
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiological functions of supramolecular assemblies of small molecules in the cellular environment
Jingyu
Wang
 *a,
Hui
Li
a and
Bing
Xu
*b
*a,
Hui
Li
a and
Bing
Xu
*b
aSchool of Biomedical Engineering and Technology, Tianjin Medical University, Tianjin 300070, P. R. China. E-mail: wangjy_026@tmu.edu.cn
bDepartment of Chemistry, Brandeis University, Waltham, Massachusetts 02454, USA. E-mail: bxu@brandeis.edu
First published on 28th January 2021
Abstract
Like biomacromolecules, certain small molecules (e.g., aggregators) are able to self-assemble in the aqueous phase to form nanoscale aggregates. Though it is well-established that the aggregates may interact with enzymes in vitro, the study of the biological activities of assemblies of small molecules in the cellular environment is only at its beginning. This review summarizes the recent progress in exploring the biological functions of supramolecular assemblies of small molecules (SASMs). We first discuss the use of SASMs to inhibit pathogenic cells, such as cancer cells and bacteria. The use of SASMs to target different parts of cancer cells, such as the pericellular space, the cytosol, and subcellular organelles, and to combine with other bioactive entities (e.g., proteins and clinically used drugs), is particularly promising for addressing the challenge of acquired multidrug resistance in cancer therapy. Then, we describe the use of SASMs to sustain physiological functions of normal cells, that is, promoting cell proliferation and differentiation for tissue regeneration. After that, we show the use of SASMs as a basic tool to research cell behaviors, for instance, identifying specific cells, improving enzyme probes, revealing membrane dynamics, imaging molecular self-assembly, and mimicking context-dependent signaling. Finally, we give an outlook on the research of SASMs. We expect that this review, by highlighting the biological functions of SASMs, provides a starting point to explore the chemical biology of SASMs.
 Jingyu Wang | Jingyu Wang received her BS from Hebei Normal University in 2008. She obtained her PhD in 2014 from Nankai University under the supervision of Professor Zhimou Yang. She has been working at Tianjin Medical University since 2014, and went to Professor Bing Xu's laboratory as a visiting scholar in 2020. Her research interests focus on applications of peptide self-assembly in biology. |
 Hui Li | Hui Li is a fourth-year-undergraduate student at Tianjin Medical University currently. He is now following Dr Jingyu Wang in the research of peptide self-assembly in biological applications. |
 Bing Xu | After receiving his BS and MS from Nanjing University in 1987 and 1990, Bing Xu obtained his PhD in 1996 from the University of Pennsylvania. Before starting his independent research at the Hong Kong University of Science and Technology in Aug 2000, he was an NIH postdoctoral fellow at Harvard University. He was tenured as an associated professor in Jan 2006 and became a full professor in July 2008 at HKUST. Bing Xu currently is a professor in the Department of Chemistry, Brandeis University. His current research focuses on the applications of enzymatic noncovalent synthesis in materials, biology, and medicine. |
1. Introduction
Molecular self-assembly – the spontaneous, noncovalent association of molecules to give higher order structures – is a fundamental cellular process in biology. As a ubiquitous process in nature, endogenous protein monomers self-organize to form supramolecular assemblies (e.g., filaments of actin and tubulin)1 in the cellular environment, which are indispensable for normal cellular functions (e.g., cell migration, mitosis, and mass transportation). Moreover, it is common for protein assemblies to exhibit emergent properties. For example, the functions of tubulin filaments (e.g., as part of the machinery of mitosis) drastically differ from the functions of a single tubulin (e.g., as a GTPase).2Like proteins, small molecules also self-assemble to form supramolecular assemblies to exhibit emergent properties that drastically differ from those of the individual molecules. For example, lipid rafts,3 an endogenous version of assemblies of small molecules (e.g., cholesterols and lipids), remain a notable mystery in cell biology. In fact, advances in several unrelated fields (e.g., biomaterials, drug screening, and neurodegenerative diseases) over the past decade have highlighted the biological importance of supramolecular assemblies of small molecules (SASMs), which are also a form of molecular self-assembly. The development of small molecule hydrogels for biomedical applications such as lanreotide autogel for treating acromegaly4 clearly validates the application of SASMs in biomedicine. Being able to sequester enzymes or unfolded proteins,5 block β-amyloid formation,6 and activate enzymes,7 SASMs are emerging as a new molecular entity in biomedical research. SASMs are capable of modulating cell differentiation,8 maintaining cell growth,9–15 inducing cell death,7,16–20 inhibiting cancer cell growth in vivo,21 or promoting the proliferation of stem cells.22 These facts indicate that SASMs are able to interact with cells and to exhibit emergent properties – that is, specific biological functions that are drastically different from their individual molecules23 – a phenomenon that is more common and important than one previously thought. Thus, it is worthwhile to review the work on the biological functions of SASMs in the cellular environment for the future development of SASMs.
Although considerable work has been published on small molecule assembly with biological applications, such as the applications of liposomes in biomedicine, we choose to review small peptides other than lipids because the structural diversity of peptides offers (almost) unlimited opportunity for designing functions via engineering the molecules and the processes of assembly, especially in the context of enzymatic reactions. In the following sections, we first discuss the use of SASMs to inhibit pathogenic cells, such as cancer cells and bacteria, particularly the potential for using SASMs to address the challenges of acquired multidrug resistance in cancer therapy and antimicrobial therapy. Then, we describe the use of SASMs to sustain physiological functions of normal cells, such as promoting cell proliferation and differentiation for tissue engineering. After that, we summarize the use of SASMs as a useful tool to study cell behaviors, for instance, identifying specific cells, improving enzyme probes, revealing membrane dynamics, imaging molecular self-assembly, and mimicking context-dependent signaling. Finally, we discuss the outlook of this emerging research area. We expect this review to provide a starting point to study the chemical biology of SASMs, which is a new direction for exploring the functions of noncovalent synthesis.24,25
2. SASMs inhibit pathogenic cells
Self-assembly of small molecules in water is a rather common observation, as evidenced by the identification of aggregators during drug screening.7,26,27 High-throughput screening is a common method for identifying drug candidates. Among the filtered products during screening, “false positive” ones that have non-drug-like traits are predominant. This observation stimulates efforts for researching these “futile” substances, consequently finding that the aggregates, not the individual molecules, are the source of the observed biological activities.28 These results have inspired the study of SASMs, neither the individual constituents29 nor the precipitates,30 for various biological functions, such as inhibition of cancer cells or bacteria.31,322.1 SASMs for killing cancer cells selectively
Cancer is a devastating disease that remains a major threat to public health. The most significant challenge in cancer therapy is adaptive drug resistance caused by genomic instability and the tumor microenvironment even among the initially effective treatments.33,34 Although cancer would, apparently, have little to do with Alzheimer's disease, an etiological study (i.e., the Framingham Heart Study) on a large population of patients concluded that the patients with Alzheimer's disease have a lower risk of cancers.35 This serendipitous discovery of the inverse comorbidity between cancer and Alzheimer's disease has a profound implication – that is, supramolecular fibrils formed by self-assembly inhibit cancer cells, and it would be unlikely for cancer cells to develop resistance towards fibrils. This hypothesis has led to selective generation of SASMs via enzymatic reactions inside cancer cells for killing the cancer cells. For example, as shown in Fig. 1A, after compound 1a diffuses into HeLa cells, intracellular esterase catalytically cut off the hydrophilic butyric diacid (2) to generate compound 1 inside the cells. The molecules of 1, having strong intermolecular aromatic–aromatic interactions, self-assemble to form nanofibers, thus increasing the intracellular viscosity and inhibiting HeLa cells.36 This work also represents the first example of modulating the intracellular phase transition by man-made molecules for controlling the cell fate. A later study shows that SASMs, indeed, minimize acquired drug resistance.37,38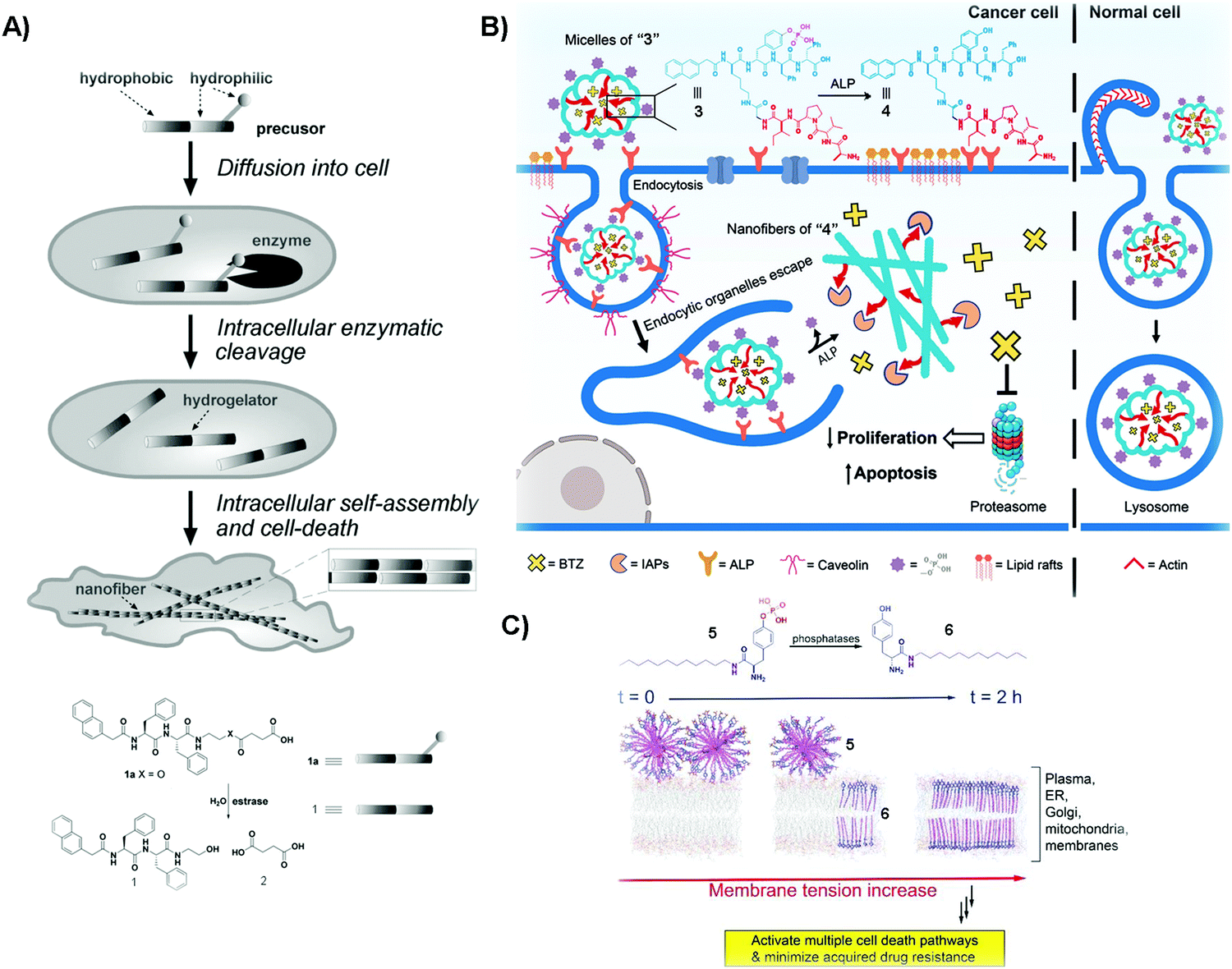 | ||
| Fig. 1 (A) Illustration of the process of killing cancer cells, and the molecular structures of 1a and its corresponding products resulting from esterase catalyzed hydrolysis. (B) Molecular structures of 3 and 4 and the diagram of compound 3 inhibiting cancer cells and rescuing normal ones. (C) Molecular structures of 5 and 6 and the illustration of SASMs increasing the membrane tension to inhibit cancer cells. Reproduced with permission from ref. 36, 38 and 40. | ||
One unique property of 1 is that 1 selectively kills cancer cells, but not normal cells. This unique feature promises SAMSs as a fundamentally new molecular entity for developing cancer therapy to increase efficacy, but without increasing toxicity. This notion turns out to be valid and general. As shown in Fig. 1B, compound 3, a phosphorylated peptide bearing peptide AVPI, an antagonist to the inhibitors of apoptotic proteins (IAPs) that impede the anticancer drug BTZ, is capable of incorporating BTZ. Normally, alkaline phosphatase (ALP), which exists in both the intra- and extracellular space,39 is overexpressed in/on cancer cells and acts as a cancer biomarker. The quantity of ALP is based on the type of cancer cells.39 Herein, the micelles of “3” interact with ALP from the cell surface to initiate endocytosis to enter cancer cells, and cytosolic overexpressed ALP in these cells cuts off the hydrophilic phosphate groups, converting the micelles of “3” to the nanofibers of “4”, antagonizing IAPs and releasing BTZ to inhibit cancer cells. Herein, ALP, being overexpressed in cancer cells, but not in normal cells, acts as a context-dependent signal. That is, the micelles of “3” enter these normal cells by macropinocytosis, and then traffic BTZ into the lysosome because normal expression of ALP is unable to drastically speed up dephosphorylation of the micelles. The trafficking of BTZ to lysosomes thus minimizes the side effects of BTZ on normal cells.40 In this study, the enzymatic reaction enables selectively generating functional SASMs in cancer cells, but not in normal cells. This approach should be applicable to the use of other enzymes to form SASMs for targeting cancer cells.
Multidrug resistance (MDR), which emerges in patients receiving long-term anticancer medication, is a major challenge in cancer therapy. In the complex tumor microenvironment, lipids are able to control multidrug-efflux pumps in cancer cells, and their dysfunction leads to MDR in cancer.41 Therefore, lipids represent an important and challenge target for countering MDR because of the large quantity and heterogeneity of lipids.42 Designing SASMs to target lipid membranes promises an effective approach against MDR cells. As shown in Fig. 1C, a lipid derivative (5) self-assembles into micelles. ALP, overexpressed on cancer cells, catalyzes the dephosphorylation of 5 to insert the self-assembled 6 into membranes. This process increases the tension of the cell membrane, thus stimulating a variety of regulated cell-death pathways and minimizing acquiring MDR.38 In the above studies, the SASMs exhibit emergent properties because individual molecules (i.e., without assembling) are innocuous to cells. Moreover, SASMs, being generated by a specific enzymatic reaction, are also able to interact with multiple targets or to disrupt multiple pathways in cancer cells,43 which makes SASMs attractive molecular entities for killing cancer cells effectively, reducing side effects, and minimizing MDR in cancer therapy.44,45
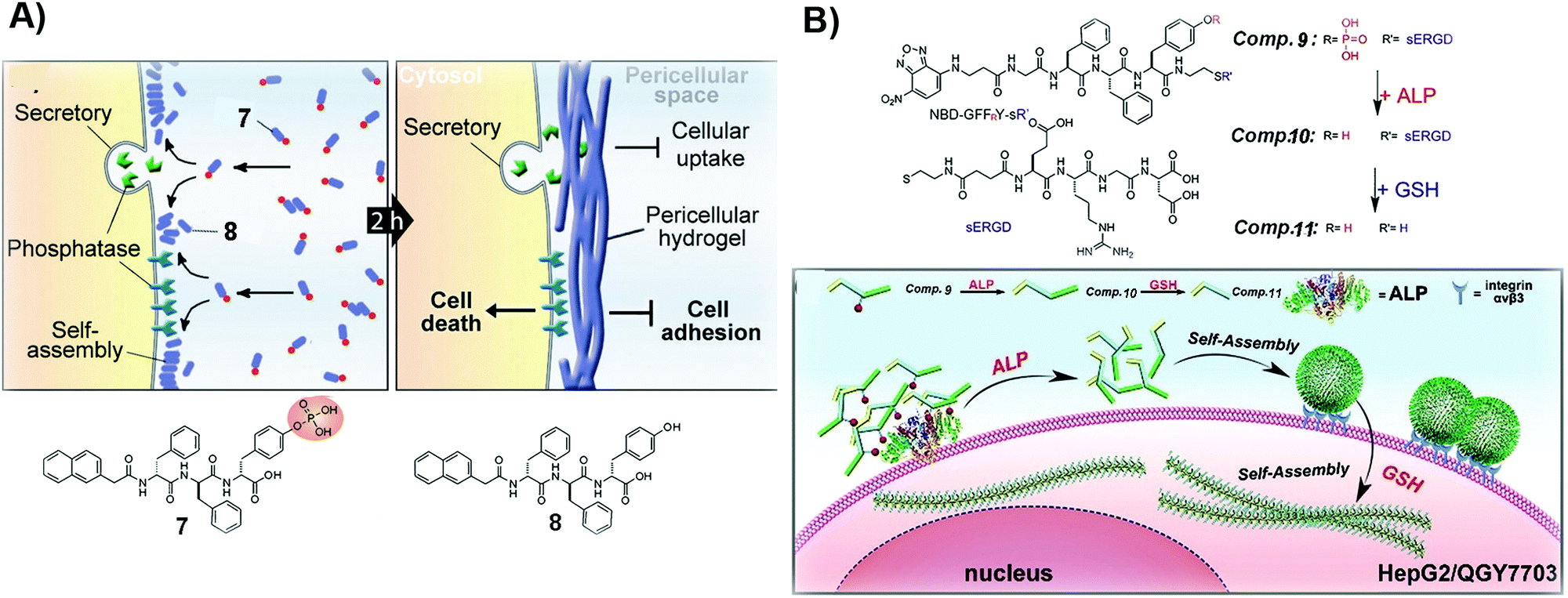 | ||
| Fig. 2 (A) Illustration of the formation of a pericellular hydrogel and the molecular structures of 7 and 8. (B) Molecular structures of 9–11 and the diagram of the process of tandem self-assembly. Reproduced with permission from ref. 39 and 46. | ||
For certain cancer cells, targeting only one extra- or intracellular location is insufficient to effectively inhibit cancer cells. Thus, tandem molecular self-assembly, acting as an approach to increase the sophistication of SASMs, is able to interact with cancer cells more extensively at multiple locations for increasing the anticancer efficacy. As shown in Fig. 2B, ALP, acting as a key factor, hydrolyzes 9 to remove the phosphate group from 9. The resulting molecule, 10, self-assembles to form nanoparticles, which adhere to cancer cells because of the RGD motif, a ligand of integrin, in 10. When the assemblies of 10 enter cells and are reduced by glutathione (GSH), the nanoparticles lose the hydrophilic part (ERGD) to generate nanofibers made of 11. Being superior to the self-assembly controlled only by ALP, this tandem molecular assembly is able to inhibit cancer cells efficiently. Because cancer cells express cancer biomarkers, such as ALP and GSH, at higher levels than normal cells do, ALP and GSH contribute to the high efficacy of 9 against cancer cells.39 This approach also provides a way to target cancer cells with more precise spatial control. The principle of tandem molecular assembly should be useful for targeting multiple other targets or locations, such as subcellular organelles, for countering MDR and reducing adverse effects in cancer therapy.
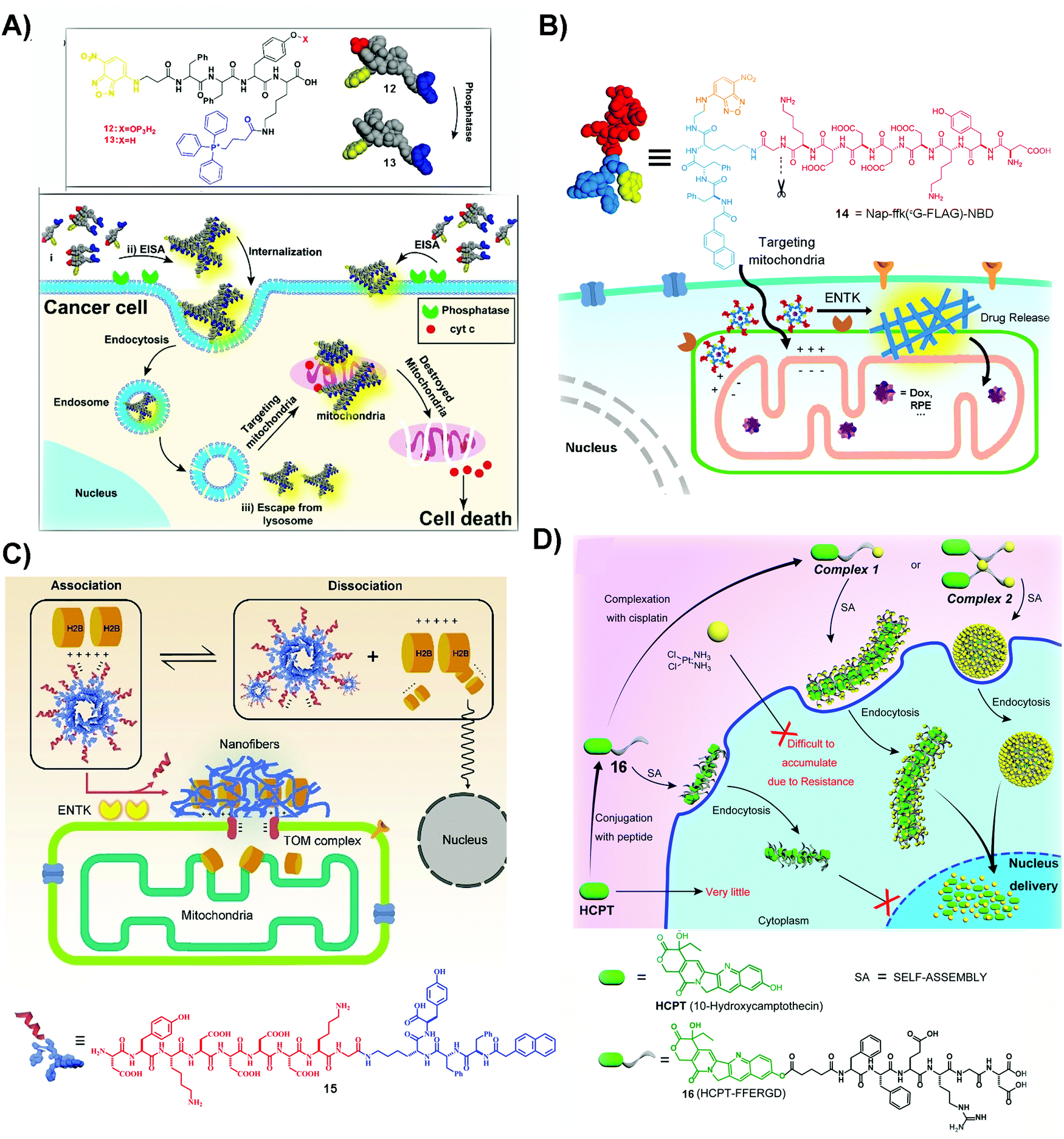 | ||
| Fig. 3 (A) The structures of 12 and 13 and the illustration of 12 inhibiting cancer cells. Reproduced with permission from ref. 56. Copyright 2016, American Chemical Society. (B) The structure of 14 and the diagram of 14 targeting mitochondria by ENTK-instructed self-assembly. Adapted with permission from ref. 57. Copyright 2018, American Chemical Society. (C) Illustration of retention of H2B on mitochondria and the molecular structure of 15. Reproduced with permission from ref. 62. (D) Illustration of a dual drug system to locate the nanofibers and nanoparticles at the nucleus and the molecular structure of 16. Adapted with permission from ref. 64. Copyright 2017, American Chemical Society. | ||
An unexpected molecule for targeting mitochondria is 14, a substrate of enterokinase (ENTK). ENTK, in fact, is a protease, which presents on the mitochondria of HeLa cells.57 ENTK-instructed self-assembly generates SASMs for targeting mitochondria of cancer cells. As shown in Fig. 3B, 14, the peptide derivative bearing a Flag tag (i.e. a protein tag),58 forms micelles. In HeLa cells, the cleavage of the Flag by ENTK turns the micelles to nanofibers, which are largely located on mitochondria. The micelles formed by the precursor (14) are capable of delivering doxorubicin (Dox), an anticancer drug, into the cancer cells, and then the Dox is mainly released to mitochondria, which causes increased cytotoxicity against HeLa cells compared to Dox only.57 This is the first report to employ enzymatic noncovalent synthesis24 for targeting mitochondria, complementing the “TPP” approach.56 This approach should be applicable to the use of other enzymes and to inhibit other cancer cells, which also stimulates efforts to design more precursors that allow enzymatic reactions to generate SASMs at mitochondria of cancer cells, as shown by the recent work of Sun et al.59 In addition, the process of forming SASMs catalyzed by ENTK also finds applications for delivering genes selectively into mitochondria of cancer cells.60
The process of generating SASMs by ENTK is also able to traffic proteins, such as histone 2B, to mitochondria. H2B normally localizes in the nucleus and is absent in mitochondria. Although a proteomic analysis reports the existence of H2B in the mitochondria of a liver cancer cell,61 the detailed mechanism remains to be elucidated. Interestingly, ENTK catalyzed SASMs traffic H2B into mitochondria, which may provide a way for better understanding inter-organelle communication in cancer cells. As shown in Fig. 3C, 15 is a molecule similar to 14, both of them having the Flag tag. The molecule of 15 is able to interact with H2B, preventing H2B into nucleus. With the catalysis of mitochondrial ENTK, 15 loses the Flag tag and self-assembles to form nanofibers, which retain H2B and traffic this protein into mitochondria. In addition, the cytotoxicity of a combination of 15 and Dox to cancer cells is higher than that of Dox alone.62 This method underscores the use of SASMs for manipulating inter-organelle crosstalk, which remains underexplored. In addition, this method should be applicable to combine other clinical drugs with SASMs for inhibiting cancer cells effectively.
The past few years have seen the exploration of SASMs for targeting different organelles in cancer therapy. Besides mitochondria, SASMs are also able to target other organelles, such as the endoplasmic reticulum63 and nucleus.64,65 One of the merits is that SASMs can deliver clinical anticancer drugs, which interact with DNA or DNA-associated proteins, into the nucleus. As shown by the work of Yang et al., SASMs traffic cisplatin and 10-hydroxycamptothecine (HCPT) into the nucleus to increase the anticancer efficacy. In that study, cisplatin, by chelation of itself with a carboxylate of an amino acid, is able to bond with 16 to form a dual drug-delivery system (Fig. 3D). Adding 1 and 1.5 equivalents of cisplatin into compound 16 forms complex 1 and 2, which self-assemble to form nanofibers and nanoparticles, respectively. These nanofibers or nanoparticles are able to accumulate in the nucleus, inhibiting cancer cells effectively because cisplatin chelates with DNA to inhibit DNA synthesis and HCPT antagonizes DNA topoisomerase I.64,66,67 Another dual drug design, in which the chelated cisplatin is up to 8 equivalents, is able to achieve superior nucleus accumulation when the actual loading of cisplatin is 4 equivalents. This approach offers excellent inhibitory efficiency against cancer cells,65 thus promising the use of SASMs to target subcellular organelles for high inhibitory activity against cancer cells and to engineer more molecules to form higher-order assemblies for countering MDR in cancer therapy.
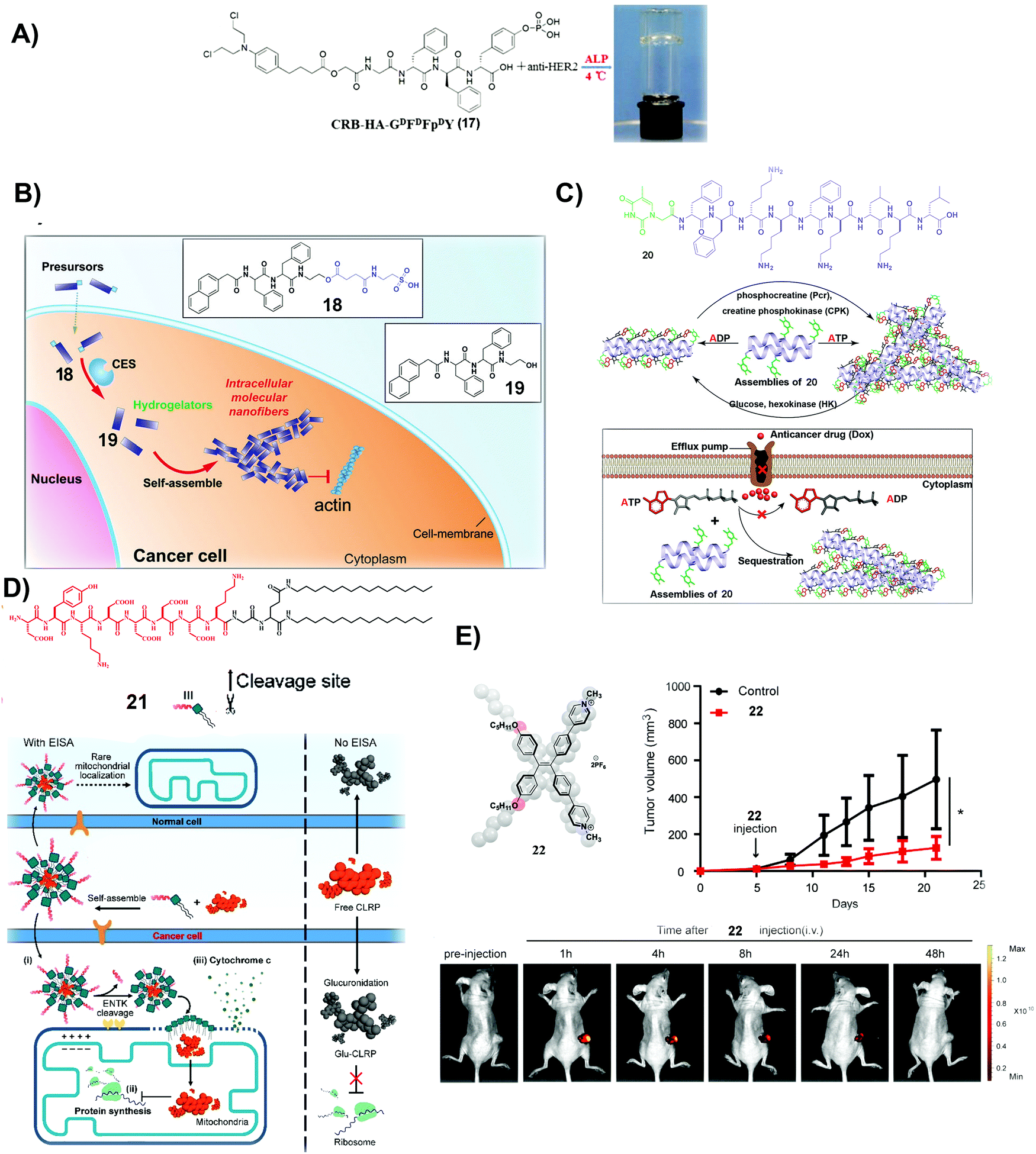 | ||
| Fig. 4 (A) Molecular structure of 17 and optical image of the co-assembled hydrogel. (B) Molecular structures of 18 and 19 and illustration of the process of self-assembly. (C) Molecular structure of 20 and the illustration of the nanostructure change by interconverting ATP and ADP and the mechanism of slowing down the efflux pumps to retain DOX. (A–C) Reproduced with permission from ref. 68–70. (D) Molecular structure of 21 and the plausible mechanism of 21 and CLRP inhibiting cancer cells over normal ones. (E) Molecular structure of 22 and measurement of tumor volume and fluorescent visualization of tumors after intravenous injection of 22 in tumor-bearing mice. (D and E) Adapted with permission from ref. 71 and 72. Copyright 2020, American Chemical Society. | ||
Besides covalently conjugating a clinically used anticancer drug (e.g., CRB) to small molecules to form SASMs (e.g., SASMs of 17), co-incubating first-line anticancer drugs and SASMs provides a simpler method to inhibit cancer cells.69,70Fig. 4B shows an amphiphile molecule 18, which is a substrate of carboxylesterase (CES), an enzyme overexpressed in ovarian cancer cells. After entering cells and being hydrolyzed by CES, 18 turns into 19, which self-assembles to form nanofibers. Co-incubation of 18 and cisplatin boosts the activity of cisplatin against SKOV3 and A2780cis cells, two kinds of drug-resistant ovarian cells.69 This method, based on SASMs, considerably increases the anticancer efficacy and reduces MDR. Thus, SASMs should be applicable to sensitize cancer cells to other first-line anticancer drugs for enhancing the efficacy of anticancer drugs when resistance to those anticancer drugs emerges.
Because efflux of anticancer drugs is a common mechanism for MDR in cancer therapy, targeting the efflux pumps is a promising approach against MDR. As shown in Fig. 4C, a nucleopeptide, 20, of which the capping group is thymine, is able to form SASMs for sequestering ATP. Because efflux pumps rely on ATP to be functional, sequestering ATP by SASMs of 20 impedes the efflux of drugs, thus reducing MDR. For example, co-incubation of 20 and doxorubicin (DOX), an anticancer drug, is capable of retaining DOX to inhibit MDR cancer cells.70 This work highlights the use of SASMs to impede MDR in cancer therapy, which contributes to the efforts to search for more SASMs to encapsulate clinical drugs for increasing the toxicity of these drugs against MDR cancer cells.
Combining antibiotics with SASMs also drastically increases the inhibitory efficacy of the antibiotics against cancer cells. Chloramphenicol (CLRP), a clinically approved antibiotic, is ineffective to inhibit live cancer cells because cytosolic glucuronidase detoxifies it.75 A peptide–lipid conjugate, 21, as a substrate of ENTK on mitochondria, forms micelles to encapsulate CLRP. Being catalytically hydrolyzed by ENTK to lose the Flag, the micelles become SASMs and release CLRP into mitochondria of cancer cells, as shown in Fig. 4D. Because mitochondria lack antagonistic glucuronidases to detoxify CLRP, CLRP inhibits mitochondrial protein synthesis and interrupts the metabolism of these liver cancer cells. Such inhibition of mitochondrial protein synthesis sensitizes the cancer cells to cisplatin.71 This intriguing phenomenon expands the applications of SASMs to repurpose clinically validated drugs for effectively suppressing cancer cells and reducing the systemic burden.
Photodynamic therapy (PDT) is a useful approach to minimize MDR. Therefore, a photosensitizer is another kind of molecule for combining with SASMs to inhibit cancer cells more efficiently. Based on the design principle of theranostics, which integrates diagnosis and therapy into the same molecular platform, fluorescent molecule assemblies are another type of SASMs for cancer theranostics. As shown in Fig. 4E, 22, which generates fluorophore SASMs formed by the aggregation of a fluorophore, is able to act as a PDT agent for imaging and inhibiting colon tumors simultaneously.72 This study expands the molecular space of SASMs to fluorescent and phototoxic small molecules that aggregate in aqueous phases.
2.2 SASMs for inhibiting bacteria specifically
Microbial infection lowers the quality of life. In some acute and severe microbial infections, such as in osteomyelitis, amputation or death usually happens. Despite the development of antibiotics for reducing the mortality and morbidity of patients, the emergence of antimicrobial drug resistance remains a threat to public health. Generally, effective targeting and increasing treatment efficacy are two main approaches to counter MDR.76,77 The unique merits of SASMs, such as acting over multiple length scales and quick responses to biological cues,78–80 make them attractive candidates for overcoming MDR. For example, lipid-based SASMs are capable of switching their anti-infectious ability “on” and “off” in response to changes of pH.81 Moreover, some enzymes, being overexpressed on or in microbial cells, are able to act as a trigger to form SASMs for specifically inhibiting bacteria.82 In addition, combination therapy, such as using antibiotics or proteins to combine with SASMs, should inhibit microbial invasion more efficiently than each single agent. In this section, we mainly discuss enzymes as the trigger for inhibiting microbial infection and combining SASMs with other bioactive entities, such as antibiotics and proteins, to inhibit infection and to reduce MDR.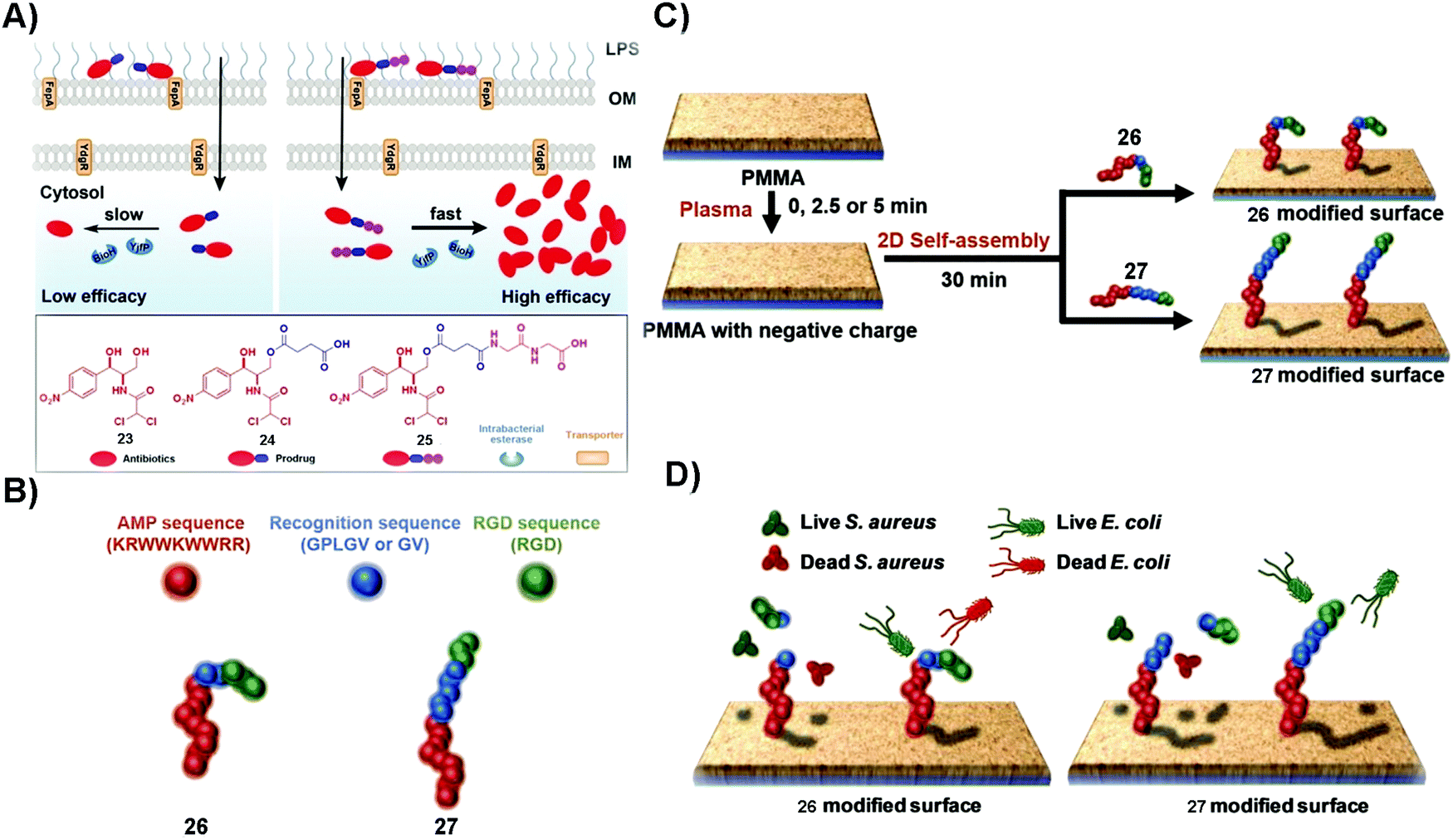 | ||
| Fig. 5 (A) Plausible mechanism of 25 to inhibit Gram-negative bacteria more effectively than 24 and the molecular structures of 23, 24 and 25. (B) The construction of the fusion peptides, 26 and 27. (C) The generation of fusion peptide (26 or 27)-modified surfaces. (D) The assemblies of 26 kill both S. aureus and E. coli and those of 27 distinguish S. aureus and E. coli strains and kill the bacteria selectively. Reproduced with permission from ref. 83 and 85. | ||
Broad-spectrum antibiotics, such as chloramphenicol (23), have the merit of killing many varieties of microorganism, but they are more likely to result in MDR, especially in that patients usually undergo long term administration of this type of antibiotics. Using SASMs to target gelatinase is able to specifically inhibit one certain kind of bacteria over another one. As shown in Fig. 5B, the fusion peptide (26 or 27) consists of an antimicrobial peptide (red part), a recognition sequence (blue parts) that is the substrate of bacterial gelatinase,86 and a cellular adhesion sequence (green part). As shown in Fig. 5C and D, 26 and 27 are able to generate 2D assemblies on a poly(methyl methacrylate) (PMMA) surface, which exert inhibitory activity against bacteria by gelatinase-catalyzed cleavage of the assemblies of 26 and 27. While the assemblies of 26 kill both S. aureus and E. coli, the assemblies of 27 are able to differentiate S. aureus and E. coli strains,85 likely due to the difference in the bacterial cell membranes. This study provides an approach to design SASMs to kill a certain kind of microbial cells over another one, which may pave the way to design SASMs as narrow-spectrum antibiotics for reducing the odds of evolving antimicrobial MDR.
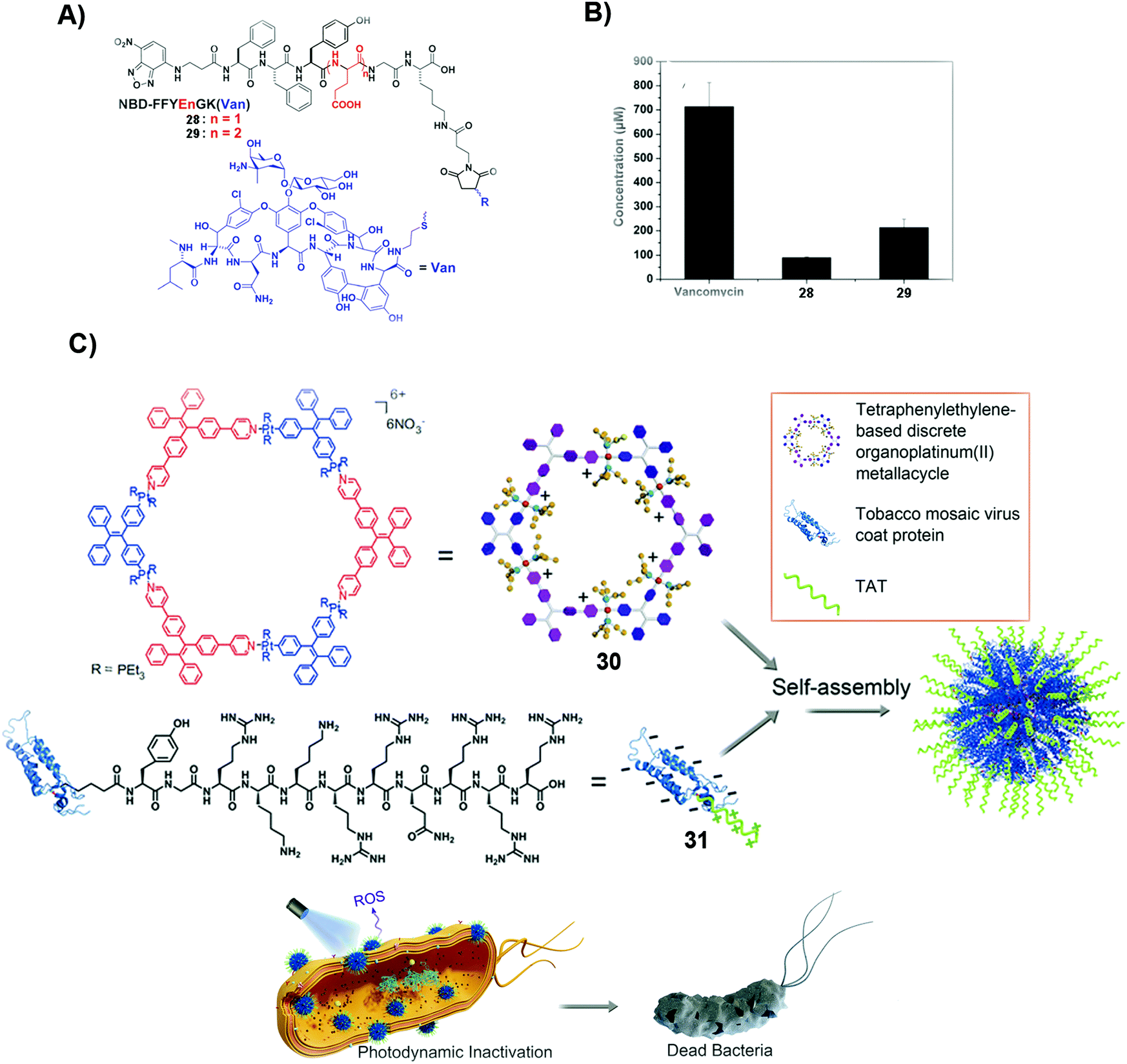 | ||
| Fig. 6 (A) Molecular structures of 28 and 29. (B) The minimum concentrations of different compounds, such as vancomycin, 28 and 29, to inhibit drug-resistant bacteria, E. faecalis. (A and B) Reproduced with permission from ref. 87. Copyright 2014, Royal Society of Chemistry. (C) Mechanism of deactivating and inhibiting bacteria by SASMs combined with proteins. Reproduced with permission from ref. 89. | ||
Photodynamic inactivation (PDI) is to damage the bacterial membrane and induce bacterial death by photoirradiation. The advantage of PDI is that it minimizes the odds of MDR. But obtaining photosensitizers that have the ability to intercalate bacterial cell membranes is challenging. Combining SASMs that incorporate the photosensitizer with a protein leads to the intercalation of cell membranes and enhanced PDI efficacy against bacteria. As shown in Fig. 6C, the photosensitizer, 30, and the transacting activator of transduction (TAT) peptide-decorated protein (31), which has the ability to insert into the membranes, self-assemble to form nanoparticles, which intercalate cell membranes, promote ROS production, and boost the PDI efficiency to inhibit bacteria.89 This study underscores the combination of SASMs with proteins to inhibit bacteria, which may stimulate the design of other SASMs to combine with proteins against MDR without the need for light.
3. SASMs for maintaining physiological functions of normal cells
SASMs find applications not only in eliminating pathogenic cells, but also in sustaining physiological functions of normal cells, such as promoting cell proliferation and differentiation for tissue engineering, as discussed in the following section.3.1 SASMs promote cell proliferation and differentiation
SASMs are able to mimic the extracellular matrix (ECM) for maintaining cell proliferation. As shown in Fig. 7A, 32, being reduced by TCEP, forms 33, which self-assembles to form a hydrogel. The hydrogel undergoes directional domain sliding motion by the hydrophobic collapse transition. The sliding motion within the hydrogel accelerates cell proliferation by mechanotransduction, which translocates yes-associated protein, YAP, from the cytosol into the nucleus.90 This work highlights SASMs to mimic the ECM to promote cell proliferation, which implies the promise of SASMs in other fields, such as in cell differentiation. | ||
| Fig. 7 (A) Molecular structures of 32 and 33 and the process of formation of the hydrogel and possible mechanism of the hyrdogel stimulating cell proliferation. Adapted with permission from ref. 90. Copyright 2020, Royal Society of Chemistry. (B) Molecular structure of 34 and illustration of neural differentiation by adding BMSCs on 34, which self-assembles into nanofibers. Adapted with permission from ref. 92. Copyright 2018, American Chemical Society. | ||
Traumatic injuries in the central nervous system (CNS) result from damaging nervous tissue. Thus, it is attractive to use hydrogels made of SASMs to mimic the ECM for the regeneration of neurons. Since laminin, as an indispensable protein of the ECM, plays a critical role in cell differentiation during nerve regeneration,91 SASMs, as a mimic of laminin in the ECM, are able to bias the differentiation of neuron progenitor cells for forming neurites, which promises a potential approach for repairing the CNS. As shown in Fig. 7B, 34, based on the laminin-mimetic peptide IKVAV, self-assembles to form nanofibers, which drive differentiation of human bone marrow mesenchymal stem cells (BMSCs) to the neural lineage.92 Besides nerve cells, mesenchymal stem cells (MSCs) are able to differentiate into other cells as therapeutics in tissue engineering. For example, SASMs are able to prompt bone and cartilage generation, which provides a guideline for discovering metabolites that may be relevant to therapeutic drugs.93 These studies illuminate a promising direction for SASMs in cell proliferation and differentiation, which, ultimately, may make applications of SASMs in tissue engineering possible.
3.2 SASMs for ischemic hind-limb salvage, sarcopenia treatment, and acute kidney injury (AKI) repair
Insulin-like growth factor-I (IGF-1) plays important regulatory functions in tissue engineering.94 GYGSSSRRAPQT, the C region of IGF-1 (named IGF-1C), which interacts specifically with the IGF-1 receptor (IGF-1R),95 emerges as a functional mimic of IGF-1. IGF-1C, however, exhibits an unstructured conformation by itself, making it less effective to activate IGF-1R.96 SASMs of IGF-1C, adopting a β-sheet conformation to act as a mimic of IGF-1, not only bind with IGF-1R, but also activate it, evidenced in the study of ischemic hind-limb salvage. As shown in Fig. 8A, 35, of which the green part is IGF-1C, self-assembles to form assemblies that adopt a β-sheet conformation. The experimental result shows that the β-sheet-conformational IGF-1C, in the form of a hydrogel, has strong affinity to IGF-1R. Moreover, it exerts superior ability to enhance ischemic hind-limb salvage than the IGF-1 protein, by activating the downstream signaling pathway.97 Encouraged by this study, other SASMs, which also mimic IGF-1, are capable of treating other diseases, such as treating sarcopenia and AKI.98,99 As shown in Fig. 8B, an ultrashort sequence SSSR substitutes IGF-1C in 35, forming compound 37. Fig. 8C and D show that assemblies of 37 reduce the myoblast damage in a dexamethasone (Dex)-instructed sarcopenia model.98 Additionally, as shown in Fig. 8E, 38, an amphiphile molecule similar to 35 but having D-formed amino acids, self-assembles to form a hydrogel. The hydrogel improves the activity of human placenta-derived mesenchymal stem cells (hP-MSCs) to rescue murine in an AKI model.99 Besides acting as a carrier medium of MSCs, SASMs are also able to incorporate other bioactive entities, such as excellular vesicles, for increasing the therapeutic effiency of the excellular vesicles in AKI.100 These studies highlight the promise of SASMs for application in tissue engineering.101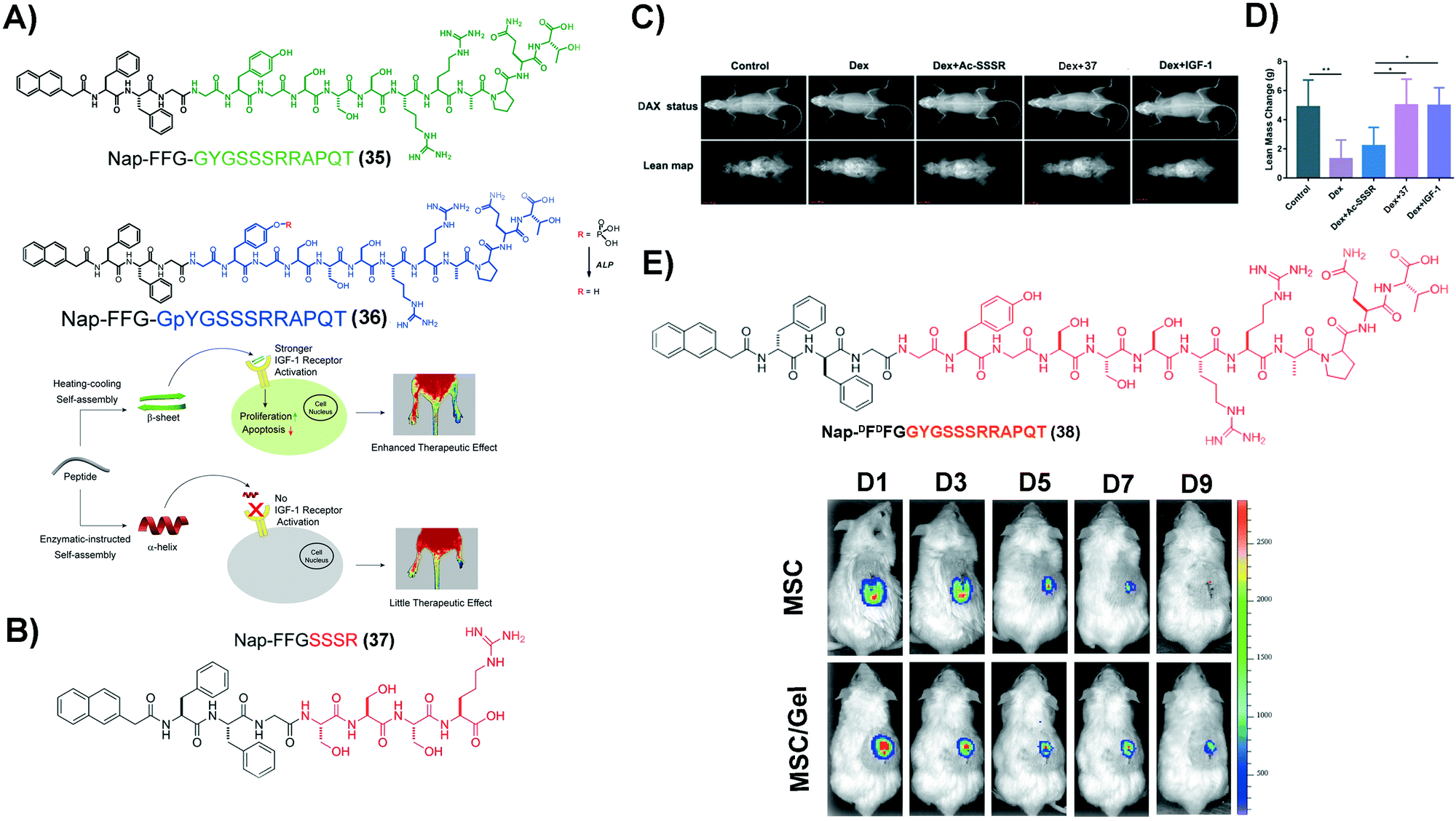 | ||
| Fig. 8 (A) Molecular structures of 35 and 36 and illustration of the corresponding hydrogels reparing the ischemic hind-limb. (B) Molecular structure of 37. (C) Lean mass changes after different treatment, using dual-energy X-ray absorptiometry (DXA). (D) Caculation of the lean mass in (C). (A–D) Adapted with permission from ref. 97 and 98. Copyright 2019 and 2020, American Chemical Society. (E) Molecular structure of 38 and tracking of hP-MSCs, which are labelled by luciferin activities, in an AKI model over a period of 9 days. Reproduced with permission from ref. 99. Copyright 2020, Dove Medical Press. | ||
4. SASMs as a tool to study cell behaviors
SASMs are not only capable of inhibiting pathogenic cells and maintaining physiological functions of normal cells, but also are able to act as a useful tool to study cell behaviors, such as revealing protein–peptide interactions. As shown in Fig. 9, 4,7-di(thiophen-2-yl)-2,1,3-benzothiadiazole (DBT), a fluorescent molecule, conjugated to two molecules of a peptide (EEGWRESAI) that binds with tax-interacting protein-1 (TIP-1) generates molecule 39. Because of the environmentally sensitive fluorescence of DBT, 39 interacts with TIP-1 or a tetramer protein, ubiquitin-like domain protein (ULD) and TIP-1 (ULD-TIP1), to exhibit far-red/near-infrared (FR/NIR) fluorescence, after forming spherical nanoaggregates or nanofibers. Besides, in solution, this interconverting fluorescent switch of “on” and “off” with high contrast is able to report peptide–protein interactions in bacteria.102 In addition to studying peptide–protein interactions, SASMs also act as an important tool to study other cell behaviors, such as identifying specific cells,103–106 improving enzyme probes,107 revealing membrane dynamics,108 enhancing molecular imaging,109,110 and mimicking context-dependent signaling.111 | ||
| Fig. 9 Molecular structure of 39 and illustration of lighting up the fluorescence of 39 to monitor peptide–protein interactions. Adapted with permission from ref. 102. Copyright 2014, American Chemical Society. | ||
4.1 SASMs for identifying specific cells, improving enzyme probes, or revealing membrane dynamics
Cellular senescence is a natural process sustaining normal physiological functions of life.112 However, excessive accumulation of senescent cells may result in some diseases, such as atherosclerosis and tissue dysfunction.113,114 Thus, it is necessary to identify and remove senescent cells in a timely manner. Forming SASMs in senescent cells turns out to be a valid approach to identify and remove the cells. As shown in Fig. 10A and B, β-galactosidase, overexpressed in senescent cells, is able to catalyze the hydrolytic removal of β-galactose in 40, a peptide containing a fluorophore, to generate 41. Self-assembly of 41 results in nanofibers, which exhibit increased fluorescence intensity for identifying senescent HeLa cells (s-HeLa). When the concentration of 40 is at 400 μM, only approximately 30% of senescent endothelial cells (s-ECs) survive, as shown in Fig. 10C.103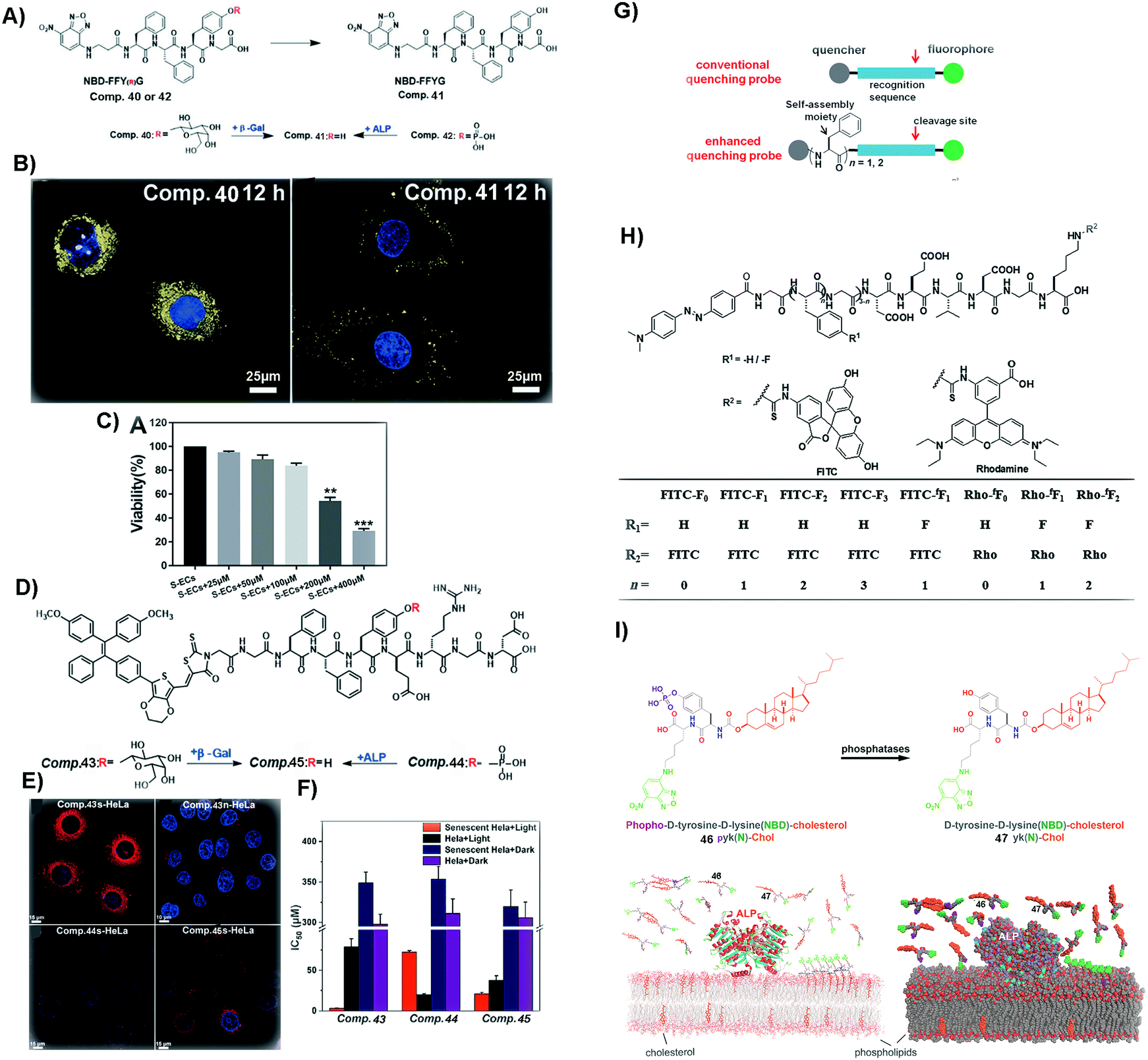 | ||
| Fig. 10 (A) Molecular structures of 40–42. (B) Images of senescent HeLa cells incubated with 40 and 41 (blue fluorescence from DAPI, representing the nucleus, and yellow from NBD, the N-terminal molecule). (C) Viability of s-ECs incubated with 40 at different concentrations. (A–C) Reproduced with permission from ref. 103. Copyright 2019, Royal Society of Chemistry. (D) Molecular structures of 43–45. (E) Images of s-HeLa incubated with different compounds (43–45) and n-HeLa incubated with 43 (blue fluorescence from DAPI, representing the nucleus, and red from methoxy-substituted tetraphenylethene (TPE), the N-capping motif). (F) IC50 of HeLa cells incubated with different compounds (43–45). (G) Illustration of enhancing quenching of the self-assembled probe. (H) Molecular structures of different probes. (D–H) Reproduced with permission from ref. 104 and 107. (I) Molecular structures of 46 and 47 and the process of revealing the membrane dynamics by incubating 46 with cells. Adapted with permission from ref. 108. Copyright 2018, American Chemical Society. | ||
Besides removing senescent normal cells, it is more urgent to eliminate senescent cancer cells. As shown in Fig. 10D and E, 43, a peptide derivative similar to 40, contains a fluorescent molecule that is able to generate reactive oxygen species (ROS). Upon β-galactosidase catalyzed deglycosylation, 43 turns into 45, which self-assembles to enhance the fluorescence intensity for distinguishing s-HeLa from the wild type HeLa cells (n-HeLa). Moreover, upon light irradiation to generate ROS, the assemblies of 45 exhibit appreciable inhibitory activity against s-HeLa (see Fig. 10F).104 In addition to identifying and inhibiting cancer cells, SASMs are also able to image106 and inhibit tumors.105
Although these methods are easy and effective, the background signal of some fluorescent probes could be too high. Thus, aggregation-causing SASMs are able to address this issue. As shown in Fig. 10H, the precursor, of which the N-terminal is dabcyl, a quencher, is a kind of traditional quenching probe. When caspases (apoptosis-associated enzymes) are present, the quencher is cleaved and the fluorescence intensity increases. To enhance the quenching effect of the precursors and lower the background signal, one approach is to incorporate different numbers of amino acids or amino acid derivatives, such as phenylalanine or 4-fluoro phenylalanine, which forms the compounds shown in Fig. 10H (n = 1/2/3). Assemblies of these compounds lower the background signals. Upon the addition of caspases, the fluorescence signal increases, which results in a high signal-to-noise ratio compared to that of traditional ones.107
Besides intracellular imaging, a method is still needed that is able to reveal the dynamics of the cell membrane. For example, as shown in Fig. 10I, compound 46, containing 4-nitro-2,1,3-benzoxadiazole (NBD) and cholesterol, upon ALP catalysis forms molecule 47. Assemblies of 47 are able to probe the nanoscale heterogeneity of membranes and reveal the membrane dynamics, thus providing a useful probe for understanding the dynamics of cell membranes.108
4.2 SASMs for imaging molecular self-assembly
SASMs are particularly useful to enhance molecular imaging for studying cell behavior. As shown in Fig. 11A and B, 48 conjugates with NBD to form compound 49. After 49 enters into cells, under the catalysis of ALP, it loses the phosphate group to form 50, which self-assembles to generate fluorescent nanofibers in nuclei, mitochondria and plasma membranes. This method, which is to image molecular self-assembly, is efficient to evaluate self-assembly and monitor the dynamics of these nanofibers.109 In addition to imaging fluorescent molecule self-assembly, this method is also applicable for imaging non-fluorescent molecule self-assembly. For example, as shown in Fig. 11C, 51, a non-fluorescent molecule, is able to co-assemble with a small amount of 53, a compound that is similar to 49 but with the fluorescent molecule changed from NBD to dansyl (DNS). Adding 200 nM (lower than the critical micelle concentration) of 53 in cell culture results in fluorescence that is uniformly distributed. But on adding 200 nM of 53 and 500 μM (higher than the critical micelle concentration) of 51 in the cell culture, the fluorescence in the cells is localized (Fig. 11D). This observation proves that the assemblies are from 51, not from 53, confirming that this assay is able to image the self-assembly of non-fluorescent molecules. The result also suggests that the process of self-assembly may be through the protein secretion pathway, which is the endoplasmic reticulum to the Golgi to lysosomes/secretion.110 These studies underscore SASMs as promising nanoprobes to enhance molecular imaging, which allows enzyme-triggered SASMs to be further developed as a unique approach for imaging cell behavior.115,116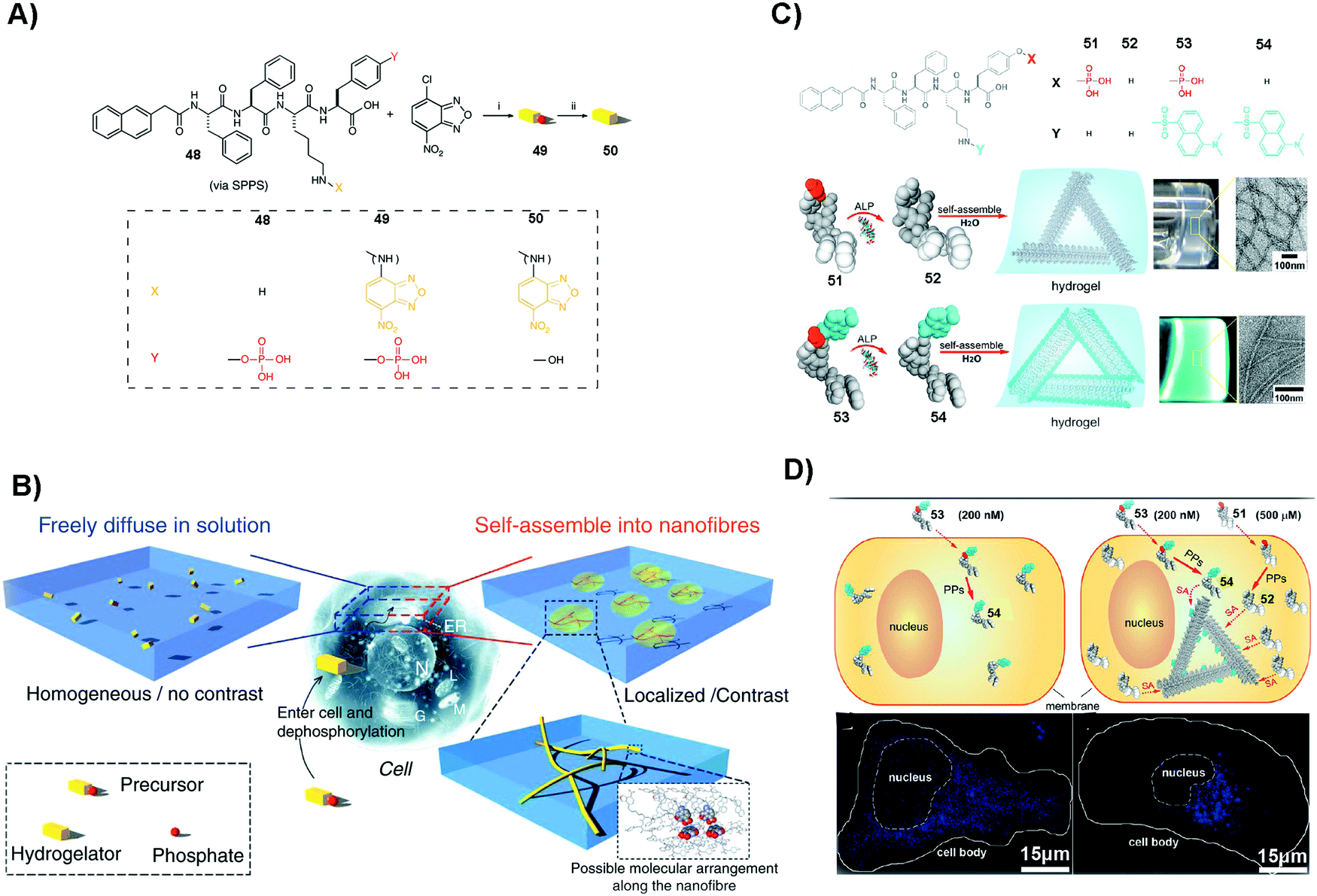 | ||
| Fig. 11 (A) Molecular structures of 48–50. (B) Illustration of molecular assembly. (A and B) Reproduced with permission from ref. 109. (C) Molecular structures of 51–54 and the corresponding hydrogels. (D) Images of HeLa cells incubated with different compounds (53 or 51/53). PP: protein phosphatase. (C and D) Adapted with permission from ref. 110. Copyright 2013, American Chemical Society. | ||
4.3 SASMs for mimicking context-dependent signaling
Context-dependent signaling plays an important role in many cellular functions. For example, in developing embryos, some WNTs (a protein family that secretes glycoproteins) can activate β-catenin to influence cell proliferation and survival, while other ones can modulate cellular movement by activating a β-catenin-independent signaling pathway.117 Thus, using active matter such as SASMs to mimic context-dependent signaling is an effective method to understand the cell behavior. As shown in Fig. 12A, since vancomycin (Van) is an antibiotic that can specifically bind with D-Ala-D-Ala, the designed phosphopeptide 55 containing D-Ala-D-Ala is able to bind to Van, for inhibiting cancer cells. As shown in Fig. 12B, tissue-specific ALP (ALPL) on Saos-2 cells is able to catalyze the formation of the supramolecular complex (55:Van), generating nanofibers and resulting in cell death. An uncompetitive inhibitor can antagonize the activity of ectophosphatases (e.g., ALPL). Thus, when adding this inhibitor to the cell culture, fewer nanofibers are generated in the cells and the cells survive and form 3D spheroids. More inhibitors are capable of resulting in the changes of the morphologies of the surviving cells, from 3D spheroids to 2D sheets.111 The use of SASMs to mimic context-dependent signaling may help develop new approaches for studying cell behaviors. | ||
| Fig. 12 (A) Illustration of 55 binding with Van. (B) SASMs mimicking context-dependent signaling. iA: instructed assembly. Reproduced with permission from ref. 111. | ||
5. Outlook
The past research on SASMs provides certain molecular and cellular understanding of the emergent properties of SASMs. Although SASMs show therapeutic effects on some diseases, many challenges still exist. For example, MDR in cancer and bacterial therapies still threatens human health. It is essential to increase the activities of SASMs to minimize the emergence of MDR. For instance, immunotherapy, a safe and efficient solution, is able to treat cancer with SASMs as the adjuvant.118 The bacterial biofilm microenvironment might be a novel target to improve the efficacy of SASMs.119 Applying SASMs in tissue engineering and regenerative medicine is at the beginning, but there is a concern of amyloidogenicity of aggregates. This issue requires careful kinetic control of the formation of SASMs to avoid inducing amyloid formation of endogenous biomolecules. Moreover, enhancing the activation of multipotent proteins is efficient to promote the use of SASMs in tissue engineering and regenerative medicine, such as in bone regeneration.120 It is likely that the further understanding of enzymatic noncovalent synthesis in biology24 will also help address current challenges of SASMs in disease treatment.With the ability to design SASMs121–123 and to determine the morphologies124 and structures of SASMs,125,126 it is time to explore the functions and applications of SASMs. While SASMs have exhibited a wide range of biological functions, the mechanistic study of SASMs remains insufficient. The elucidation of the molecular mechanisms of the biological functions of SASMs, undoubtedly, will contribute to the development of supramolecular assemblies for ultimately treating life-threatening diseases.
The small molecules used for forming SASMs have been largely centered on peptides, but the principles revealed by the study of SASMs should be applicable to other types of small molecules, such as nucleobases127 or carbohydrates,120 for discriminating the protein targets of supramolecular assemblies. Further research on SASMs will establish the knowledge foundation for engineering supramolecular assemblies to regulate protein–protein interaction networks128 for controlling the fate of cells. In addition, the knowledge obtained in the research on SASMs will provide new insights for helping understand other challenging problems related to molecular self-assembly, such as the molecular mechanism of the aggregates of aberrant proteins that are considered as a plausible cause of neurodegenerative diseases (e.g., Alzheimer's disease129). In addition, SASMs also represent a new direction of supramolecular chemical biology,130 a promising field of chemistry that remains to be explored by molecular scientists.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This article is partially supported by China Scholarship Council (to JYW) and NIH (CA142746 and CA252364 to BX).References
- B. Alberts, Essential Cell Biology, Garland Science, New York, 3rd edn, 2009 Search PubMed.
- A. A. Hyman, S. Salser, D. N. Drechsel, N. Unwin and T. J. Mitchison, Mol. Biol. Cell, 1992, 3, 1155–1167 CrossRef CAS PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- P. Caron, A. Beckers, D. R. Cullen, M. I. Goth, B. Gutt, P. Laurberg, A. M. Pico, M. Valimaki and W. Zgliczynski, J. Clin. Endocrinol. Metab., 2002, 87, 99–104 CrossRef CAS PubMed.
- S. L. McGovern, B. T. Helfand, B. Feng and B. K. Shoichet, J. Med. Chem., 2003, 46, 4265–4272 CrossRef CAS PubMed.
- B. Y. Feng, B. H. Toyama, H. Wille, D. W. Colby, S. R. Collins, B. C. H. May, S. B. Prusiner, J. Weissman and B. K. Shoichet, Nat. Chem. Biol., 2008, 4, 197–199 CrossRef CAS PubMed.
- J. A. Zorn, H. Wille, D. W. Wolan and J. A. Wells, J. Am. Chem. Soc., 2011, 133, 19630–19633 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103–13107 CrossRef CAS PubMed.
- K. A. Mosiewicz, K. Johnsson and M. P. Lutolf, J. Am. Chem. Soc., 2010, 132, 5972–5974 CrossRef CAS PubMed.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS PubMed.
- S. W. Liao, T. B. Yu and Z. B. Guan, J. Am. Chem. Soc., 2009, 131, 17638–17646 CrossRef CAS PubMed.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- Y. Kuang, Y. Gao and B. Xu, Chem. Commun., 2011, 47, 12625–12627 RSC.
- Y. Kuang, J. F. Shi, J. Li, D. Yuan, K. A. Alberti, Q. B. Xu and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 8104–8107 CrossRef CAS PubMed.
- Z. M. Yang, G. L. Liang, Z. F. Guo, Z. H. Guo and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 8216–8219 CrossRef CAS PubMed.
- Z. M. Yang, K. M. Xu, Z. F. Guo, Z. H. Guo and B. Xu, Adv. Mater., 2007, 17, 3152–3156 CrossRef.
- Y. Kuang and B. Xu, Angew. Chem., Int. Ed., 2013, 52, 6944–6948 CrossRef CAS PubMed.
- Y. Kuang, X. W. Du, J. Zhou and B. Xu, Adv. Healthcare Mater., 2014, 3, 1217–1221 CrossRef CAS PubMed.
- X. Du, J. Zhou, O. Guvench, F. O. Sangiorgi, X. Li, N. Zhou and B. Xu, Bioconjugate Chem., 2014, 25, 1031–1035 CrossRef CAS PubMed.
- Y. Wang, W. Du, T. Zhang, Y. Zhu, Y. Ni, C. Wang, F. M. Sierra Raya, L. Zou, L. Wang and G. Liang, ACS Nano, 2020, 14, 9585–9593 CrossRef CAS PubMed.
- H. He, W. Tan, J. Guo, M. Yi, A. N. Shy and B. Xu, Chem. Rev., 2020, 120, 9994–10078 CrossRef CAS PubMed.
- G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS.
- D. Duan, A. K. Doak, L. Nedyalkova and B. K. Shoichet, ACS Chem. Biol., 2015, 10, 978–988 CrossRef CAS PubMed.
- A. N. Ganesh, E. N. Donders, B. K. Shoichet and M. S. Shoichet, Nano Today, 2018, 19, 188–200 CrossRef CAS PubMed.
- S. L. McGovern, E. Caselli, N. Grigorieff and B. K. Shoichet, J. Med. Chem., 2002, 45, 1712–1722 CrossRef CAS PubMed.
- Y. Kuang and B. Xu, Angew. Chem. Int. Ed., 2013, 52, 6944–6948 CrossRef CAS PubMed.
- C. Liang, D. Zheng, F. Shi, T. Xu, C. Yang, J. Liu, L. Wang and Z. Yang, Nanoscale, 2017, 9, 11987–11993 RSC.
- S. C. Owen, A. K. Doak, P. Wassam, M. S. Shoichet and B. K. Shoichet, ACS Chem. Biol., 2012, 7, 1429–1435 CrossRef CAS PubMed.
- L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler-Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. DeGrado and E. Gazit, Nat. Commun., 2017, 8, 1365 CrossRef PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- J. A. Driver, A. Beiser, R. Au, B. E. Kreger, G. L. Splansky, T. Kurth, D. P. Kiel, K. P. Lu, S. Seshadri and P. A. Wolf, Br. Med. J., 2012, 344, e1442 CrossRef PubMed.
- Z. M. Yang, K. M. Xu, Z. F. Guo, Z. H. Guo and B. Xu, Adv. Mater., 2007, 19, 3152–3156 CrossRef CAS.
- H. Wang, Z. Feng, C. Yang, J. Liu, J. E. Medina, S. A. Aghvami, D. M. Dinulescu, J. Liu, S. Fraden and B. Xu, Mol. Cancer Res., 2019, 17, 907–917 CrossRef CAS PubMed.
- J. Wang, W. Tan, G. Li, D. Wu, H. He, J. Xu, M. Yi, Y. Zhang, S. A. Aghvami, S. Fraden and B. Xu, Chemistry, 2020, 26, 15116–15120 CrossRef CAS PubMed.
- J. Zhan, Y. Cai, S. He, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2018, 57, 1813–1816 CrossRef CAS PubMed.
- H. He, S. Liu, D. Wu and B. Xu, Angew. Chem., Int. Ed., 2020, 59, 16445–16450 CrossRef CAS PubMed.
- J. Kopecka, P. Trouillas, A. C. Gasparovic, E. Gazzano, Y. G. Assaraf and C. Riganti, Drug Resist. Updates, 2020, 49, 100670 CrossRef PubMed.
- N. Koundouros and G. Poulogiannis, Br. J. Cancer, 2020, 122, 4–22 CrossRef CAS PubMed.
- W. Chen, S. Li, J. C. Lang, Y. Chang, Z. Pan, P. Kroll, X. Sun, L. Tang and H. Dong, Small, 2020, 16, e2002780 CrossRef PubMed.
- W. Du, Y. Chong, X. Hu, Y. Wang, Y. Zhu, J. Chen, X. Li, Q. Zhang, G. Wang, J. Jiang and G. Liang, Adv. Funct. Mater., 2020, 30, 1908073 CrossRef CAS.
- F. Wang, H. Su, R. Lin, R. W. Chakroun, M. K. Monroe, Z. Wang, M. Porter and H. Cui, ACS Nano, 2020, 14, 10083–10094 CrossRef CAS PubMed.
- Y. Kuang, J. Shi, J. Li, D. Yuan, K. A. Alberti, Q. Xu and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 8104–8107 CrossRef CAS PubMed.
- Y. Wang, J. Zhan, Y. Chen, S. Ai, L. Li, L. Wang, Y. Shi, J. Zheng and Z. Yang, Nanoscale, 2019, 11, 13714–13719 RSC.
- D. R. Green and G. Kroemer, Science, 2004, 305, 626–629 CrossRef CAS PubMed.
- H. M. McBride, M. Neuspiel and S. Wasiak, Curr. Biol., 2006, 16, R551–560 CrossRef CAS PubMed.
- G. C. Kujoth, A. Hiona, T. D. Pugh, S. Someya, K. Panzer, S. E. Wohlgemuth, T. Hofer, A. Y. Seo, R. Sullivan, W. A. Jobling, J. D. Morrow, H. Van Remmen, J. M. Sedivy, T. Yamasoba, M. Tanokura, R. Weindruch, C. Leeuwenburgh and T. A. Prolla, Science, 2005, 309, 481–484 CrossRef CAS PubMed.
- R. S. Balaban, S. Nemoto and T. Finkel, Cell, 2005, 120, 483–495 CrossRef CAS PubMed.
- S. Pattingre, A. Tassa, X. Qu, R. Garuti, X. H. Liang, N. Mizushima, M. Packer, M. D. Schneider and B. Levine, Cell, 2005, 122, 927–939 CrossRef CAS PubMed.
- C. J. Green and D. R. Reed, Science, 1998, 281, 1309–1312 CrossRef PubMed.
- J. Nunnari and A. Suomalainen, Cell, 2012, 148, 1145–1159 CrossRef CAS PubMed.
- R. J. Burns, R. A. Smith and M. P. Murphy, Arch. Biochem. Biophys., 1995, 322, 60–68 CrossRef CAS PubMed.
- H. Wang, Z. Feng, Y. Wang, R. Zhou, Z. Yang and B. Xu, J. Am. Chem. Soc., 2016, 138, 16046–16055 CrossRef CAS PubMed.
- H. He, J. Wang, H. Wang, N. Zhou, D. Yang, D. R. Green and B. Xu, J. Am. Chem. Soc., 2018, 140, 1215–1218 CrossRef CAS PubMed.
- T. Hopp, K. Prickett, V. Price, R. Libby, C. March, D. Cerretti, D. Urdal and P. Conlon, Bio/Technology, 1988, 6, 1204–1210 CrossRef CAS.
- L. Yang, R. Peltier, M. Zhang, D. Song, H. Huang, G. Chen, Y. Chen, F. Zhou, Q. Hao, L. Bian, M. L. He, Z. Wang, Y. Hu and H. Sun, J. Am. Chem. Soc., 2020, 142, 18150–18159 CrossRef CAS PubMed.
- H. He, X. Lin, D. Wu, J. Wang, J. Guo, D. R. Green, H. Zhang and B. Xu, Cell Rep.: Phys. Sci., 2020, 1, 100270 Search PubMed.
- Y. S. Choi, J. Hoon Jeong, H. K. Min, H. J. Jung, D. Hwang, S. W. Lee and Y. Kim Pak, Mol. BioSyst., 2011, 7, 1523–1536 RSC.
- H. He, J. Guo, X. Lin and B. Xu, Angew. Chem., Int. Ed., 2020, 59, 9330–9334 CrossRef CAS PubMed.
- B. J. Kim, Y. Fang, H. He and B. Xu, Adv. Healthcare Mater., 2020, 2000416 Search PubMed.
- Y. Cai, H. Shen, J. Zhan, M. Lin, L. Dai, C. Ren, Y. Shi, J. Liu, J. Gao and Z. Yang, J. Am. Chem. Soc., 2017, 139, 2876–2879 CrossRef CAS PubMed.
- T. Xu, C. Liang, D. Zheng, X. Yan, Y. Chen, Y. Chen, X. Li, Y. Shi, L. Wang and Z. Yang, Nanoscale, 2020, 12, 15275–15282 RSC.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188–200 CrossRef CAS PubMed.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed.
- C. Liang, L. Zhang, W. Zhao, L. Xu, Y. Chen, J. Long, F. Wang, L. Wang and Z. Yang, Adv. Healthcare Mater., 2018, 7, e1800899 CrossRef PubMed.
- J. Li, Y. Kuang, J. Shi, J. Zhou, J. E. Medina, R. Zhou, D. Yuan, C. Yang, H. Wang, Z. Yang, J. Liu, D. M. Dinulescu and B. Xu, Angew. Chem., Int. Ed., 2015, 54, 13307–13311 CrossRef CAS PubMed.
- H. Wang, Z. Feng, Y. Qin, J. Wang and B. Xu, Angew. Chem., Int. Ed., 2018, 57, 4931–4935 CrossRef CAS PubMed.
- H. He, X. Lin, J. Guo, J. Wang and B. Xu, ACS Nano, 2020, 14, 6947–6955 CrossRef CAS PubMed.
- H. T. Feng, S. Zou, M. Chen, F. Xiong, M. H. Lee, L. Fang and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 11442–11450 CrossRef CAS PubMed.
- D. Schrama, R. A. Reisfeld and J. C. Becker, Nat. Rev. Drug Discovery, 2006, 5, 147–159 CrossRef CAS PubMed.
- F. Wang, D. Xu, H. Su, W. Zhang, X. Sun, M. K. Monroe, R. W. Chakroun, Z. Wang, W. Dai, R. Oh, H. Wang, Q. Fan, F. Wan and H. Cui, Sci. Adv., 2020, 6, eaaz8985 CrossRef PubMed.
- M. Chen, B. LeDuc, S. Kerr, D. Howe and D. A. Williams, Drug Metab. Dispos., 2010, 38, 368–375 CrossRef CAS PubMed.
- W. Chen, S. Yang, S. Li, J. C. Lang, C. Mao, P. Kroll, L. Tang and H. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 28681–28689 CrossRef CAS PubMed.
- W. Chen, S. Li, P. Renick, S. Yang, N. Pandy, C. Boutte, K. T. Nguyen, L. Tang and H. Dong, J. Mater. Chem. B, 2019, 7, 2915–2919 RSC.
- W. Tanaka, H. Shigemitsu, T. Fujisaku, R. Kubota, S. Minami, K. Urayama and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 4997–5004 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed.
- T. Yoshii, M. Ikeda and I. Hamachi, Angew. Chem., Int. Ed., 2014, 53, 7264–7267 CrossRef CAS PubMed.
- M. Gontsarik, A. Yaghmur, Q. Ren, K. Maniura-Weber and S. Salentinig, ACS Appl. Mater. Interfaces, 2019, 11, 2821–2829 CrossRef CAS PubMed.
- Z. Yang, G. Liang, Z. Guo, Z. Guo and B. Xu, Angew. Chem., 2007, 119, 8364–8367 CrossRef.
- J. Wang, D. L. Cooper, W. Zhan, D. Wu, H. He, S. Sun, S. T. Lovett and B. Xu, Angew. Chem., Int. Ed., 2019, 58, 10631–10634 CrossRef CAS PubMed.
- H. Han, Y. Gao, M. Chai, X. Zhang, S. Liu, Y. Huang, Q. Jin, A. Grzybowski, J. Ji and K. Yao, J. Controlled Release, 2020, 327, 676–687 CrossRef CAS PubMed.
- L. Wang, J. Chen, X. Zeng, P. P. Cheung, X. Zheng, L. Xie, X. Shi, L. Ren, X. Huang and Y. Wang, Adv. Sci., 2019, 6, 1801827 CrossRef PubMed.
- L. L. Li, G. B. Qi, F. Yu, S. J. Liu and H. Wang, Adv. Mater., 2015, 27, 3181–3188 CrossRef CAS PubMed.
- C. Ren, H. Wang, X. Zhang, D. Ding, L. Wang and Z. Yang, Chem. Commun., 2014, 50, 3473–3475 RSC.
- L. Yang, C. Zhang, F. Huang, J. Liu, Y. Zhang, C. Yang, C. Ren, L. Chu, B. Liu and J. Liu, J. Controlled Release, 2020, 324, 354–365 CrossRef CAS PubMed.
- S. Gao, X. Yan, G. Xie, M. Zhu, X. Ju, P. J. Stang, Y. Tian and Z. Niu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23437–23443 CrossRef CAS PubMed.
- S. Song, J. Wang, Z. Cheng, Z. Yang, L. Shi and Z. Yu, Chem. Sci., 2020, 11, 1383–1393 RSC.
- A. Faissner and J. Reinhard, Glia, 2015, 63, 1330–1349 CrossRef PubMed.
- W. Ji, Z. Alvarez, A. N. Edelbrock, K. Sato and S. I. Stupp, ACS Appl. Mater. Interfaces, 2018, 10, 41046–41055 CrossRef CAS PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Chem, 2016, 1, 298–319 CAS.
- C. Borselli, H. Storrie, F. Benesch-Lee, D. Shvartsman, C. Cezar, J. W. Lichtman, H. H. Vandenburgh and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3287–3292 CrossRef CAS PubMed.
- F. F. Vajdos, M. Ultsch, M. L. Schaffer, K. D. Deshayes, J. Liu, N. J. Skelton and A. M. de Vos, Biochemistry, 2001, 40, 11022–11029 CrossRef CAS PubMed.
- N. Yamada, R. Yanai, M. Nakamura, M. Inui and T. Nishida, Invest. Ophthalmol. Vis. Sci., 2004, 45, 1125–1131 CrossRef PubMed.
- Y. Shang, D. Zhi, G. Feng, Z. Wang, D. Mao, S. Guo, R. Liu, L. Liu, S. Zhang, S. Sun, K. Wang, D. Kong, J. Gao and Z. Yang, Nano Lett., 2019, 19, 1560–1569 CrossRef CAS PubMed.
- Y. Shang, M. Kuang, Z. Wang, Y. Huang, L. Liu, X. Zhao, R. Zhang, Y. Zhao, R. Peng, S. Sun, Q. Yang and Z. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 34678–34688 CrossRef CAS PubMed.
- H. Wang, Y. Shang, X. Chen, Z. Wang, D. Zhu, Y. Liu, C. Zhang, P. Chen, J. Wu, L. Wu, D. Kong, Z. Yang, Z. Li and X. Chen, Int. J. Nanomed., 2020, 15, 4311–4324 CrossRef CAS PubMed.
- C. Zhang, Y. Shang, X. Chen, A. C. Midgley, Z. Wang, D. Zhu, J. Wu, P. Chen, L. Wu, X. Wang, K. Zhang, H. Wang, D. Kong, Z. Yang, Z. Li and X. Chen, ACS Nano, 2020, 14, 12133–12147 CrossRef CAS PubMed.
- J. Tan, M. Zhang, Z. Hai, C. Wu, J. Lin, W. Kuang, H. Tang, Y. Huang, X. Chen and G. Liang, ACS Nano, 2019, 13, 5616–5622 CrossRef CAS PubMed.
- H. Wang, J. Liu, A. Han, N. Xiao, Z. Xue, G. Wang, J. Long, D. Kong, B. Liu, Z. Yang and D. Ding, ACS Nano, 2014, 8, 1475–1484 CrossRef CAS PubMed.
- T. Xu, Y. Cai, X. Zhong, L. Zhang, D. Zheng, Z. Gao, X. Pan, F. Wang, M. Chen and Z. Yang, Chem. Commun., 2019, 55, 7175–7178 RSC.
- Z. Gao, H. Gao, D. Zheng, T. Xu, Y. Chen, C. Liang, L. Wang, D. Ding and Z. Yang, Sci. China Chem., 2020, 63, 398–403 CrossRef CAS.
- X. Chen, H. Gao, Y. Deng, Q. Jin, J. Ji and D. Ding, ACS Nano, 2020, 14, 5121–5134 CrossRef CAS PubMed.
- Z. Ding, H. Sun, S. Ge, Y. Cai, Y. Yuan, Z. Hai, T. Tao, J. Hu, B. Hu, J. Wang and G. Liang, Adv. Funct. Mater., 2019, 29, 1903860 CrossRef CAS.
- C. Ren, H. Wang, D. Mao, X. Zhang, Q. Fengzhao, Y. Shi, D. Ding, D. Kong, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2015, 54, 4823–4827 CrossRef CAS PubMed.
- H. Wang, Z. Feng, S. J. Del Signore, A. A. Rodal and B. Xu, J. Am. Chem. Soc., 2018, 140, 3505–3509 CrossRef CAS PubMed.
- Y. Gao, J. Shi, D. Yuan and B. Xu, Nat. Commun., 2012, 3, 1033 CrossRef PubMed.
- Y. Gao, C. Berciu, Y. Kuang, J. Shi, D. Nicastro and B. Xu, ACS Nano, 2013, 7, 9055–9063 CrossRef CAS PubMed.
- H. Wang, Z. Feng and B. Xu, Angew. Chem., Int. Ed., 2019, 58, 5567–5571 CrossRef CAS PubMed.
- D. Munoz-Espin and M. Serrano, Nat. Rev. Mol. Cell Biol., 2014, 15, 482–496 CrossRef CAS PubMed.
- T. Minamino, H. Miyauchi, T. Yoshida, Y. Ishida, H. Yoshida and I. Komuro, Circulation, 2002, 105, 1541–1544 CrossRef CAS PubMed.
- S. He and N. E. Sharpless, Cell, 2017, 169, 1000–1011 CrossRef CAS PubMed.
- Z. Hai, Y. Ni, D. Saimi, H. Yang, H. Tong, K. Zhong and G. Liang, Nano Lett., 2019, 19, 2428–2433 CrossRef CAS PubMed.
- X. Liu, X. Sun and G. Liang, Biomater. Sci., 2020 10.1039/d0bm01020k.
- R. T. Moon, A. D. Kohn, G. V. De Ferrari and A. Kaykas, Nat. Rev. Genet., 2004, 5, 691–701 CrossRef CAS PubMed.
- Z. C. Luo, Q. J. Wu, C. B. Yang, H. M. Wang, T. He, Y. Z. Wang, Z. Y. Wang, H. Chen, X. Y. Li, C. Y. Gong and Z. M. Yang, Adv. Mater., 2017, 29, 1601776 CrossRef PubMed.
- H. J. Han, Y. F. Gao, M. Y. Chai, X. B. Zhang, S. R. Liu, Y. Huang, Q. Jin, A. Grzybowski, J. Ji and K. Yao, J. Controlled Release, 2020, 327, 676–687 CrossRef CAS PubMed.
- S. S. Lee, T. Fyrner, F. Chen, Z. Alvarez, E. Sleep, D. S. Chun, J. A. Weiner, R. W. Cook, R. D. Freshman, M. S. Schallmo, K. M. Katchko, A. D. Schneider, J. T. Smith, C. Yun, G. Singh, S. Z. Hashmi, M. T. McClendon, Z. Yu, S. R. Stock, W. K. Hsu, E. L. Hsu and S. I. Stupp, Nat. Nanotechnol., 2017, 12, 821–829 CrossRef CAS PubMed.
- J. Rodon Fores, M. Criado-Gonzalez, A. Chaumont, A. Carvalho, C. Blanck, M. Schmutz, F. Boulmedais, P. Schaaf and L. Jierry, Angew. Chem., Int. Ed., 2020, 59, 14558–14563 CrossRef CAS PubMed.
- C. Vigier-Carriere, F. Boulmedais, P. Schaaf and L. Jierry, Angew. Chem., Int. Ed., 2018, 57, 1448–1456 CrossRef CAS PubMed.
- J. Rodon Fores, M. Criado-Gonzalez, M. Schmutz, C. Blanck, P. Schaaf, F. Boulmedais and L. Jierry, Chem. Sci., 2019, 10, 4761–4766 RSC.
- H. Cui, E. T. Pashuck, Y. S. Velichko, S. J. Weigand, A. G. Cheetham, C. J. Newcomb and S. I. Stupp, Science, 2009, 327, 555–559 CrossRef PubMed.
- Z. Feng, H. Wang, F. Wang, Y. Oh, C. Berciu, Q. Cui, E. H. Egelman and B. Xu, Cell. Rep. Phys. Sci., 2020, 1, 10085 Search PubMed.
- A. Lampel, S. A. McPhee, H. A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. Frederix, T. D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS PubMed.
- C. G. Evans and E. Winfree, Chem. Soc. Rev., 2017, 46, 3808–3829 RSC.
- M. A. Yildirim, K. I. Goh, M. E. Cusick, A. L. Barabasi and M. Vidal, Nat. Biotechnol., 2007, 25, 1119–1126 CrossRef CAS PubMed.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed.
- K. Petkau-Milroy and L. Brunsveld, Org. Biomol. Chem., 2013, 11, 219–232 RSC.
| This journal is © The Royal Society of Chemistry 2021 |