
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduced axial chirality by a tight belt: naphthalene chromophores fixed in a 2,5-substituted cofacial para-phenylene–ethynylene framework†
Eric
Sidler
 a,
Juraj
Malinčík
a,
Alessandro
Prescimone
a and
Marcel
Mayor
*abc
a,
Juraj
Malinčík
a,
Alessandro
Prescimone
a and
Marcel
Mayor
*abc
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, Basel 4056, Switzerland. E-mail: Marcel.Mayor@unibas.ch; Web: https://www.chemie1.unibas.ch/~mayor/
bInstitute for Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), P. O. Box 3640, Karlsruhe 76021, Germany
cLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-Sen University (SYSU), Guangzhou 510275, China
First published on 28th July 2021
Abstract
We report the design of a synthetically easy accessible axial chirality-inducing framework for a chromophore of choice. The scaffold consists of two basic para-phenylene-ethynylene backbones separated by laterally placed corner units. Substitution with an inherently achiral chromophore at the 2 and 5 positions of the central phenylene excitonically couples the chromophore associated transition and thereby results in chiroptical properties. Using 6-methoxynaphthalene as a model chromophore, we present the synthesis, structural analysis and spectroscopic investigation of the framework. The chiral framework was synthesized in three straightforward synthetic steps and fully characterized. The obtained racemic compounds were resolved using HPLC and assignment of the absolute configuration was performed using the exciton chirality method, crystallography and DFT calculations. This simple yet potent framework might prove useful to enrich the structural diversity of chiral materials.
Introduction
Chiral chromophores belong to a group of molecules that have caught the attention of chemists for years. Combining spectroscopic properties of chromophores (e.g., absorption and luminescence) with chirality induces remarkable properties such as circular dichroism (CD)1 and circularly polarized luminescence (CPL).2,3 The differential absorption (CD) and emission (CPL) of left- and right-handed circularly polarized light are quantum mechanical phenomena and materials bearing these features have found application in a wide range of fields. CD- and/or CPL-active materials are used for display technologies,4–6 structural analysis,7–10 spin filters,11,12 sensors13,14 or photoswitches15–17 and therefore remain attractive targets for synthetic chemists.Chiroptical materials that have found application in technology are often self-assembled nanostructures or polymeric aggregates. This way, powerful chromophores such as rylenes,18–20 porphyrins21,22 or other polyaromatic hydrocarbons,23,24 which are usually planar and therefore inherently achiral, can be assembled in chiral superstructures, and thereby gain chiroptical properties. Compared to higher ordered structures, single chiral molecules25,26 have some important advantages, such as higher solubility and easier synthetic access. Prominent examples thereof are chiral helicenes featuring exceptional chiroptics due to their distorted π-system.27–29 Although there are many contributions to induce chirality also into planar chromophores, these strategies require intense synthetic investigations and are usually not suitable for a large variety of chromophore types.30–32 Having a simple framework at hand that allows to fix an achiral chromophore in a chiral environment would provide chiroptical properties without requiring the formation of chiral superstructures. A few of such dedicated axially chiral frameworks have already been reported. For example, Nakashima and coworkers reported a chiral binaphthylene moiety that was substituted with two perylenebiscarboxydiimide (PDI).33 The resulting dimer is highly emissive and CPL-active, and although the emissive state stems from achiral chromophore, the model compounds demonstrates the power of chiral organization of achiral chromophores. Applying a similar strategy, the group of Siegel reported the synthesis and investigation of a diaminobiphenyl substituted with PDIs.34 The bichromophoric system shows strong CD and CPL activity induced by the chiral backbone and excitonically couples the chromophoric transitions. Recently, an unprecedented structural design of a chiral molecule bearing an inherently achiral chromophore was reported.35 Using a simple substitution reaction, a chiral binaphthol (BINOL) was attached to an achiral boron dipyrromethene (BODIPY), by which the strong luminophore was enriched with chiroptical properties showing CPL levels similar to single molecules with much more complex structures.
Inspired by these simple yet powerful chirality inducing structural frameworks, we envisioned to complement the structural diversity of chiral chromophores by designing a much more rigid framework that allows bringing four chromophores in an optically stable chiral environment. In this work, we show the synthesis, structural analysis, chiral resolution and spectroscopic investigation of our framework design. Additionally, spectroscopic data are supported by density-functional theory (DFT) calculations.
Results & discussion
Design
The design concept is sketched in Fig. 1. Profiting from a simple rectangular scaffold, the four chromophores (red) are fixed in a chiral environment. Following along the yellow arrow of the illustration, the chromophores above the plane of the framework form a right-handed helix, and likewise do the chromophores below the plane. A pair of laterally placed corner units (brown wooden parts) separates the two opposed frameworks (metal bar with central wooden log) from each other and fixes the scaffold in its position. Rotation around the framework axis (metal bars) of one of the opposed sides would result in the achiral isomer depicted in Fig. 1b. If the chromophore of choice is of sufficient dimension, rotation around the framework axis is sterically hindered. The dimensionality of the chromophore not only rigidifies the entire architecture, it might even favor the formation of helical chiral species during macrocyclization. Note that the spatial distance between both pairs of cofacially arranged chromophores is larger in the chiral arrangement (Fig. 1a) compared with their achiral parallel arrangement (Fig. 1b). Choosing an adequate size of the chromophore and corner unit should inhibit rotation, and should render the entire tetra-chromophore architecture optically stable. Such a helical arrangement not only results in chiroptical properties from achiral chromophores, their spatial proximity combined with their nonparallel transition dipole moments (TDMs) might result in Davydov splittings of the associated transitions, which is particularly interesting for singlet fission.36–38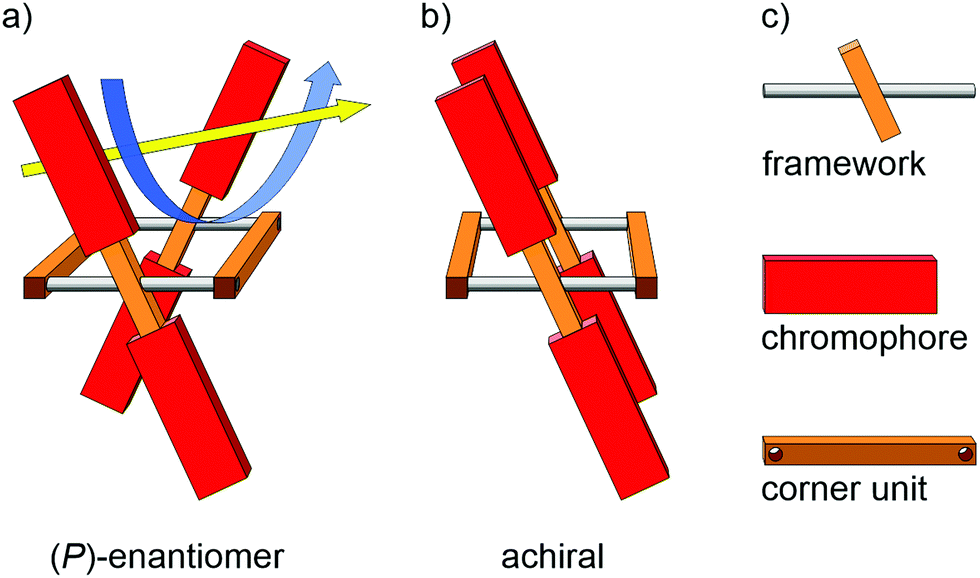 | ||
| Fig. 1 Concept to fix the spatial arrangement of four chromophores by a tight rectangular scaffold. (a) The (P)-enantiomer is depicted to display the helical chirality emerging from the fixed chromophores. The yellow arrow is the view direction helping to spot the helically arranged chromophores (visualized by the blue arrow). (b) Illustration of the achiral configuration with parallel arranged chromophores. (c) Legend explaining the various subunits of the scaffold. | ||
The molecular subunits considered to realize the intended concept are displayed in Fig. 2a. As rigid framework mounting two chromophores with an angle across the main axis a 2,5-substituted para-phenylene–ethynylene (PE) was chosen. The 2 and 5 positions were substituted with the chromophore of choice. Beside the rigidity and the rotational degree of freedom along the main axis, the PE additionally provides electronic communication between both chromophores though the central π-system, a favorable feature which was reported to effectively enhance the CPL properties of axially chiral compounds.39 A 1,4-substituted cyclohexa-2,5-diene was identified as the optimal corner unit structure, as they are generally stable towards the intended reaction conditions, provide an ideal angle and have been shown to be applicable for a plethora of macrocyclization reactions. In order to introduce more flexibility into the system, 1,4-substituted cyclohexane was also considered as potential corner unit. Moreover, it precludes any through-bond communication between the two opposed PEs that might be present to some extent in the cyclohexadiene moieties due to homo-conjugation through the sp3-hybridized carbons. 6-Methoxynaphthalene was chosen as a model chromophore since the electronic configuration and optical properties of naphthalene have been studied intensively,40,41 and its oblongness fulfills the dimensional criteria guaranteeing the structural integrity and thus also the optical stability of the resulting architecture. Furthermore, naphthalenes are prominent chromophores in excitionically coupled systems.42–44
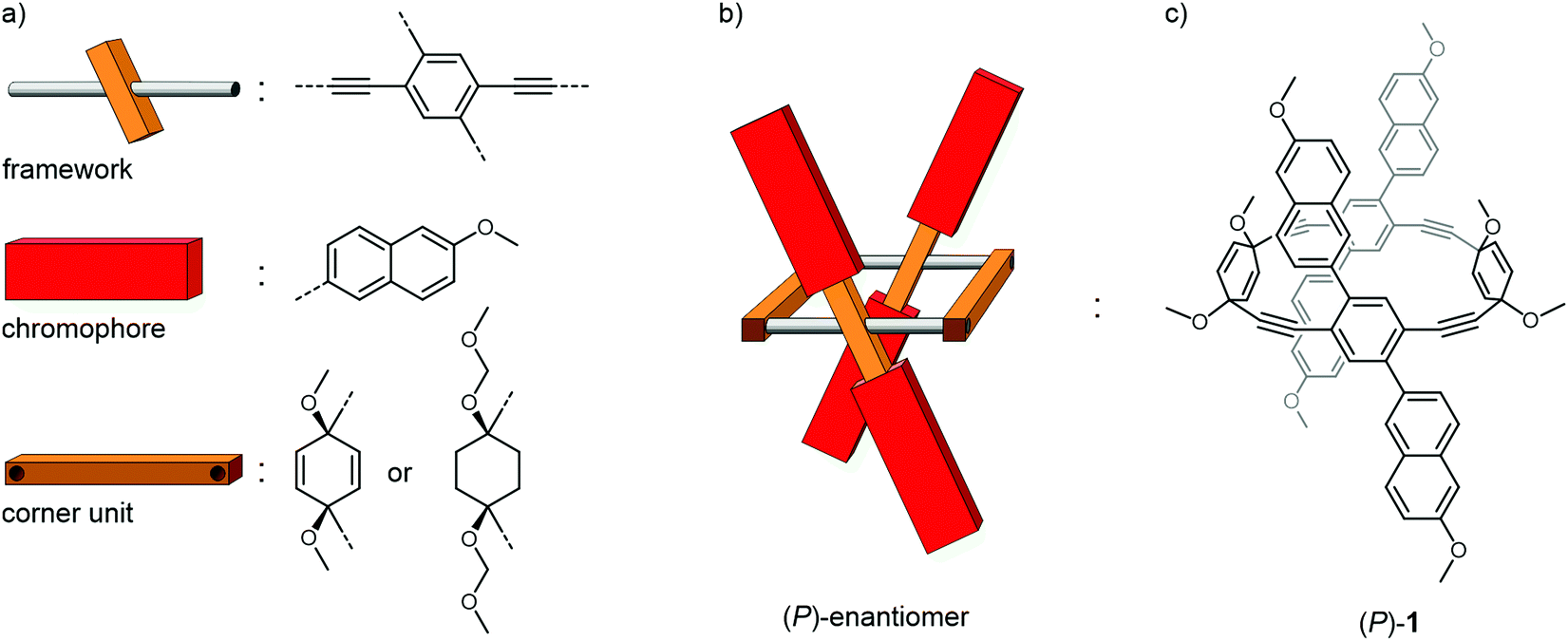 | ||
| Fig. 2 (a) The molecular subunits representing the functional building blocks. (b) Conceptual picture of the (P)-enantiomer and (c) its realization with the molecular subunits using 1,4-cyclohexadiene corner units resulting in the target structure (P)-1. | ||
Retrosynthesis
Retrosynthetically, the frameworks with either cyclohexa-2,5-diene or cyclohexane as corner units can be treated analogously. The elaborated retrosynthetic analysis is shown in Scheme 1. A first disconnection of the racemic dimeric macrocycles rac-1 or rac-2 relies on a twofold Sonogashira cross coupling of 3 or 4 after removal of the (triisopropyl)silyl (TIPS) protecting group. High-dilution conditions should minimize the oligo- and polymerization reactions associated with non-templated macrocyclization reactions and hence increase the obtained yield. Monomer 3 is conveniently disconnected to the literature known ethynylene substituted cyclohexa-2,5-diene 645 bearing two orthogonal silyl alkyne protecting groups and 2,5-naphthyl substituted 1,4-dibromobenzene 5. Correspondingly, 4 can be divided into a suitably protected 1,4-ethynylene substituted cyclohexane 7 and 5. Corner unit 7 should be accessible by 1,2-carbonyl additions of the respective lithium acetylides on 1,4-cyclohexandione, in an analogous manner to the reported synthesis of 6. In a final retrosynthetic step, 5 should be accessible by a classical Suzuki cross coupling between commercially available 1,4-diiodo-2,5-dibromobenzene and a suitable boronic acid derivative of the naphthalene building block.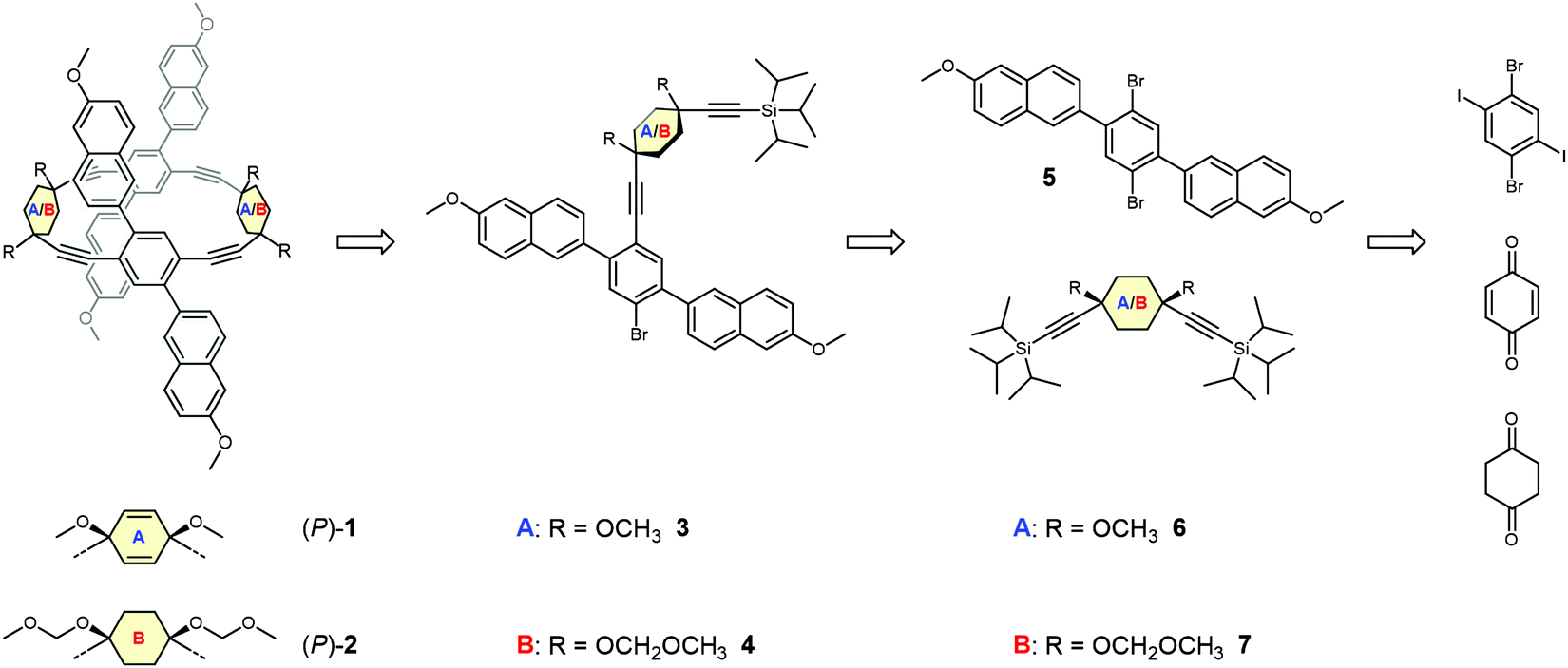 | ||
| Scheme 1 The retrosynthetic analysis of the target structures (P)-1 and (P)-2, which represent rac-1 and rac-2, respectively. | ||
Synthesis and characterization
The synthesis of both macrocyclic dimers, rac-1 and rac-2, is displayed in Scheme 2. It started with the commercially available 1,4-diiodo-2,5-dibromobenzene which underwent a twofold Suzuki coupling with 6-methoxy-2-naphthaleneboronic acid profiting from the reactivity difference between iodine and bromine. Employing a toluene/ethanol/water solvent mixture allowed the isolation of the hardly soluble 2,5-naphthyl substituted 1,4-dibromobenzene 5 by a simple filtration of the reaction mixture and a subsequent washing step with acetone in 80% yield. Corner unit 6 was prepared according to the reported procedures by successive 1,2-carbonyl additions of in situ prepared lithium trimethylsilylacetylide and lithium (triisopropylsilyl)acetylide on p-benzoquinone. Corner 8 was prepared accordingly from 1,4-cyclohexanedione and the respective organolithium species. In order to increase the ratio of the (Z)-isomer relative to the undesired (E)-isomer, a reported procedure by Itami and coworkers using CeCl3 as Lewis-acid for the synthesis of similar structures was adapted.46 The following protection of the alcohol moieties with methoxymethyl ether yielded 7 in 75%. With the corner units in hand, compound 5 then underwent a statistically controlled Sonogashira coupling with 7 or 6. Mono-reacted products 3 and 4 were isolated by flash column chromatography and then subjected to a macrocyclization at high dilution conditions (1 mM or 4 mM, respectively) to favor dimerization by a twofold Sonogashira coupling reaction to yield the PE dimers rac-1 and rac-2 in yields of 14% and 17%, respectively. The isolation of the dimeric PEs rather than higher ordered macrocyclic structures was confirmed by high-resolution-mass-spectrometry (HR-MS), where the expected exact masses of 1171.4180 (m/z, [M + Na]+) for rac-1 and 1299.5195 (m/z, [M + Na]+) for rac-2 were detected.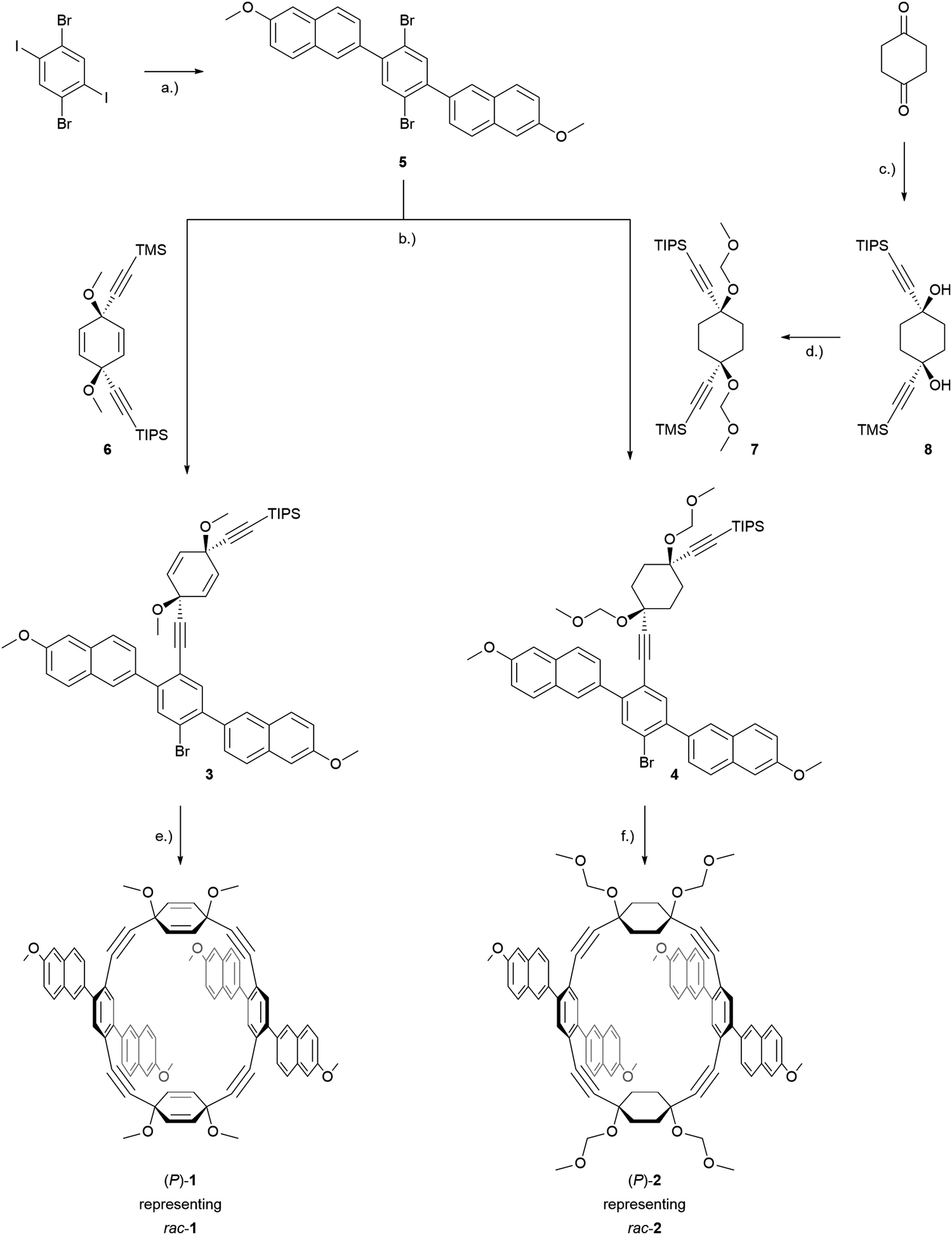 | ||
Scheme 2 Synthesis of rac-1 and rac-2. Reagents and conditions: (a) 6-methoxy-2-naphthaleneboronic acid, K2CO3, Pd(PPh3)4, toluene/ethanol/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1), 90 °C, 16 h, 80%; (b) (1) K2CO3, MeOH, 1–2 h; (2) Pd(PPh3)4, CuI, THF/piperidine (3:1), 80 °C, 3–16 h, 37% for 3, 22% for 4 (c) (1) CeCl3, LiCl, THF, rt, 2 h; (2) (Triisopropylsilyl)acetylene, n-BuLi, THF, −78 °C, 4 h; (3) Ethynyltrimethylsilane, n-BuLi, THF, −78 °C, 16 h, 25%; (d) iPr2NEt, methoxymethyl chloride, CH2Cl2, 16 h, 71%; (e) (1) TBAF, THF, 1 h; (2) Pd(PPh3)4, CuI, THF/NEt3 (3:1, 4 mM), 14%; (f) (1) TBAF, THF, 1 h; (2) Pd(PPh3)4, CuI, THF/piperidine (3:1, 1 mM), 17%. :1:1), 90 °C, 16 h, 80%; (b) (1) K2CO3, MeOH, 1–2 h; (2) Pd(PPh3)4, CuI, THF/piperidine (3:1), 80 °C, 3–16 h, 37% for 3, 22% for 4 (c) (1) CeCl3, LiCl, THF, rt, 2 h; (2) (Triisopropylsilyl)acetylene, n-BuLi, THF, −78 °C, 4 h; (3) Ethynyltrimethylsilane, n-BuLi, THF, −78 °C, 16 h, 25%; (d) iPr2NEt, methoxymethyl chloride, CH2Cl2, 16 h, 71%; (e) (1) TBAF, THF, 1 h; (2) Pd(PPh3)4, CuI, THF/NEt3 (3:1, 4 mM), 14%; (f) (1) TBAF, THF, 1 h; (2) Pd(PPh3)4, CuI, THF/piperidine (3:1, 1 mM), 17%. | ||
The high symmetry of the isolated macrocycles became obvious in the respective 1H-NMR spectra of rac-1 and rac-2, where for each of the naphthalenes only one set of signals was observed (see ESI†). Furthermore, the proton signals of the 1,4-cycohexadiene in rac-1 displayed a signal splitting corroborating exclusively the formation of the desired chiral compound. As sketched in Fig. 3, in the case of the desired chiral macrocycle (left side of Fig. 3), the cyclohexadiene protons Ha and Hb are in a different chemical environment since Ha is in closer proximity to the chromophore and Hb further apart. Thus, a doublet of a doublet multiplicity (3JHH and 4JHH) is observed for both Ha and Hb. In contrast, if the macrocycle with the correct mass would be the achiral dimer (right side of Fig. 3), the symmetry of the molecule would differ and render each pair of protons above and below the framework plane chemically and magnetically identical and only a small 4JHH coupling constant would be expected. In case of the cyclohexane connected dimer rac-2, the same analysis by 1H-NMR is less suited due to the large broad signals observed, attributed to the diastereotopicity of the cyclohexane CH2 protons. However, the formation of the desired chiral compound was confirmed in both cases by their solid state structures analyzed by X-ray diffraction experiments.
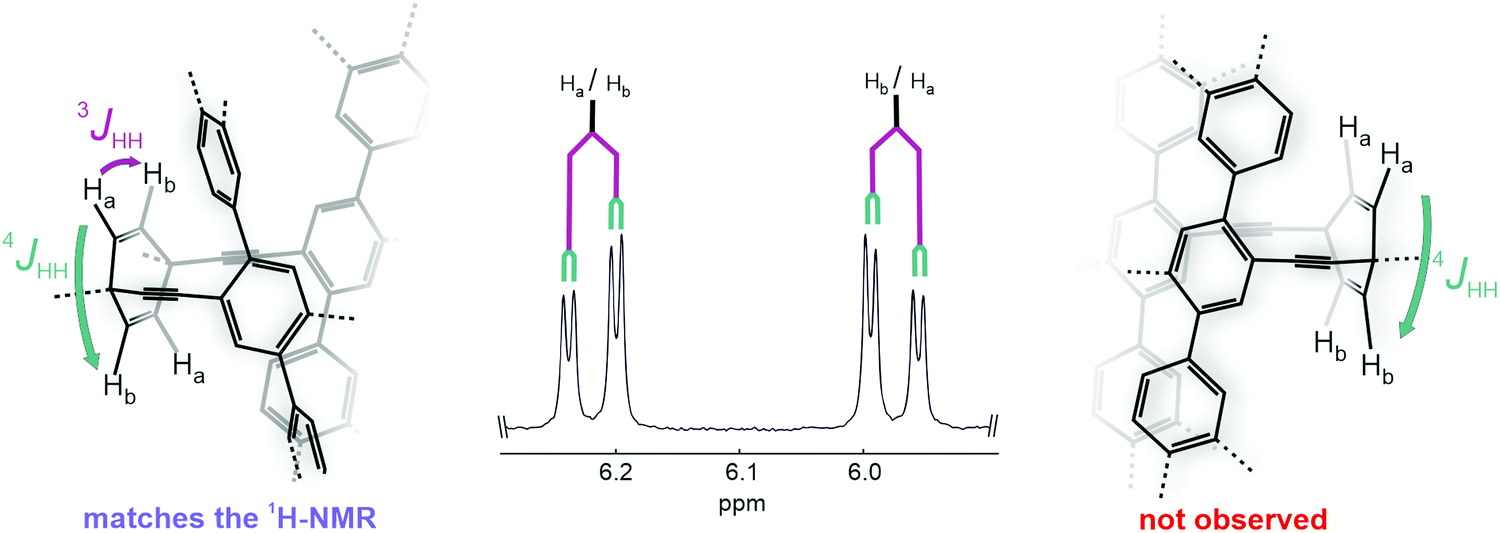 | ||
| Fig. 3 The segment of the 1H NMR spectrum of rac-1 displaying the splitting of the 1,4-cyclohexadiene protons. The observed signal splitting correlates only with the structural features of the chiral macrocycle (left side), while only a small 4JHH coupling would be recorded for the achiral isomer (right side). | ||
Single crystals suitable for X-ray analysis were obtained by top-layering a chloroform solution of either rac-1 or rac-2 with methanol. Both crystals belong to the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) spacegroup and were found to crystallize in a unit cell containing both enantiomers (M)-1 and (P)-1, or (M)-2 and (P)-2, respectively once (Fig. 4).
spacegroup and were found to crystallize in a unit cell containing both enantiomers (M)-1 and (P)-1, or (M)-2 and (P)-2, respectively once (Fig. 4).
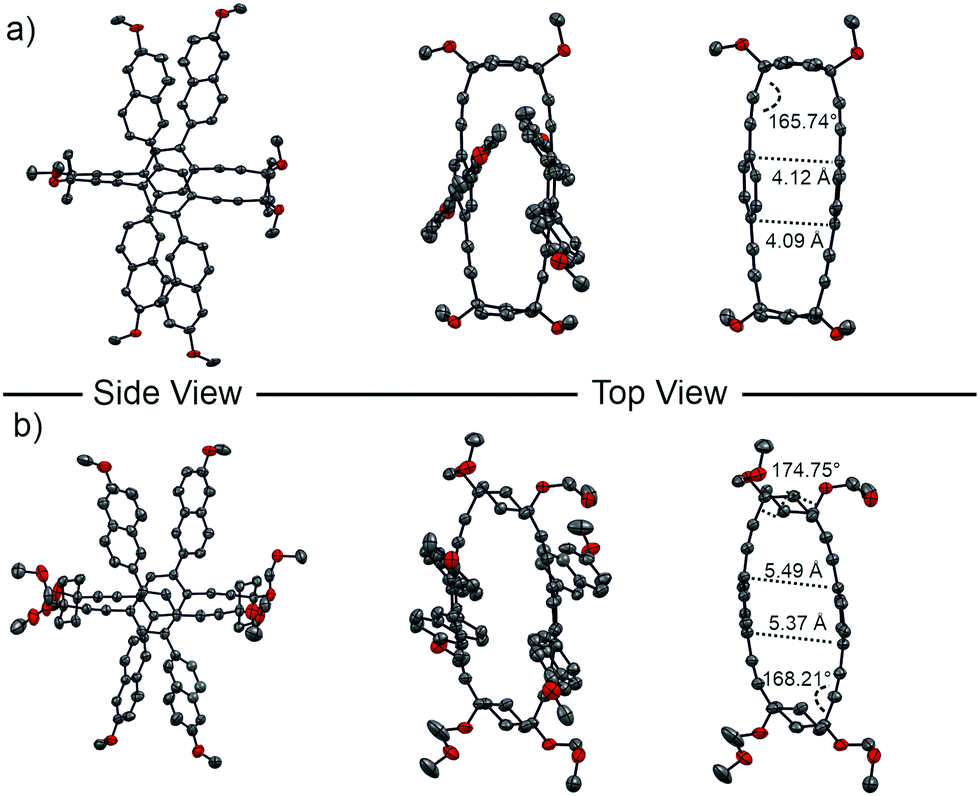 | ||
| Fig. 4 Side and top view of the solid-state structures of rac-1 (a) and rac-2 (b) as ORTEP-plots. Exclusively the (P)-enantiomers of both compounds are displayed. The unit cell comprises both enantiomers. Hydrogen atoms are omitted for clarity and to improve the visibility of the macrocyclic structure, the naphthalenes are omitted in both pictures on the right. Thermal ellipsoids are plotted on a 50% probability level. | ||
Analysis of the solid state structure of (P)-1 revealed a carbon–carbon distance of the directly opposed carbons in the central para-phenylene ring of 4.09 and 4.12 Å, showcasing a slight distortion of the framework. With an angle of 165.74°, the alkyne bonds show only a small deviation from a straight geometry. In the case of (P)-2, the four naphthalene chromophores are pointing more outwards than in (P)-1, spanning a more star-shaped geometry. The difference in geometry can be attributed to the chair conformation of the cyclohexane that is maintained in the macrocyclic structure, as seen by the dihedral angle of 174.75° between the two axially placed substituents at the 1 and 4 position of cyclohexane. The cyclohexane corner also results in a larger distance of the opposed framework sides leading to a carbon–carbon distance of 5.37 and 5.49 Å, while the alkyne bond angle of 168.21° stays comparable to (P)-1.
The UV-Vis spectra of the macrocyclic dimers rac-1 and rac-2 are displaying redshifts of the highest wavelength transition and generally a much stronger absorption for all the transitions, as compared to the open precursors 3 and 4. The absorption maxima of macrocycle rac-1 (λmax) at 272 nm has an extinction coefficient (ε) of 147965 L mol−1 cm−1, whereas the one of 3 at 263 nm is with ε = 81202 L mol−1 cm−1 smaller (see Table 1 for all values). Since the extinction coefficient of the absorption maxima of both macrocycles is less than twice the extinction value of the open precursors, we concluded that the chromophores of the opposed sides are excitonically coupled (vide infra). If the chromophores were non-interacting, one would expect a simple doubling of absorption intensities upon dimerization of 3 and 4. Additionally, while for rac-1 the redshift of the absorption spectra might possibly be explained by an increased conjugated system by homo-conjugation through the 1,4-cyclohexa-2,5-diene moiety, it is precluded in rac-2 due to the insulating cyclohexane unit, further supporting the hypothesis of an excitonically coupled system.
| Compound | λ max [nm] | ε [L mol−1 cm−1] | λ em (excitation) [nm] |
|---|---|---|---|
| 3 | 263 | 81202 |
403 (315) |
| rac-1 | 272 | 147965 |
423 (340) |
| 4 | 260 | 73064 |
389 (313) |
| rac-2 | 270 | 112052 |
418 (322) |
The open precursors 3 and 4, and their respective macrocycles rac-1 and rac-2 are fluorescent in solution as well as in solid state. The fluorescence is in both macrocycles redshifted, as compared to the precursors (Table 1 and Fig. 5). The bathochromic shift of the emission peak is attributed to the emission from the low-lying exciton coupled state, fulfilling Kasha's rule.47
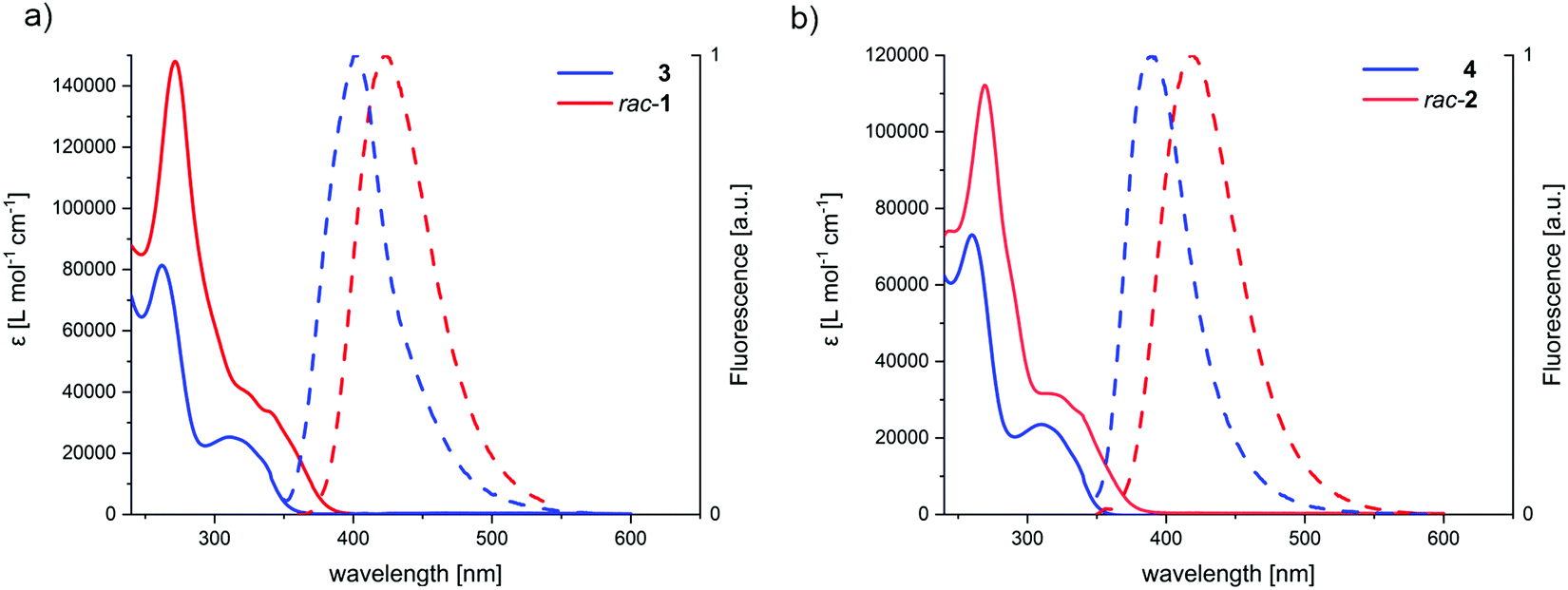 | ||
| Fig. 5 Absorption (solid line) and emission (dashed line) spectra of (a) rac-1 (red) and its precursor 3 (blue), (b) rac-2 (red) and its precursor 4 (blue) in CH2Cl2 (c ∼ 10−6 M and 10−7 M for absorption and emission, respectively). Compound 4, 3, rac-2 and rac-1 were excited at 313, 315, 322 and 340 nm, respectively. | ||
Separation of the racemic macrocycles into enantiomers was achieved by subjecting rac-1 and rac-2 to high-pressure-liquid-chromatography (HPLC) on a chiral stationary phase (see ESI† for more details). To our delight, the enantiomers of both macrocycles were separable using the same mobile phase and column, thus simplifying the search for conditions. The successful separation of the enantiomers was confirmed by the mirrored image of the CD spectra (Fig. 6). For macrocycle 1, the highest molar dichroism is observed at 297 nm (Δε = 300 and −286 L mol−1 cm−1 for (M)-1 and (P)-1, respectively), while for 2 the molar dichroism is less intense and slightly hypsochromically shifted to 292 nm (Δε = 187 and −214 L mol−1 cm−1 for (M)-2 and (P)-2, respectively). The macrocycles both feature an excitonic–coupled dichroism with a center around 310 nm, which was made use of for the assignment of the absolute configuration. Both obtained frameworks are optically stable, as they show no evidence of racemization upon stirring at 110 °C (see ESI†).
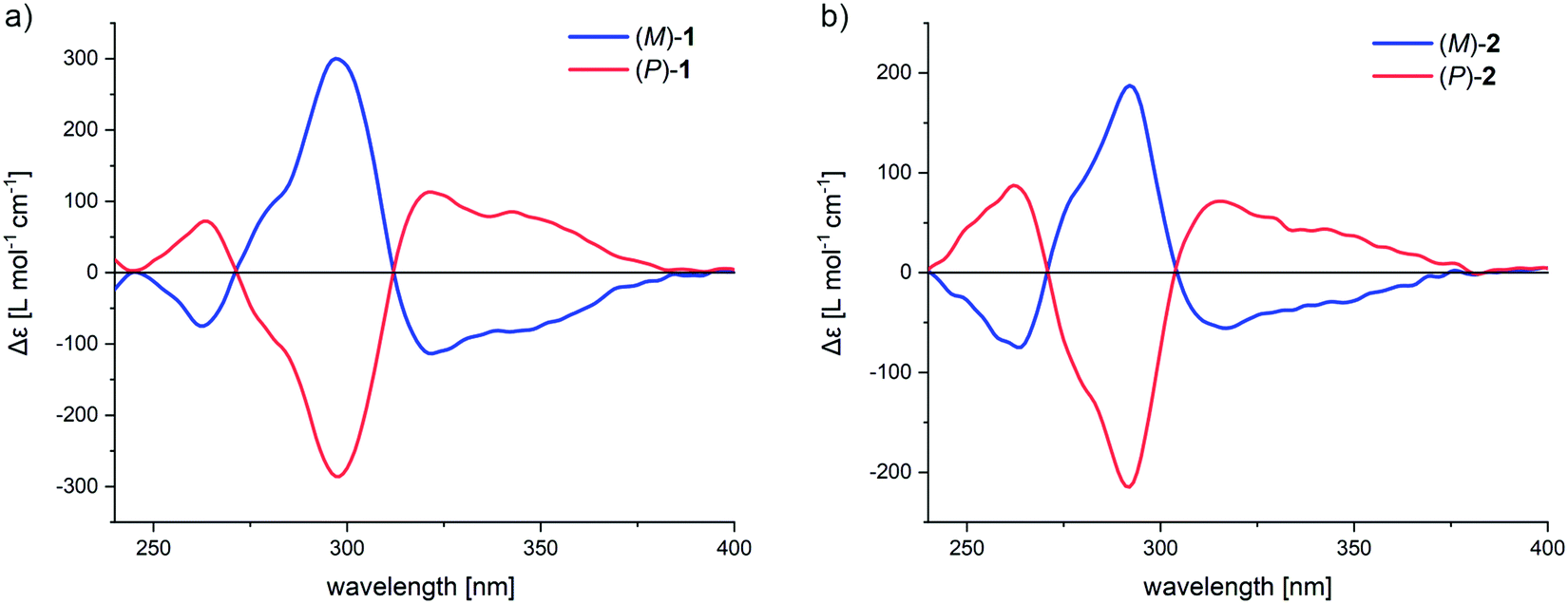 | ||
| Fig. 6 Circular dichroism spectra of (a) (P)-1 (red) and (M)-1 (blue), (b) (P)-2 (red) and (M)-2 (blue) in CH2Cl2 (c ∼ 10−6 M). Absolute configuration was assigned according to exciton chirality method, DFT calculations and X-ray crystallography. | ||
Assignment of absolute configuration
The exciton chirality method (ECM) is convenient to determine the absolute configuration of multichromophoric systems by the analysis of a bisignate CD signal emerging due to excitonically coupled transitions.48,49 Similar chromophores in spatial proximity and chiral configuration usually bear non-parallel transition dipole moments, which leads to the so-called Davydov splitting, as often referred to in H- and J-aggregates.50 The splitting arises due to a head-to-head and head-to-tail type arrangement of the chromophores associated excitons, which comes hand in hand with an opposite sign of the rotational strength, thus leading to the bisignate CD signal. The signal is referred to a positive couplet when the higher wavelength component is a positive CD signal and to a negative couplet if the higher wavelength component is negative. As non-empirically determined, although with known exceptions,51 a positive couplet arises when a clockwise orientation of the TDMs is present. Likewise, a negative couplet occurs for counter-clockwise orientation. Such couplings of oscillators were first discovered based on optical rotatory dispersion experiments and later in CD spectroscopy by Kuhn and co-workers.52,53 These findings led to the application of such couplings for the determination of absolute configuration of biaryl compounds already in the 1960s.54,55Fig. 7a shows the established transition dipole moment orientation of unsubstituted naphthalene.40,42 The transition dipole moment that is involved in the exciton coupling in 1 and 2 is the 1Lb transition, oriented along the long axis of the naphthalene. Since TDM orientation can differ in macromolecular structures and the ECM only reliably works with proper knowledge of its orientation, as has been shown by many corrections of reported assignments,56,57 assignment by the ECM has to be considered with caution. In substituted naphthalenes it has been shown that the TDM is oriented along the substitution axis, which in the case of 1 and 2 is further supported by the conjugation to the central phenylene and adjacent naphthalene.58 Accordingly, the TDMs involved in the exciton coupling has the orientation displayed in Fig. 7b by bold red arrows. When projecting the two TDMs in a Newman-type projection (Fig. 7b, top), the rotation of the TDM from front to back occurs in a clockwise fashion for the (P)-enantiomer, thus according to the theory results in a positive exciton couplet. Applying this exciton chirality rule to the CD spectra in Fig. 6 leads to the assignment of the blue line of the CD spectra to the (M)-enantiomer and the red line to the (P)-enantiomer for both 1 and 2.
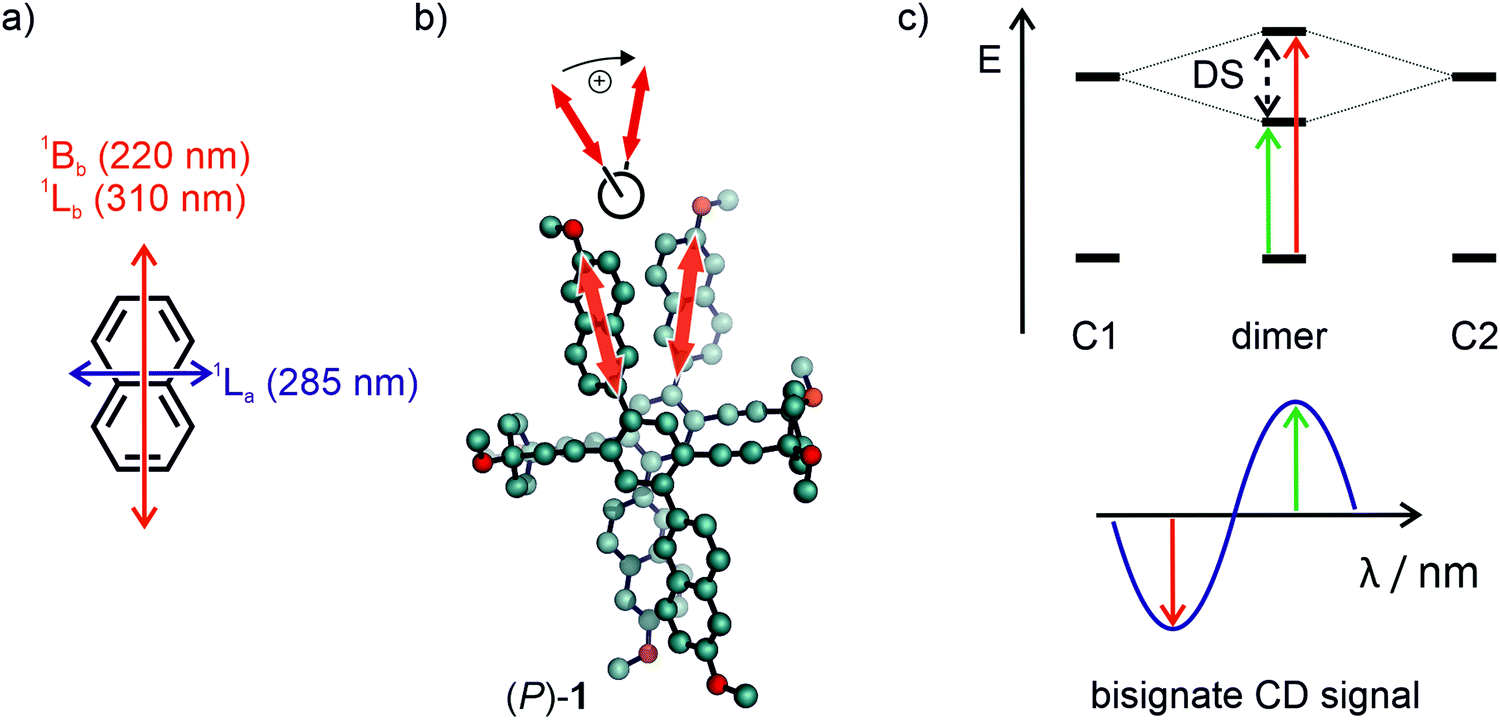 | ||
| Fig. 7 (a) Representation of the main electronic transitions in naphthalene.42 (b) Representation of the clockwise oriented naphthalenes and their transition dipole moment (bold red arrow) from front to back. (c) Top: Energy diagram of the transitions of isolated chromophores (C1 and C2) and the resulting Davydov splitting (DS) upon dimerization in a chiral configuration. Bottom: The resulting positive exciton couplet of the CD experiment for a clockwise transition dipole moment orientation due to the Davydov splitting. | ||
The assignment of the enantiomers was further supported by DFT calculations. With the crystal structure as starting point, first the molecular geometry was optimized at D3BJ-B3LYP/6-31g(d) level of theory followed by TD-DFT calculation at CAM-B3LYP/6-31g(d) level of theory (ESI†). In the case of 2, the methoxymethyl ether groups were replaced with methoxy in order to reduce the required calculation resources. The calculated CD spectra nicely coincides with the experimental data (Fig. 8), and also predicts the positive CD couplet for both (P)-enantiomers. Analysis of the natural transition orbitals (see ESI†) involved in the transitions of the bisignate signals indeed show that they are located on the naphthalene chromophore, supporting the assignment by the ECM.
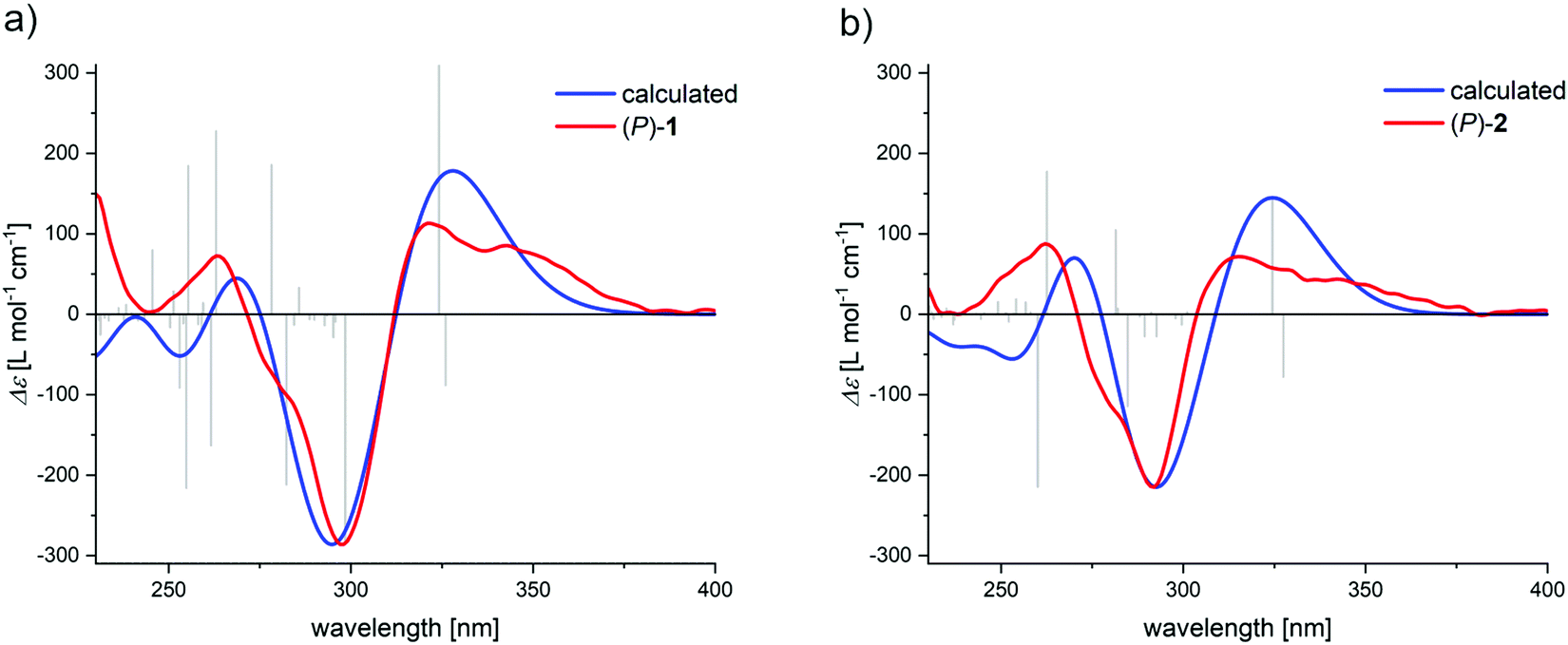 | ||
| Fig. 8 Comparison of the experimental and calculated spectra for (P)-1 in (a) and (P)-2 in (b). | ||
Although weakly diffracting, single crystals suitable for X-ray diffraction measurements were obtained for the (M)-enantiomer of 1 by top-layering a chloroform solution of the first eluting peak on the HPLC with methanol, confirming the assignment of the enantiomers obtained by ECM and DFT calculations (see ESI†). Although single crystals were grown also for (P)-1/2 and (M)-2, they were too small and too weakly diffracting and thus not suitable for proper analysis, despite several attempts.
Conclusion
In summary, a scaffold consisting of two para-phenylene–ethynylene frameworks rigidified by laterally placed cyclohexa-2,5-diene or cyclohexane corner units is presented. The framework bears four mounting points for a chromophore of choice, which places them in an axially chiral configuration. The configuration is optically stable due to a hindered rotation around the framework axis when the chromophore is of large enough dimension. The arrangement of the chromophore excitonically couples its associated transitions, which might be useful in singlet fission applications. With 6-methoxynaphthalene as model chromophore, the axial chirality-inducing framework has been assembled in three steps. The frameworks were analyzed by mass spectrometry, NMR spectroscopy, UV-Vis and emission spectroscopy, and X-ray crystallography. NMR and X-ray analysis revealed the formation of the desired chiral configuration and chiral resolution was performed using HPLC with a chiral stationary phase. The obtained enantiopure scaffolds displayed intense molar dichroism with Δε of up to 300 L mol−1 cm−1. Assignment of the absolute configuration was accomplished by the exciton chirality method and DFT calculations, and was corroborated by solid-state structure analysis. Using this simple chirality-inducing scaffold, inherently achiral chromophores can be arranged in a helical chiral manner and be enriched by chiroptical properties. We are currently exploring the potential of the approach for a variety of stable but achiral chromophores and also envision to analyze their CPL properties.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The article is dedicated to our friend and steady source of inspiration Kees Hummelen. The authors acknowledge the Swiss National Science Foundation for financial support (grant Nr. 200020-178808). M. M. acknowledges support by the 111 project (90002-18011002).References
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, John Wiley & Sons, Inc., 2nd edn, 2000 Search PubMed.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- T. Tanabe, T. Sato, K. Fukaishi and Y. Kakubari, Dig. Tech. Pap. - Soc. Inf. Disp. Int. Symp., 2015, 46, 987–990 CrossRef.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- P. L. Polavarapu, Chirality, 2016, 28, 445–452 CrossRef CAS PubMed.
- O. Kotova, S. Blasco, B. Twamley, J. O’Brien, R. D. Peacock, J. A. Kitchen, M. Martínez-Calvo and T. Gunnlaugsson, Chem. Sci., 2015, 6, 457–471 RSC.
- A. Gergely, J. Pharm. Biomed. Anal., 1989, 7, 523–541 CrossRef CAS.
- N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- M. Suda, Y. Thathong, V. Promarak, H. Kojima, M. Nakamura, T. Shiraogawa, M. Ehara and H. M. Yamamoto, Nat. Commun., 2019, 10, 2455 CrossRef.
- P. C. Mondal, N. Kantor-Uriel, S. P. Mathew, F. Tassinari, C. Fontanesi and R. Naaman, Adv. Mater., 2015, 27, 1924–1927 CrossRef CAS.
- Z. Dai, J. Lee and W. Zhang, Molecules, 2012, 17, 1247–1277 CrossRef CAS PubMed.
- A. Ozcelik, R. Pereira-Cameselle, N. Poklar Ulrih, A. G. Petrovic and J. L. Alonso-Gómez, Sensors, 2020, 20, 974 CrossRef CAS PubMed.
- C. Petermayer and H. Dube, J. Am. Chem. Soc., 2018, 140, 13558–13561 CrossRef CAS.
- P. Ravat, T. Šolomek and M. Juríček, ChemPhotoChem, 2019, 3, 180–186 CrossRef CAS.
- P. Ravat, T. Šolomek, D. Häussinger, O. Blacque and M. Juríček, J. Am. Chem. Soc., 2018, 140, 10839–10847 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- G. Echue, I. Hamley, G. C. Lloyd Jones and C. F. J. Faul, Langmuir, 2016, 32, 9023–9032 CrossRef CAS PubMed.
- X. Yang, J. Han, Y. Wang and P. Duan, Chem. Sci., 2019, 10, 172 RSC.
- T. Harada, N. Kajiyama, K. Ishizaka, R. Toyofuku, K. Izumi, K. Umemura, Y. Imai, N. Taniguchi and K. Mishima, Chem. Commun., 2014, 50, 11169–11172 RSC.
- D.-M. Lee, J.-W. Song, Y.-J. Lee, C.-J. Yu and J.-H. Kim, Adv. Mater., 2017, 29, 1700907 CrossRef.
- Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, Angew. Chem., 2018, 130, 9111–9116 CrossRef.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chemistry, 2015, 21, 13488–13500 CrossRef.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS.
- K. Yavari, W. Delaunay, N. De Rycke, T. Reynaldo, P. Aillard, M. Srebro-Hooper, V. Y. Chang, G. Muller, D. Tondelier, B. Geffroy, A. Voituriez, A. Marinetti, M. Hissler and J. Crassous, Chem. – Eur. J., 2019, 25, 5303–5310 CrossRef CAS PubMed.
- C. Schaack, L. Arrico, E. Sidler, M. Górecki, L. Di Bari and F. Diederich, Chem. – Eur. J., 2019, 25, 8003–8007 CrossRef CAS.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- M. Ball, B. Fowler, P. Li, L. A. Joyce, F. Li, T. Liu, D. Paley, Y. Zhong, H. Li, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2015, 137, 9982–9987 CrossRef CAS.
- N. J. Schuster, R. Hernández Sánchez, D. Bukharina, N. A. Kotov, N. Berova, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2018, 140, 6235–6239 CrossRef CAS PubMed.
- H. Lu, Coord. Chem. Rev., 2016, 15 CAS.
- T. Kawai, K. Kawamura, H. Tsumatori, M. Ishikawa, M. Naito, M. Fujiki and T. Nakashima, ChemPhysChem, 2007, 8, 1465–1468 CrossRef CAS.
- H. Langhals, A. Hofer, S. Bernhard, J. S. Siegel and P. Mayer, J. Org. Chem., 2011, 76, 990–992 CrossRef CAS.
- E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef PubMed.
- B. S. Basel, C. Hetzer, J. Zirzlmeier, D. Thiel, R. Guldi, F. Hampel, A. Kahnt, T. Clark, D. M. Guldi and R. R. Tykwinski, Chem. Sci., 2019, 10, 3854–3863 RSC.
- D. Sun, G.-H. Deng, B. Xu, E. Xu, X. Li, Y. Wu, Y. Qian, Y. Zhong, C. Nuckolls, A. R. Harutyunyan, H.-L. Dai, G. Chen, H. Chen and Y. Rao, iScience, 2019, 19, 1079–1089 CrossRef CAS PubMed.
- K. Broch, J. Dieterle, F. Branchi, N. J. Hestand, Y. Olivier, H. Tamura, C. Cruz, V. M. Nichols, A. Hinderhofer, D. Beljonne, F. C. Spano, G. Cerullo, C. J. Bardeen and F. Schreiber, Nat. Commun., 2018, 9, 954 CrossRef CAS PubMed.
- Y. Kitayama, T. Amako, N. Suzuki, M. Fujiki and Y. Imai, Org. Biomol. Chem., 2014, 12, 4342–4346 RSC.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef CAS.
- M. Rubio, M. Merchán, E. Ortí and B. O. Roos, Chem. Phys., 1994, 179, 395–409 CrossRef CAS.
- L. Di Bari, G. Pescitelli and P. Salvadori, J. Am. Chem. Soc., 1999, 121, 7998–8004 CrossRef CAS.
- G. D. Scholes, G. O. Turner, K. P. Ghiggino, M. N. Paddon-Row, J. J. Piet, W. Schuddeboom and J. M. Warman, Chem. Phys. Lett., 1998, 292, 601–606 CrossRef CAS.
- G. D. Scholes, K. P. Ghiggino, A. M. Oliver and M. N. Paddon-Row, J. Am. Chem. Soc., 1993, 115, 4345–4349 CrossRef CAS.
- S. Sankararaman and M. Srinivasan, Org. Biomol. Chem., 2003, 1, 2388–2392 RSC.
- H. Omachi, S. Matsuura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 10202–10205 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- N. Harada and K. Nakanishi, Acc. Chem. Res., 1972, 5, 257–263 CrossRef CAS.
- N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Comprehensive Chiroptical Spectroscopy: Volume 2: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules, John Wiley & Sons, Inc., 2012 Search PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- T. Bruhn, G. Pescitelli, S. Jurinovich, A. Schaumlöffel, F. Witterauf, J. Ahrens, M. Bröring and G. Bringmann, Angew. Chem., Int. Ed., 2014, 53, 14592–14595 CrossRef CAS PubMed.
- W. Kuhn, Trans. Faraday Soc., 1930, 26, 293 RSC.
- W. Kuhn and R. Rometch, Helv. Chim. Acta, 1944, 27, 1346 CrossRef CAS.
- S. F. Mason and G. W. Vane, J. Chem. Soc. B, 1966, 370–374 RSC.
- S. F. Mason, R. H. Seal and D. R. Roberts, Tetrahedron, 1974, 30, 1671–1682 CrossRef CAS.
- G. Pescitelli, Mar. Drugs, 2018, 16, 388 CrossRef CAS PubMed.
- G. Pescitelli and L. Di, Bari, J. Nat. Prod., 2017, 80, 2855–2859 CrossRef CAS.
- N. Berova, G. A. Ellestad and N. Harada, Comprehensive Natural Products II, Elsevier Science, 2010, vol. 9, pp. 91–146 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082783–2082785. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc02180j |
| This journal is © The Royal Society of Chemistry 2021 |