
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and melt-spinning of partly bio-based thermoplastic poly(cycloacetal-urethane)s toward sustainable textiles†
Niklas
Warlin
 a,
Erik
Nilsson
bc,
Zengwei
Guo
c,
Smita V.
Mankar
a,
Nitin G.
Valsange
a,
Nicola
Rehnberg
ad,
Stefan
Lundmark
e,
Patric
Jannasch
*a and
Baozhong
Zhang
*a
a,
Erik
Nilsson
bc,
Zengwei
Guo
c,
Smita V.
Mankar
a,
Nitin G.
Valsange
a,
Nicola
Rehnberg
ad,
Stefan
Lundmark
e,
Patric
Jannasch
*a and
Baozhong
Zhang
*a
aCentre of Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: Baozhong.zhang@chem.lu.se; Patric.jannasch@chem.lu.se
bPlasman, Molndalsvagen 36, 412 63 Gothenburg, Sweden
cDepartment of Chemistry, Biomaterials and Textile, RISE - Research Institutes of Sweden, Mölndal, SE-43153, Sweden
dStrategic R&D, Bona AB, Box 210 74, 200 21 Malmö, Sweden
ePerstorp AB, Innovation, Perstorp Industrial Park, 284 80 Perstorp, Sweden
First published on 10th August 2021
Abstract
A rigid diol with a cyclic acetal structure was synthesized by facile acetalation of fructose-based 5-hydroxymethyl furfural (HMF) and partly bio-based di-trimethylolpropane (di-TMP). This diol (Monomer T) was copolymerized with potentially bio-based flexible polytetrahydrofuran and diisocyanates to prepare thermoplastic poly(cycloacetal-urethane)s. A modified one-step solution polymerization protocol resulted in relatively high molecular weights (Mn ∼ 41.5–98.9 kDa). All the obtained poly(cycloacetal-urethane)s were amorphous with tuneable glass transition temperatures up to 104 °C. Thermogravimetric analysis indicated that these polymers were thermally stable up to 253 °C and had a relatively high pyrolysis char residue, which may indicate potential inherent flame resistance. Melt rheology measurements were performed to determine a suitable processing window between 165–186 °C, after which the polymer was successfully melt-spun into ∼150 meters of homogeneous fibres at 185 °C. The resulting fibres could be readily hydrolysed under acidic conditions, resulting in partial recovery of the original chemical building blocks.
Introduction
Conventional fossil-based plastics (polymers) have given rise to many environmental challenges such as greenhouse gas emissions, global warming, plastics littering, as well as various health issues.1–5 Many of these challenges can be potentially mitigated by the transition toward sustainable polymers from, e.g., biomass resources.3,6 During the past decade, tremendous efforts have been made to facilitate the transition toward sustainable bio-based polymers.7–9 Recently, the end-of-life recycling aspects of bio-based polymers have received increasing attention,10–14 which demands newly designed bio-based polymers to enable and facilitate their recycling.15–19 This demand is particularly important in applications where the polymer materials are complex and challenging to recycle.20,21 Textiles are among such applications, where fibres and yarns usually contain complex materials and additives and are thus not usually recycled.22 Therefore, new bio-based polymer fibres with improved recyclability are attractive for sustainable textiles.23,24To recycle complex polymer mixtures as textiles, chemical recycling is considered more practical than conventional mechanical recycling.20,22,25 In this context, polymers with acid-sensitive acetal bonds (polyacetals) are particularly attractive,26–29 because they can be conveniently acid-hydrolyzed which can enable chemical recycling even from complex textiles.25 Polyacetals exist widely in nature, including cellulose, hemicellulose, starch, chitin, etc. Synthetic polyacetals are also commercially available, such as polyoxymethylene (POM) and polyvinyl butyral (PVB). In addition, linear aliphatic polyacetals were also prepared by acetal metathesis polymerizations, which partially mimic the properties of polyethylene.30,31 Rigid polycycloacetals have also been reported,32–39 which often exhibit enhanced Tg, thermal stability, and mechanical strength.40–42 Recently, Perstorp AB has commercialized Akestra™, a PET-like poly(cycloacetal-ester), which shows promising application potential for hot-filling food packaging.41 Many other poly(acetal-ester)s,43,44 poly(acetal-carbonate)s,45,46 and poly(acetal-urethane)s have also been reported in literature.40,47 To our knowledge, the potential of polyacetals toward sustainable textiles has not been investigated.
In the context of sustainable textiles, thermoplastic poly(acetal-urethane)s (PAUs) are particularly interesting, because in general thermoplastic polyurethanes (TPUs) offer an attractive alternative to non-recyclable polyurethane thermosets in textiles.48,49 For instance, Spandex is a thermoset polyurethane material that is widely used in fabrics to gain elasticity, but its crosslinked structures inevitably prevent textile recycling.50,51 Recently, a TPU namely Elastollan™ (by BASF) has been spun into fibres with tuneable mechanical properties, which can be potentially a recyclable substitute for Spandex. When TPUs contain acid-labile acetal structures (i.e. PAUs), it is anticipated that they can be conveniently hydrolysed under acidic conditions, which may enable chemical recycling.
To develop bio-based polyacetals, sugars are particularly attractive resources because their aldehyde and hydroxyl groups can form acetals conveniently, as well as their relatively low cost and abundance in nature.52,53 There has been a rapid development of sugar-sourced polymers, such as commercial bio-PE (e.g. I'm green™ by Braskem), bio-PET (e.g. PlantBottle™ by Coca Cola), polylactic acid (PLA),54,55 as well as many other sugar-based new polymers.38,52,56–68 Recently, a sugar-based platform molecule, 5-hydroxymethylfurfural (HMF),69,70 was used to fabricate PAUs toward coating applications.47 However, these PAUs were not readily suitable for textile investigations due to the relatively low thermal stability of the monomer, the presence of crosslinking, and the lack of flexible segments (as in Elastane fibres).
Herein we present a facile synthesis of a new tailor-designed diol monomer with cyclic acetal units using sugar-based HMF and partly bio-based di-trimethylolpropane with enhanced hydrophobicity and thermal stability. This diol was polymerized together with flexible polytetrahydrofuran (polyTHF) in various ratios, yielding a series of PAUs with relatively high molecular weight and tuneable glass transition temperatures. The processing window for a selected polymer was evaluated by melt-rheology measurements, and ∼150 meters of homogeneous PAU fibre was prepared by melt-spinning. Finally, chemical recyclability of the obtained PAU fibre by acidic hydrolysis was preliminarily investigated. To our knowledge, this is the first attempt toward bio-based polyacetal fibres for sustainable textiles.
Experimental section
Materials
Dicyclohexylmethane-4,4′-diisocyanate (12MDI, 99.5%) was purchased from Evonik, polyTHF (stab. 250 mg kg−1 BHT, Mn = 2000 g mol−1) and dibutylamine (>99%) were purchased from BASF. di-Trimethylolpropane (97%), 2-propanol (99.8%), dimethyl acetamide (DMAc, 99%), LiBr (99%), 2-butanone (99%), methylene diphenyl diisocyanate (MDI, 98%), p-toluenesulfonic acid (p-TsOH, 98.5%), methyl tert-butyl ether (MTBE, HPLC grade, 99.8%) and DMSO-d6 (99.9% atom D) were purchased from Sigma-Aldrich. HMF (98%) was purchased from Nanjing Confidence Chemical Co., Ltd. Methanol was purchased from VWR Chemicals. Tetrahydrofuran (THF) was purchased from Honeywell. Dibutyltin dilaurate (DBTDL, Metatin katalysator 712 ES) was purchased from Dow Chemicals. All solvents, reagents and chemicals were used without further purification.Analytical methods
1H and 13C NMR measurements were performed on a Bruker DR X400 spectrometer at 400.13 MHz and 100.61 MHz, respectively. Chemical shifts were reported as δ values (ppm). HRMS was taken on a Micromass QTOF mass spectrometer (ESI). Size exclusion chromatography (SEC) measurements for the poly(urethane-urea)s were carried out using Agilent 1100/1200 Infinity HPLC System equipped with three columns (GPC column PSS GRAM 3000 Å, 10 μm; GPC column PSS GRAM 1000 Å, 10 μm; GPC column PSS GRAM 30 Å, 10 μm) connected in sequence at 40 °C in DMAc with LiBr (5 g L−1) at a flow rate of 1 mL min−1. Calibration was carried out with ReadyCal-Kit poly(methyl methacrylate) standards Mp = 202–2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200000 Da. TGA measurements were carried out on a TA instrument mode TGA Q500. The samples were first heated to 120 °C for 20 min to remove any trace of water, and the measurement was performed by heating from 50–850 °C with a heating rate of 10 °C min−1. DSC measurements were performed on a DSC Q2000 analyzer from TA instruments. Data was recorded from −70 to 200 °C, and Tg was determined from the second heating cycle. The synthesized polyurethanes (PU70 and PU85) were made into films of dimensions (17.5 mm × 6 mm × 1 mm) for dynamic mechanical analysis (DMA). PU films were made by hot pressing the polymers at 140 °C. The mold was fixed in the press and the polymer was allowed to melt for 1 min after which the pressure was increased to 300 kg for 1 min, 600 kg for 1 min, 1000 kg for 1 min and finally 2000 kg for 1 min. DMA measurements were performed in a stretching mode using TA instruments Q800 analyzer. The measured samples were heated from −60 °C to 140 °C at a heating rate of 3 °C min−1 and frequency of 1 Hz with strain of 0.05%. Rheology measurements were carried out on Advanced Rheometer AR2000 ETC from TA Instruments, using parallel plates with a diameter of 20 mm under nitrogen atmosphere to prevent oxidative degradation. The temperature sweep experiment was performed in the interval 140–220 °C with a heating rate of 3 °C min−1. The measurement was performed with a strain of 1% and a frequency of 1 Hz. The cross-polarized light microscopy images were taken with an Olympus BX50 microscope.
200000 Da. TGA measurements were carried out on a TA instrument mode TGA Q500. The samples were first heated to 120 °C for 20 min to remove any trace of water, and the measurement was performed by heating from 50–850 °C with a heating rate of 10 °C min−1. DSC measurements were performed on a DSC Q2000 analyzer from TA instruments. Data was recorded from −70 to 200 °C, and Tg was determined from the second heating cycle. The synthesized polyurethanes (PU70 and PU85) were made into films of dimensions (17.5 mm × 6 mm × 1 mm) for dynamic mechanical analysis (DMA). PU films were made by hot pressing the polymers at 140 °C. The mold was fixed in the press and the polymer was allowed to melt for 1 min after which the pressure was increased to 300 kg for 1 min, 600 kg for 1 min, 1000 kg for 1 min and finally 2000 kg for 1 min. DMA measurements were performed in a stretching mode using TA instruments Q800 analyzer. The measured samples were heated from −60 °C to 140 °C at a heating rate of 3 °C min−1 and frequency of 1 Hz with strain of 0.05%. Rheology measurements were carried out on Advanced Rheometer AR2000 ETC from TA Instruments, using parallel plates with a diameter of 20 mm under nitrogen atmosphere to prevent oxidative degradation. The temperature sweep experiment was performed in the interval 140–220 °C with a heating rate of 3 °C min−1. The measurement was performed with a strain of 1% and a frequency of 1 Hz. The cross-polarized light microscopy images were taken with an Olympus BX50 microscope.
Synthesis of Monomer T
To a well-stirred mixture of partially dissolved HMF (11.5 g, 91.2 mmol) and di-TMP (11.4 g, 45.5 mmol) in 2-propanol (40 mL) was added at room temperature a solution of p-TsOH (215 mg) in 2-propanol (20 mL). The reaction mixture was stirred at room temperature overnight under N2. Afterward, the reaction was quenched by the addition of solid NaHCO3 (204 mg). The precipitate was then collected by vacuum filtration and washed with 2-PrOH (2 × 20 mL), NaHCO3 aqueous solution (15 mL, 0.1 M), and distilled water (3 × 20 mL) to yield a white solid as crude Monomer T (20.9 g). The crude product was dissolved in boiling MeOH (200 mL), and precipitated into deionized water (1.4 L). The solid was then collected by vacuum filtration and washed with water (3 × 100 mL) to give Monomer T (15.7 g, 73.9%). Tm: 160 °C (DSC), 1H NMR (400.13 MHz, DMSO-d6, δ, ppm): 0.78 (t, 6H, J = 7.6 Hz, CH3, 1.17 (q, 4H, J = 7.6 Hz, CCH2), 3.59 (d, 4H, J = 11.5 Hz, OCH2-Hax, 3.63 (s, 4H, OCH2, 3.91 (d, 4H, J = 11.5 Hz, OCH2-Heq), 4.35 (d, 2H, J = 5.6 Hz, ArCH2), 5.23 (t, 2H, J = 5.7 Hz, -OH), 5.48 (s, 2H, ArCH), 6.24 (d, 2H, J = 3.2 Hz, ArH), 6.34 (d, 2H, J = 3.2 Hz, ArH) 13C NMR (100.61 MHz, DMSO-d6, δ, ppm) 7.2, 24.1, 36.9, 56.0, 70.2, 72.0, 95.9, 107.8, 108.4, 150.5, 155.7. HRMS (ESI+, m/z): exact mass calculated for C24H35O9+: 467.2279, found 467.2281. FT-IR: 3463, 2962, 2929, 2886 2860, 2161, 1568, 1488, 1462, 1419, 1413, 1386, 1358, 1323, 1252, 1239, 1185, 1163, 1126, 1096, 1058, 1023, 1005, 970, 960, 937, 925, 892, 848, 798, 776, 741, 666, 634, 541, 512, 491, 456, 425, 413 cm−1. T5 = 278 °C (TGA).Synthesis of poly(cycloacetal-urethane)s (PAUs)
The synthesis of PAU-51 (51 describes the mass% T respective to diols) is described as a typical example. To a 50 mL round bottom flask with a magnetic stirrer at 30 °C was added Monomer T (3.81 g, 8.17 mmol), polyTHF (3.80 g, 1.90 mmol), MDI (2.50 g, 9.99 mmol) and 2-butanone (10 mL). The mixture was stirred for 10 min until a heterogeneous slurry was formed. Afterward, a diluted solution of DBTDL in 2-butanone (0.20 mL, 10 v%, 0.034 mmol) was added dropwise, and then the temperature was raised to 40 °C. After being stirred for 10 min, a second fraction of the diluted DBTDL solution in 2-butanone (0.20 mL, 10 v%, 0.034 mmol) was added, followed by a further increase of the reaction temperature (to 50 °C). After stirring at 50 °C for 5 min, the reaction mixture turned transparent and a significantly increased viscosity was observed. To reduce the viscosity, 2-butanone (10 mL) was added immediately. Another two portions of 2-butanone (5 mL each) were added after 5 and 15 min respectively to reduce the viscosity. Afterward, a droplet of the reaction mixture was taken out and analysed by FT-IR to confirm the complete consumption of isocyanates. To ensure the reaction was completed, the reaction was continued for another hour after no isocyanate signal became invisible in the FT-IR spectrum. Afterward, dibutylamine solution (1 mL, 2 M in 2-butanone) was added to quench the reaction. The polymer was collected by suction filtration and rinsed with 2-butanone (5 mL). The collected polymer was then re-dissolved in THF (80 mL), precipitated into MTBE (1.2 L), and dried overnight at 70 °C under vacuum to give the final polymer as a white powder (7.83 g, 77.4%). 1H NMR (400.13 MHz, THF-d8, δ, ppm): 0.83 (t, 6H, J = 7.6 Hz, CH3, 1.23 (q, 4H, J = 7.6 Hz, CCH2), 1.58 (s, 4H, (THF)β-CH2), 3.36 (s, 4H, (THF)α-CH2), 3.54 (d, 4H, J = 11.4 Hz, OCH2-Hax), 3.72 (s, 4H, OCH2), 3.81 (s, 2H, ArCH2), 3.99 (d, 2H, J = 11.4 Hz, OCH2-Heq), 4.08 (t, 2H, J = 6.5 Hz, (THF)αurethane-CH2), 5.01 (s, 2H, FurCH2), 5.40 (s, 2H, FurCH), 6.32 (d, 2H, J = 3.2 Hz, FurH), 6.37 (d, 2H, J = 3.2 Hz, FurH), 7.03 (d, 2H, J = 8.4 Hz, ArH), 7.35 (d, 2H, J = 8.4 Hz, ArH) 13C NMR (100.61 MHz, DMSO-d6, δ, ppm) 7.2, 27.6, 37.7, 41.2, 58.7, 71.1, 71.3, 73.1, 97.1, 108.4, 111.3, 118.9, 129.8, 136.5, 138.4, 150.9, 153.3, 153.8. FT-IR: 3286, 2939, 2855, 1731, 1600, 1535, 1464, 1413, 1367, 1310, 1217, 1101, 1050, 1017, 982, 930, 799, 770, 655, 512, 452 cm−1Melt-spinning of PAU fibres
Prior to spinning, the material was dried for 16 h at 60 °C in a vacuum oven. Fibre spinning was performed on a CEAST Rheoscope 1000 capillary rheometer. A 10 mm long capillary with a diameter (dcap) of 0.6 mm was used. Diameter of the barrel (dbar) was 9.55 mm, and fibres were produced at a piston speed (vp) of 5 mm min−1. Exit velocity of the melt (vm) was calculated to 5 m min−1 using eqn (1). Fibres were produced with two different draw speeds. The initial take up speed (vw) was about 50 m min−1, and in the end it was about 90 m min−1. Melt draw ratio (MDR) was 5 in the start and 18 in the end of the spinning, as calculated using eqn (2). The temperature of the barrel was 185 °C. Attempts to draw the fibres were done over a hot pin at different temperatures between 50–90 °C.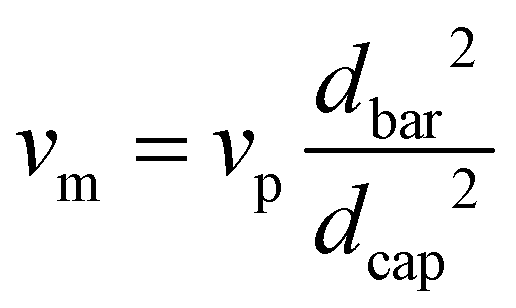 | (1) |
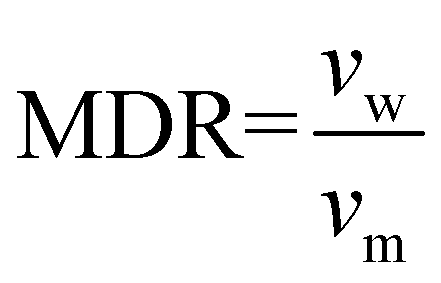 | (2) |
The tensile properties of the produced fibers were measured by the Vibrodyn/Vibroscope device from Lenzing Technique GmbH & Co KG, Austria according to methodology standardized by the International Organization of Standardization (ISO 5079:2020 and ISO 1973:2021).71,72 The testing speed was 20 mm min−1 and gauge length 20 mm. The fibres were conditioned for at least 24 hours in the standard climate RH 65 ± 2% and temperature 20 ± 2 °C before testing.
Chemical recycling
The melt-spun fibre of PAU-86 (58.1 mg) was cut into pieces (∼2 cm) and added to a 25 mL round bottom flask with HCl (6 M, 15 mL) at 60 °C. The mixture was stirred for 24 h until the fibres fully dissolved and a brown slurry was obtained. The slurry was diluted with distilled water (15 mL) and quenched by slow addition of K2CO3 powder (6.25 g, 45.2 mmol) until no more bubbles formed. Afterward, the slurry was filtered to give a brown solid and a clear filtrate. The brown solid was extracted with THF (15 mL) and then dried to give a brown crude product (13 mg). The brown crude product was dissolved in THF-d8 for 1H NMR analyses. The clear filtrate was extracted with acetonitrile (3 × 15 mL), and the combined organic phase was concentrated in vacuo to give a beige crude residue (158 mg). The beige crude residue was then dissolved again in acetonitrile (15 mL), filtered (to remove salt), and concentrated in vacuo to give the crude product (15.6 mg). The crude product was dissolved in DMSO-d6 for 1H NMR analyses.Results and discussion
Synthesis
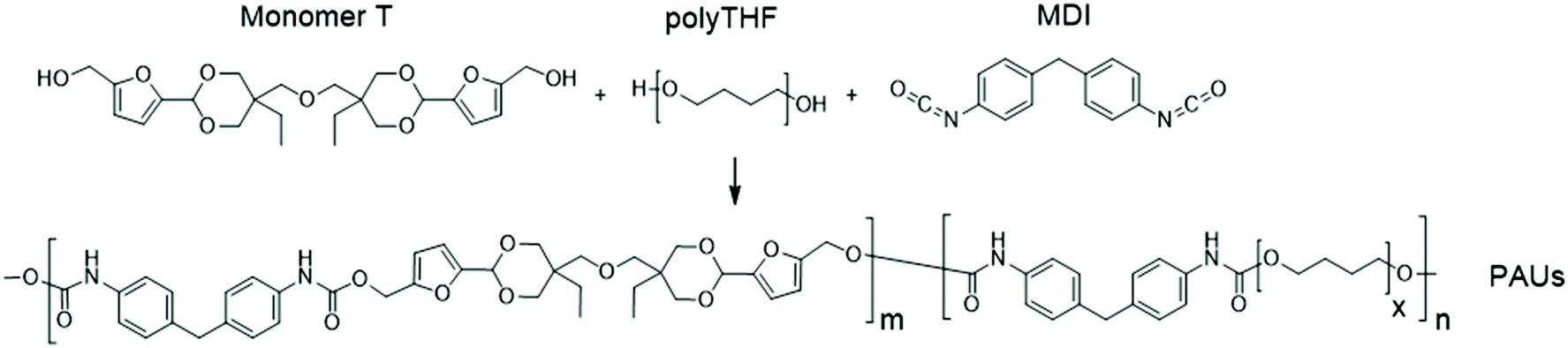
Monomer T was synthesized using bio-based HMF and di-TMP (Scheme 1), according to a modified mild acid-catalysed procedure reported earlier for the synthesis of another spirocyclic acetal monomer.47 Di-TMP has two hydrophobic ethyl groups, which increases the hydrophobicity of the resulting monomer T so it could be easily extracted after synthesis to improve the yields. In addition, the mildly hydrophobic di-TMP could be extracted relatively efficiently from the aqueous phase after acidic hydrolysis (discussed later in the chemical recycling section). An environmentally friendly solvent, 2-PrOH, was used for the reaction.73 It is known that HMF is unstable at elevated temperatures forming dark substances (humins), particularly in the presence of acid.74 Therefore, the acetalation reaction was kept at room temperature overnight, which gave Monomer T in satisfying yield (74%) and purity (according to 1H NMR spectrum, Fig. S1†) after straightforward purification by precipitation in water. The reactivity of Monomer T in polyurethane synthesis was first investigated by reactions with two commercial diisocyanates, MDI and H12MDI (Scheme 1). The former is the most widely used aromatic isocyanate in the industry, accounting for more than two thirds of all isocyanates.75 The latter is an aliphatic analog to MDI, which is commonly used for outdoor applications. According to literature, polyurethanes are commonly synthesized by using a slightly excess of diisocyanate to diol to form a low molecular weight pre-polymer with isocyanate end groups, which is then subjected to chain extension using a diamine or diol to reach high molecular weight.49 However, this prepolymer method may lead to non-random distribution of different blocks if more than one diols are used. We therefore investigated direct solution polymerizations of Monomer T and diisocyanates (i.e. MDI and H12MDI), aiming to achieve high molecular weight without crosslinking. A modified solution polymerization procedure was subsequently adopted,47 using dibutyltin dilaurate catalyst. During the polymerization, the catalyst was added dropwise in two portions over 20 min at relatively low temperatures (30 and 40 °C) in order to avoid uncontrollable temperature increase due to the exothermic urethane formation. It was observed that the viscosity increased rapidly after the final addition of catalyst. Hence, more solvent (10 mL) was added to prevent coagulation. We observed that a 5 mol% excess of isocyanate (with respect to Monomer T) significantly increased the molecular weights (Table 1). This can be attributed to the reaction of moisture with the excess of isocyanate groups,76 which helps to reach the stoichiometric balance between the isocyanate and alcohol groups. For both diisocyanates, this procedure resulted in moderate molecular weights, while higher molecular weights were obtained when MDI was used (Mn ∼ 62.6 kDa, Table 1) compared to H12MDI (Mn ∼ 26.3 kDa). This demonstrated the effectiveness of this modified synthetic protocol for Monomer T without chain extender. Next, copolymerizations of MDI, Monomer T and polyTHF was investigated (Scheme 1). PolyTHF is a flexible polymeric diol that is frequently used for the synthesis of TPUs.77–80 According to the homopolymerization conditions discussed earlier, 5 mol% excess of MDI with respect to the sum of Monomer T and polyTHF was used. It was observed that the viscosity of the reaction mixture increased more rapidly for these copolymerizations, compared to the homopolymerizations of PAU-MDI. Therefore, a larger amount of solvent (10 mL) was used to reduce the viscosity and prevent coagulation. In addition, the temperature was increased to 50 °C after the addition of all catalysts until the reaction mixture turned clear and transparent. The isocyanate consumption, and thus the progress of the polymerization, was monitored by FTIR measurements. After the disappearance of the isocyanate peak at 2253 cm−1 in the FTIR spectrum of the reaction mixture, the reaction was continued for 60 min to ensure the complete consumption of the isocyanates. Finally, the reaction was quenched with dibutylamine, and the resulting PAU copolymer was purified by a straightforward precipitation, yielding high molecular weights (Mn ∼ 41.5–98.9 kDa) and low to medium polydispersity (PDI∼ 1.6–3.4). The molecular structure of Monomer T and the resulting polymers were confirmed by 1H NMR-spectroscopy (Fig. 1). First, all the proton signals of Monomer T were unambiguously assigned (Fig. 1A) with the help of 2D-NMR spectroscopy (COSY, HMBC, HMQC and NOESY, Fig. S1–S6†). The triplet at 4.18 ppm (a) and doublet at 4.40 ppm (b) were assigned to the hydroxyl and CH2- protons connected to the furan ring, respectively. The doublets at 6.14 and 6.27 ppm (c, d) were assigned to the aromatic protons of furan. The singlet at 5.39 ppm (e) was attributed to the acetal proton. The two doublets at 4.00 and 3.54 ppm were assigned to the protons of the cyclic acetal units at equatorial and axial positions (feq and fax respectively). The quartet and triplet at 1.84 (g) and 0.85 ppm (h) were assigned to the ethyl protons. Finally, the singlet at 3.74 ppm (i) was assigned to the CH2 attached to the central ether unit.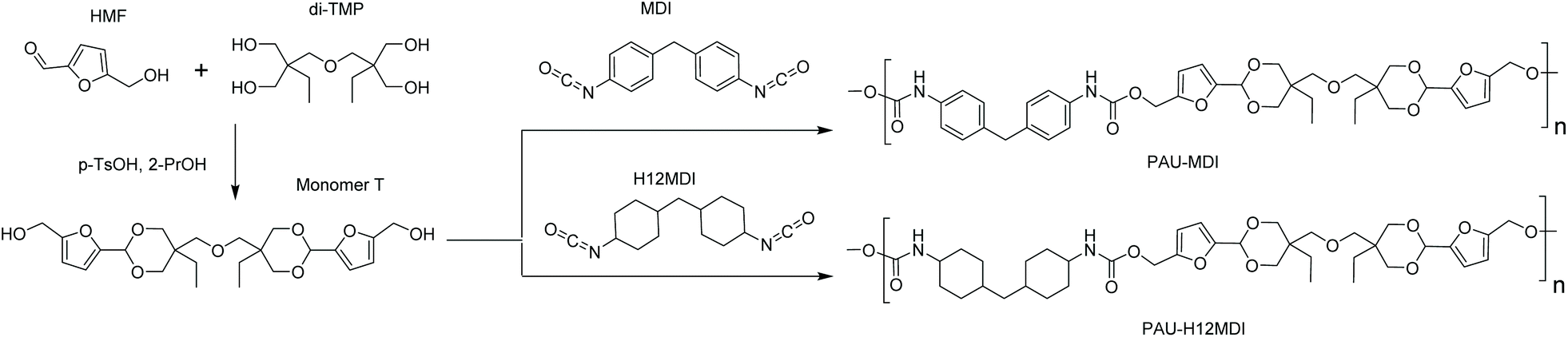 | ||
| Scheme 1 Synthesis of Monomer T with cyclic acetal structure and its homopolymerizations with two diisocyanates, H12MDI and MDI. | ||
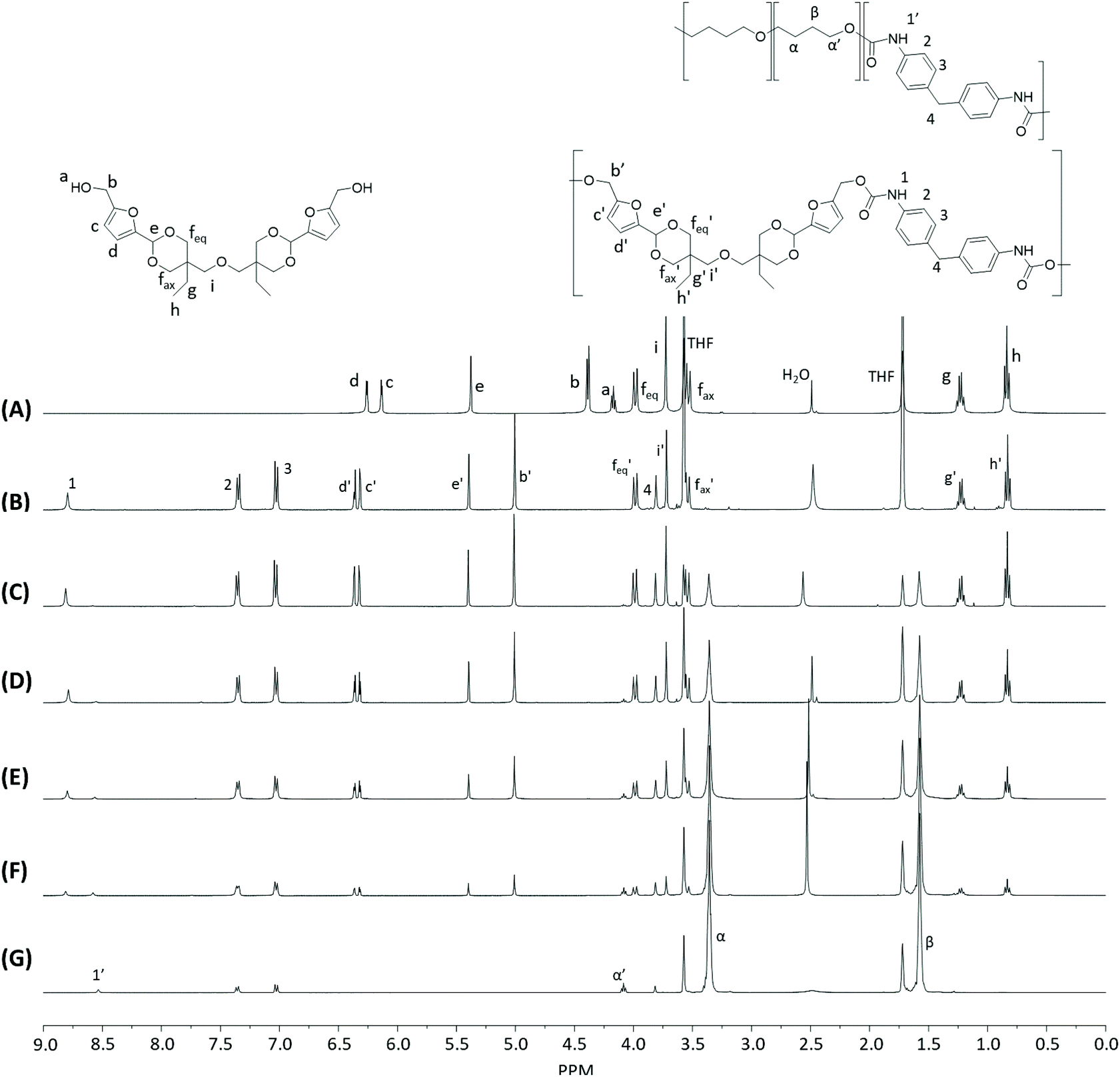 | ||
| Fig. 1 1H NMR spectra of Monomer T (A), PAU-MDI (B), PAU-86 (C), PAU-71 (D), PAU-51 (E), PAU-25 (F), PAU-0 (G). THF-d8 were used as the solvent for all measurements. | ||
| Isocyanate | Feed isocyanate (mol%) | M n (kDa) | M w (kDa) | Đ | |
|---|---|---|---|---|---|
| PAU-MDI | MDI | 100 | 37.7 | 65.2 | 1.73 |
| MDI | 105 | 62.6 | 97.3 | 1.55 | |
| PAU-H12MDI | H12MDI | 100 | 22.0 | 43.9 | 2.00 |
| H12MDI | 105 | 26.3 | 49.2 | 1.87 |
After polymerizations, the hydroxyl proton signal (a) in the 1H NMR spectrum of Monomer T completely disappeared (Fig. 1B–F), indicating the consumption of Monomer T. In the meantime, a new singlet at 8.81 ppm (1) was observed in the 1H NMR spectra of all the PAUs except for PAU-0 (Fig. 1B–F). This peak was assigned to urethane NH proton formed between Monomer T and MDI units. In the meantime, a different urethane NH proton (1′) formed between polyTHF and MDI units was clearly discerned at slightly upper field (8.58 ppm). The relative ratio between the two urethane NH signals (1 and 1′) was consistent with the chemical composition of the polymers. Other signals from the MDI units were also observed (2–4). The CH2 protons attached to furan (b) and the aromatic protons (c, d) shifted downfield, which was consistent with their locations near the reaction moieties (electron withdrawing urethane groups). The chemical shifts of the protons relatively far away from the reaction moieties (e, feq, fax, g, h) remained almost unchanged after the polymerizations. The proton of the CH2 group of polyTHF units after urethane formation was observed at 4.08 ppm (α′), which showed increasing relative intensity as the increased content of incorporated polyTHF units (i.e., decreased content of Monomer T units from Fig. 1C–G).
Thermal properties
Thermal stability is a major concern when hydroxymethylfuran is part of the monomer structure.81,82 It was noteworthy that Monomer T exhibited relatively high thermal stability according to TGA results (Fig. 2), as shown by the high initial thermal decomposition temperature (T5∼278 °C). This enhanced thermal stability makes Monomer T superior comparing to other hydroxymethylfuran-based diols with or without acetal units.47,82–84 After polymerizations, the obtained PAUs also showed relatively high thermal stability (T5∼253–381 °C, Table 2), compared to other polyurethanes prepared using hydroxymethylfuran-based diols (e.g. furan-2,5-diyldimethanol).82–84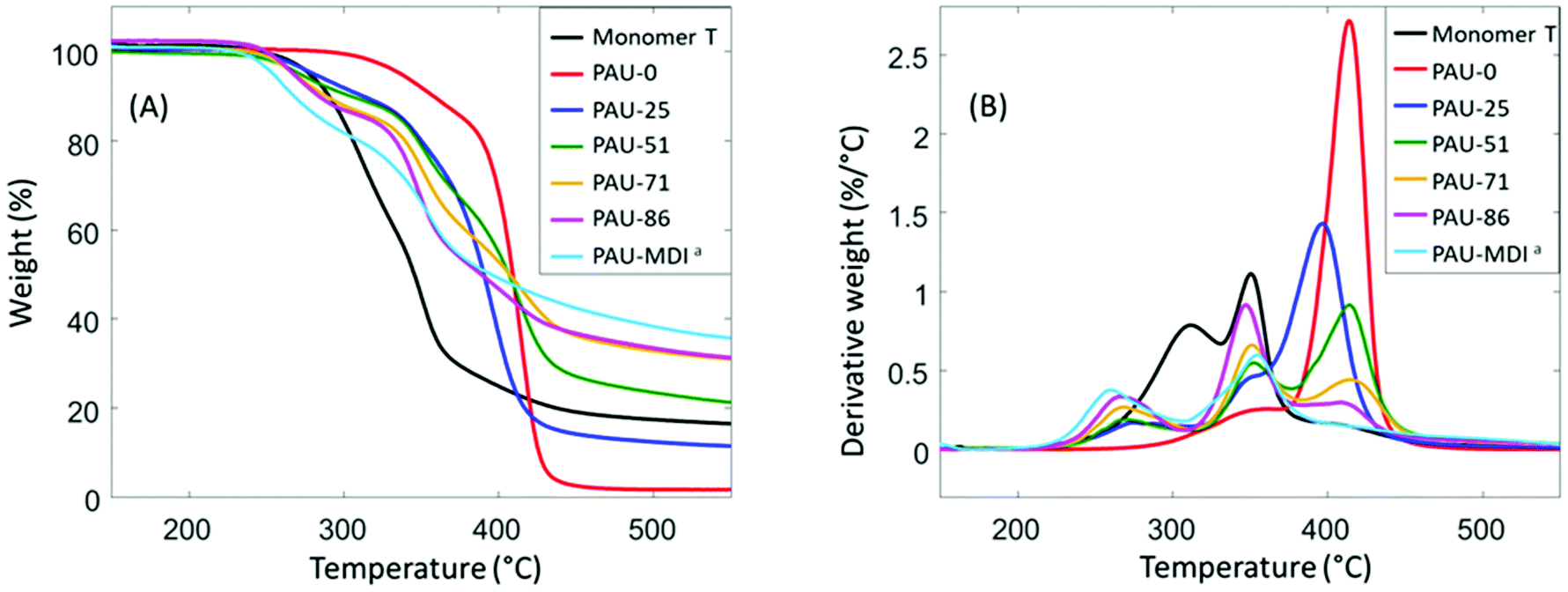 | ||
| Fig. 2 TGA (A) weight loss and (B) first derivative weight loss curves of Monomer T and the polymers. aSince PAU-MDI and PAU-100 were identical; the name PAU-MDI was used due to consistency with Table 1. | ||
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mol % Ta | Mass % Ta | M n (kDa) | M w (kDa) | Đ | T g (°C) | T g (°C) | T d (°C) | T 5 (°C) | CYg (%) | LOIh | |
| a Incorporated mol % and mass % of Monomer T units with respect to the total amount of diols, according to 1H NMR calculations. b M n, Mw and polydispersity index (Đ) were measured by SEC. c Measured from the second heating DSC curves. d Measured by DMA as the peak loss moduli. e The temperature for maximum degradation rate in TGA. f The temperature for 5% weight loss in TGA curves. g Residual mass in percentage at 850 °C in TGA. h Calculated according to van Krevelen's method.90 In the nomenclature of the polymers, the number indicated the mass per cent of Monomer T units with respect to all diol units (i.e., the sum of Monomer T units and polyTHF units). i Since PAU-MDI and PAU-100 were identical, the name PAU-MDI was used due to consistency with Table 1. | |||||||||||
| PAU-0 | 0 | 0 | 85.7 | 201 | 2.35 | — | — | 414 | 337 | 2.0 | 0.18 |
| PAU-25 | 59.0 | 25.1 | 98.9 | 333 | 3.37 | — | — | 274, 400 | 281 | 11.0 | 0.22 |
| PAU-51 | 81.6 | 50.8 | 49.5 | 94.9 | 1.92 | — | — | 271, 357, 409 | 275 | 18.8 | 0.25 |
| PAU-71 | 91.1 | 70.5 | 41.5 | 99.6 | 2.40 | 91 | 79 | 270, 352, 416 | 268 | 23.3 | 0.27 |
| PAU-86 | 96.3 | 85.7 | 48.2 | 94.4 | 1.96 | 104 | 99 | 266, 349, 411 | 268 | 26.0 | 0.28 |
| PAU-MDIi | 100 | 100 | 62.6 | 97.3 | 1.55 | 104 | — | 262, 355 | 253 | 32.9 | 0.31 |
It was also observed that the T5 values consistently decreased as more Monomer T units were present. Since urethane groups are rather thermally unstable,85 it is therefore concluded that the urethane groups formed between Monomer T and MDI started to degrade at ∼250 °C, while the urethane adjacent to the polyTHF diol were more thermally stable. From the derivative curves (Fig. 2B), three decomposition rate maxima were observed. The first decomposition rate maximum (∼250–260 °C) was observed in all copolymers with Monomer T units, which showed increasing intensity with increased content of Monomer T units. This maximum was attributed to the thermal decomposition of the urethane linkage between MDI and Monomer T units, which was consistent with their corresponding T5 values discussed above. The other two decomposition rate maxima at higher temperatures (∼350–360 °C, and ∼420 °C) were due to the decomposition of the “other” urethane group formed by polyTHF and MDI units and decomposition of polyether structures from polyTHF units, respectively, which was consistent with literature.85–89
Interestingly, relatively high char yields (CY) were observed after pyrolysis of the PAUs by TGA at 850 °C (Table 2), which showed an increasing trend with increased content of incorporated Monomer T units. This may indicate a potential inherent flame resistance of the polymers.90 According to literature, the limiting oxygen index (LOI), a measure of the oxygen content in air to maintain stable flame, can be calculated using the CY values at 850 °C. An LOI value greater than 0.26 is commonly an indication for inherent flame resistance.90 As shown in Table 2, the calculated LOI values for the PAUs with relatively high Monomer T contents (e.g. PAU-71, PAU-85 and PAU-MDI) were all greater than 0.26, which indicated that they could be inherently flame-resistant. These polymers would be of particular interest for textiles used for furniture applications.90–93 DSC was used to evaluate the thermal behaviour of the obtained PAUs (Fig. 3). PAU-0 without Monomer T units was semi-crystalline with a melting endotherm at 20 °C (second heating cycle). When Monomer T was used, all the resulting PAUs were fully amorphous. This may be explained by the fact that soft segment polyTHF can facilitate crystallite formation, while the bulky rigid Monomer T segments disrupt the crystal packing. The PAUs with relatively high Monomer T content (≥71 mass %) showed discernible glass transitions with increasing Tg values (91 °C to 104 °C), which was consistent with the increased backbone rigidity. It was noted that the glass transition of PAU-71 was broadened (Fig. 3B). For PAUs with even less Monomer T contents (PAU-0 to PAU-51), the glass transition became even more broadened, and Tg values could not be meaningfully measured. Such broadened glass transition of copolymers has been reported before for other polymer structures.94,95
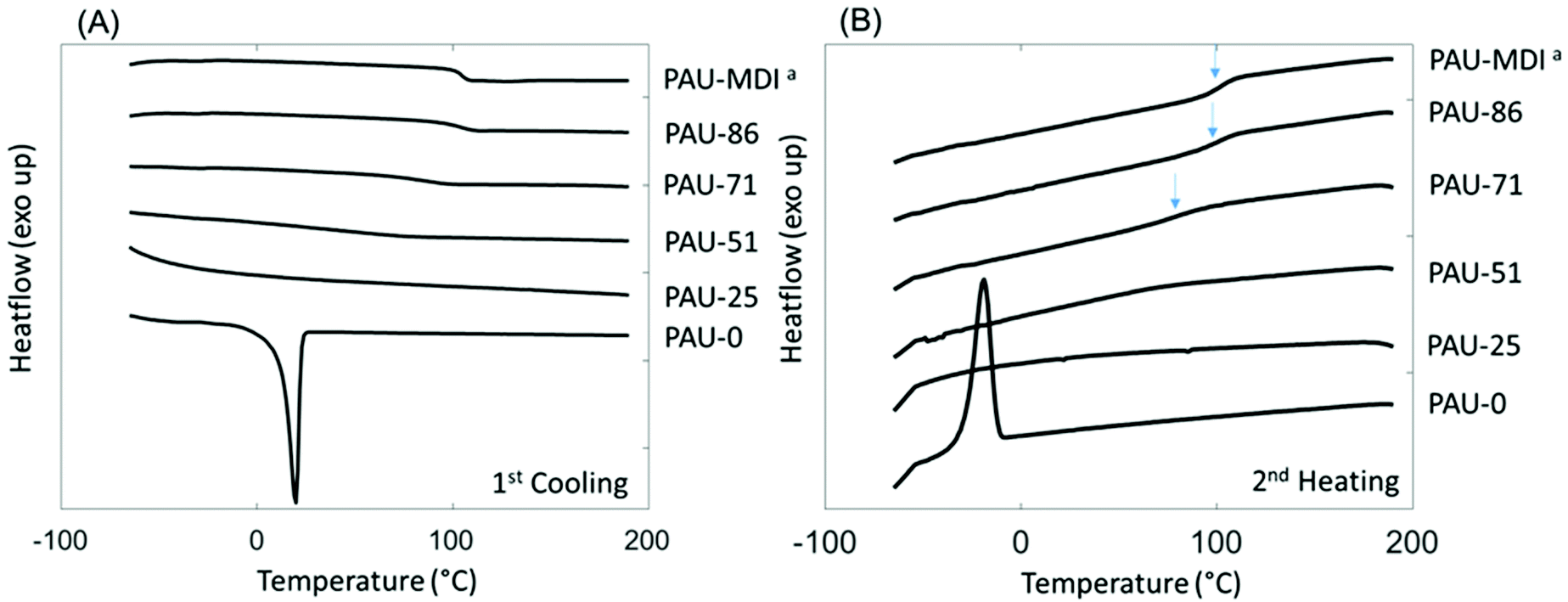 | ||
| Fig. 3 DSC (A) first cooling and (B) second heating curves of PAUs. The glass transition of PAU-MDI, PAU-86 and PAU-71 was indicated by blue arrows in (B). aSince PAU-MDI and PAU-100 were identical; the name PAU-MDI was used for consistency with Table 1. | ||
The glass transition of two polymers (PAU-71 and PAU-86) was further investigated using temperature-sweeping DMA (Fig. S38, ESI†). The other polymers failed to form homogenous films with proper mechanical stability for DMA measurements. PAU-MDI films were too brittle to measure. PAU-0 to PAU-51 samples were sticky and formed bubbles during hot pressing. For the two samples that were successfully measured (PAU-71 and PAU-86), the temperature at the maximum loss modulus values were 79 °C and 99 °C, respectively. For PAU-86, the DMA-based Tg value was consistent with the DSC-based value (ΔTg ∼ 5 °C). However, for PAU-71 the DMA-based Tg value was significantly lower (ΔTg ∼ 12 °C) than the DSC-based value (Table 2). In principle, the different Tg values measured by both methods may be attributed to the different processes that were measured.96 In addition, for PAU-71 the loss modulus curve displayed a broad maximum (Fig. S38, ESI†), which was consistent with the broader glass transition in the DSC curve (Fig. 3B). This contributed to the relatively large difference between the Tg values of PAU-71 measured by the two methods.
Melt-spinning of PAU fibre
PAU-86 was selected to further evaluate the melt-spinnability toward potential sustainable textiles. First, melt processing temperature was investigated using temperature sweep rheology. The sample was subjected to a constant strain of 1% (in the viscoelastic region) during a temperature increase from 140 °C to 220 °C at 3 °C min−1 (Fig. 4). As a result, G′ and G′′ intersect at 165 °C, indicating that the material starts to flow. This temperature is therefore considered as the lower limit for melt processing. On the other hand, the tanδ curve showed a peak at 186 °C, at which the material exhibited the most viscous character. This was considered the highest suitable processing temperature where degradation could be avoided. Therefore, the processing window of PAU-86 was set between 165 and 186 °C. In addition, another intersect between G′ and G′′ curves was observed at higher temperature (∼210 °C), which indicated degradation and gel formation. This was confirmed by the observation that the sample after the melt rheology measurement significantly expanded with bubbles contained, and the sample became only partially soluble in THF (while the pristine sample was fully soluble in THF). The 1H NMR spectrum of the soluble fraction of the sample after the measurement showed the signals for Monomer T (Fig. S39, ESI†), indicating the degradation of urethane units at 220 °C. This was also consistent with the previously discussed TGA results.
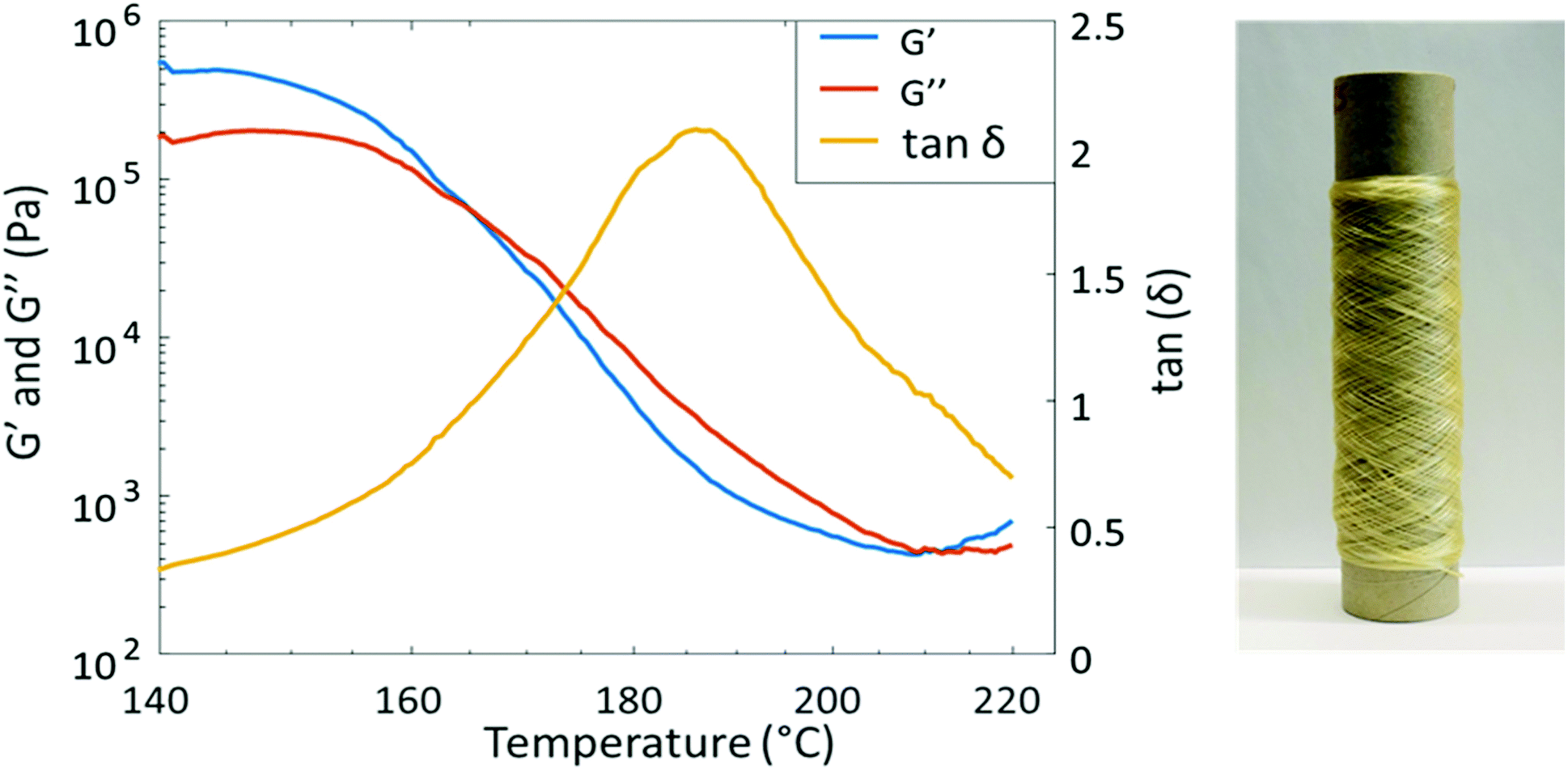 | ||
| Fig. 4 Temperature sweep rheological data of PAU-86 (left) and a roll of PAU-86 fibre melt-spun at 185 °C (right). | ||
The purpose of the spinning test was to explore if the material can be spun into fibres, and if the fibres could be drawn in the solid state to impart mechanical strength. Therefore, relatively thick fibres were produced with a linear density of 37 and 67 dtex, respectively. Melt-spinning was carried out according to the schematic representation in Scheme S40, ESI.† First, melt spinning at relatively low temperature (175 °C) was attempted. However, it was observed that the melt viscosity at this temperature was too high to sustain the flow of molten polymer. Therefore, a processing temperature at 185 °C was attempted, which resulted in significantly reduced melt viscosity and thus a stable molten polymer flow. By using ∼10 g of polymers, a ∼150 m long fibre with a 0.24 ± 0.044 mm diameter was successfully prepared (Fig. 4). The fibre could be spun without any failure to consume approximately 10 g of the material. This indicated the material was relatively homogenous and no gel particles or lumps can be seen in the fibres. Under cross-polarized optical microscopy, the extended crystallites in the direction of the fibre could be clearly observed (Fig. S41, ESI†). It was also observed that the fibres at the end of the spinning process showed more regular crystallite orientations.
Fibre characterization
The obtained PAU-86 fibre was fully soluble in THF-d8, indicating the absence of crosslinking and gelation. In the 1H NMR spectrum of the dissolved fibre (Fig. S42, ESI†), the signals corresponding to Monomer T were observed with relatively low intensity. This indicated minor chain scissions due to the decomposition of urethane groups between MDI and Monomer T, which was consistent with the TGA results. The same 1H NMR signals were also observed after the rheology measurements (Fig. S39, ESI†). The formed small amount of Monomer T after thermal treatment (melt-spinning or rheology measurements) may undergo further degradation at relatively high temperatures forming dark humins, which may cause the observed light yellow coloration of the obtained fibres (PAU polymers were white after synthesis, Fig. S43, ESI†). The molecular weight of the fibre was estimated by SEC to Mn = 16.7 kDa and Mw = 48.5 kDa. The reduction in molecular weight was most likely caused by thermal degradation due to the high temperatures necessary for the melt spinning.The PAU-86 fibre was further analysed by TGA and DSC measurements (Fig. S44 and 45, ESI†) and the results were compared to that of the unprocessed PAU-86 powder. It was observed that the PAU-86 fibre showed slightly decreased thermal stability (T5 = 244 °C) compared to the unprocessed PAU-86 powder (T5 = 268 °C), which may be related to the decreased molecular weight.
Tensile testing of the fibre after 2 weeks’ storage indicated that the fibre was generally brittle with some elasticity. The tensile strain at maximum load was higher for the fibres produced with a higher MDR, causing higher orientation in the material. The higher orientation is also reflected in the tenacity, 7.8 cN per tex for the fibre with high MDR compared to 6.7 cN per tex for the fibre spun at lower take-up speed. These values are comparable to that of commercial Elastane, which have a tenacity of 5–13 cN per tex.97 These preliminary results are encouraging and demonstrate the possibility of using melt spinning to prepare fibres using the new PAU polymers. However, more investigations specifically regarding how to reduce the thermal degradation and coloration during melt spinning need to be carried out before scaling up the production. This could potentially be mitigated by adding thermal stabilizers such as antioxidants which are often used to prevent coloration in Elastane.98 Another possibility would be to carry out processing under nitrogen atmosphere during processing to eliminate oxidation by air. Finally, solution spinning could be an alternative process, which has been already used to prepare commercial Elastane fibres. Unfortunately, solution spinning is considerably more expensive and less environmentally friendly than melt spinning.
Chemical recycling of fibres
The potential of chemical recycling of the PAU-86 fibre under acidic conditions was preliminarily investigated and compared to that of a commercial Elastane fibre. Both fibres were cut into pieces and submerged in HCl (6 M) at 60 °C. After 10 days, the Elastane fibre remained intact and the solution remained clear. After filtration, the starting mass was almost fully recovered, showing that Elastane fibre could not be hydrolysed under such acidic conditions.By contrast, the PAU-86 fibres gradually turned darker and became smaller during the hydrolysis experiment, until fully dissolved after 24 h (Fig. S45, ESI†). In the meantime, the solution turned from clear transparent to brown. Afterward, the heterogeneous solution was neutralized with K2CO3 and filtered to give a brown solid and a transparent filtrate. The brown solid was extracted with THF and analysed by 1H-NMR, showing the presence of polyTHF, MDA and humins (Fig. S47, ESI†). Unfortunately, the yield could not be quantified due to the unknown structure of humins. The filtrate was extracted with acetonitrile and concentrated to give a pale-yellow solid as the crude product. 1H NMR spectroscopy (Fig. S48–50, ESI†) showed that the obtained crude product contained relatively pure di-TMP and 4,4-diaminodiphenylmethane (MDA) with the yields of 69% and 17%, respectively. Further increasing the hydrolysis time to 48 h and then 52 days only slightly increased the recycled yields up to 77% of di-TMP (by 48 h hydrolysis) 22% of MDA (by 52 d of hydrolysis, see Table S2, ESI†). This indicated that 24–48 hours’ hydrolysis under acidic conditions were sufficient for chemical recycling of di-TMP and MDA. It was noted that even after 52 days of hydrolysis, the recycled product did not show any other chemical besides di-TMP and MDA according to its 1H NMR spectrum, which indicated the stability of the recycled building blocks under such conditions. To further improve the yields, extraction of the aqueous phase after hydrolysis was attempted with three other solvents: DCM, methanol and THF (Table S3, ESI†). The use of DCM slightly increased the MDA recovery to 25% but significantly lowered the recovery of di-TMP (∼15%), possibly because DCM has too low polarity to extract di-TMP out from the aqueous phase. When methanol was used, no phase separation was observed, even when the aqueous phase was saturated with KCl salt. Finally, extraction with THF yielded ∼84% of di-TMP and ∼20% of MDA, which was slightly higher than the use of acetonitrile.
Unfortunately, no HMF was recovered by this method, which could be due to its low stability under such conditions forming humins.47,99,100 To confirm the occurrence of acid hydrolysis of acetal units, the initial stage of hydrolysis during the first 8 h was monitored by analysing the residue fibre using 1H NMR spectroscopy (Fig. 5). As a result, the acetal signal in the polymer backbone (signal e) showed continuously reduced intensity during the first 4 h (Fig. 5A–E), which became completely invisible after 6 h (Fig. 5F and G). In the meantime, the aliphatic signals of the di-TMP units in the polymers (g and h) also reduced intensity. It was also clearly observed that new signals corresponded to the CH2-protons connected to the furan ring (b′), aromatic protons of the furan unit (c′ and d′), and the aldehyde proton (e′) appeared during the measurement time, which confirmed the occurrence of the hydrolysis of cyclic acetal units under the acidic conditions, yielding HMF or HMF-terminated oligomers. Further optimization of the chemical recycling conditions, particularly toward HMF recycling will be worth exploring in the future.
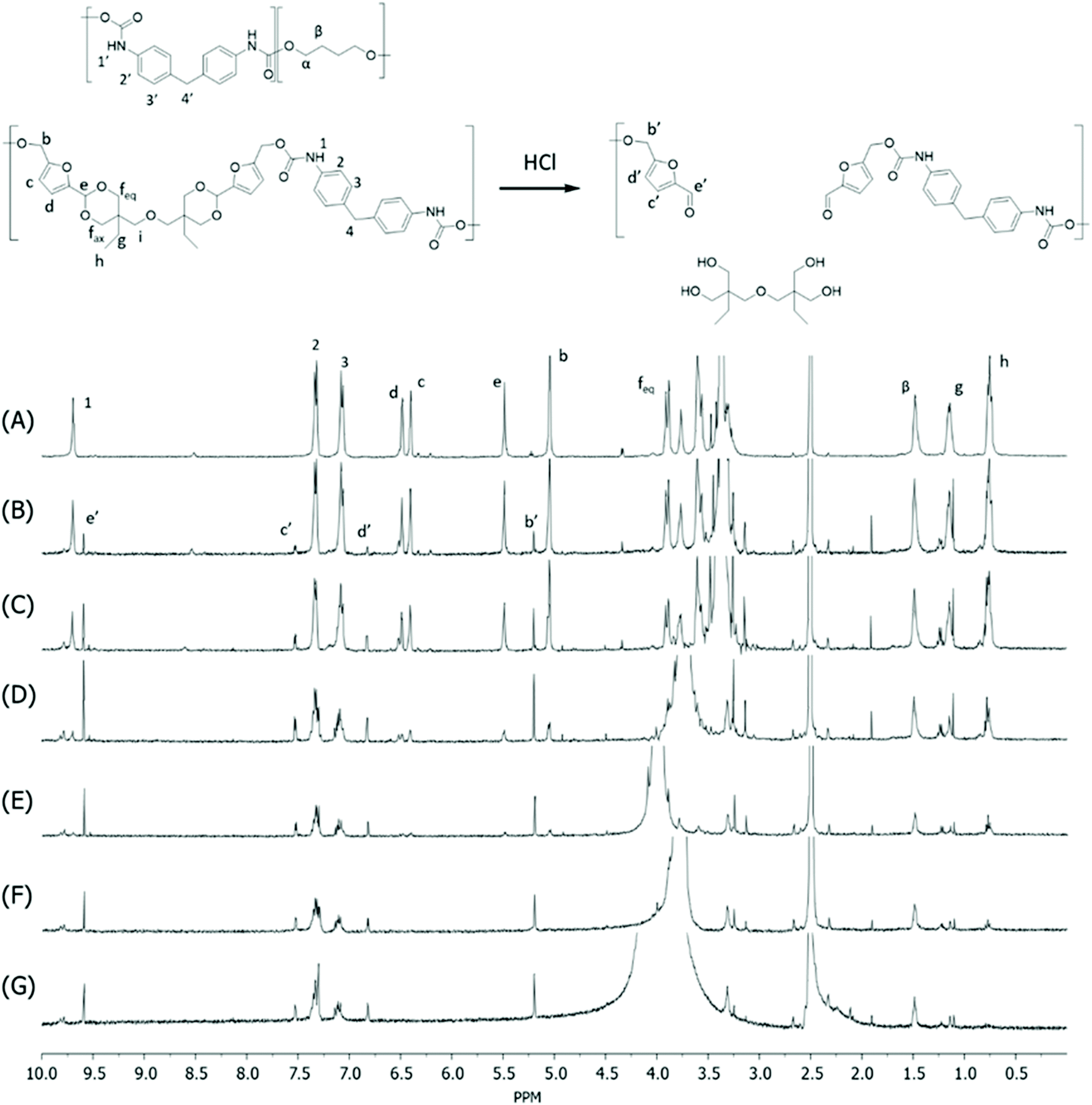 | ||
| Fig. 5 1H NMR spectra in DMSO-d6 of the fibre residue during the acid hydrolysis of the PAU-86 fibre after (A) 0 h, (B) 1 h, (C) 2 h, (D) 3 h, (E) 4 h, (F) 6 h, and (G) 8 h, respectively. | ||
Conclusion
We have presented the first investigations on bio-based synthetic polyacetal fibres toward sustainable textiles. A new bio-based diol (Monomer T) with a cyclic acetal structure and hydrophobic ethyl groups was conveniently synthesized from bio-sourced molecules, HMF and di-TMP. This monomer showed relatively high thermal stability among other hydroxyfuran-based monomers, which facilitated their polyurethane polymerization at elevated temperatures. Polymerizations of this rigid acetal monomer with isocyanate (MDI) and flexible diol polyTHF using a modified solution polymerization protocol yielded a series of poly(cycloacetal-urethane)s (PAUs) with moderate molecular weights and tuneable Tg. According to TGA results, the incorporation of Monomer T units reduced the thermal stability of the resulting PAUs at high temperatures (above 250 °C), but increased char yields after pyrolysis at 850 °C. This indicated potential inherent flame resistance. Temperature sweep rheological properties of a selected PAU containing 86 mass% Monomer T units were investigated, which revealed a processing window between 165–186 °C. Melting spinning of this polymer at 185 °C resulted in a long fibre with homogeneous thickness and low coloration. The resulting fibre was not mechanically tough, which motivates further optimization of the molecular structure, synthesis and processing. Finally, the polyacetal fibre was completely acid-hydrolysed after 24 h, giving relatively pure molecular building blocks, which indicates its potential for chemical recycling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Mistra Foundation (the “STEPS” project, No. 2016/1489), the Crafoord Foundation (No. 20160774 and 20180939), and the Royal Physiographic Society in Lund. Nilson Group AB is acknowledged for financial support. We thank Åsa Halldén Björklund and Linda Zellner from Perstorp AB and John P. Jensen from Nordzucker Technology for valuable discussions, Bartosz Schmidt for the SEC analysis, Sofia Essén for the mass spectrometry measurements.References
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- R. W. Howarth and A. Ingraffea, Nature, 2011, 477, 271–273 CrossRef CAS PubMed.
- E. MacArthur, D. Waughray and M. R. Stuchtey, The New Plastics Economy Rethinking the future of plastics, 2016 Search PubMed.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS.
- I. Wojnowska-Baryła, D. Kulikowska and K. Bernat, Sustain., 2020, 12, 1–12 CrossRef.
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551–2571 CrossRef CAS.
- J. P. Brutman, G. X. De Hoe, D. K. Schneiderman, T. N. Le and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11097–11106 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Appl. Polym. Mater., 2020, 2, 1917–1924 CrossRef CAS.
- L. Cederholm, P. Olsén, M. Hakkarainen and K. Odelius, Polym. Chem., 2020, 11, 4883–4894 RSC.
- X. Tang and E. Y. X. Chen, Chem, 2019, 5, 284–312 CAS.
- J. Myung, N. I. Strong, W. M. Galega, E. R. Sundstrom, J. C. A. Flanagan, S. G. Woo, R. M. Waymouth and C. S. Criddle, Bioresour. Technol., 2014, 170, 167–174 CrossRef CAS PubMed.
- C. Jönsson, R. Wei, A. Biundo, J. Landberg, L. Schwarz Bour, F. Pezzotti, A. Toca, L. M. Jacques, U. T. Bornscheuer and P.-O. Syrén, ChemSusChem, 2021, 14, 1–14 CrossRef.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- Z. Guo, M. Eriksson, H. de la Motte and E. Adolfsson, J. Cleaner Prod., 2020, 124579 Search PubMed.
- O. Avinc and A. Khoddami, Fibre Chem., 2010, 42, 68–78 CrossRef CAS.
- D. Battegazzore, T. Abt, M. L. Maspoch and A. Frache, Composites, Part B, 2019, 163, 761–768 CrossRef CAS.
- M. Sanchis-Sebastiá, E. Ruuth, L. Stigsson, M. Galbe and O. Wallberg, Waste Manage., 2021, 121, 248–254 CrossRef PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS.
- J. E. Park, D. Y. Hwang, G. H. Choi, K. H. Choi and D. H. Suh, Biomacromolecules, 2017, 18, 2633–2639 CrossRef CAS PubMed.
- H. Hu, Y. Tian, Z. Kong, W. B. Ying, C. Chen, F. Li, R. Zhang and J. Zhu, ACS Sustainable Chem. Eng., 2021, 9, 2280–2290 CrossRef CAS.
- H. Otsuka and T. Endo, Macromolecules, 1999, 32, 9059–9061 CrossRef CAS.
- A. G. Pemba, J. A. Flores and S. A. Miller, Green Chem., 2013, 15, 325–329 RSC.
- S. Chikkali, F. Stempfle and S. Mecking, Macromol. Rapid Commun., 2012, 33, 1126–1129 CrossRef CAS PubMed.
- R. A. Awl, W. E. Neff, D. Welsleder, E. H. Pryde and N. Regional, J. Am. Oil Chem. Soc., 1976, 53, 20–26 CrossRef CAS.
- B. S. Rajput, S. R. Gaikwad, S. K. Menon and S. H. Chikkali, Green Chem., 2014, 16, 3810–3818 RSC.
- A. Sudo, T. Sano, M. Harada and D. Ishida, ACS Macro Lett., 2014, 3, 808–812 CrossRef CAS.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214–3221 RSC.
- G. Akbulut, H. B. Sonmez and F. Wudl, J. Polym. Res., 2013, 20, 1–8 CAS.
- C. Lavilla, A. Martínez De Ilarduya, A. Alla and S. Muñoz-Guerra, Polym. Chem., 2013, 4, 282–289 RSC.
- C. Lavilla, A. Alla, A. M. De Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1591–1604 CrossRef CAS.
- H. B. Sonmez, F. G. Kuloglu, K. Karadag and F. Wudl, Polym. J., 2012, 44, 217–223 CrossRef CAS.
- S. Lingier, P. Espeel, S. S. Suarez, O. Türünç, S. De Wildeman and F. E. Du Prez, Eur. Polym. J., 2015, 70, 232–239 CrossRef CAS.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- A. C. Law, D. S. Stankowski, B. H. Bomann, S. Suhail, K. H. Salmon, S. W. Paulson, M. J. Carney and N. J. Robertson, J. Appl. Polym. Sci., 2020, 137, 1–6 CrossRef.
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- C. Lavilla, A. M. De Ilarduya, A. Alla, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Macromolecules, 2012, 45, 8257–8266 CrossRef CAS.
- G.-H. Choi, D. Y. Hwang and D. H. Suh, Macromolecules, 2015, 48, 6839–6845 CrossRef CAS.
- S. Lingier, Y. Spiesschaert, B. Dhanis, S. De Wildeman and F. E. Du Prez, Macromolecules, 2017, 50, 5346–5352 CrossRef CAS.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S. H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- M. Khalifa, S. Anandhan, G. Wuzella, H. Lammer and A. R. Mahendran, Polym. Technol. Mater., 2020, 59, 1751–1769 CAS.
- J. Datta and P. Kasprzyk, Polym. Eng. Sci., 2018, 58, E14–E35 CrossRef CAS.
- S. Schrüfer, D. Sonnleitner, G. Lang and D. W. Schubert, Polymers, 2020, 12, 1276–1299 CrossRef.
- J. Schmidt, R. Wei, T. Oeser, L. A. Dedavid e Silva, D. Breite, A. Schulze and W. Zimmermann, Polymers, 2017, 9, 65 CrossRef PubMed.
- J. A. Galbis, M. D. G. García-Martín, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS.
- United States Department of Agriculture, Sugar: World Markets and Trade, 2020 Search PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, 2004, vol. 1 Search PubMed.
- Coca-Cola Expands Access to PlantBottle IP - News & Articles, https://www.coca-colacompany.com/news/coca-cola-expands-access-to-plantbottle-ip, (accessed 20 April 2020).
- J. A. Galbis, M. D. G. Garc, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- R. T. Duan, Q. X. He, X. Dong, D. F. Li, X. L. Wang and Y. Z. Wang, ACS Sustainable Chem. Eng., 2016, 4, 350–362 CrossRef CAS.
- C. Japu, A. Martínez De Ilarduya, A. Alla, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Polym. Chem., 2014, 5, 3190–3202 RSC.
- Z. Yang, L. Liu, H. An, C. Li, Z. Zhang, W. Fang, F. Xu and S. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 9968–9979 CrossRef CAS.
- B. S. Rajput, S. B. Pawal, D. V. Bodkhe, I. N. Rao, A. V. S. Sainath and S. H. Chikkali, Eur. Polym. J., 2020, 134, 109775 CrossRef CAS.
- E. Slavko and M. S. Taylor, Macromolecules, 2020, 53, 8192–8201 CrossRef CAS.
- Z. Yang, X. Li, F. Xu, W. Wang, Y. Shi, Z. Zhang, W. Fang, L. Liu and S. Zhang, Green Chem., 2021, 23, 447–456 RSC.
- A. Rosengren, S. J. Butler, M. Arcos-Hernandez, K. E. Bergquist, P. Jannasch and H. Stålbrand, Green Chem., 2019, 21, 2104–2118 RSC.
- S. Maiti, S. Manna, N. Banahene, L. Pham, Z. Liang, J. Wang, Y. Xu, R. Bettinger, J. Zientko, A. P. Esser-Kahn and W. Du, Angew. Chem., 2020, 59, 18943–18947 CrossRef CAS PubMed.
- L. Su, S. Khan, J. Fan, Y. N. Lin, H. Wang, T. P. Gustafson, F. Zhang and K. L. Wooley, Polym. Chem., 2017, 8, 1699–1707 RSC.
- J. H. Kamps, V. Ramakrishnan, T. Hoeks, B. J. P. Jansen, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2019, 52, 3187–3198 CrossRef CAS.
- H. Marubayashi, T. Ushio and S. Nojima, Macromolecules, 2019, 52, 4624–4633 CrossRef CAS.
- W. C. Shearouse, L. M. Lillie, T. M. Reineke and W. B. Tolman, ACS Macro Lett., 2015, 4, 284–288 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- ISO 1973:2021—Textile fibres—Determination of linear density—Gravimetric method and vibroscope method, 2021.
- ISO 5079:2020—Textile fibres—Determination of breaking force and elongation at break of individual fibres, 2020.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- H. Shen, H. Shan and L. Liu, ChemSusChem, 2020, 13, 513–519 CrossRef CAS PubMed.
- Isocyanates Market Forecast, Trend Analysis & Competition Tracking - Global Market Insights 2019 to 2027, https://www.factmr.com/report/4174/isocyanates-market, (accessed 16 July 2020).
- DOW, Pure, Modified and Polymeric MDI Handling & Storage Guide, 2009 Search PubMed.
- T. Sui, N. Baimpas, I. P. Dolbnya, C. Prisacariu and A. M. Korsunsky, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- H. Y. Mi, X. Jing, B. N. Napiwocki, B. S. Hagerty, G. Chen and L. S. Turng, J. Mater. Chem. B, 2017, 5, 4137–4151 RSC.
- A. Magnin, E. Pollet, R. Perrin, C. Ullmann, C. Persillon, V. Phalip and L. Avérous, Waste Manage., 2019, 85, 141–150 CrossRef CAS PubMed.
- E. Głowińska, W. Wolak and J. Datta, J. Polym. Environ., 2021, 1–10 Search PubMed.
- L. Zhang, F. C. Michel and A. C. Co, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1495–1499 CrossRef CAS.
- J. F. Wilson and E. Y. X. Chen, ACS Sustainable Chem. Eng., 2019, 7, 7035–7046 CrossRef CAS.
- C. Oh, E. H. Choi, E. J. Choi, T. Premkumar and C. Song, ACS Sustainable Chem. Eng., 2020, 8, 4400–4406 CrossRef CAS.
- Z. Mou, S. Feng and E. Y. X. Chen, Polym. Chem., 2016, 7, 1593–1602 RSC.
- Y. Shieh, H. Chen, K. Liu and Y. Twu, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4126–4134 CrossRef.
- Y. Zhang, Z. Xia, H. Huang and H. Chen, J. Anal. Appl. Pyrolysis, 2009, 84, 89–94 CrossRef CAS.
- F. Gaboriaud and J. P. Vantelon, J. Polym. Sci. A1, 1982, 20, 2063–2071 CAS.
- L. Costa, M. P. Luda, G. G. Cameron and M. Y. Qureshi, Polym. Degrad. Stab., 2000, 67, 527–533 CrossRef CAS.
- J. A. Hiltz and I. A. Keough, Characterization of Poly(ether)urethanes - High Resolution Thermogravimetric Analysis, 2004 Search PubMed.
- D. W. van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- A. Cyriac, S. H. Lee, J. K. Varghese, J. H. Park, J. Y. Jeon, S. J. Kim and B. Y. Lee, Green Chem., 2011, 13, 3469–3475 RSC.
- S. Bourbigot and S. Duquesne, J. Mater. Chem., 2007, 17, 2283–2300 RSC.
- Y. Qi, Z. Weng, Y. Kou, L. Song, J. Li, J. Wang, S. Zhang, C. Liu and X. Jian, Chem. Eng. J., 2021, 406, 126881 CrossRef CAS.
- J. Kim, M. M. Mok, R. W. Sandoval, D. J. Woo and J. M. Torkelson, Macromolecules, 2006, 39, 6152–6160 CrossRef CAS.
- M. D. Lefebvre, M. Olvera De La Cruz and K. R. Shull, Macromolecules, 2004, 37, 1118–1123 CrossRef CAS.
- A. Rudin and P. Choi, The Elements of Polymer Science and Engineering - 3rd Edition, 3rd edn, 2012 Search PubMed.
- Spandex fiber - CAMEO, http://cameo.mfa.org/wiki/Spandex_fiber, (accessed 3 June 2021).
- J. Hu, J. Lu and Y. Zhu, Polym. Rev., 2008, 48, 275–301 CrossRef CAS.
- F. Menegazzo, E. Ghedini and M. Signoretto, Molecules, 2018, 23, 1–18 CrossRef PubMed.
- M. Sayed, N. Warlin, C. Hulteberg, I. Munslow, S. Lundmark, O. Pajalic, P. Tunå, B. Zhang, S.-H. Pyo and R. Hatti-Kaul, Green Chem., 2020, 22, 5402–5413 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00450f |
| This journal is © The Royal Society of Chemistry 2021 |