
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of acrolein in ambient air and in the atmosphere of environmental test chambers†
Alexandra
Schieweck
 *,
Erik
Uhde
and
Tunga
Salthammer
*,
Erik
Uhde
and
Tunga
Salthammer
Fraunhofer WKI, Department of Material Analysis and Indoor Chemistry, Bienroder Weg 54E, 38108 Braunschweig, Germany. E-mail: alexandra.schieweck@wki.fraunhofer.de; Tel: +49 531 2155 924
First published on 2nd September 2021
Abstract
Acrolein (2-propenal) is a reactive substance undergoing multiple reaction pathways and an airborne pollutant with known corrosive, toxic and hazardous effects to the environment and to human health. So far, investigating the occurrence of acrolein in indoor air has been challenging due to analytical limitations. The classic DNPH-method has proven to be error-prone, even though it is still recommended in specific testing protocols. Thus, different approaches for an accurate determination of ambient acrolein have been introduced. In this work, an overview of already published data regarding emission sources and air concentrations is provided. In addition, a new method for the quantitative determination of acrolein in environmental test chambers and in indoor air is presented. Analysis is carried out using thermal desorption and coupled gas chromatography/mass spectrometry (TD-GC/MS) after sampling on the graphitized carbon black (GCB) Carbograph™ 5TD. All analytical steps have been carefully validated and compared with derivatization techniques (DNPH and DNSH) as well as online detection using PTR-QMS. The sampling time is short due to the low air collection volume of 4 L. Although derivatization is not applied, a detection limit of 0.1 μg m−3 can be achieved. By increasing the sampling volume to 6 L, the limit of detection can be lowered to 0.08 μg m−3. No breakthrough during sampling or analyte loss during storage of the acrolein laden sampling tubes was found. Therefore, the presented method is robust, easy-to-handle and also very suitable for routine analyses and surveys.
Environmental significanceAcrolein is considered a priority indoor air pollutant. It is formed and released by several chemical processes and is known to have a negative impact on human health. Consequently, the accurate determination of acrolein in the indoor environment is of considerable importance. This paper introduces a new analytical method based on solid sorbent sampling and subsequent analysis by TD-GC/MS. It is easy-to-handle, fast and robust with a low limit of detection (LOD) at small sampling volumes. Validation was performed using recovery and breakthrough tests as well as chamber experiments. The reliability of the method makes it particularly suitable for routine analyses and surveys. |
1. Introduction
Acrolein (2-propenal) is the simplest monounsaturated aldehyde and has two theoretically possible conformers.1 The substance, which is liquid at room temperature, is classified as corrosive, toxic and hazardous to the environment. The International Agency for Research on Cancer (IARC) lists acrolein as a group 3 carcinogen. The toxic effect is mainly based on the formation of protein and DNA adducts.2 Irritant effects,3 decreased respiratory function4 and cardiovascular diseases5,6 were also reported. The various exposure paths (inhalation, dermal, and oral) are discussed by Faroon et al.7 The Office of Environmental Health Hazard Assessment (OEHHA) gives the following Reference Exposure Limits (REL) for the inhalation path: acute REL = 2.5 μg m−3; 8 h REL = 0.7 μg m−3; chronic REL = 0.35 μg m−3.8 Trantallidi and co-workers9 derived a short-term health-based Critical Exposure Limit (CEL) of 21 μg m−3 from the results of a chamber study. Health Canada has released Residential Indoor Air Quality Guidelines (RIAQG) for contaminants which impact indoor air. For acrolein, a long-term exposure limit (24 hours) of 0.44 μg m−3 and a short-term exposure limit (1 hour) of 38 μg m−3 are recommended.10In industrial use, acrolein is an intermediate in the production of acrylic acid and methionine. Acrolein synthesis routes are the aldol condensation of formaldehyde and acetaldehyde, the dehydrogenation of glycerol and, practically most important, the catalyzed oxidation of propene.11,12 First, Esterbauer et al.13 and later, Stevens and Maier14 reported the exogenous and endogenous pathways of acrolein formation (see Fig. 1).
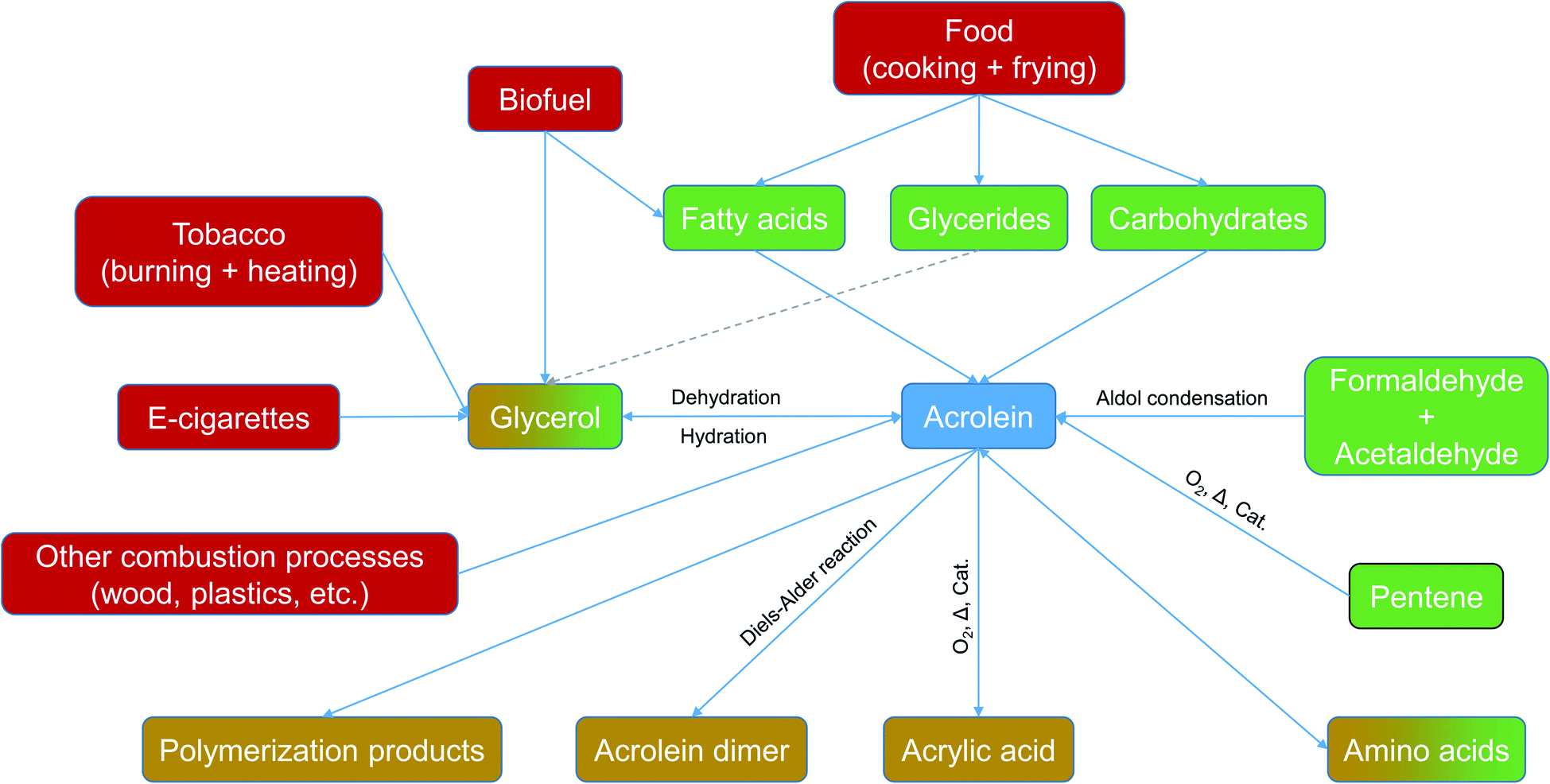 | ||
| Fig. 1 Exogenous and endogenous pathways of acrolein formation and reaction. | ||
Acrolein is ubiquitous in the environment and an intermediate in atmospheric chemistry.15,16 Although various sources have been identified, human indoor exposure to acrolein has rarely been studied so far, even though Logue et al.17 identified acrolein as one of nine priority hazards in their review on chemical air contaminants in residences. The comparatively small number of publications on acrolein in indoor air may be due to the fact that VVOCs (very volatile organic compounds) have only rarely been included in systematic surveys.18 Moreover, as an α,β-unsaturated carbonyl compound, acrolein is highly reactive and therefore poses a particular challenge from an analytical point of view. Thus, various methods for determining acrolein in ambient air have been developed.
1.1 Study objectives
With regard to the importance of acrolein in the environment, the current paper discusses published data and already applied analytical techniques. Furthermore, facing the integration of acrolein into the routine analysis of VVOCs19 and surveys, we are introducing a new method for its quantitative determination in environmental test chambers and in indoor air. By sampling on a solid adsorber without the need of derivatization, only small sampling volumes of air are required to reach sufficiently low detection limits. The analysis is carried out using thermal desorption followed by coupled gas chromatography/mass spectrometry (TD-GC/MS). The method has been tested and validated within the framework of various chamber experiments. During these test series, currently available and published methods based on DNPH- and DNSH-derivatization, respectively, were included for comparison. In addition, sampling on Tenax® TA according to ISO 16000-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 was considered as it can be applied during sampling protocols if no additional sampling on low-molecular carbonyls is foreseen, even though it is not recommended for the reliable determination of acrolein in air.
20 was considered as it can be applied during sampling protocols if no additional sampling on low-molecular carbonyls is foreseen, even though it is not recommended for the reliable determination of acrolein in air.
2. Acrolein emission sources and published levels in ambient air
2.1 Indoor emission sources
In indoors, acrolein is known to be mainly formed through thermal processes, which include the decomposition of wood,21,22 fuel,23–25 and plastics,26,27 incense burning,28 cooking and deep-frying,29–34 smoking,35 tobacco heating36 and vaping.37,38 The cause of the formation of acrolein during food preparation are the decomposition processes of fatty acids,39 glycerides and carbohydrates contained in food. Acrolein was identified to be emitted from solid wood and wood-based products in low concentrations, including insulation materials and flooring, as shown in Table 1.40,41 Several studies have focused on the release of acrolein due to cooking processes. Depending on the oil type and the temperature, elevated concentrations can be reached.39,42,43 Studies performed in hotel kitchens and restaurants revealed indoor levels in the range of 10 μg m−3 to 590 μg m−3.29,31 The sometimes high acrolein concentrations in the smoke and steam of tobacco products and e-cigarettes can be explained by the addition of glycerol and triacetin.44 Heat-not-burn cigarettes have been found to be minor emission sources compared to conventional cigarettes.37 A comprehensive study targeting gaseous emissions from burning scented and unscented candles measured unit specific emission rates between 8 μg unit−1 h−1 and 20 μg unit−1 h−1 and highlighted the fact that acrolein is released as a by-product which cannot be avoided.45| Category | Value | Unit | Reference |
|---|---|---|---|
| Wood-based products | |||
|
|||
| Softwood | |||
| Douglas fir | 2 | μg m−2 h−1 | Schieweck40 |
| 8.1 ± 0.90 | ng g−1 | Seaman et al.41 | |
| Pine | 8 | μg m−2 h−1 | Schieweck40 |
| 5.9 ± 0.55 | ng g−1 | Seaman et al.41 | |
| 2 | μg m−2 h−1 | Fraunhofer WKI, unpublished results | |
| Spruce | 2 | μg m−2 h−1 | Schieweck40 |
| 2.5 | μg m−2 h−1 | Fraunhofer WKI, unpublished results | |
|
|||
| Hardwood | |||
| Oak | <0.5 | μg m−2 h−1 | Schieweck40 |
| Red oak | 1.0 ± 0.14 | ng g−1 | Seaman et al.41 |
| Larch | 2 | μg m−2 h−1 | Schieweck40 |
| 15 | μg m−2 h−1 | Fraunhofer WKI, unpublished results | |
| Beech | <0.5 | μg m−2 h−1 | Schieweck40 |
| Yellow poplar | 1.0 ± 0.14 | ng g−1 | Seaman et al.41 |
| Redwood lumber | 1.3 ± 0.41 | ng g−1 | Seaman et al.41 |
|
|||
| Laminated veneer lumber | |||
| Hardwood/formaldehyde-resin | <0.5 | μg m−2 h−1 | Schieweck40 |
| Hardwood/polyvinyl acetate-resin | <0.5 | μg m−2 h−1 | Schieweck40 |
| Hardwood, dried without adhesive | <0.5 | μg m−2 h−1 | Schieweck40 |
| Hardwood, cooked | 1 | μg m−2 h−1 | Schieweck40 |
|
|||
| Solid structural timber | |||
| Softwood/melamine-resin | 2 | μg m−2 h−1 | Schieweck40 |
| Softwood/polyurethane | 1 | μg m−2 h−1 | Schieweck40 |
|
|||
| Laminated timber | |||
| Softwood/melamine-resin | 1.5 | μg m−2 h−1 | Schieweck40 |
| Softwood/isocyanate | 1.5 | μg m−2 h−1 | Schieweck40 |
|
|||
| Plywood panel | |||
| Three-layer panels | 2.5 | μg m−2 h−1 | Schieweck40 |
| Plywood | 3 | μg m−2 h−1 | Fraunhofer WKI, unpublished results |
|
|||
| Wooden composite board | |||
| Oriented strand board (OSB) | <0.5 | μg m−2 h−1 | Schieweck40 |
| 2 | μg m−2 h−1 | Fraunhofer WKI, unpublished results | |
| Particle board | <0.5 | μg m−2 h−1 | Schieweck40 |
| 1.0 ± 0.045 | ng g−1 | Seaman et al.41 | |
| 6.5 | μg m−2 h−1 | Fraunhofer WKI, unpublished results | |
| Acoustic panels | 2.5 | μg m−2 h−1 | Schieweck40 |
|
|||
| Insulation | |||
| Wood fibre insulation | 1 | μg m−2 h−1 | Schieweck40 |
| 6.5 | μg m−2 h−1 | Fraunhofer WKI, unpublished results | |
| Wooden fibreboard | 1 | μg m−2 h−1 | Schieweck40 |
| Rigid foam | 0.5 | μg m−2 h−1 | Fraunhofer WKI, unpublished results |
|
|||
| Flooring | |||
| Parquet flooring | <1.25 | μg m−2 h−1 | Schieweck40 |
| Laminate flooring | <1.25 | μg m−2 h−1 | Schieweck40 |
|
|||
| Paint, construction materials | |||
| Latex paint | 0.35 ± 0.08 | ng g−1 | Seaman et al.41 |
| Plaster board | 3 | μg m−2 h−1 | Fraunhofer WKI, unpublished results |
|
|||
| Plastics | |||
| Foam plastic building materials applied on plywood, burning (max. 600–700 °C) | max. 400 | ppm | Morikawa and Yanai102 |
| PE-foil | <0.5 | μg m−2 h−1 | Fraunhofer WKI, unpublished results |
|
|||
| Cooking | |||
| Canola oil, heated to 180 °C | 52.6 ± 2.5 | mg h−1 L−1 | Fullana et al.42 |
| (Extra-virgin) olive oil, heated to 180 °C | 9.3 ± 1.2–9.6 ± 0.9 | mg h−1 L−1 | Fullana et al.42 |
| Soybean oil, heated to 180 °C | 11.8 ± 67.4 | mg h−1 L−1 | da Silva and Pereira43 |
| Palm oil, heated to 180 °C | 2.58 ± 41.5 | mg h−1 L−1 | da Silva and Pereira43 |
| Oil cooking events, 5 min post-cooking events | 10.68–27.04 | mg h−1 kg−1 | Seaman et al.30 |
| Different oil types, heated to 150–220 °C | max. 400–4160 at 220 °C | ppb | Endo et al.39 |
| Stir-fry cooking in a test house | 0.56 | mg per person meal | Arata et al.34 |
|
|||
| Smoking | |||
| Conventional cigarettes | 6.5 ± 4.5 | μg unit−1 min−1 | Ruprecht et al.37 |
| Heat-not-burn cigarettes (iQOS) | 0.09 ± 0.03 | μg unit−1 min−1 | Ruprecht et al.37 |
|
|||
| Candles | |||
| Different fuels, no fragrance | <8 | μg unit−1 h−1 | Salthammer et al.45 |
| Palm, fruit fragrance | 12 | μg unit−1 h−1 | Salthammer et al.45 |
| Paraffin, fresh fragrance | 13 | μg unit−1 h−1 | Salthammer et al.45 |
| Paraffin, fruit fragrance | 12 | μg unit−1 h−1 | Salthammer et al.45 |
| Paraffin, spice & edibles fragrance | 20 | μg unit−1 h−1 | Salthammer et al.45 |
| Soy, fresh fragrance | 10 | μg unit−1 h−1 | Salthammer et al.45 |
| Soy, fruit fragrance | 8 | μg unit−1 h−1 | Salthammer et al.45 |
| Soy, spice & edibles fragrance | 14 | μg unit−1 h−1 | Salthammer et al.45 |
| Stearin, fruit fragrance | 10 | μg unit−1 h−1 | Salthammer et al.45 |
2.2 Acrolein levels in outdoor and indoor air
Table 2 summarizes published acrolein data measured in outdoor and indoor air. In the Oakland-San Francisco Bay area, acrolein concentrations between 0.032 μg m−3 and 0.100 μg m−3 were measured.46 In further measurements in California, the concentrations in urban areas were significantly higher than in remote and coastal areas.47 As part of “The Relationship of Indoor, Outdoor and Personal Air (RIOPA)” study, the indoor/outdoor ratio in 234 U.S. non-smoking homes, located in California, New Jersey and Texas, was found to be ≈1.3 with median values of 0.59 μg m−3 and 0.46 μg m−3 for indoor and outdoor air, respectively.48 Acrolein concentrations in the range 0.1 ppb to 1.0 ppb (0.2–2.7 μg m−3, at P = 1013 mbar and T = 298 K) were detected in the rural air of Portugal.49 Seaman and co-workers41 investigated acrolein in the air of California apartments and found concentrations of up to 12.2 μg m−3. The values indoors were always significantly higher than outdoors (3–40 times). For the apartments examined, they calculated emission rates between 0.31 mg h−1 and 1.46 mg h−1. Recently, indoor exposure to a large range of volatile organic compounds (VOCs) was investigated with high time resolution both during summer and winter in two Californian residences.50 Due to continuous indoor sources (e.g. lumber), the main daily occupant exposure was ∼4.6 ppb-h d−1 (mean value). Concentration peaks were observed in dependence of cooking events as specific emission sources. Both oven cooking and stovetop cooking were identified to produce the largest increase in indoor acrolein exposure. Enhancements in acrolein levels caused by an electric oven were higher than those associated with the use of a natural gas stovetop. The results confirmed the fact that heated cooking oil is a major acrolein source indoors.50 In Canadian homes, measured acrolein concentrations were between 0.1 μg m−3 and 4.9 μg m−3.51 During the “House Observations of Microbial and Environmental Chemistry (HOMEChem)” study,52 the emissions of scripted everyday activities were measured in a test house in Texas. The amount of released acrolein during stir-fry cooking was 0.56 mg per person meal. As measurements were performed by PTR-TOF-MS, the recorded ion C3H5O+ can be assigned both to acrolein and to a propionic acid fragment. The formation of acrolein is attributed to high-temperature degradation of cooking oil and heated vegetables, respectively.34 In a Chinese temple, the acrolein level was up to 3 times higher during incense burning than on the outside.28 Williams et al.53 determined an average acrolein concentration of 5–6 μg m−3 in London smoking rooms exceeding those measured in comparison rooms. A comprehensive study targeting carbonyl emissions in the exhaust of commercial kitchens in China was performed by Ho et al.54 revealing substantial amounts of acrolein in dependence of different cooking ingredients. Schieweck40 published acrolein data obtained by indoor measurements in wooden prefabricated houses in Germany. Depending on the ventilation type and the construction stage, the concentrations varied between 5.1 μg m−3 and 18.3 μg m−3.3. Analytical approaches
Table 3 provides an overview of analytical methods and procedures for the detection of ambient acrolein. As a rule, the substance is derivatized directly on the collection phase. The classic DNPH method according to ISO 16000-355 and ASTM D519756 is applied in different testing protocols, but has been criticized to be unsuitable.57–59 Alternative techniques have therefore been developed including derivatization with dansylhydrazine (DNSH),60–62 sampling on solid sorbents,63 thermal desorption gas chromatography,64 online monitoring using proton-transfer-reaction mass spectrometry (PTR-MS),50,65,66 solid phase microextraction67 and derivatization with o-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine (PFBHA).68
3.1 DNPH-coated sorbents
The most applied method is a solid phase extraction technique and corresponds to ISO 16000-355 and ASTM D5197.56 Sorbent cartridges are filled with silica gel as the substrate which is impregnated with 2,4-dinitrophenylhydrazine (DNPH) as a derivatization agent. Rarely, DNPH was used as a solution filled in impinger bottles according to the US EPA Method TO-5.69 After extraction with acetonitrile (ACN), the analysis of the formed hydrazone was performed by HPLC-separation and subsequent detection by UV. Williams et al.53 performed comparative measurements of both approaches, but without identifying acrolein after sampling on DNPH-cartridges even though it was highlighted that this method is less expensive and time-consuming with lower material consumption than the wet chemical technique. Several publications have discussed the limitations and uncertainties associated with the determination of airborne unsaturated carbonyls when using DNPH-coated solid sorbent cartridges.57–59 Airborne oxidants, such as ozone (O3), nitrogen oxide (NO) and nitrogen dioxide (NO2) can interfere with unsaturated carbonyls.70–72 The reaction of acrolein and DNPH forming acrolein-2,4-DNP-hydrazone leads also to several dimerization and trimerization products with similar chromatographic properties and overlapping peaks which might lead to an over-estimation of acrolein concentrations.58 Using fluorescence detection techniques, different absorption maxima of these side products were observed.57,73,74 The quantification is more complicated due to varying formation rates of the dimers and trimers in dependence of environmental factors during sampling and analytical conditions and also due to varying response factors under single-wavelength detection.59,73 An additional drawback is the fact that only acrolein-2,4-DNPH is commercially available as external standard solution and is therefore commonly used for calibration. However, this neither allows comparative measurements nor calibration of all other reaction products formed. Also, due to reaction processes and a resulting variation of formed peaks, the acrolein-DNP-hydrazone may not be correctly identified in real air samples. A summation of the acrolein-DNP-hydrazone and associated adduct peak areas might provide an estimation of the detected acrolein concentration, but seems to be limited to a storage period of approximately 5 h.58,59 However, an advantage of this established method is that it allows the detection of a broad range of carbonyls in one analytical step. The determination of acrolein can therefore be easily integrated into daily routine.
3.2 DNSH-coated sorbents
Due to the limitations of the DNPH-method, a silica-based-bonded C18 sorbent coated with dansylhydrazine (DNSH) was introduced as a reagent for derivatizing unsaturated carbonyls such as acrolein.57,60–62,75 Herrington and co-workers61 analyzed the formed DNSH-derivatives after ACN-extraction with HPLC-fluorescence techniques; the peaks should be identified and quantified on the basis of individual standard substances.61,62 Corresponding to the acrolein-DNSH derivatives formed, three chromatographic peaks can be observed.61,76 Two peaks are attributed to E- and Z-isomers of mono-derivatized acrolein with a molecular weight of 303.38 g mol−1. The third peak is described as di-derivatized acrolein (dimer) with a molecular mass of 568.71 g mol−1 and is recommended to be used for acrolein analysis to obtain stable results.61 The DNSH-based technique is just recommended for acrolein with a low limit of detection (LOD) of 0.24 μg m−3 at low sampling volumes (7.5 L) and short sampling times (30 min), as the classic DNPH-method works well for discontinuous sampling of other low-weight carbonyls. The DNSH-method was further modified to increase the sensitivity by using a di-derivatized acrolein-hydrazone for quantification and to extend the analytical sensitivity to crotonaldehyde. Sampling is performed by using modified solid phase extraction tubes as diffusive, tube-type samplers (the Passive Aldehydes and Ketones Sampler (PAKS)).613.3 PFPH-coated sorbents
Ho and Yu64 also developed a technique based on a derivatization agent, but using coupled gas chromatography/mass spectrometry after thermal desorption (TD-GC/MS) for analysis. Pentafluorophenyl hydrazine (PFPH) was introduced as a coating for Tenax® TA (60/80 mesh) as a solid sorbent. Even though the authors describe the method as highly sensitive with a LOD of ∼0.8 μg m−3 for a sampling volume of 24 L, which led to the detection of higher background values, the approach has not been adopted widely.643.4 PFBHA sampling
In order to trap ambient acrolein at trace levels (ppt) with short sampling times, a method based on a mist chamber was developed which is filled with a sodium bisulfite solution to absorb volatile acrolein. After dissociation of the sulfonates, the formed free carbonyls are derivatized with o-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine (PFBHA) resulting in thermally stable oxime adducts which can be separated by GC followed by electron capture negative ionization mass spectrometry (GC/MS). Short term fluctuations of acrolein shall be detected by this technique at very short sampling times and a high sensitivity (LOD: 0.012 μg m−3). Drawbacks are the facts that this approach requires intensive work and custom-made mist chambers as well as new calibration at each sampling day.30,41,68,77 Destaillats et al.46 sampled indoor air in impingers filled with a PFBHA solution (0.25 mg mL−1). The formed PFBHA derivatives were extracted in dichloromethane after acidification with H2SO4. Analysis was performed by high-resolution gas chromatography coupled with ion trap mass spectrometry (HRGC/ITMS). The method offers low limits of detection (LOD) at low sampling volumes, see Table 3.46 Ho et al.54 compared PFPH and PFBHA sampling in their study of acrolein concentrations in exhaust air of Chinese restaurants, but without highlighting similarities and differences of these methods.3.5 Headspace and SPME-techniques
Headspace-based techniques have been published already in 1987 and later in 2013. In the 1987 study, the morpholine derivative of acrolein, 3-morpholinopropanal, was used to determine acrolein emissions. Analysis of the headspace was performed by GC coupled with a thermionic detector (TID),32 and in the 2013 experiments a GC/MS system was used.39 In addition, solid phase microextraction (SPME fibers) was also used with DNPH or PFPH as the derivatization agent and subsequent GC/MS-analysis.67,78 Fullana and co-workers42 collected fumes of heated oil in Tedlar bags and analyzed them by GC/MS without a prior derivatization step. Identification of acrolein was confirmed by laboratory standards, and quantification was performed using MS response factors. A combination of headspace and needle trap microextraction (NTME) with subsequent GC/MS-analysis was applied by Lomonaco et al.273.6 Solid sorbent sampling
Solid sorbents were just applied in very few cases without prior derivatization. Singer et al.79 used Tenax® TA filled sorbent tubes with an extra section of Carbosieve® S-III, a carbon molecular sieve (CMS). Due to its very strong sorbent strength, Carbosieve® S-III is recommended to sample very volatile organic compounds (VVOCs) in the range C2 to C5.35,79 However, the results indicated a possible sample breakthrough or incomplete recovery of acrolein. In addition, acrolein concentrations happened to be below the LOQ and, thus, complicated the quantification of background values.353.7 On-line monitoring
As a fast on-line technique with high resolution, proton-transfer-reaction time-of-flight (PTR-TOF) mass spectrometry was introduced to detect and quantify acrolein in air samples with a LOD of 21 pptv, based on a 1 min integration period, and a precision of 1.5%.65 For acrolein, the nominal target M + 1 ion is m/z 57, the proton affinity of acrolein is 797.0 kJ mol−180 and the calculated proton transfer reaction rate constant between H3O+ and acrolein is 3.35 × 10−9 cm3 s−1.65,66,81 Lunderberg et al.50 used a proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS) to investigate occupant exposure patterns indoors in two California residences. During occupancy the mean acrolein concentrations were below 1 ppb.
4. Experimental methodology
4.1 Solid sorbent sampling without derivatization
A VVOC method developed earlier and published in 201819 served as the basis for the experiments described here. Single-bed stainless steel desorption tubes (Markes International Ltd., 89 mm length, 6.4 mm O.D.) were filled with the graphitized carbon black (GCB) Carbograph™ 5TD (20/40 mesh, Markes International Ltd.) as a solid sorbent. ∼300 mg of the sorbent was placed between glass wool end plugs. Environmental test chamber air was sampled actively by means of a pump with a flow rate of 125 mL min−1 and a total sampling volume of 4 L. After sampling, the tubes were sealed with Swagelok brass end caps fitted with PTFE ferrules. For analysis, the tubes were thermally desorbed (TD-100, Markes International Ltd.) into a coupled GC/MS system (Agilent 7890A/5975C). The conditions for thermal desorption were prepurge 3 min with a flow rate of 50 mL min−1, primary desorption at 300 °C for 6 min at a flow rate of 20 mL min−1, no inlet split, cold trap low 25 °C, pretrap fire purge 3 min at 50 mL min−1, trap heating rate 40 °C s−1, cold trap high at 300 °C for 6 min, outlet split 10 mL min−1, and the flow path temperature at 200 °C. The cold trap contained quartz wool/Carbograph™ 1TD (40/60 mesh) and Carboxen® 1000 (80/100 mesh) with a ratio of 1:4.
The compounds were separated on a fused silica capillary column (6%/94% cyanopropylphenyl/dimethylpolysiloxane) of medium polarity (DB 624, 60 m, 0.32 mm, 1.8 μm, Agilent J&W). The initial column oven temperature was 30 °C (6 min), increased in a first step to 45 °C (1 °C min−1) and in a second step to 240 °C with an increasing rate of 40 °C min−1 (25.8 min run). The GC was operated in scan mode with a mass range of 20–450 amu, the MS source temperature was 230 °C, and the quadrupole temperature was 150 °C.
Further information regarding calibration and breakthrough of acrolein is provided (ESI, Section S1†).
4.2 Comparison with other analytical methods
For preparing DNSH-cartridges, 100 mg DNSH (98%, 500 mg; Sigma Aldrich) were diluted with 100 mL acetonitrile (Rotisolv Ultra LC-MS; Carl Roth GmbH + Co. KG). After shaking, the solution was placed into an ultrasonic bath for 5 min until the hydrazine was fully dissolved. 100 μL acetic acid (100%; Merck) were added to the solution. The cartridges (S10; Supelco, Inc.) were cleaned with acetonitrile and subsequently eluted with the DNSH-solution for coating the silica gel bed. The prepared cartridges were dried in a nitrogen stream (100 mL min−1) for one hour. Afterwards, the cartridges were closed with end caps and stored under laboratory conditions. After extraction with acetonitrile, samples were measured by HPLC (Zorbax RRHD Eclipse Plus, C18, 95 Å, 1.8 μm, 2.1 × 50 mm, Agilent Technologies) followed by MS-detection (Agilent 6470 Triple Quad LCMS). Quantification was performed against a liquid standard solution of DNSH in acetonitrile (50 ng mL−1).
The mass spectra and retention data of acrolein were identified with its reference compound. For quantification, its own response factor was used. The limit of quantitation (LOQ) was ≤1 μg m−3.
Adsorbent media, analytical methods and sampling parameters chosen are also summarized in ESI, Section S4†.
4.3 Experimental set-up
:1 (0.825 L min−1:0.275 L min−1). Before the air flow passed the sampling sorbent tube, the smaller fraction was diluted with nitrogen (N2; 5000 mL min−1). For verification of the method, the test was also carried out with a toluene permeation tube (G-Cal, VICI Metronics) as a reference. The permeation rate of toluene was experimentally determined by the manufacturer and given with 2590 ± 648 ng min−1 at a temperature of 60 °C (333.15 K). The permeation rate qd1 = 992 ng min−1 of toluene at 45 °C was calculated according to empirical eqn (1):85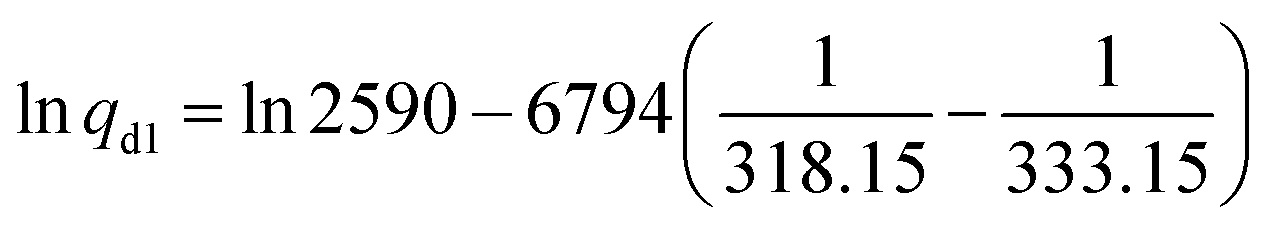 | (1) |
Additionally, the permeation rate was checked by measuring the weight loss of the permeation tubes over the experimental period. The target concentration [μg m−3] of each substance in the gas flow was calculated by considering the permeation rate given by the manufacturer and calculated on the weight loss [ng min−1], the carrier gas flow [mL min−1] and the split ratio [1:3]. The test lasted 28 days. For safety reasons, the experiments were performed under an exhaust hood.
The test method was repeated in a second test series in order to verify if contaminants in the chamber air might influence the results due to chemical interactions. Therefore, the 24 m3 chamber was loaded with upholstered furniture and furnishings. The loading factor was not determined. The 1 m3 test chamber was loaded with different materials (i.e. leather, foam, wood-based materials, and bamboo) with a loading factor of 4.8 m2 m−3.
Environmental conditions in the test chambers during both test series were set as follows: 23 °C ± 1 °C temperature, 50% ± 3% relative humidity and an air exchange rate of 1 h−1 (1 m3 test chamber) and 0.5 h−1 (24 m3 test chamber), respectively (see also the ESI, Table S4†).
The background value was determined before each experiment. If this value was above the LOQ, the measurement data were corrected accordingly. In order to avoid memory effects, the respective chamber was heated to 80 °C for 12–16 h after each experiment.
The experiments were performed under conditions which are representative for the indoor environment (23 °C, ∼50% RH). As can be seen in Tables 1 and 2, cooking is a major emission source of acrolein indoors. Moreover, many cooking processes cause a relative humidity higher than 50%. The potential problem of water retention by solid sorbents has been addressed by several studies with a main focus on carbon molecular sieves (CMS) which are known to retain water due to their high sorbent strength.86–89 Thus, water management options are often needed when using CMS for indoor air sampling.90 Carbograph™ 5TD as graphitized carbon black (GCB) is classified as a hydrophobic adsorbent medium by different manufacturers. It is also used together with Tenax® TA as a hydrophobic combination for solid sorbent sampling.90 Richter et al.91 determined recovery rates of 85 ± 14% and 116 ± 8% under dry (0% RH) and humid (50% RH) sampling conditions, respectively. The water adsorption capacities of different solid sorbents were comprehensively examined by Helmig and Vierling89 including different types of Tenax, Carbotrap and Carbosieve® S-III as well as Carboxen® 569. Regarding the two CMS Carbosieve® S-III and Carboxen® 569, they proposed several steps to reduce the water uptake during sampling. However, the Carbotrap types (20/40 mesh), which are comparable to Carbograph™ 5TD, showed no water uptake rates so they were recommended for air sampling even at high humidities. Fastyn et al.88 also come to the conclusion that the water uptake of Carbograph™ 5TD is low in comparison to other sorbents.
| Country, location | T [°C]/RH [%] | N | C [μg m−3] | References | |
|---|---|---|---|---|---|
| a Conversion of ppb to μg m−3 at T = 23 °C. | |||||
| California, Oakland-San Francisco Bay area | Outdoor | — | 12 | 0.032–0.100 | Destaillats et al.46 |
| California, non-smoking residences | Outdoor | 12–25/49–100 | — | 0.09–1.7 | Seaman et al.41 |
| Indoor | 17–22/52–79 | — | 2.1–12.2 | Seaman et al.41 | |
| California, coastal | Outdoor | — | 20 | 0.041–0.13 | Cahill47 |
| California, remote | Outdoor | — | 20 | 0.041–0.11 | Cahill47 |
| California, urban | Outdoor | — | 14 | 0.101 | Cahill47 |
| California, urban, LA Basin | Outdoor | — | 8 | 0.32 | Cahill47 |
| California, single-family residences, summer | Indoor | — | 1 | 1.3 (0.57 ppb) | Lunderberg et al.50 |
| California, single-family residences, winter | Indoor | — | 2 | 0.8–1.0 (0.36–0.44 ppb) | Lunderberg et al.50 |
| United states, urban areas and private homes | Outdoor | — | 353 | 0.46 | Liu et al.48 |
| Indoor | — | 353 | 0.59 | Liu et al.48 | |
| Canada | Indoor | — | 59 | 1 ± 1 | Gilbert et al.51 |
| Portugal | Outdoor | 25 (298 K)/— | 51 | 0.2–2.3 (0.1–1.0 ppb) | Cerqueira et al.49 |
| UK, Northern London, Mall, non-smoking | Indoor | 20/— | — | 0.5 ± 1.1 (0.2 ± 0.5 ppb) | Williams et al.53 |
| UK, Northern London, student lounge | Indoor | 20/— | — | 6.2 ± 2.5 (2.7 ± 1.1 ppb) | Williams et al.53 |
| Germany, prefabricated wooden house, naturally ventilated, after shell completion | Indoor | 15–16/67–71 | 8 | 8.6 ± 6.3 | Schieweck40 |
| Germany, prefabricated wooden house, naturally ventilated, at delivery and in use | Indoor | 23–26/58–63 | 15 | 18.3 ± 13.7 | Schieweck40 |
| Germany, prefabricated wooden house, mechanically ventilated, at delivery | Indoor | 20–25/31–56 | 8 | 5.1 ± 2.0 | Schieweck40 |
| China, temple during incense burning | Outdoor | — | — | 75 (32.5 ppbv) | Ho and Yu28 |
| Indoor | — | — | 63–214 (27.3–92.9 ppbv) | Ho and Yu28 | |
| China, home during incense burning | Indoor | — | — | 39 (16.8 ppbv) | Ho and Yu28 |
| China, general Chinese restaurants (large, medium and small sized), exhaust | Indoor | 21.0–36.6/38.5–62.1 | 9 | ∼15.7–800.5 (6.8–347 ppb)a | Ho et al.54 |
| China, Sichuan spicy food, exhaust | Indoor | 43.0/36.3 | 2 | ∼387.6–567.5 (168–246 ppb)a | Ho et al.54 |
| China, HK style fast food, exhaust | Indoor | 29.3/46.0 | 2 | ∼58.8–100.1 (25.5–43.4 ppb)a | Ho et al.54 |
| China, Chinese BBQ manufacturer, exhaust | Indoor | 24.4–33.0/43.7–82.0 | 4 | ∼7.4–218.9 (3.2–94.9 ppb)a | Ho et al.54 |
| China, Korean BBQ, exhaust | Indoor | 34.4/64.4 | 2 | ∼148.3–168.2 (64.3–72.9 ppb)a | Ho et al.54 |
| China, Western fast-food shops, exhaust | Indoor | 24.0–32.2/28.5–67.9 | 12 | ∼21.9–205.1 (9.5–88.9 ppb)a | Ho et al.54 |
| China, Western Restaurant, exhaust | Indoor | 22.2/51.0 | 2 | ∼107.3–111.9 (46.5–48.5 ppb)a | Ho et al.54 |
| Finland, restaurant kitchens and bakeries | Indoor | 180–300 °C cooking temperature | 10 | 10–590 | Vainiotalo and Matveinen31 |
| Norway, restaurant kitchens | Indoor | — | 44 | 10 ± 10 | Svendsen et al.29 |
| Sorbent type/media | Derivatization agent | Analytical technique | Column | Minimum detection limit (MDL) | Sampling volume (flow rate) | Reference |
|---|---|---|---|---|---|---|
| Mist chamber/bisulfite solution (0.1 M) | Hydrogen peroxide, pentafluorobenzylhydroxyl amine (PFBHA), sulfuric acid | GC/MS | — | — | 190 L (19 L min−1) | Seaman et al.30,41,68 |
| DB-5 (30 m, 0.25 mm, 0.25 μm) | — | — | Cahill and Okamoto25 | |||
| — | 41 ng m−3 | 200 L (10 L min−1) | Cahill47 | |||
| Tedlar bags | — | GC/MS and GC/FID | Supel-Q Plot column (30 m, 0.53 mm) | — | 0.5 L | Fullana et al.42 |
| Impinger | Hydrogen peroxide, pentafluorobenzylhydroxyl amine (PFBHA), 20 mL (0.25 mg mL−1) | GC/MS | DB-XLB HRGC (30 m, 0.25 mm, 0.25 μm) | 0.91 ± 0.26 ng m−3 | 1.8–4 L (0.45–1 L min−1) | Destaillats et al.46 |
| Silica gel | 2,4-Dinitrophenylhydrazine (DNPH) | HPLC/UV | C18 reverse phase | — | — | Svendsen et al.29 |
| Supelcosil LC-18 (250 mm, 4.6 mm, 5 μm) | — | - (1 L min-1) | Cerqueira et al.49 | |||
| ChromSep OmniSpher 5, C18 (150 mm × 5.6 μm) | — | — | Gilbert et al.51 | |||
| — | — | 30 L (0.5 L min−1) | Wang and Morrison103 according to TO-11A104 | |||
| Novapak C18 (150 mm, 3.9 mm, 4 μm) | — | 5–10 L (50 mL min−1) | Machado Corrêa and Arbilla23 | |||
| C18 reverse phase | — | 60–120 L | Jiang and Zhang105 according to TO-11A104 | |||
| — | — | — | Ruprecht et al.37 according to TO-11A104 | |||
| LC-DAD-MS | Xterra MS C18 (250 mm, 2.1 mm, 5 μm) | 5 mL | da Silva and Pereira43 | |||
| Impinger | 2,4-Dinitrophenylhydrazine (DNPH), solution (10 mL) | HPLC/UV | L1, C18 (250 mm, 4.6 mm, 5 μm) | — | Max. 80 L (0.5 L min-1) | Williams et al.53 according to TO-569 |
| x-Terra MS column (250 mm, 2.1 mm, 5 μm) | — | — | Guarieiro et al.24 | |||
| Solid/personal aldehydes and ketones sampler (PAKS) | Silica-based bonded C18 packing material, coated with DNSH | HPLC-fluorescence | Waters Nova-Pak C18 column (3.9 × 150 mm) | 26 pg | — | Zhang et al.60 |
| 0.25 μg m−3 | Herrington et al.61 | |||||
| Tenax® TA/Carbosieve® S-III | — | TD-GC/MS | — | — | Max. 3 L (5 mL min−1) | Singer et al.,79 according to TO-1106 |
| Tenax® TA | Pentafluorophenyl hydrazine (PFPH) | TD-GC/MS | HP-5 MS (30 m, 0.25 mm, 0.25 μm) | LOD: ∼0.8 μg m−3 (<0.3 ppb) | 0.72–3.89 L (20–30 mL min−1) | Ho and Yu64 |
| LOD: ∼13.2–3.2 μg m−3 (5.7–1.4 ppb) | 0.3–1.2 L (20 mL min−1) | Ho et al.54 | ||||
| Pentafluorobenzylhydroxyl amine (PFBHA) | PFPH, LOD: ∼3 μg m−3 (1.3 ppb) and ∼1.6 μg m−3 (0.7 ppb) | 1.5 L and 3 L (20 mL min−1) | Ho et al.54 | |||
| Solid phase microextraction (SPME, 85 μm polyacrylate) | 2,4-Dinitrophenylhydrazine (DNPH) | GC/MS | HP-5 MS (30 m, 0.25 mm, 0.25 μm) | — | — | Osório and de Lourdes Cardeal 78 |
| Solid phase microextraction (SPME, 65 μm polydimethylsiloxane-divinylbenzene) | Pentafluorophenyl hydrazine (PFPH) | GC/MS | 5% diphenyl, 95% dimethylpolysiloxane (30 m, 0.25 mm, 0.25 μm) | — | — | Dias et al.67 |
| Headspace | — | HS-GC/TID | DB-1 (30 m, 0.25 mm) | — | — | Umano and Shibamoto32 |
| HS-GC/FID | DB-5 or WAX fused silica capillary columns (18 m, 0.18 mm) | — | — | Endo et al.39 | ||
| Headspace/needle trap microextraction (NTME) | — | GC/MS | DB-624 ultra inert (60 m, 0.25 mm, 1.4 μm) | — | — | Lomonaco et al.27 |
| Carbograph™ 5TD | — | TD-GC/MS | DB 624 (60 m, 0.32 mm, 1.8 μm) | LOQ: 0.4 μg m−3 | 4 L (0.125 mL min−1) | This work |
| LOQ: 0.3 μg m−3 | 6 L (0.125 mL min−1) | |||||
| PTR-QMS (scrubber) | LOD: ∼2 ppb | Knighton et al.66 | ||||
| PTR-TOF-MS | LOD: 21 ppt | Graus et al.65 |
5. Results and discussion
5.1 Recovery of acrolein on the solid sorbent
As shown in Fig. 2 and supported by the measurement data, a constant target concentration in the carrier gas flow was achieved after approx. 14 days. Mean values obtained by active air sampling on Carbograph™ 5TD and Tenax® TA, respectively, were 20 ± 1 μg m−3 and 7 ± 1 μg m−3. The permeation rates of acrolein and toluene, the calculated target concentrations and the measurement data (mean ± standard deviation) from active sampling on Carbograph™ 5TD and Tenax® TA are summarized in the ESI, Table S6†. The target concentrations for acrolein calculated on the basis of the manufacturer information and on the basis of the weight differed to a small extent of ∼9.4% (24 μg m−3vs. 22 μg m−3). Measurement data obtained by active sampling on Carbograph™ 5TD were in the same range (20 ± 2 μg m−3) and quite good considering the tolerance given for the permeation rate (578 ± 29 ng min−1). Therefore, the findings show that the recovery of acrolein on Carbograph™ 5TD is good and reproducible with a low standard deviation. The recovery of acrolein on Tenax® TA was poor and a factor of 3 lower (7 ± 1 μg m−3) than on Carbograph™ 5TD.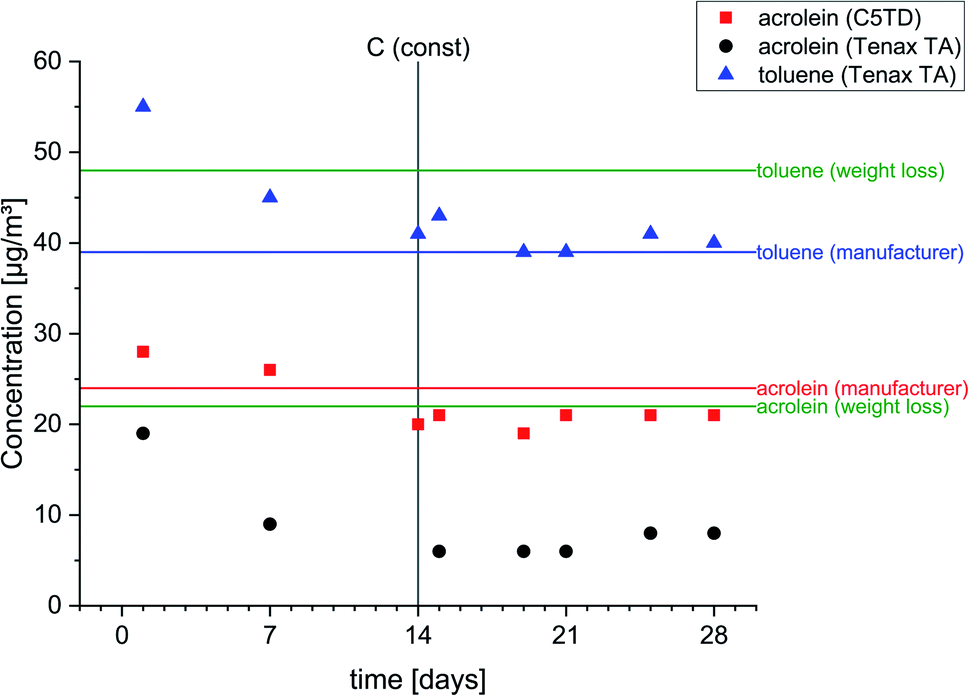 | ||
| Fig. 2 Concentrations of acrolein and toluene obtained by permeation tubes and active air sampling on Carbograph™ 5TD (C5TD) and Tenax® TA. | ||
However, the obtained data on Tenax® TA for toluene (mean value: 40 ± 1 μg m−3) were in the range of the target concentration based on the calculated permeation rate according to the manufacturer information (39 μg m−3), but differed from the permeation rate calculated using the weight loss (48 μg m−3). The difference between both permeation rates might be explained by a less exact weight loss measurement. In general, it also cannot be excluded that the permeation rate might be influenced by external parameters during the experiment due to different materials and designs of the permeation tubes.
5.2 Recovery of acrolein concentrations dosed into chamber air
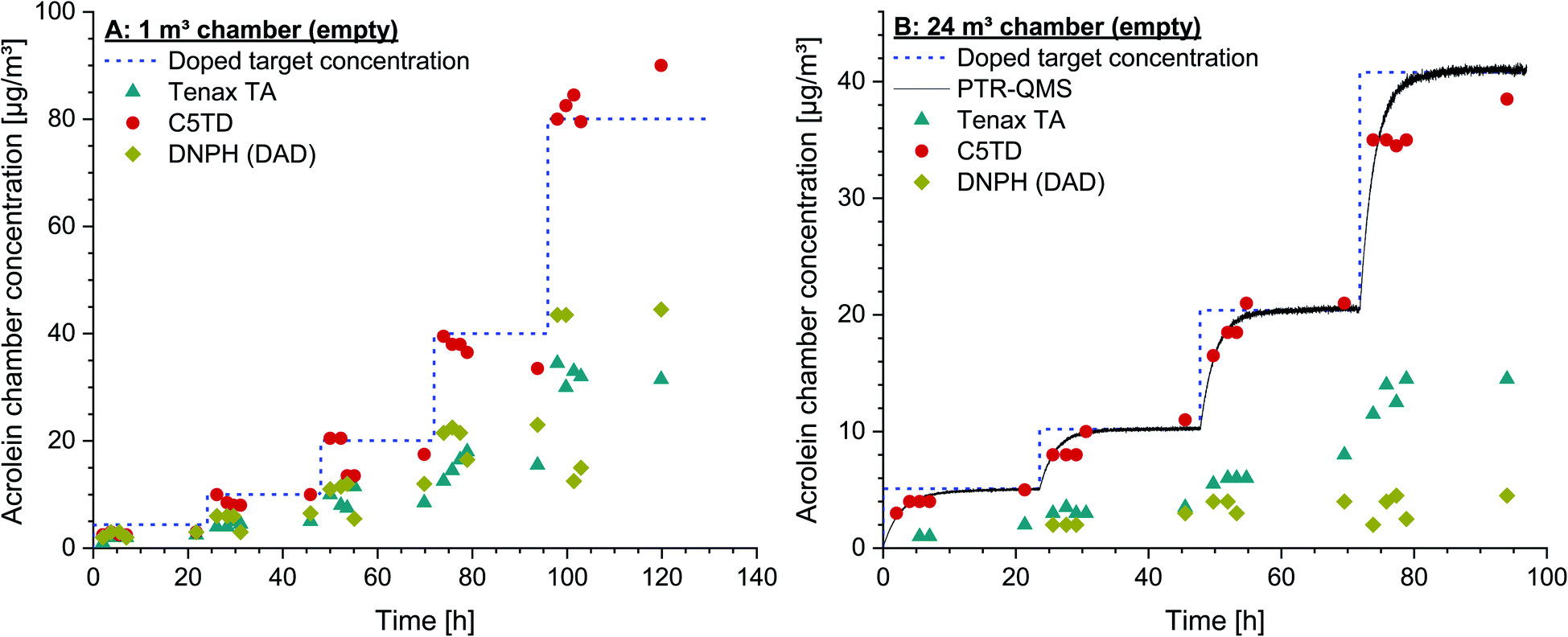 | ||
| Fig. 3 (A) Acrolein target concentrations in chamber air of an empty 1 m3 glass test chamber. Concentration range 4.4–80.1 μg m−3. Measurement data obtained by active air sampling on Carbograph™ 5TD (C5TD), Tenax® TA and DNPH (analysis by HPLC/DAD). (B) Dosed acrolein target concentrations in chamber air of an empty 24 m3 stainless steel test chamber. Concentration range 5.1–40.8 μg m−3. Measurement data obtained by active air sampling on Carbograph™ 5TD (C5TD), Tenax® TA and DNPH (analysis by HPLC/DAD). Continuous online-monitoring by PTR-QMS. | ||
The poor recovery on Tenax® TA was again underlined by the chamber test results. Both the data obtained by sampling on Tenax® TA and on DNPH-cartridges showed a broad variation and no correlation with the dosed acrolein concentrations. Analysing the cartridges by MS-detection did not lead to better results. In contrast, a reliable quantification was not possible due to the fact that the formed peaks could neither be exactly identified nor their formation was reproducible. By applying DNSH-cartridges, the di-derivatized acrolein (dimer) with a molecular mass of 568.71 g mol−1, as recommended for quantification by Herrington and co-workers,61 could not be detected in the present study. The areas of the two stereoisomers were summed for quantification, but did not lead to reliable results. Also, calibration was not possible, as neither a reproducible peak distribution nor a reproducible peak ratio were obtained. The differences between the findings achieved in this study and those published might be due to the different analytical set-up. Herrington et al.61 used tube-type diffusive samplers, which were analyzed by HPLC and subsequent fluorescence detection. Moreover, their HPLC system was equipped with a different analytical C18 column (3.9 × 300 mm, 60 Å, 4 μm). The set-up of the system was not modified to that used by Herrington et al.61,62 as the DNSH-method was tried to be included into daily routine work according to ISO 16000-3,55 which is even more difficult as one analytical run lasts 80 min. Due to these analytical difficulties, analysis of both DNPH-cartridges and DNSH-cartridges by HPLC/MS was not possible so that no results are included in the figures.
In order to check the reproducibility of the results, the experiment in the 1 m3 glass chamber was repeated by using a new test gas with the same specifications and from the same manufacturer. The same concentrations could also be set with this new test gas under identical conditions, which indicates a high degree of accuracy of the given specifications. The results of the C5TD analyses were also completely analogous to the data shown in Fig. 3A.
A further test series was carried out in an empty 1 m3 glass chamber to examine the recovery of low acrolein concentrations (0.5–5 μg m−3). In Fig. 4 the acrolein concentrations measured after active sampling on Carbograph™ 5TD are plotted versus the dosed target concentration. The data were corrected for the background concentration in the empty chamber (duplicate measurements, 0.83 ± 0.02 μg m−3). The figure visualizes a linear dependency between the measurement data and the target concentrations. The respective data and standard deviations are provided in the figure captions.
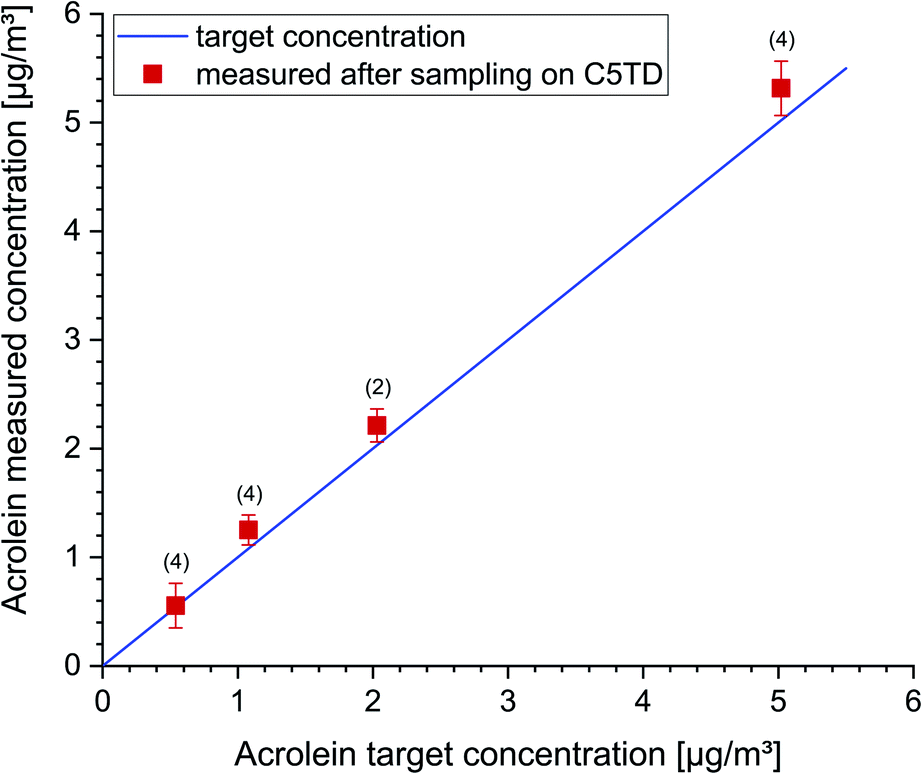 | ||
| Fig. 4 Acrolein target concentrations in the air of an empty 1 m3 glass test chamber versus acrolein concentration measured after active sampling on Carbograph™ 5TD (C5TD). The whiskers show the respective standard deviations, the numbers in brackets indicate the number of repeated measurements. Individual data (target/measured in μg m−3): 0.54/0.56 ± 0.21; 1.08/1.25 ± 0.14; 2.03/2.21 ± 0.15; 5.02/5.31 ± 0.25. | ||
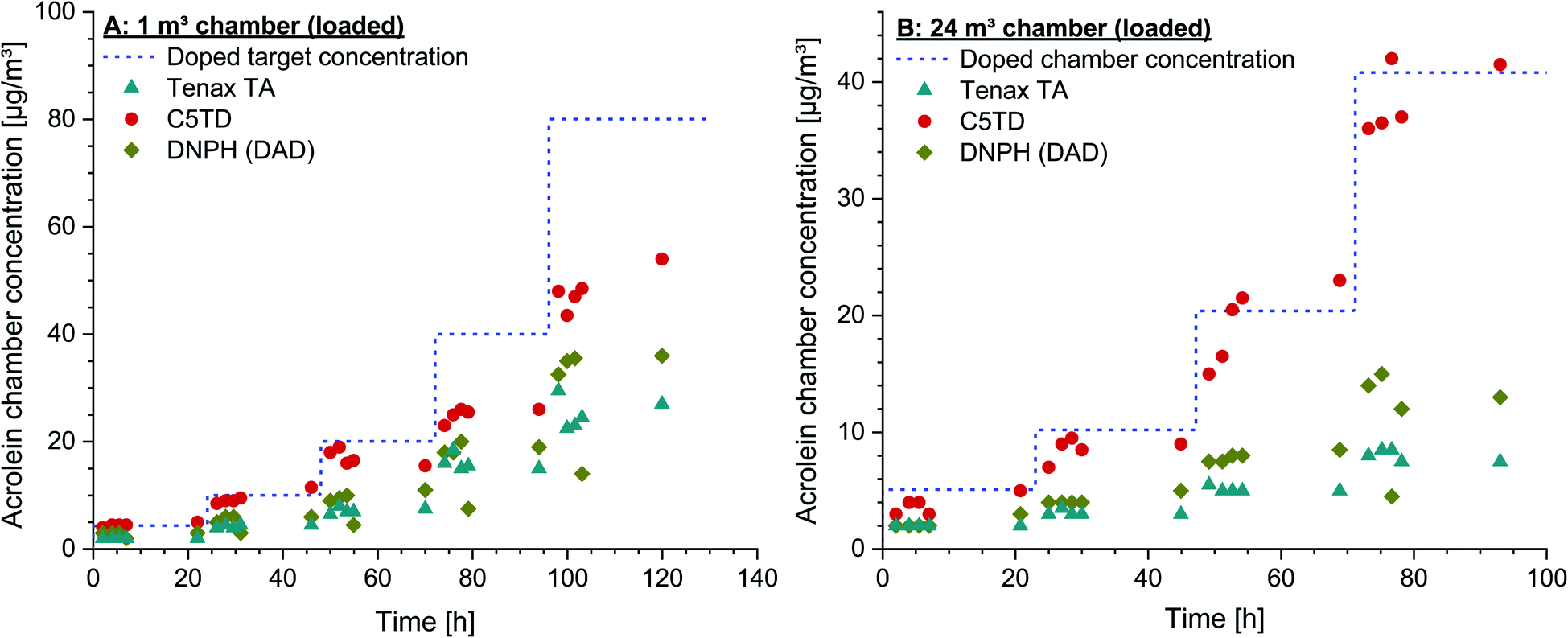 | ||
| Fig. 5 (A) Acrolein target concentrations in the air of a 1 m3 glass test chamber, loaded with indoor materials. The concentration range is 4.4–80.1 μg m−3. Measurement data obtained by active air sampling on Carbograph™ 5TD (C5TD), Tenax® TA and DNPH (analysis by HPLC/DAD). (B) Dosed acrolein target concentrations in chamber air of a 24 m3 stainless steel test chamber, loaded with furniture. Concentration range 5.1–40.8 μg m−3. Measurement data are obtained by active air sampling on Carbograph™ 5TD (C5TD), Tenax® TA and DNPH (analysis by HPLC/DAD). | ||
5.3 Monitoring of acrolein by PTR-QMS
The experimental design was also used to examine the suitability of the PTR-QMS for indoor air measurements. The mass of the mainly occurring product ion C3H5O+ is m/z 57.033,92 while the low-resolution quadrupole only allows the setting of the nominal mass m/z 57. Zhao and Zhang81 published a reaction rate constant of k = 3.35 × 10−9 cm3 s−1 for acrolein and estimated an uncertainty of ±15%. They used the theoretical approach of Su and Bowers,93 which is based on the polarizability α and the dipole moment μD of the respective compound. However, Zhao and Zhang81 used αcis = 6.15 × 10−9 cm3 and μD,cis = 2.636 D for the cis conformation of acrolein, which is significantly more unstable at room temperature than that of the trans conformation.92,94 Both parameters α and μD are significantly higher for the trans conformer.95,96 Cappellin et al.97 used the values αcis = 6.48 × 10−9 cm3 and μD,trans = 3.41 D to calculate reaction constants between 3.20 × 10−9 cm3 s−1 and 3.85 × 10−9 cm3 s−1 for different settings of their PTR-TOF-MS.The PTR-QMS experiments took place in the 24 m3 chamber. If only acrolein was dosed (empty chamber), the theoretical curve could be accurately fitted to the PTR-QMS signal (see Fig. 3B). The calibration was carried out using the steady-state concentrations; the reaction constant determined according to eqn (2) was k = 2.95 × 10−9 cm3 s−1.
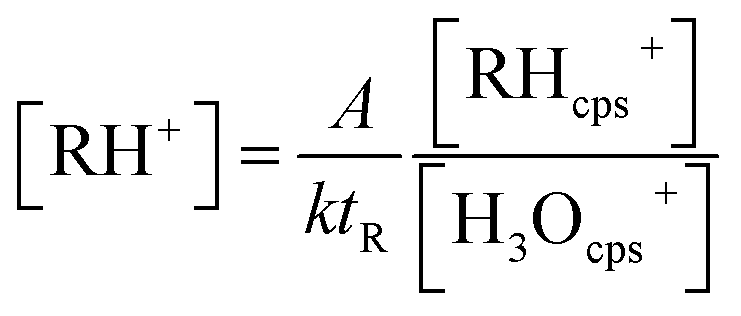 | (2) |
[RH+] is the concentration in μg m−3, k is the reaction rate constant, tR is the reaction time, [RHcps+] and [H3Ocps+] are the ion signals in counts per second and A is a system specific parameter, which involves the pressure in the drift tube, the temperature, the ion transmission coefficients and factors for the conversion from ppb to μg m−3 (see the ESI, Section S3,† for details).
This experimentally determined k for acrolein is at the lower end but still in good agreement with previously reported data. Taking into account the experimental uncertainties, a reaction constant of 3–4 × 10−9 cm3 s−1 appears to be sufficiently accurate for the standard applications of PTR-QMS.
In the loaded chamber, however, there were clear positive deviations from the respective concentration (not shown). Although there are few substances like butene and its isomers with an M + 1 mass m/z 57, it can be assumed that fragments of propionic acid, 1-butanol or alkyl residues interfere with the signal for acrolein.98,99 For this reason, the PTR-QMS technology appears to be hardly suitable for monitoring acrolein in indoor air. Therefore, Knighton et al.66 developed a scrubber system specifically for the detection of acrolein by PTR-QMS. They reported a reaction constant of k = 4.2 × 10−9 cm3 s−1, which is applicable for their particular instrument. However, a PTR-TOF-MS, as used by Lunderberg et al.50 is much better suited to correct for interferences and, thus, for detecting acrolein in real indoor environments.
6. Conclusions and outlook
The analytical method described here enables the selective determination of low acrolein concentrations in the air of test chambers. Other VVOCs and VOCs do not interfere, so that the method is also suitable for the analysis of indoor air. No breakthrough occurs with a total sampling volume of 4 L and an air flow rate of 125 mL min−1. Storing of sampled C5TD tubes under laboratory conditions over a time period of 7 days is possible without analyte loss, even at a higher sampling volume of 10 L. A limit of quantitation (LOQ) of 0.4 μg m−3 could be achieved for the low concentration range when calibrating with a liquid standard solution for a sampling volume of 4 L and can be lowered to a LOQ of 0.3 μg m−3 when sampling 6 L. Even though the liquid analytical grade standard of acrolein was discontinued in Europe, acrolein dissolved in methanol can be used for analytical purposes in future. The DNPH-method according to ISO 16000-355 with sampling on cartridges showed the previously reported problems, making its application for acrolein analysis impossible, even with detection by mass spectrometry. Trapping the target substance on DNSH-coated silica gel cartridges was tested as an alternative approach, but without success under routine conditions. In order to obtain a method which can be integrated into daily routine, an existing HPLC system was used, whose settings complied with ISO 16000-3.55 The preparation of DNSH-cartridges was sophisticated and the restrictions during sampling might be unacceptable when using a heavier weight sorbent bed. Moreover, not all of the three chromatographic peaks (acrolein-DNSH-stereoisomers and acrolein-DNSH-dimer) which are described in the literature58,61 could be detected. Due to interfering substances, the PTR-QMS technology at m/z 57 appears to be hardly suitable for monitoring acrolein in indoor environments. PTR-TOF-MS has been shown to be better suited to overcome interference problems.50
In summary, collecting airborne acrolein on GCB Carbograph™ 5TD followed by TD-GC/MS has proven to be a promising approach regarding discontinuous sampling and analysis. It offers a fast, easy-to-handle and robust solution for routine analyses and indoor surveys in the relevant concentration range.19 The recovery of acrolein on Carbograph™ 5TD is good and reproducible with a low standard deviation. As even on freshly packed and conditioned tubes acrolein can be found in trace amounts, it is strongly recommended to document carefully the contamination substances and their amounts detected by the first analysis of a fresh tube. The method is suitable for assessing indoor air quality with regard to the acute and chronic Reference Exposure Limits (RELs) published by OEHHA.8 However, assessment using the U.S. EPA Reference Concentration for Inhalation Exposure of 2 × 10−5 mg m−3100 is beyond the scope of this method. In general, such a low detection limit in the range of 0.02 μg m−3 can only be achieved with specially designed methods,46 which, however, are hardly suitable for routine measurements. This underlines the impracticability of non-accessible guide values as discussed before.45,101
Author contributions
The authors designed and planned the test series presented in this manuscript. All authors read and approved the final manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors are grateful to C. Fauck, S. Kaps, N. Schulz and S. Wientzek (Fraunhofer WKI) for technical support during the test series. We are also grateful to J. Gunschera (Fraunhofer WKI) for providing unpublished data.References
- C. E. Blom, G. Grassi and A. Bauder, Molecular structure of s-cis- and s-trans-acrolein determined by microwave spectroscopy, J. Am. Chem. Soc., 1984, 106, 7427–7431, DOI:10.1021/ja00336a022.
- A. Moghe, S. Ghare, B. Lamoreau, M. Mohammad, S. Barve, C. McClain and S. Joshi-Barve, Molecular mechanisms of acrolein toxicity: relevance to human disease, Toxicol. Sci., 2015, 143, 242–255, DOI:10.1093/toxsci/kfu233.
- A.-S. Claeson and N. Lind, Human exposure to acrolein: time-dependence and individual variation in eye irritation, Environ. Toxicol. Pharmacol., 2016, 45, 20–27, DOI:10.1016/j.etap.2016.05.011.
- T. J. Woodruff, E. M. Wells, E. W. Holt, D. E. Burgin and D. A. Axelrad, Estimating risk from ambient concentrations of acrolein across the United States, Environ. Health Perspect., 2007, 115, 410–415, DOI:10.1289/ehp.9467.
- R. J. Henning, G. T. Johnson, J. P. Coyle and R. D. Harbison, Acrolein can cause cardiovascular disease: a review, Cardiovasc. Toxicol., 2017, 17, 227–236, DOI:10.1007/s12012-016-9396-5.
- K. E. McGraw, D. W. Riggs, S. Rai, A. Navas-Acien, Z. Xie, P. Lorkiewicz, J. Lynch, N. Zafar, S. Krishnasamy, K. C. Taylor, D. J. Conklin, A. P. DeFilippis, S. Srivastava and A. Bhatnagar, Exposure to volatile organic compounds – acrolein, 1,3-butadiene, and crotonaldehyde – is associated with vascular dysfunction, Environ. Res., 2021, 196, 110903, DOI:10.1016/j.envres.2021.110903.
- O. Faroon, N. Roney, J. Taylor, A. Ashizawa, M. Lumpkin and D. Plewak, Acrolein health effects, Toxicol. Ind. Health, 2008, 24, 447–490, DOI:10.1177/0748233708094188.
- Office of Environmental Health Hazard Assessment (OEHHA), Air Toxics Hot Spots Program. Risk Assessment Guidelines. Guidance Manual for Preparation of Health Risk Assessments, Air, Community, and Environmental Research Branch Office of Environmental Health Hazard Assessment, 2015 Search PubMed.
- M. Trantallidi, C. Dimitroulopoulou, P. Wolkoff, S. Kephalopoulos and P. Carrer, EPHECT III: health risk assessment of exposure to household consumer products, Sci. Total Environ., 2015, 536, 903–913, DOI:10.1016/j.scitotenv.2015.05.123.
- Health Canada, Residential Indoor Air Quality Guidelines: Acrolein, Her Majesty the Queen in Right of Canada, as Represented by the Minister of Health Ottawa, 2021 Search PubMed.
- D. Arntz, A. Fischer, M. Höpp, S. Jacobi, J. Sauer, T. Ohara, T. Sato, N. Shimizu and H. Schwind, Acrolein and methacrolein, in Ullmann’s, Encyclopedia of Industrial Chemistry, 2012, DOI:10.1002/14356007.a01_149.pub2.
- L. Liu, X. P. Ye and J. J. Bozell, A comparative review of petroleum-based and bio-based acrolein production, ChemSusChem, 2012, 5, 1162–1180, DOI:10.1002/cssc.201100447.
- H. Esterbauer, R. J. Schaur and H. Zollner, Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes, Free Radical Biol. Med., 1991, 11, 81–128, DOI:10.1016/0891-5849(91)90192-6.
- J. F. Stevens and C. S. Maier, Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease, Mol. Nutr. Food Res., 2008, 52, 7–25, DOI:10.1002/mnfr.200700412.
- D. Grosjean, Atmospheric chemistry of toxic contaminants. 3. Unsaturated aliphatics: acrolein, acrylonitrile, maleic anhydride, J. Air Waste Manage. Assoc., 1990, 40, 1664–1669, DOI:10.1080/10473289.1990.10466814.
- E. Grosjean, E. L. Williams and D. Grosjean, Atmospheric chemistry of acrolein, Sci. Total Environ., 1994, 153, 195–202, DOI:10.1016/0048-9697(94)90198-8.
- J. M. Logue, T. E. McKone, M. H. Sherman and B. C. Singer, Hazard assessment of chemical air contaminants measured in residences, Indoor Air, 2011, 21, 92–109, DOI:10.1111/j.1600-0668.2010.00683.x.
- T. Salthammer, Very volatile organic compounds: an understudied class of indoor air pollutants, Indoor Air, 2016, 26, 25–38, DOI:10.1111/ina.12173.
- A. Schieweck, J. Gunschera, D. Varol and T. Salthammer, Analytical procedure for the determination of very volatile organic compounds (C3-C6) in indoor air, Anal. Bioanal. Chem., 2018, 410, 3171–3183, DOI:10.1007/s00216-018-1004-z.
- ISO 16000-6, Indoor Air-Part 6: Determination of volatile organic compounds in indoor and test chamber air by active sampling on Tenax TA® sorbent, thermal desorption and gas chromatography using MS or MS-FID, Beuth Verlag, Berlin, 2012 Search PubMed.
- K. Gaeggeler, A. S. H. Prevot, J. Dommen, G. Legreid, S. Reimann and U. Baltensperger, Residential wood burning in an Alpine valley as a source for oxygenated volatile organic compounds, hydrocarbons and organic acids, Atmos. Environ., 2008, 42, 8278–8287, DOI:10.1016/j.atmosenv.2008.07.038.
- B. Languille, V. Gros, J.-E. Petit, C. Honoré, A. Baudic, O. Perrussel, G. Foret, V. Michoud, F. Truong, N. Bonnaire, R. Sarda-Estève, M. Delmotte, A. Feron, F. Maisonneuve, C. Gaimoz, P. Formenti, S. Kotthaus, M. Haeffelin and O. Favez, Wood burning: a major source of volatile organic compounds during wintertime in the Paris region, Sci. Total Environ., 2020, 711, 135055, DOI:10.1016/j.scitotenv.2019.135055.
- S. Machado Corrêa and G. Arbilla, Carbonyl emissions in diesel and biodiesel exhaust, Atmos. Environ., 2008, 42, 769–775, DOI:10.1016/j.atmosenv.2007.09.073.
- L. L. N. Guarieiro, A. F. de Souza, E. A. Torres and J. B. de Andrade, Emission profile of 18 carbonyl compounds, CO, CO2, and NOx emitted by a diesel engine fuelled with diesel and ternary blends containing diesel, ethanol and biodiesel or vegetable oils, Atmos. Environ., 2009, 43, 2754–2761, DOI:10.1016/j.atmosenv.2009.02.036.
- T. M. Cahill and R. A. Okamoto, Emissions of acrolein and other aldehydes from biodiesel-fueled heavy-duty vehicles, Environ. Sci. Technol., 2012, 46, 8382–8388, DOI:10.1021/es301659u.
- M. Paabo and B. C. Levin, A literature review of the chemical nature and toxicity of the decomposition products of polyethylenes, Fire Mater., 1987, 11, 55–70, DOI:10.1002/fam.810110203.
- T. Lomonaco, E. Manco, A. Corti, J. La Nasa, S. Ghimenti, D. Biagini, F. Di Francesco, F. Modugno, A. Ceccarini, R. Fuoco and V. Castelvetro, Release of harmful volatile organic compounds (VOCs) from photo-degraded plastic debris: a neglected source of environmental pollution, J. Hazard. Mater., 2020, 394, 122596, DOI:10.1016/j.jhazmat.2020.122596.
- S. S. H. Ho and J. Z. Yu, Concentrations of formaldehyde and other carbonyls in environments affected by incense burning, J. Environ. Monit., 2002, 4, 728–733, 10.1039/b200998f.
- K. Svendsen, H. N. Jensen, I. Sivertsen and A. K. Sjaastad, Exposure to cooking fumes in restaurant kitchens in Norway, Ann. Occup. Hyg., 2002, 46, 395–400, DOI:10.1093/annhyg/mef045.
- V. Y. Seaman, D. H. Bennett and T. M. Cahill, Indoor acrolein emission and decay rates resulting from domestic cooking events, Atmos. Environ., 2009, 43, 6199–6204, DOI:10.1016/j.atmosenv.2009.08.043.
- S. Vainiotalo and K. Matveinen, Cooking fumes as a hyienic problem in the food and catering industries, Am. Ind. Hyg. Assoc. J., 1993, 54, 376–382, DOI:10.1080/15298669391354838.
- K. Umano and T. Shibamoto, Analysis of acrolein from heated cooking oils and beef fat, J. Agric. Food Chem., 1987, 35, 909–912, DOI:10.1021/jf00078a014.
- P. G. Shields, G. X. Xu, W. J. Blot, J. F. Fraumeni Jr, G. E. Trivers, E. D. Pellizzari, Y. H. Qu, Y. T. Gao and C. C. Harris, Mutagens from heated chinese and U.S. cooking oils, JNCI, J. Natl. Cancer Inst., 1995, 87, 836–841, DOI:10.1093/jnci/87.11.836.
- C. Arata, P. K. Misztal, Y. Tian, D. M. Lunderberg, K. Kristensen, A. Novoselac, M. E. Vance, D. K. Farmer, W. W. Nazaroff and A. H. Goldstein, Volatile organic compound emissions during HOMEChem, Indoor Air, 2021, 1–19, DOI:10.1111/ina.12906.
- B. C. Singer, A. T. Hodgson, K. S. Guevarra, E. L. Hawley and W. W. Nazaroff, Gas-phase organics in environmental tobacco smoke. 1. Effects of smoking rate, ventilation, and furnishing level on emission factors, Environ. Sci. Technol., 2002, 36, 846–853, DOI:10.1021/es011058w.
- L. Cancelada, M. Sleiman, X. Tang, M. L. Russell, V. N. Montesinos, M. I. Litter, L. A. Gundel and H. Destaillats, Heated tobacco products: volatile emissions and their predicted impact on indoor air quality, Environ. Sci. Technol., 2019, 53, 7866–7876, DOI:10.1021/acs.est.9b02544.
- A. A. Ruprecht, C. De Marco, A. Saffari, P. Pozzi, R. Mazza, C. Veronese, G. Angellotti, E. Munarini, A. C. Ogliari, D. Westerdahl, S. Hasheminassab, M. M. Shafer, J. J. Schauer, J. Repace, C. Sioutas and R. Boffi, Environmental pollution and emission factors of electronic cigarettes, heat-not-burn tobacco products, and conventional cigarettes, Aerosol Sci. Technol., 2017, 51, 674–684, DOI:10.1080/02786826.2017.1300231.
- J. M. Logue, M. Sleiman, V. N. Montesinos, M. L. Russell, M. I. Litter, N. L. Benowitz, L. A. Gundel and H. Destaillats, Emissions from electronic cigarettes: assessing vapers' intake of toxic compounds, secondhand exposures, and the associated health impacts, Environ. Sci. Technol., 2017, 51, 9271–9279, DOI:10.1021/acs.est.7b00710.
- Y. Endo, C. Hayashi, T. Yamanaka, K. Takayose, M. Yamaoka, T. Tsuno and S. Nakajima, Linolenic acid as the main source of acrolein formed during heating of vegetable oils, J. Am. Oil Chem. Soc., 2013, 90, 959–964, DOI:10.1007/s11746-013-2242-z.
- A. Schieweck, Very volatile organic compounds (VVOC) as emissions from wooden materials and in indoor air of new prefabricated wooden houses, Build. Environ., 2021, 190, 107537, DOI:10.1016/j.buildenv.2020.107537.
- V. Y. Seaman, D. H. Bennett and T. M. Cahill, Origin, occurrence, and source emission rate of acrolein in residential indoor air, Environ. Sci. Technol., 2007, 41, 6940–6946, DOI:10.1021/es0707299.
- A. Fullana, A. A. Carbonell-Barrachina and S. Sidhu, Comparison of volatile aldehydes present in the cooking fumes of extra virgin olive, olive, and canola oils, J. Agric. Food Chem., 2004, 52, 5207–5214, DOI:10.1021/jf035241f.
- T. O. da Silva and P. A. d. P. Pereira, Influence of time, surface-to-volume ratio, and heating process (continuous or intermittent) on the emission rates of selected carbonyl compounds during thermal oxidation of palm and soybean oils, J. Agric. Food Chem., 2008, 56, 3129–3135, DOI:10.1021/jf0734525.
- S. Vreeke, D. H. Peyton and R. M. Strongin, Triacetin enhances levels of acrolein, formaldehyde hemiacetals, and acetaldehyde in electronic cigarette aerosols, ACS Omega, 2018, 3, 7165–7170, DOI:10.1021/acsomega.8b00842.
- T. Salthammer, J. Gu, S. Wientzek, R. Harrington and S. Thomann, Measurement and evaluation of gaseous and particulate emissions from burning scented and unscented candles, Environ. Int., 2021, 155, 106590, DOI:10.1016/j.envint.2021.106590.
- H. Destaillats, R. S. Spaulding and M. J. Charles, Ambient air measurement of acrolein and other carbonyls at the Oakland-San Francisco Bay Bridge toll plaza, Environ. Sci. Technol., 2002, 36, 2227–2235, DOI:10.1021/es011394c.
- T. M. Cahill, Ambient acrolein concentrations in coastal, remote, and urban regions in California, Environ. Sci. Technol., 2014, 48, 8507–8513, DOI:10.1021/es5014533.
- W. Liu, J. Zhang, L. Zhang, B. J. Turpin, C. P. Weisel, M. T. Morandi, T. H. Stock, S. Colome and L. R. Korn, Estimating contributions of indoor and outdoor sources to indoor carbonyl concentrations in three urban areas of the United States, Atmos. Environ., 2006, 40, 2202–2214, DOI:10.1016/j.atmosenv.2005.12.005.
- M. A. Cerqueira, C. A. Pio, P. A. Gomes, J. S. Matos and T. V. Nunes, Volatile organic compounds in rural atmospheres of central Portugal, Sci. Total Environ., 2003, 313, 49–60, DOI:10.1016/S0048-9697(03)00250-X.
- D. M. Lunderberg, P. K. Misztal, Y. Liu, C. Arata, Y. Tian, K. Kristensen, R. J. Weber, W. W. Nazaroff and A. H. Goldstein, High-resolution exposure assessment for volatile organic compounds in two California residences, Environ. Sci. Technol., 2021, 55(10), 6740–6751, DOI:10.1021/acs.est.0c08304.
- N. L. Gilbert, M. Guay, J. David Miller, S. Judek, C. C. Chan and R. E. Dales, Levels and determinants of formaldehyde, acetaldehyde, and acrolein in residential indoor air in Prince Edward Island, Canada, Environ. Res., 2005, 99, 11–17, DOI:10.1016/j.envres.2004.09.009.
- D. K. Farmer, M. E. Vance, J. P. D. Abbatt, A. Abeleira, M. R. Alves, C. Arata, E. Boedicker, S. Bourne, F. Cardoso-Saldaña, R. Corsi, P. F. DeCarlo, A. H. Goldstein, V. H. Grassian, L. Hildebrandt Ruiz, J. L. Jimenez, T. F. Kahan, E. F. Katz, J. M. Mattila, W. W. Nazaroff, A. Novoselac, R. E. O’Brien, V. W. Or, S. Patel, S. Sankhyan, P. S. Stevens, Y. Tian, M. Wade, C. Wang, S. Zhou and Y. Zhou, Overview of HOMEChem: house observations of microbial and environmental chemistry, Environ. Sci.: Processes Impacts, 2019, 21, 1280–1300, 10.1039/c9em00228f.
- I. D. Williams, D. M. Revitt and R. S. Hamilton, A comparison of carbonyl compound concentrations at urban roadside and indoor sites, Sci. Total Environ., 1996, 189–190, 475–483, DOI:10.1016/0048-9697(96)05248-5.
- S. S. H. Ho, J. Z. Yu, K. W. Chu and L. L. Yeung, Carbonyl emissions from commercial cooking sources in Hong Kong, J. Air Waste Manage. Assoc., 2006, 56, 1091–1098, DOI:10.1080/10473289.2006.10464532.
- ISO 16000-3, Indoor Air-Part 3: Determination of formaldehyde and other carbonyl compounds in indoor air and test chamber air - Active sampling method, Beuth Verlag, Berlin, 2013 Search PubMed.
- ASTM D5197-16, Standard test method for determination of formaldehyde and other carbonyl compounds in air (active sampler methodology), ASTM International, West Conshohocken, PA, 2016, DOI:10.1520/d5197-16.
- J. S. Herrington and M. D. Hays, Concerns regarding 24-h sampling for formaldehyde, acetaldehyde, and acrolein using 2,4-dinitrophenylhydrazine (DNPH)-coated solid sorbents, Atmos. Environ., 2012, 55, 179–184, DOI:10.1016/j.atmosenv.2012.02.088.
- R. Schulte-Ladbeck, R. Lindahl, J.-O. Levin and U. Karst, Characterization of chemical interferences in the determination of unsaturated aldehydes using aromatic hydrazine reagents and liquid chromatography, J. Environ. Monit., 2001, 3, 306–310, 10.1039/b101354h.
- S. S. H. Ho, K. F. Ho, W. D. Liu, S. C. Lee, W. T. Dai, J. J. Cao and H. S. S. Ip, Unsuitability of using the DNPH-coated solid sorbent cartridge for determination of airborne unsaturated carbonyls, Atmos. Environ., 2011, 45, 261–265, DOI:10.1016/j.atmosenv.2010.09.042.
- J. Zhang, L. Zhang, Z. Fan and V. Ilacqua, Development of the personal aldehydes and ketones sampler based upon DNSH derivatization on a solid sorbent, Environ. Sci. Technol., 2000, 34, 2601–2607, DOI:10.1021/es9911869.
- J. S. Herrington, L. Zhang, D. Whitaker, L. Sheldon and J. J. Zhang, Optimizing a dansylhydrazine (DNSH) based method for measuring airborne acrolein and other unsaturated carbonyls, J. Environ. Monit., 2005, 7, 969–976, 10.1039/b502063h.
- J. S. Herrington and J. J. Zhang, Development of a method for time-resolved measurement of airborne acrolein, Atmos. Environ., 2008, 42, 2429–2436, DOI:10.1016/j.atmosenv.2007.12.019.
- A. Gold, C. E. Dube and R. B. Perni, Solid sorbent for sampling acrolein in air, Anal. Chem., 1978, 50, 1839–1841, DOI:10.1021/ac50035a029.
- S. S. H. Ho and J. Z. Yu, Determination of airborne carbonyls: comparison of a thermal desorption/GC method with the standard DNPH/HPLC method, Environ. Sci. Technol., 2004, 38, 862–870, DOI:10.1021/es034795w.
- M. Graus, M. Müller and A. Hansel, High resolution PTR-TOF: quantification and formula confirmation of VOC in real time, J. Am. Soc. Mass Spectrom., 2010, 21, 1037–1044, DOI:10.1016/j.jasms.2010.02.006.
- W. B. Knighton, S. C. Herndon, J. H. Shorter, R. C. Miake-Lye, M. S. Zahniser, K. Akiyama, A. Shimono, K. Kitasaka, H. Shimajiri and K. Sugihara, Laboratory evaluation of an aldehyde scrubber system specifically for the detection of acrolein, J. Air Waste Manage. Assoc., 2007, 57, 1370–1378, DOI:10.3155/1047-3289.57.11.1370.
- C. M. Dias, H. C. Menezes and Z. L. Cardeal, Environmental and biological determination of acrolein using new cold fiber solid phase microextraction with gas chromatography mass spectrometry, Anal. Bioanal. Chem., 2017, 409, 2821–2828, DOI:10.1007/s00216-017-0226-9.
- V. Y. Seaman, M. J. Charles and T. M. Cahill, A sensitive method for the quantification of acrolein and other volatile carbonyls in ambient air, Anal. Chem., 2006, 78, 2405–2412, DOI:10.1021/ac051947s.
- U.S. Environmental Protection Agency (U.S. EPA), Method TO-5: Determination of aldehydes and ketones in ambient air using High Performance Liquid Chromatography (HPLC), in Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, Center for Environmental Research Information, Office of Research and Development, 2nd edn, 1999 Search PubMed.
- E. Grosjean, D. Grosjean, M. P. Fraser and G. R. Cass, Air quality model evaluation data for organics. 2. C1−C14 carbonyls in Los Angeles air, Environ. Sci. Technol., 1996, 30, 2687–2703, DOI:10.1021/es950758w.
- A.-P. Sirju and P. B. Shepson, Laboratory and field investigation of the DNPH cartridge technique for the measurement of atmospheric carbonyl compounds, Environ. Sci. Technol., 1995, 29, 384–392, DOI:10.1021/es00002a014.
- S. M. van Leeuwen, L. Hendriksen and U. Karst, Determination of aldehydes and ketones using derivatization with 2,4-dinitrophenylhydrazine and liquid chromatography–atmospheric pressure photoionization-mass spectrometry, J. Chromatogr. A, 2004, 1058, 107–112, DOI:10.1016/j.chroma.2004.08.149.
- R. Pal and K.-H. Kim, Experimental choices for the determination of carbonyl compounds in air, J. Sep. Sci., 2007, 30, 2708–2718, DOI:10.1002/jssc.200700206.
- S. Achatz, G. Lörinci, N. Hertkorn, I. Gebefügi and A. Kettrup, Disturbance of the determination of aldehydes and ketones: Structural elucidation of degradation products derived from the reaction of 2,4-dinitrophenylhydrazine (DNPH) with ozone, Fresenius. J. Anal. Chem., 1999, 364, 141–146, DOI:10.1007/s002160051313.
- D. R. Rodier, L. Nondek and J. W. Birks, Evaluation of ozone and water vapor interferences in the derivatization of atmospheric aldehydes with dansylhydrazine, Environ. Sci. Technol., 1993, 27, 2814–2820, DOI:10.1021/es00049a022.
- E. A. Pereira, E. Carrilho and M. F. M. Tavares, Laser-induced fluorescence and UV detection of derivatized aldehydes in air samples using capillary electrophoresis, J. Chromatogr. A, 2002, 979, 409–416, DOI:10.1016/S0021-9673(02)01258-X.
- T. M. Cahill, M. J. Charles and V. Y. Seaman, Development and Application of a Sensitive Method to Determine Concentrations of Acrolein and Other Carbonyls in Ambient Air, Report Research Report 149, Boston, Massachusetts, 2010 Search PubMed.
- V. M. Osório and Z. de Lourdes Cardeal, Determination of acrolein in french fries by solid-phase microextraction gas chromatography and mass spectrometry, J. Chromatogr. A, 2011, 1218, 3332–3336, DOI:10.1016/j.chroma.2010.11.068.
- B. C. Singer, A. T. Hodgson and W. W. Nazaroff, Gas-phase organics in environmental tobacco smoke: 2. Exposure-relevant emission factors and indirect exposures from habitual smoking, Atmos. Environ., 2003, 37, 5551–5561, DOI:10.1016/j.atmosenv.2003.07.015.
- E. P. L. Hunter and S. G. Lias, Evaluated gas phase basicities and proton affinities of molecules: an update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656, DOI:10.1063/1.556018.
- J. Zhao and R. Zhang, Proton transfer reaction rate constants between hydronium ion (H3O+) and volatile organic compounds, Atmos. Environ., 2004, 38, 2177–2185, DOI:10.1016/j.atmosenv.2004.01.019.
- J. W. Einax, H. W. Zwanziger and S. Geiß, Chemometrics in Environmental Analysis, WILEY-VCH, Weinheim, 1997 Search PubMed.
- DIN 32645, Chemical Analysis – Decision Limit, Detection Limit and Determination Limit under Repeatability Conditions – Terms, Methods, Evaluation.Beuth Verlag, Berlin, 2008 Search PubMed.
- T. Schripp, S. Etienne, C. Fauck, F. Fuhrmann, L. Märk and T. Salthammer, Application of proton-transfer-reaction-mass-spectrometry for indoor air quality research, Indoor Air, 2014, 24, 178–189, DOI:10.1111/ina.12061.
- B. Boyle, The Complete Guide to Testing Chemical Sensors, Owlstone Whitepaper Search PubMed.
- P. Fastyn, W. Kornacki, M. Kardas, J. Gawlowski and J. Niedzielski, Adsorption of water vapour from humid air in carbon molecular sieves: Carbosieve S-III and Carboxens 569, 1000 and 1001, Analyst, 2003, 128, 198–203, 10.1039/b209296d.
- M. Gawryś, P. Fastyn, J. Gawłowski, T. Gierczak and J. Niedzielski, Prevention of water vapour adsorption by carbon molecular sieves in sampling humid gases, J. Chromatogr. A, 2001, 933, 107–116, DOI:10.1016/s0021-9673(01)01250-x.
- P. Fastyn, W. Kornacki, T. Gierczak, J. Gawłowski and J. Niedzielski, Adsorption of water vapour from humid air by selected carbon adsorbents, J. Chromatogr. A, 2005, 1078, 7–12, DOI:10.1016/j.chroma.2005.04.096.
- D. Helmig and L. Vierling, Water Adsorption Capacity of the Solid Adsorbents Tenax TA, Tenax GR, Carbotrap, Carbotrap C, Carbosieve SIII, and Carboxen 569 and Water Management Techniques for the Atmospheric Sampling of Volatile Organic Trace Gases, Anal. Chem., 1995, 67, 4380–4386, DOI:10.1021/ac00119a029.
- E. Woolfenden, Sorbent-based sampling methods for volatile and semi-volatile organic compounds in air. Part 2. Sorbent selection and other aspects of optimizing air monitoring methods, J. Chromatogr. A, 2010, 1217, 2685–2694, DOI:10.1016/j.chroma.2010.01.015.
- M. Richter, E. Juritsch and O. Jann, Determination of recovery rates of adsorbents for sampling very volatile organic compounds (C1C6) in dry and humid air in the sub-ppb range by use of thermal desorption gas chromatography-mass spectrometry, J. Chromatogr. A, 2020, 1626, 461389, DOI:10.1016/j.chroma.2020.461389.
- G. R. D. Maré, Abinitio study of rotational isomerism in acrolein, Can. J. Chem., 1985, 63, 1672–1680, DOI:10.1139/v85-280.
- T. Su and M. T. Bowers, Ion-polar molecule collisions. Effect of molecular size on ion-polar molecule rate constants, J. Am. Chem. Soc., 1973, 95, 7609–7610, DOI:10.1021/ja00804a011.
- W. Li, A. Maris, C. Calabrese, I. Usabiaga, W. D. Geppert, L. Evangelisti and S. Melandri, Atmospherically relevant acrolein–water complexes: spectroscopic evidence of aldehyde hydration and oxygen atom exchange, Phys. Chem. Chem. Phys., 2019, 21, 23559–23566, 10.1039/c9cp04910j.
- N. B. Hannay and C. P. Smyth, The dipole moments and structures of ketene and of several polar molecules containing conjugated systems, J. Am. Chem. Soc., 1946, 68, 1357–1360, DOI:10.1021/ja01211a079.
- R. Wagner, J. Fine, J. W. Simmons and J. H. Goldstein, Microwave spectrum, structure, and dipole moment of s-trans acrolein, J. Chem. Phys., 1957, 26, 634–637, DOI:10.1063/1.1743359.
- L. Cappellin, T. Karl, M. Probst, O. Ismailova, P. M. Winkler, C. Soukoulis, E. Aprea, T. D. Märk, F. Gasperi and F. Biasioli, On quantitative determination of volatile organic compound concentrations using proton transfer reaction time-of-flight mass spectrometry, Environ. Sci. Technol., 2012, 46, 2283–2290, DOI:10.1021/es203985t.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Proton-transfer-reaction mass spectrometry: applications in atmospheric sciences, Chem. Rev., 2017, 117, 13187–13229, DOI:10.1021/acs.chemrev.7b00325.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, R. J. Weber, W. W. Nazaroff and A. H. Goldstein, Characterizing sources and emissions of volatile organic compounds in a northern California residence using space- and time-resolved measurements, Indoor Air, 2019, 29, 630–644, DOI:10.1111/ina.12562.
- U.S. EPA, Integrated Risk Information System (IRIS), Acrolein - chemical assessment summary. Last revised 06/03/2003, https.//iris.epa.gov/static/pdfs/0364_summary.pdf, accessed 02 June 2021 Search PubMed.
- T. Salthammer, Critical evaluation of approaches in setting indoor air quality guidelines and reference values, Chemosphere, 2011, 82, 1507–1517, DOI:10.1016/j.chemosphere.2010.11.023.
- T. Morikawa and E. Yanai, Toxic gases and smoke evolution from foam plastic building materials burning in fire environments, J. Fire Sci., 1989, 7, 131–141, DOI:10.1177/073490418900700204.
- H. Wang and G. C. Morrison, Ozone-initiated secondary emission rates of aldehydes from indoor surfaces in four homes, Environ. Sci. Technol., 2006, 40, 5263–5268, DOI:10.1021/es060080s.
- U.S. Environmental Protection Agency (U.S. EPA), Method TO-11A: Determination of formaldehyde in ambient air using adsorbent cartridge followed by High Performance Liquid Chromatography (HPLC), in, Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, Center for Environmental Research Information, Office of Research and Development, 2nd edn, 1999 Search PubMed.
- C. Jiang and P. Zhang, Indoor carbonyl compounds in an academic building in Beijing, China: concentrations and influencing factors, Front. Environ. Sci. Eng., 2012, 6, 184–194, DOI:10.1007/s11783-011-0309-3.
- U.S. Environmental Protection Agency (U.S. EPA), Method TO-1: Method for the determination of volatile organic compounds (VOCs) in ambient air using Tenax® adsorption and gas chromatography/mass spectrometry (GC/MS), in, Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, Center for Environmental Research Information, Office of Research and Development, 2nd edn, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1em00221j |
| This journal is © The Royal Society of Chemistry 2021 |