
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-performance all-organic aqueous batteries based on a poly(imide) anode and poly(catechol) cathode†
Nagaraj
Patil
 *a,
Andreas
Mavrandonakis
a,
Christine
Jérôme
b,
Christophe
Detrembleur
b,
Nerea
Casado
c,
David
Mecerreyes
cd,
Jesus
Palma
a and
Rebeca
Marcilla
*a
*a,
Andreas
Mavrandonakis
a,
Christine
Jérôme
b,
Christophe
Detrembleur
b,
Nerea
Casado
c,
David
Mecerreyes
cd,
Jesus
Palma
a and
Rebeca
Marcilla
*a
aElectrochemical Processes Unit, IMDEA Energy, Avda. Ramón de la Sagra 3, 28935 Móstoles, Madrid, Spain. E-mail: rebeca.marcilla@imdea.org; nagaraj.patil@imdea.org
bCentre for Education and Research on Macromolecules (CERM), CESAM Research Unit, Department of Chemistry, University of Liege, Allée de la Chimie B6A, 4000 Liége, Belgium
cPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda. Tolosa 72, 20018 Donostia-San Sebastián, Spain
dIkerbasque, Basque Foundation for Science, E-48011, Bilbao, Spain
First published on 16th December 2020
Abstract
Aqueous all-polymer batteries (AqPBs) are foreseen as promising solutions for safe, sustainable, and high-performance energy storage applications. Nevertheless, their development is still challenging as it demands precise optimization of both electrodes and the electrolyte composition to be able to sustain a stable redox activity, while delivering an optimal voltage output. Herein, we report AqPBs based on a poly(imide) (PI) anode and poly(catechol) (PC) cathode that exhibit tunable cell voltage depending on the salt used in the aqueous electrolyte, i.e., 0.58, 0.74, 0.89, and 0.95 V, respectively, when Li+, Zn2+, Al3+, and Li+/H+ were utilized as charge carriers. The PI–PC full-cell delivers the best rate performance (a sub-second charge/discharge) and cycling stability (80% capacity retention over 1000 cycles at 5 A g−1) in Li+. Furthermore, a maximum energy/power density of 80.6 W h kganode+cathode−1/348 kW kganode+cathode−1 is achieved in Li+/H+, superior to most of the previously reported AqPBs.
Introduction
The proper integration of affordable, scalable and eco-friendly electrochemical energy storage (EES) systems is critical to facilitate the development of a safe, secure, and sustainable energy landscape.1,2 However, commercial Li-ion batteries exclusively include insertion-type inorganic anodes, 3d transition metal cathodes and organic electrolytes, which are unsafe, scarce, expensive and energy intensive, and hardly meet the requirements of a sustainable society.3 This calls for a radical change in research and development beyond Li-ion battery chemistries to meet the ever-increasing sustainability demands.4Aqueous all-polymer batteries (AqPBs) that incorporate redox-active polymers (RAPs) as organic electrode materials (OEMs) and aqueous solutions as safe and cost-effective electrolytes can be promising alternatives for the development of sustainable energy storage systems.5–7 Although a plethora of RAPs have been successfully applied as OEMs in numerous rechargeable battery technologies (mostly, in metal-ion–polymer configuration),8–17 examples of AqPBs sporadically appeared in the literature.18,19 This is partly due to the formidable challenge that requires careful designing of both anode and cathode RAP partners to be not only able to sustain their redox activity in aqueous media, but also deliver a high voltage output within a relatively narrow electrochemical window of aqueous electrolytes (∼1.23 and ∼2 V for pure water and typical salt-in-water electrolytes, respectively).20–23
Based on the charge storage mechanisms of RAPs, they can be generally classified into: n-type undergoing N↔N−, p-type undergoing P↔P+, and bipolar exhibiting B−↔B↔B+ redox reactions, with the simultaneous shuttling of electroneutralizing cations, anions, and dual ions, respectively.8–15,18,19 Depending on the RAP (n- or p-type) and the chemical nature of the charge carriers, three kinds of AqPBs were realized: (i) n|n-type combination is generally preferred for the capacity-oriented design (20–65 mA h gcell−1, <1 V),24–26 p|p-type combination mostly preferred for the voltage-oriented design (1.1–1.3 V, <45 mA h gcell−1),27–29 and n|p-type combination offers a compromise between the capacity and the voltage (10–52 mA h gcell−1 and 0.9–1.3 V)29–33 (Fig. S1 and Table S1, see the ESI†). From these limited examples, it is evident that the development of AqPBs is at a slow pace and further it is necessary to simultaneously improve their capacity and voltage in order to be competitive with other aqueous-based battery technologies.18,19
Among different varieties of n-type Redox-Active Polymers (RAPs), poly(imide)s and poly(quinone)s have been attracting tremendous interest in AqPBs owing to their (i) high theoretical specific capacity, tunable redox potentials and fast kinetics, (ii) high chemical and electrochemical stability, and (iii) good compatibility with different types of cationic charge carriers (e.g., Li+, Na+, Mg2+, Zn2+, Al3+, etc.) (vide supra). Particularly, their last merit is highly encouraging, which enables them to be used as universal organic electrodes in numerous polymer-based battery configurations.34–40 So far, their n-type redox behavior was mainly exploited to design various metal-ion–polymer mono- and multivalent batteries, but interactions between the polymeric host and the charge carriers were largely overlooked. Of note, our recent work emphasized the importance such interactions from the perspective of electrochemical performance tunability, but in half-cells.41
In this article, we demonstrate the construction of AqPBs, comprising a poly(imide) anode and poly(catechol) cathode. First, we studied the electrochemical performance of individual electrodes in different aqueous electrolytes containing Li+, Zn2+, Al3+, and Li+/H+ charge carriers. Then, all-organic full-cells were tested denoting an increase of cell voltage depending on the charge carriers from Li+ to Zn2+, to Al3+ and to Li+/H+, respectively. Finally, the performance of full-cells was optimized including cyclability studies and compared favorably with the state-of-the-art all-polymer aqueous stationary batteries.
Experimental section
Materials
Lithium nitrate (LiNO3, ≥ 99.0%, Sigma-Aldrich), zinc sulfate heptahydrate (ZnSO4·7H2O, ≥ 99.5%, Sigma-Aldrich), aluminum nitrate nonahydrate (Al(NO3)3, 9H2O, ≥98.0%, Sigma-Aldrich), sulfuric acid (H2SO4, ACS reagent, 95–98%, Sigma-Aldrich), anhydrous 1-methyl-2-pyrrolidone (NMP, ≥ 99.5%, Sigma-Aldrich), lithium(I) bis(trifluoromethanesulfonyl)imide (LiTFSI; 99.9%, Solvionic), zinc(II) bis(trifluoromethanesulfonyl)imide (Zn(TFSI)2; 99.5%, Solvionic), aluminum(III) tris(trifluoromethanesulfonyl)imide (Al(TFSI)3; 99.5%, Solvionic), polyvinylidene fluoride (PVDF; Sigma Aldrich), thin multi-walled carbon nanotubes (CNTs; Elicarb® MW, Thomas-Swan) and carbon paper (Toray TP-060, QUINTECH) were used as received. Anhydrous N,N-dimethylformamide (DMF, ≥ 99.8%, Sigma-Aldrich) was dried over 4 Å molecular sieves for at least 48 h, distilled, and stored under argon prior to use.Synthesis of redox-active polymers
The synthesis of the cathode active-material, P(DA70-stat-AMPS30) (termed simply poly(catechol) = PC, Mn ≈ 12.0 kg mol−1 and Mw/Mn ≈ 1.19) bearing catechol pendants has been previously reported.42 The anode organic partner, poly(1,4,5,8-naphthalenetetracarboxylic diimide-1,2-diethoxyethane) (poly(imide) = PI) with imide functionalities in the polymer backbone, was synthesized by a polycondensation reaction as described previously.43Electrode preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CNT was 6:4 wt%), proceeding to sonication for 2 h in a bath sonicator (Branson 2510, 100 W, 42 kHz) and overnight stirring to prepare the electrode slurry. This viscous smooth slurry was then uniformly coated onto a carbon paper current collector by a doctor blade method, dried overnight at 50 °C under vacuum and cut into 12-mm disks. The mass loading of polymers on the current collector was controlled between 1.1 and 1.5 mg cm−2.
:CNT:PVDF was 6:3:1, and the loading of PI was about 1.5 mg cm−2.
:CNT was 6:4 wt%), proceeding to sonication for 2 h in a bath sonicator (Branson 2510, 100 W, 42 kHz) and overnight stirring to prepare the electrode slurry. This viscous smooth slurry was then uniformly coated onto a carbon paper current collector by a doctor blade method, dried overnight at 50 °C under vacuum and cut into 12-mm disks. The mass loading of polymers on the current collector was controlled between 1.1 and 1.5 mg cm−2.
:CNT:PVDF was 6:3:1, and the loading of PI was about 1.5 mg cm−2.
PI–PC full-cell fabrication
PI–PC full-cells were assembled using circular discs (12 mm diameter) of PI/CNTs as the anode, PC/CNTs as the cathode, and a pair of porous Whatman® glass microfiber filters (Grade GF/B) soaked with ∼200 μL of electrolyte in CR2032 coin cells. The mass ratio between PI and PC was 1:1, 1:0.9, 1:0.7, and 1:0.72 in Li+, Zn2+, Al3+, and Li+/H+ configurations, respectively, according to the capacities of individual electrodes in the three-electrode investigations. The aqueous electrolytes for Li+, Zn2+ and Al3+ ion batteries were prepared by dissolving LiNO3, ZnSO4 or Al(NO3)3 in degassed milli-Q water to a 2.5 M concentration. The mixed aqueous electrolyte (Li+/H+) was composed of 2.5/0.25 M LiNO3/H2SO4. The cells were assembled in a high-purity argon-filled glovebox (MBraun; O2 < 0.5 ppm) to avoid any possible contamination by oxygen.
Electrochemical characterization
The electrochemical performance of individual electrodes and full-cells was investigated by cycling voltammetry (CV) and galvanostatic charge–discharge (GCD) experiments using a Bio-logic VMP3 multichannel Potentiostat/Galvanostat (Biologic SP-150). The CV and GCD profiles of redox-active polymers (RAPs) were obtained using a flooded three-electrode electrochemical cell with RAP/CNT ink (80/20 wt%) modified glassy carbon (GC, with an area of 0.07 cm2), Ag/AgCl (KCl sat.) and platinum wire as the working, reference and counter electrodes, respectively, in different aqueous electrolytes. The electrolyte was degassed with argon, and CV/GCD was performed at room temperature under a positive pressure of argon atmosphere. The real-time evolution of individual voltages of electrode partners in full-cells was assessed with Swagelok™ T-cells, in which PI, PC, and Ag/AgCl (KCl sat.) were used as the anode, cathode and reference electrode, respectively. As control experiments, the CV analysis of PC electrodes was also carried out in aqueous citrate buffer solutions (pH = 5) and organic electrolytes (anhydrous DMF) containing Li+ (from LiTFSI), Zn2+ (from Zn(TFSI)2), and Al3+ (from Al(TFSI)3) charge carriers. Ag wire was used as a pseudo-reference electrode in organic electrolytes.Theoretical specific capacity (mA h g−1) was calculated using the equation = (26801 × n × wRA)/MWRA, where n is the number of electrons exchange per unit of catechol/o-quinone (n = 2), and wRA and MWRA and are the weight fraction and the molecular weight of redox-active (RA) units, respectively.42 The theoretical specific capacity of PC and PI is about 180 and 166 mA h g−1, respectively. The energy density (W h kg−1) of the full-cell was determined by integrating the area under the galvanostatic specific capacityanode+cathode–voltage discharge profiles. Power density (W kg−1) was calculated as (energy density/discharge time) × 3600.
Computational method
The structure of the PI and PC polymers is truncated to a simple monomer structure that is composed of naphthalenetetracarboxylic diimide (NDI) substituted with a –CH3 group at the nitrogen position and a catecholate (CAT) moiety, respectively. The lithium, zinc and aluminum cations are considered to have their first coordination sphere filled with 4, 6 and 6 explicit water molecules, respectively. The binding free energies of the solvated metal cations are calculated with respect to the reduced form of the catecholate/imide monomer moieties from the following equation: [MON]2− + y[M(H2O)x]m+ → [MON–My(H2O)nx−z]ym−2 + z(H2O), where MON is the catecholate or imide species. The absolute redox potentials are calculated from: E = −ΔGrxn/(2F), where ΔGrxn is the free energy change for the 2e−-reduction reaction: [MON] + 2e− + y[M(H2O)x]m+ → [MON-My(H2O)nx−z]ym−2 + z(H2O). Full details of the computational protocol are presented in the ESI† and are taken from our previous work.41Results and discussion
Effect of charge carriers on the electrochemical response
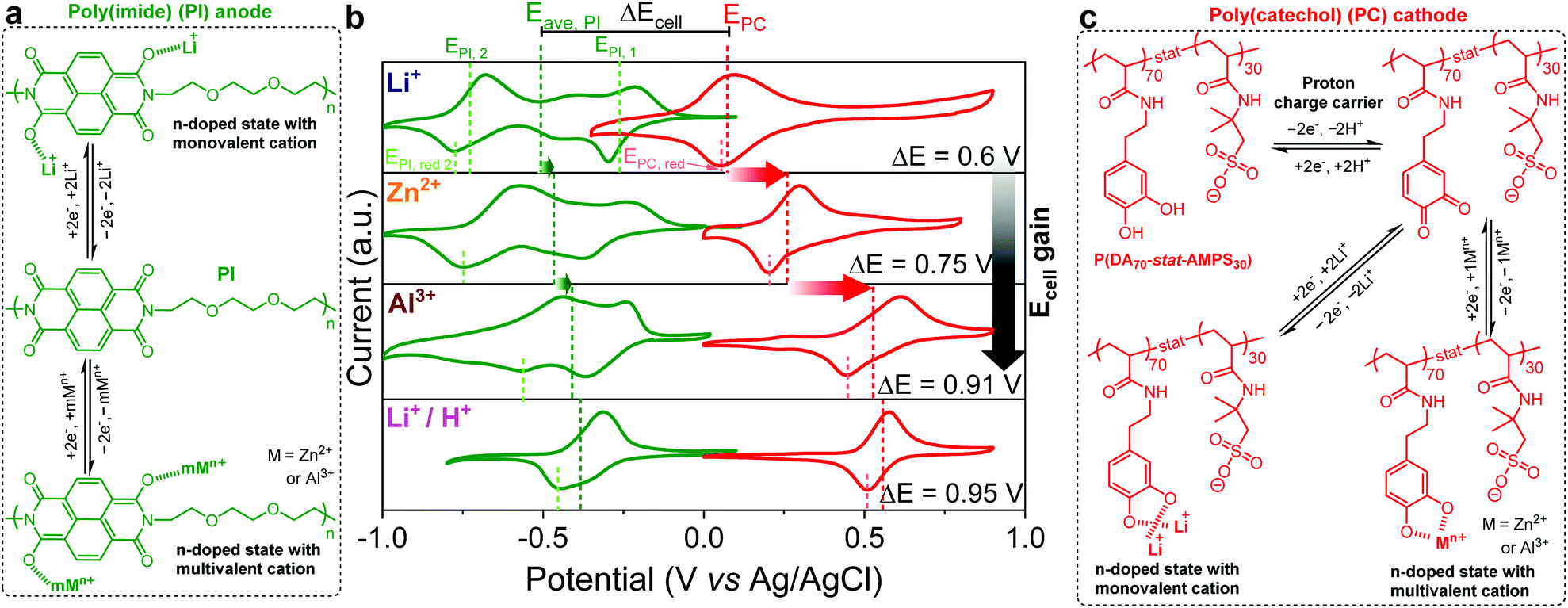 | ||
| Fig. 1 (a and c) Schematic representation of the n-type redox behavior of PI (a) and PC (c) with both mono- and multivalent cations. (b) Cyclic voltammograms of PI (profiles in green) and PC (profiles in red) half-cells in unbuffered aqueous electrolytes containing different charge carriers, i.e., Li+, Zn2+, Al3+, and Li+/H+ (top to bottom). The CVs were recorded at 10 mV s−1 in a standard three-electrode configuration. | ||
On the other hand, the poly(catechol) cathode (PC) (see Fig. 1c for the chemical structure) featured a single redox process corresponding to an anodic peak of PC2−[2Li+, 1Zn2+ or 1Al3+] to PQ (PQ refers to poly(ortho-quinone) transformation) and a cathodic peak of PQ to PC2−[2Li+, 1Zn2+ or 1Al3+] conversions. The CVs in Fig. 1b evidenced the quasi-reversible nature of this two-Li+ (one-Zn2+ or one-Al3+) and two-electron process.41 Importantly, differently from the PI, the average peak potentials of PC were dramatically shifted positive (i.e., EPC, Li+ ≈ 0.1 V, EPC, Zn2+ ≈ 0.28 V, and EPC, Al3+ ≈ 0.51 V), in the same order of electroneutralizing cation valences (Li+ < Zn2+ < Al3+), as previously reported by our group.41
Although the electrochemical analysis was carried out in unbuffered aqueous electrolytes containing different charge carriers, it was recently demonstrated by our group that the reduction/oxidation of ortho-quinones/catecholates in similar electrolytes occurs via the coordination/uncoordination of cations of the supporting salts.41 Very recently, Yan et al.47 and Wang et al.48 have demonstrated a similar redox mechanism with anthraquinone and quinone compounds, respectively. Additionally, this redox reaction mechanism with the coordination/uncoordination of designated metal cations has been also well proven for both PC and PI elsewhere in the context of metal-ion aqueous batteries.34–40
The reduction of ortho-quinones into catecholates and the subsequent n-type coordination reaction depend not only on the pH of the aqueous electrolyte, but also on the nature and concentration of cationic charge carriers.49 As control experiments, we accomplished CV analysis of PC in both aqueous citrate buffer solutions (pH = 5; Fig. 2a) and aprotic organic electrolyte media (Fig. 2b) containing Li+, Zn2+, and Al3+ charge carriers, separately, which revealed a similar shift of redox potential in the Li+ < Zn2+ < Al3+ order. Notably, the latter results are in line with the recent study by Svensson et al., who elucidated the redox mechanism of quinone and catechol redox polymers in organic electrolytes containing different charge carriers.50 It is worth mentioning that besides the main redox peak associated with the coordination/uncoordination of Li+, the CV of PC in a slightly acidic Li+ buffer solution (Fig. 2a) also shows a small peak at higher potential which might be attributed to the contribution of protons to the redox mechanism. Interestingly, this second peak does not appear in the CV of PC in a neutral Li+ containing aqueous electrolyte (see Fig. 1b). These results suggest that the neutral pH favours Li+ cycling, whereas under slightly acidic conditions both protons and lithium ions might be involved. Furthermore, the involvement of different charge carriers (Li+, Zn2+ or Al3+) in the redox reaction of PC was confirmed here by ex situ ATR FTIR experiments (Fig. 2c), where the stretching vibration of the catecholic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond at ∼1650 cm−1 becomes discernible upon electrochemical oxidation but its intensity significantly decreased upon reduction. Parallelly, the evolution of a characteristic catecholate stretching band below 1200 cm−1 upon the formation of catecholic C–O−⋯Mn+ (Mn+ = Li+, Zn2+ or Al3+) during the reduction was also evident and different from proton reduction. Interestingly, its red shift in the Li+ (1202 cm−1) < Zn2+ (1106 cm−1) < Al3+ (1029 cm−1) order indicates stronger interaction of Mn+ with C–O− in the same order of metal cation valences.40,51 Taken together, these results confirm that the cations of the supporting electrolytes are mainly involved in the electrochemical reaction. However, despite all those control experiments, we cannot completely exclude the involvement of protons on the redox mechanism in acidic electrolytes and further studies would be necessary to undoubtedly elucidate this aspect.
O bond at ∼1650 cm−1 becomes discernible upon electrochemical oxidation but its intensity significantly decreased upon reduction. Parallelly, the evolution of a characteristic catecholate stretching band below 1200 cm−1 upon the formation of catecholic C–O−⋯Mn+ (Mn+ = Li+, Zn2+ or Al3+) during the reduction was also evident and different from proton reduction. Interestingly, its red shift in the Li+ (1202 cm−1) < Zn2+ (1106 cm−1) < Al3+ (1029 cm−1) order indicates stronger interaction of Mn+ with C–O− in the same order of metal cation valences.40,51 Taken together, these results confirm that the cations of the supporting electrolytes are mainly involved in the electrochemical reaction. However, despite all those control experiments, we cannot completely exclude the involvement of protons on the redox mechanism in acidic electrolytes and further studies would be necessary to undoubtedly elucidate this aspect.
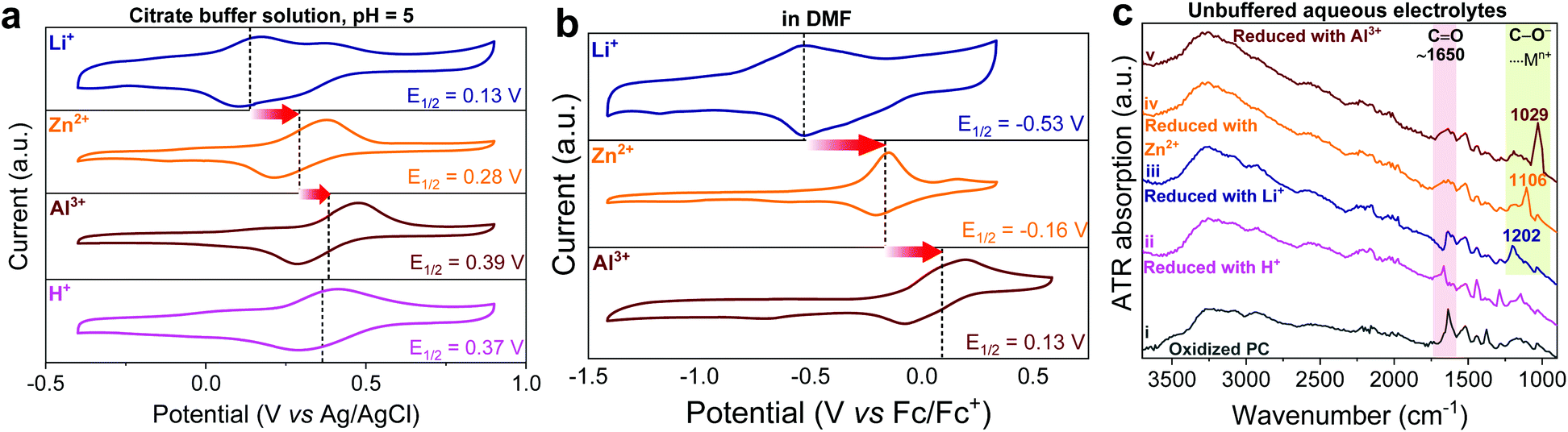 | ||
| Fig. 2 (a) Cyclic voltammograms of PC in (a) aqueous citrate buffer solutions (pH = 5) and (b) organic electrolytes (anhydrous DMF). The CVs were recorded at 5 mV s−1 in a standard three-electrode configuration. Potentials in Fig. 2b were later corrected with the ferrocene/ferrocenium (Fc/Fc+) redox couple. (c) Vertically offset ex situ ATR-FTIR spectra of oxidized PC(i) and reduced PC with H+ (ii), Li+ (iii), Zn2+ (iv), and Al3+ (v) in the aqueous unbuffered electrolytes. | ||
| Cation | PI anode | PC cathode | E cell (V) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| E ave (V) | E red 2, expt (V) | E red 2, calc (V) | E bind/cationd (kcal mol−1) | E 1/2 (V) | E red, expt (V) | E red 2, calc (V) | E bind (kcal mol−1) | ||
| a Average conversion potential (Eave) = (EPI,1 + EPI,2)/2 measured at 10 mV s−1. b E red 2, exp obtained experimentally at 10 mV s−1. c Refer to the ESI to see the full details of the computational method to calculate Ered 2, calc. d Refer to the ESI to see the full details of the computational method to calculate Ebinding per cation. e Average peak potential (E1/2) = Eanodic peak + Ecathodic peak/2 measured at 10 mV s−1. f E red, exp obtained experimentally at 10 mV s−1. g E cathode (E1/2) − Eanode (Eave) at 10 mV s−1. All the potentials (in CV and DFT calculations) are referenced against Ag/AgCl. h Relative reduction potential gain of active materials for binding with Zn2+ or Al3+ compared to Li+. i Change in the binding energy of active materials with Zn2+ or Al3+ in the reduced state compared to that with Li+. n d = not determined. | |||||||||
| Li+ | −0.50 | −0.78 | −0.71 | +1.2 | +0.1 | +0.05 | −0.06 | −7.3 | 0.6 |
| Zn2+ | −0.47 | −0.73 | −0.60 (+0.11)h | −2.8 (−4.0)i | +0.28 | +0.2 | +0.17 (+0.23)h | −25.2 (−17.9)i | 0.75 |
| Al3+ | −0.40 | −0.54 | −0.49 (+0.22)h | −7.9 (−9.0)i | +0.51 | +0.43 | +0.69 (+0.76)h | −49.0 (−41.7)i | 0.91 |
| Li+/H+ | −0.38 | −0.45 | nd | nd | +0.57 | +0.52 | n d | n d | 0.95 |
In this sense, it is well-known that catechols also undergo ortho-quinone/catechol redox transformations via a proton-coupled two-electron process (Fig. 1c) at significantly high redox potentials.61–63 Therefore, to boost the voltage output of the PI–PC full-cell further we explored the mixed electrolyte concept (containing Li+ and H+).25Fig. 1b (lower panel) shows the CVs of PI and PC in such a mixed electrolyte anticipating a maximum voltage output of 0.95 V with H+ and Li+ involved in the electrochemistry of the PC cathode and PI anode, respectively. It should be noted that achieving AqPBs with an output voltage above 0.9 V is not that simple task, particularly for the n|n-type combination (Table S1†). We did not consider proton cycling in the case of PI because to the best of our knowledge, imides are not known to undergo proton coupled redox reactions. Evidently, Gheytani et al. demonstrated the pH independent redox activity of PI in aqueous electrolytes.35
Analogues to the CV observations, a distinctive small and huge increase of Ered 2, calc in the order of Li+ < Zn2+ < Al3+ was accounted for PI and PC, respectively. Assuming the similar redox mechanism of both PI and PC based on their n-type nature, the difference in redox potentials should be ascribed to the differences in strength of metal cation-reduced redox core interactions (Ebind). The computed Ebind between enolate (of PI) and Li+, Zn2+ and Al3+ was +1.2, −2.8, and −7.9 kcal mol−1 per cation, respectively (Table 1). However, a higher Ebind of −7.3, −25.2, and −49.0 kcal mol−1 per cation was obtained for metal cation–catecholate (of PC) with Li+, Zn2+ and Al3+, respectively. In general, a higher value of Ebind is associated with larger positive shifts in reduction potentials due to the enhanced thermodynamic stabilization of the reduced species into the complexes,41,47,50 and thus their re-oxidation becomes increasingly difficult and occurs at more positive potentials in the Li+ < Zn2+ < Al3+ order. Therefore, a small enhancement by −4.0 and −9.0 kcal mol−1 in Ebind induced a minute increase of redox potential for enolate (PI) with Zn2+ and Al3+, respectively, compared to Li+, whereas, a huge increase in redox potential for catecholate (PC) was linked to the large increment of Ebind (by −17.9 and −41.7 kcal mol−1 for Zn2+ and Al3+, respectively, compared to Li+). This distinctive trend in Ebind and thus redox potential between PI and PC are presumably due to their different electron density. Localized electron density on the catecholate ring and the ortho-nature of quinone redox sites lead to strong metal–polymer interactions in PC,52,53 while for PI the para-oriented redox-active enolates and more diffused electron density over the extended naphthalenetetracarboxylic diimide (NDI) ring result in weaken metal–polymer interactions.54
Electrochemical performance of individual electrodes
Fig. 3 shows the rate performance of PI and PC half-cells by GCD experiments at progressively increasing C-rates in different aqueous electrolytes containing Li+, Zn2+, Al3+, and Li+/H+ charge carriers.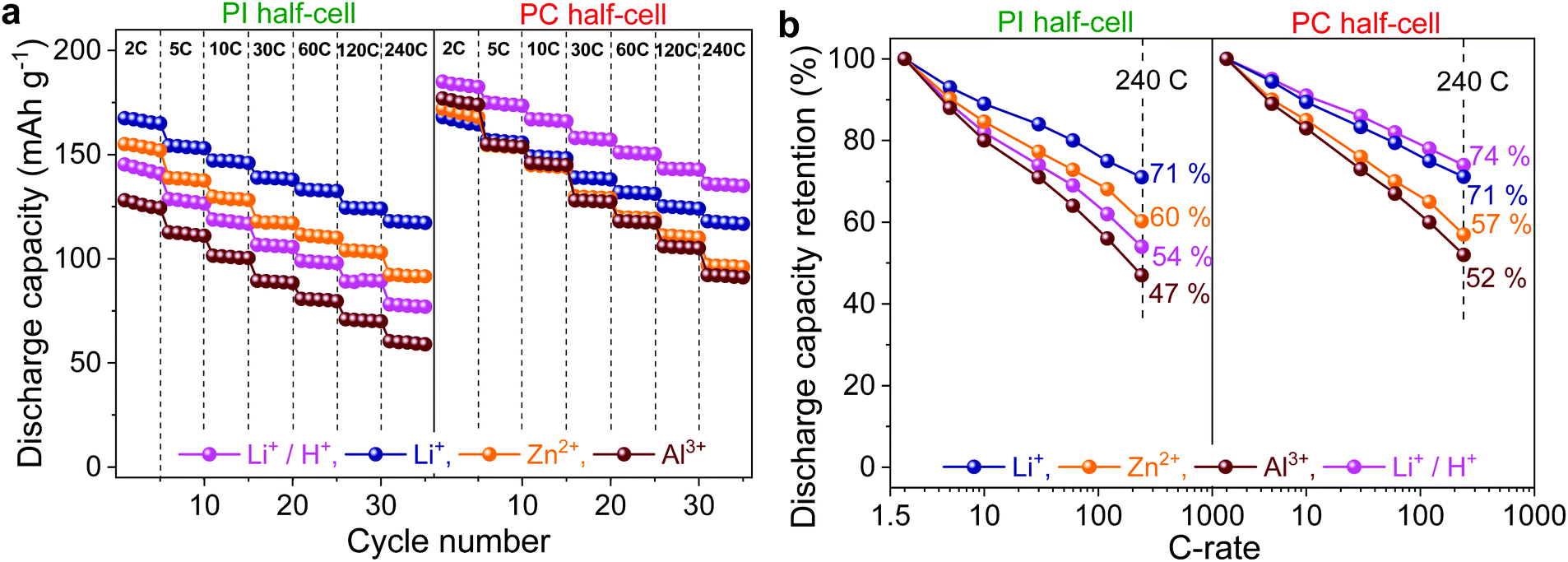 | ||
| Fig. 3 (a) Discharge capacity vs. cycle number at increasing C-rates. (b) Discharge capacity retention at different C-rates. The discharge capacities at higher C-rates are normalized with respect to the discharge capacity at 2C (1C = 166 and 180 mA g−1 for PI and PC, respectively). | ||
The representative specific capacity–potential profiles are given in the ESI (Fig. S4 and S5†). It is also evident from these potential profiles that despite their sloping nature, the charge/discharge plateaus of PI and PC were shifted to higher values in the Li+ < Zn2+ < Al3+ order, but to a different extent, which is in good agreement with CV studies and DFT calculations. Both PI and PC delivered reversible capacities in the range of 128–164 mA h g−1 and 167–183 mA h g−1, respectively, at 2C in the aforementioned electrolytes (Fig. 3a). PI in Li+ provided the highest discharge capacity of 164 mA h g−1, close to the theoretical limit (166 mA h g−1). However, lower capacity utilization values of 93, 86 and 77% were obtained in Zn2+, Al3+, and Li+/H+, respectively. Notably, the coulombic efficiencies were close to 100% in all the cases (Fig. S4†). On the other hand, PC demonstrated a better active-material utilization in the range of 93–100% (theoretical specific capacity of 180 mA h g−1) but lower coulombic efficiencies at low C-rates, and subsequently recovered to the quantitative values at higher C-rates (Fig. S5†).
With increasing C-rates from 2C to 240C, both the discharge capacities (Fig. 3a) and consequently the capacity retentions (capacities at higher C-rates w.r.t. the capacity at 2C; Fig. 3b) of PI and PC decreased monotonically in the Li+ < Zn2+ < Al3+ order. As demonstrated in our previous publication,41 this diminished rate performance should be ascribed to the sluggish ion mobilities on account of stronger metal cation–polymer interactions in the same order of charge carrier valences. Remarkably, even at a very high C-rate of 240C, PI and PC still retained 71, 60 and 47% and 71, 57 and 52% of their initial capacities in Li+, Zn2+ and Al3+, respectively. Additionally, both the electrode partners of the all-organic cell also demonstrated excellent dynamic performance in the Li+/H+ electrolyte, but the overall performance of PC was superior to that of PI (Fig. 3).
In order to understand the origin of the outstanding rate performance of PI and PC, the electrochemical kinetics of redox reactions in polymer working electrodes were investigated by CV at different scan rates (v) in the aqueous electrolyte containing Li+ as the representative charge carrier. The Laviron method was used to determine the transfer coefficient (α) and apparent reaction rate constant (k0) parameters (Fig. 4a, d and S6†).64 The value of α was close to 0.5 for both systems, indicating almost symmetric energy barriers for the oxidation and reduction reactions. Furthermore, the calculated k0 was found to be 1.5 and 2.6 s−1 for PI and PC, respectively. Additionally, considering that the peak currents (ip) in the CV curves obey the power-law relationship as: ip = avb, where a and b are adjustable coefficients, the slope (b-value) of the log(ip) vs. log(v) plot provides insights into the underlying redox mechanism.65 Ideally, the b-value of 0.5 indicates a diffusion-controlled process, whereas the b-value of 1.0 is the signature of a capacitive-controlled behavior.
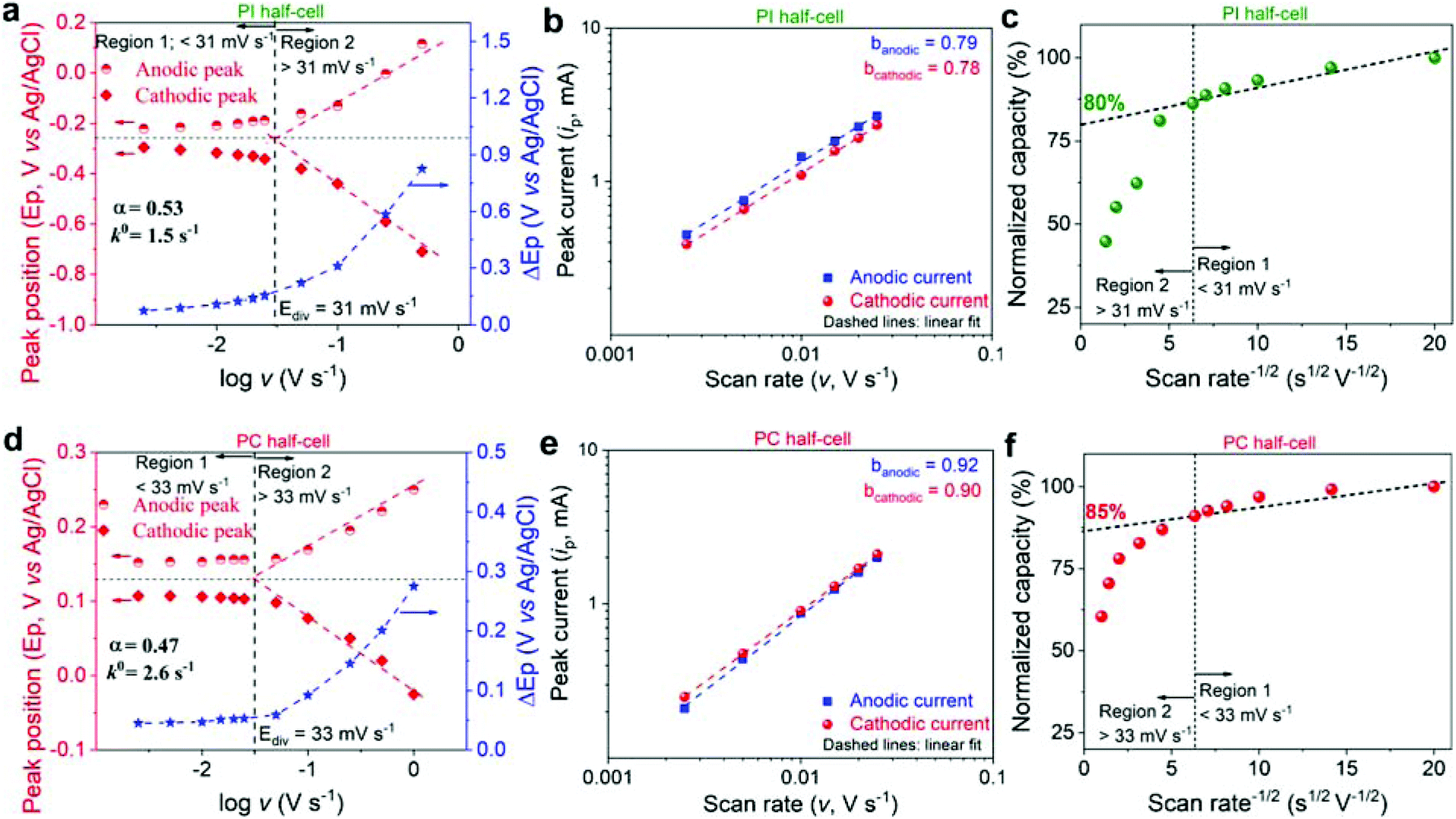 | ||
| Fig. 4 Kinetic evaluation of PI and PC half-cells. (a and d) Laviron plots, indicating the variation of anodic and cathodic peak positions (Ep), and ΔEp as a function of the scan rate on a logarithmic scale. (b and e) Plots of peak current vs. scan rate on a logarithmic scale. (c and f) Normalized capacity vs. scan rate−1/2 plots. The dashed vertical line in (a, c, d and f) separates two distinct kinetic regions: Region 1 below Ediv where the capacity is mostly independent of the scan rate, and region 2 above Ediv where the capacity is limited by semi-infinite linear diffusion. The y-intercept in Fig. 4c and f corresponds to the extrapolation of the infinite scan rate capacity (% capacitynon-diffusion-controlled). This dashed diagonal line is drawn based on the two distinct kinetic regions. | ||
High b-values of 0.79/0.78 and 0.92/0.90 for the anodic/cathodic peaks were obtained for PI and PC (Fig. 4b and e), respectively, suggesting a mixed electrochemical response but tending to be less diffusion limited. Here, the capacitive-type electrochemical response is assumed to have mainly originated from bulk electrochemical reaction sites (similar to intercalation pseudocapacitance that is observed in some inorganic intercalation compounds, which is different from conventional surface pseudocapacitance in nanomaterials)65–67 that are less limited by the diffusion processes on account of polymer's high reaction rates and superior ion mobilities.41–43 To further quantify the contributions of diffusion- and non-diffusion-controlled charge storage to the total capacity, the correlation between normalized capacity and v−1/2 was established (Fig. 4c and f). The capacitycapacitive-controlled, which is non-diffusion limited, was obtained as the y-intercept,65 resulting in 80 and 85% for PI and PC, respectively, resembling a fast pseudocapacitive behavior. Thanks to the smart design of both the electrode partners, they are engineered to exhibit pseudo-capacitive like energy storage behavior on account of fast redox reactions and facilitated ion-mobility in the bulk of the material. In fact, the incorporation of ion conducting 2-acrylamido-2-methylpropane sulfonate42 and poly(ethylene oxide)43 comonomer moieties in PC and PI, respectively, assisted the ion transport. Therefore, superior rate performance is anticipated for the PI–PC full-cells.
It is worth noting that both PI and PC demonstrated satisfactory stable cycling over 100 GCD cycles at 10C, retaining 98, 97, 91, and 98% (Fig. S7†) and 96, 95, 94, and 94% (Fig. S8†) of their initial capacities, respectively, with quantitative coulombic efficiencies (>99%) in Li+, Zn2+, Al3+, and Li+/H+.
Electrochemical performance evaluation of PI–PC all-organic batteries
Based on the excellent electrode performance of PI and PC individual electrodes, along with their tunable voltage behaviors, all-organic aqueous rechargeable batteries were assembled with PI and PC serving as the anode and cathode, respectively. A schematic illustration of the full-cell structure and the overall electrode reactions of the cells in two different sets of aqueous electrolytes are given in Scheme 1.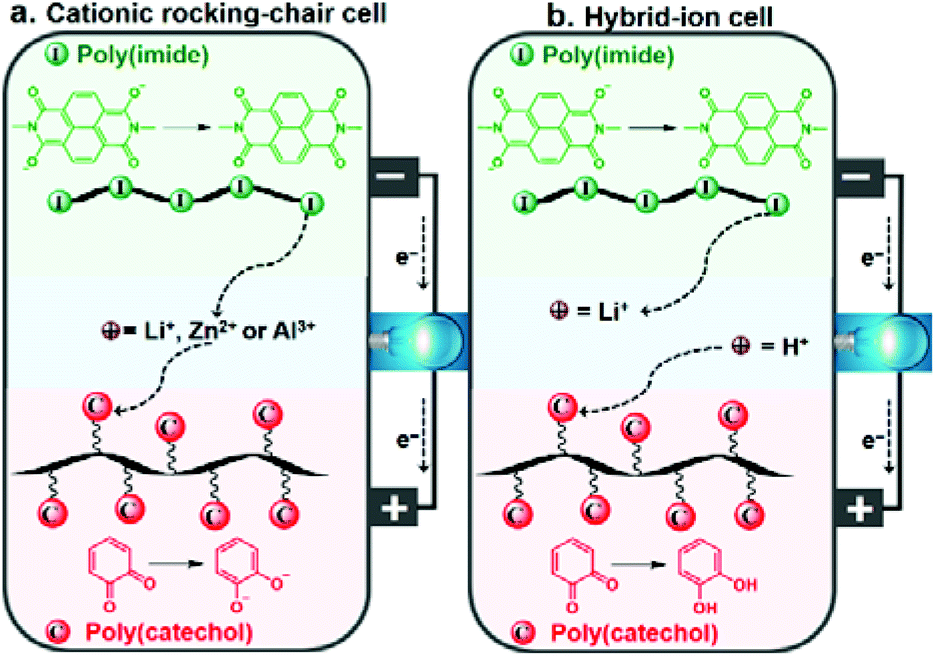 | ||
| Scheme 1 Schematic of the working principle of the PI–PC full-cell during the discharge process in a cationic rocking-chair mode with Li+, Zn2+ or Al3+ (a), and hybrid-ion configuration with Li+/H+ (b). | ||
The full-cells operate in a cationic rocking-chair mode in Li+, Zn2+ or Al3+ based electrolytes, with unidirectional shuttling of charge carriers from the anode to the cathode during discharge (Scheme 1a) and from the cathode to the anode during charge.18 On the other hand, the PI–PC full-cell is assumed to operate in a non-rocking chair cationic hybrid-ion configuration with a mixed Li+/H+ electrolyte: during the discharging process, enolates are oxidized to imides while releasing Li+ into the electrolyte reservoir, and ortho-quinones are reduced to the classical catechols with the uptake of H+ from the electrolyte (Scheme 1b).25 Conversely, catechols are oxidized to ortho-quinones, while H+ reversibly breaks away from the cathode and return to the electrolyte, and the imides are reduced to enolates with the concomitant coordination of Li+. However, the interaction of H+ with PI in the Li+/H+ mixed electrolyte can be argued, and we partially discard this through the following controlled experiment. When PI was cycled in 0.25 M H2SO4, the electrochemical stability rapidly diminished upon cycling, indicating its poor compatibility with protons (Fig. S9†), which was already reported by Wang et al.68 Encouragingly, the cycling stability of the PI half-cell (Fig. S7e†) and PI–PC full-cell (discussed later, shown in Fig. 5c) in Li+/H+ significantly improved compared to only in H+, once again suggesting that Li+ is involved in the redox reaction of PI.
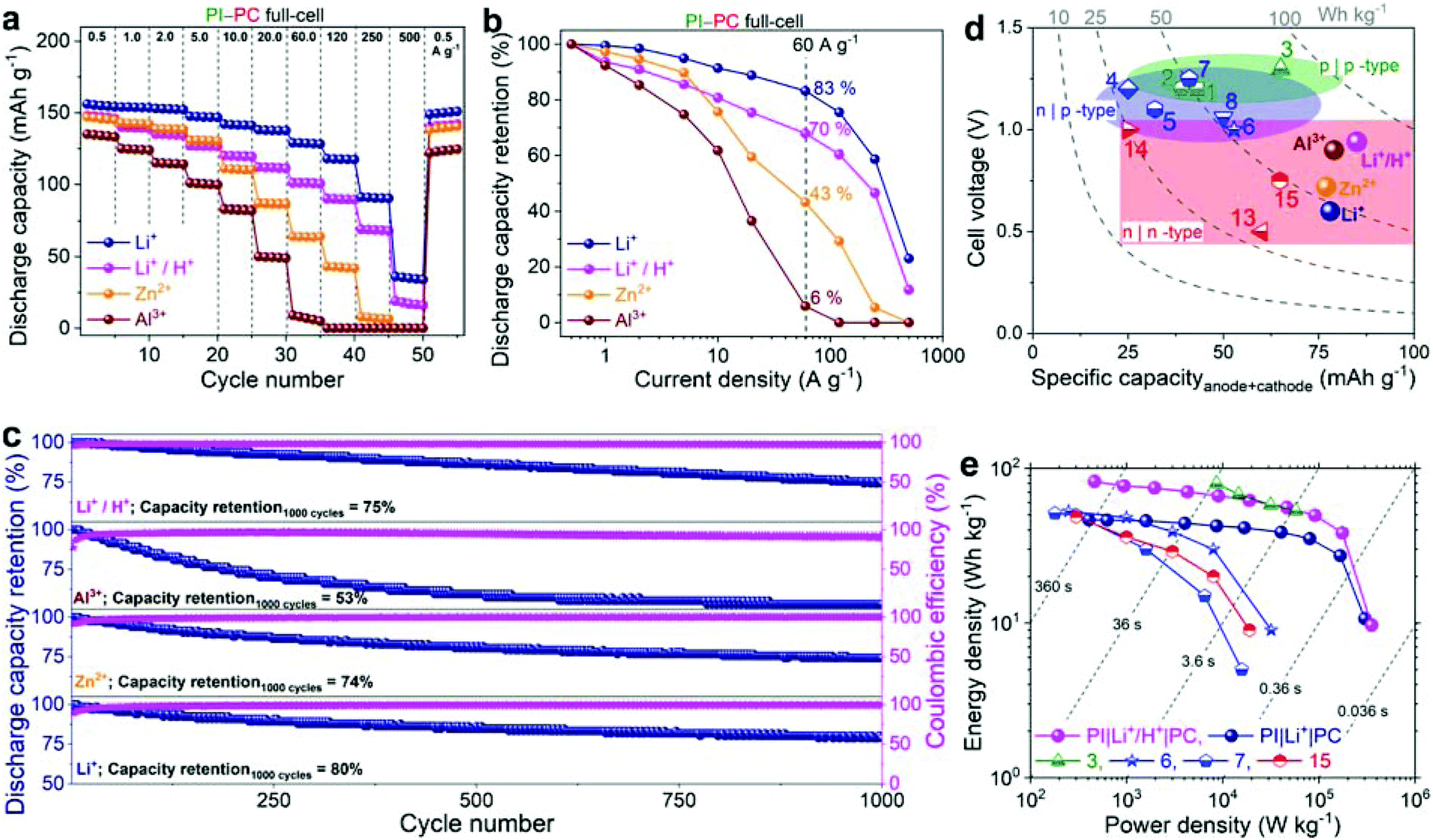 | ||
| Fig. 5 (a and b) Rate performance of PI–PC full-cells: discharge capacity vs. cycle number at different current densities (a), and discharge capacity retention (b). (c) Cyclic performance: discharge capacity retention (%) and coulombic efficiencies measured at 5 A g−1. The capacity and current density in experiments a–c were based on the PI component of the full-cell. (d and e) Comparison of full-cell performance with the state-of-the-art all-polymer aqueous stationary batteries: cell voltage vs. specific capacity (d) and Ragone plot (e). The specific capacity and gravimetric energy/power density were evaluated based on the total mass of the anode and cathode. Both these plots for various all-organic full-cells (mostly, all-polymer) are computed by considering some of the best performing full-cells in their class (see Table S1†). p|p-type: half-filled green symbols in the light green region, n|p-type: half-filled blue symbols in the light blue region, n|n-type: half-filled red symbols in the light red region, and filled spheres represent this work. | ||
The mass ratio between PI and PC in the full-cell was calculated to be 1:1, 1:0.9, 1:0.7, and 1:0.72 in Li+, Zn2+, Al3+, and Li+/H+ configurations, respectively, based on the capacities of individual electrodes in the three-electrode investigations. To validate the different mass balance, the overall voltage of PI–PC full-cells and the individual voltages of the PI anode and PC cathode were monitored during electrochemical experiments in Swagelok™ T-cells (see Fig. S10 and the ESI for detailed discussion†). Fig. S10a–c† show that the average cell potential (taken as half the sum of mid-point voltages of charge and discharge branches) increases from 0.58 to 0.74 and further to 0.89 V when the type of charge carrier changed from Li+ to Zn2+, and further to Al3+. This is in good agreement with the CVs of individual electrodes (see Fig. 1b). Accordingly, a higher voltage output of 0.95 V was exhibited by the full-cell with a mixed Li+/H+ electrolyte (Fig. S10d†).
Then, the rate capability of full-cells in the aforementioned electrolytes was assessed in a two-electrode configuration by GCD studies at progressively increasing current densities. At a low current density of 0.5 A g−1, full-cells were able to deliver 155, 145, 134, and 146 mA h g−1 discharge capacities in Li+, Zn2+, Al3+, and Li+/H+, respectively (Fig. 5a). The rate performance of PI–PC full-cells is once again hampered in the Li+ < Zn2+ < Al3+ order, which is in accordance with the half-cell studies. For instance, when the applied current density was increased to 60 A g−1, full-cells achieved discharge capacities of 129, 64, and 7 mA h g−1, corresponding to 83, 43, and 6% of their initial capacities in Li+, Zn2+, and Al3+, respectively (Fig. 5b). Remarkably, full-cells in Li+ and Li+/H+ can still deliver discharge capacities of 35 and 17 mA h g−1, respectively, at even an extremely high current density of 500 A g−1 (corresponding to a sub-second charge or discharge), reflecting the unprecedented rate capabilities of both PI and PC in these electrolytes. Furthermore, when the current density was brought back to 0.5 A g−1, nearly quantitative capacity recovery was observed in all the cases, discarding irreversible capacity fade during the high current experiments. It is also worth noting here that the average coulombic efficiencies were close to 100% in Li+ and Zn2+ aqueous electrolytes at all the current densities, while slightly lower efficiencies (ca., 94–97%) were obtained below 1 A g−1 with Al3+ and Li+/H+ and then subsequently improved to > 99% (Fig. S11†).
The cycling stability of PI–PC full-cells was evaluated at a current density of 5 A g−1. Full-cells were able to maintain 80, 74, 53, and 75% of their initial capacities over 1000 GCD cycles in Li+, Zn2+, Al3+, and Li+/H+, respectively (Fig. 5c and S12†). Additionally, the coulombic efficiencies were stabilized at >99% after the initial few catechol-to-ortho-quinone activation cycles.
Finally, to demonstrate the availability of our PI–PC full-cells as sustainable high-performance energy storage systems, we compared their performance with that of the state-of-the-art all-polymer aqueous stationary batteries. The maximum attained specific capacities in the range of 78–85 mA h g−1 were higher than that of the reported AqPBs, and the average cell voltage of 0.95 V in Li+/H+ was also notable for the n|n-type configuration (Fig. 5d and Table S1†). As shown in the Ragone plot (Fig. 5e), full-cells in Li+ and Li+/H+ containing aqueous electrolytes can deliver a maximum energy/power density of 46.5 W h kg−1/302 kW kg−1 and 82.2 W h kg−1/355 kW kg−1, respectively. The concurrent high specific capacity, high cell voltage, and ultrafast kinetics, particularly in the mixed Li+/H+ aqueous electrolyte, endowed the PI–PC full-cell with a steady and high energy density over a wide range of power values that clearly rivals most of the state-of-the-art AqPBs, and also comparable to the poly(viologen)–poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl acrylamide) aqueous battery (example 3 in Fig. 5e and Table S1†),28 which is based on the p|p-type combination. Yet another advantage of n|n-type combination is that it can make use of lighter weight shuttling cations (e.g., H+, Li+etc.), in contrast to the p|p- and n|p-type configurations, which require bulky anions and dual ions, respectively, as the electroneutralizing charge carriers. Consequently, when the energy and power density were evaluated based on the total mass of both electrodes and the consumed salt, a small decrease of energy/power density (<1%) was noted for PI–PC full-cells in Li+ and Li+/H+, while a substantial lowering of 19, 32, and 30% energy/power density for the examples 3,28 6,30 and 7,33 respectively, is noticeable from their corresponding Ragone plots (Fig. 5e, S13 and Table S1†).
Conclusions
In this work, we have developed all-organic aqueous batteries comprising a poly(imide) (PI) anode and poly(catechol) (PC) cathode that demonstrated tunable electrochemical performance. We realized this by exploiting the distinctive strength of metal cation–redox core ionic interactions of electrode partners with different charge carriers based on their n-type redox behavior, resulting in various cationic rocking-chair cells. The increase of cell voltage from 0.58 to 0.74 and further to 0.89 V was achieved, when the type of charge carrier changed from Li+ to Zn2+ and to Al3+. Adversely, this gain also comes with the penalty of rate capability and cycling stabilities in the Li+ < Zn2+ < Al3+ order. The PI–PC full-cell also worked in hybrid-ion mode with a mixed Li+/H+ aqueous electrolyte, exhibiting the highest cell voltage of 0.95 V with good cycling stability and excellent rate capability. The concurrent high specific capacity (85 mA h g−1), high cell voltage (0.95 V), and ultrafast kinetics (working at 500 A g−1, which corresponds to 0.1 s charge or discharge), particularly in Li+/H+, provided an impressive energy density of ∼80.6 W h kg−1, where a high value of ∼10 W h kg−1 was still achieved at an unprecedented power density of ∼348 kW kg−1 based on the total mass of both electrodes and the consumed salt. This overall performance is far superior to that of most of the reported all-polymer aqueous stationary batteries.Based on the CV, GCD results and DFT calculations, we postulate that PI and PC can be used as universal organic electrodes in numerous Li-ion and post-Li-ion energy storage technologies, including proton and all-polymer batteries, stemming from their versatile n-type charge storage mechanism. The development of such universal organic electrodes is particularly intriguing and gaining popularity among the battery community due to the fact that it demands minimal electrode/device engineering efforts. We hypothesize that the combination of high safety aqueous electrolytes and high-performance PI and PC based organic electrodes could offer a practical option as a truly sustainable energy storage system for wide use in large-scale applications.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
RM, NP and AM thank the Spanish Ministry of Science, Innovation and Universities through the SUSBAT project (Ref. RTI2018-101049-B-I00) (MINECO/FEDER, UE) and the European Research Council (ERC) through the “MFreeB” project (grant agreement No. 726217) for financial support. NP also appreciates Spanish MINECO for the Juan de la Cierva-formation fellowship [FJC2018-037781-I] received to carry out this work. AM acknowledges the TALENTO grant (2017-T1/AMB-5264) from the Comunidad de Madrid for financial support. The authors acknowledge the computing facilities of CSUC for providing resources that contributed to the research results reported within this paper. DM and NC are grateful to the financial support of the European Research Council by the Proof of Concept Grant Innovative Polymeric Batteries by 3D Printing (iPes-3DBat) 789875. CD and CJ thank the “Fonds de la Recherche Scientifique” (FRS-FNRS) for funding.Notes and references
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- K. Turcheniuk, D. Bondarev, V. Singhal and G. Yushin, Nature, 2018, 559, 467–470 CrossRef CAS PubMed.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- R. Demir-Cakan, M. R. Palacin and L. Croguennec, J. Mater. Chem. A, 2019, 7, 20519–20539 RSC.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef PubMed.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S.-K. Jung, H.-D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef PubMed.
- K. Amin, L. Mao and Z. Wei, Macromol. Rapid Commun., 2019, 40, 1800565 CrossRef PubMed.
- J. Xie and Q. Zhang, Small, 2019, 15, 1805061 CrossRef PubMed.
- Y. Y. Lai, X. Li and Y. Zhu, ACS Appl. Polym. Mater., 2020, 2(2), 113–128 CrossRef CAS.
- N. Casado, D. Mantione, D. Shanmukaraj and D. Mecerreyes, ChemSusChem, 2019, 13(9), 2464–2470 CrossRef PubMed.
- Y. Chen, S. Zhuo, Z. Li and C. Wang, EnergyChem, 2020, 100030 CrossRef.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120(14), 6490–6557 CrossRef CAS PubMed.
- P. Poizot, F. Dolhem and J. Gaubicher, Curr. Opin. Electrochem., 2018, 9, 70–80 CrossRef CAS.
- C. Friebe, A. Lex-Balducci and U. S. Schubert, ChemSusChem, 2019, 12, 4093–4115 CrossRef CAS PubMed.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- D. P. Tabor, R. Gómez-Bombarelli, L. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, J. Mater. Chem. A, 2019, 7, 12833–12841 RSC.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed.
- G. Hernández, N. Casado, A. M. Zamarayeva, J. K. Duey, M. Armand, A. C. Arias and D. Mecerreyes, ACS Appl. Energy Mater., 2018, 1, 7199–7205 CrossRef.
- Y. Zhang, Y. An, L. Wu, H. Chen, Z. Li, H. Dou, V. Murugadoss, J. Fan, X. Zhang, X. Mai and Z. Guo, J. Mater. Chem. A, 2019, 7, 19668–19675 RSC.
- N. Chikushi, H. Yamada, K. Oyaizu and H. Nishide, Sci. China, Ser. B: Chem., 2012, 55, 822–829 CrossRef CAS.
- N. Sano, W. Tomita, S. Hara, C.-M. Min, J.-S. Lee, K. Oyaizu and H. Nishide, ACS Appl. Mater. Interfaces, 2013, 5, 1355–1361 CrossRef CAS PubMed.
- K. Sato, R. Katagiri, N. Chikushi, S. Lee, K. Oyaizu, J.-S. Lee and H. Nishide, Chem. Lett., 2017, 46, 693–694 CrossRef CAS.
- X. Dong, H. Yu, Y. Ma, J. L. Bao, D. G. Truhlar, Y. Wang and Y. Xia, Chem. - Eur. J., 2017, 23, 2560–2565 CrossRef CAS PubMed.
- H. Long, W. Zeng, H. Wang, M. Qian, Y. Liang and Z. Wang, Adv. Sci., 2018, 5, 1700634 CrossRef PubMed.
- K. Hatakeyama-Sato, H. Wakamatsu, K. Yamagishi, T. Fujie, S. Takeoka, K. Oyaizu and H. Nishide, Small, 2019, 15, 1805296 CrossRef PubMed.
- Y. Zhang, Y. An, B. Yin, J. Jiang, S. Dong, H. Dou and X. Zhang, J. Mater. Chem. A, 2019, 7, 11314–11320 RSC.
- H. Qin, Z. P. Song, H. Zhan and Y. H. Zhou, J. Power Sources, 2014, 249, 367–372 CrossRef CAS.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- X. Fan, F. Wang, X. Ji, R. Wang, T. Gao, S. Hou, J. Chen, T. Deng, X. Li, L. Chen, C. Luo, L. Wang and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 7146–7150 CrossRef CAS PubMed.
- J. Bitenc, K. Pirnat, T. Bančič, M. Gaberšček, B. Genorio, A. Randon-Vitanova and R. Dominko, ChemSusChem, 2015, 8, 4128–4132 CrossRef CAS PubMed.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- T. Liu, K. C. Kim, B. Lee, Z. Chen, S. Noda, S. S. Jang and S. W. Lee, Energy Environ. Sci., 2017, 10, 205–215 RSC.
- S. Zhang, W. Zhao, H. Li and Q. Xu, ChemSusChem, 2020, 13, 188–195 CrossRef CAS PubMed.
- N. Patil, A. Mavrandonakis, C. Jérôme, C. Detrembleur, J. Palma and R. Marcilla, ACS Appl. Energy Mater., 2019, 2, 3035–3041 CrossRef CAS.
- N. Patil, A. Aqil, F. Ouhib, S. Admassie, O. Inganäs, C. Jérôme and C. Detrembleur, Adv. Mater., 2017, 29, 1703373 CrossRef PubMed.
- G. Hernández, N. Casado, R. Coste, D. Shanmukaraj, L. Rubatat, M. Armand and D. Mecerreyes, RSC Adv., 2015, 5, 17096–17103 RSC.
- L. J. Krause, J. Electrochem. Soc., 1988, 135, 1137 CrossRef CAS.
- L. J. Krause, J. Electrochem. Soc., 1989, 136, 1379 CrossRef CAS.
- W. Deng, Y. Shen, J. Qian and H. Yang, Chem. Commun., 2015, 51, 5097–5099 RSC.
- L. Yan, C. Zhao, Y. Sha, Z. Li, T. Liu, M. Ling, S. Zhou and C. Liang, Nano Energy, 2020, 73, 104766 CrossRef CAS.
- H. Wang, R. Emanuelsson, A. Banerjee, R. Ahuja, M. Strømme and M. Sjödin, J. Phys. Chem. C, 2020, 124, 13609–13617 CrossRef CAS.
- P. S. Guin, S. Das and P. C. Mandal, Int. J. Electrochem., 2011, 2011, 1–22 CrossRef.
- M. Svensson, Conducting polymers for battery applications, Uppsala universitet, 2020 Search PubMed.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Adv. Mater., 2014, 26, 6572–6579 CrossRef CAS PubMed.
- N. Patil, C. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2018, 82, 34–91 CrossRef CAS.
- M. Krogsgaard, V. Nue and H. Birkedal, Chem. - Eur. J., 2016, 22, 844–857 CrossRef CAS PubMed.
- Y. Geng, S.-X. Wu, H. Li, X. Tang, Y. Wu, Z.-M. Su and Y. Liao, J. Mater. Chem., 2011, 21, 15558 RSC.
- G. S. Vadehra, R. P. Maloney, M. A. Garcia-Garibay and B. Dunn, Chem. Mater., 2014, 26, 7151–7157 CrossRef CAS.
- T. Yokoji, H. Matsubara and M. Satoh, J. Mater. Chem. A, 2014, 2, 19347–19354 RSC.
- N. Patil, M. Aqil, A. Aqil, F. Ouhib, R. Marcilla, A. Minoia, R. Lazzaroni, C. Jérôme and C. Detrembleur, Chem. Mater., 2018, 30, 5831–5835 CrossRef CAS.
- K. H. Jung, G. S. Jeong, C. Y. Go and K. C. Kim, Energy Storage Mater., 2020, 24, 237–246 CrossRef.
- G. Dai, Y. Gao, Z. Niu, P. He, X. Zhang, Y. Zhao and H. Zhou, ChemSusChem, 2020, 13(9), 2264–2270 CrossRef CAS PubMed.
- A. Jouhara, N. Dupré, A.-C. Gaillot, D. Guyomard, F. Dolhem and P. Poizot, Nat. Commun., 2018, 9, 4401 CrossRef PubMed.
- Q. Lin, Q. Li, C. Batchelor-McAuley and R. G. Compton, J. Phys. Chem. C, 2015, 119, 1489–1495 CrossRef CAS.
- N. Patil, D. Cordella, A. Aqil, A. Debuigne, S. Admassie, C. Jérôme and C. Detrembleur, Macromolecules, 2016, 49, 7676–7691 CrossRef CAS.
- K. Pirnat, N. Casado, L. Porcarelli, N. Ballard and D. Mecerreyes, Macromolecules, 2019, 52, 8155–8166 CrossRef CAS.
- M. Sterby, R. Emanuelsson, X. Huang, A. Gogoll, M. Strømme and M. Sjödin, Electrochim. Acta, 2017, 235, 356–364 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- Y. Gogotsi and R. M. Penner, ACS Nano, 2018, 12, 2081–2083 CrossRef CAS PubMed.
- Y. Jiang and J. Liu, Energy Environ. Mater., 2019, 2, 30–37 CrossRef.
- Y. Wang, X. Cui, Y. Zhang, L. Zhang, X. Gong and G. Zheng, Adv. Mater., 2016, 28, 7626–7632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI Table, computational procedure and figures. See DOI: 10.1039/d0ta09404h |
| This journal is © The Royal Society of Chemistry 2021 |