
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CsPbI3 nanocrystal films: towards higher stability and efficiency
Yingying
Tang†
 ,
Arnon
Lesage†
and
Peter
Schall
*
,
Arnon
Lesage†
and
Peter
Schall
*
Institute of Physics, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: P.Schall@uva.nl
First published on 26th November 2020
Abstract
Perovskite nanocrystals (NCs) have seen unprecedented interest in recent years for their numerous applications in optoelectronic devices. Out of the all-inorganic perovskites, CsPbI3 NCs possess a bandgap closest to the ideal one for solar cells (SCs). CsPbI3 NC-based devices have developed rapidly, while at the same time they show the most instability of the three halide perovskites. Therefore, there is urgent need to find treatments that improve their stability and enhance their efficiency, thus maximizing their applicability. This review summarizes recent developments of CsPbI3 NC films focusing on the treatments to enhance their stability, as well as on photophysical processes that could help enhance their efficiency in optoelectronic devices.
 Yingying Tang | Yingying Tang received her PhD degree from the University of the Chinese Academy of Sciences (Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences), China. In 2016–2018, she was honored as a Villum postdoc fellowship at Department of Chemistry, Technical University of Denmark, Denmark. She was involved in the development of photoelectrochemical and optical properties of perovskites as well as application as photodetectors. She is currently a postdoc at University of Amsterdam. Her research interests are in design, synthesis, assembly and optical properties of all-inorganic metal halide perovskite nanomaterials. |
 Arnon Lesage | Arnon Lesage received his PhD in Physics from the University of Amsterdam. His research revolves around optical properties of quantum dots and nano-structured materials, and ways to modify their optical properties. Currently he explores applications of QD's as photoactive layers to tune optical spectra, using inorganic perovskites and other inorganic semiconductor NCs. |
 Peter Schall | Peter Schall is full professor in Soft matter and Nanomaterials at the University of Amsterdam. He received his PhD in physics from RWTH Aachen (Germany) and joined the University of Amsterdam after a postdoc at Harvard University (USA). His current research spans the soft condensed matter and nanomaterials fields, including the assembly of nanocrystals such as perovskites into supercrystals. His research focuses on assembly principles governed by equilibrium and nonequilibrium statistical physics, and the mechanical and optical properties of the resulting micro- and nano-structured materials (research webpage: http://www.peterschall.de). |
1. Introduction
Inorganic perovskite nanocrystals (IP-NCs) are currently a hot topic in the research community due to their superior optical properties, facile synthesis and improved stability.1–6 The NCs are distinct from the bulk perovskites due to the quantum confinement effect giving rise to properties such as size-dependent photoluminescence (PL), high surface/volume ratio, high photoluminescence quantum yield (PLQY) and enhanced phase stability. Application of perovskite NCs in solar cell (SC) devices has resulted in reported efficiencies of up to ∼17.39% from 10.77% in 2016 (Fig. 1a).7–12 Meanwhile, extremely high color purity and easily tunable emission of perovskites also make them a promising material for light-emitting devices (LEDs).13–16 For instance, Zn-alloyed CsPbI3 NC-based LEDs showed improved performances in the past few years, reaching an external quantum efficiency (EQE) of 15.1%.17 Evidently, these materials have great potential for next-generation photovoltaics (PV).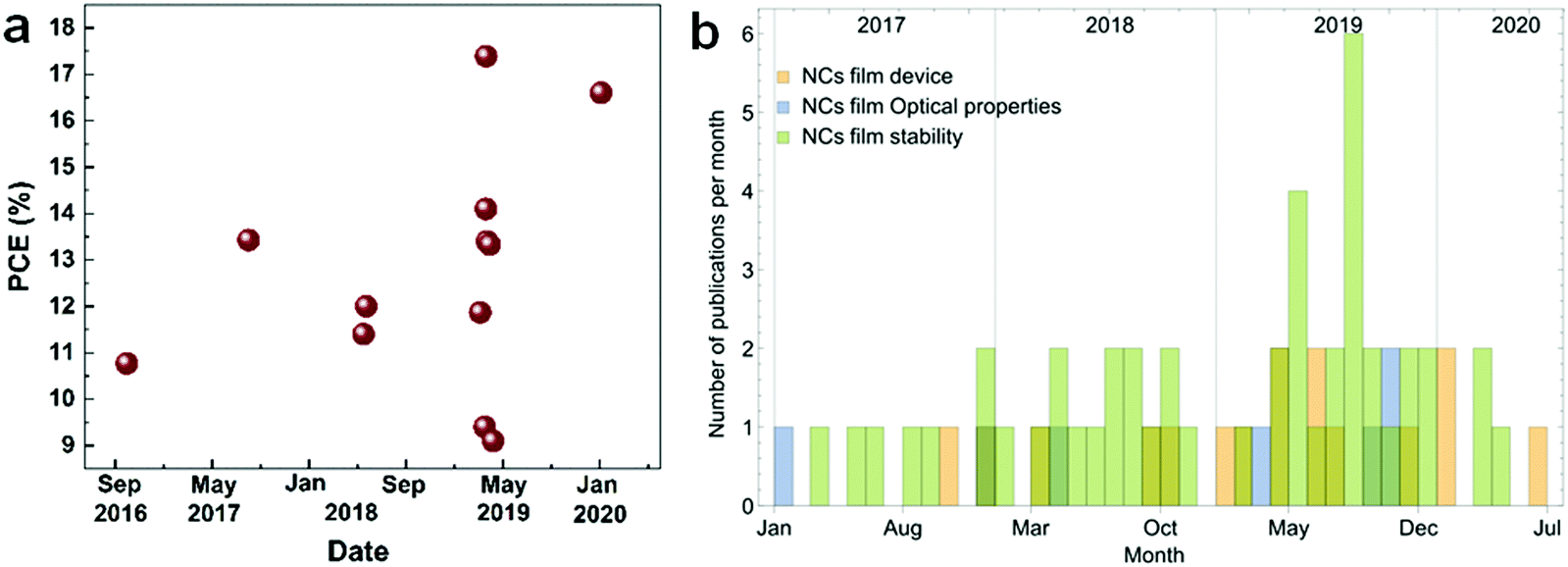 | ||
| Fig. 1 Research on CsPbI3 NC films. (a) Recently reported PV cell efficiency for CsPbI3 NC films based SCs. (b) Recently reported publications for CsPbI3 NC film developments (stability, photon recycling, carrier multiplication and exciton effect). Source: Web of Science. | ||
Among the inorganic perovskite halides, the red perovskite, CsPbI3, has a bandgap which is closest to the ideal one for SCs, ∼1.3 eV. At the same time, iodide-based perovskites are proving the most challenging to be stabilized for the long term. It is known that metal-halide perovskites are not stable in water, polar solvents, oxygen and light, which hinders their commercial applications. Several strategies have been suggested to enhance the NCs’ stability and delay the degradation of their properties. For example, while CsPbI3 NC films immersed in water degrade within hours, the treated NC films show stable optical properties for up to one month. Similar improvements are obtained for NCs stored at high humidity and high temperature. In addition to stability, power conversion efficiency (PCE) is a crucial factor for PV applications. For a high-efficiency device, the NCs must be assembled into dense, ordered films with superior structural and charge transport properties. Application of diverse treatments can, besides improving the stability, also enhance the PCE, which is limited by poor electronic coupling of the NCs or by scattering at interfaces or defects in the NC lattice. A number of chemical treatments have been devised to enhance the NC coupling and allow for higher charge carrier mobilities. As an example, SCs based on NaOAc treated CsPbI3 NCs have been shown to retain 90% PCE for 22 days.18
Finally, the assembly and treatment of NC films offer new opportunities to implement efficiency-enhancing optoelectronic effects such as photon recycling (PR) and multiexciton generation (MEG); if realized in practice, these could provide significant enhancements to the efficiency of optoelectronic devices.
There are several reviews on perovskite NCs focusing on the other lead-halides,19–23 whereas the red CsPbI3 perovskite NCs have received less attention. Yet the number of publications reporting on their treatments and stability has increased recently (Fig. 1b). This review will expound the research trend towards red-perovskite NC film-based devices, highlighting recent effective ways towards high stability and efficiency. In particular, we review works investigating stability improvement in Section 2; this part will highlight the most effective treatments and techniques to produce stable CsPbI3 films. Some of these techniques could be also relevant for the other lead-halides and perovskites. We then discuss dedicated photophysical processes, which could offer additional efficiency enhancement of the NC films in Section 3. We hope this review may inspire new directions in the development of NC-based optoelectronic devices. Meanwhile, much of the discussion and techniques apply to other semiconductor materials as well, so that it may be useful beyond the scope of the perovskite community.
2. Stability of CsPbI3 NC films
Among the lead-halide inorganic perovskite NCs (based on Br, Cl, or I), the iodide-based ones are the most unstable. It is easily observed that the NC films change to yellow color or degrade partially within one month, leading to a deterioration of the optical properties including decreased PLQY, spectral changes and decreased device performance. Improving the stability of CsPbI3 NC films is therefore a crucial challenge on the way towards applications.Structural and environmental causes are at the root of this instability, and a number of studies aim to reveal the underlying degradation mechanism and resolve the phase instability of CsPbI3.24–28 Halide perovskites are ionic materials that are easily dissolved in aqueous solutions. Hence, water and polar solvents are detrimental to CsPbI3 NCs due to their ability to dissolve the perovskite crystals. NC films suffer from the same problem, as they absorb water; however, encapsulation can help to delay this process by protecting the NCs and slowing down humidity exchange with the environment.
Structural instability
Depending on the temperature, four phases (α, β, γ and δ) can be identified in CsPbI3 NCs. The α-phase, a high temperature cubic phase, forms at around ∼620 K and is metastable. Upon cooling it will transform into the tetragonal β-phase at ∼510 K, and then finally to the orthorhombic γ-phase at ∼325 K.29 Below that, a phase transition to the δ-phase takes place. The α, β and γ-phases are all optically active, and can be used for PV and other devices. They are collectively referred to as the black phase. The δ-phase, however, is a non-luminous material, which is not useful for devices and is often termed yellow phase, referring to its yellow appearance.To assess the structural stability of the perovskite system, the Goldschmidt tolerance factor (t) and octahedral factor (μ) have been used as key parameters. They are based on the radii of the atomic constituents30 and are expressed as, 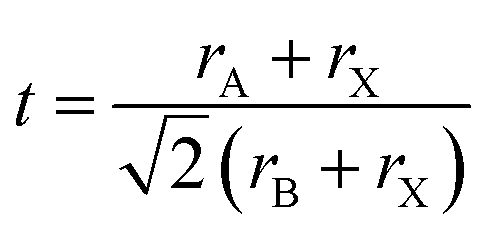 and μ = rB/rA, where rA, rB and rX are the effective ionic radii of the A, B, and X atoms of the general ABX3 perovskite structure. The rule of thumb is that the higher the values of these parameters, the more stable the structure is. For CsPbI3 in the α-phase, t and μ are calculated to be 0.893 and 0.47, respectively, which indicates that the iodide-based perovskite in the black phase is metastable. In contrast, for the most stable halide, CsPbBr3, t = 0.92 and μ = 0.50 are found.31
and μ = rB/rA, where rA, rB and rX are the effective ionic radii of the A, B, and X atoms of the general ABX3 perovskite structure. The rule of thumb is that the higher the values of these parameters, the more stable the structure is. For CsPbI3 in the α-phase, t and μ are calculated to be 0.893 and 0.47, respectively, which indicates that the iodide-based perovskite in the black phase is metastable. In contrast, for the most stable halide, CsPbBr3, t = 0.92 and μ = 0.50 are found.31
In addition to the tolerance factor, the thermodynamic instability of CsPbI3 perovskites is related to structural distortions. It is known that the “black” phase, or α-CsPbI3, is stable at high temperature, ∼620 K, and the “yellow” phase (δ-CsPbI3) is stable at room temperature.32 Therefore, the black α-phase is intrinsically unstable at room temperature, even in a moisture-free atmosphere, and will transform spontaneously from the δ-phase only above 321 K. The small absolute value of enthalpy of CsPbI3 compared to Br and Cl-based perovskites indicates that the entropic contribution to their Gibbs free energy of formation will be significant and may outweigh the contribution of enthalpy. This indicates that the functional black α-CsPbI3 phase is entropically favored.33–36 Theoretical calculations further prove that vibrational instabilities exist, induced by octahedral tilting in their high-temperature cubic phase from lattice-dynamics calculations.32 Investigation of single-crystal γ-CsPbI3 indicates that the rattling of the Cs cation, lower coordination of the Cs site, and local octahedral distortion contribute to the thermodynamic instability at room temperature.35
Environmental instability
The environmental instability is the result of oxygen absorption and light illumination. Photo-generated electrons in perovskites can react with oxygen in the air, yielding O2− and leading to degradation of the MAPbI3 films.37,38 As a result, the photoluminescence displays a strong decrease in intensity, indicating that O2 acts as a scavenger of photoexcited electrons. Yuan et al. further mentioned that in contrast to MAPbI3, decomposition of CsPbI3 usually happens under a combined condition of oxygen, light illumination and water, while the decomposition does not take place with oxygen alone.39 Likewise, light illumination can decrease the photoluminescence intensity, leading to detachment of the capping agent, collapse of the CsPbI3 quantum dot surface, and finally formation of surface Pb0. The consequence is PL quenching with a dramatic decrease of the PLQY.40To address these issues and improve the stability of CsPbI3 NCs, a number of methods have been introduced. For example, substitution of Pb2+ by other cations such as Mn2+ through doping provides a possibility to increase the parameter t by increasing the effective radius ratio, and therefore the corresponding stability. For the fabrication of films, many strategies have been suggested, classified according to the type of materials (Table 1) and the processing step at which the treatment is employed. While the above example of Mn2+ is used to stabilize the structure, other methods such as surface ligand passivation, polymer encapsulation or salt treatments mainly interact with the surface of the NCs or films without changing the intrinsic structure. An overview of the different treatments classified by the processing step, during which they are applied, is given in Fig. 2.
| Materials | Advantages | Disadvantages |
|---|---|---|
| Aliphatic ligands | Easy to apply, leads to higher stability and superior optical properties | Weak binding and easy to detach from NCs |
| Inorganic materials | Stable and strongly bound passivation | Difficult to apply and optimize, affects optical properties; not flexible |
| Polymers | Flexible and transparent; effective protection from the environment | Easy to decompose under high temperature; insulating |
| Salts | Larger coupling interaction with NCs; improve conductivity | Difficult to obtain uniform treatment of layers |
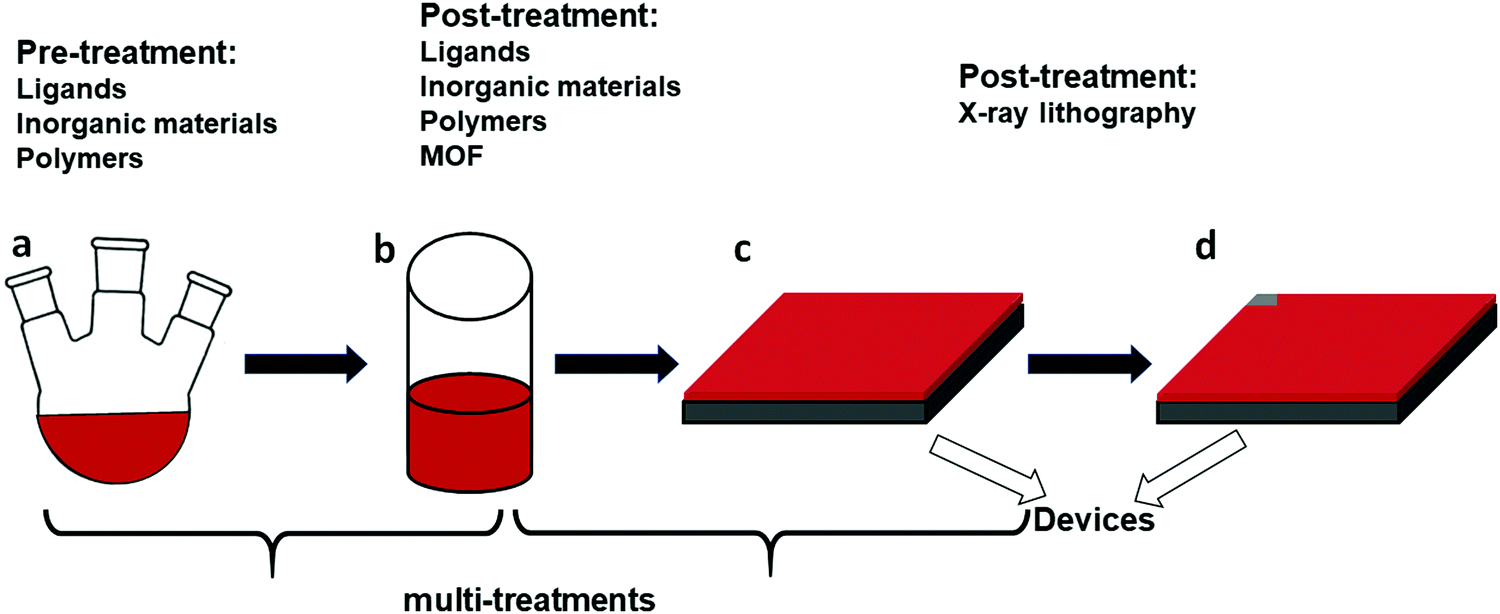 | ||
| Fig. 2 Methods to enhance the stability of CsPbI3 NC films: (a) pre-treatments using precursors for the CsPbI3 NCs, such as ligands, polymers, and salts. (b) Post-treatments of the CsPbI3 NCs by ligands, inorganic materials, polymers and salts. Ligands are typically attached to the surface of the NCs through physical absorption or Coulomb attraction. Polymers can encapsulate the NCs, binding through active sites such as N or O, which can form bonds with the Pb2+ of perovskites. Salt treatments are usually used to mitigate defects of perovskite NCs or films, in which charged defects can be passivated by the salts. (c and d) Fabricated CsPbI3 NC films and their further post-treatment by X-ray lithography. | ||
A distinction is made between pre-treatment and post-treatment, referring to the synthesis step of the NCs. For the pre-treatments, the materials are introduced into the synthesis precursors. For post-treatments – after the formation of the NCs – two options exist: they can be applied before or after the NCs are assembled into a film. Furthermore, a recent strategy to stabilize the NCs is based on the metal–organic framework (MOF), where lead-based frameworks constructed by large organic molecules take advantage of the strong ionic bonding between the organic ions and Pb2+ in perovskites in order to serve as ligands and enhance the stability of the NCs.
2.1 Passivation by surface ligands
Surface ligands are crucial to the NCs for several reasons. First, they arrest the growth of the NCs during synthesis, thus defining the resulting NC size. Second, the ligands are important to passivate the surface. Incomplete coverage of the NC surface can lead to the formation of surface traps or defects, which would result in lower quality NCs. Third, the choice of surface ligands has a crucial effect on the NC stability: passivation using hydrophobic ligands can prevent or delay the degradation from humidity or oxygen. In this regard, different types of surface ligands have been investigated for their ability to enhance the stability and carrier transport of CsPbI3 NC films. The typical ligands, oleic acid (OA) and oleylamine (OLA), have proven to be useful in passivating the NC surface defects; however, the long linear chains of these insulating ligands block efficient carrier transport between the NCs. Instead, mixtures of short and long carbon-chain ligands, such as octanoic acid and OA, or conductive ligands like thiol or phosphine-based ligands offer alternative choices to provide good carrier transport while stabilizing the NCs. In the following, we will classify them according to their role and treatment steps.(i) Pre-treatment. Pre-treatment implies that the surface ligands are added to the perovskite precursors before the crystallization of the CsPbI3 NCs. Here, several of those treatments are reviewed in detail. Both cation ligands such as didodecyldimethylammonium bromide (DDAB),41 benzoyl iodide,42 and anion ligands such as aluminum distearate (AIDS)43 have been reported to be useful for the passivation of the NCs.
DDAB was introduced into the precursors of CsPbI3 NCs during the hot-injection reaction,41 and the stability was studied after spin-coating of the treated NCs into films (Table 2). It was found that both the stability towards polar solvents and the thermal stability improved dramatically: upon submerging the films in water for 50 minutes, the PL intensity decreased by merely 5%. Furthermore, even upon heating to 160 °C, the PL kept 89% of its original intensity. Meanwhile, the untreated OA/OLA capped NCs lost 93% of PL intensity during the same treatment, resulting in a yellow film. It is speculated that DDA+ has a stronger affinity to negative sites (e.g. I−) of the perovskite lattice as well as exhibits short branched chains, thus providing an effective surface passivation. LEDs based on DDAB-stabilized CsPbI3 NCs displayed a maximum EQE of 1.25% and a luminance of 468 cd m−2, which is ∼100 times improved compared to OA/OLA-CsPbI3 LEDs.
| Material | Treatment | Deposition technique | Film/device | Stability | Ref. |
|---|---|---|---|---|---|
| Note: pre means pre-treatment and post means post-treatment; neat represents neat films. | |||||
| DDAB | Pre- | Spin-coating | Neat/LEDs | At 160 °C, PL kept 90%; EQE of 1.25% and a luminance of 468 cd m−2 | 41 |
| Benzoyl iodide | Pre- | Drop-casted | Neat | 20 days in air | 42 |
| AIDS | Pre- | Drop-casted | Neat | 85 °C, 16 hours | 43 |
| AET | Post | Spin-coating | Neat/photo-detectors | Neat: in water after 1 hour, 33% PL left (untreated 14%); high photoresponsivity (105 mA W−1) and detectivity (5 × 1013 Jones at 450 nm and 3 × 1013 Jones at 700 nm); 95% of the initial responsivity in ambient air for 40 h without any encapsulation | 45 |
| IDA | Pre-/post | Spin-coating | Neat/LEDs | Phase stable after 40 days, EtOAc wash, PL no changes; EQE of 5.02% and 748 cd m−2 luminance | 47 |
| Mn-Doping | Pre- | Drop-casted | Neat | Ambient condition, one month (untreated 5 days) | 47 |
| PbS | Pre- | Spin-casted | Neat/LEDs | Neat: 33 days in air; EQE of 11.8%, the EL intensity of CsPbI3 LEDs showed a negligible change | 58 |
| Iodine modified graphene oxide | Pre- | Neat | Under ambient conditions (dark, low temperature, ∼4 °C and low humidity, ∼1% RH) for 4 weeks | 61 | |
| Mn-doping | Post | Drop-casted | Neat | 28 days ambient condition | 66 |
| μ-Graphene | Post | Spin-coating | Neat/SCs | 60% humidity and 100 °C, phase unchanged by XRD; PCE of 11.40 | 69 |
| Chloride doping | Post | Spin-coating | Neat | 96 hours, after exposing the samples to RH of 11% at 23 °C; 18 hours in a 11% RH atmosphere at 23 °C | 70 |
| Ag doping/passivation | Post | Drop-casted | Neat/LEDs | Neat: 80% PL after 48 hours in ambient (50%); EQE of 11.2%; EL intensity of Ag-based CsPbI3 LEDs maintained 80% of their initial value after 10 days of storage in a glovebox under nitrogen | 67 |
| PMMA | Post | Drop-casted | Neat/LEDs | Neat: 25 days in air and 4 days in water; the position and emission intensity did not change under lighting for 24 h, at room temperature, RH 65% | 75 |
| PMMA-co-P(MA-POSS)-co-P(MA-NC) | Pre-/post | Spin-coating | Neat/WLEDs | Neat: water: 60 days; thermal: 120 °C, 80% PL left, UV: 156 hours, 81% PL left; a low correlated color temperature of 4325 K, and a luminous efficacy of 26.3 lm W−1 | 78 |
| FPEAI/Mn/H2PbI4 | Pre- | Spin-coating | Neat/SCs | Neat: 10 days in air PL and phase; PCE of 13.4%; retaining 92% after 500 h without encapsulation in ambient air | 51 |
| NaOAc | Post after film | Spin-coating | SCs | PCE of 13.3%; SCs: 90% PCE, 22 days in dark, 20% humidity | 18 |
| CsNO3 | Post after film | Spin-coating | SCs | PCE 14.10%; SCs: 87% PCE in N2 after 7 days; after 54 hours of storage in air, 70% PCE | 48 |
| SiO2 coating/PMMA | Pre- and post | Spin-coating | Neat/WLEDs | Neat: at least 5 days for only film under UV light; a power efficacy of 61.2 lm W−1 and little variation of the emission spectrum was observed after worked 10 h | 62 |
| ZIF-8, mixed with PMMA | Post | Drop-casted | WLEDs | QDs: ∼43% PL intensity after 30 days at ambient condition and ∼35% at 90 °C; luminescence efficiency 12.85 lm W−1 | 79 |
| X-ray lithography | — | — | Neat | 3 days in water | 80 |
Higher thermal stability was also observed for AIDS-capped CsPbI3 NCs deposited on a glass substrate.43 After heating to 85 °C for 8 hours at ambient conditions, the film still exhibited bright emission. Even after heating for 16 hours, the NCs were still in a black phase as confirmed by X-Ray diffraction (XRD). The AIDS ligands can suppress the defects generated by the detachment of OLA and OA ligands.
(ii) Post-treatment of NCs. The surface ligands can also be introduced at a later stage, after formation of the NCs. The reason to do so is quite practical, considering the limited solubility of some of the surface ligands in the reaction oil (octadecene). Especially when using the hot-injection method, it is easier to introduce the ligands after the reaction oil is replaced. In this case, the ligands can be added in the form of solution or powder. After the treatments of the CsPbI3 NCs, films can be prepared and studied. The most popular post-treatment of NCs is ligand exchange. A typical way to treat the NC films is to rinse them by methyl acetate (MeOAc), which will decompose into acetic acid and methanol as intermediate substances to substitute the long-chain ligands of the NCs and further improve the carrier transport properties. However, acetic acid is detrimental to the perovskite NCs; therefore alternative treatments are explored that maintain the perovskite crystal quality.
Kim et al. reported the use of NaOAc, after spin-coating CsPbI3 NCs onto a substrate, the NC film was soaked in a NaOAc/MeOAc solution for ligand exchange, where no fusion appeared between the NCs.18 As indicated by X-ray photoelectron spectroscopy (XPS), a small amount of Pb–OH was observed, confirming the successful bonding between the Pb2+ host and the OAc− ligands. This is further confirmed by an IR signal at 3650 cm−1 corresponding to the O–H stretching and Pb–OH bending peaks. Concomitantly, the IR signal of aliphatic hydrocarbon weakened, suggesting the successful removal of long-chain oleates. The SCs based on the NaOAc-treated NC films showed a PCE of 13.3% and a high stability, with 90% PCE remaining after 22 days in darkness and at 20% relative humidity (RH).
(i) Pre-treatment. Different from adding ligands together with precursors, the pre-treatment of ligands could also be performed after the injection of Cs-oleate. Recently, Lin et al. reported the use of hexamethyldisilathiane (HMS) for pre-treatment of CsPbI3 NCs during hot injection synthesis.44 The HMS-CsPbI3 NC films retained the main black cubic phase and high PL brightness when exposed to water vapor (RH: 98 ± 1%) even after 32 hours, while only a small amount of δ-phase was observed by XRD (Fig. 3a). As a reference, untreated films turned into the δ-phase completely after 60 min. Density functional theory (DFT) calculations were used to calculate the bond strength of the ligands to the Pb2+ site. A much higher binding energy for HMS (−0.89 eV) than that for OLA ligands (−0.37 eV) was found, confirming the strong absorption ability of HMS to the NCs. Therefore, a strong Lewis-base bond on the lead-rich perovskite surface is formed due to the lone pair electrons by the sulfur in the HMS, prohibiting the generation of surface defects.
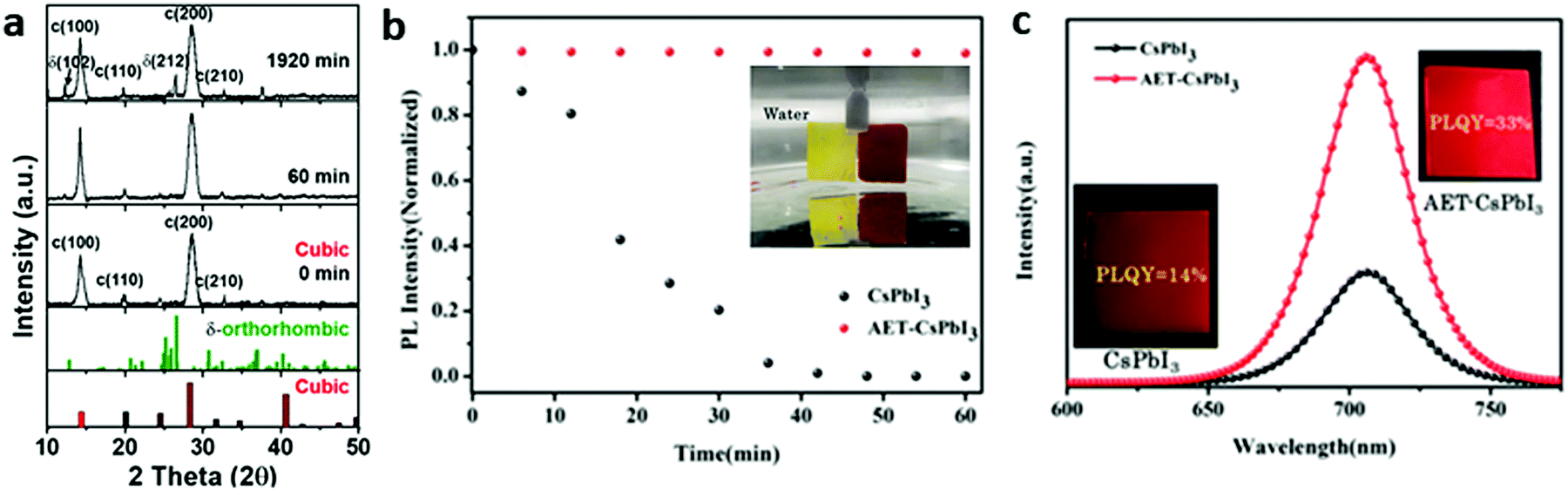 | ||
| Fig. 3 Stability of treated CsPbI3 NCs. (a) In situ XRD measurements of HMS-CsPbI3 NC films. Reprinted with permission from ref. 44. Copyright 2020, American Chemical Society. (b) PL intensity of CsPbI3 and AET-CsPbI3 NC films immersed in water for different times. Inset: Photographs of the CsPbI3 and AET-CsPbI3 samples in water. (c) PL curves of the treated and untreated NC films shown in (b). Insets show samples and corresponding PLQY. (b and c) Reprinted with permission from ref. 45. Copyright 2016, Wiley-VCH. | ||
(ii) Post-treatment of NCs. The most popular post-treatment of NCs is ligand exchange. For example, 2-aminoethanethiol (AET)45 and bidentate 2,2′-iminodibenzoic acid (IDA)46 have been reported to improve the stability of the NC films. Recently, a bidentate ligand, IDA-passivated CsPbI3 NC film retained a stable phase after 40 days and after ethyl acetate wash, while the PL displayed no changes. After fabricating into SCs, 5% EQE and 748 cd m−2 luminance were observed.47 The AET ligands appear to passivate the surface defects and improve the quality as shown by PLQY and charge transport measurements. AET treated CsPbI3 NC films displayed higher stability in water, where the film kept a dark-red color and maintained 90% of its PL intensity after it was immersed into water for 60 minutes. Furthermore, a two-times increase in radiative recombination rate (1.45 × 107 s−1), compared to the untreated films, as well as high PLQY of 33% were observed as shown in Fig. 3b and c. Photodetectors based on the AET-CsPbI3 QD films exhibit remarkable performance compared to CsPbI3 QD films, high photoresponsivity (105 mA W−1) and detectivity (5 × 1013 Jones at 450 nm and 3 × 1013 Jones at 700 nm) without an external bias. Without any encapsulation, more than 95% of the initial responsivity in ambient air was retained for 40 h, showing excellent stability of the photodetectors.
Another choice for the post-treatment is CsNO3.48 In this case, a saturated surface treatment solution is first prepared by dissolving anhydrous CsNO3 into EtOAc. A NC film is then prepared by repeated spin-coating of the NCs/octane and MeOAc solution, respectively. After that, the film is treated by the saturated surface solution for 10 seconds. As evidenced by scanning electron microscopy (SEM) and atomic force microscopy (AFM), the NC morphology remained intact, confirming the treatment had negligible effect on the grain structure of the NCs. The corresponding film-based SCs showed an impressive efficiency of 14.10% and displayed 87% PCE after 7 days in N2 (control cells, 64%) and 70% PCE (control cells, 64%) after 54 hours of storage in air (RH, 40%). The enhanced stability is attributed to the structure stabilization of the NCs, while the high efficiency of 14.1%, is due to the filling of vacancies and traps in the NC lattice. The Cs-salt treatment doubled the carrier transport from 0.7 cm2 V−1 s−1 to 1.54 cm2 V−1 s−1, while the salt/MeOAc saturated solution facilitated the ligand exchange of the films with the assistance of the hydrolysis of MeOAc (Fig. 4a).49
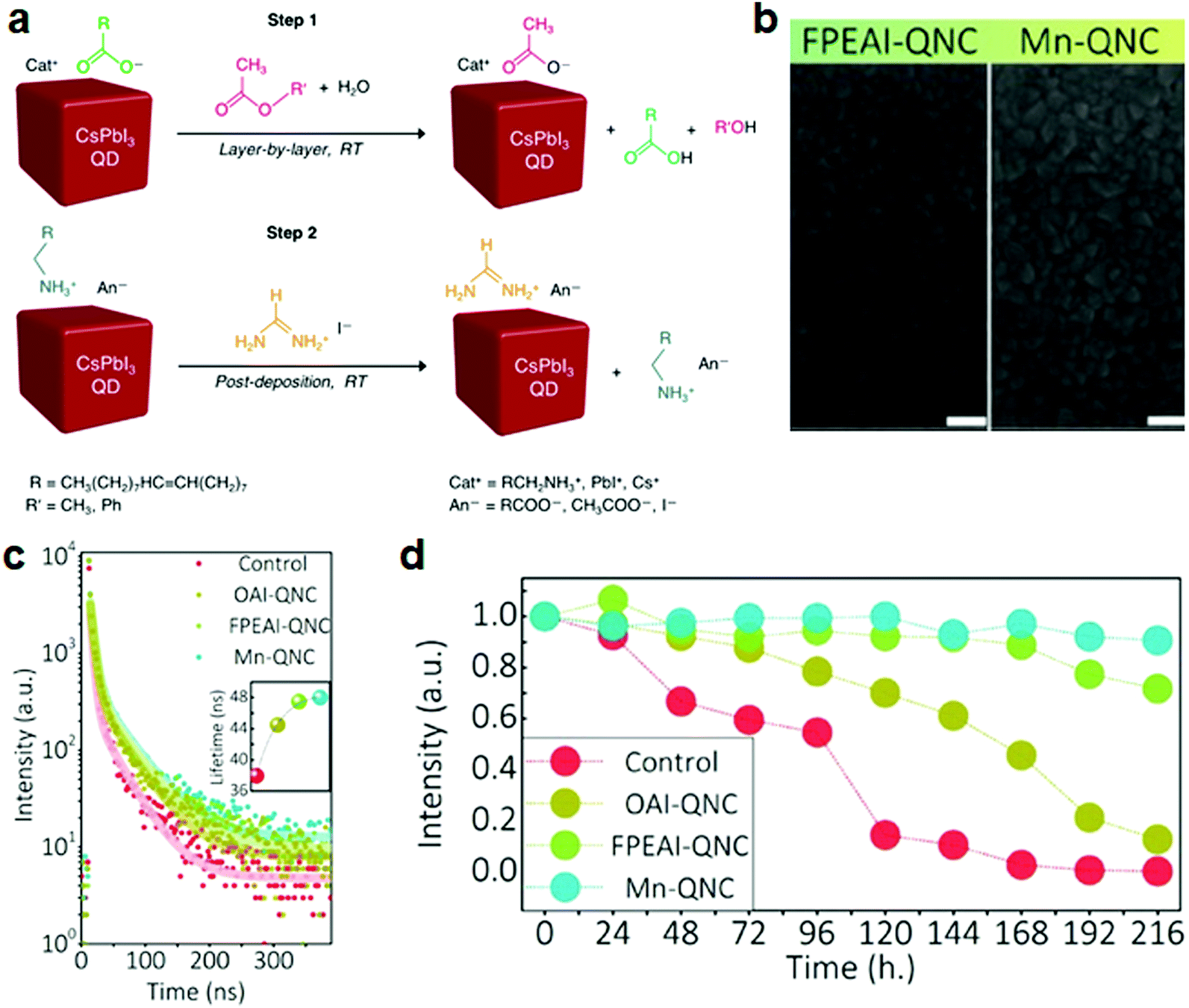 | ||
| Fig. 4 Stability improvement of CsPbI3 NC films by ligand exchange and doping. (a) Schematic of the ligand-exchange reaction of CsPbI3 NCs in films. (a) Reprinted with permission from ref. 49. Copyright 2018, American Chemical Society. (b) SEM images of FPEAI and FPEAI-Mn-based CsPbI3 NC films. (c) PL decay of pristine, OAI-, FPEAI-, and FPEAI-Mn-based NC films. Inset: Corresponding lifetime of the initial short-time decay. (d) PL stability at ambient condition for 10 days. (b–d) Reprinted with permission from ref. 51. Copyright 2019, Wiley-VCH. | ||
Treatments by multiple salts can further tune the coupling between the NCs and thus further enhance the electrical properties of the films. For example, an AX post-treatment, where A = formamidinium (FA+), methylammonium (MA+), or cesium (Cs+) and X = I−, was applied to improve the electronic coupling between the CsPbI3 NCs, which, was shown to enhance the carrier mobility.50 In this study, saturated Pb(NO3)2 was prepared to treat the NC films. The films were then fabricated by three to five cycles of NC deposition followed by treatments with Pb(NO3)2, and finally soaked in the AX salt in EtOAc. The resulting mobility of the films showed significant improvement from 0.23 to 0.50 cm2 V−1 s. In these steps, it is necessary to choose a solvent that dissolves the ligand-treating salt well, but does not affect the NCs. In this post-treatment, the process is always performed after film fabrication; therefore, the NCs should be resistant to the chosen solvents (MeOAc or EtOAc) and should not be redispersed. The salt treatment enables tuning of the interparticle distance and coupling interaction between the NCs, which enhances the conductivity and carrier mobility towards device requirements. However, a challenge of these post-treatments is that they can lead to a non-uniform salt distribution inside the films as a result of the limited salt penetration and diffusion into the layers. The most common way to address this challenge is the use of multiple cycles of thin-film deposition followed by a salt treatment. In this way, each deposited layer is treated more uniformly.
(iii) Multiple treatments. Since the ligand detachment from the NCs can cause instability of the CsPbI3 NC films, the ligand properties are directly related to the long-term performance stability of devices. To boost the stability and efficiency at the same time, multistep treatments are a possible solution. For instance, a CsPbI3 NC film was prepared by spin-coating the mixture of CsI and 4-fluorophenethylammonium iodide (FPEAI) in dimethylformamide (DMF) onto a substrate at room temperature, while doping with Mn2+ to replace the Pb2+ (Fig. 4b–d).51 H2PbI4 was added to increase the solubility of Mn2+ and thus its amount of doping. Due to the strong binding between the FPEA+ ligands (the conjugated aromatic rings terminated with strong electronegative fluoride in FPEAI molecules) and the perovskite lattice, a homogenous film without pinholes was obtained (Fig. 4b). The CsPbI3 NCs retained the α-phase and exhibited unchanged PL intensity at ambient conditions for 10 days (Fig. 4c and d).
The presented surface-ligand passivation is a facile approach to introduce different functions directly onto the surface of the NCs, as ligands control the inter-particle distance, coupling interaction and NC stability. Nevertheless, the challenge of the surface ligands (including both pre- and post-treatments) is that they are weakly coupled to the NCs, and can therefore easily detach from NCs and dissolve in solvents (i.e., toluene) leading to the aggregation or degradation of the NCs. This calls for additional passivation methods as discussed below.
2.2 Inorganic materials encapsulation or doping
Besides ligand treatments, passivation of inorganic NCs can be achieved using doping or encapsulation. These treatments include: doping of ions (Zn2+, Sn2+, Mn2+, Sr2+) into the NC structure by partial replacement of Pb2+, core–shell structures (SiO2, Al2O3, etc.), surface passivation using inorganic ions such as Cl−, or embedding the NCs into an inorganic matrix. Unlike the surface-ligand passivation, most of the passivation treatments using inorganic materials are accompanied by a chemical reaction. For example, the doping of ions into the Pb2+ sites is accompanied by the formation of bonds between the charged dopants and the I− host ions. This leads to treatments that are more stable in comparison to the weakly-bound ligands. Again, the processes can be divided into pre- and post-treatments, based on when the treatment is applied.Similarly, rare-earth ions are expected to be another potential dopant.53 By partial replacement of Pb2+ with Gd3+, the Gd-doped NCs exhibited prolonged phase stability of up to 11 days under ambient condition, in contrast to 5 days for undoped NCs, as evidenced by XRD. This may again be attributed to the increased tolerance factor of the perovskite structure, and the decreased defect density in NCs. The latter has been confirmed by the decrease of the Urbach energy, which indicates electronic or structural disorder, from 14.1 to 11.4 meV upon doping.
Both experimental and theoretical works have reported the incorporation of alkali metal ions to improve the phase stability and device performance of perovskites.54–57 After incorporating Rb/K dopants, an effective improvement of moisture-resistance of the films was observed, attributed to the formation of a water-repelling surface. This was also confirmed by theoretical calculation with a mitigated charge shift, alleviated lattice distortion and a higher activation barrier in Cs0.92Rb0.08PbI3/Cs0.92Rb0.08PbI3.54 Nam et al. have also reported that in Cs0.925K0.075PbI2Br SCs, the PCE maintained 80% of the initial value for 120 h after stored at ambient condition (RH 20%; 20 °C), while a CsPbI2Br device gradually decreased and reached half of its initial value in 72 h.57
Besides doping, core–shell structures have been used for stability enhancement. In these, perovskite NCs act as the core, and another type of inorganic material serves as the shell protecting the NCs. This protective shell is very different from surface ligands, which cannot easily form a dense layer surrounding the NCs. Materials that have been used as shells include Cs4PbI6, SiO2, and PbS. Among those, the latter is particularly compatible with the CsPbI3 NC lattice due to their minimal lattice mismatch (<5%). Hence, the matching lattices of the PbS shell and the CsPbI3 core58–60 show great possibility for high-quality CsPbI3/PbS NCs. The corresponding films were stable for 33 days under ambient condition. Alternatively, iodine-modified graphene oxide61 offers a chance for strong binding as well tensile stress between the graphene oxide and the NCs. Under ambient conditions (dark, ∼4 °C and ∼1% RH), the modified films retained PL stability for up to 4 weeks. Xi et al. employed (3-aminopropyl)triethoxysilane (APTES) as a SiO2 precursor and mixed it with the PbI2 and Cs-oleate. The solution was left in air for 3 hours to form a silica matrix by silanization. Upon the formation of a silica layer, the NCs showed a stability of at least 5 days compared to the OA/OLA-NCs, which lost PL in 1 day. On longer time scales, the PLQY of the silica-coated NCs only dropped from 70% to 60% during three months.62 The luminance efficiency has reached 61.2 lm W−1 for the above white-light emitting diode (WLED) with a half-life of 227 h.
The insulating perovskite phase, Cs4PbI6, can also be used as a shell,63 but more notably, it serves as a matrix to embed the NCs.64 Grandhi et al. developed a hybrid CsPbI3/Cs4PbI6 NC structure, in which they embedded CsPbI3 NCs into the hexagonal Cs4PbI6 matrix. The hybrid NC films could be stored at ambient conditions for a few weeks without losing too much red luminescence (Fig. 5a); after this time, the red luminescence intensity was similar to that of an untreated CsPbI3 film stored for 5 days. Moreover, thermal stability tests showed that the hybrid NC films retained 60% of their original PL efficiency after 6 hours of annealing at 100 °C. Even after seven heating–cooling cycles, still more than 60% of the original PL intensity was retained. Furthermore, besides thermal stability, the stability in aqueous solvents is also improved: upon immersion in water, the emission disappeared only after 4 days, while for an untreated NC film, the emission ceased after 1–2 hours. As a possible stabilization mechanism, it is suggested that the lowering of the Gibbs free energy due to the incorporation of the NCs in the matrix prevents the phase transformation from the emissive γ (black) to the non-emissive δ (yellow) phase (Fig. 5b).
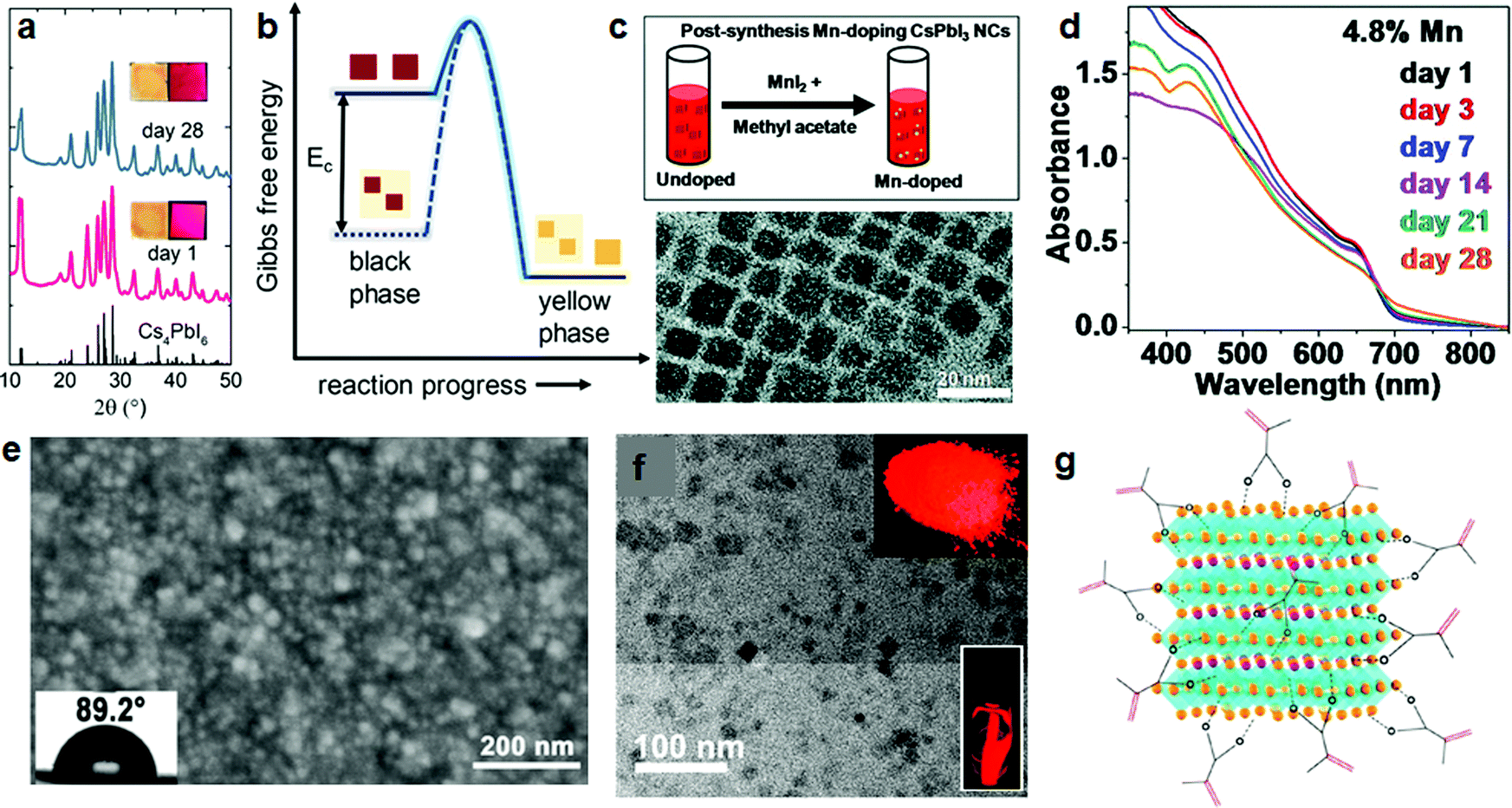 | ||
| Fig. 5 Stability improvements of CsPbI3 NCs by treatment with inorganic materials and polymers. (a) XRD patterns of CsPbI3–Cs4PbI6 hybrid NCs, fresh and after 28 days. Inset: Photographs of the NC films under room light (left) and 365 nm light (right). (b) Schematic of the Gibbs free energy barrier for converting black to yellow phases of CsPbI3, with and without encapsulation. (a and b) Reprinted with permission from ref. 64. Copyright 2020, American Chemical Society. (c) Post-treatment by MnI2 to obtain Mn-doped CsPbI3 NCs (top), and transmission electron microscope (TEM) image of the Mn-doped CsPbI3 NCs (bottom). (d) Absorption spectra of the corresponding Mn-doped CsPbI3 NC films at different days. (c and d) Reprinted with permission from ref. 66. Copyright 2019, The Royal Society of Chemistry. (e) Scanning electron microscope (SEM) images of CsPbI3 and μGR/CsPbI3 films. Insets: Photograph of water drops deposited on the films, indicating the contact angles. (e) Reprinted with permission from ref. 69. Copyright 2018, Wiley-VCH. (f) TEM image of PMPOPNC-CsPbI3 NCs, inset: photographs of PMPOPNC-CsPbI3 NCs film and powders under UV light. (g) Schematic of the cubic CsPbI3 NCs coated by polymerizable ligands. (f and g) Reprinted with permission from ref. 78. Copyright 2018, American Chemical Society. | ||
Enhanced stability of CsPbI3 can also be achieved by pre-treating with silica. Silica pre-treatment of CsPbI3 was obtained by so called capped silica (CA-SiO2) nanoparticles in a two-step method. First, CA-SiO2 nanoparticles of around 115 nm were prepared from the mixture of 3-aminopropyltriethoxysilane (APTES) and trimethoxy-(octadecyl)silane (TMODS) precursors. Then the CA-SiO2 particles were mixed with the perovskite precursors, after which nucleation took place on the surface of the CA-SiO2 NPs. The CsPbI3 NCs grew in situ on the surface of the CA-SiO2 particles. Uniform and strong red emission was observed under UV light for films made of the CsPbI3 NCs on CA-SiO2 particles. A WLED made from CsPbX3 (X = Cl, Br, I) NCs on CA-SiO2 was stable after operating 20 min at 20 mA with the intensity of electroluminescence (EL) almost unchanged.65
The stability offered by SiO2 encapsulation was also explored as a post-treatment. Tetraethyl orthosilicate was mixed with CsPbI3 NCs to initiate the encapsulation of SiO2 to obtain CsPbI3/SiO2 composites with a diameter of 120 nm. After coating, the water-solubility and water stability were improved, after which the nanocomposites could disperse into water homogeneously. Strong emission was still observed after immersing into water for 4 h. CsPbI3 composite films showed no red emission after storage in air for 36 h, while CsPbI3/SiO2 composite films remained stable after two months.68
Alternatively, a mixture with an appropriate ratio between CsPbCl3 and CsPbI3 NCs was used to form 100–200 nm thick films of CsPbI3−xClx NCs.70 The films were subsequently soaked in a NH4SCN/isopropanol solution to remove surface ligands. After exposing to ambient conditions (RH, 11% at 23 °C), the films were stable for 96 hours without phase changes, in contrast to 18 hours for untreated films, as determined by XRD and absorption spectra.
Another multiple treatment employed Sr(OAc)2 and trimethylsilyl iodide (TMSI) or SrI2 with excess PbI2 which were directly added into the mixture of the precursors.71 Different from the former two methods, a salt treatment was performed after the synthesis of the NCs but prior to the film fabrication. By substituting Pb2+ with Sr2+, the formation energy of α-CsPbI3 QDs increased, resulting in reduced structural distortion and a stable cubic perovskite structure. With the synergistic effect of Sr2+ substitution and I− passivation, photoirradiation-induced degradation was largely delayed to up to 20 days for the NC film.
While the above treatments provide some improved properties, several issues need to be addressed: doping ions into the sites of Pb2+ requires careful optimization of the doping concentration to avoid lattice collapse and resulting deterioration of the optical properties. Concerning the core–shell structures, the poor flexibility of the shell materials can limit their potential application. To this end, the shell thickness is a key factor determining the structural stability and optical properties, where thick shells can largely passivate the NCs but also block the absorption and emission of the core materials.
2.3 Polymer matrix or encapsulation
Considering the poor flexibility of inorganic materials, polymers have been used as an alternative material for encapsulation of the perovskite NCs. Polymer materials are usually transparent and have little effect on the light absorption of the CsPbI3 NC film. Furthermore, polymers are flexible allowing flexible devices. Importantly, after coating with appropriate polymers, the films show increased protection against moisture. Meanwhile, the coating also provides a good way to avoid the leakage of lead into the environment.To illustrate such a copolymer multiple treatment, methacrylic acid (MA) was directly added into the perovskite precursors to form MA-CsPbI3 NCs. These were then introduced into a mixture of methyl methacrylate (MMA) or a mixture of MMA and methacrylisobutyl POSS (MA-POSS) as monomers and 2,2-azobis(isobutyronitrile) (AIBN) as the initiator for UV irradiation, as displayed in Fig. 5f and g.78 The final composite films, PMMA-co-P(MA-POSS)-co-P(MA-NC), were stable upon water immersion for 60 days, with the PL intensity retained 80% even at 120 °C. Furthermore, under UV light irradiation for 156 hours, 81% of the PL intensity remained. MA ligands can effectively passivate the perovskite NCs by coordinating surface atoms with the ligand's terminal carboxylate group, while the polymer matrix provides a permeation barrier for water and UV light. A WLEDs device exhibited a luminous efficacy of 26.3 lm W−1 at an operating current of 20 mA. The color of the device was stable at currents between 5 and 50 mA, indicating that the device is stable at high current densities.
A crucial point of the polymer treatments is to find an appropriate polymer that can be well dissolved in the non-polar solvents, in which the perovskite NCs are dispersed. Another important consideration is that due to the limited solubility of the polymers in the solvents (toluene or hexane), a proper ratio between the polymers and NCs should be carefully investigated to avoid cluster formation or jamming of the samples.
2.4 Others
In addition to the above-mentioned treatments by doping, encapsulation or ligand exchange, other specific treatments have been suggested to improve the stability of CsPbI3 NCs, including metal organic frameworks (MOFs)79 and X-ray irradiation techniques.80 MOFs are a class of porous compounds consisting of metal ions or clusters coordinated to organic ligands to form one-, two-, or three-dimensional structures. The pores, which are hydrophobic, are constructed from aromatic ligands and could be used to confine the perovskite NCs. In addition, the large size of the pores and their good thermostability further offers improved stability of the perovskite-MOF structure as well as functional properties such as catalysis and gas storage.81–83Concerning the former, ZIF-8 (zeolitic imidazolate frameworks) powder, a subset of MOF, was mixed with CsPbI3 NCs, and injected into a PMMA-in-toluene solution, which was subsequently spin-coated to produce a film.79 The films were studied for their long-term storage and thermal stability. They retained ∼43% PL intensity after 30 days at ambient condition and ∼35% at 90 °C. The fabricated WLEDs exhibited a color temperature of 8461 K, and luminescence efficiency of 12.85 lm W−1.
X-ray irradiation treatments, on the other hand, can form intermolecular C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding of the organic ligands coating the surface of the NCs.80 This was shown by X-ray irradiation in vacuum using the Al Kα monochromatic source of an XPS system. The irradiated film showed improved stability, as confirmed by its stable color under UV light. Furthermore, it showed stability of several weeks in air, 1 day in ultrapure Milli-Q water, and 3 days in a biological buffer (PBS 1×, phosphate buffer solution), while samples without irradiation lost PL emission after 5 days in air. The enhanced stability was associated with the transformation of the ligand shell into intermolecular CC bonding due to the X-ray irradiation.
C bonding of the organic ligands coating the surface of the NCs.80 This was shown by X-ray irradiation in vacuum using the Al Kα monochromatic source of an XPS system. The irradiated film showed improved stability, as confirmed by its stable color under UV light. Furthermore, it showed stability of several weeks in air, 1 day in ultrapure Milli-Q water, and 3 days in a biological buffer (PBS 1×, phosphate buffer solution), while samples without irradiation lost PL emission after 5 days in air. The enhanced stability was associated with the transformation of the ligand shell into intermolecular CC bonding due to the X-ray irradiation.
3. Optical mechanisms towards higher efficiency of CsPbI3 NC films
In this part, we review photophysical effects that may offer further efficiency increase and improved functionality of perovskite NC films and devices. These include carrier multiplication, optical gain and photon recycling (PR). They are well known from other semiconductor NC systems, and have recently been studied in some depth in perovskite NCs. Due to the yet limited number of studies on CsPbI3 NC films, we include essential studies of the optical properties and phenomena of other perovskite NCs as well, which are also applicable to the iodide-based perovskites.3.1 Efficient carrier multiplication
The absorption of a photon usually leads to the creation of a single electron–hole pair, or exciton. However, it is in principle possible to generate two or more excitons by absorption of a single photon. When the electron is excited to the conduction band with some excess energy, by a photon with energy greater than the bandgap energy, it is promoted to a higher state producing a hot carrier. Normally, the excess energy is quickly lost as the carrier cools down to the band-edge within a few picoseconds; however, if the excess energy is sufficient to excite a second exciton, then the formation of two excitons may result from the absorption of a single photon. This process is known as multiple exciton generation (MEG). Such photoexcitation of multiple e–h pairs could significantly improve the conversion efficiency of converters and PV devices, as it allows for PCE beyond the Shockley–Queisser (SQ) limit.84 The minimum energy required to excite two excitons, i.e. the sum of the two exciton energies and additional energy, which is lost due to inefficiencies, is termed the MEG threshold energy. The amount of energy lost depends on the material, but in either case the MEG threshold energy must be at least twice the bandgap energy of the material (hν > 2Eg).A process similar to MEG has been observed in many bulk semiconductors where it is known as impact ionization.85–87 Nevertheless, its importance in bulk materials is limited due to the high threshold energy required for the process in many materials.88 In NCs, the MEG process becomes more prominent as MEG threshold energies are lower.89 Among the lead-halide perovskite NCs, the iodide-based ones have the lowest bandgap energy and are therefore the most suitable to observe and ultimately utilize the MEG effect. With a bandgap energy of around 1.7 eV, MEG could already be observable at the UV part of the solar spectrum.
MEG was observed in a number of NC perovskite systems, most notably in hybrid perovskites. Li et al.90 observed MEG in colloidal FAPbI3 NCs using pump–probe spectroscopy, in which they observed the signal of reverse-Auger recombination (Fig. 6a) for pump energies above the threshold energy, and no signature for energies below that threshold. Here, the hot carrier should be generated by a single photon, meaning that the average number of absorbed photons per NC, 〈N〉, should be kept well below 1. In this study, the MEG threshold energy was determined to be relatively low, at 2.25Eg, as shown in Fig. 6b. The threshold energy also relates to a quantity called the MEG efficiency, which is a measure of the number of additional excitons generated per additional energy absorbed above the MEG threshold energy in units of the bandgap energy.62 The MEG threshold energy and MEG efficiency are thus directly related, and the low threshold energies observed for perovskite NCs translate into high MEG efficiencies, well above the MEG efficiency of other NC systems such as PbSe and PbS.
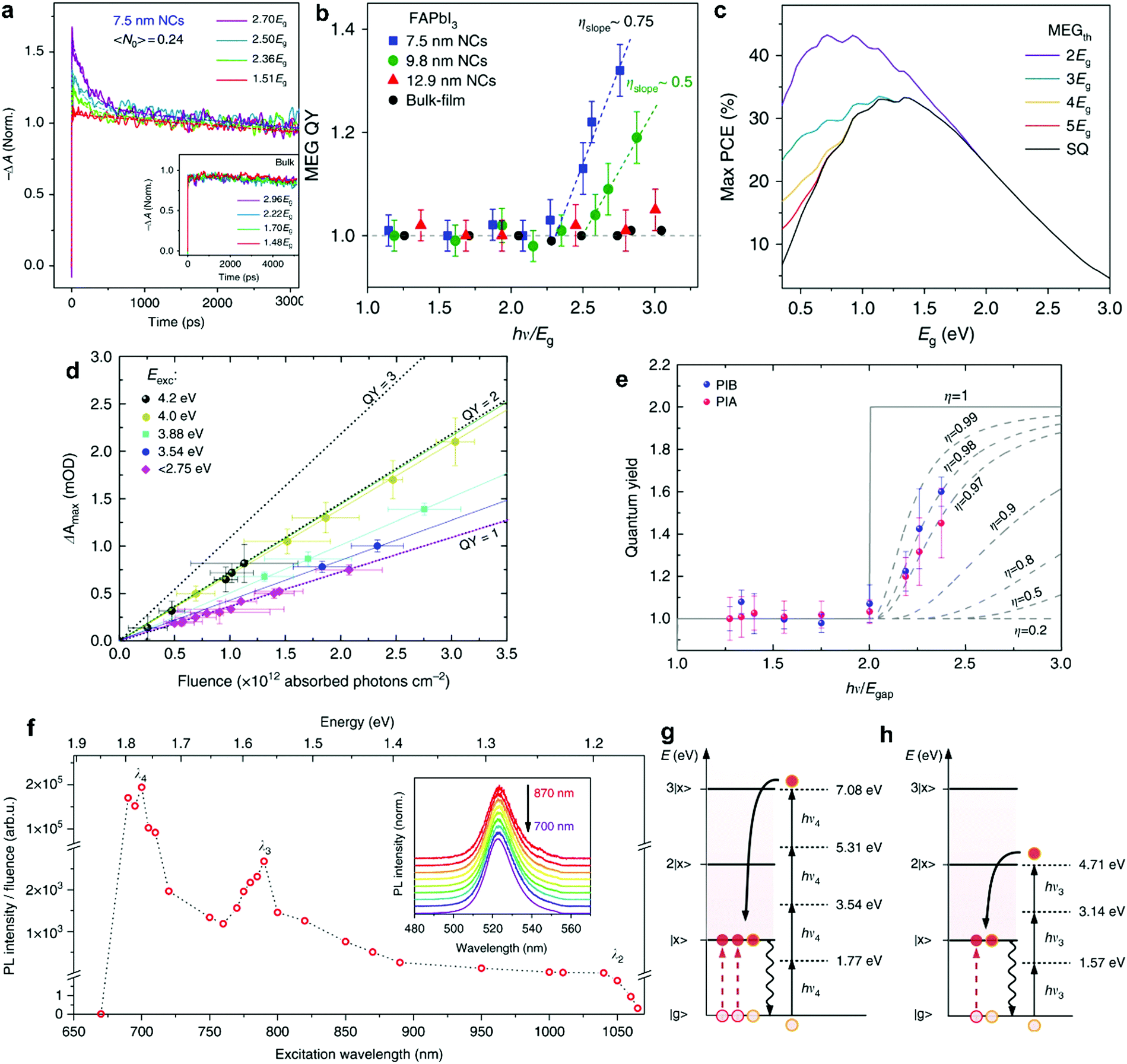 | ||
| Fig. 6 MEG in perovskite NCs. (a) TA signal of 7.5 nm NCs, indicating extra charge carriers generated when pumping with photon energies above the MEG threshold. (b) the QY of MEG as a function of excitation energy hν/Eg, indicates a MEG threshold of 2.25Eg for these NCs, and MEG efficiency of 75%. (c) MEG enables PCE beyond the SQ limit, here the maximum theoretical PCE is calculated as function of bandgap energy, without MEG (SQ line) and with MEG at different threshold energies. (a–c) Reprinted with permission from ref. 90. Copyright 2018, Springer Nature. (d) MEG related TA signal as a function of pump fluence, showing linear behavior with fluence, and change in slope due to MEG for photon energies above threshold. (e) the QY of MEG as a function of excitation energy, hν, indicates a threshold close to 2 × Eg for these NCs, and MEG efficiency of approximately 97%. (d and e) Reprinted with permission from ref. 91. Copyright 2018, Springer Nature. (f) Photoluminescence excitation (PLE) spectrum, PL intensity as a function of excitation wavelength, showing peaks due to multiple photon absorption resonances. The peaks λ4 and λ3 corresponding with the schemes shown in (g) and (h), corresponding to 4 and 3 photons absorbed to combine into 3 and 2 excitons excited, respectively. (f–h) Reprinted with permission from ref. 92. Copyright 2018, Springer Nature. | ||
The MEG effect can significantly enhance the theoretical PCE provided sufficiently low threshold energy. This is shown in Fig. 6c, in which the maximum theoretical efficiency is calculated for different bandgap energies.91 The different lines represent different MEG thresholds energies. For example, a threshold energy of 2Eg can increase the theoretical PCE from around 30% (SQ limit, black line) to 44% (violet curve) for sufficiently low band gap energies (Eg ≤ 1 eV). Very high MEG efficiency and low threshold energy were determined in CsPbI3 NCs by de Weerd et al.91 The transient absorption (TA) signal as a function of pump fluence revealed a QY between 1 and 2, approaching 2 for sufficiently high photon energy, as shown in Fig. 6d. A plot of the extracted QY versus photon energy (Fig. 6e) shows that MEG occurs with not only a remarkably low threshold energy close to 2Eg, but also with high MEG efficiency of approximately 90%. Yet, the high bandgap energy of 1.7 eV prevents significant PCE enhancement in CsPbI3 (Fig. 6c). Clearly, CsPbI3 NCs have the best properties compared to the other lead-halide perovskites, with uniquely efficient MEG. Nevertheless, they are not yet ideal for PV devices. To take full advantage of the MEG effect and to increase the PCE in devices, the perovskite bandgap energy needs to be decreased. Perhaps modified CsPbI3 NCs with lower bandgap can fully exploit the superior MEG properties.
In another study employing films of perovskite NCs, the formation of several excitons from multiple sub-bandgap photons was reported. Manzi et al. studied CsPbBr3 close-packed NC films using sub-band gap photon excitation (hν < Eg), and showed that with multiple photons below the bandgap they could observe exciton emission (Fig. 6f).92 The resonant absorption peaks corresponded to multiple photon absorption. Energy conservation implies that the number p of absorbed photons with energy hν is related to the number N of excitons with energy Ex according to p × hν = N × Ex, as shown in Fig. 6g and h. This remarkable finding demonstrates that both multiple photon absorption as well as MEG can take place simultaneously. Moreover, in contrast to the previous studies, the generated excitons recombined radiatively, indicating that the generated excitons separated into adjacent NCs. Notably, this resonant MEG was only observed in dense films and not in colloidal dispersions like in the former two studies. The question arises whether also in iodide-based perovskite NC films, the excitons produced by MEG can be split into excitons in adjacent NCs, as has also been observed in other NC films such as Si NCs.93 Depending on the application at hand, such behavior would be highly beneficial for applications.
3.2 Optical gain, exciton effect
One of the most interesting optoelectronic applications in which lead-halide perovskite NCs could play a role is lasers. Already in 2015, amplified spontaneous emission (ASE) has been shown to take place in lead-halide perovskites.94 Lead-halide perovskite NCs can provide a platform on which full tunability of wavelengths can be achieved across the visible spectrum, while providing ASE capabilities. More recent works have studied the threshold values for ASE under different conditions to uncover the underlying mechanism.Of central importance to ASE is the role of the exciton and biexciton states. Yakunin et al. and later works identified ASE in PL spectra as a narrow peak which emerges upon sufficiently high excitation fluence.94 Interestingly, the ASE peak is red-shifted compared to the PL peak, which leads to two possible interpretations: (a) either the ASE originates from the biexcitonic state, which is lower in energy than the excitonic state, or (b) the ASE undergoes reabsorption in the Urbach tail, and therefore appears at lower energy.
Yumoto et al. studied the behavior of hot and cold (ground) excitons by optical pumping in NCs95 according to the schematic in Fig. 7a. Their results indicate that the biexciton binding energy, Δxx, increases initially with the exciton excess energy, as shown in Fig. 7b. The deeper-bound biexciton state causes enhanced induced absorption of the biexciton suppressing the optical gain for a short period after the excitation (up to ∼3 ps). These results indicate that efficiently exciting excitons with a small Δxx would be beneficial for optical gain, and for applications such as ultrafast switching devices and femtosecond pulse generation. A more recent study by Kobiyama et al. has expanded on these results. Using similar pump–probe experiments and additional pump–pump–probe experiments, they could separate several excitonic configurations,96 including excitation of a single hot exciton (h), a hot–hot bi-exciton (hh), and a hot–ground biexciton (hg), as shown in Fig. 7a. They find that for the asymmetric (hg) excitation, Δxx is the smallest. This smaller shift is also evident in the enhanced optical gain as shown in Fig. 7c and d. These results confirm the importance of minimizing the biexciton binding energy to enhance ASE.
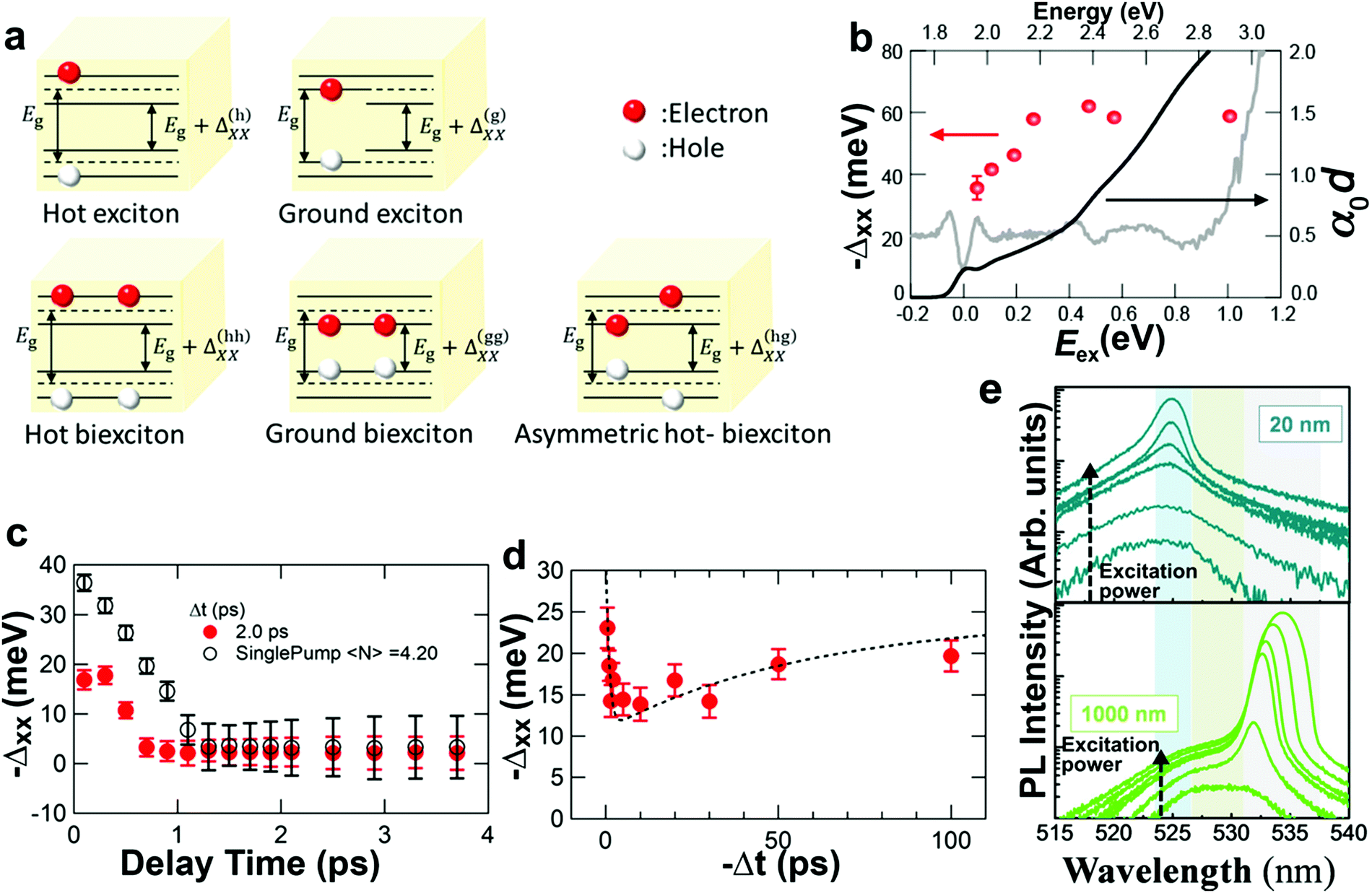 | ||
| Fig. 7 Optical gain, exciton effect. (a) Schematic of different exciton types and associated biexciton binding energy, Δxx. (b) Δxx for different excess energies of the excitation (red). Rapid initial increase of Δxx with excess energy is observed. (b) Reprinted with permission from ref. 95. Copyright 2018, American Chemical Society. (c and d) Biexciton binding energy as a function of delay time for different configurations in pump–pump–probe experiments. The binding energy and biexciton-related ASE can be influenced. (a, c and d) Reprinted with permission from ref. 96. Copyright 2020, American Chemical Society. (e) PL spectra of CsPbBr3 NCs for different excitation powers (increase indicate by black arrow), showing the onset of sharp peaks attributed to ASE at higher excitation powers. The panels show sample thickness of 20 and 1000 nm, showing a stronger redshift of the ASE peak for the thicker layer, this effect is attributed to reabsorption in thicker layers (e) reprinted with permission from ref. 97. Copyright 2019, American Chemical Society. | ||
While the double degeneracy of the ground state and the redshift of the ASE peak suggest that ASE is dominated by the biexciton-to-exciton transition, other interpretations have suggested the involvement of single excitons only,97 as concluded from PL spectroscopy measurements. For the excitation fluency range 〈N〉, between 1 and 2, they observe a decrease in the ASE QY, which cannot be understood if it is caused by biexciton-to-exciton transitions; rather, the decrease in QY is probably the result of Auger recombination. The band edge shift is then ascribed to reabsorption, as validated by different thicknesses of IP-NC films (20 nm up to 1000 nm). The thicker the film the more shift was observed in the ASE peak, indicating reabsorption of the ASE emission (Fig. 7e), which could be wrongly interpreted as a lower transition energy. These results firmly support the theory of single exciton ASE instead of biexciton-to-exciton mediated ASE.
This apparent discrepancy can perhaps be understood, if we consider the different nature of the experiments. The former probes the TA dynamics at room temperature, while the latter explores the PL properties at cryogenic temperatures, possibly giving rise to different NC properties. More research is needed to resolve this discrepancy.
3.3 Photon recycling
PR is the repeated photon emission and absorption inside a material. It requires overlap between the emission and absorption bands, and relies on high PL QY as well as strong absorption to sustain the recurring re-absorption and emission cycles. In bulk crystals, fluctuations in material composition might induce enough variation in the bands to broaden the PL peak and increase the overlap. A similar effect is expected to occur in dense NC ensembles, due to polydispersity of the NC size, composition, and defects in the NCs, which all create variations in the electronic bands, thus enhancing the overlap in absorption and emission.For perovskites, the high PLQY, strong absorption, and small Stokes shift make them good candidates to exhibit PR. Indeed, reabsorption and re-emission were observed in bulk CsPbI3 films98 by direct measurement of the emission at different positions of the sample, as shown in Fig. 8a. The increasing separation between excitation and collection positions results in increasing contribution of PR to the signal. Calculations that predict the expected emission as a function of distance, taking into account only the reabsorption of the emitted photons following the Beer–Lambert law cannot reproduce the experimental emission, as shown in Fig. 8b at 765 nm and 800 nm as well as in Fig. 8c and d. If one takes into account only changes to the spectrum caused by Beer–Lambert, then the spectrum is affected only by the absorption spectrum, which will affect shorter wavelengths more than longer wavelengths. Indeed, the difference between experiment and Beer–Lambert prediction is especially pronounced for the shorter wavelength, which is strongly absorbed by the material. PR, however, leads to re-emission at this wavelength, strongly enhancing the actual measured intensity. This discrepancy between experiment and the Beer–Lambert model illustrates that the model lacks the full description of the system, and suggests re-emission. This study further demonstrates that by repeated photon absorption and emission, the PR effect can lead to increased effective carrier diffusion lengths. Evidently, high PR rates due to the effectively increased carrier diffusion have long been identified as beneficial for solar devices.99
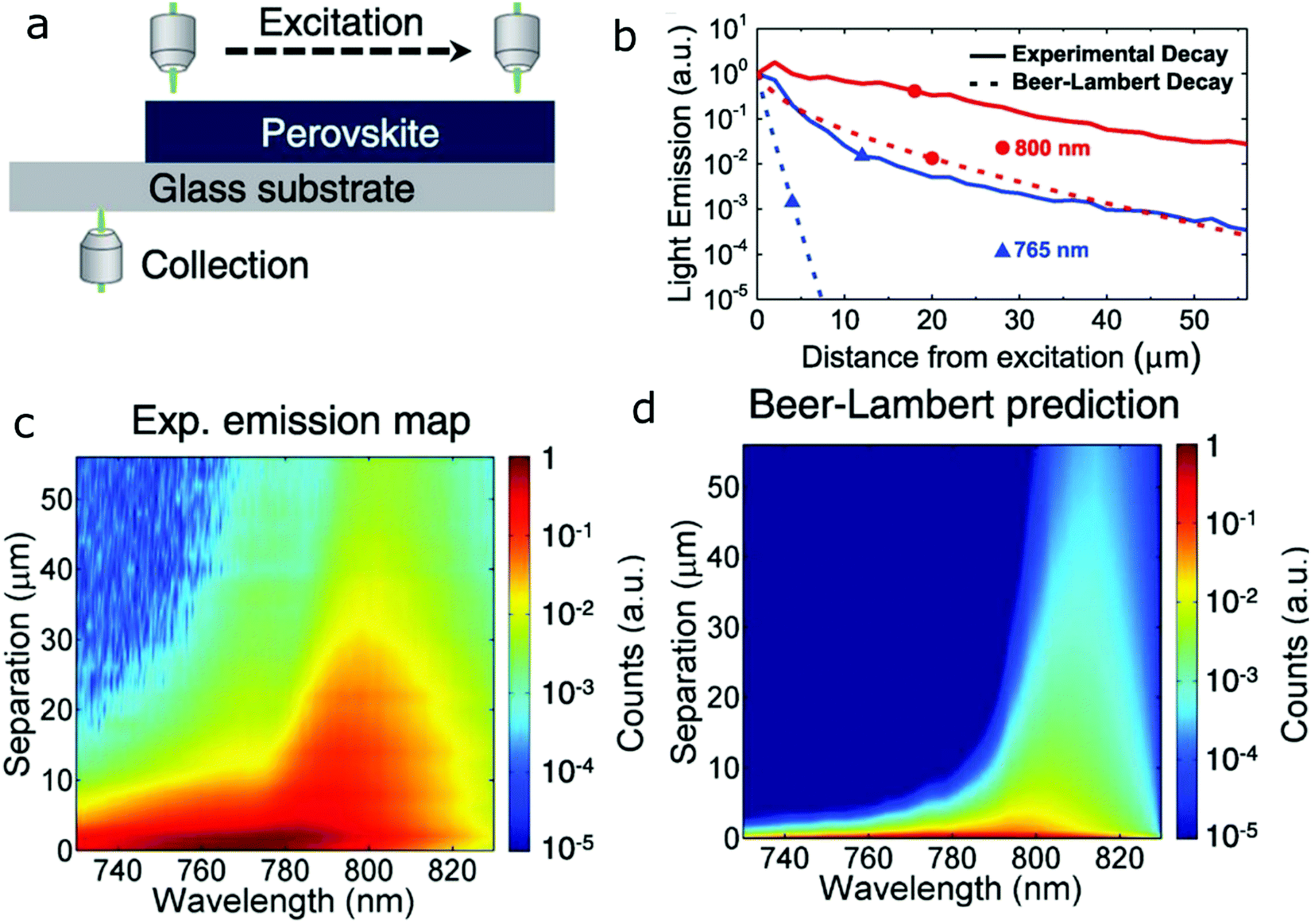 | ||
| Fig. 8 PR. (a) Schematic of the experiment showing the varying excitation and collection positions. (b) Measured and calculated light emission at 765 and 800 nm, as a function of distance. (c) Experimentally observed PL spectra as a function of separation distance. (d) Calculated emission spectra as a function of separation distance in which the simple Beer–Lambert law is used to predict the change of emission with separation distance, without taking into account PR. Reprinted with permission from ref. 98. Copyright 2016, AAAS. | ||
Besides bulk perovskite layers, PR should also occur in colloidal perovskite NCs and NC films. Here, it should be possible to modify the extent of PR by controlling the concentration of the NCs. Additionally, NCs have higher absorption cross-section than their bulk counterpart, and possibly more controllable bandgap variability due to their size and composition, affecting directly their emission linewidth and Stokes shift. Clearly, the benefits of using NCs to study PR are numerous. By employing a mixture of CsPbCl3 and CsPbI3 NCs, Davis et al. found that the emission of the Cl-based NCs could be strongly absorbed and re-emitted by the iodide based particles.100 Yet, this effect can be considered as a mere energy transfer between two distinct populations, lacking the recurring (cascade) nature characteristic of PR.
Thus, while studies so far have demonstrated the occurrence of PR in bulk perovskite crystal films, the proof of PR in perovskite NCs, both in dispersions and deposited NC films is still lacking. Especially for CsPbI3 NCs, PR could provide a means to increase the conversion efficiency of NC films in PV devices. The use of different treatments as discussed in Section 2 can also offer additional possibilities to modify the PR and diffusion rates. In this way, PR in perovskite NC films can be studied and exploited in detail and with a high degree of tunability not offered in bulk films.
4. Summary and future prospect
This review has discussed recent developments in stability and efficiency enhancement of CsPbI3 perovskite NCs. Compared to bulk perovskite films, the NC film-based devices are still in an early stage of development, but have recently seen a rapid development. Out of these, the iodide-based perovskite NC films provide the best (lowest) band gap energy, but also face the challenge of the highest instability.A variety of strategies has been suggested to improve their stability, and the carrier mobility of the NC films, including ligand exchange, encapsulation, and salt treatments. While a single treatment improving on all aspects has yet to be developed, the stability has been dramatically improved, most notably from a few weeks for untreated films at room temperature up to 4 months for films treated by phenylethylammonium additives.43 In more demanding conditions, encapsulation of the NC film in a polymer has proven to work well, and up to 60 days of storage in water was reached by co-polymer (PMMA-co-P(MA-POSS)) encapsulation.78 Salt treatments mainly contribute to improve the film conductivity, but certain salt post-treatment, i.e. by NaOAc, can improve both the conductivity and stability by passivating surface traps of the films.
Furthermore, a number of photophysical effects can boost the conversion efficiency of perovskite NC films, most notably MEG and PR. While MEG has been confirmed in several lead-halide NC systems including CsPbI3, PR has so far been only confirmed for perovskite bulk films. However, to make a device that benefits from MEG, a dense film would require that the multiple excitons are stable long enough to be extracted from the device. While this may in principle be possible by separation of the excitons onto adjacent NCs, it has yet to be demonstrated in practice. Similarly, while PR has been identified to contribute to and improve efficiencies of NC devices, the exact behavior of PR in NC films is yet to be identified. In this regard, red perovskite NC-based films, which have been relatively poorly explored, offer great opportunities to discover novel phenomena as well as significant efficiency increase in devices. However, several critical issues are still great challenges in the future. The key issue for the instability of CsPbI3 NCs is the phase transition from black to yellow phase at room temperature. For NCs produced by the hot-injection method, a maximum of 4 months was reported to preserve the black CsPbI3 NCs at room temperature. NCs obtained by room-temperature synthesis suffer from even lower stability, which poses a central challenge. Hence, more studies on the passivation, encapsulation or doping are essential for improving their stability. In addition to stability, there is great potential for efficiency enhancement by optical phenomena, which however needs more exploration. For example, investigation of PR on CsPbI3 colloidal NCs and NC films have not been reported yet, but offer good opportunities to further boost PV efficiency.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful for the Dutch Technology Foundation STW, The Netherlands Organization for Scientific Research (NWO), and the Joint Solar Program (JSP III, 680-91-011) of The NWO for financial support. We acknowledge Marco van der Laan for his kind help in the explanation of photon recycling.References
- Q. S. Chen, J. Wu, X. Y. Ou, B. L. Huang, J. Almutlaq, A. A. Zhumekenov, X. W. Guan, S. Y. Han, L. L. Liang, Z. G. Yi, J. Li, X. J. Xie, Y. Wang, Y. Li, D. Y. Fan, D. B. L. Teh, A. H. All, O. F. Mohammed, O. M. Bakr, T. Wu, M. Bettinelli, H. H. Yang, W. Huang and X. G. Liu, Nature, 2018, 561, 88–93 CrossRef CAS.
- M. S. Kirschner, B. T. Diroll, P. J. Guo, S. M. Harvey, W. Helweh, N. C. Flanders, A. Brumberg, N. E. Watkins, A. A. Leonard, A. M. Evans, M. R. Wasielewski, W. R. Dichtel, X. Y. Zhang, L. X. Chen and R. D. Schaller, Nat. Commun., 2019, 10, 504 CrossRef CAS.
- Q. G. Zhang, B. Wang, W. L. Zheng, L. Kong, Q. Wan, C. Y. Zhang, Z. C. Li, X. Y. Cao, M. M. Liu and L. Li, Nat. Commun., 2020, 11, 31 CrossRef CAS.
- M. R. Yang, P. Moroz, E. Miller, D. Porotnikov, J. Cassidy, C. Ellison, X. Medvedeva, A. Klinkova and M. Zamkov, ACS Photonics, 2020, 7, 154–164 CrossRef CAS.
- S. G. Motti, F. Krieg, A. J. Ramadan, J. B. Patel, H. J. Snaith, M. V. Kovalenko, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2020, 30, 1909904 CrossRef CAS.
- J. C. Beimborn, L. R. Walther, K. D. Wilson and J. M. Weber, J. Phys. Chem. Lett., 2020, 11, 1975–1980 CrossRef CAS.
- Q. Zhao, A. Hazarika, X. Chen, S. P. Harvey, B. W. Larson, G. R. Teeter, J. Liu, T. Song, C. Xiao, L. Shaw, M. Zhang, G. Li, M. C. Beard and J. M. Luther, Nat. Commun., 2019, 10, 2842 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS.
- G. R. Li, F. W. R. Rivarola, N. J. L. K. Davis, S. Bai, T. C. Jellicoe, F. de la Peña, S. C. Hou, C. Ducati, F. Gao, R. H. Friend, N. C. Greenham and Z.-K. Tan, Adv. Mater., 2016, 28, 3528–3534 CrossRef CAS.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS.
- Y.-H. Kim, H. Cho, J. H. Heo, T.-S. Kim, N. Myoung, C.-L. Lee, S. H. Im and T.-W. Lee, Adv. Mater., 2015, 27, 1248–1254 CrossRef CAS.
- X. Y. Shen, Y. Zhang, S. V. Kershaw, T. S. Li, C. C. Wang, X. Y. Zhang, W. Y. Wang, D. G. Li, Y. H. Wang, M. Lu, L. J. Zhang, C. Sun, D. Zhao, G. S. Qin, X. Bai, W. W. Yu and A. L. Rogach, Nano Lett., 2019, 19, 1552–1559 CrossRef CAS.
- J. Kim, B. Koo, W. H. Kim, J. Choi, C. Choi, S. J. Lim, J.-S. Lee, D.-H. Kim, M. J. Ko and Y. Kim, Nano Energy, 2019, 66, 104130 CrossRef CAS.
- J. L. Shi, Y. Wang and Y. X. Zhao, Energy Environ. Mater., 2019, 2, 73–78 CrossRef CAS.
- Q. F. Ye, Y. Zhao, S. Q. Mu, P. Q. Gao, X. W. Zhang and J. B. You, Sci. China: Chem., 2019, 62, 810–821 CrossRef CAS.
- J. Gong, P. J. Guo, S. E. Benjamin, P. G. Van Patten, R. D. Schaller and T. Xu, J. Energy Chem., 2018, 27, 1017–1039 CrossRef.
- A. Dutta and N. Pradhan, ACS Energy Lett., 2019, 4, 709–719 CrossRef CAS.
- J. T. Gan, J. X. He, R. L. Z. Hoye, A. Mavlonov, F. Raziq, J. L. MacManus-Driscoll, X. Q. Wu, S. Li, X. T. Zu, Y. Q. Zhan, X. Y. Zhang and L. Qiao, ACS Energy Lett., 2019, 4, 1308–1320 CrossRef CAS.
- J. A. Steele, H. D. Jin, I. Dovgaliuk, R. F. Berger, T. Braeckevelt, H. F. Yuan, C. Martin, E. Solano, K. Lejaeghere, S. M. J. Rogge, C. Notebaert, W. Vandezande, K. P. F. Janssen, B. Goderis, E. Debroye, Y.-K. Wang, Y. T. Dong, D. X. Ma, M. Saidaminov, H. R. Tan, Z. H. Lu, V. Dyadkin, D. Chernyshov, V. Van Speybroeck, E. H. Sargent, J. Hofkens and M. B. J. Roeffaers, Science, 2019, 365, 679–684 CrossRef CAS.
- D. B. Straus, S. Guo and R. J. Cava, J. Am. Chem. Soc., 2019, 141, 11435–11439 CrossRef CAS.
- A. Marronnier, H. Lee, B. Geffroy, J. Even, Y. Bonnassieux and G. Roma, J. Phys. Chem. Lett., 2017, 8, 2659–2665 CrossRef CAS.
- U.-G. Jong, C.-J. Yu, Y.-H. Kye, Y.-S. Kim, C.-H. Kim and S.-G. Ri, J. Mater. Chem. A, 2018, 6, 17994–18002 RSC.
- Y. Z. Lin, B. Chen, Y. J. Fang, J. J. Zhao, C. X. Bao, Z. H. Yu, Y. H. Deng, P. N. Rudd, Y. F. Yan, Y. B. Yuan and J. S. Huang, Nat. Commun., 2018, 9, 4981 CrossRef.
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J.-M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS.
- H. Kronmller and S. Parkin, Handbook of magnetism and advanced magnetic materials, John Wiley & Sons, Hoboken, NJ, 2007 Search PubMed.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- R. X. Yang, J. M. Skelton, E. L. da Silva, J. M. Frost and A. Walsh, J. Phys. Chem. Lett., 2017, 8, 4720–4726 CrossRef CAS.
- S. Dastidar, C. J. Hawley, A. D. Dillon, A. D. Gutierrez-Perez, J. E. Spanier and A. T. Fafarman, J. Phys. Chem. Lett., 2017, 8, 1278–1282 CrossRef CAS.
- D. S. Tsvetkov, M. O. Mazurin, V. V. Sereda, I. L. Ivanov, D. A. Malyshkin and A. Y. Zuev, J. Phys. Chem. C, 2020, 124, 4252–4260 CrossRef CAS.
- D. B. Straus, S. Guo, A. M. M. Abeykoon and R. J. Cava, Adv. Mater., 2020, 32, 2001069 CrossRef CAS.
- B. Wang, N. Novendra and A. Navrotsky, J. Am. Chem. Soc., 2019, 141, 14501–14504 CrossRef CAS.
- A. Senocrate, T. Acartürk, G. Y. Kim, R. Merkle, U. Starke, M. Grätzel and J. Maier, J. Mater. Chem. A, 2018, 6, 10847–10855 RSC.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- G. C. Yuan, C. Ritchie, M. Ritter, S. Murphy, D. E. Gómez and P. Mulvaney, J. Phys. Chem. C, 2018, 122, 13407–13415 CrossRef CAS.
- R. An, F. Y. Zhang, X. S. Zou, Y. Y. Tang, M. L. Liang, I. Oshchapovskyy, Y. C. Liu, A. Honarfar, Y. Q. Zhong, C. S. Li, H. F. Geng, J. S. Chen, S. E. Canton, T. Pullerits and K. B. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 39222–39227 CrossRef CAS.
- Y. H. Huang, W. L. Luan, M. K. Liu and L. Turyanska, J. Mater. Chem. C, 2020, 8, 2381–2387 RSC.
- M. Imran, V. Caligiuri, M. J. Wang, L. Goldoni, M. Prato, R. Krahne, L. D. Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS.
- W. N. Xue, X. Y. Wang, W. Wang, F. F. He, W. Zhu and Y. Li, CCS Chem., 2020, 2, 13–23 CrossRef CAS.
- C.-C. Lin, S.-K. Huang, C.-E. Hsu, Y.-C. Huang, C.-Y. Wei, C.-Y. Wen, S.-S. Li, C.-W. Chen and C.-C. Chen, J. Phys. Chem. Lett., 2020, 11, 3287–3293 CrossRef CAS.
- C. H. Bi, S. V. Kershaw, A. L. Rogach and J. J. Tian, Adv. Funct. Mater., 2019, 29, 1902446 CrossRef.
- C. X. Zhang, L. Turyanska, H. C. Cao, L. X. Zhao, M. W. Fay, R. Temperton, J. O'Shea, N. R. Thomas, K. Y. Wang, W. L. Luan and A. Patanè, Nanoscale, 2019, 11, 13450–13457 RSC.
- J. Pan, Y. Q. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. J. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS.
- X. F. Ling, S. J. Zhou, J. Y. Yuan, J. W. Shi, Y. L. Qian, B. W. Larson, Q. Zhao, C. C. Qin, F. C. Li, G. Z. Shi, C. Stewart, J. X. Hu, X. L. Zhang, J. M. Luther, S. Duhm and W. L. Ma, Adv. Energy Mater., 2019, 9, 1900721 CrossRef.
- L. M. Wheeler, E. M. Sanehira, A. R. Marshall, P. Schulz, M. Suri, N. C. Anderson, J. A. Christians, D. Nordlund, D. Sokaras, T. Kroll, S. Harvey, J. J. Berry, L. Y. Lin and J. M. Luther, J. Am. Chem. Soc., 2018, 140, 10504–10513 CrossRef CAS.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, 4204 CrossRef.
- J. Xi, C. C. Piao, J. Byeon, J. Yoon, Z. X. Wu and M. Choi, Adv. Energy Mater., 2019, 9, 1901787 CrossRef.
- Q. A. Akkerman, D. Meggiolaro, Z. Y. Dang, F. D. Angelis and L. Manna, ACS Energy Lett., 2017, 2, 2183–2186 CrossRef CAS.
- C. M. Guvenc, Y. Yalcinkaya, S. Ozen, H. Sahin and M. M. Demir, J. Phys. Chem. C, 2019, 123, 24865–24872 CrossRef CAS.
- M. H. Zhang, X. J. Chen, J. W. Xiao, M. Q. Tai, D. Legut, J. C. Shi, J. L. Qu, Q. Zhang, X. Li, L. Chen, R. F. Zhang, H. Lin and Q. F. Zhang, Nanoscale, 2020, 12, 6571 RSC.
- S.-Q. Wang, S. Y. Shen, X.-X. Xue, Y. Q. He, Z.-W. Xu, K. Q. Chen and Y. X. Feng, Appl. Phys. Express, 2019, 12, 051017 CrossRef CAS.
- D. W. Liu, W. Y. Zha, Y. M. Guo and R. J. Sa, ACS Omega, 2020, 5, 893–896 CrossRef CAS.
- J. K. Nam, S. U. Chai, W. Cha, Y. J. Choi, W. Kim, M. S. Jung, J. Kwon, D. Kim and J. H. Park, Nano Lett., 2017, 17, 2028–2033 CrossRef CAS.
- X. Y. Zhang, M. Lu, Y. Zhang, H. Wu, X. Y. Shen, W. Zhang, W. T. Zheng, V. L. Colvin and W. W. Yu, ACS Cent. Sci., 2018, 4, 1352–1359 CrossRef CAS.
- X. Gong, Z. Yang, G. Walters, R. Comin, Z. Ning, E. Beauregard, V. Adinolfi, O. Voznyy and E. H. Sargent, Nat. Photonics, 2016, 10, 253–257 CrossRef CAS.
- Z. Ning, X. Gong, R. Comin, G. Walters, F. Fan, O. Voznyy, E. Yassitepe, A. Buin, S. Hoogland and E. H. Sargent, Nature, 2015, 523, 324–328 CrossRef CAS.
- Q. Zhang, Y. Y. Zhou, Y. X. Wei, M. Q. Tai, H. Nan, Y. C. Gu, J. H. Han, X. W. Yin, J. B. Li and H. Lin, J. Mater. Chem. C, 2020, 8, 2569–2578 RSC.
- C. Sun, Y. Zhang, C. Ruan, C. Y. Yin, X. Y. Wang, Y. D. Wang and W. W. Yu, Adv. Mater., 2016, 28, 10088–10094 CrossRef CAS.
- C. Jia, H. Li, X. W. Meng and H. B. Li, Chem. Commun., 2018, 54, 6300–6303 RSC.
- G. K. Grandhi, N. S. M. Viswanath, J. H. In, H. B. Cho and W. B. Im, J. Phys. Chem. Lett., 2020, 11, 3699–3704 CrossRef CAS.
- A. Z. Pan, Y. N. Li, Y. S. Wu, K. Yan, M. J. Jurow, Y. Liu and L. He, Mater. Chem. Front., 2019, 3, 414 RSC.
- W. J. Mir, A. Swarnkar and A. Nag, Nanoscale, 2019, 11, 4278–4286 RSC.
- M. Lu, X. Y. Zhang, X. Bai, H. Wu, X. Y. Shen, Y. Zhang, W. Zhang, W. T. Zheng, H. W. Song, W. W. Yu and A. L. Rogach, ACS Energy Lett., 2018, 3, 1571–1577 CrossRef CAS.
- N. Ding, D. L. Zhou, X. K. Sun, W. Xu, H. W. Xu, G. C. Pan, D. Y. Li, S. Zhang, B. Dong and H. W. Song, Nanotechnology, 2018, 29, 345703 CrossRef.
- Q. Wang, Z. W. Jin, D. Chen, D. L. Bai, H. Bian, J. Sun, G. Zhu, G. Wang and S. Z. (Frank) Liu, Adv. Energy Mater., 2018, 8, 1800007 CrossRef.
- Z. L. Zhang, L. L. Shen, Y. Zhao, Y. J. Zhang, H. S. Yang, W. D. Xiang, X. J. Liang, G. X. Chen and H. T. Yu, Chem. Eng. J., 2020, 385, 123415 CrossRef CAS.
- J.-S. Yao, J. Ge, K.-H. Wang, G. Z. Zhang, B.-S. Zhu, C. Chen, Q. Zhang, Y. Luo, S.-H. Yu and H.-B. Yao, J. Am. Chem. Soc., 2019, 141, 2069–2079 CrossRef CAS.
- Y. L. Wang, X. Cheng, K. Yuan, Y. Wan, P. Li, Y. H. Deng, H. R. Yu, X. L. Xu, Y. Zeng, W. J. Xu, Y. P. Li, R. M. Ma, K. Watanabe, T. Taniguchi, Y. Ye and L. Dai, Sci. Bull., 2018, 63, 1576–1582 CrossRef CAS.
- V. A. Hintermayr, C. Lampe, M. Löw, J. Roemer, W. Vanderlinden, M. Gramlich, A. X. Böhm, C. Sattler, B. Nickel, T. Lohmüller and A. S. Urban, Nano Lett., 2019, 19, 4928–4933 CrossRef CAS.
- T. Ye, B. Zhou, F. Zhan, F. L. Yuan, S. Ramakrishna, D. Golberg and X. Wang, Solar RRL, 2020, 4, 2000014 CrossRef CAS.
- C. S. Chen, D. Li, Y. H. Wu, C. Chen, Z.-G. Zhu, W. Y. Shih and W.-H. Shih, Nanotechnology, 2020, 31, 20 Search PubMed.
- A. N. AleshinaIgor, P. Shcherbakov, E. V. Gushchina, L. B. Matyushkin and V. A. Moshnikov, Org. Electron., 2017, 50, 213–219 CrossRef.
- J. J. Wu, J. Y. Tong, Y. Gao, A. F. Wang, T. Zhang, H. R. Tan, S. M. Nie and Z. T. Deng, Angew. Chem., Int. Ed., 2020, 59, 7738–7742 CrossRef CAS.
- A. Z. Pan, J. L. Wang, M. J. Jurow, M. J. Jia, Y. Liu, Y. S. Wu, Y. F. Zhang, L. He and Y. Liu, Chem. Mater., 2018, 30, 2771–2780 CrossRef CAS.
- S. L. Mei, B. B. Yang, X. Wei, H. Q. Dai, Z. H. Chen, Z. J. Cui, G. L. Zhang, F. X. Xie, W. L. Zhang and R. Q. Guo, Nanomaterials, 2019, 9, 832 CrossRef CAS.
- F. Palazon, Q. A. Akkerman, M. Prato and L. Manna, ACS Nano, 2016, 10, 1224–1230 CrossRef CAS.
- J.-H. Cha, K. Noh, W. P. Yin, Y. Lee, Y. Park, T. K. Ahn, A. Mayoral, J. Kim, D.-Y. Jung and O. Terasaki, J. Phys. Chem. Lett., 2019, 10, 2270–2277 CrossRef CAS.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- J. J. Ren, T. R. Li, X. P. Zhou, X. Dong, A. V. Shorokhov, M. B. Semenov, V. D. Krevchik and Y. H. Wang, Chem. Eng. J., 2019, 358, 30–39 CrossRef CAS.
- M. C. Hanna, M. C. Beard and A. J. Nozik, J. Phys. Chem. Lett., 2012, 3, 2857–2862 CrossRef CAS.
- K. G. McKay and K. B. McAfee, Phys. Rev., 1953, 91, 1079–1084 CrossRef CAS.
- J. Tauc, J. Phys. Chem. Solids, 1959, 8, 219–223 CrossRef CAS.
- D. J. Robbins, Phys. Status Solidi B, 1980, 98, 11–36 CrossRef CAS.
- O. E. Semonin, J. M. Luther, S. Choi, H.-Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS.
- M. C. Beard, J. M. Luther, O. E. Semonin and A. J. Nozik, Acc. Chem. Res., 2012, 46, 1252–1260 CrossRef.
- M. Li, R. Begum, J. Fu, Q. Xu, T. M. Koh, S. A. Veldhuis, M. Grätzel, N. Mathews, S. Mhaisalkar and T. C. Sum, Nat. Commun., 2018, 9, 4197 CrossRef.
- C. de Weerd, L. Gomez, A. Capretti, D. M. Lebrun, E. Matsubara, J. Lin, M. Ashida, F. C. M. Spoor, L. D. A. Siebbeles, A. J. Houtepen, K. Suenaga, Y. Fujiwara and T. Gregorkiewicz, Nat. Commun., 2018, 9, 4199 CrossRef.
- A. Manzi, Y. Tong, J. Feucht, E.-P. Yao, L. Polavarapu, A. S. Urban and J. Feldmann, Nat. Commun., 2018, 9, 1518 CrossRef.
- M. T. Trinh, R. Limpens, W. D. A. M. de Boer, J. M. Schins, L. D. A. Siebbeles and T. Gregorkiewicz, Nat. Photonics, 2012, 6, 316–321 CrossRef CAS.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. D. Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS.
- G. Yumoto, H. Tahara, T. Kawawaki, M. Saruyama, R. Sato, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2018, 9, 2222–2228 CrossRef CAS.
- E. Kobiyama, H. Tahara, R. Sato, M. Saruyama, T. Teranishi and Y. Kanemitsu, Nano Lett., 2020, 20, 3905–3910 CrossRef CAS.
- J. Navarro-Arenas, I. Suárez, V. S. Chirvony, A. F. Gualdrón-Reyes, I. Mora-Seró and J. Martinez-Pastor, J. Phys. Chem. Lett., 2019, 10, 6389–6398 CrossRef CAS.
- L. M. Pazos-Outón, M. Szumilo, R. Lamboll, J. M. Richter, M. Crespo-Quesada, M. Abdi-Jalebi, H. J. Beeson, M. Vrućinić, M. Alsari, H. J. Snaith, B. Ehrler, R. H. Friend and F. Deschler, Science, 2016, 351, 1430–1433 CrossRef.
- A. Marti, J. L. Balenzategui and R. F. Reyna, J. Appl. Phys., 1997, 82, 4067–4075 CrossRef CAS.
- N. J. Davis, F. J. de la Peña, M. Tabachnyk, J. M. Richter, R. D. Lamboll, E. P. Booker, F. Wisnivesky Rocca Rivarola, J. T. Griffiths, C. Ducati, S. M. Menke and F. Deschler, J. Phys. Chem. C, 2017, 121, 3790–3796 CrossRef CAS.
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2020 |