
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitochondria-targeted inhibitors of the human SIRT3 lysine deacetylase†
Kathrin S.
Troelsen
 ,
Michael
Bæk
,
Alexander L.
Nielsen
,
Andreas S.
Madsen‡
,
Nima
Rajabi§
and
Christian A.
Olsen
*
,
Michael
Bæk
,
Alexander L.
Nielsen
,
Andreas S.
Madsen‡
,
Nima
Rajabi§
and
Christian A.
Olsen
*
Center for Biopharmaceuticals & Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, DK-2100, Copenhagen, Denmark. E-mail: cao@sund.ku.dk
First published on 1st February 2021
Abstract
Sirtuin 3 (SIRT3) is the major protein lysine deacetylase in the mitochondria. This hydrolase regulates a wide range of metabolically involved enzymes and has been considered as a potential drug target in certain cancers. Investigation of pharmacological intervention has been challenging due to a lack of potent and selective inhibitors of SIRT3. Here, we developed a strategy for selective inhibition of SIRT3 in cells, over its structurally similar isozymes that localize primarily to the nucleus (SIRT1) and the cytosol (SIRT2). This was achieved by directing the inhibitors to the mitochondria through incorporation of mitochondria-targeting peptide sequences into the inhibitor structures. Our inhibitors exhibited excellent mitochondrial localization in HeLa cells as indicated by fluorophore-conjugated versions, and target engagement was demonstrated by a cellular thermal shift assay of SIRT3 using western blotting. The acetylation state of documented SIRT3 target MnSOD was shown to be increased in cells with little effect on known targets of SIRT1 and SIRT2, showing that our lead compound exhibits selectivity for SIRT3 in cells. We expect that the developed inhibitor will now enable a more detailed investigation of SIRT3 as a potential drug target and help shed further light on the diverse biology regulated by this enzyme.
Introduction
The sirtuin (SIRT) enzymes are evolutionarily conserved hydrolases of the class III lysine deacetylases (KDACs), cleaving acyl-based posttranslational modifications (PTMs) on lysine side chains in the proteome. The human genome encodes seven sirtuin isoforms, SIRT1–7, which have different substrate specificities, cellular localization, and tissue-dependent expression levels.1 The major deacetylases [i.e., targeting the ε-N-acetyllysine (Kac) PTM] are SIRT1–3, SIRT6, and SIRT7, of which the class I sirtuins, SIRT1–3, share the highest similarity in substrate specificity.2,3 Thus, SIRT1–3 and 6 efficiently hydrolyze Kac residues as well as longer hydrocarbon-based ε-N-acyllysine PTMs such as ε-N-myristoyllysine (Kmyr)4–8 to regulate diverse biological function, including metabolic homeostasis and health span.9 However, the class I sirtuins have also been implicated in the pathogenesis of various diseases and, depending on the condition, either activation or inhibition of SIRT1–3 have been considered as potential therapeutic strategies to treat several cancers and neurodegenerative disorders.10–12 The SIRT3 isoform is the only mitochondrially localized sirtuin that exhibits potent deacetylase activity, with other mitochondrial sirtuins, SIRT4 and SIRT5, mainly targeting negatively charged PTMs.13–19 Here, SIRT3 regulates a number of metabolic enzymes involved in the respiratory chain,20 TCA cycle,21,22 fatty acid β-oxidation,23 and ketogenesis.24 Furthermore, it controls mitochondrial oxidative pathways by regulating the production of reactive oxygen species (ROS),25e.g. through activation of manganese superoxide dismutase (MnSOD).26,27The sirtuins share a common deacylase mechanism, which is dependent on the co-substrate nicotinamide adenine dinucleotide (NAD+). This mechanism has been utilized to develop so-called mechanism-based inhibitors, by the use of substrate-mimicking chemotypes that form stalled intermediates in the active site of the sirtuin.28–35 Many mechanism-based inhibitors exhibit high potency and, in several cases, high selectivity toward specific sirtuin subtypes. However, due to the shared mechanism and similar substrate preferences between SIRT1–3, it has been difficult to target SIRT3 selectively. Therefore, we envisioned adopting a strategy to achieve selective targeting of the enzyme through specific subcellular localization of the inhibitor, rather than solely relying on selective enzyme recognition. Among several demonstrated examples of mitochondrial targeting of various payloads,36 a particularly appealing approach for our strategy was the mitochondria-targeting peptides developed by Kelley and co-workers.37–39 Based on recent investigations of mechanism-based peptide inhibitors of other sirtuins,32,35 we hypothesized that mitochondria-targeting peptide tags could be elaborated into potent inhibitors of the SIRT3 that would exhibit selectivity in cells (Fig. 1). By designing such chemotypes and optimizing their selectivity profiles to inhibit only SIRT1–3 and not inhibit other sirtuins or HDACs, we could demonstrate selective inhibition of SIRT3 in cultured cells as well as target engagement illustrated by cellular thermal shift assays.
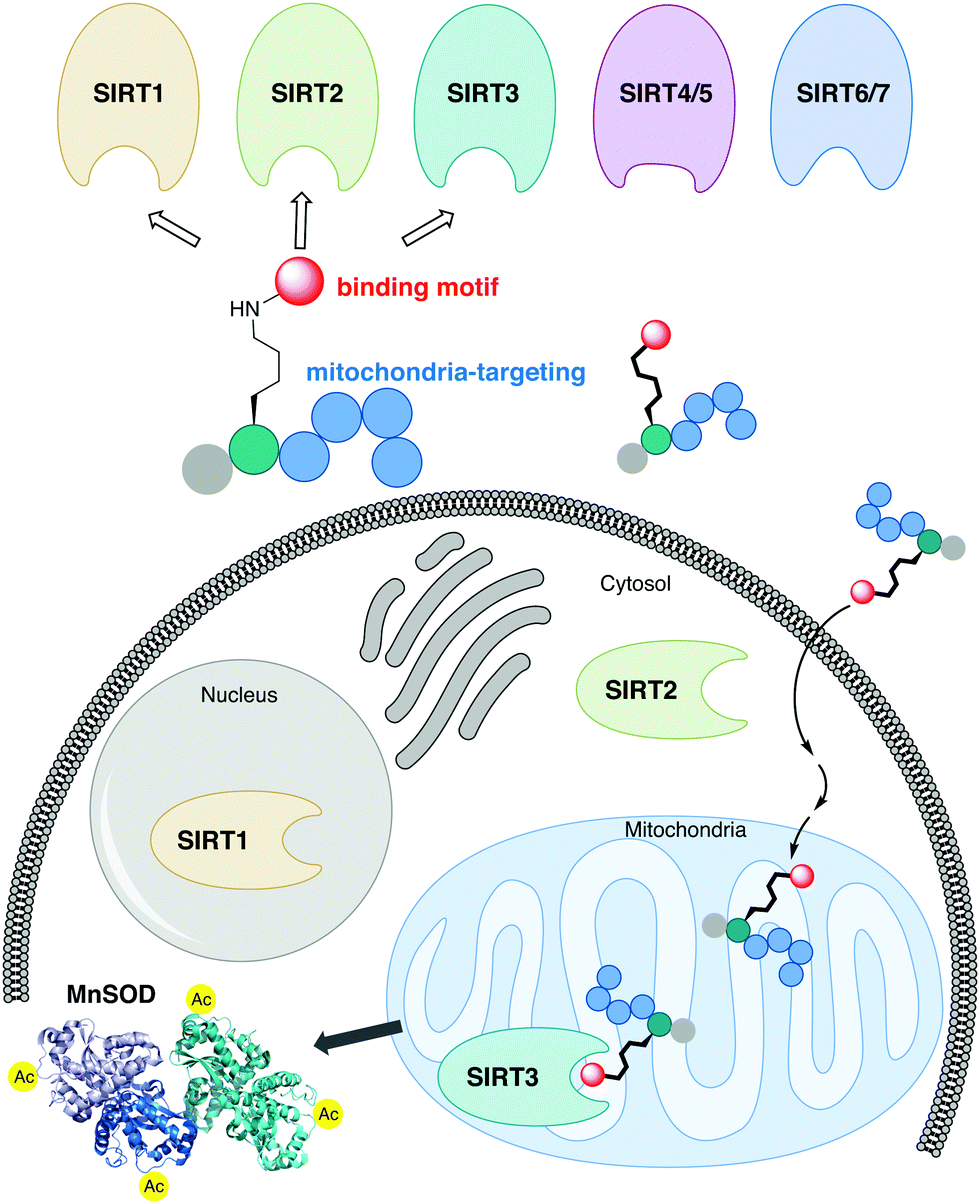 | ||
| Fig. 1 Schematic representation of the developed concept for inhibition of SIRT3. A potent inhibitor that exhibit class I sirtuin selectivity, inhibiting SIRT1–3, is targeted to the mitochondria to inhibit SIRT3 selectively in cells by specific localization. | ||
Results and discussion
Structure–activity relationship study
We envisioned that the mitochondria-targeting peptides developed by Kelley and co-workers37–39 could be starting points for mechanism-based inhibitors of sirtuins. It was hypothesized that such inhibitors could be designed by incorporating a thiocarbonyl-containing lysine residue known to enable sirtuin inhibition.3,29,40,41 Based on insight from previous structure–activity relationship (SAR) studies targeting SIRT2 and SIRT5, including selectivity profiling and co-crystal structures,32,35 we positioned the thiocarbonyl-containing lysine residue as the N-terminal amino acid. With a preliminary series of compounds, we addressed the length of the mitochondria-targeting peptide combined with ε-N-thioacetylated or ε-N-thiomyristoylated N-terminal lysine residues (see Schemes S1 and S2 for syntheses, ESI†). These inhibitors were evaluated for their ability to inhibit the deacetylation activities of SIRT1–3, applying previously described fluorescence-based assay protocols.14,42 Gratifyingly, this revealed that the peptide scaffolds allowed for potent inhibition of SIRT1–3 (Fig. S1, ESI†). Alternation of the hydrophobic and cationic residues in the mitochondria-targeting sequence, resulted in slightly increased selectivity for SIRT1 and further analogues with this architecture were not pursued (Fig. S1, ESI†). Based on this initial compound series, the thiomyristoylated analogs were abandoned due to their apparent selectivity towards SIRT2, which was not surprising based on previous literature.43,44 A small series of substitutions of the N-terminal functional group were selected based on previous SAR studies and X-ray co-crystal structures, which indicated a high degree of freedom for the selection of functionalities at this position (Fig. S2, ESI†).32,35 Based on this series, we proceeded with the 3-phenylpropionyl group (c; Fig. 2) and the alkyne-containing group (a, Fig. 2 and Scheme S6, ESI†), which is amenable to incorporation of fluorophores or other tags using Cu(I)-catalyzed azide–alkyne Huisgen 3+2 cycloaddition “click” chemistry.45,46 Inspired by previous studies of SIRT1–329,35,47–51 and the structures of the active sites in SIRT1–3 (Fig. S3, ESI†), we next analyzed a number of thiocarbonyl binding motifs (1–10; Fig. 2A). The most potent inhibitors of SIRT3, which did not at the same time inhibit the other two isoforms to a more substantial extent, were compounds 1 and 4 (Fig. 2B). Because ε-N-thioacetyllysine residues have been shown to be processed by SIRT1–349 and HDAC8,52 the ε-N′-methylthiourea functionality (4) was chosen for further investigations.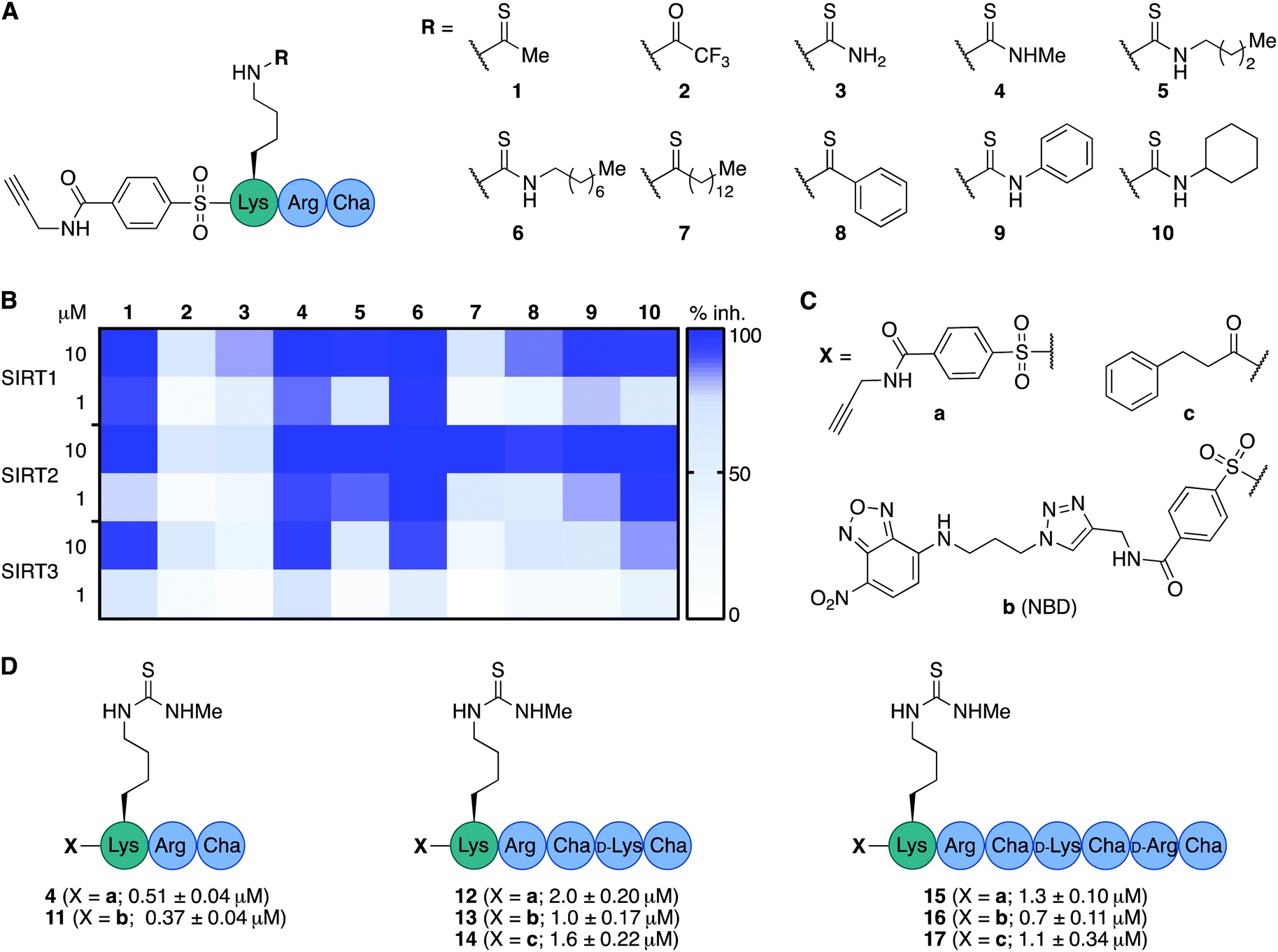 | ||
| Fig. 2 Structure–activity relationship of inhibitors against SIRT1–3. (A) Structures of compounds 1–10. (B) Heatmap summarizing potencies of compounds 1–10 against SIRT1–3 based on %-inhibition. All assays were performed at least twice in duplicate and the values can be found in Table S1 (ESI†). (C) Structures of N-terminal functional groups introduced in compounds 11–17. (D) Structures and IC50 values and standard deviations recorded for compounds 11–17 based on a minimum of two individual assays performed in duplicate. Further information and selectivity data can be found in Fig. S5 and Table S1 (ESI†). | ||
Satisfied that potent inhibition of SIRT3 could be achieved with compound 4 without significant selectivity towards either SIRT1 or SIRT2 (Fig. S4A and S5, ESI†), we synthesized the compound series 11–17 (Fig. 2C and D). This series combines (a) alkyne tag-, (b) fluorescent nitrobenzoxadiazole (NBD)-, and (c) phenylpropionyl-containing N-termini with peptide scaffolds of trimer, pentamer, and heptamer length. Additional potency profiling against recombinant KDAC enzymes was performed for the non-fluorescent compounds 12, 14, 15, and 17 of this series, showing similar effects across SIRT1–3 and excellent selectivity compared to SIRT5–7 and HDAC1–3 (Fig. S5, ESI†).
The NBD fluorophore-containing series (11, 13, 16), for evaluation of mitochondrial localization, was further supplemented with analogs of the heptamer containing Abz, BODIPY, ATTO, and EDANS to evaluate the effect of the fluorophore on cell entry (see Scheme S2B for structures, ESI†). Before analyzing the mitochondria-targeting ability of the fluorophore-containing analogs we demonstrated excellent stability of selected inhibitors in DMEM cell culture medium (Fig. S6, ESI†). Additionally, toxicity was evaluated for selected compounds against a series of immortalized cell lines to inform us about appropriate dosing during the cellular fluorescence experiments. Across our panel of cell lines tested (HEK293T, HeLa, Jurkat, and MCF-7), the most potent compound was 17, which did not exhibit EC50 values below 10–20 μM (Fig. S7 and Table S2, ESI†).
Mitochondrial localization
The cellular localization of compounds 11, 13, and 16 was first evaluated in HeLa cells (Fig. 3 and Fig. S8, ESI†). Not surprisingly, the trimeric scaffold 11 did not display satisfactory cellular or mitochondrial uptake (Fig. 3A), which is in line with a previous report where tetramers were the smallest motifs shown to induce targeting of the mitochondria.37 Both the pentameric (13) and heptameric (16) probes were taken up by the cells and showed excellent overlap with the MitoTrackerTM co-staining dye (Fig. 3B and C). When analyzing the fraction of cells with overlap, it was evident that the heptameric inhibitor (16) was superior to the pentameric inhibitor (13). However, all three NBD-conjugated inhibitors were photo-bleaching within minutes, making the analysis challenging. Therefore, we investigated the more efficient heptameric inhibitor, using a selection of alternative fluorophores, chosen based on their reported bleaching properties, size, charge, and commercial availability. The aim being to identify a fluorophore with better performance, which could be readily incorporated without altering the properties of the inhibitor too drastically. Unfortunately, poor cellular uptake was observed for the ATTO-containing analog (S11) (Fig. S8, ESI†). Similarly, the EDANS-conjugated analog (S12) showed low permeability, perhaps due to the negative charge present in this fluorophore (Fig. S8, ESI†). An analog containing the widely used BODIPY fluorophore (S13) exhibited excellent photostability and good mitochondrial targeting as judged by the co-localization with MitoTracker dye (Fig. S8, ESI†). However, significant perturbation of a large fraction of the cells was observed, including vesicle formation, possibly through disruption of the mitochondrial membranes. Finally, 2-aminobenzoyl (Abz), which offers a minimal structural modification of the inhibitor due to its small size, was introduced to give compound S14. Although this fluorophore is less powerful with a quantum yield of 0.6 for the free acid,53 compared to 0.94 for BODIPY,54 it exhibited cellular uptake and did show overlap with the MitoTracker dye, indicating mitochondrial targeting of compound S14 (Fig. S8, ESI†).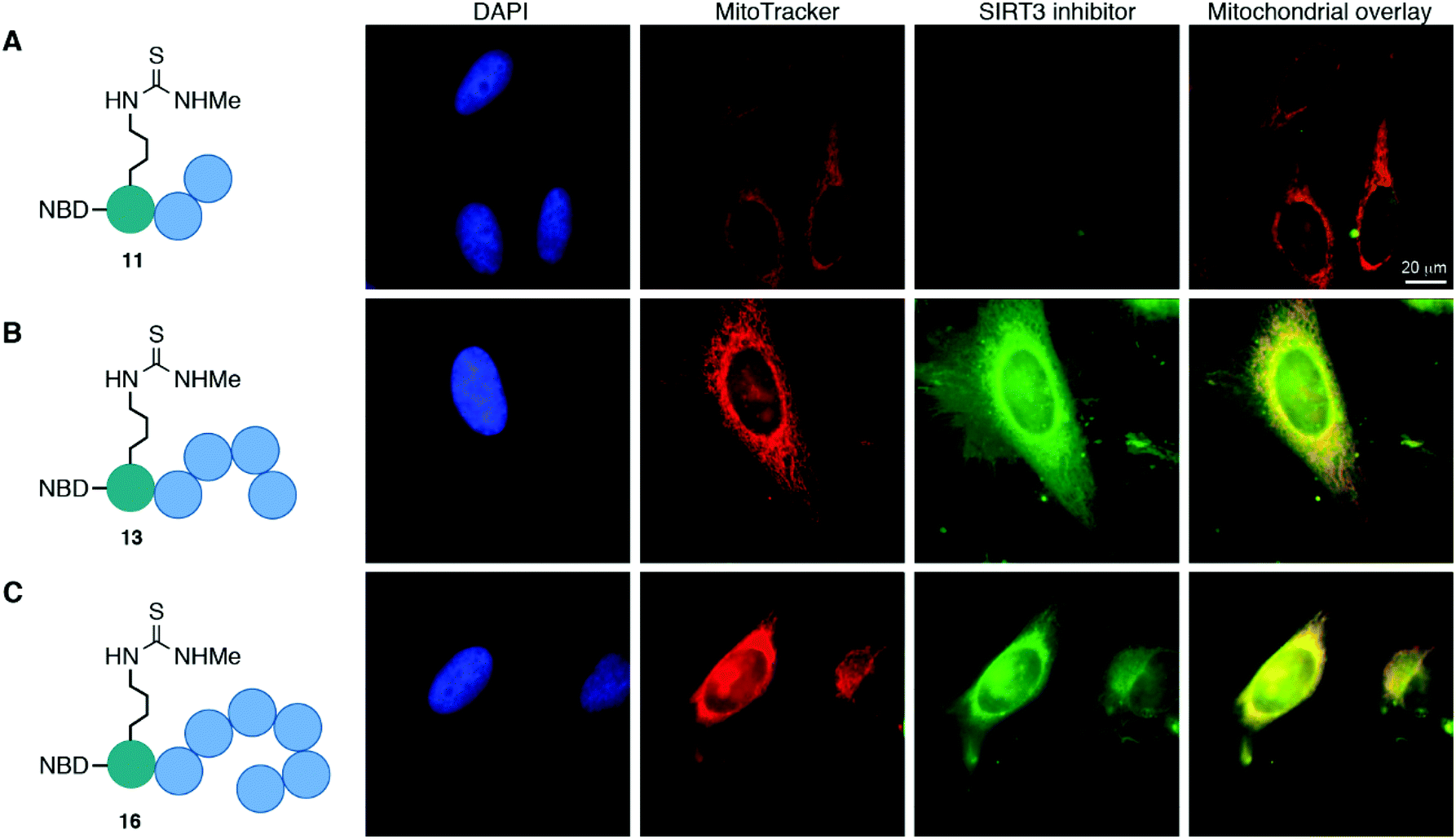 | ||
| Fig. 3 Mitochondrial targeting of fluorophore labeled inhibitors in HeLa cells, determined by co-staining with MitoTrackerTM. (A) NBD-labeled trimer (11); pearson correlation coefficient (r) = −0.66. (B) NBD-labeled pentamer (13); pearson correlation coefficient (r) = 0.59. (C) NBD-labeled heptamer (16); pearson correlation coefficient (r) = 0.46. | ||
Brightfield images indicated primarily healthy HeLa cells after treatment with most fluorophore-conjugated (Fig. S8, ESI†) as well as the non-fluorophore-conjugated compounds (14 and 17; Fig. S9, ESI†). However, slightly increased toxicity was observed for the heptameric scaffolds and substantial changes to the cell morphology were visible for the BODIPY-conjugated compound as also indicated by the fluorescence images discussed above. As expected, the control peptide NBD-TAT (S15) was distributed evenly within the cells with no significant co-localization to the mitochondria and no indication of toxicity in the brightfield images (Fig. S8, ESI†).
Taken together, the data strongly suggest that inhibitors, based on both pentameric and heptameric mitochondria-targeting peptides, are indeed shuttled to the mitochondria in HeLa cells in culture.
Selective targeting of SIRT3 in HEK293T cells in culture
With the compounds based on the longer heptapeptide mitochondria-targeting sequence showing convincing localization to the mitochondria, we were interested in investigating whether SIRT3 was inhibited selectively in cells. Cellular studies were performed with the non-fluorescent compound 17, due to its low cytotoxicity against HEK293T, HeLa, Jurkat, and MCF-7 cells (EC50 values >10 μM) (Fig. S7 and Table S2, ESI†), its low extent of compromising the cell morphology according to brightfield microscopic images of treated HeLa cells (Fig. S9, ESI†), as well as its stability in growth medium (Fig. S6, ESI†). First, we chose the documented mitochondrial protein target MnSOD, for which the degree of acetylation of lysine 68 (K68) has been shown to be regulated by SIRT3.26,27,55,56 Inspired by the work of Meier and co-workers on non-enzymatic acylation,57 we developed a novel mitochondria-targeted acetylating agent (18; Scheme 1) to be used as a positive control (see Scheme S4 for synthesis and structure, ESI†). Gratifyingly, compound 18 showed a significant increase in MnSOD (K68) acetylation in mitochondria enriched fractions of HEK293T lysates. Similarly, cells treated with compound 17 showed a significant increase in MnSOD (K68) acetylation, using 10 μM of the inhibitor (Fig. 4A and Fig. S10, S11, ESI†).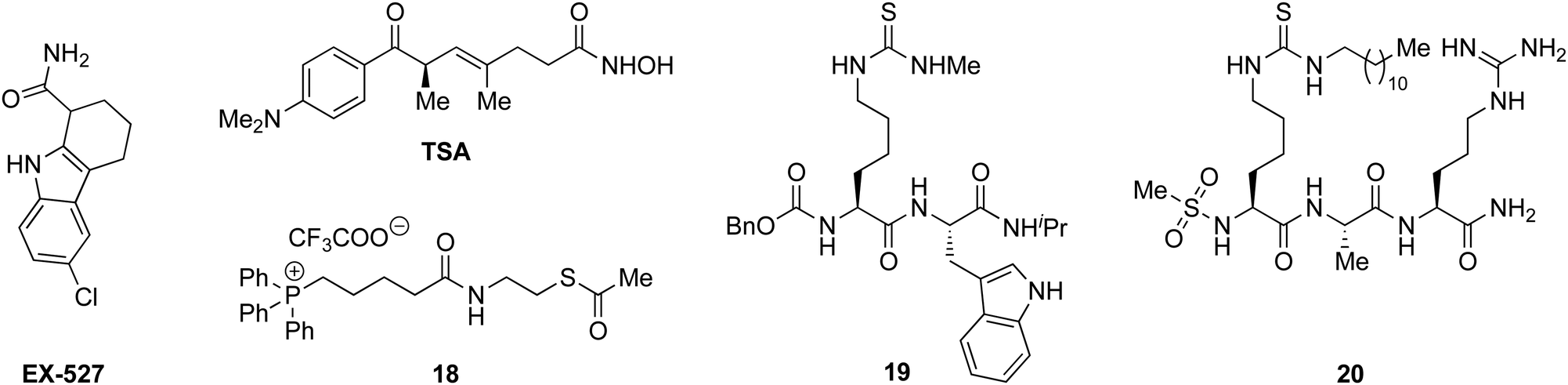 | ||
| Scheme 1 Structures of EX527, TSA, and compounds 18–20.33,35,49 | ||
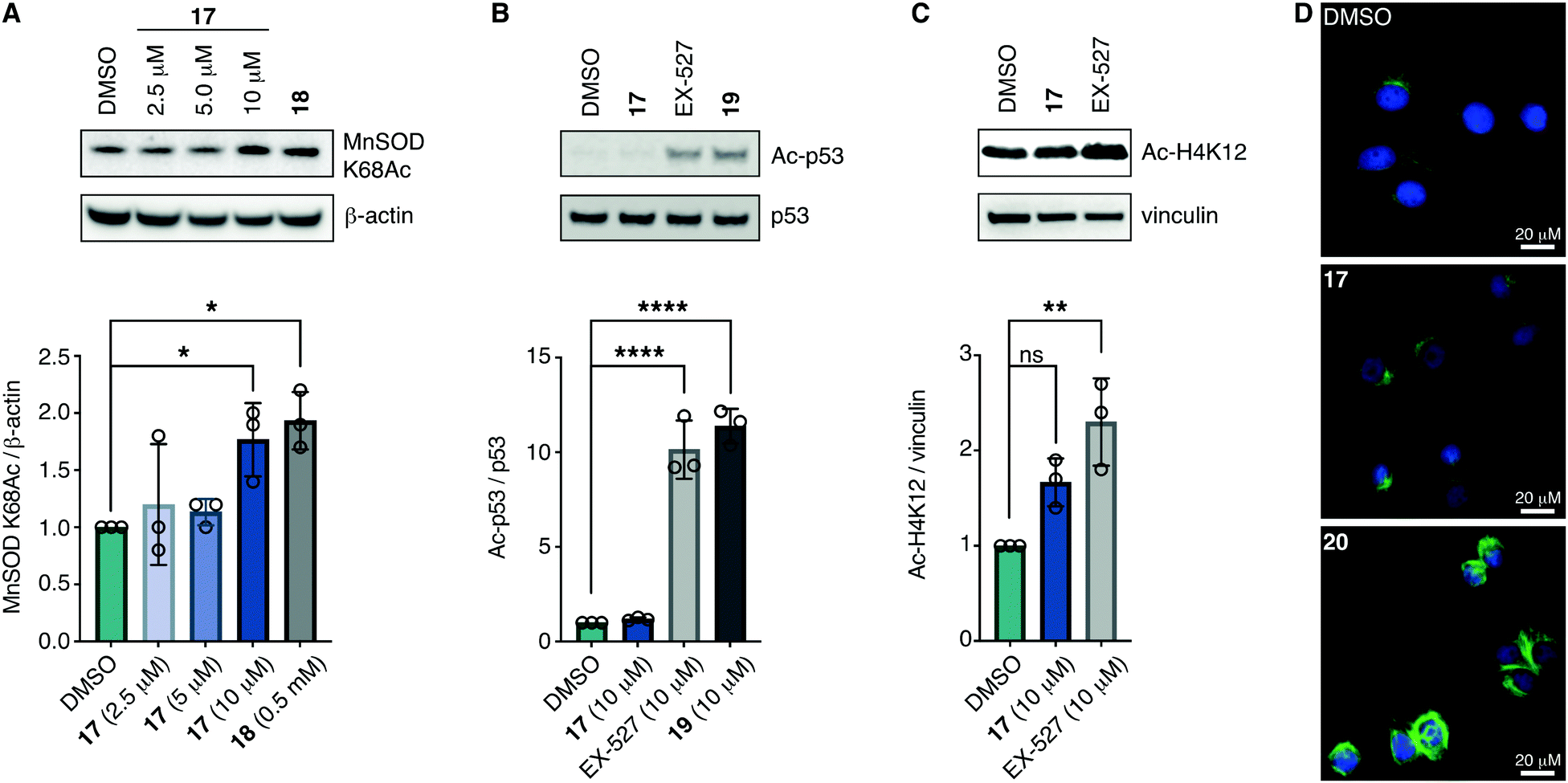 | ||
| Fig. 4 Inhibition of SIRT3 in HEK293T cells. (A) Representative western blot and quantification data showing dose-dependent effect of 17 on acetylation of the documented mitochondrial SIRT3 target MnSOD compared to the positive control 18 (0.5 mM) (n = 3; for full blots and replicates, see Fig. S10 and S11, ESI†). (B) Evaluation of off-target effect of 17 (10 μM) on p53 acetylation compared to positive controls EX-527 (10 μM) and 19 (10 μM) by western blot. Cells were cotreated with TSA (1 μM) to abolish interfering activity of Zn2+-dependent HDACs (n = 3; for conditions, full blots, and replicates, see Fig. S12, ESI†). (C) Evaluation of off-target effect of 17 (10 μM) on H4K12 acetylation compared to positive control EX-527 (10 μM) by western blot. Cells were cotreated with TSA (1 μM) to abolish interfering activity of Zn2+-dependent HDACs (n = 3; for conditions, full blots, and replicates, see Fig. S13, ESI†). (D) Immunofluorescence investigation of the effect of 17 (10 μM) on α-tubulin acetylation compared to DMSO and 20 (5 μM) as the positive control. The data are representative images from two individual experiments. For conditions and additional images, see Fig. S14 (ESI†). Significance of the levels of Ac-p53, H4K12Ac, and MnSOD K68Ac were calculated using one-way ANOVA test. Adjusted p values: ns denotes p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to DMSO treated control. | ||
Finally, we addressed whether the targeting of SIRT3 was also selective over SIRT1 and 2 in HEK293T cells. For SIRT1, we chose the well documented target p5351,58,59 and analyzed the levels of acetylated p53 (K382) in HEK293T cells treated with compound 17, compared to the known SIRT1 inhibitors EX-52760 and 1933,49 as positive controls (Scheme 1, Fig. 4B and Fig. S12, ESI†). Although, compounds 17, EX-527, and 19 are equipotent against SIRT1 in vitro,49 the effect on p53 acetylation (K382) by compound 17 is significantly lower than the positive control compounds (Fig. 4B), suggesting a high degree of selectivity for SIRT3 over SIRT1 in cells.
We furthermore, tested the potential effect of 17 on histone acetylation. The cell treatments were performed as described for the p53 acetylation above and immunoblots were performed for global histone 4 acetylation (Ac-H4), histone 3-lysine 9 acetylation (Ac-H3K9), and histone 4-lysine 12 acetylation (Ac-H4K12) (Fig. S13, ESI†). Only blots for the latter mentioned modification showed acetylation levels above vehicle treatment (DMSO), normalized to vinculin as the loading control. For this modification, a small but statistically insignificant effect of 17 on acetylation was observed, while the effect of known SIRT1 inhibitor EX-527 was more prominent (Fig. 4C). For SIRT2, we and others have recently reported on the challenges of determining cellular effects on lysine acetylation by performing western blots on whole cell extracts.61 We therefore investigated the effect of 17, on α-tubulin acetylation (K40) qualitatively by performing immunofluorescence experiments, comparing to DMSO and 20 (Scheme 1) as negative and positive controls, respectively (Fig. 4D and Fig. S14, ESI†). While treatment with compound 20 produced a significant increase in α-tubulin acetylation compared to the DMSO control, no change was observed when treating cells with compound 17, indicating high selectivity towards SIRT3 over SIRT2 in cells.
Target engagement in HEK293T cells by cellular thermal shift assay
To further evaluate whether compound 17 is directly targeting SIRT3 in living cells, we performed cellular thermal shift assays in HEK293T cells in culture, using immunoblotting for the subsequent analysis (Fig. 5 and Fig. S15–S19, ESI†).62–64 A homologue of compound 17, containing a free lysine residue (17-K), which does not inhibit sirtuin activity was synthesized and tested as a negative control (for synthesis and profiling of 17-K, please consult Scheme S3, Fig. S5 and Table S1, ESI†). | ||
| Fig. 5 Representative blots, plots, and Tagg values from cellular thermal shift assay evaluation of 17vs.17-K, with DMSO control. (A) Investigation of SIRT1 levels from HEK293T cells upon treatment with DMSO, 17 (10 μM) or 17-K (10 μM) and heating to the specified temperatures. (n = 3; for full dataset see Fig. S15 and for full blots and replicates, see full western blot section, ESI†). (B) Investigation of SIRT2 levels from HEK293T cells upon treatment with DMSO, 17 (10 μM) or 17-K (10 μM) and heating to the specified temperatures. (n = 3; for full dataset see Fig. S16 and ESI,† for full blots and replicates). (C) Investigation of SIRT3 levels from HEK293T cells upon treatment with DMSO, 17 (10 μM) or 17-K (10 μM) and heating to the specified temperatures (n = 5 for DMSO and compound 17 treatments. n = 3 for compound 17-K treatments; for full dataset see Fig. S17, S18 and ESI,† for full blots and replicates). Significance of the CETSA shifts were calculated using unpaired t-test of Tagg values from independent experiments. Adjusted p values: *p < 0.05, **p < 0.01 compared to DMSO treated control. | ||
Treatment with the selected compounds at various temperatures was followed by cell lysis to provide whole cell lysates, which were subjected to SDS-PAGE gel electrophoresis and western blotting. Analysis of the normalized data led to a statistically significant (p < 0.01) shift in thermal stability of SIRT3 for 17 and not 17-K compared to the DMSO control, strongly suggesting specific target engagement of compound 17 with SIRT3 in HEK293T cells in culture. The SIRT2 enzyme did not appear to be stabilized by neither 17 nor 17-K, while positive control compound SirReal2 exhibited stabilization of this enzyme (Fig. S16, ESI†). These findings were in agreement with the evaluation of ability to affect α-tubulin acetylation in the immunohistochemical assays discussed above. For SIRT1, on the other hand, compound 17 appeared to stabilize the enzyme up to a similar temperature as the positive control SIRT1 inhibitor 19 (Fig. 5 and Fig. S15, S19, ESI†). This was surprising because we did not record an effect of 17 comparable to 19 in the p53 acetylation assay, which indirectly reports on SIRT1 inhibition in the cells. It should therefore be noted that the thermal shift assay does not report on compound potency or ability to inhibit the enzyme in question, but simply whether the inhibitor binds to the enzyme in a fashion that causes an increase in thermal stability. All the data taken together, therefore shows that both SIRT1 and SIRT3 bind compound 17 in cultured HEK293T cells but only SIRT3 is inhibited by 17 to an extent where significant downstream effect on known deacetylation targets are recorded.
Conclusion
It has been a major challenge to develop inhibitors that selectively target SIRT3 of the class I sirtuin enzymes (of the class III KDACs). Due to the high structural similarity of the active sites of SIRT1–3, small molecule chemotypes have largely failed in producing selective inhibition of SIRT3 over SIRT1 and 2. The differences in the structures of the extended substrate-binding pockets among these three enzymes have enabled the development of selective inhibitors of SIRT2 but have not been successfully harnessed to target SIRT3. Here, we developed compounds based on an alternative strategy that takes advantage of the differential sub-cellular localization of the three class I sirtuins. In work reported during the course of our study, a commonly used mitochondrial targeting motif (the triphenylphosphonium group) was attached to a SIRT2 inhibitor, to inhibit SIRT3 in the mitochondria rather than SIRT1 and SIRT2 in the nucleus and cytosol, respectively.65 The chemotypes developed in the present study, however, have a fundamentally different architecture and includes optimization of their selectivity profiles to dial down affinity for SIRT1 and SIRT2. Our design is predicated on the fusion of attributes from mechanism-based class I sirtuin inhibitors with mitochondria-targeting peptides. After succeeding in the achievement of potent enzyme inhibition in vitro, we optimized the mitochondria-targeting properties and secured compound stability together with limited toxicity. The resulting probe compound exhibited direct engagement of SIRT3 in the mitochondria of cells in culture by an inhibitor molecule for the first time and the downstream acetylation level of the documented SIRT3 target MnSOD was increased.We expect that this novel probe will enable investigation of the function of SIRT3 with unprecedented precision and thus help uncover the potential for development of future therapeutics targeting this enzyme. Finally, our results provide a framework that may be exploited for the targeting of other mitochondrial proteins, including the mitochondrial sirtuin isoforms, SIRT4 and SIRT5, by incorporating alternative acyl group mimics that are selectively targeted by these enzymes.
Author contributions
C. A. O. conceived the study; K. S. T., M. B. and N. R. performed chemical synthesis; K. S. T., M. B. and A. L. N. performed biochemical characterization; K. S. T., M. B. and A. L. N. performed cell-based assays; C. A. O wrote the manuscript with input from all authors; A. S. M., N. R. and C. A. O. supervised the study; C. A. O. acquired funding.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Julie E. Bolding for assistance with mitochondrial enrichment and donation of recombinant SIRT7. This work was supported by the Lundbeck foundation (PhD Fellowship grant R218-2016-1277 and Running Cost grant R289-2018-2074), the Danish Independent Research Council-Natural Sciences (Grant No. 6108-00166B), the Carlsberg Foundation (2013-01-0333, CF15-011, and CF18-0442), the Novo Nordisk Foundation (NNF17OC0029464), and the European Research Council (ERC-CoG-725172–SIRFUNCT).References
- M. C. Haigis and D. A. Sinclair, Annu. Rev. Pathol., 2010, 5, 253–295 CrossRef CAS
.
- A. Chalkiadaki and L. Guarente, Nat. Rev. Cancer, 2015, 15, 608–624 CrossRef CAS
-
N. Rajabi, I. Galleano, A. S. Madsen and C. A. Olsen, in Sirtuins in Health and Disease, ed. W. Zheng, Academic Press, 2018, vol. 154, pp. 25–69 Search PubMed
- H. Jiang, S. Khan, Y. Wang, G. Charron, B. He, C. Sebastian, J. Du, R. Kim, E. Ge, R. Mostoslavsky, H. C. Hang, Q. Hao and H. Lin, Nature, 2013, 496, 110 CrossRef CAS
- J. L. Feldman, J. Baeza and J. M. Denu, J. Biol. Chem., 2013, 288, 31350–31356 CrossRef CAS
- A. S. Madsen, C. Andersen, M. Daoud, K. A. Anderson, J. S. Laursen, S. Chakladar, F. K. Huynh, A. R. Colaco, D. S. Backos, P. Fristrup, M. D. Hirschey and C. A. Olsen, J. Biol. Chem., 2016, 291, 7128–7141 CrossRef CAS
- D. Rauh, F. Fischer, M. Gertz, M. Lakshminarasimhan, T. Bergbrede, F. Aladini, C. Kambach, C. F. Becker, J. Zerweck, M. Schutkowski and C. Steegborn, Nat. Commun., 2013, 4, 2327 CrossRef
- L. L. Yang, H. L. Wang, Y. H. Yan, S. Liu, Z. J. Yu, M. Y. Huang, Y. Luo, X. Zheng, Y. Yu and G. B. Li, Eur. J. Med. Chem., 2020, 192, 112201 CrossRef CAS
- R. H. Houtkooper, E. Pirinen and J. Auwerx, Nat. Rev. Mol. Cell Biol., 2012, 13, 225–238 CrossRef CAS
- M. Schiedel, D. Robaa, T. Rumpf, W. Sippl and M. Jung, Med. Res. Rev., 2018, 38, 147–200 CrossRef CAS
- M. S. Bonkowski and D. A. Sinclair, Nat. Rev. Mol. Cell Biol., 2016, 17, 679–690 CrossRef CAS
- Y. Jiang, J. Liu, D. Chen, L. Yan and W. Zheng, Trends Pharmacol. Sci., 2017, 38, 459–472 CrossRef CAS
- J. Du, Y. Zhou, X. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. Lin, Science, 2011, 334, 806 CrossRef CAS
- A. S. Madsen and C. A. Olsen, J. Med. Chem., 2012, 55, 5582–5590 CrossRef CAS
- K. A. Anderson, F. K. Huynh, K. Fisher-Wellman, J. D. Stuart, B. S. Peterson, J. D. Douros, G. R. Wagner, J. W. Thompson, A. S. Madsen, M. F. Green, R. M. Sivley, O. R. Ilkayeva, R. D. Stevens, D. S. Backos, J. A. Capra, C. A. Olsen, J. E. Campbell, D. M. Muoio, P. A. Grimsrud and M. D. Hirschey, Cell Metab., 2017, 25(838–855), e815 Search PubMed
- M. Pannek, Z. Simic, M. Fuszard, M. Meleshin, D. Rotili, A. Mai, M. Schutkowski and C. Steegborn, Nat. Commun., 2017, 8, 1513 CrossRef
- M. Tan, C. Peng, K. A. Anderson, P. Chhoy, Z. Xie, L. Dai, J. Park, Y. Chen, H. Huang, Y. Zhang, J. Ro, G. R. Wagner, M. F. Green, A. S. Madsen, J. Schmiesing, B. S. Peterson, G. Xu, O. R. Ilkayeva, M. J. Muehlbauer, T. Braulke, C. Muhlhausen, D. S. Backos, C. A. Olsen, P. J. McGuire, S. D. Pletcher, D. B. Lombard, M. D. Hirschey and Y. Zhao, Cell Metab., 2014, 19, 605–617 CrossRef CAS
- C. Roessler, C. Tuting, M. Meleshin, C. Steegborn and M. Schutkowski, J. Med. Chem., 2015, 58, 7217–7223 CrossRef CAS
- C. Roessler, T. Nowak, M. Pannek, M. Gertz, G. T. Nguyen, M. Scharfe, I. Born, W. Sippl, C. Steegborn and M. Schutkowski, Angew. Chem., Int. Ed., 2014, 53, 10728–10732 CrossRef CAS
- B. H. Ahn, H. S. Kim, S. Song, I. H. Lee, J. Liu, A. Vassilopoulos, C. X. Deng and T. Finkel, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14447–14452 CrossRef CAS
- C. Schlicker, M. Gertz, P. Papatheodorou, B. Kachholz, C. F. Becker and C. Steegborn, J. Mol. Biol., 2008, 382, 790–801 CrossRef CAS
- H. Cimen, M. J. Han, Y. Yang, Q. Tong, H. Koc and E. C. Koc, Biochemistry, 2010, 49, 304–311 CrossRef CAS
- M. D. Hirschey, T. Shimazu, E. Goetzman, E. Jing, B. Schwer, D. B. Lombard, C. A. Grueter, C. Harris, S. Biddinger, O. R. Ilkayeva, R. D. Stevens, Y. Li, A. K. Saha, N. B. Ruderman, J. R. Bain, C. B. Newgard, R. V. Farese, Jr., F. W. Alt, C. R. Kahn and E. Verdin, Nature, 2010, 464, 121–125 CrossRef CAS
- T. Shimazu, M. D. Hirschey, L. Hua, K. E. Dittenhafer-Reed, B. Schwer, D. B. Lombard, Y. Li, J. Bunkenborg, F. W. Alt, J. M. Denu, M. P. Jacobson and E. Verdin, Cell Metab., 2010, 12, 654–661 CrossRef CAS
- A. S. Bause and M. C. Haigis, Exp. Gerontol., 2013, 48, 634–639 CrossRef CAS
- X. Qiu, K. Brown, M. D. Hirschey, E. Verdin and D. Chen, Cell Metab., 2010, 12, 662–667 CrossRef CAS
- R. Tao, M. C. Coleman, J. D. Pennington, O. Ozden, S. H. Park, H. Jiang, H. S. Kim, C. R. Flynn, S. Hill, W. Hayes McDonald, A. K. Olivier, D. R. Spitz and D. Gius, Mol. Cell, 2010, 40, 893–904 CrossRef CAS
- B. M. Hirsch, C. A. Gallo, Z. Du, Z. Wang and W. Zheng, MedChemComm, 2010, 1, 233–238 RSC
- B. C. Smith and J. M. Denu, Biochemistry, 2007, 46, 14478–14486 CrossRef CAS
- B. C. Smith and J. M. Denu, J. Biol. Chem., 2007, 282, 37256–37265 CrossRef CAS
- B. C. Dancy, S. A. Ming, R. Papazyan, C. A. Jelinek, A. Majumdar, Y. Sun, B. M. Dancy, W. J. Drury, 3rd, R. J. Cotter, S. D. Taverna and P. A. Cole, J. Am. Chem. Soc., 2012, 134, 5138–5148 CrossRef CAS
- N. Rajabi, M. Auth, K. R. Troelsen, M. Pannek, D. P. Bhatt, M. Fontenas, M. D. Hirschey, C. Steegborn, A. S. Madsen and C. A. Olsen, Angew. Chem., Int. Ed., 2017, 56, 14836–14841 CrossRef CAS
- S. V. Olesen, N. Rajabi, B. Svensson, C. A. Olsen and A. S. Madsen, Biochemistry, 2018, 57, 3903–3915 CrossRef CAS
- M. Bæk, P. Martin-Gago, J. S. Laursen, J. L. H. Madsen, S. Chakladar and C. A. Olsen, Chem. – Eur. J., 2020, 26, 3862–3869 CrossRef
- A. L. Nielsen, N. Rajabi, N. Kudo, K. Lundø, C. Moreno-Yruela, M. Bæk, M. Fontenas, A. Lucidi, A. S. Madsen, M. Yoshida and C. A. Olsen, RSC Chem. Biol., 2021, 2 10.1039/d0cb00036a
- M. C. Frantz and P. Wipf, Environ. Mol. Mutagen., 2010, 51, 462–475 CAS
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 CrossRef CAS
- L. F. Yousif, K. M. Stewart and S. O. Kelley, ChemBioChem, 2009, 10, 1939–1950 CrossRef CAS
- K. L. Horton, M. P. Pereira, K. M. Stewart, S. B. Fonseca and S. O. Kelley, ChemBioChem, 2012, 13, 476–485 CrossRef CAS
- D. G. Fatkins, A. D. Monnot and W. Zheng, Bioorg. Med. Chem. Lett., 2006, 16, 3651–3656 CrossRef CAS
- B. Chen, W. Zang, J. Wang, Y. Huang, Y. He, L. Yan, J. Liu and W. Zheng, Chem. Soc. Rev., 2015, 44, 5246–5264 RSC
- D. Wegener, F. Wirsching, D. Riester and A. Schwienhorst, Chem. Biol., 2003, 10, 61–68 CrossRef CAS
- T. Kosciuk, I. R. Price, X. Zhang, C. Zhu, K. N. Johnson, S. Zhang, S. L. Halaby, G. P. Komaniecki, M. Yang, C. J. DeHart, P. M. Thomas, N. L. Kelleher, J. C. Fromme and H. Lin, Nat. Commun., 2020, 11, 1067 CrossRef CAS
- A. S. Farooqi, J. Y. Hong, J. Cao, X. Lu, I. R. Price, Q. Zhao, T. Kosciuk, M. Yang, J. J. Bai and H. Lin, J. Med. Chem., 2019, 62, 4131–4141 CrossRef CAS
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS
- J. L. Feldman, K. E. Dittenhafer-Reed, N. Kudo, J. N. Thelen, A. Ito, M. Yoshida and J. M. Denu, Biochemistry, 2015, 54, 3037–3050 CrossRef CAS
- X. Bao, Y. Wang, X. Li, X. M. Li, Z. Liu, T. Yang, C. F. Wong, J. Zhang, Q. Hao and X. D. Li, eLife, 2014, 3, e02999 CrossRef
- N. Rajabi, A. L. Nielsen and C. A. Olsen, ACS Med. Chem. Lett., 2020, 11, 1886–1892 CrossRef CAS
- B. Chen, J. Wang, Y. Huang and W. Zheng, Bioorg. Med. Chem. Lett., 2015, 25, 3481–3487 CrossRef CAS
- H. Jing, J. Hu, B. He, Y. L. Negron Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y. L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS
- M. Zessin, Z. Kutil, M. Meleshin, Z. Nováková, E. Ghazy, D. Kalbas, M. Marek, C. Romier, W. Sippl, C. Bařinka and M. Schutkowski, Biochemistry, 2019, 58, 4777–4789 CrossRef CAS
- A. S. Culf, H. Yin, S. Monro, A. Ghosh, D. A. Barnett, R. J. Ouellette, M. Cuperlovic-Culf and S. A. McFarland, Bioorg. Med. Chem., 2016, 24, 929–937 CrossRef CAS
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS
- Y. Chen, J. Zhang, Y. Lin, Q. Lei, K. L. Guan, S. Zhao and Y. Xiong, EMBO Rep., 2011, 12, 534–541 CrossRef CAS
- C. He, J. M. Danes, P. C. Hart, Y. Zhu, Y. Huang, A. L. de Abreu, J. O'Brien, A. J. Mathison, B. Tang, J. M. Frasor, L. M. Wakefield, D. Ganini, E. Stauder, J. Zielonka, B. N. Gantner, R. A. Urrutia, D. Gius and M. G. Bonini, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23534–23541 CrossRef CAS
- R. A. Kulkarni, A. J. Worth, T. T. Zengeya, J. H. Shrimp, J. M. Garlick, A. M. Roberts, D. C. Montgomery, C. Sourbier, B. K. Gibbs, C. Mesaros, Y. C. Tsai, S. Das, K. C. Chan, M. Zhou, T. Andresson, A. M. Weissman, W. M. Linehan, I. A. Blair, N. W. Snyder and J. L. Meier, Cell Chem. Biol., 2017, 24, 231–242 CrossRef CAS
- T. Suzuki, T. Asaba, E. Imai, H. Tsumoto, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2009, 19, 5670–5672 CrossRef CAS
- H. Vaziri, S. K. Dessain, E. N. Eaton, S.-I. Imai, R. A. Frye, T. K. Pandita, L. Guarente and R. A. Weinberg, Cell, 2001, 107, 149–159 CrossRef CAS
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J. F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, J. Med. Chem., 2005, 48, 8045–8054 CrossRef CAS
- T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Olah, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovadi, M. Schutkowski, W. Sippl, O. Einsle and M. Jung, Nat. Commun., 2015, 6, 6263 CrossRef CAS
- D. Martinez Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef
- D. Martinez Molina and P. Nordlund, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 141–161 CrossRef CAS
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundback, P. Nordlund and D. Martinez Molina, Nat. Protoc., 2014, 9, 2100–2122 CrossRef CAS
- M. Li, Y. L. Chiang, C. A. Lyssiotis, M. R. Teater, J. Y. Hong, H. Shen, L. Wang, J. Hu, H. Jing, Z. Chen, N. Jain, C. Duy, S. J. Mistry, L. Cerchietti, J. R. Cross, L. C. Cantley, M. R. Green, H. Lin and A. M. Melnick, Cancer Cell, 2019, 35(916–931), e919 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00216j |
| ‡ Present address A. S. M. is now at Novo Nordisk A/S, DK-2880, Bagsværd, Denmark. |
| § Present address N. R. is now at Red Glead Discovery, 223 63, Lund, Sweden. |
| This journal is © The Royal Society of Chemistry 2021 |