
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct targeting of TDP-43, from small molecules to biologics: the therapeutic landscape
Liberty
Francois-Moutal
ab,
David Donald
Scott
ab and
May
Khanna
 *abc
*abc
aDepartment of Pharmacology, College of Medicine, University of Arizona, 1501 North Campbell Drive, P.O. Box 245050, Tucson, AZ 85724, USA. E-mail: maykhanna@email.arizona.edu; Fax: +520-626-2204; Tel: +520-626-2147
bCenter of Innovation in Brain Science, Tucson, AZ 85721, USA
cBio5 Institute, University of Arizona, Tucson, USA
First published on 21st June 2021
Abstract
Tar DNA binding (TDP)-43 proteinopathy, typically described as cytoplasmic accumulation of highly modified and misfolded TDP-43 molecules, is characteristic of several neurodegenerative diseases such as Amyotrophic Lateral Sclerosis (ALS) and limbic-predominant age-related TDP-43 encephalopathy (LATE). TDP-43 proposed proteinopathies include homeostatic imbalance between nuclear and cytoplasmic localization, aggregation of ubiquitinated and hyper-phosphorylated TDP-43, and an increase in protein truncation of cytoplasmic TDP-43. Given the therapeutic interest of targeting TDP-43, this review focuses on the current landscape of strategies, ranging from biologics to small molecules, that directly target TDP-43. Antibodies, peptides and compounds have been designed or found to recognize specific TDP-43 sequences but alleviate TDP-43 toxicity through different mechanisms. While two antibodies described here were able to induce degradation of pathological TDP-43, the peptides and small molecules were primarily designed to reduce aggregation of TDP-43. Furthermore, we discuss promising emerging therapeutic targets.
Introduction
Tar DNA binding protein (TDP)-43 is a nucleic acid binding protein consisting of three domains, a folded N-terminal domain, two RNA Recognition Motifs (RRM1 and 2) and a mostly unstructured C-terminal domain where most of the known disease-associated mutations occur (reviewed in ref. 1). Mostly localized to the nucleus, TDP-43 is a ribonucleic acid-binding protein of the hnRNP family,2 that can translocate to the cytoplasm and mitochondria.3–5 As a nucleic acid binding protein, TDP-43 regulates RNA processing, including mRNA splicing-mainly by binding to UG-rich intronic regions-, RNA stability and transport (reviewed in ref. 6 and 7).TDP-43 proteinopathies are usually characterized by cytoplasmic accumulation of misfolded and heavily modified TDP-43, accompanied by nuclear clearance.8–10 The origin of these aggregates is debatable, but stress granule and dynamics are thought to play a role.11–13 Even though the exact mechanisms remain largely unknown, pathological TDP-43 is thought to exert a plethora of deleterious effects ranging from nuclear transport inhibition by sequestering transport factors,14 to impaired autophagy and mitochondrial dysfunction,15 among others. The accumulation of those insults is thought to ultimately lead to neurite loss and subsequent neuronal death.
TDP-43 proteinopathies (review in ref. 16) include Amyotrophic Lateral Sclerosis – Frontotemporal lobar degeneration (ALS-FTLD) spectrum, facial onset sensory and motor neuronopathy (FOSMN), Guam Parkinson-dementia complex (G-PDC) with ALS (G-ALS), multisystem proteinopathy (MSP) and Perry disease. Limbic-predominant age-related TDP-43 encephalopathy (LATE) as well as cerebral age-related TDP-43 with sclerosis (CARTS), recently discovered in older patients (≥85 years old), have also been characterized as TDP-43 proteinopathies. Moreover, Alzheimer's disease and chronic traumatic encephalopathy (CTE) have exhibited concomitant TDP-43 pathology.
Given the interest in targeting TDP-43 in neurodegenerative diseases for therapeutic development, this report focuses on the existing strategies, ranging from biologics to small molecules, able to directly recognize or modulate TDP-43 pathology. Structural insights on targeted regions of TDP-43 will aid our understanding and may instruct future targeting. A more general overview of compounds has been recently written that describes how to mitigate pathological characteristics of TDP-43 such as expression or mislocalization.17,18
Biologics
Antibodies targeting TDP-43
In 2012, Shodai and collaborators developed an antibody against misfolded TDP-43 based on the assumption that RRM2 aberrant conformation is linked to pathological TDP-43.19 They specifically focused their efforts on residues E246 and D247 (Fig. 1A). Intriguingly, these residues are mostly buried within the natively folded TDP-43 structure but they are (i) part of an essential cleavage site in FTLD-U brains,10 (ii) localized in a defined amyloidogenic core19,20 and (iii) implicated in dimerization of RRM2 which is thought to lead to TDP-43 aggregation,21 similar to the dimer interface residues of SOD1 observed in familial ALS.22 Shodai and colleagues used a subconstruct of TDP-43 consisting of amino acids 241–260, to generate hybridomas which were later selected for their ability to bind TDP-43 RRM2 but not E246G/D247G RRM2. The resulting antibody was called 3B12A and recognized only highly dense TDP-43 structures in the nucleus, cytoplasmic TDP-43 with an impaired NLS as well as nearly 80% of TDP-43 inclusions in anterior horn cells of ALS patients. This led to the conclusion that exposed E246/D247 residues on TDP-43 may be a marker of TDP-43 inclusions in ALS/FTLD.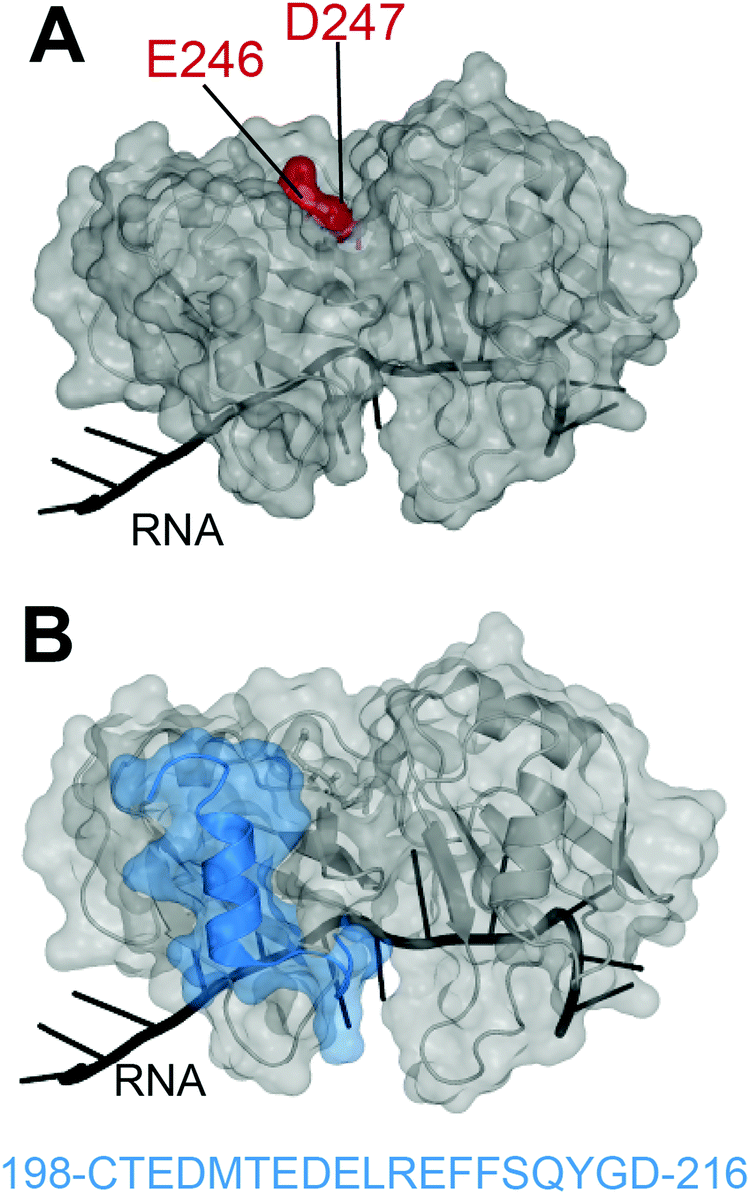 | ||
| Fig. 1 Targeting of TDP-43 by antibodies. (A) The 3B12A antibody is able to recognize aberrantly exposed E246 and D247 (red) in misfolded TDP-43 found in ALS tissues.23 (B) Two other antibodies, 2G11 and 2H1, developed by ref. 31 to recognize a TDP-43 sequence specific to humans (blue) and were able to recognize aggregates in samples from ALS-FTLD and LATE patients (PDB ID: 4bs254). | ||
The same group later discovered the ability of the 3B12A antibody to induce proteasome degradation via a RIDPEDGETK sequence found in the 3B12A heavy chain.23 That sequence exhibits a high PEST score, which typically defines sequences rich in proline, glutamic acid, serine, and threonine that act as degradation signals.24,25 Further addition of a CMA (chaperone-mediated autophagy) sequence to the 3B12A antibody, resulted in efficient cell clearance of misfolded TDP-43 and reversed TDP-43 induced toxicity in HEK293A cells overexpressing cytoplasmic misfolded TDP-43 (TDP-43mNLS,C173S/C175S) as well as in cerebral cortex of newborn mice transduced with TDP-43mNLS,C173S/C175S.23 Even though further testing of this strategy needs to be considered, especially in adult models and/or patient derived induced stem cells, this was the first example of direct targeting of TDP-43 by an antibody leading to cellular clearance of pathological aggregates with therapeutic promise.
Another antibody was developed by Pozzi et al.,26 and is described as targeting the TDP-43 RRM1 domain. The rationale for this approach was based on TDP-43 RRM1 being implicated in aggregation via its misfolding,27 similar to RRM2, and/or by its oxidation.28 Another reason to target TDP-43-RRM1 was to disrupt its interaction with p65 NF-κB, which binds TDP-43 NTD and RRM1 and leads to inflammation in glial cells and sensitizes neurons to toxic insults.29 Although it should be noted that the binding between TDP-43 and p65 was shown through immunoprecipitation and not direct biophysical interactions. To generate hybridomas, they used TDP-43 encompassing part of the N-terminal region, the nuclear localization signal (NLS), and the entire RRM1 domain, which presumably spans amino acids (aa) 31 to 176 as this is the interface with p65 they describe.29 Then, they selected antibodies based on their ability to detect the input TDP-43 protein and to disrupt the p65 interaction, which led to the E6 antibody. Transfection of E6-derived vectors encoding single-chain fragment variable (scFv) antibodies induced proteasome and autophagy-driven degradation of TDP-43 in HEK293 cells. Viral-mediated expression of a scFv antibody (named VH7Vk9), further reduced LPS-induced inflammation markers in mouse cortex. Finally, treatment with VH7Vk9 improved cognitive function in TDP-43G348C mice, a well-known model of familial ALS/FTD30 as well as motor performance in another model of familial ALS/FTD, namely TDP-43A315T mice.30
Even though the exact binding site of the antibody remains obscure and could very well implicate TDP-43-NTD, VH7Vk9's ability to induce TDP-43 degradation by promoting polyubiquitination of TDP-43 without addition of a degradation signal is striking. Analysis of the VH7Vk9 sequence might reveal a PEST sequence, similar to ref. 23. Alternatively, the possibility that VH7Vk9 disrupts aggregates leading to TDP-43 being accessible to degradation was speculated by the authors.
Very recently, Trejo-Lopez developed two novel antibodies against TDP-43 RRM2, specific to the human form.31 These antibodies were raised against residues 198–216 of RRM2 (Fig. 1B), chosen based on the weaker conservation of this sequence between human and mouse (68%) compared to the rest of the sequence (97%). Antibodies 2G11 and 2H1 efficiently recognized human TDP-43 over the mouse version of the protein and were able to detect TDP-43 aggregates of ALS-FTLD and LATE, in a comparable manner to a commercially available anti-TDP-43 antibody. The authors describe that the novelty of these antibodies resides in their preference for cytoplasmic inclusions of TDP-43 over nuclear TDP-43, reminiscent of the anti-pSer409/410, which recognizes phosphorylated-TDP43. Structural analysis of the TDP-43 epitope recognized by 2G11 and 2H1 (Fig. 1B) reveals a sequence encompassing a loop and alpha-helix, with no known modifications or mutations attributable to disease state1 and this sequence is not at the interface with nucleic acids. The recognition of this sequence by the antibodies in cytoplasmic inclusions only, raises interesting questions. It would be fascinating to investigate the antibodies on normal patient samples to ascertain whether the site of recognition does indeed contain yet undiscovered PTMs that might not be detected by traditional mass spectrometry techniques.
In addition to the discovery of possible therapeutic strategies or diagnostic tools, these antibodies have uncovered unique surfaces on TDP-43, that may be differentially exposed in the disease state and relevant in TDP-43 cytoplasmic pathology.
Peptides targeting TDP-43
In 2013, Liu and collaborators developed peptides targeting TDP-43-CTD to decrease TDP-43 aggregation and reduce subsequent toxicity in cells.32 The assumption was that TDP-43 aggregation occurs through self-interaction and TDP-43 derived peptides could mitigate aggregation. They defined these peptides using a peptide array, where overlapping TDP-43 peptides were tiled and incubated with full-length TDP-43. The interaction peptides, named A, B, C, D and E, were all sequences from the C-terminus region of TDP-43, but their exact sequences were not disclosed. All five peptides were able to reduce TDP-43 interaction with the peptide array, compared to a scrambled version of peptide C. Tat conjugated peptides inhibited TDP-43 aggregation of overexpressed TDP-43 as well as arsenite-induced TDP-43 inclusions in HeLa cells. The peptides were non cytotoxic but were not able to rescue TDP-43 related toxicity either. The authors hence concluded that inhibition of TDP-43 aggregation was not sufficient in itself to rescue subsequent toxicity.In another recent effort to promote clearance of TDP-43 aggregates, Gao et al. designed several multifunctional peptides comprising a combination of three parts33 (Fig. 2A). The first part is a hydrophobic motif consisting of adamantane, a tricyclodecane cage-shaped compound. Adamantane-based hydrophobic tagging has been used to mimic the unfolded state of the target protein leading to its efficient degradation,34,35 and has the advantage of increasing blood–brain–barrier penetration of conjugated molecules.36,37 The second part is a TDP-43 “recognition motif” consisting of TDP-43 peptides, either 311-MNFGAFSINP-320 or 246-EDLIIKGISV-255. While the 246–255 sequence has been heavily described as an amyloidogenic core,19,20 the sequence 311–320 is less studied but was still reported as aggregation prone.20 It spans part of the GaroS1 region (aa. 273–317), that contributes to the formation of hydrogels38 and the very beginning of the hydrophobic region (residues 318–340) that can adopt both helical and thioflavin-T-positive filaments reminiscent of β-amyloid.39–42 The third part is a cell-penetrating peptide: Tat (RRRQRRKKRG) or Poly-D-Arginine (D-Arg)8. The C-terminal derived peptides were unsurprisingly not further pursued because of low solubility. The peptide D4, which contains E246/D247 residues, showed the strongest ability to reduce TDP-43 levels in an N2a cell line overexpressing TDP-43, while peptides lacking the hydrophobic adamantine moiety had no effect. The results were recapitulated in Drosophila overexpressing TDP-43. While enhanced degradation of TDP-43 by the D4 peptide is implied because of the adamantane moiety, the authors did not assess the effect of the D4 peptide on TDP-43 ability to autoregulate its own RNA or on cytoplasmic inclusions. Nevertheless, this study highlights again the seemingly central role of E246/D247 in TDP-43 misfolding and aggregation, making it an attractive therapeutic target in neurodegeneration.
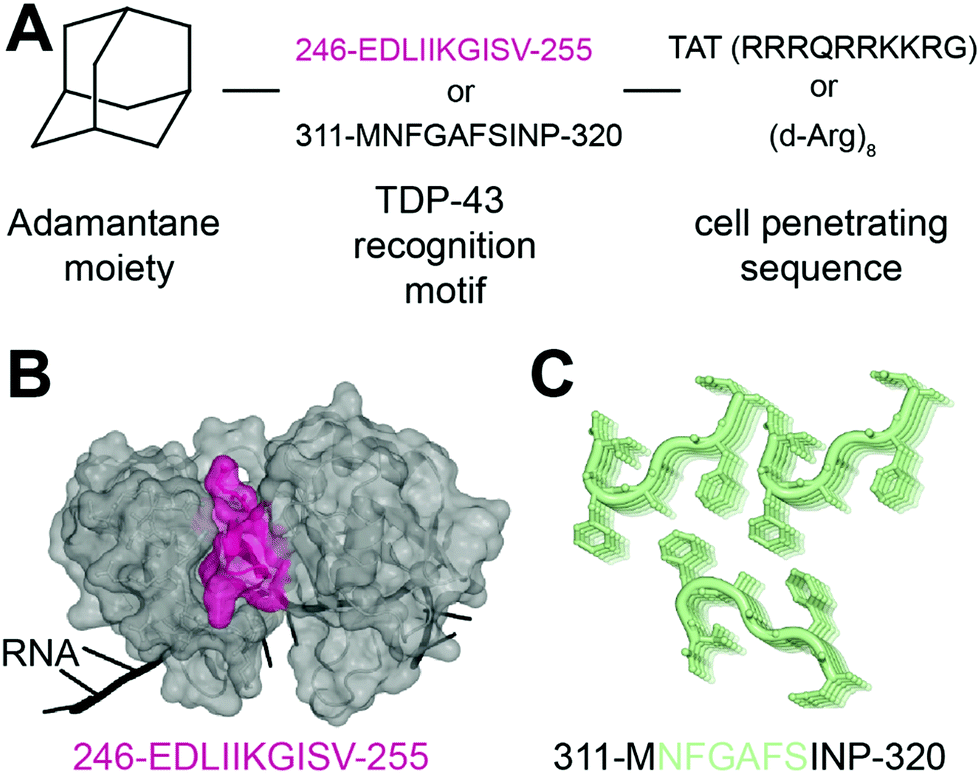 | ||
| Fig. 2 Peptides targeting TDP-43. (A) Structure of the peptides used in ref. 33. (B) The RRM2 sequence used for peptide D4 (pink) is mapped on TDP-43 structure (PDB ID: 4bs254). (C) The sequence MNFGAFSINP is known to form aggregates (PDB ID: 5whn59). | ||
Small molecules
There are very few studies that focus on directly targeting TDP-43, probably due to the lack of pockets traditionally considered as druggable. The very first study targeting TDP-43, in 2013, used a high-throughput alpha-screen to find inhibitors of DNA binding.43 The emerging aminoquinoline compounds augmented caspase-7 degradation of TDP-43 in human neuroglioma H4 although the exact binding site and mechanism of action of those compounds were never elucidated.In 2016, Prasad and collaborators44 examined the effect of acridine derivatives potential to mitigate TDP-43 aggregation, since acridine-based compounds were previously shown to inhibit amyloid formation of PrPSc, among others45 (Fig. 3A). The authors thus report AIM4, [4,5-bis{(N-carboxy methyl imidazolium)methyl}acridine] dibromide, able to significantly reduce in vitro TDP-43 amyloid-like aggregation as well as TDP-43 inclusions in a yeast model cell. More recently, Babinchak et al. showed the ability of 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (bis-ANS), and other similar compounds such as Congo Red, to modulate the liquid–liquid phase separation (LLPS) of TDP-43.46 While those studies demonstrate the potential of planar molecules in binding and mitigating TDP-43 aggregation/liquid phase separation, those compounds will require extensive validation in terms of toxicity and off-targeting properties since most of them are related to intercalant molecules or are known to be extremely toxic.
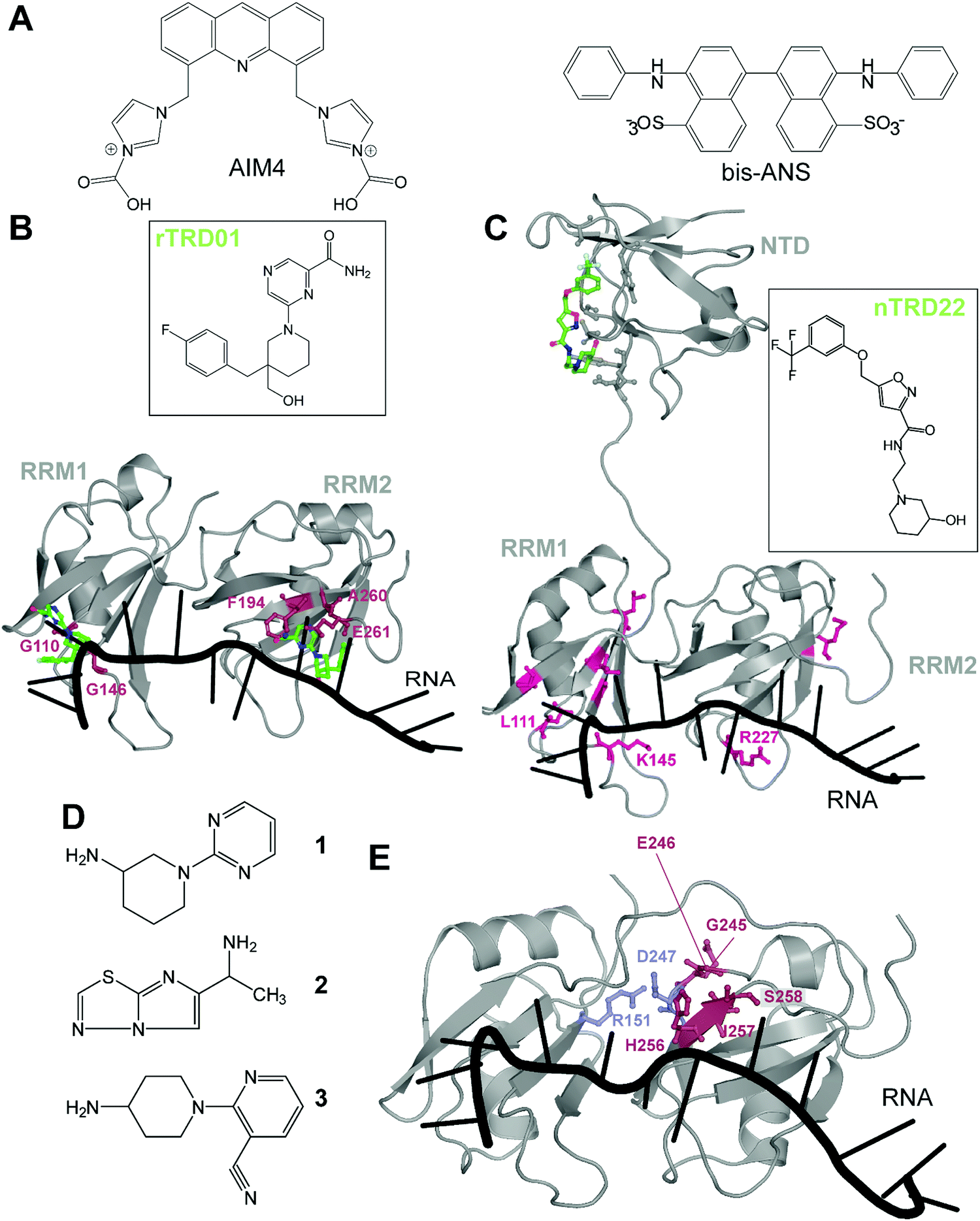 | ||
| Fig. 3 Small molecules targeting TDP-43. (A) Planar molecules able to bind TDP-43 CTD and mitigate LLPS formation or CTD driven aggregation. (B) rTRD01 (green, stick and balls representation) is thought to be able to bind both RRM domains of TDP-43 and displace nucleic acid binding (PDB ID: 4bs2).54 Residues shifted by rTRD01 binding to TDP-43 in HSQC-NMR are displayed in pink, stick and balls representation. rTRD01 is thought to bind RRM1 and RRM2. (C) Composite figure of TDP-43 NTD (PDB ID: 5mdi63) with TDP-43 RRM domains (PDB ID: 4bs254). nTRD22 (green, stick and balls representation) binds to TDP-43 NTD and allosterically modulates residues implicated in nucleic acid binding (pink, stick and balls representation) to disrupt the binding. (D) Fragment compounds found to bind TDP-43.53 (E) Fragments are able to bind a pocket in TDP-43 RRM2 domain (red, stick and balls representation). The pocket is near the residues D247 and R151 (blue, stick and balls), shown to form a salt bridge, necessary for nucleic acid binding (PDB ID: 4bs254). | ||
In recent years, our laboratory has focused on rational, structure-based targeting of TDP-43. Our first discovery used an in silico approach targeting the RRMs,47 more specifically the nucleic acid binding interface of RRM1, since mutations inhibiting nucleic acid binding completely reversed ALS phenotypes in TDP-43 overexpressed Drosophila models.48 We validated one compound, rTRD01, through NMR spectroscopy and Microscale Thermophoresis (MST), obtaining a binding constant in the micromolar range (kD = 89.4 ± 0.8 μM), a relatively weak affinity that will be improved using structure activity relationship (SAR) studies. Interestingly, this compound was able to displace (GGGGCC)4 RNA, but not the native binding of (UG)6 repeats, probably due to lower affinity of TDP-43 for (GGGGCC)4 (nanomolar) compared to the canonical sequence (sub-nanomolar). Lastly, investigation of this compound in an ALS Drosophila model resulted in reduced locomotor defects.
These experiments characterized the first small-molecule directly targeting TDP-43 to identify therapeutics for ALS, even though RNA-recognition pockets are more shallow than conventional kinase ATP-binding pockets. It is important to note that although rTRD01 was obtained from a screen against RRM1, it could potentially also bind to RRM2 (Fig. 3B). HSQC-NMR shifts were observed in both RRM domains and it was not clear if this was due to allostery or direct binding to RRM2; the simplest explanation is direct binding. Further experiments will be used to determine if rTRD01 modulates these RRM domains independently. Our findings also display selective binding of TDP-43 to different RNA sequences. Interference with binding of specific RNA sequences could provide a therapeutic advantage by not altering native functions of TDP-43, as well as the possible identification of pathological biomarkers in neurodegenerative disease.
Another molecule we recently discovered is nTRD22 (Fig. 3C), found using the same pipeline as previously described, but targeting the N-terminal domain (NTD).49 The NTD is implicated in oligomer formation and support of nucleotide binding,50–52 among other functions. Interestingly, nTRD22 was shown to bind specifically to the NTD, with an affinity of 145 ± 3 μM, but was also observed to (i) shift residues in the RRM domains, as demonstrated by HSQC-NMR and (ii) displace canonical UG6 binding, unlike rTRD01 that directly targeted the RNA binding domains. We hypothesized that nTRD22 could affect the RRM domain through allostery, although current studies are ongoing to define how the NTD portion of TDP-43 influences the RRM domains. nTRD22 was further able to reduce TDP-43 protein levels in rodent primary motor neurons, recapitulating TDP-43 RNA binding-deficient mutants and supporting impaired TDP-43 binding to nucleic acids. Finally, nTRD22 mitigated motor impairment in a Drosophila model of ALS. This study not only shows the possibility of TDP-43 NTD to be targeted by small molecules, but also a possible therapeutic interest for such a strategy.
Through these works, potential allosteric interactions between TDP-43 domains were brought to light, even though further investigation is needed to understand the precise mechanism. This insight proposes new intrinsic regulatory roles of TDP-43, as well as a new landscape of therapeutics. As stated before, targeting the NTD resulted in a better inhibition of canonical UG6 RNA binding which might help achieve higher specificity with fewer off-target effects than rTRD01.
Other than our studies, an effort to find molecules targeting TDP-43 using a fragment-based NMR study identified hits that bound to the RRM domain.53 Three different NMR techniques were used, saturation transfer difference (STD), water ligand observed via gradient spectroscopy (WaterLOGSY) and Carr–Purcell–Meiboom–Gill (CPMG), to screen 89 cocktails, each containing 10 small molecules. A secondary screen on individual compounds, using the same NMR techniques, revealed four hits that were further examined using Chemical Shift Perturbations (CSPs) of the 15N-labeled tandem RRMs of TDP-43. Three of these hits, 1, 2 and 3, were able to induce significant CSPs on RRM2 residues G245, E246, H256, I257 and S258 (Fig. 3D and E). This group further validated the binding of these 3 compounds to TDP-43 RRM2 only using HSQC. Hit 1 was docked on TDP-43-RRM (PDB ID: 4bs254) using the data driven approach HADDOCK and was predicted to form a hydrogen bond with the side chain of S258.
While the authors note that their hits 1–3 bind near a nucleic acid binding site on TDP-43 and that a fragment growth strategy can be used to target residues Asn259, and Glu261, implicated in RNA/DNA recognition, it should be noted that RRM2 binding to nucleic acid by itself is very weak, as previously shown by mutations in RRM2 domain,21,55 which questions the efficiency of this approach. An alternative strategy to inhibit nucleic acid binding could be to target D247, which makes a salt bridge with R151, stabilizing the RRM1-RRM2 orientation when RNA is bound56 (Fig. 3D). It is also interesting to note that the pathologically exposed and cleaved E246 is being targeted by compounds 1–3. Given the role of E246/D247 in ALS, as described before, this avenue represents an interesting alternative therapeutic option.
Conclusion
Given the attractiveness of targeting TDP-43 directly to treat neurodegenerative diseases, such as ALS-FTLD, the last few years have seen several different strategies emerge with variable success (Table 1). Antibodies, peptides and compounds have been designed or found to recognize specific TDP-43 sequences but alleviate TDP-43 toxicity through different mechanisms. While two antibodies described here were able to induce degradation of pathological TDP-43, the peptides and small molecules were primarily designed to target aggregation of TDP-43.| Name | Therapeutic type | Targeted site | Cell | Fly | Mice |
|---|---|---|---|---|---|
| 3B12A | Antibody | Misfolded RRM2 | Clearance of pathological TDP-43 in HEK293A cells | N/A | Clearance of TDP-43 aggregates in newborn mice transduced with TDP-43mNLS,C173S/C175S |
| VH7Vk9 | Antibody | Misfolded RRM1 | Induced proteasome and autophagy-driven degradation of TDP-43 in HEK293 cells | N/A | Improved cognitive function in TDP-43G348C mice |
| Liu “A, B, C, D, E” | Peptide | CTD | Reduced aggregation of TDP-43 in HeLa cells but not toxicity | N/A | N/A |
| Gao “D4” | Peptide | CTD | Reduced TDP-43 levels in an N2a cell line overexpressing TDP-43 | Recapitulated in Drosophila overexpressing TDP-43 | N/A |
| AIM4 | Small molecule | CTD | Reduced TDP-43 inclusions in a yeast S. cerevisiae model | N/A | N/A |
| rTRD01 | Small molecule | RRM1/2 | N/A | ALS Drosophila model results in reduced locomotor defects | N/A |
| nTRD22 | Small molecule | NTD | Reduced TDP-43 protein levels in rodent primary motor neurons | Mitigated motor impairment in Drosophila model of ALS | N/A |
Despite its lack of traditional targetable pockets, we and others have shown the ability of small molecules to bind TDP-43, albeit with a rather low affinity.47,49,53 SAR studies focusing on improving the affinity of existing compounds will reveal how effective these strategies are by increasing potency without increasing off-targeting. Alternatively, exploiting conjugation of proteolysis targeting chimera (PROTAC) to these small molecules is a potential new avenue. PROTAC relies on two covalently combined molecules, one recognizing the protein target and the other able to recruit an E3 ubiquitin ligase for ubiquitination and subsequent degradation of the protein target.57 Even though TDP-43 is already ubiquitinated in pathological aggregates, antibodies targeting TDP-43 were able to direct TDP-43 towards degradation with and without direct ubiquitination.23,26 PROTAC targeting of TDP-43 is thus a potential new strategy, with the caveat that the approach does not target well-folded physiological TDP-43 over inclusions. From this review, it is clear that targeting residues E246 and especially D247 is of high interest, since they seem to be exposed in TDP-43 inclusions.23 The coupling of targeting this pathological surface with a degradation strategy led to efficient clearing of aggregates.
In defining therapeutics that can target the predominantly unstructured C-terminus of TDP-43, there are two plausible modes of action: the peptides could either modulate or disrupt protein–protein (PPI) interactions that occur through the C-terminus. There are also other mechanisms we do not consider, such as modulation of condensate, either directly or through condensate formation, or binding to other non-proteinaceous entities. On the other hand, targeting the TDP-43 CTD seems highly challenging. Peptides targeting the CTD failed;32,33 it is highly unstructured in the soluble form and forms polymorphic fibrils,58 making rational design of molecules difficult. Further, there is disease and tissue heterogeneity of TDP-43 C-terminal fragments in terms of composition and structure. Nevertheless, recent efforts, led by the Eisenberg laboratory among others, in structurally characterizing TDP-43 CTD peptides,42,58–61 with and without mutations, will help tremendously in targeting this region. Based on similar efforts to structurally characterize tau segments driving aggregation by MicroED, inhibitors of tau aggregation were developed by targeting VQIINK steric zipper interfaces with peptides derived from the same sequence (VQIINK).62 Based on these studies, they suggest tau amyloid polymorphism with distinct structures of tau fibrils. Similar efforts on structurally defining patient derived TDP-43 inclusions, which are possibly polymorphic, are very likely to provide in the future the mean to design small molecules or biologics that might specifically target these structures.
Although none of the strategies described here have led to compounds that have reached clinical trials, several promising targeting options have been developed. Given the challenges consistently faced in neurodegenerative disease drug development, there needs to be extreme care in choosing appropriate: (i) animal models, potentially in combination with patient derived stem cells, to translate specific disease hallmarks being targeted, (ii) formulation, delivery route and ability to eventually cross the blood–brain–barrier by the drug candidates and (iii) better clinical trial design, for optimal and successful development of therapeutic candidates.
Further experiments are needed to define efficiency and/or utilize combinations of therapies to achieve clearance of toxic aggregates as well as inhibition of TDP-43 pathology.
Conflicts of interest
There are no conflicts to declare.References
- L. François-Moutal, S. Perez-Miller, D. D. Scott, V. G. Miranda, N. Mollasalehi and M. Khanna, Structural Insights Into TDP-43 and Effects of Post-Translational Modifications, Front. Mol. Neurosci., 2019, 12, 301 CrossRef PubMed.
- M. D. Purice and J. P. Taylor, Linking HnRNP Function to ALS and FTD Pathology, Front. Neurosci., 2018, 12, 326 CrossRef PubMed.
- Y. M. Ayala, P. Zago, A. D’Ambrogio, Y. F. Xu, L. Petrucelli, E. Buratti and F. E. Baralle, Structural Determinants of the Cellular Localization and Shuttling of TDP-43, J. Cell Sci., 2008, 121, 3778–3785 CrossRef CAS PubMed.
- S. A. Davis, S. Itaman, C. M. Khalid-Janney, J. A. Sherard, J. A. Dowell, N. J. Cairns and M. A. Gitcho, TDP-43 Interacts with Mitochondrial Proteins Critical for Mitophagy and Mitochondrial Dynamics, Neurosci. Lett., 2018, 678, 8–15 CrossRef CAS PubMed.
- W. Wang, L. Wang, J. Lu, S. L. Siedlak, H. Fujioka, J. Liang, S. Jiang, X. Ma, Z. Jiang and E. L. Da Rocha, et al., The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity, Nat. Med., 2016, 22, 869–878 CrossRef CAS.
- C. Lagier-Tourenne, M. Polymenidou and D. W. Cleveland, TDP-43 and FUS/TLS: Emerging Roles in RNA Processing and Neurodegeneration, Hum. Mol. Genet., 2010, 19, R46–R64 CrossRef CAS.
- A. Ratti and E. Buratti, Physiological Functions and Pathobiology of TDP-43 and FUS/TLS Proteins, J. Neurochem., 2016, 138, 95–111 CrossRef CAS PubMed.
- M. Neumann, D. M. Sampathu, L. K. Kwong, A. C. Truax, M. C. Micsenyi, T. T. Chou, J. Bruce, T. Schuck, M. Grossman and C. M. Clark, et al., Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis, Science, 2006, 314, 130–133 CrossRef CAS PubMed.
- M. Neumann, L. K. Kwong, E. B. Lee, E. Kremmer, A. Flatley, Y. Xu, M. S. Forman, D. Troost, H. A. Kretzschmar and J. Q. Trojanowski, et al., Phosphorylation of S409/410 of TDP-43 Is a Consistent Feature in All Sporadic and Familial Forms of TDP-43 Proteinopathies, Acta Neuropathol., 2009, 117, 137–149 CrossRef CAS PubMed.
- T. Nonaka, F. Kametani, T. Arai, H. Akiyama and M. Hasegawa, Truncation and Pathogenic Mutations Facilitate the Formation of Intracellular Aggregates of TDP-43, Hum. Mol. Genet., 2009, 18, 3353–3364 CrossRef CAS PubMed.
- Y. Khalfallah, R. Kuta, C. Grasmuck, A. Prat, H. D. Durham and C. Vande Velde, TDP-43 Regulation of Stress Granule Dynamics in Neurodegenerative Disease-Relevant Cell Types/631/80/304/631/378/87/13/1/13/31/13/51/13/109/13/106/13/89/14/19/14/32/82/80 Article, Sci. Rep., 2018, 8, 1–49 CAS.
- D. Gordon, R. Dafinca, J. Scaber, J. Alegre-Abarrategui, L. Farrimond, C. Scott, D. Biggs, L. Kent, P. L. Oliver and B. Davies, et al., Single-Copy Expression of an Amyotrophic Lateral Sclerosis-Linked TDP-43 Mutation (M337V) in BAC Transgenic Mice Leads to Altered Stress Granule Dynamics and Progressive Motor Dysfunction, Neurobiol. Dis., 2019, 121, 148–162 CrossRef CAS PubMed.
- Y. Chen and T. J. Cohen, Aggregation of the Nucleic Acid– Binding Protein TDP-43 Occurs via Distinct Routes That Are Coordinated with Stress Granule Formation, J. Biol. Chem., 2019, 294, 3696–3706 CrossRef CAS PubMed.
- C. C. Chou, Y. Zhang, M. E. Umoh, S. W. Vaughan, I. Lorenzini, F. Liu, M. Sayegh, P. G. Donlin-Asp, Y. H. Chen and D. M. Duong, et al., TDP-43 Pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD, Nat. Neurosci., 2018, 21(2), 228–239 CrossRef CAS PubMed.
- J. Gao, L. Wang, T. Yan, G. Perry and X. Wang, TDP-43 Proteinopathy and Mitochondrial Abnormalities in Neurodegeneration, Mol. Cell. Neurosci., 2019, 100, 103396 CrossRef CAS PubMed.
- E. M. J. De Boer, V. K. Orie, T. Williams, M. R. Baker, H. M. De Oliveira, T. Polvikoski, M. Silsby, P. Menon, M. Van Den Bos and G. M. Halliday, et al., TDP-43 Proteinopathies: A New Wave of Neurodegenerative Diseases, J. Neurol., Neurosurg. Psychiatry, 2021, 92(1), 86–95 CrossRef PubMed.
- E. Buratti, Targeting TDP-43 Proteinopathy with Drugs and Drug-like Small Molecules, Br. J. Pharmacol., 2021, 1, 1298–1315 CrossRef PubMed.
- J. Fernández-Ruiz; E. de Lago; C. Rodríguez-Cueto and M. A. Moro, Recent Advances in the Pathogenesis and Therapeutics of Amyotrophic Lateral Sclerosis.British Journal of Pharmacology. John Wiley and Sons Inc, March 1, 2021, pp. 1253–1256 Search PubMed.
- A. Shodai, A. Ido, N. Fujiwara, T. Ayaki, T. Morimura, M. Oono, T. Uchida, R. Takahashi, H. Ito and M. Urushitani, Conserved Acidic Amino Acid Residues in a Second RNA Recognition Motif Regulate Assembly and Function of TDP-43, PLoS One, 2012, 7 Search PubMed.
- A. Saini and V. S. Chauhan, Delineation of the Core Aggregation Sequences of TDP-43 C-Terminal Fragment, ChemBioChem, 2011, 12, 2495–2501 CrossRef CAS PubMed.
- P. H. Kuo, L. G. Doudeva, Y. T. Wang, C. K. J. Shen and H. S. Yuan, Structural Insights into TDP-43 in Nucleic-Acid Binding and Domain Interactions, Nucleic Acids Res., 2009, 37, 1799–1808 CrossRef CAS PubMed.
- R. Rakhit, J. Robertson, V. C. Vande, P. Horne, D. M. Ruth, J. Griffin, D. W. Cleveland, N. R. Cashman and A. Chakrabartty, An Immunological Epitope Selective for Pathological Monomer-Misfolded SOD1 in ALS, Nat. Med., 2007, 13(6), 754–759 CrossRef CAS PubMed.
- Y. Tamaki, A. Shodai, T. Morimura, R. Hikiami, S. Minamiyama, T. Ayaki, I. Tooyama, Y. Furukawa, R. Takahashi and M. Urushitani, Elimination of TDP-43 Inclusions Linked to Amyotrophic Lateral Sclerosis by a Misfolding-Specific Intrabody with Dual Proteolytic Signals, Sci. Rep., 2018, 8(1), 6030 CrossRef PubMed.
- S. Rogers, R. Wells and M. Rechsteiner, Amino Acid Sequences Common to Rapidly Degraded Proteins: The PEST Hypothesis, Science, 1986, 234(4774), 364–368 CrossRef CAS PubMed.
- A. Sandoval, N. Oviedo, A. Tadmouri, T. Avila, M. De Waard and R. Felix, Two PEST-like Motifs Regulate Ca2+/Calpain-Mediated Cleavage of the CaVβ3 Subunit and Provide Important Determinants for Neuronal Ca2+ Channel Activity, Eur. J. Neurosci., 2006, 23(9), 2311–2320 CrossRef PubMed.
- S. Pozzi, S. S. Thammisetty, P. Codron, R. Rahimian, K. V. Plourde, G. Soucy, C. Bareil, D. Phaneuf, J. Kriz and C. Gravel, et al., Virus-Mediated Delivery of Antibody Targeting TAR DNA-Binding Protein-43 Mitigates Associated Neuropathology, J. Clin. Invest., 2019, 129(4), 1581–1595 CrossRef PubMed.
- A. Shodai, T. Morimura, A. Ido, T. Uchida, T. Ayaki, R. Takahashi, S. Kitazawa, S. Suzuki, M. Shirouzu and T. Kigawa, et al., Aberrant Assembly of RNA Recognition Motif 1 Links to Pathogenic Conversion of TAR DNA-Binding Protein of 43 KDa (TDP-43), J. Biol. Chem., 2013, 288, 14886–14905 CrossRef CAS PubMed.
- C. K. Chang, M. H. Chiang, E. K. W. Toh, C. F. Chang and T. H. Huang, Molecular Mechanism of Oxidation-Induced TDP-43 RRM1 Aggregation and Loss of Function, FEBS Lett., 2013, 587, 575–582 CrossRef CAS PubMed.
- V. Swarup, D. Phaneuf, N. Dupré, S. Petri, M. Strong, J. Kriz and J. P. Julien, Deregulation of TDP-43 in Amyotrophic Lateral Sclerosis Triggers Nuclear Factor KB-Mediated Pathogenic Pathways, J. Exp. Med., 2011, 208(12), 2429–2447 CrossRef CAS PubMed.
- V. Swarup, D. Phaneuf, C. Bareil, J. Robertson, G. A. Rouleau, J. Kriz and J. P. Julien, Pathological Hallmarks of Amyotrophic Lateral Sclerosis/Frontotemporal Lobar Degeneration in Transgenic Mice Produced with TDP-43 Genomic Fragments, Brain, 2011, 134(Pt 9), 2610–2626 CrossRef PubMed.
- J. A. Trejo-Lopez, Z. A. Sorrentino, C. J. Riffe, G. M. Lloyd, S. A. Labuzan, D. W. Dickson, A. T. Yachnis, S. Prokop and B. I. Giasson, Novel Monoclonal Antibodies Targeting the RRM2 Domain of Human TDP-43 Protein, Neurosci. Lett., 2020, 738, 135353 CrossRef CAS PubMed.
- R. Liu, G. Yang, T. Nonaka, T. Arai, W. Jia and M. S. Cynader, Reducing TDP-43 Aggregation Does Not Prevent Its Cytotoxicity, Acta Neuropathol. Commun., 2013, 1, 49 CrossRef PubMed.
- N. Gao, Y. P. Huang, T. T. Chu, Q. Q. Li, B. Zhou, Y. X. Chen, Y. F. Zhao and Y. M. Li, TDP-43 Specific Reduction Induced by Di-Hydrophobic Tags Conjugated Peptides, Bioorg. Chem., 2019, 84, 254–259 CrossRef CAS PubMed.
- H. S. Tae, T. B. Sundberg, T. K. Neklesa, D. J. Noblin, J. L. Gustafson, A. G. Roth, K. Raina and C. M. Crews, Identification of Hydrophobic Tags for the Degradation of Stabilized Proteins, ChemBioChem, 2012, 538–541 CrossRef CAS PubMed.
- B. Coll-Martínez, A. Delgado and B. Crosas, The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents, Molecules, 2020, 25(24), 5956 CrossRef PubMed.
- R. Spector, Transport of Amantadine and Rimantadine through the Blood–Brain Barrier, J. Pharmacol. Exp. Ther., 1988, 244(2), 516–519 CAS.
- N. Tsuzuki, T. Hama, M. Kawada, A. Hasui, R. Konishi, S. Shiwa, Y. Ochi, S. Futaki and K. Kitagawa, Adamantane as a Brain-directed Drug Carrier for Poorly Absorbed Drug. 2. AZT Derivatives Conjugated with the 1-adamantane Moiety, J. Pharm. Sci., 1994, 83(4), 481–484 CrossRef CAS PubMed.
- M. Kato, T. W. Han, S. Xie, K. Shi, X. Du, L. C. Wu, H. Mirzaei, E. J. Goldsmith, J. Longgood and J. Pei, et al., Cell-Free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels, Cell, 2012, 149, 753–767 CrossRef CAS PubMed.
- L. L. Jiang, M. X. Che, J. Zhao, C. J. Zhou, M. Y. Xie, H. Y. Li, J. H. He and H. Y. Hu, Structural Transformation of the Amyloidogenic Core Region of TDP-43 Protein Initiates Its Aggregation and Cytoplasmic Inclusion, J. Biol. Chem., 2013, 288, 19614–19624 CrossRef CAS PubMed.
- M. Mompeán, E. Buratti, C. Guarnaccia, R. M. M. Brito, A. Chakrabartty, F. E. Baralle and D. V. Laurents, Structural Characterization of the Minimal Segment of TDP-43 Competent for Aggregation, Arch. Biochem. Biophys., 2014, 545, 53–62 CrossRef PubMed.
- L. Lim, Y. Wei, Y. Lu and J. Song, ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43, PLoS Biol., 2016, 14 Search PubMed.
- L. L. Jiang, J. Zhao, X. F. Yin, W. T. He, H. Yang, M. X. Che and H. Y. Hu, Two Mutations G335D and Q343R within the Amyloidogenic Core Region of TDP-43 Influence Its Aggregation and Inclusion Formation, Sci. Rep., 2016, 6 Search PubMed.
- J. A. Cassel, M. E. McDonnell, V. Velvadapu, V. Andrianov and A. B. Reitz, Characterization of a Series of 4-Aminoquinolines That Stimulate Caspase-7 Mediated Cleavage of TDP-43 and Inhibit Its Function, Biochimie, 2012, 94, 1974–1981 CrossRef CAS PubMed.
- A. Prasad, G. Raju, V. Sivalingam, A. Girdhar, M. Verma, A. Vats, V. Taneja, G. Prabusankar and B. K. Patel, An Acridine Derivative, [4,5-Bis{(N-Carboxy Methyl Imidazolium)Methyl}acridine] Dibromide, Shows Anti-TDP-43 Aggregation Effect in ALS Disease Models, Sci. Rep., 2016, 6, 39490 CrossRef CAS PubMed.
- C. Korth, B. C. H. May, F. E. Cohen and S. B. Prusiner, Acridine and Phenothiazine Derivatives as Pharmacotherapeutics for Prion Disease, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(17), 9836–9841 CrossRef CAS.
- W. M. Babinchak, B. K. Dumm, S. Venus, S. Boyko, A. A. Putnam, E. Jankowsky and W. K. Surewicz, Small Molecules as Potent Biphasic Modulators of Protein Liquid-Liquid Phase Separation, Nat. Commun., 2020, 11(1), 5574 CrossRef CAS PubMed.
- L. François-Moutal, R. Felemban, D. D. Scott, M. R. Sayegh, V. G. Miranda, S. Perez-Miller, R. Khanna, V. Gokhale, D. C. Zarnescu and M. Khanna, Small Molecule Targeting TDP-43's RNA Recognition Motifs Reduces Locomotor Defects in a Drosophila Model of Amyotrophic Lateral Sclerosis (ALS), ACS Chem. Biol., 2019, 14, 2006–2013 CrossRef PubMed.
- R. Ihara, K. Matsukawa, Y. Nagata, H. Kunugi, S. Tsuji, T. Chihara, E. Kuranaga, M. Miura, T. Wakabayashi and T. Hashimoto, et al., RNA Binding Mediates Neurotoxicity in the Transgenic Drosophila Model of TDP-43 Proteinopathy, Hum. Mol. Genet., 2013, 22, 4474–4484 CrossRef CAS PubMed.
- N. Mollasalehi, L. Francois-Moutal, D. D. Scott, J. A. Tello, H. Williams, B. Mahoney, J. M. Carlson, Y. Dong, X. Li and V. G. Miranda, et al., An Allosteric Modulator of RNA Binding Targeting the N-Terminal Domain of TDP-43 Yields Neuroprotective Properties, ACS Chem. Biol., 2020, 15(11), 2854–2859 CrossRef CAS PubMed.
- M. Mompeán, V. Romano, D. Pantoja-Uceda, C. Stuani, F. E. Baralle, E. Buratti and D. V. Laurents, The TDP-43 N-Terminal Domain Structure at High Resolution, FEBS J., 2016, 283, 1242–1260 CrossRef PubMed.
- H. Qin, L. Z. Lim, Y. Wei and J. Song, TDP-43 N Terminus Encodes a Novel Ubiquitin-like Fold and Its Unfolded Form in Equilibrium That Can Be Shifted by Binding to SsDNA, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18619–18624 CrossRef CAS.
- C. K. Chang, T. H. Wu, C. Y. Wu, M. H. Chiang, E. K. W. Toh, Y. C. Hsu, K. F. Lin, Y. H. Liao, T. H. Huang and J. J. T. Huang, The N-Terminus of TDP-43 Promotes Its Oligomerization and Enhances DNA Binding Affinity, Biochem. Biophys. Res. Commun., 2012, 425, 219–224 CrossRef CAS PubMed.
- G. Nshogoza, Y. Liu, J. Gao, M. Liu, S. A. Moududee, R. Ma, F. Li, J. Zhang, J. Wu and Y. Shi, et al., NMR Fragment-Based Screening against Tandem RNA Recognition Motifs of TDP-43, Int. J. Mol. Sci., 2019, 20(13), 3230 CrossRef CAS.
- P. J. Lukavsky, D. Daujotyte, J. R. Tollervey, J. Ule, C. Stuani, E. Buratti, F. E. Baralle, F. F. Damberger and F. H. T. Allain, Molecular Basis of UG-Rich RNA Recognition by the Human Splicing Factor TDP-43, Nat. Struct. Mol. Biol., 2013, 20, 1443–1449 CrossRef CAS.
- Y. Furukawa, Y. Suzuki, M. Fukuoka, K. Nagasawa, K. Nakagome, H. Shimizu, A. Mukaiyama and S. Akiyama, A Molecular Mechanism Realizing Sequence-Specific Recognition of Nucleic Acids by TDP-43, Sci. Rep., 2016, 6 Search PubMed.
- B. N. Flores, X. Li, A. M. Malik, J. Martinez, A. A. Beg and S. J. Barmada, An Intramolecular Salt Bridge Linking TDP43 RNA Binding, Protein Stability, and TDP43-Dependent Neurodegeneration, Cell Rep., 2019, 27, 1133–1150.e8 CrossRef CAS.
- K. Cyrus, M. Wehenkel, E. Y. Choi, H. Swanson and K. B. Kim, Two-Headed PROTAC: An Effective New Tool for Targeted Protein Degradation, ChemBioChem, 2010, 11(11), 1531–1534 CrossRef CAS PubMed.
- Q. Cao, D. R. Boyer, M. R. Sawaya, P. Ge and D. S. Eisenberg, Cryo-EM Structures of Four Polymorphic TDP-43 Amyloid Cores, Nat. Struct. Mol. Biol., 2019, 26, 619–627 CrossRef CAS PubMed.
- E. L. Guenther, Q. Cao, H. Trinh, J. Lu, M. R. Sawaya, D. Cascio, D. R. Boyer, J. A. Rodriguez, M. P. Hughes and D. S. Eisenberg, Atomic Structures of TDP-43 LCD Segments and Insights into Reversible or Pathogenic Aggregation, Nat. Struct. Mol. Biol., 2018, 25, 463–471 CrossRef CAS PubMed.
- Q. Li, W. M. Babinchak and W. K. Surewicz, Cryo-EM Structure of Amyloid Fibrils Formed by the Entire Low Complexity Domain of TDP-43, Nat. Commun., 2021, 12 Search PubMed.
- E. L. Guenther, P. Ge, H. Trinh, M. R. Sawaya, D. Cascio, D. R. Boyer, T. Gonen, Z. H. Zhou and D. S. Eisenberg, Atomic-Level Evidence for Packing and Positional Amyloid Polymorphism by Segment from TDP-43 RRM2, Nat. Struct. Mol. Biol., 2018, 25, 311–319 CrossRef CAS PubMed.
- P. M. Seidler, D. R. Boyer, J. A. Rodriguez, M. R. Sawaya, D. Cascio, K. Murray, T. Gonen and D. S. Eisenberg, Structure-Based Inhibitors of Tau Aggregation, Nat. Chem., 2018, 10 Search PubMed.
- T. Afroz, E. M. Hock, P. Ernst, C. Foglieni, M. Jambeau, L. A. B. Gilhespy, F. Laferriere, Z. Maniecka, A. Plückthun and P. Mittl, et al., Functional and Dynamic Polymerization of the ALS-Linked Protein TDP-43 Antagonizes Its Pathologic Aggregation, Nat. Commun., 2017, 8, DOI:10.1038/s41467-017-00062-0.
| This journal is © The Royal Society of Chemistry 2021 |