
Advances in transition metal-free deborylative transformations of gem-diborylalkanes
Woohyun
Jo
 a,
Jun Hee
Lee
*b and
Seung Hwan
Cho
*a
a,
Jun Hee
Lee
*b and
Seung Hwan
Cho
*a
aDepartment of Chemistry, Pohang University of Science and Technology, Pohang, Republic of Korea. E-mail: seungwhan@postech.ac.kr
bDepartment of Advanced Materials Chemistry, Dongguk University – Gyeongju Campus, Gyeongju, Gyeongsangbuk-do, Republic of Korea. E-mail: leejunhee@dongguk.ac.kr
First published on 19th March 2021
Abstract
Carbanions serve as key intermediates in a variety of chemical transformations. Particularly, α-borylcarbanions have received considerable attention in recent years because of their peculiar properties, including the ability of boron atom resonance to stabilise the adjacent negatively charged carbon atom. This feature article summarises recent progress in the synthetic utilisation of α-borylcarbanions, including carbon–carbon bond formation with alkyl halides, alkenes, N-heteroarenes, and carbonyls. Carbon–boron bond formation in organohalides mediated by α-borylcarbanions is also summarised.
 Woohyun Jo | Woohyun Jo received his BS degree from Pohang University of Science and Technology (POSTECH) in 2016 and PhD degree at the same university under the guidance of Prof. Seung Hwan Cho in 2021. He is now working as a postdoctoral researcher at POSTECH in the same group and simultaneously pursuing mandatory military service. His current research is focusing on the development of transition-metal-free carbon–carbon bond forming reactions using gem-diborylalkanes as alkylating reagents. |
 Jun Hee Lee | Jun Hee Lee received his BS (1995) degree in chemistry from Dongguk University and MS (1997) and PhD (2001) degrees in chemistry from Sogang University under the guidance of the late Prof. Jahyo Kang. After completing mandatory military service, he moved abroad to work with Professors Dean Toste (UC Berkeley) and Samuel J. Danishefsky (Columbia University) as a postdoctoral fellow between 2005 and 2010. After several research careers in Korea, he joined Dongguk University Gyeongju Campus as an assistant professor in 2016. His current research interest focuses on highly chemoselective reactions for the synthesis of biologically active compounds. |
 Seung Hwan Cho | Seung Hwan Cho received his BS degree from Korea Advanced Institute of Science and Technology (KAIST) in 2006 and PhD degree at the same university under the guidance of Prof. Sukbok Chang in 2011. After postdoctoral experience with Prof. John F. Hartwig at University of California, Berkeley, he joined Pohang University of Science and Technology (POSTECH) as an assistant professor in 2014, and then promoted to associate professor in 2018. His research interests are the design, synthesis and application of multi-organometallic compounds. |
1. Introduction
Since the discovery of the Grignard reagent in 1900, carbanions have occupied a central position in organic synthesis together with carbocations and free radicals. Such electron-rich species have been used as key intermediates in a diverse range of chemical transformations such as substitution, addition, elimination, and rearrangement reactions, thereby revolutionising synthetic organic chemistry. Consequently, various strategies have been devised to generate carbanions, including the cleavage of the relatively acidic C–H bond with an appropriate base, metal–halogen exchange in organohalides, transmetalation, and decarboxylation.1 However, the carbanions formed by these classical routes require strictly controlled conditions (e.g. an inert gas atmosphere, the use of Schlenk techniques, and/or cryogenic conditions) when reacting with electrophiles due to their unstable nature.2Owing to their unique properties such as stability and reactivity, α-borylcarbanions that bear an organoboron unit adjacent to an anionic carbon atom have recently received considerable attention from the synthetic chemistry community.3 The properties of α-borylcarbanions can be rationalised by their resonance stabilisation, which originates from the partial filling of the vacant p-orbital of sp2-hybridised boron with the electron pair from the adjacent negatively charged carbon, thus enabling various electrophiles to react under relatively mild conditions (Scheme 1a). Typically, α-borylcarbanions have been generated by the deprotonation of the α-C–H bond of alkylborons with a sterically hindered amide base [e.g. lithium diisopropylamide (LDA) and lithium 2,2,6,6-tetramethylpiperidide (LiTMP)],4 by the transmetalation of α-stannyl5 boronate esters, or by the lithium–halogen exchange of α-haloborons (Scheme 1b).5,6 While these procedures offer routes to generate α-borylcarbanions, they still suffer from several limitations, including harsh conditions, low functional group compatibility, and narrow substrate scope. In this context, the base-promoted deborylation of gem-diborylalkanes has emerged as a superb alternative to conventional methods for gaining access to α-borylcarbanions.7 The easy accessibility of gem-diborylalkanes from readily available starting materials renders this strategy an attractive approach to generate α-borylcarbanions.
This feature article focuses on recent advances in the usage of α-borylcarbanions, mainly those generated from gem-diborylalkanes in the presence of a base, for the selective forging of carbon–carbon bonds. We also cover carbon–boron bond formation in aryl halides with gem-diborylalkanes under transition metal-free conditions as α-borylcarbanions play a pivotal role in these reactions.
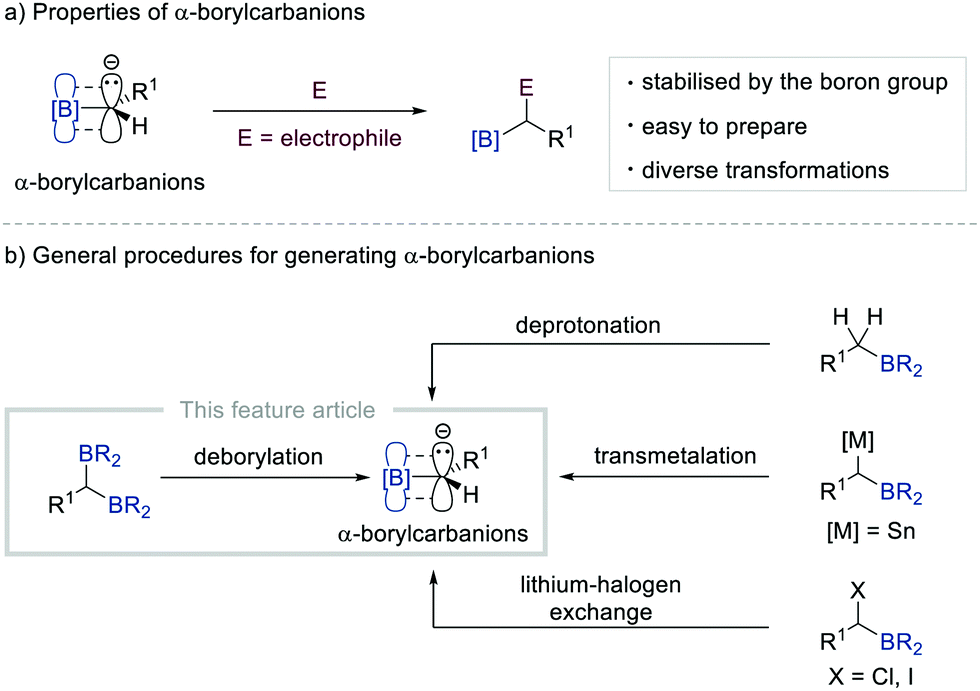 | ||
| Scheme 1 Approaches for the generation of α-borylcarbanions. | ||
2. Carbon–carbon bond formation
2.1 Generation of α-borylcarbanions containing alkyl-substituted borane moieties and their applications
Brown and Zweifel first reported the generation of α-borylcarbanions from gem-diborylalkanes in 1961 (Scheme 2).8 They found that gem-diborylalkanes 1, synthesised via the double hydroboration of terminal alkynes with diborane (B2H6), could be activated chemoselectively in the presence of NaOH as a base. The subsequent dissociation of the resulting boronate species gave α-borylcarbanions 2, which were then trapped by H2O as a proton source. The oxidation of the remaining borane by H2O2 afforded aliphatic alcohols 3. Subsequently, Cainelli and Mukaiyama reported that one of the borane groups in gem-diborylalkanes could be selectively removed by employing alkyllithium as an activator to provide α-borylcarbanions 2. These α-borylcarbanions could be captured by CO2, carbonyls, alkyl halides, and esters to provide access to an assortment of useful building blocks such as gem-dicarboxylic acids (4),9 olefins (5),10 secondary carbinols (6),11 alkyl(aryl)ketones (7),12 and trans-α-alkyl-β-hydroxyketones (8).12 However, all of these studies used gem-diborylalkanes possessing alkyl-substituted borane moieties, thereby limiting their synthetic applicability because of their narrow substrate scope and instability during column chromatography.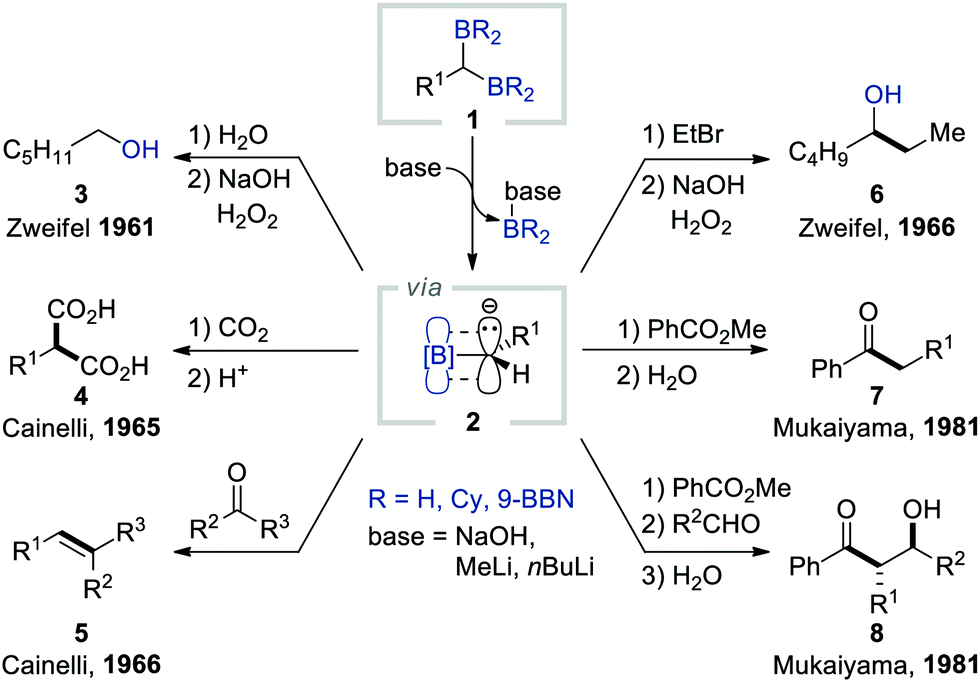 | ||
| Scheme 2 Early examples of the deborylative functionalisation of gem-diborylalkanes. | ||
2.2 Alkylation of alkyl halides with α-[(pinacolato)boryl]alkylanions
α-Borylcarbanion chemistry has experienced a renaissance since 2014 due to the development of many useful technologies for synthesising gem-diborylalkanes with a pinacolato group as a boron substituent, which are more stable than gem-diborylalkanes possessing alkyl-substituted borane moieties. Representative synthetic approaches include the hydroboration of alkynes and alkenes with pinacolborane or bis(pinacolato)diboron, C–H borylation with bis(pinacolato)diboron, cross-coupling with bis(pinacolato)diboron, insertion of bis(pinacolato)diboron in diazo compounds, and the SN2 reaction of alkyl halides with [bis(pinacolato)boryl]lithium.7a–c,13During an investigation into the utility of quaternary gem-bis[(pinacolato)boryl]alkanes 12, prepared via the Pt-catalysed Srebnik diborylation of diazoalkanes, Kingsbury and Wammack serendipitously achieved the deborylative alkylation reaction of 12 in 2014.14 When they attempted the Matteson homologation15,16 of 9 (R1 = Me, R2 = Ph) using a cold mixture of bromochloromethane (1.0 equiv.) and n-butyllithium (1.0 equiv.) in THF, a tertiary carbinol having an n-butyl substituent instead of the anticipated homologated quaternary 1,2-diol was isolated in a decent yield of 86% after oxidative workup with alkaline H2O2. They reasoned that the deborylation of the ate species 10 derived from the complexation of 9 with chloromethyl lithium delivered a boron-stabilised carbanion 11. This scenario was confirmed by the further bimolecular alkylation of the same α-[(pinacolato)boryl]alkylanion, generated in situ from 9 (R1 = Me, R2 = Ph) by the action of a solution of MeLi, with several primary alkyl bromides such as ethyl bromide, n-butyl bromide, allyl bromide, and benzyl bromide (Scheme 3). The corresponding tertiary alkylboronate esters 14a and 14b or, when necessary, tertiary carbinols 15a and 15b were isolated after a basic peroxide workup.
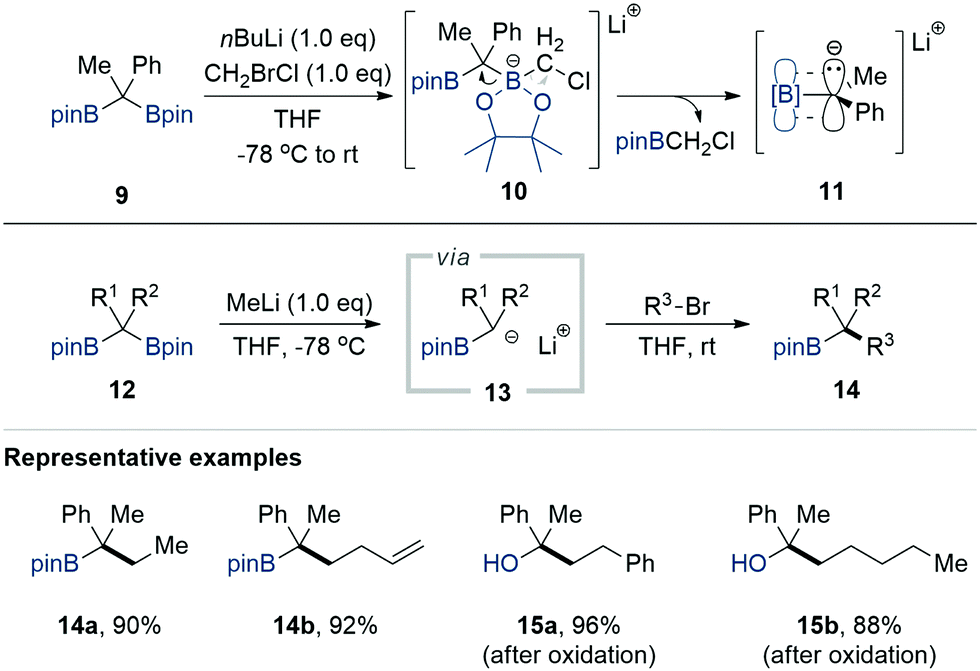 | ||
| Scheme 3 Methyllithium-mediated deborylative alkylation of gem-bis[(pinacolato)boryl]alkanes. | ||
After Kingsbury's work, Morken and co-workers independently reported an alkoxide-mediated deborylative alkylation of gem-bis[(pinacolato)boryl]alkanes that allowed the formation of carbon–carbon bonds between geminal boronates and alkyl halides, which could furnish a wide range of primary, secondary, and tertiary alkylboronate esters, including cyclopropyl derivatives 16c–d, in good to excellent yield with high chemo- and diastereoselectivity (Scheme 4).17 Notably, five-membered carbocyclic organoboronate 19, which is hardly accessible by other synthetic means, has been successfully prepared using an intramolecular version of this deborylative alkylation. Mechanistic studies have suggested α-[(pinacolato)boryl]alkylanion as an intermediate that can be generated in situ upon the treatment of 12 with NaOtBu via the selective mono-deborylation of an ate complex accompanied by the generation of tert-BuOBpin. Spectroscopic studies have also supported the intermediary presence of a boron alkylidene, which likely forms because of the significant sideways overlap between the Lewis-acidic trivalent boron atom and the negatively charged carbon centre.
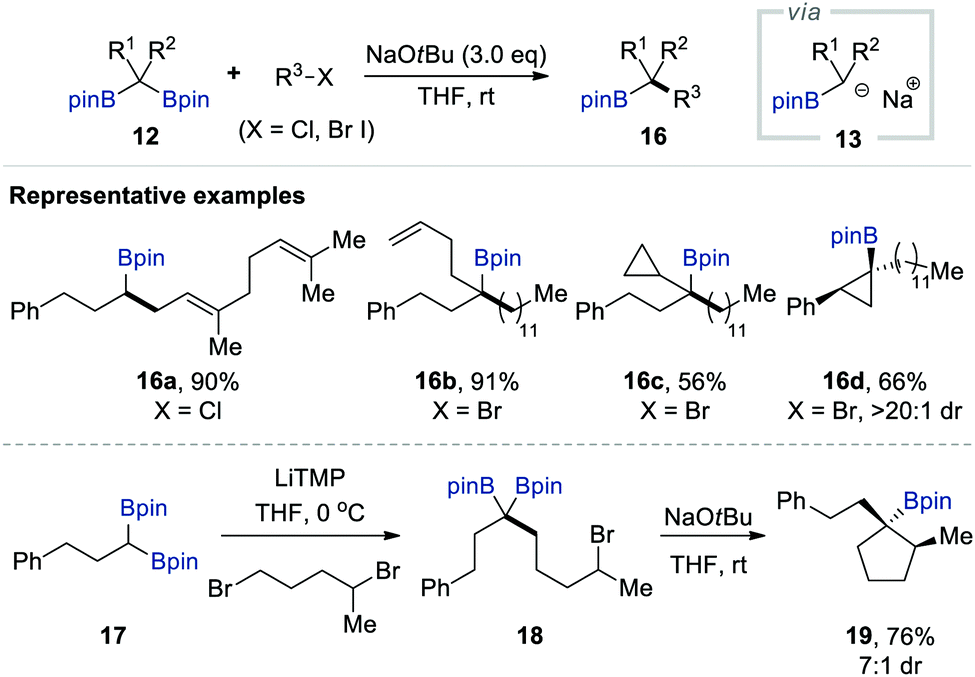 | ||
| Scheme 4 NaOtBu-mediated deborylative alkylation of gem-bis[(pinacolato)boryl]alkanes. | ||
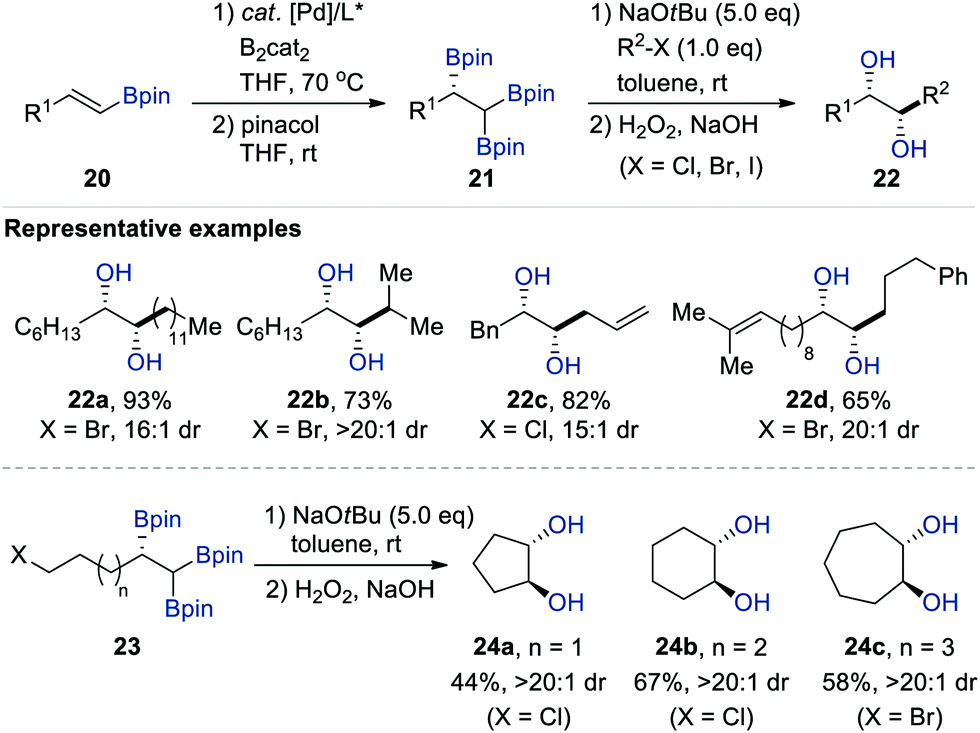
Meanwhile, the Morken group also reported the NaOtBu-mediated diastereoselective deborylative alkylation of chiral enantioenriched 1,1,2-tris[(pinacolato)boryl]alkanes 21, prepared via the Pt-catalysed enantioselective diboration of vinyl boronates 20, with not only primary alkyl halides (X = Cl, Br, I), but also secondary electrophiles (Scheme 5).18 This transformation exclusively provided the syn diastereomers of 1,2-diols 22 in all cases upon oxidative workup with H2O2/NaOH. Moreover, an intramolecular version of this sequence enabled the synthesis of carbocyclic anti-1,2-diols 24a–c containing five-, six-, and seven-membered rings with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr).
:1 dr).
 | ||
| Scheme 5 Diastereoselective alkylation of enantioenriched 1,1,2-tris[(pinacolato)boryl]alkanes. | ||
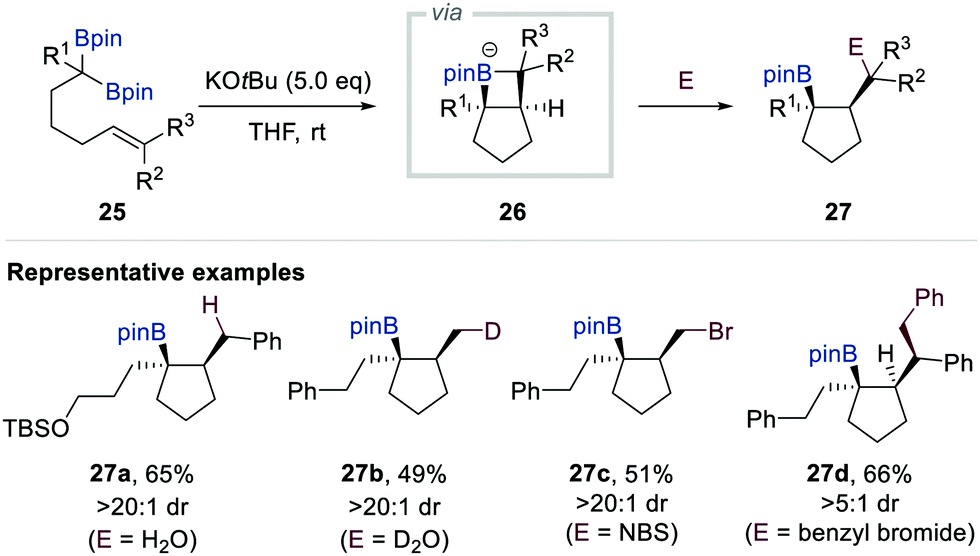
In 2017, Morken et al. reported a new reactivity mode of boron alkylidenes, which are an alternative resonance hybrid of α-[(pinacolato)boryl]alkylanions, based on intramolecular [2+2]-cycloaddition with alkene (Scheme 6).19 Whereas the SN2 alkylation product was obtained when gem-bis[(pinacolato)boryl]alkane 25 bearing a tethered alkene was treated with additional alkyl halide in the presence of KOtBu, the unusual intramolecular deborylative [2+2] cycloadducts were formed when the reaction was conducted without the alkyl halide electrophile. Based on detailed mechanistic studies including isotope labelling experiments, radical clock experiments, and density functional theory (DFT) calculations, the authors proposed that an ate complex of 5-borata[3.2.0]bicycloheptane 26 is the key intermediate in this deborylative cyclisation. Moreover, they found that such intermediates could be trapped by various electrophiles including D2O, N-bromosuccinimide (NBS), I2, benzyl bromide, allyl bromide, and Eschenmoser salt to deliver a wide range of cyclopentane-containing products 27 (Scheme 6).
 | ||
| Scheme 6 Intramolecular net [2+2] cycloaddition and functionalisation. | ||
2.3 Alkylation of N-heteroarenes with α-[(pinacolato)boryl]alkylanions
In 2016, Cho and co-workers reported the deoxygenative alkylation of N-heteroaromatic N-oxides 28 employing gem-bis[(pinacolato)boryl]alkanes 1 as versatile alkylating reagents; this reaction proceeds with perfect regioselectivity at the 2-positions of various N-heterocycles under transition metal-free conditions (Scheme 7).20a The optimal reaction parameters (alkylation in toluene at 80 °C with NaOMe as the base) provided the deoxygenated C2-alkylated N-heteroarene 29. Given the importance of the site-selective methylation of N-heteroaromatic compounds (i.e. the so-called magic methyl effect) in drug discovery and medicinal chemistry,21 this alkylation enabled the incorporation of a wide range of primary alkyl groups, including methyl, ethyl, 3-phenylpropyl, 3-(tert-butyldimethylsilyloxy)propyl, and 5-methyl-4-hexenyl groups, into the cores of N-heterocycles with complete regiocontrol. The same group subsequently succeeded in the convenient secondary alkylation of N-heteroaromatic N-oxides using internal gem-bis[(pinacolato)boryl]alkanes 12.20b Fixing all other conditions such as the base and solvent, an elevated temperature of 120 °C was necessary for these more challenging alkylation reactions. A wide range of internal alkylating reagents and N-heteroaromatic N-oxides were tolerated under the optimal conditions, offering an operationally convenient process to synthesise secondary alkylated N-heteroarenes without an additional deoxygenation step.22 Based on mechanistic studies including deuterium-labelling, cross-over, and NMR experiments, the authors proposed that the alkylation could proceed via the following three-step process (Scheme 7). First, the nucleophilic addition of in situ-generated α-[(pinacolato)boryl]alkylanion 13 to the 2-position of N-oxide 28 occurred. The subsequent migration of the (pinacolato)boryl group of 30 to the anionic oxygen of the N-oxide gave carbanion 31 accompanied by the spontaneous release of NaOBpin via carbanion-induced intramolecular proton transfer, resulting in the formation of the C2-alkylated N-heteroaromatic compound 29.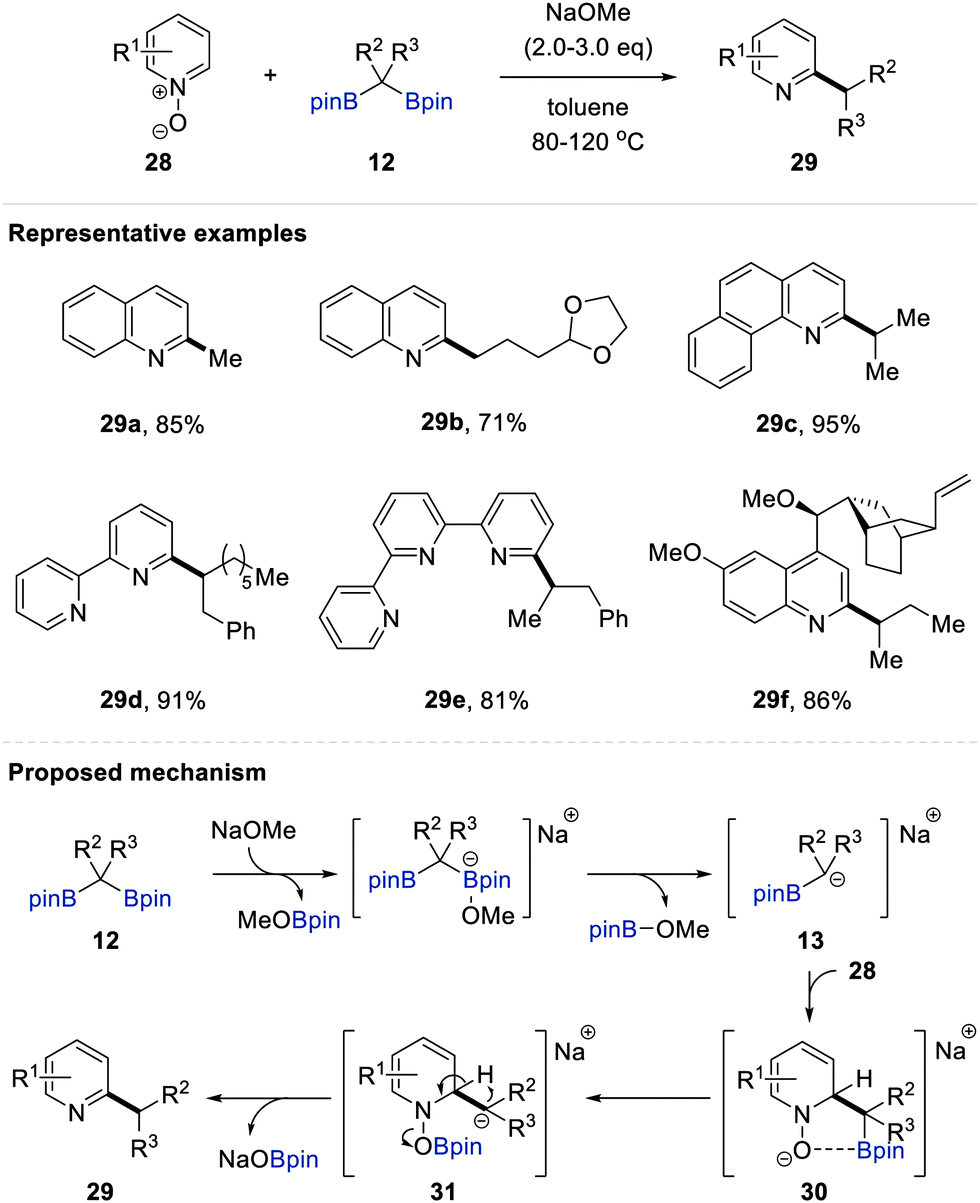 | ||
| Scheme 7 C2-selective primary and secondary alkylation of N-heteroaromatic N-oxides with gem-bis[(pinacolato)boryl]alkanes. | ||
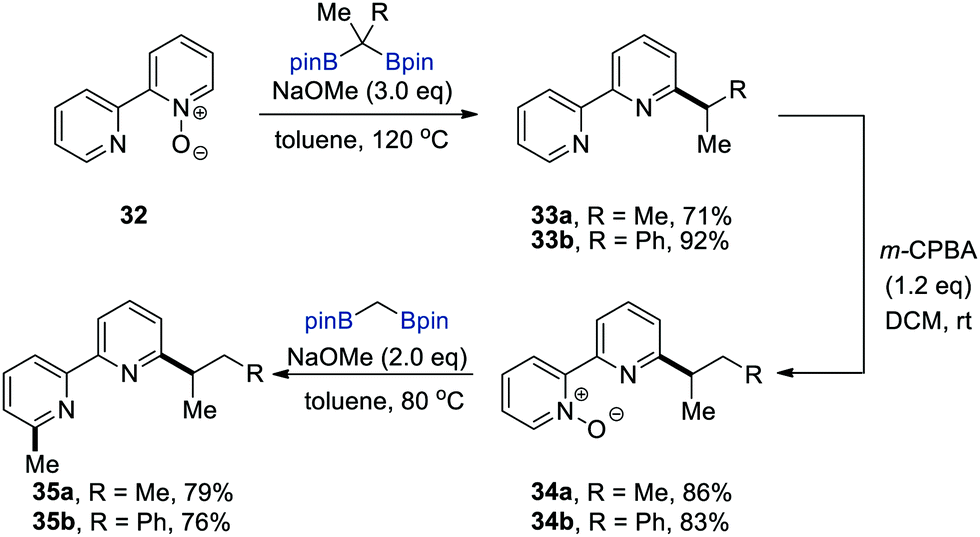
To demonstrate the synthetic utility of this deoxygenative alkylation, the authors performed the sequential functionalisation of 2,2′-bipyridyl N-oxide (32) to prepare N,N-chelating ligands 35, which can be constructed in principle via sequential alkylations on each of the pyridine rings in 2,2′-bipyridyl (Scheme 8). The sequence commenced with the deoxygenative alkylation of 32 followed by the chemoselective oxidation of m-CPBA (m-chloroperbenzoic acid) at the less hindered pyridine moiety in the resulting bipyridines 33 to afford bipyridine mono-N-oxides 34. The second deoxygenative alkylation of 34 led to asymmetrically 3,3′-disubstituted 2,2′-bipyridines 35, which are valuable N,N-bidentate ligands in coordination chemistry.23
 | ||
| Scheme 8 Sequential alkylations of 2,2′-bipyridyl. | ||
The use of oxygenated N-heterocycle substrates is the main drawback of the NaOMe-mediated C2-selective primary and secondary alkylation reactions developed by Cho and co-workers because minimising the number of redox steps is essential for streamlined organic synthesis from the viewpoint of modern redox economy.24 In fact, the direct alkylation of pyridines and their derivatives, which are privileged scaffolds in natural products, pharmaceuticals, agrochemicals, and organic materials, is among the most promising strategies.25 However, these methods, which are mostly based on transition metal-catalysed C–H activation, often have limited substrate scope and proceed under harsh conditions.26 In particular, the incorporation of a methyl group into the N-heterocyclic core has proven challenging using these procedures. In principle, ideal methods for incorporating alkyl groups including methyl groups in pyridines and quinolines involve adding an appropriate carbon nucleophile at the electrophilic sites of N-heteroarenes followed by the rearomatisation of the resulting alkylated dihydro-N-heteroarenes.27
Very recently, Baik and Cho reported the direct ZnMe2-mediated C2- or C4-selective primary and secondary alkylation of electron-deficient N-heteroaromatics 36 (e.g. pyridines and quinolines) using gem-bis[(pinacolato)boryl]alkanes 12 as alkylating reagents (Scheme 9).28 This single-step alkylation protocol features a remarkably broad substrate scope of N-heteroarenes 36 and gem-bis[(pinacolato)boryl]alkanes 12, including derivatives bearing pharmaceutically relevant motifs, as exemplified by 29i and 37c, with complete regioselectivity. While substituted pyridines and quinolines exclusively provided C2-alkylated N-heterocycles, simple pyridine furnished C4-alkylated pyridine derivatives with high regioselectivity.
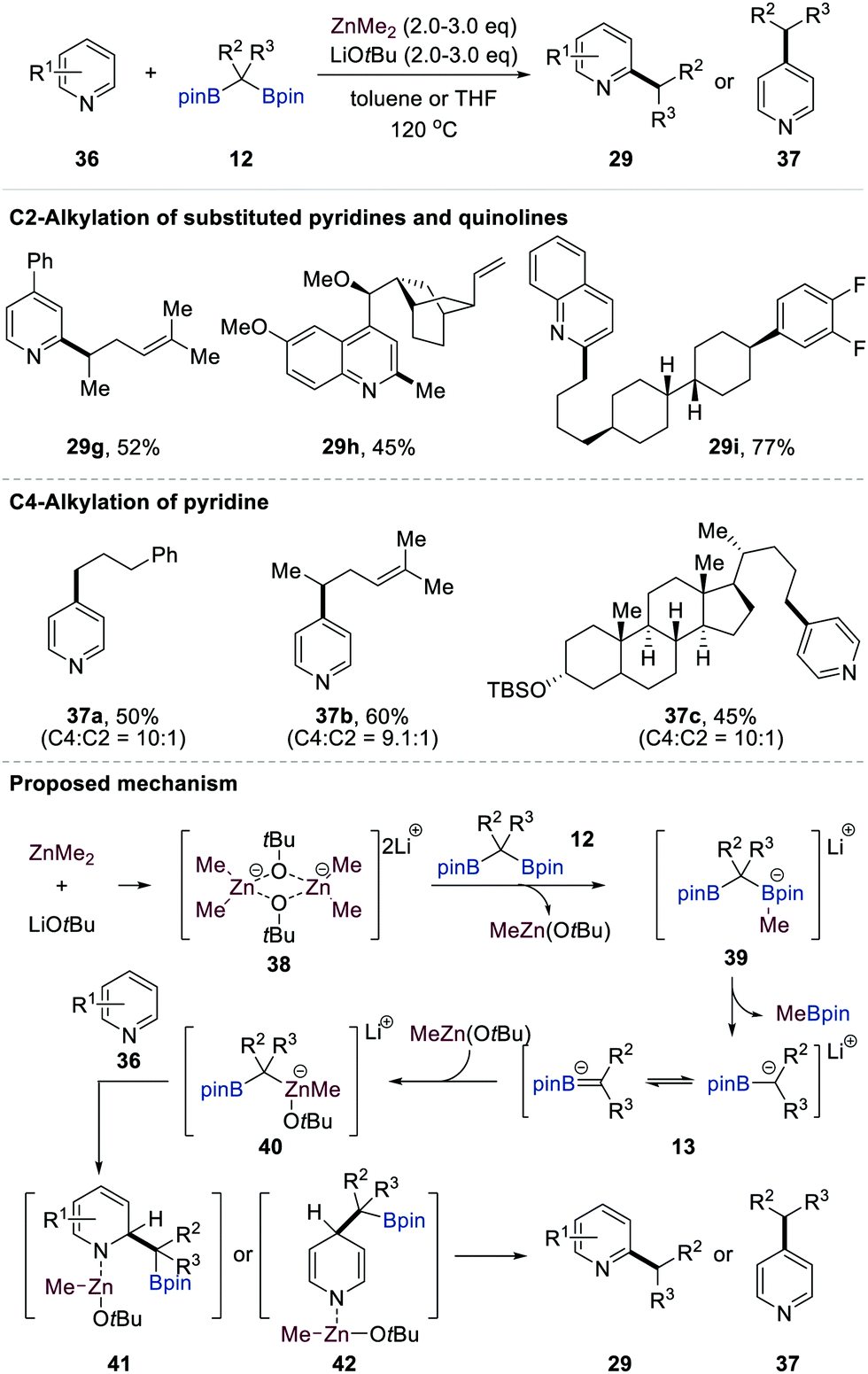 | ||
| Scheme 9 ZnMe2-Mediated direct alkylation of N-heteroarenes with gem-bis[(pinacolato)boryl]alkanes. | ||
Based on a thorough investigation by various experimental methods combined with DFT calculations and NMR spectroscopic analysis, Baik and Cho proposed a plausible mechanism for the direct ZnMe2-mediated C2- or C4-selective alkylation of pyridine and quinoline derivatives (Scheme 9). The formation of dimeric intermediate 3829 facilitated a methyl transfer from ZnMe2 to one of the boron atoms of gem-bis[(pinacolato)boryl]alkanes 12 to give MeZnOtBu and methyl boronate complexes 39. The latter could generate transient anionic intermediates 13 with the liberation of MeBpin. The binding of 13 to MeZnOtBu species yielded stable zincates 40. The MeZnOtBu fragment in 41 could bind to the nitrogen atom of N-heterocycles 36, and this Lewis acid–base interaction increased the electrophilicity of 36. The borylalkyl anion in 40 then attacked the C2-carbon as a nucleophile to afford dearomatised intermediates 41. The DFT calculations showed that C2-alkylation is kinetically favoured over C4-alkylation. In sharp contrast, C4-alkylation is more feasible when pyridine is employed as the substrate, which agrees with the experimental outcomes. The aromatisation of intermediates 41 and 42 accompanied by the liberation of Zn(0) and tBuOH (or CH4) followed by the protodeborylation of the resulting 2- or 4-(borylalkyl)-N-heterocycles provides C2-alkylated (29) or C4-alkylated (37) products.
2.4 Carbon–carbon bond-forming reaction of carbonyl derivatives with α-[(pinacolato)boryl]alkylanions
In 2018, Liu and co-workers reported the first example of the dual functionalisation of both carbon–boron bonds of gem-bis[(pinacolato)boryl]alkanes 12via deoxygenative enolisation with carboxylic acids followed by electrophilic trapping (Scheme 10).30 The authors found that methyl lithium plays a pivotal role as an efficient activating reagent in this transformation. Careful monitoring of the reaction mixture by in situ infrared spectroscopy supported the initial interaction between the carboxylic acid 43 and gem-bis[(pinacolato)boryl]alkanes 12 to generate the chelating complex 46. The acidic proton of 46 could then be removed by the addition of methyl lithium to afford dianion 47, in which one of the boron centres is occupied by a methyl group, as a key intermediate. The fragmentation of 47 was promoted by heating the reaction mixture at 100 °C, affording α-[(pinacolato)boryl]alkylanion 13 along with carboxylate 48. The combination of these reactive species generated boron enolate 44, presumably via intermediate 49. A diverse range of electrophiles could quench this reactive enolate, finally resulting in the formation of various α-mono-, di-, and trisubstituted ketones 45a–f (Scheme 10).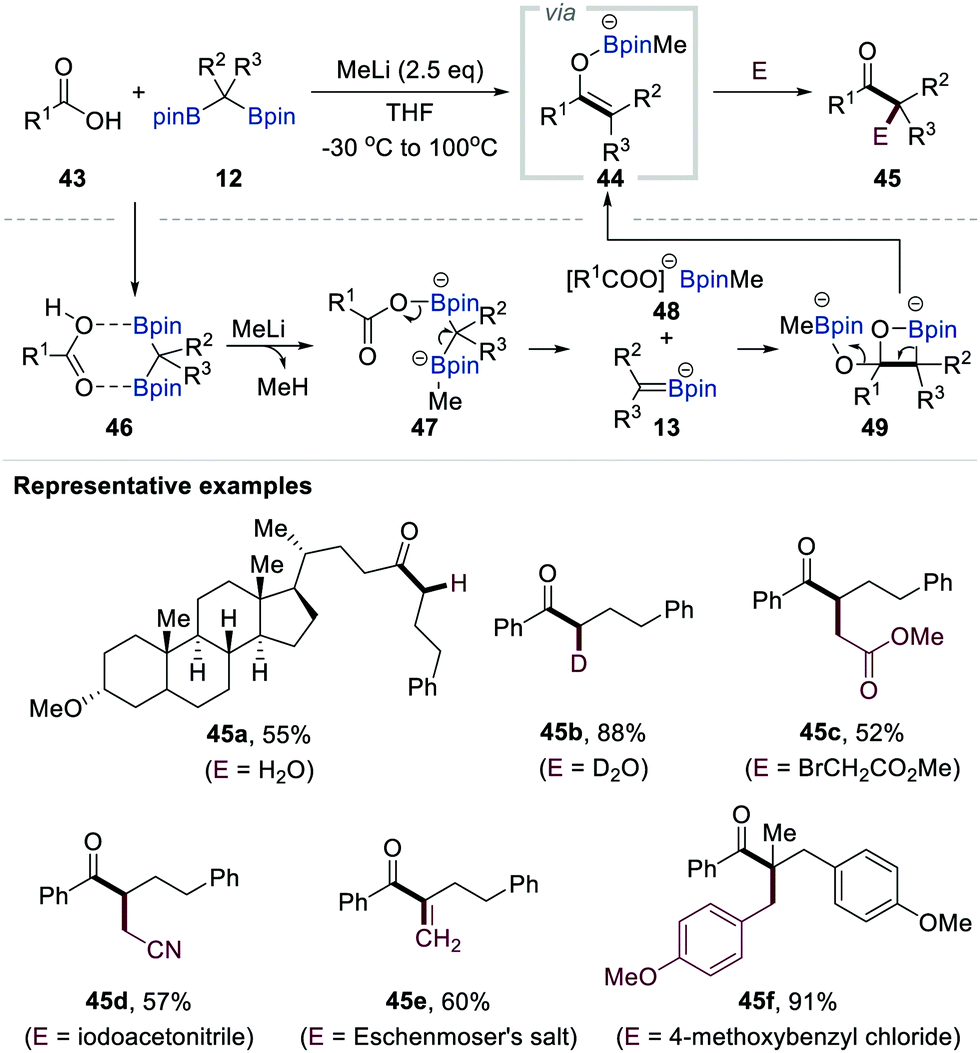 | ||
| Scheme 10 Functionalisation of gem-bis[(pinacolato)boryl]alkanes with carboxylic acids. | ||
Chirik et al. reported the chemoselective conversion of esters into ketones via the LiOtBu-promoted deborylative addition of gem-bis[(pinacolato)boryl]alkanes 12 (R2 = Ar, R3 = H) possessing aryl or heteroaryl substituents to the esters (Scheme 11).31 Extensive NMR spectroscopic studies supported the formation of α-[(pinacolato)boryl]alkylanions upon the release of tBuO-Bpin. These α-[(pinacolato)boryl]alkylanions then reacted with esters (50) to generate boron enolates 51 (R2 = Ar, R3 = H). Having identified boron enolates 51 (R2 = Ar, R3 = H) as the reactive intermediate, the authors also demonstrated three protocols to prepare ketones 45g–j bearing tertiary and quaternary carbon centres by changing the electrophile, starting material, and additional quenching reagent of an alkylation step.
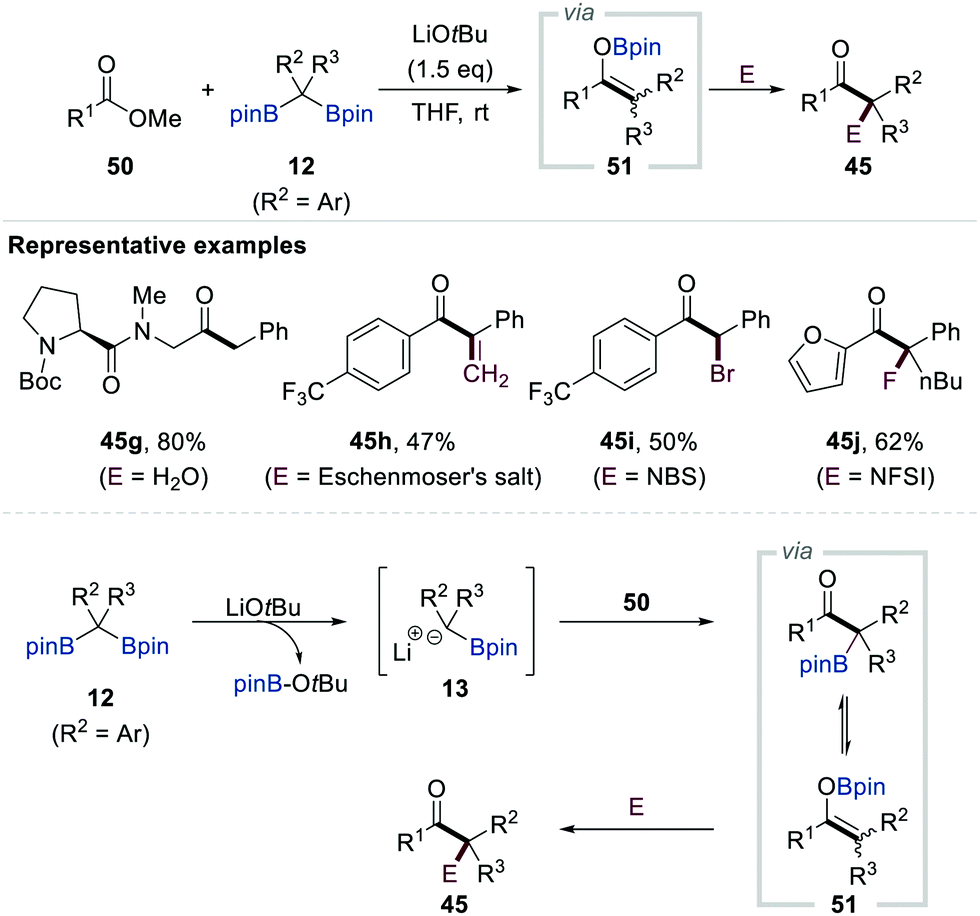 | ||
| Scheme 11 Functionalisation of gem-bis[(pinacolato)boryl]alkanes with esters. | ||
Based on their previous study on carboxylic acids (Scheme 10), in 2020, Liu and co-workers reported the chemodivergent transformation of primary, secondary, and tertiary amides 52 with gem-bis[(pinacolato)boryl]alkanes 12 as pronucleophiles (Scheme 12).32 For primary and secondary amides, the B–O elimination pathway was predominant in the generation of metallo-enamine intermediates 53 after the initial addition of α-[(pinacolato)boryl]alkylanions to amides 52. Furthermore, the authors found that n-butyllithium (4.0 equiv.) was the optimal base for primary amides, while a mixture of n-butyllithium (3.0 equiv.) and Al(OEt)3 (1.5 equiv.) was necessary for the activation of secondary amides. In stark contrast, B–N elimination occurred predominantly for tertiary amides to generate boron enolate intermediates 51 using methyl lithium for the activation of gem-bis[(pinacolato)boryl]alkanes 12. The in situ trapping of either metallo-enamines or enolates with various electrophiles generated various functionalised ketones 45k–r, enamides, α-alkylated cyclic amines, and β-ketoamides.
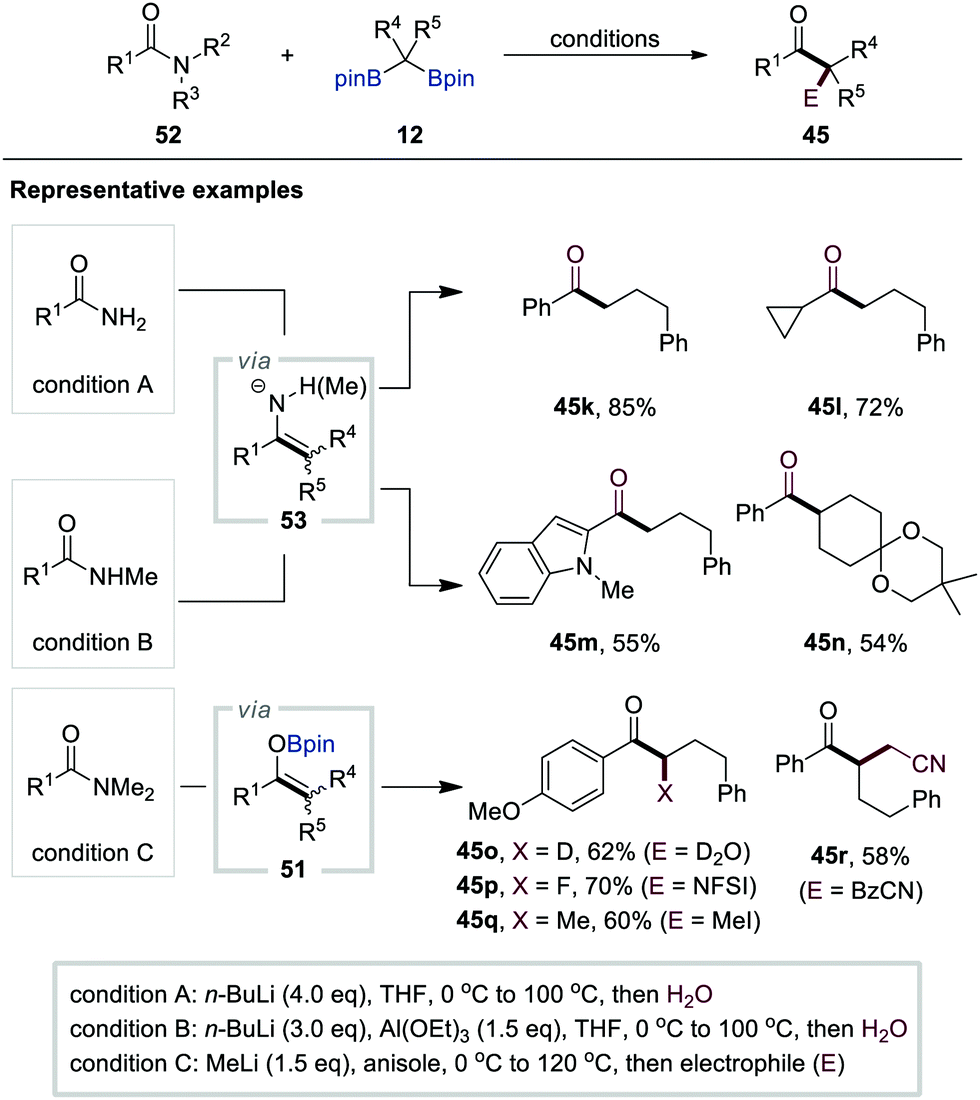 | ||
| Scheme 12 Functionalisation of gem-bis[(pinacolato)boryl]alkanes with esters. | ||
3. Carbon–boron bond formation
In contrast to the notable progress in the utilisation of gem-bis[(pinacolato)boryl]alkanes 12 for constructing carbon–carbon bonds via α-[(pinacolato)boryl]alkylanion 13 species, the alternative use of gem-bis[(pinacolato)boryl]alkanes for the formation of carbon–boron bonds in aryl halides was only recently reported by Cho et al. in 2017 (Scheme 13).33 When aryl and vinyl iodides 54 were treated with bis[(pinacolato)boryl]methane 12 (R2 = R3 = H) in the presence of NaOtBu as an activator in an equal mixture of toluene and THF at elevated temperature, the borylation of aryl and vinyl iodides occurred to afford aryl and vinylboronate esters 55 in good yields.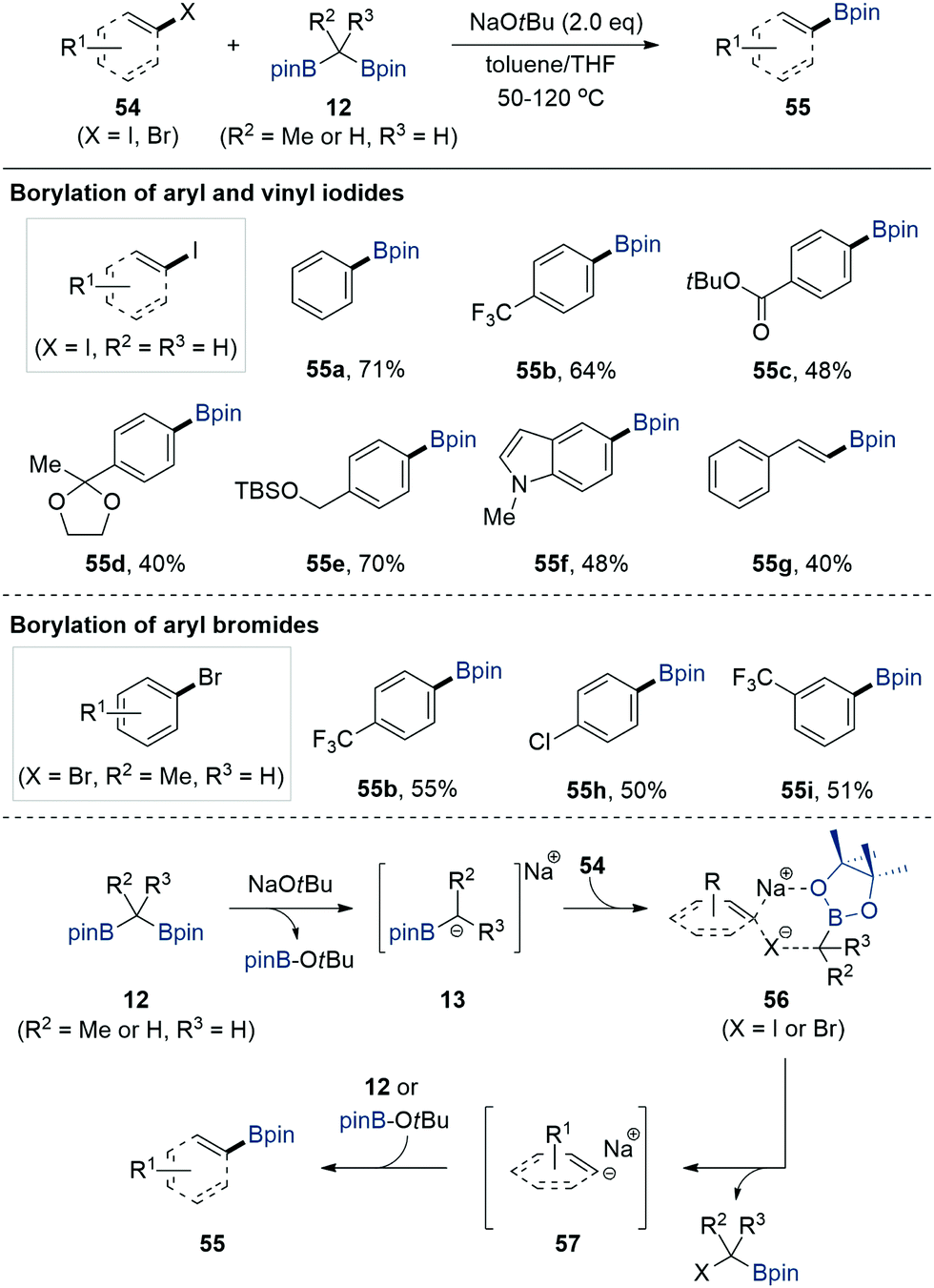 | ||
| Scheme 13 Borylation of aryl halides with gem-bis[(pinacolato)boryl]alkanes. | ||
Experimental and theoretical investigations revealed that the formation of Lewis acid/base adducts between α-[(pinacolato)boryl]alkylanions and organohalides facilitated the formation of six-membered coordinated intermediate 56 and released sodium aryl anion species 57, which subsequently underwent carbon–boron bond formation reaction upon the adoption of [(pinacolato)boryl]iodoalkanes or tert-butoxyboronate ester as a boron source. Based on an understanding of the reaction mechanism, the authors showed that aryl bromides also participated in the developed transition metal-free borylation reaction when gem-bis[(pinacolato)boryl]ethane (R2 = Me, R3 = H) was used as a borylating reagent in the presence of NaOtBu in THF at 120 °C.
4. Conclusion
This feature article focuses on the transition metal-free deborylative functionalisation of gem-diborylalkanes via α-borylcarbanions as the key intermediates. As exemplified by our independent works and the works of others, upon treatment with metal alkoxide or alkyl lithium, α-borylcarbanions generated from gem-diborylalkanes are sufficiently nucleophilic to react with various electrophiles, providing a diverse range of useful building blocks. Although α-borylcarbanions have been successfully applied to various carbon–carbon and carbon–boron bond-forming reactions, there are still multiple challenges. For example, a stoichiometric amount of alkoxide or alkyl lithium as an activator of gem-diborylalkanes makes the reaction less practical as these strong bases typically show low functional group compatibility. Consequently, the development of alternative methods for the activation of gem-diborylalkanes under milder conditions is highly desirable to encompass a wide range of substrates in given reactions. Radical-based approach might be an attractive alternative to the based-promoted activation of gem-diborylalkanes.34,35 Moreover, carbon–boron bond-forming reactions of aryl(vinyl) chlorides as electrophiles is another challenge to be overcome. Regardless of these issues, the last half-decade has seen significant advances in the deborylative functionalisation of gem-diborylalkanes via α-borylcarbanions as versatile intermediates and fascinating new developments in this area of research are continuously being developed. We hope that this feature article provides helpful guidance for future research in this promising, ever-growing research field.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF-2019R1A2C2004925). J. H. L. is grateful for the National Research Foundation of Korea (NRF-2020R1F1A1076460) and the Dongguk University Research Fund of 2021.Notes and references
- (a) Advances in Carbanion Chemistry, ed. V. Snieckus, JAI Press, Greenwich, CT, 2nd edn, 1996 Search PubMed; (b) E. Buncel and J. M. Dust, Carbanion Chemistry: Structures and Mechanisms, American Chemical Society, Washington, DC, 2003 Search PubMed; (c) K. Vollhardt and N. Schore, Organic Chemistry: Structure and Function, W. H. Freeman, New York, 2014 CrossRef; (d) The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (e) S. J. Blanksby and J. H. Bowie, “Carbanions: formation, structure and thermochemistry”, The encyclopedia of mass spectrometry, ed. M. L. Gross and R. M. Caprioli, Elsevier, Amsterdam, 1st edn, 2005 Search PubMed.
- (a) G. A. Russell and A. G. Bemis, J. Am. Chem. Soc., 1966, 88, 5491–5497 CrossRef CAS; (b) M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1985, 107, 2174–2175 CrossRef CAS; (c) P. P. Power, Acc. Chem. Res., 1988, 21, 147–153 CrossRef CAS; (d) S. Gronert, Chem. Rev., 2001, 101, 329–360 CrossRef CAS PubMed; (e) S. Harder, Chem. – Eur. J., 2002, 8, 3229–3232 CrossRef CAS PubMed; (f) S. Harder, F. Feil and T. Repo, Chem. – Eur. J., 2002, 8, 1991–1999 CrossRef CAS PubMed.
- (a) J. W. Wilson, J. Organomet. Chem., 1980, 186, 297–300 CrossRef; (b) I. Marek and J.-F. Normant, Chem. Rev., 1996, 96, 3241–3268 CrossRef CAS PubMed; (c) T. Klis, S. Lulinski and J. Serwatowski, Curr. Org. Chem., 2010, 14, 2549–2566 CrossRef CAS; (d) R. J. Maza, J. J. Carbó and E. Fernández, Adv. Synth. Catal., 2021, 363 DOI:10.1002/adsc.202100192.
- For the initial discovery for the generation of α-borylcarbanions via deprotonation method, see: (a) M. W. Rathke and R. Kow, J. Am. Chem. Soc., 1972, 94, 6854–6856 CrossRef CAS; (b) D. S. Matteson and K. Arne, J. Am. Chem. Soc., 1978, 100, 1325–1326 CrossRef CAS; (c) D. S. Matteson and K. H. Arne, Organometallics, 1982, 1, 280–288 CrossRef CAS; (d) D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20–28 CrossRef CAS; (e) D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 230–236 CrossRef CAS.
- D. S. Matteson and J. W. Wilson, Organometallics, 1985, 4, 1690–1692 CrossRef CAS.
- For the zinc-stablised α-borylcarbanions from α-haloboronic esters, see: (a) P. Knochel, J. Am. Chem. Soc., 1990, 112, 7431–7433 CrossRef CAS; (b) M. J. Rozema, A. Sidduri and P. Knochel, J. Org. Chem., 1992, 57, 1956–1958 CrossRef CAS.
- For the recent reviews on gem-diborylalkanes, see: (a) C. Wu and J. Wang, Tetrahedron Lett., 2018, 59, 2128–2140 CrossRef CAS; (b) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306–1327 CrossRef CAS; (c) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC; (d) K. D. Kim and J. H. Lee, Bull. Korean Chem. Soc., 2018, 39, 5–7 CrossRef CAS.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 3834–3840 CrossRef CAS.
- G. Cainelli, G. D. Bello and G. Zubiani, Tetrahedron Lett., 1965, 6, 3429–3432 CrossRef.
- G. Cainelli, G. D. Bello and G. Zubiani, Tetrahedron Lett., 1966, 7, 4315–4318 CrossRef.
- (a) G. Zweifel and H. Arzoumanian, Tetrahedron Lett., 1966, 7, 2535–2538 CrossRef; (b) G. Zweifel and H. Arzoumanian, J. Am. Chem. Soc., 1967, 89, 291–295 CrossRef CAS.
- T. Mukaiyama, M. Murakami, T. Oriyama and M. Yamaguchi, Chem. Lett., 1981, 1193–1196 CrossRef CAS.
- (a) K. Endo, M. Hirokami and T. Shibata, Synlett, 2009, 1331–1335 CrossRef CAS; (b) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894–899 CrossRef CAS PubMed; (c) X. Feng, H. Jeon and J. Yun, Angew. Chem., Int. Ed., 2013, 52, 3989–3992 CrossRef CAS PubMed; (d) H. Li, X. Shangguan, Z. Zhang, S. Huang, Y. Zhang and J. Wang, Org. Lett., 2014, 16, 448–451 CrossRef CAS PubMed; (e) S. H. Cho and J. F. Hartwig, Chem. Sci., 2014, 5, 694–698 RSC; (f) A. Morinaga, K. Nagao, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2015, 54, 15859–15862 CrossRef CAS PubMed; (g) W. N. Palmer, J. V. Obligacion, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 766–769 CrossRef CAS PubMed; (h) S. K. Bose, S. Brand, H. O. Omoregie, M. Haehnel, J. Maier, G. Bringmann and T. B. Marder, ACS Catal., 2016, 6, 8332–8335 CrossRef CAS; (i) T. C. Atack and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 6139–6142 CrossRef CAS PubMed; (j) L. Li, T. Gong, X. Lu, B. Xiao and Y. Fu, Nat. Commun., 2017, 8, 345–351 CrossRef PubMed; (k) L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257–5264 CrossRef CAS PubMed; (l) G. Gao, Z. Kuang and Q. Song, Org. Chem. Front., 2018, 5, 2249–2253 RSC; (m) Z. He, Q. Zhu, X. Hu, L. Wang, C. Xia and C. Liu, Org. Chem. Front., 2019, 6, 900–907 RSC; (n) N. Kumar, R. R. Reddy and A. Masarwa, Chem. – Eur. J., 2019, 25, 8008–8012 CrossRef CAS PubMed; (o) Z. He, J. Xu, Y. Hu, L. Wang, X. Wu, C. Xia and C. Liu, Chin. J. Org. Chem., 2019, 39, 3438–3445 CrossRef CAS; (p) T. Yamamoto, A. Ishibashi and M. Suginome, Org. Lett., 2019, 21, 6235–6240 CrossRef CAS PubMed; (q) B. Zhao, Z. Li, Y. Wu, Y. Wang, J. Qian, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 9448–9452 CrossRef CAS PubMed; (r) J. H. Docherty, K. Nicholson, A. P. Dominey and S. P. Thomas, ACS Catal., 2020, 10, 4686–4691 CrossRef CAS; (s) M. Hu and S. Ge, Nat. Commun., 2020, 11, 765–774 CrossRef CAS PubMed; (t) D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2020, 142, 9119–9123 CrossRef CAS PubMed; (u) X. Wang, X. Cui, S. Li, Y. Wang, C. Xia, H. Jiao and L. Wu, Angew. Chem., Int. Ed., 2020, 59, 13608–13612 CrossRef CAS PubMed; (v) L. Zhang, X. Si, F. Rominger and A. S. K. Hashmi, J. Am. Chem. Soc., 2020, 142, 10485–10493 CrossRef CAS PubMed; (w) X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, ACS Catal., 2021, 11, 1–18 CrossRef CAS; (x) J.-X. Xu, F.-P. Wu and X.-F. Wu, Catal. Commun., 2021, 149, 106205 CrossRef CAS.
- A. J. Wommack and J. S. Kingsbury, Tetrahedron Lett., 2014, 55, 3163–3166 CrossRef CAS.
- (a) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588–7590 CrossRef CAS; (b) D. S. Matteson and R. Ray, J. Am. Chem. Soc., 1980, 102, 7590–7591 CrossRef CAS; (c) D. S. Matteson and K. M. Sadhu, J. Am. Chem. Soc., 1983, 105, 2077–2078 CrossRef CAS; (d) D. S. Matteson, K. M. Sadhu and M. L. Peterson, J. Am. Chem. Soc., 1986, 108, 810–819 CrossRef CAS; (e) D. S. Matteson, R. Soundararajan, O. C. Ho and W. Gatzweiler, Organometallics, 1996, 15, 152–163 CrossRef CAS.
- For the selected reviews on Matteson homologation, see: (a) D. S. Matteson, Acc. Chem. Res., 1988, 21, 294–300 CrossRef CAS; (b) D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed; (c) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed; (d) H. Wang, C. Jing, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 16859–16872 CrossRef CAS PubMed.
- K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581–10584 CrossRef CAS PubMed.
- J. R. Coombs, L. Zhang and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 16140–16143 CrossRef CAS PubMed.
- X. Liu, T. M. Deaton, F. Haeffner and J. P. Morken, Angew. Chem., Int. Ed., 2017, 56, 11485–11489 CrossRef CAS PubMed.
- (a) W. Jo, J. Kim, S. Choi and S. H. Cho, Angew. Chem., Int. Ed., 2016, 55, 9690–9694 CrossRef CAS PubMed; (b) C. Hwang, W. Jo and S. H. Cho, Chem. Commun., 2017, 53, 7573–7576 RSC.
- (a) H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed; (b) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- For recent examples and references therein, see: (a) K. D. Kim and J. H. Lee, Org. Lett., 2018, 20, 7712–7716 CrossRef CAS PubMed; (b) J. H. An, K. D. Kim and J. H. Lee, J. Org. Chem., 2021, 86, 2876–2894 CrossRef CAS PubMed.
- (a) C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS PubMed; (b) N. C. Fletcher, J. Chem. Soc., Perkin Trans. 1, 2002, 1831–1842 RSC.
- (a) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed; (b) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC; (c) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed.
- (a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627–646 RSC; (b) K. Turner, Org. Process Res. Dev., 2009, 13, 381–390 CrossRef CAS; (c) K.-H. Lam, C.-H. Chui, R. Gambari, R. S.-M. Wong, G. Y.-M. Cheng, F.-Y. Lau, P. B.-S. Lai, S.-W. Tong, K.-W. Chan, W.-Y. Wong, A. S.-C. Chan and J. C.-O. Tang, Eur. J. Med. Chem., 2010, 45, 5527–5530 CrossRef CAS PubMed; (d) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed; (e) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) Y. Nakao, Synthesis, 2011, 3209–3219 CrossRef CAS; (b) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS PubMed.
- (a) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed; (b) J. Kim and S. H. Cho, Synlett, 2016, 2525–2529 CAS; (c) Y. Chen, Chem. – Eur. J., 2019, 25, 3405–3439 CrossRef CAS PubMed; (d) F.-Y. Zhou and L. Jiao, Synlett, 2021, 159–178 CAS.
- W. Jo, S.-Y. Baek, C. Hwang, J. Heo, M. H. Baik and S. H. Cho, J. Am. Chem. Soc., 2020, 142, 13235–13245 CrossRef CAS PubMed.
- (a) R. M. Fabicon, M. Parvez and H. G. Richey, J. Am. Chem. Soc., 1991, 113, 1412–1414 CrossRef CAS; (b) A. P. Purdy and C. F. George, Polyhedron, 1994, 13, 709–712 CrossRef CAS; (c) R. M. Fabicon and J. H. G. Richey, J. Chem. Soc., Dalton Trans., 2001, 783–788 RSC.
- (a) W. Sun, L. Wang, C. Xia and C. Liu, Angew. Chem., Int. Ed., 2018, 57, 5501–5505 CrossRef CAS PubMed; (b) L.-H. Zou, M. Fan, L. Wang and C. Liu, Chin. Chem. Lett., 2020, 31, 1911–1913 CrossRef CAS.
- B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed.
- W. Sun, L. Wang, Y. Hu, X. Wu, C. Xia and C. Liu, Nat. Commun., 2020, 11, 3113–3123 CrossRef CAS PubMed.
- Y. Lee, S.-y. Baek, J. Park, S.-T. Kim, S. Tussupbayev, J. Kim, M.-H. Baik and S. H. Cho, J. Am. Chem. Soc., 2017, 139, 976–984 CrossRef CAS PubMed.
- N. Kumar, R. R. Reddy, N. Eghbarieh and A. Masarwa, Chem. Commun., 2020, 56, 13–25 RSC.
- C. Wu, Z. Bao, B. Dou and J. Wang, Chem. – Eur. J., 2021, 27, 2294–2298 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |