Lee and D. J. Siegwart, Adv. Mater., 2021, 33, e2006619 CrossRef PubMed.
- A. P. French, S. Mills, R. Swarup, M. J. Bennett and T. P. Pridmore, Nat. Protoc., 2008, 3, 619–628 CrossRef CAS PubMed.
- S.-T. Yang, E. Zaitseva, L. V. Chernomordik and K. Melikov, Biophysi. J., 2010, 99, 2525–2533 CrossRef CAS PubMed.
- Y. Sato, H. Hatakeyama, Y. Sakurai, M. Hyodo, H. Akita and H. Harashima, J. Controlled Release, 2012, 163, 267–276 CrossRef CAS PubMed.
- I. Ermilova and J. Swenson, Phys. Chem. Chem. Phys., 2020, 22, 28256–28268 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting Fig. S1–S11 and additional references. See DOI: 10.1039/d1bm01454d |
| This journal is © The Royal Society of Chemistry 2022 |