
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new route to porous metal–organic framework crystal–glass composites†
Shichun
Li
 ab,
Shuwen
Yu
ac,
Sean M.
Collins
bd,
Duncan N.
Johnstone
b,
Christopher W.
Ashling
b,
Adam F.
Sapnik
b,
Philip A.
Chater
e,
Dean S.
Keeble
e,
Lauren N.
McHugh
b,
Paul A.
Midgley
b,
David. A.
Keen
f and
Thomas D.
Bennett
*b
ab,
Shuwen
Yu
ac,
Sean M.
Collins
bd,
Duncan N.
Johnstone
b,
Christopher W.
Ashling
b,
Adam F.
Sapnik
b,
Philip A.
Chater
e,
Dean S.
Keeble
e,
Lauren N.
McHugh
b,
Paul A.
Midgley
b,
David. A.
Keen
f and
Thomas D.
Bennett
*b
aInstitute of Chemical Materials, China Academy of Engineering Physics, Mianyang 621900, P. R. China
bDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge CB3 0FS, UK. E-mail: tdb35@cam.ac.uk
cXiamen Institute of Rare Earth Materials, Haixi Institute, Chinese Academy of Sciences, Xiamen 361021, P. R. China
dSchool of Chemical and Process Engineering and School of Chemistry, University of Leeds, LS2 9JT, UK
eDiamond House, Harwell Science & Innovation Campus, Diamond Light Source, Ltd., Didcot, Oxfordshire OX11 0DE, UK
fISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxon OX11 0QX, UK
First published on 28th August 2020
Abstract
Metal–organic framework crystal–glass composite (MOF CGC) materials consist of a crystalline MOF embedded within a MOF–glass matrix. In this work, a new synthetic route to these materials is demonstrated through the preparation of two ZIF-62 glass-based CGCs, one with crystalline ZIF-67 and the other with crystalline UiO-66. Previous attempts to form these CGCs failed due to the high processing temperatures involved in heating above the melting point of ZIF-62. Annealing of the ZIF-62 glass above the glass transition with each MOF however leads to stable CGC formation at lower temperatures. The reduction in processing temperatures will enable the formation of a greatly expanded range of MOF CGCs.
Introduction
Metal–organic frameworks (MOFs) contain metal nodes and organic ligands connected into networks. They possess extremely high internal surface areas and chemically tunable functionalities.1–3 Over 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 crystalline MOFs have been recorded in the Cambridge Structural Database.4 Of these, several are now sold commercially, primarily for gas storage applications, though additional uses in catalysis,5 water harvesting,6 and molecular separations7 have also been proposed. The physical forms and morphologies adopted by MOFs are of great interest in commercialization because traditional processing techniques, such as milling and sintering, are often hindered by the low mechanical stability of crystalline MOFs.8,9 Considerable progress has been made, however, in the production of binder-free commercial structures which contain appreciable porosity.10
000 crystalline MOFs have been recorded in the Cambridge Structural Database.4 Of these, several are now sold commercially, primarily for gas storage applications, though additional uses in catalysis,5 water harvesting,6 and molecular separations7 have also been proposed. The physical forms and morphologies adopted by MOFs are of great interest in commercialization because traditional processing techniques, such as milling and sintering, are often hindered by the low mechanical stability of crystalline MOFs.8,9 Considerable progress has been made, however, in the production of binder-free commercial structures which contain appreciable porosity.10
A further complication results from the susceptibility of MOFs to chemical degradation.9 Zeolitic imidazolate frameworks (ZIFs) for example, have been shown to be highly prone to degradation via acid catalysed Zn–N bond hydrolysis, which causes progressive collapse of the crystal structure accompanied by a reduction in internal surface area.11,12 Other MOFs that are relatively stable in acid, such as UiO-66 and MOF-545, are unstable to basic conditions.13
Various strategies have been proposed to enhance the processability and stability of crystalline MOFs, including modulated synthesis and post-synthetic modification,13 sol–gel processing,14 and fabrication of MOF based composites such as mixed matrix membranes (MMMs).15,16 In this latter example, crystalline MOF particles are dispersed in a polymer matrix,7,17 though poor interfacial compatibility between the polymer and crystalline MOF component leads to several related issues such as the formation of interfacial mesoporous voids and aggregation of the MOF component.18,19 Matrices possessing both good processability and compatibility with crystalline MOFs are therefore highly sought after.
The recent discovery of MOFs which form glasses20–22 opens up possible opportunities to solve processability and compatibility issues, by using a MOF–glass as a binder. ZIFs are a subset of MOFs containing tetrahedral metal ions, which are linked by imidazolate (Im, C3H3N2−) derived bidentate ligands and have structures closely related to zeolitic silica polymorphs. In particular, ZIF-62, [Zn(Im)1.75(bIm)0.25] (bIm, benzimidazolate, C7H5N2−), has been shown to form a glass of identical composition (referred to as agZIF-62), by quenching from the liquid at ca. 437 °C in an argon (Ar) atmosphere. Bulk, transparent and bubble-free forms have previously been prepared by remelting and annealing agZIF-62 above its glass transition temperature (Tg) of 318 °C.23 The absence of recrystallisation of the glass upon reheating, or slow cooling, is ascribed to the volume collapse from open crystalline to dense glass.24
We have previously fabricated MOF crystal–glass composites (MOF CGCs), in which crystalline MIL-53 [Al(OH)(O2C–C6H4–CO2)] or UiO-66 [Zr6O4(OH)4(O2C–C6H4–CO2)6] were dispersed within an agZIF-62 matrix.25 The composites were fabricated by heating a mixture of crystalline ZIF-62 and MIL-53 or UiO-66 under Ar to 450 °C, i.e. above the melting point (Tm) of ZIF-62, before cooling back down to room temperature. The crystallinity was preserved in MIL-53 CGCs but UiO-66 was found to undergo partial decomposition due to the high temperatures involved in composite formation.25
MOF CGCs may have promise in applications in gas adsorption, membrane separation, and photocatalysis, though the range of MOF CGCs which may be synthesized by this high-temperature route is however limited at present. Major problems are that the high processing temperatures result in (i) the partial or complete thermal decomposition of the crystalline component, or (ii) the dissolution of chemically compatible ZIFs in the liquid at high temperature (i.e. flux melting).
Motivated to develop lower temperature methods for the synthesis of MOF CGCs, we present here a new strategy for MOF CGC fabrication. Specifically, mixtures of pre-synthesized agZIF-62 and crystalline MOF were annealed at 400 °C, a temperature which is sufficiently above Tg (318 °C) but still below Tm (ca. 440 °C) such that ZIF-62 can be considered within its supercooled liquid regime and the glass has effectively melted.
To highlight the advantages of this method of preparation, ZIF-67 (the isostructural cobalt(II) analogue of ZIF-8), and UiO-66 were chosen as the crystalline components. The former has been reported to undergo flux-melting within the liquid state of ZIF-62,26,27 whilst the latter undergoes partial decomposition.25 Thus far, stable MOF crystal-glass composites have therefore not been formed from either ZIF-67 or UiO-66. These two crystalline MOFs therefore represent ideal candidates on which to attempt to form MOF CGCs using this new processing route.
Here, we use powder X-ray diffraction and X-ray total scattering to show the retention of crystallinity among the crystalline component of MOF–CGCs upon annealing with agZIF-62 above its Tg. Morphology and component distribution within the CGCs are characterized using scanning electron microscopy (SEM), scanning transmission electron microscopy (STEM), X-ray energy dispersive spectroscopy (EDS), and scanning electron diffraction (SED). Thermomechanical analysis (TMA) and chemical stability testing demonstrated improved thermo-mechanical stability compared to the pure crystalline precursors.
Results and discussion
Structural integrity
Samples of ZIF-62, ZIF-67 and UiO-66 were synthesized following previously reported methods (full details in Methods).28–30 A sample of agZIF-62 was obtained by melting ZIF-62 at 450 °C for one minute under Ar, followed by ball milling the cooled sample with 2 × 7 mm stainless steel balls at 25 Hz for 5 minutes to reduce particle size (Fig. S1†). Samples of crystalline ZIF-67 or UiO-66 were then mixed with the prefabricated agZIF-62 by grinding in a mortar, with 20%, 50%, and 80% weight fractions (wt%) of the crystalline component. The mixtures were pressed into pellets with a diameter of 13 mm and a thickness of 1 mm. Pressures of 10 tons (750 MPa) for CGCs containing ZIF-67, and 2 tons pressure (151 MPa) for CGCs containing UiO-66 were used, to avoid the reported Zr–OCOO bond breakage in UiO-66 upon compression.31 The formation of bulk pellets prior to heating was necessary to promote close contact between grains, and resultant coalescence of the highly viscous ZIF-62 liquid at the treatment temperatures.The pellets formed prior to heating are referred to as (‘MOF’)(agZIF-62)(X/Y) in keeping with prior literature, where X and Y indicate the percentage by mass of the crystalline and glass components respectively. For example, a non-heat treated pellet of 80 wt% ZIF-67 and 20 wt% agZIF-62 is referred to as (ZIF-67)(agZIF-62)(80/20).
MOF CGCs were then fabricated by heating the pelleted mixtures at 400 °C for 5 hours under Ar to remelt the agZIF-62 component (Fig. 1a). The CGCs obtained upon cooling are referred to as (‘MOF’)X(agZIF-62)Y. For example, a CGC with 80 wt% ZIF-67 and 20 wt% agZIF-62 is referred to as (ZIF-67)0.8(agZIF-62)0.2. The preservation of crystallinity in both ZIF-67 and UiO-66 was confirmed by X-ray diffraction (Fig. 1b, S2, and Table S1†), in contrast to previous efforts at CGC fabrication using these systems.25,26 The samples of (ZIF-67)0.8(agZIF-62)0.2 and (UiO-66)0.8(agZIF-62)0.2 (Fig. 1c and d) were macroscopic solid pieces, demonstrating the bulk samples obtained using this approach.
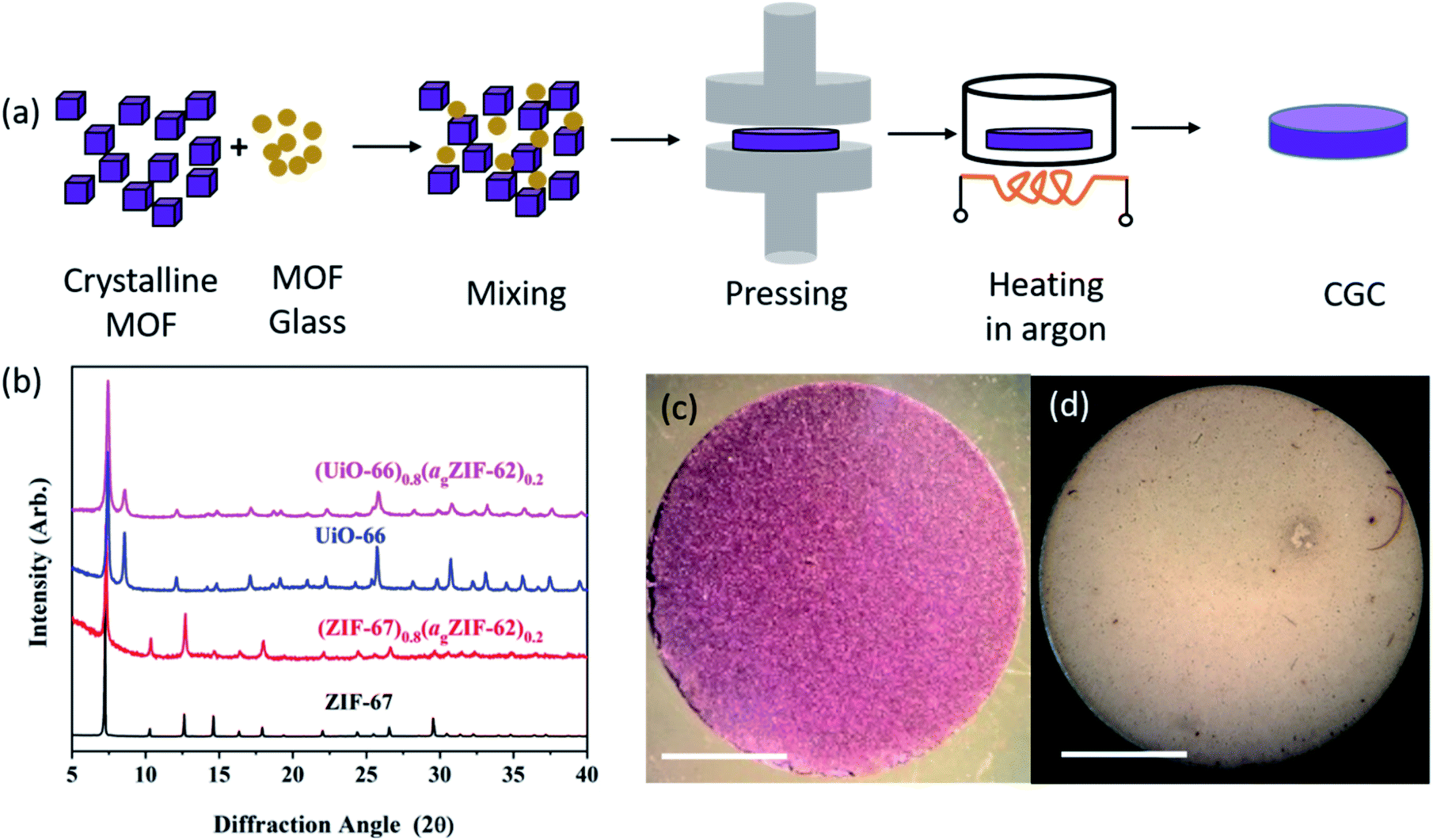 | ||
| Fig. 1 Fabrication of bulk monolithic CGCs. (a) Schematic diagram of fabrication procedure. (b) X-ray diffraction patterns of as synthesized ZIF-67 and UiO-66, alongside patterns from the CGCs. Data normalized for each sample using the intensity of the most intense peak. Optical images of (c) (ZIF-67)0.8(agZIF-62)0.2 and (d) (UiO-66)0.8(agZIF-62)0.2, scale bar of 5 mm. | ||
Synchrotron X-ray total scattering measurements were performed on (i) pure samples of agZIF-62, UiO-66 and ZIF-67, (ii) the physical mixtures after pressing, though prior to remelting; and (iii) the three MOF CGCs formed after melting (Fig. 2 and S3†). The structure factors, S(Q), for ZIF-67, UiO-66, physical mixtures and CGCs contain extensive Bragg scattering as expected. The intensity of Bragg scattering from ZIF-67 decreased in the pelletized mixture, consistent with reported partial pressure induced amorphization,32 and then again after CGC formation (Fig. 2a, b and S3†). The results are consistent with those in Fig. 1b, confirming retention of crystallinity within the CGCs, though this appears slightly reduced in the MOF–CGCs compared to the pressed physical mixtures. The pair distribution functions (PDFs), D(r), of agZIF-62, ZIF-67, (ZIF-67)(agZIF-62)(80/20) and (ZIF-67)0.8(agZIF-62)0.2 are similar below 7.5 Å (Fig. 2c, e, S3c†), though contain subtle differences such as the position of peak i.26 Long range oscillations beyond 8 Å persist in the CGC sample, as expected, though weaken with decreasing weight fraction of ZIF-67 in CGCs (Fig. S3c†). The PDFs of the UiO-66 CGCs display characteristic peaks of both agZIF-62 and UiO-66, (Fig. 2d, e).25 Like those for the ZIF-67 CGC, long range correlations in the CGCs (ascribed to Zr–Zr correlations) also weaken with decreasing weight fraction of UiO-66 (Fig. S3d†).
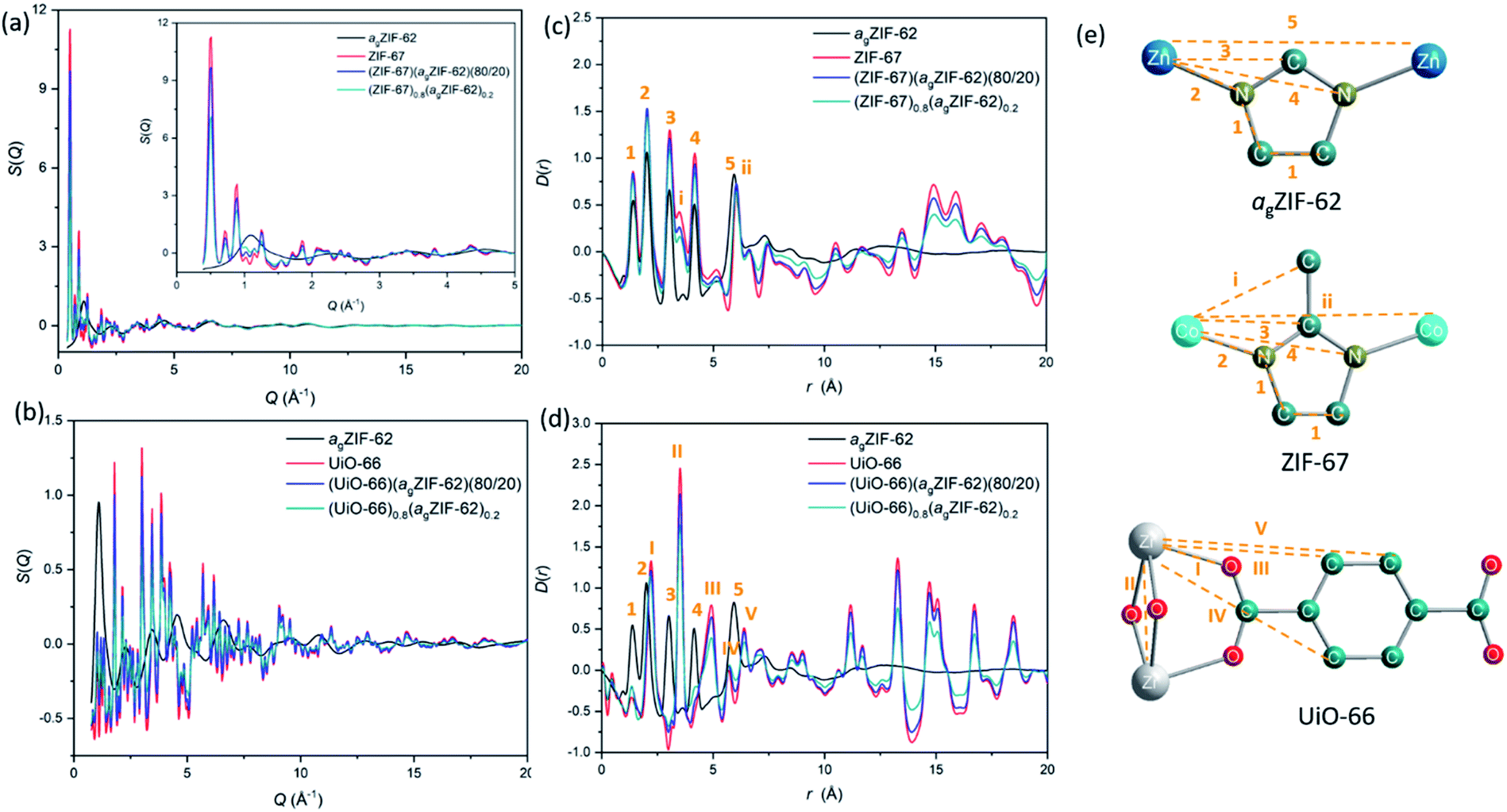 | ||
| Fig. 2 X-ray total scattering. (a and b) structure factors, S(Q), of agZIF-62, ZIF-67, UiO-66 physical mixtures and CGCs. (c and d) Corresponding pair distribution functions, D(r). (e) Scheme of peak assignment of PDF patterns. Zn, blue; C, dark green; N dark yellow; Co, light blue; Zr, grey; O, red; H, omitted for clarity. | ||
Phase distribution
The surface morphologies of both physical mixtures and CGC samples were observed by SEM, and the corresponding distribution of metal centres on the surface measured by EDS (Fig. 3, S4–S7†). Both (ZIF-67)(agZIF-62)(X/Y) and (ZIF-67)X(agZIF-62)Y exhibit relatively dense surfaces (Fig. 3, S4–S6†), indicating close contact between ZIF-67 and agZIF-62. The EDS maps of Zn and Co show separated phases of agZIF-62 and ZIF-67 in both (ZIF-67)(agZIF-62)(X/Y) and (ZIF-67)X(agZIF-62)Y. Surface cracks are apparent in regions dominated by the ZIF-67 in the case of all non-heat treated pressed samples mixtures (Fig. S5 and S6†), though appear far less prevalent in the heat treated 50% sample (Fig. 3). | ||
| Fig. 3 Surface morphology and elemental distribution of CGCs. SEM images and EDS maps of metal centres on the surface of CGCs (a and b) (ZIF-67)0.5(agZIF-62)0.5 and (c and d) (UiO-66)0.5(agZIF-62)0.5. | ||
The surfaces of (UiO-66)(agZIF-62)(X/Y) and (UiO-66)X(agZIF-62)Y were far less uniform, consistent with the lower pressures applied for pelletisation being insufficient to force close contact between the viscous ZIF-62 liquid and UiO-66 particles during remelting. Cracks at the boundaries between agZIF-62 and UiO-66 in both (UiO-66)(agZIF-62)(X/Y) and (UiO-66)X(agZIF-62)Y were located in representative EDS mappings (Fig. 3, S5, S7†). These were not observed between ZIF-67 and agZIF-62, suggesting better interfacial contact for ZIF-67 CGCs.
The (ZIF-67)0.5(ZIF-62)0.5 sample was ground into a powder and characterized using scanning transmission electron microscopy (STEM) to investigate the CGC microstructure (Fig. 4). Scanning Electron Diffraction (SED) was used to map the number of detected Bragg diffraction peaks measured at each probe position as the electron probe was scanned across the sample to reveal the location of crystalline phases in the MOF-CGCs, as shown in Fig. 4c and d. These are in effect qualitative maps of crystallinity as determined by the presence of Bragg peaks in crystalline domains and absence of Bragg peaks in non-crystalline regions (Fig. S8†).
 | ||
| Fig. 4 Scanning transmission electron microscopy. MOF CGC particles in a (ZIF-67)0.5(ZIF-62)0.5 sample. (a and b) ADF images showing particle morphology, (c and d) crystallinity maps showing the number of Bragg peaks as a function of probe position in SED data. (e and f) compositional maps of Zn and Co metal centres from STEM-EDS mapping. Arrows used to indicate correlation between the presence/absence of diffraction with that of Co/Zn. | ||
The maps of the crystalline regions demonstrate close contact between crystalline and non-crystalline regions. Comparison with compositional maps showing the distribution of metal centres, obtained via STEM-EDS mapping of the same particles and shown in Fig. 4e and f, confirms that the crystalline regions, indicated with red arrows in Fig. 4b, d and f, correspond to cobalt-rich regions, as expected for ZIF-67. Conversely, amorphous regions, indicated with blue arrows in Fig. 4a, c and e, also correspond to the presence of Zn-centres, consistent with the agZIF-62 domains in (ZIF-67)0.5(ZIF-62)0.5.
Gas uptake properties
N2 sorption isotherm experiments were performed to investigate the porosity of the CGCs (Fig. 5, S9–S11†). The BET surface area of a pure sample of ZIF-67 decreased by 27%, from 1744 to 1269 m2 g−1 upon pelletisation (Fig. 5a, S9a†). For UiO-66, the corresponding drop was 16%, from 1120 to 941 m2 g−1 (Fig. 5b, S9b†). A near-identical percentage decrease in BET surface area for the physical mixtures was observed upon pelletization, with a decrease of 29% in the case of (ZIF-67)(agZIF-62)(80/20) (1389 m2 g−1 to 981 m2 g−1), and 16% for (UiO-66)(agZIF-62)(80/20) (817 m2 g−1 to 683 m2 g−1). These decreases are ascribed to the known partial collapse of both frameworks under pressure.31 The effect of heat treatment on the pure sample pellets was also investigated, where in the case of ZIF-67, heating at 400 °C for 5 hours under Ar, i.e. the same condition for CGC formation, resulted in a very slight increase in gas sorption capacity (Fig. 5a), attributed to the removal of remaining solvent molecules in the framework. For UiO-66, a drastic decrease was noted (Fig. 5b). This is consistent with previous reports, which ascribed the decrease to the dehydroxylation of the inorganic cluster at ca. 300 °C.33,34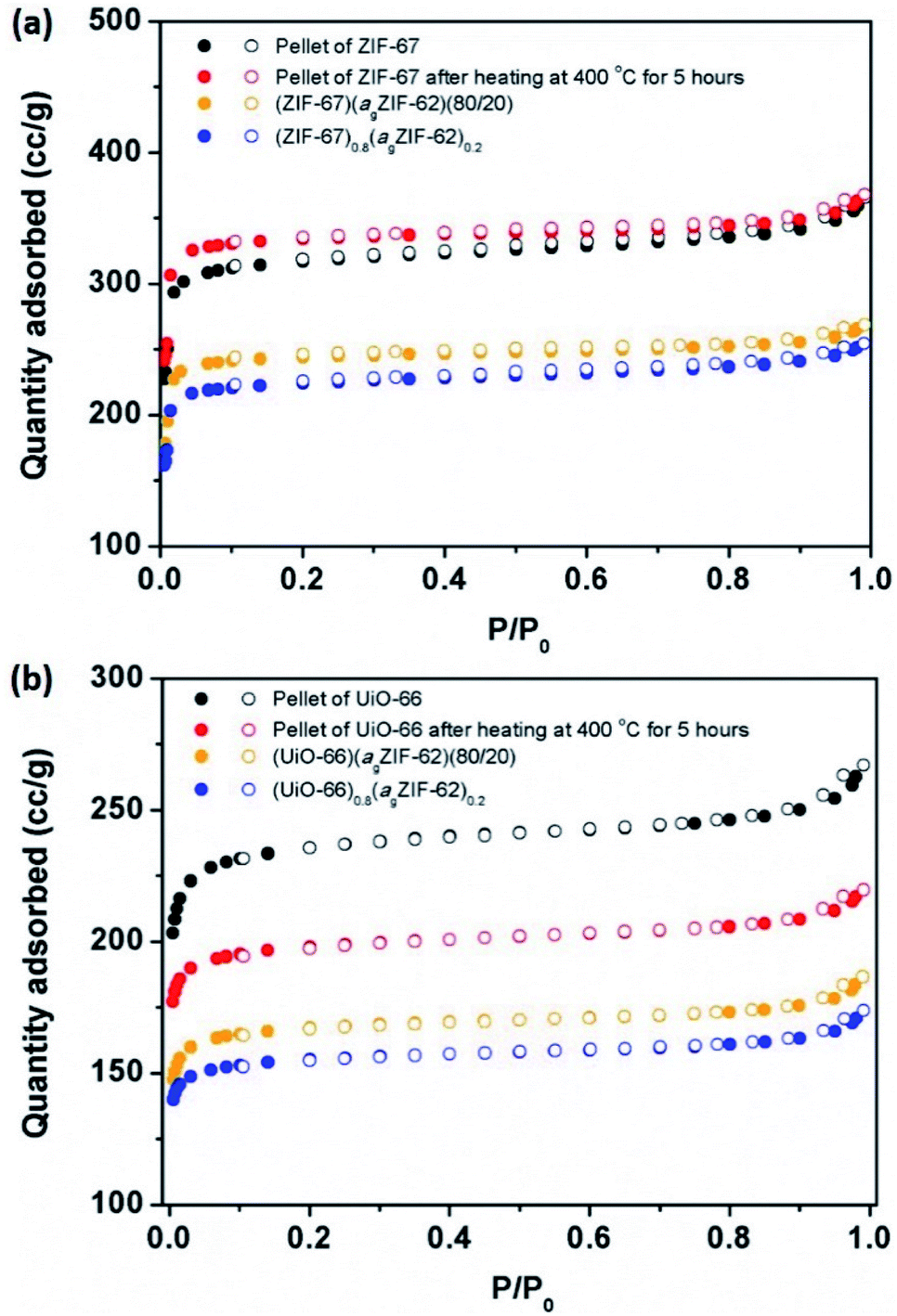 | ||
| Fig. 5 Gas sorption. N2 gas isotherms at 77 K of (a) ZIF-67 series, and (b) UiO-66 series. Solid circles represent adsorption, and hollow circles represent desorption. | ||
In contrast, CGC formation resulted in only a slight decrease in gas sorption capacity in both cases (Fig. 5). For example, BET surface areas decreased from 981 m2 g−1 (ZIF-67)(agZIF-62)(80/20) to 901 m2 g−1 (ZIF-67)0.8(agZIF-62)0.2, and from 683 m2 g−1 to 632 m2 g−1 from (UiO-66)(agZIF-62)(80/20) to (UiO-66)0.8(agZIF-62)0.2.
As expected, the gas sorption quantity of CGCs increases with the weight fraction of MOF crystals in the CGCs (Fig. S11†). The decrease in BET surface area of the CGC relative to that of the mixture for ZIF-67 is non-linear with the weight fraction of the ZIF-67 (Fig. S11c†). This implies a strong interaction between ZIF-67 and agZIF-62 in the CGC, causing partial collapse or blockage of the pores of ZIF-67. The equivalent BET surface area decrease for UiO-66 on CGC formation is approximately proportional to the weight fraction of UiO-66.
Macroscopic thermal expansion
The thermal expansivities of the unit cell of numerous crystalline MOF structures have been reported from changes to the crystal unit cell determined by refinement of variable temperature X-ray data.35–37 However, it can be argued that the macroscopic thermal expansion of the formed bulk material is just as important.38 This has been rarely reported for MOFs, due to problems in forming bulk samples for measurement by thermomechanical analysis (TMA).Here, the linear thermal expansion of a sample of agZIF-62 was measured for the first time, alongside the expansivities of pellets of MOF crystals, physical mixtures and CGCs (Fig. 6 and S12†). The glass sample, agZIF-62, was found to possess an average coefficient of thermal expansion (CTE) of 35 × 10−6 K−1 between 35 °C and 275 °C. This is much higher than that of silica-based inorganic glasses (0.59–8.5 × 10−6 K−1), and lower than that of most polymers (45–200 × 10−6 K−1), although we note that some polymers have a lower CTE, such as Kapton (20 × 10−6 K−1).39,40 This intermediate thermal expansion is perhaps expected given the inorganic–organic bonding within the system, but is nevertheless a beautiful example of the hybrid nature of the physical properties of bulk MOF systems, alongside the hybrid chemical properties often touted. A softening point, equivalent to the glass transition, was noted at 320 °C (Fig. 6a).
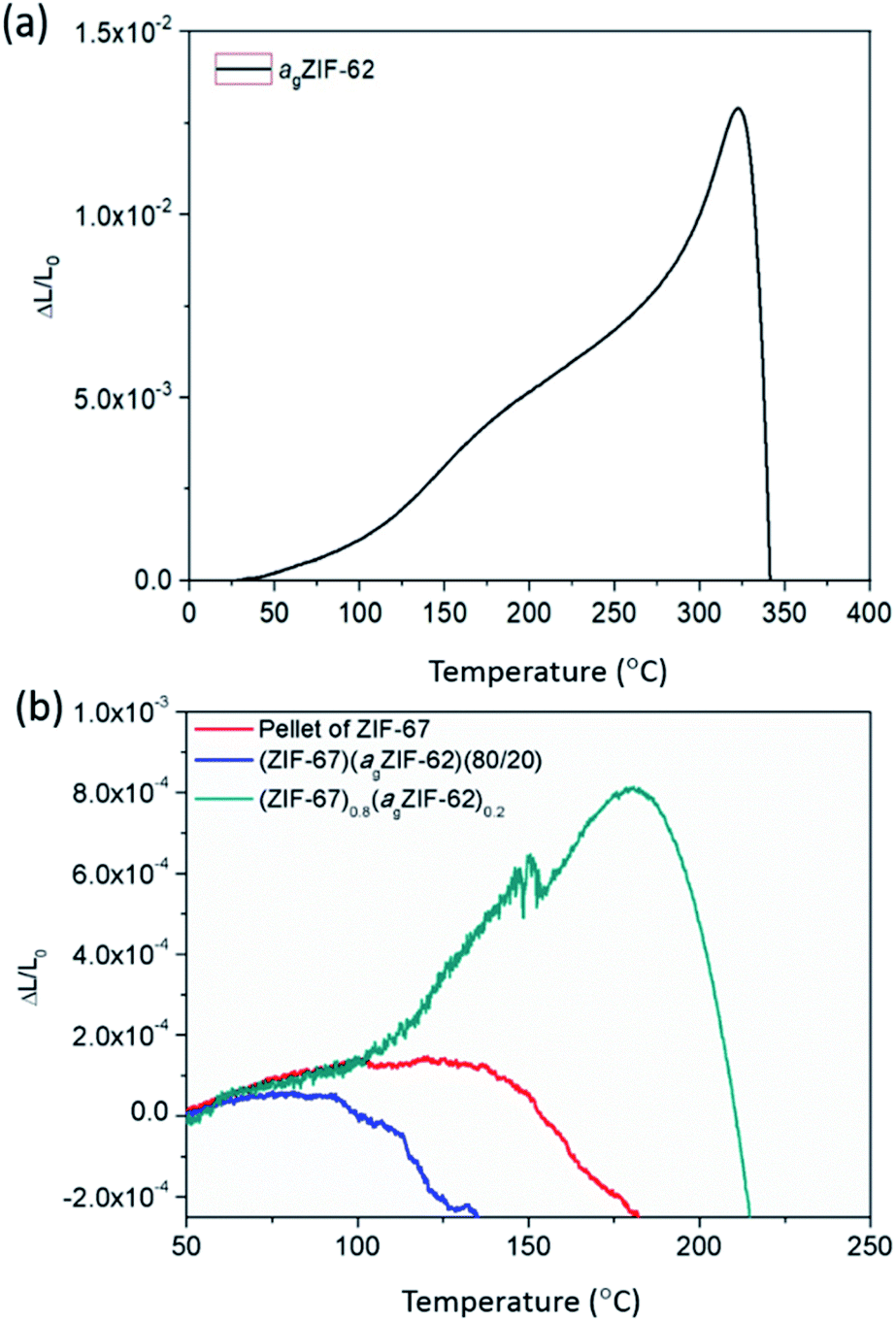 | ||
| Fig. 6 Thermo-mechanical properties. Linear thermal expansion of (a) agZIF-62 and (b) ZIF-67 series measured by TMA. | ||
The pellets of physical mixtures and CGCs (Fig. 6b, S12†) display a lower thermal expansivity than agZIF-62 over 100 °C, which is in agreement with the non-uniform nature of the sample and the presence of macroscale defects within the bulk solid. The absence of deformation (inferred from the scatter in data points) at low temperatures of the (ZIF-67)0.8(agZIF-62)0.2 sample is mainly attributed to the strong interaction between agZIF-62 and ZIF-67 in the CGC. However, in the case of UiO-66, deformation of (UiO-66)0.8(agZIF-62)0.2 was inferred from the start of measurement (Fig. S12†), which is caused by the dehydroxlyation of UiO-66 during remelting.
Chemical stability
The chemical stability of MOFs, particularly with respect to humid conditions or aqueous solutions, is also of great concern given proposed applications in catalysis and gas separation containing acidic or basic components in gas streams e.g. SO2, NH3.9,12 In the field of glasses, chemical stability is highly sought after for display technologies and protective screens.41 The chemical stability of both agZIF-62 and MOF–CGCs in aqueous solutions was therefore investigated (Fig. 7).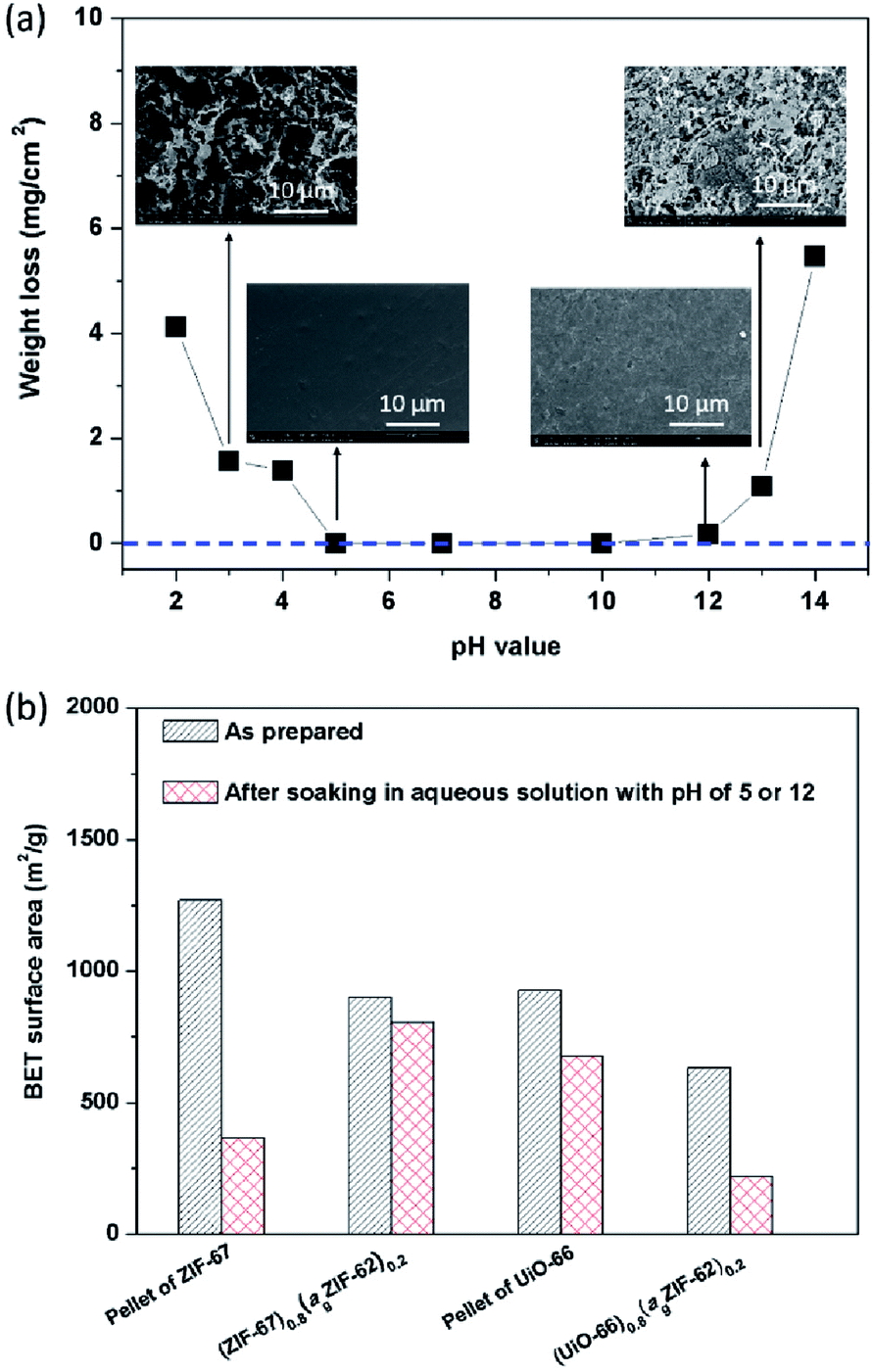 | ||
| Fig. 7 Chemical stability in aqueous acid and base. (a) Weight loss and (insets) surface SEM images of agZIF-62 after soaking in aqueous solutions with pH 2–14 at room temperature for 7 days. (b) BET surface areas of pellets of crystalline–glass mixtures and CGCs before, and after soaking in aqueous solution at pH 5 for ZIF-67 series or pH 12 for UiO-66 series at room temperature for 7 days. | ||
Specifically, the mass loss per surface area of agZIF-62 was measured after soaking a pellet of agZIF-62 in a given aqueous solution at room temperature for 7 days (Fig. S13†), which is a widely used method for assessing the stability of glasses.40 No obvious weight loss of agZIF-62 was observed in the pH range of 5–12 (Fig. 7a), indicating agZIF-62 is stable in this pH range. Apparent corrosion on the surface was however observed by SEM at pHs lower than 5, and higher than 12 (Fig. 7a), though no re-crystallization was found after the soaking experiments (Fig. S14†). The stability of agZIF-62, i.e. stable in all but very basic conditions and unstable in strongly acidic conditions, is consistent with previous observations on crystalline ZIFs, which is caused by the relatively high pKa values of azoles.9
The chemical stability of CGCs was judged by comparing the N2 adsorption quantity, e.g. BET surface area (Fig. 7b, S15†), XRD pattern (Fig. S16†), and SEM images (Fig. S17†) before, and after soaking them in aqueous solutions. Pellets of pure MOF crystals were also investigated for comparison. The samples containing ZIF-67 were soaked in aqueous acid at pH 5 for 7 days, and UiO-66 containing samples were soaked in an aqueous base at pH 12 for 7 days. A dramatic decrease in the BET surface area from 1269 m2 g−1 to 365 m2 g−1 was observed for a pellet of pure ZIF-67 after soaking, whilst only a slight decrease in BET surface area from 901 m2 g−1 to 805 m2 g−1 was found in the case of (ZIF-67)0.8(ZIF-62)0.2 after soaking (Fig. 7b).
Hysteresis in the N2 gas isotherm of pellet of ZIF-67 (Fig. S15a†) is broadly consistent with the degradation inferred by decreases in the intensity of Bragg peaks in the XRD pattern (Fig. S16a†), and the sheet-like morphology on the surface observed by SEM (Fig. S17a†). This is also consistent with the literature and is associated with a reduction in surface area.11,12 Strikingly, the degradation of ZIF-67 in the CGC was far less pronounced than that in the pellet of pure ZIF-67, indicating that (ZIF-67)0.8(ZIF-62)0.2 possesses better stability in aqueous acid than the pellet of pure ZIF-67. This may be attributed to the lower water uptake of (ZIF-67)0.8(ZIF-62)0.2 than that of the pellet of pure ZIF-67 (Fig. S18a†), which slows down the rate of hydrolysis of Co–N coordination bonds. The lower water uptake of (ZIF-67)0.8(agZIF-62)0.2 is attributed to the strongly hydrophobic nature of agZIF-62 and the unique interfacial structure between agZIF-62 and ZIF-67 in the CGC.
In the case of UiO-66, however, a severe decrease in BET surface area was found for (UiO-66)0.8(ZIF-62)0.2 after soaking, in comparison to a pellet of pure UiO-66 (Fig. 7b). This decrease in surface area is consistent with the decrease in intensity of Bragg peaks in the corresponding XRD pattern (Fig. S16b†). This may be caused by the dehydroxylation of UiO-66 in the CGC during remelting that leads to defects in UiO-66. The presence of existing defects in UiO-66 in the CGC favour further defect formation,12 leading to amorphization, although (UiO-66)0.8(agZIF-62)0.2 possesses lower water uptake than the pellet of UiO-66.
Conclusions
The new strategy for fabrication of MOF–CGCs presented here requires lower temperatures than previous methods. The work on UiO-66 also highlights the need for more in-depth studies on the thermal stability of crystalline MOFs,42 in order to fully understand mechanisms of thermal collapse. The fabrication of CGCs containing UiO-66 and ZIF-67 are however evidence of the wider applicability of the synthetic route proposed here, as previous attempts resulted in the thermal decomposition of the crystalline component or dissolution within the glass matrix. Further suitable crystalline MOF candidates might also be identified using a recent review on the thermal stability of MOFs, which lists temperatures of decomposition of several MOF families.42The lower temperatures also lead to better preservation of porosity within the formed CGCs toward N2. The thermal expansion of a MOF–glass was investigated for the first time and found to be intermediate between inorganic and organic polymers – showing that it is not just the chemical properties of MOFs which are hybrid in nature. A significant improvement in thermo-mechanical properties and chemical stabilities of the CGCs compared with pellets of the pure crystalline MOFs was noted, and bulk monolithic CGCs of the type fabricated here may show promising applications in membrane separation and photocatalysis.
Methods
Synthesis
Fabrication of bulk samples
The agZIF-62 powders were fabricated by heating ZIF-62 under Ar to 450 °C at a ramping rate of 20 °C min−1, without isothermal segment, in a tube furnace, and then naturally cooling to room temperature. The agZIF-62 powders were then ball milled along with 2 × 7 mm diameter stainless steel balls at 25 Hz for 5 min to homogenize the particle size.The bulk agZIF-62 pellet was fabricated by remelting of agZIF-62 powders.23 Specifically, 150 mg of agZIF-62 powder was pressed into a pellet with a diameter of 13 mm under 10 tons for 1 min, and then heated under Ar to 400 °C at a ramping rate of 20 °C min−1 and held for 5 hours in a tube furnace, followed by naturally cooling to room temperature.
The bulk ZIF-67 pellet was fabricated by pressing 150 mg evacuated ZIF-67 powder under 10 tons for 1 min. The bulk UiO-66 pellets were fabricated by pressing 150 mg evacuated UiO-66 powder under 2 tons and 10 tons at first, though the loss of porosity was significant due to the bond breakage of UiO-66 after compression. Thus, the bulk UiO-66 pellet was fabricated by pressing 150 mg of unevacuated UiO-66 powder under 2 tons for 1 min.
A series of MOF CGCs with different mass fraction, e.g. 20 wt%, 50 wt% and 80 wt%, of ZIF-67 or UiO-66 were prepared. The MOF crystal and agZIF-62 powders were mixed by grinding in a mortar for 5 min. Subsequently, the 150 mg mixture was pressed at 10 tons (for ZIF-62 containing mixture) or 2 tons (for UiO-66 containing mixture) for 1 min, and then heated under Ar to 400 °C at a ramping rate of 20 °C min−1 and held for 5 hour in a tube furnace, followed by naturally cooling to room temperature.
Powder X-ray diffraction (XRD) analysis
Room temperature powder XRD analysis (2θ = 5° to 40°) were collected with a Bruker-AXS D8 diffractometer using Cu Kα (λ = 1.540598 Å) radiation and a LynxEye position-sensitive detector in Bragg–Brentano parafocusing geometry. The 2θ step size was 0.02°, with 1 s per step. Pawley refinements were performed using TOPAS-Academic Version 6.43Synchrotron X-ray total scattering and pair distribution function analysis
X-ray total scattering data were collected on the I15-1 beamline at the Diamond Light Source, UK using an X-ray wavelength of 0.161669 Å (76.7 keV). Samples were loaded into borosilicate glass capillaries of 1.17 mm (inner) diameter. Data from the samples, empty instrument and empty capillary were collected in the region of ∼0.4<Q<∼26 Å−1 and processed using the GudrunX software.44,45
Scanning electron microscopy (SEM) and energy dispersive X-ray spectrometer (EDS)
The surface morphologies and element mapping of samples were observed by using a high-resolution scanning electron microscope, FEI Nova Nano SEM 450 equipped with a Bruker silicon drift EDS spectrometer. All samples were coated with gold prior to imaging.Scanning transmission electron microscopy (STEM)
Analytical STEM was used to perform crystallinity mapping based on scanning electron diffraction (SED) and compositional mapping based EDS. Data was acquired using a JEOL ARM300F at the Diamond Light Source, UK fitted with a high-resolution pole piece, cold field emitter, and JEOL spherical aberration correctors in both the probe forming and image forming optics. The instrument was operated at 200 kV and aligned in an uncorrected nanobeam configuration and using a 10 μm condenser aperture to obtain a convergence semi-angle of <1 mrad and a diffraction limited probe diameter of ca. 5 nm. Data was acquired with a scan step size of ca. 5.2 nm and a camera length of 20 cm. The probe current was ca. 2 pA. A Merlin-medipix direct electron detector, which is a counting type detector, was used to record the electron diffraction pattern at each probe position with an exposure time of 1 ms per probe position leading to a total electron fluence of ca. 5 e Å−2 based on the probe current, exposure time, and assuming a disk-like probe of the diameter above. SED data was acquired over a raster scan comprising 256 × 256 probe positions and each diffraction pattern comprised 512 × 512 pixels. EDS maps were acquired from the same regions, following SED acquisition, using a larger probe current, obtained using a 100 μm condenser aperture, to generate sufficient X-ray counts.SED data were processed using an open source Python library, pyxem-0.10.0,46 to find diffraction peaks in every measured diffraction pattern using a difference of Gaussians method, which involves subtracting a blurred version of the diffraction pattern from a less blurred version of the diffraction pattern. EDS data were processed using the open-source HyperSpy Python library to produce maps for each X-ray emission line of interest (Zr Kα, Zn Kα), which were extracted by integrating an energy window and background subtracted by linear interpolation from adjacent regions of the spectrum without other X-ray peaks present.47
Gas adsorption
N2 gas adsorption isotherm measurements were conducted on a Micromeritics ASAP 2020 instrument. Around 50 mg sample was used for each measurement. All samples were degassed at 200 °C for 6 hours prior to the adsorption/desorption test.Thermomechanical analysis (TMA)
TMA was performed by using TMA Q400 system of TA instruments at heating rate of 5 °C min−1 under 0.05 N static force in the temperature range from 30 °C to 400 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TDB thanks the Royal Society for both a University Research Fellowship (UF150021) and a research grant (RSG\R1\180395). TDB also gratefully acknowledges the EPSRC (EP/R015481/1), and the University of Canterbury Te Whare Wānanga o Waitaha, New Zealand, for a University of Cambridge Visiting Canterbury Fellowship. TDB and LNM thank the Leverhulme Trust for a Philip Leverhulme Prize. AFS acknowledges the EPSRC for a PhD studentship award under the industrial CASE scheme, along with Johnson Matthey PLC (JM11106). SMC acknowledges support from the Henslow Research Fellowship at Girton College, Cambridge. CWA would like to thank the Royal Society for a PhD studentship (RG160498) and the Commonwealth Scientific and Industrial Research Council for additional support (C2017/3108). PAM was supported by the EPSRC(EP/R008779/1). We extend our gratitude to Diamond Light Source, Rutherford Appleton Laboratory, U.K., for access to beamline I15-1 (EE20038) and for access and support in the use of the electron Physical Sciences Imaging Centre (MG22632, EM21979). SL acknowledges Natural Science Foundation of China (No. 21606212) and China Scholarship Council (CSC).Notes and references
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- M. Kondo, T. Yoshitomi, H. Matsuzaka, S. Kitagawa and K. Seki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1727 CrossRef CAS.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS.
- M. S. Denny, J. C. Moreton, L. Benz and S. M. Cohen, Nat. Rev. Mater., 2016, 1, 16078 CrossRef CAS.
- Z. Su, Y.-R. Miao, S.-M. Mao, G.-H. Zhang, S. Dillon, J. T. Miller and K. S. Suslick, J. Am. Chem. Soc., 2015, 137, 1750–1753 CrossRef CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- T. Faust, Nat. Chem., 2016, 8, 990 CrossRef CAS.
- C. Zhang, C. Han, D. S. Sholl and J. R. Schmidt, J. Phys. Chem. Lett., 2016, 7, 459–464 CrossRef CAS.
- R. Han, N. Tymińska, J. R. Schmidt and D. S. Sholl, J. Phys. Chem. C, 2019, 123, 6655–6666 CrossRef CAS.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef.
- T. Tian, Z. Zeng, D. Vulpe, M. E. Casco, G. Divitini, P. A. Midgley, J. Silvestre-Albero, J.-C. Tan, P. Z. Moghadam and D. Fairen-Jimenez, Nat. Mater., 2018, 17, 174–179 CrossRef CAS.
- T. Kitao, Y. Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- W. J. Koros and C. Zhang, Nat. Mater., 2017, 16, 289 CrossRef CAS.
- G. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, eaab0530 CrossRef.
- S. Yu, S. Li, S. Huang, Z. Zeng, S. Cui and Y. Liu, J. Membr. Sci., 2017, 540, 155–164 CrossRef CAS.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- T. D. Bennett, J.-C. Tan, Y. Yue, E. Baxter, C. Ducati, N. J. Terrill, H. H. M. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 CrossRef CAS.
- S. S. Nagarkar, H. Kurasho, N. T. Duong, Y. Nishiyama, S. Kitagawa and S. Horike, Chem. Commun., 2019, 55, 5455–5458 RSC.
- S. Li, R. Limbach, L. Longley, A. A. Shirzadi, J. C. Walmsley, D. N. Johnstone, P. A. Midgley, L. Wondraczek and T. D. Bennett, J. Am. Chem. Soc., 2019, 141, 1027–1034 CrossRef CAS.
- A. Qiao, T. D. Bennett, H. T. Tao, A. Krajnc, G. Mali, C. M. Doherty, A. W. Thornton, J. C. Mauro, G. N. Greaves and Y. Z. Yue, Sci. Adv., 2018, 4, eaao6827 CrossRef.
- J. Hou, C. W. Ashling, S. M. Collins, A. Krajnc, C. Zhou, L. Longley, D. N. Johnstone, P. A. Chater, S. Li, M.-V. Coulet, P. L. Llewellyn, F.-X. Coudert, D. A. Keen, P. A. Midgley, G. Mali, V. Chen and T. D. Bennett, Nat. Commun., 2019, 10, 2580 CrossRef.
- L. Longley, S. M. Collins, S. Li, G. J. Smales, I. Erucar, A. Qiao, J. Hou, C. M. Doherty, A. W. Thornton, A. J. Hill, X. Yu, N. J. Terrill, A. J. Smith, S. M. Cohen, P. A. Midgley, D. A. Keen, S. G. Telfer and T. D. Bennett, Chem. Sci., 2019, 10, 3592–3601 RSC.
- S. M. Collins, K. E. MacArthur, L. Longley, R. Tovey, M. Benning, C.-B. Schönlieb, T. D. Bennett and P. A. Midgley, APL Mater., 2019, 7, 091111 CrossRef.
- M. Gustafsson and X. Zou, J. Porous Mater., 2013, 20, 55–63 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- Z. Su, Y.-R. Miao, G. Zhang, J. T. Miller and K. S. Suslick, Chem. Sci., 2017, 8, 8004–8011 RSC.
- Z. Su, Y. R. Miao, S. M. Mao, G. H. Zhang, S. Dillon, J. T. Miller and K. S. Suslick, J. Am. Chem. Soc., 2015, 137, 1750–1753 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- A. D. Wiersum, E. Soubeyrand-Lenoir, Q. Yang, B. Moulin, V. Guillerm, M. B. Yahia, S. Bourrelly, A. Vimont, S. Miller, C. Vagner, M. Daturi, G. Clet, C. Serre, G. Maurin and P. L. Llewellyn, Chem.–Asian J., 2011, 6, 3270–3280 CrossRef CAS.
- D. Dubbeldam, K. S. Walton, D. E. Ellis and R. Q. Snurr, Angew. Chem., Int. Ed., 2007, 46, 4496–4499 CrossRef CAS.
- L. B. d. Bourg, A. U. Ortiz, A. Boutin and F.-X. Coudert, APL Mater., 2014, 2, 124110 CrossRef.
- S. J. Baxter, A. Schneemann, A. D. Ready, P. Wijeratne, A. P. Wilkinson and N. C. Burtch, J. Am. Chem. Soc., 2019, 141, 12849–12854 CrossRef CAS.
- R. N. Widmer, G. I. Lampronti, B. Kunz, C. Battaglia, J. H. Shepherd, S. A. T. Redfern and T. D. Bennett, ACS Appl. Nano Mater., 2018, 1, 497–500 CrossRef CAS.
- R. A. Orwoll, Densities, Coefficients of Thermal Expansion, and Compressibilities of Amorphous Polymers, Springer New York, New York, NY, 2007 Search PubMed.
- J. E. Shelby, Introduction to Glass Science and Technology, The Royal Society of Chemistry, 2005 Search PubMed.
- J. C. Mauro, C. S. Philip, D. J. Vaughn and M. S. Pambianchi, Int. J. Appl. Glass Sci., 2014, 5, 2–15 CrossRef.
- C. Healy, K. M. Patil, B. H. Wilson, L. Hermanspahn, N. C. Harvey-Reid, B. I. Howard, C. Kleinjan, J. Kolien, F. Payet, S. G. Telfer, P. E. Kruger and T. D. Bennett, Coord. Chem. Rev., 2020, 419 Search PubMed.
- A. Coelho, TOPAS-Academic (Version 6), Coelho Software, Brisbane, 2016 Search PubMed.
- A. K. Soper and E. R. Barney, J. Appl. Crystallogr., 2011, 44, 714–726 CrossRef CAS.
- A. K. Soper, Sci. Technol. Facil. Counc. Tech. Rep., RAL-TR-2011-013, 2011 Search PubMed.
- D. N. Johnstone, P. Crout, J. Laulainen, S. Høgås, B. Martineau, T. Bergh, S. Smeets, S. Collins, J. Morzy, H. Ånes, E. Prestat, T. Doherty, T. Ostasevicius, M. Danaie and R. Tovey, Pyxem/Pyxem: Pyxem 0.10.0, Zenodo, 2019, DOI:10.5281/zenodo.3533653.
- F. de la Pena, T. Ostasevicius, V. Tonaas Fauske, P. Burdet, P. Jokubauskas, M. Nord, M. Sarahan, E. Prestat, D. N. Johnstone, J. Taillon, u. Jan Caron, T. Furnival, K. E. MacArthur, A. Eljarrat, S. Mazzucco, V. Migunov, T. Aarholt, M. Walls, F. Winkler, G. Donval, B. Martineau, A. Garmannslund, L.-F. Zagonel and I. Iyengar, Microsc. Microanal., 2017, 23, 214–215 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction, electron microscopy, gas sorption and chemical stability measurements. See DOI: 10.1039/d0sc04008h |
| This journal is © The Royal Society of Chemistry 2020 |