
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTIPS-pentacene triplet exciton generation on PbS quantum dots results from indirect sensitization†
Christopher M.
Papa
 a,
Sofia
Garakyaraghi
a,
Devin B.
Granger
b,
John E.
Anthony
b and
Felix N.
Castellano
*a
a,
Sofia
Garakyaraghi
a,
Devin B.
Granger
b,
John E.
Anthony
b and
Felix N.
Castellano
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695-8204, USA. E-mail: fncastel@ncsu.edu
bDepartment of Chemistry, University of Kentucky, Lexington, Kentucky 40506-0055, USA
First published on 18th May 2020
Abstract
Many fundamental questions remain in the elucidation of energy migration mechanisms across the interface between semiconductor nanomaterials and molecular chromophores. The present transient absorption study focuses on PbS quantum dots (QDs) of variable size and band-edge exciton energy (ranging from 1.15 to 1.54 eV) post-synthetically modified with a carboxylic acid-functionalized TIPS-pentacene derivative (TPn) serving as the molecular triplet acceptor. In all instances, selective excitation of the PbS NCs at 743 nm leads to QD size-dependent formation of an intermediate with time constants ranging from 2–13 ps, uncorrelated to the PbS QD valence band potential. However, the rate constant for the delayed formation of the TPn triplet excited state markedly increases with increasing PbS conduction band energy, featuring a parabolic Marcus free energy dependence in the normal region. These observations provide evidence of an indirect triplet sensitization process being inconsistent with a concerted Dexter-like energy transfer process. The collective data are consistent with the generation of an intermediate resulting from hole trapping of the initial PbS excited state by midgap states, followed by formation of the TPn triplet excited state whose rate constant and yield increases with decreasing quantum dot size.
Introduction
Inorganic–organic hybrid materials constructed from semiconductor nanocrystals (NCs) with appended chromophoric molecules have found increasingly widespread use for a number of light-harvesting and light-emitting functions.1–3 Most of these photonic processes rely on formally spin-triplet excited states, which are generally long-lived in organic molecules owing to their singlet ground states making radiative transitions spin-forbidden while possessing relatively low intrinsic spin–orbit coupling.4,5 Meanwhile, triplet excited states are more easily accessed in inorganic semiconductor NCs due to their ill-defined (nearly isoenergetic) spin states following light absorption.6 The earliest reports of triplet exciton transfer in such hybrid materials incorporated PbS or PbSe NCs coated with a thin film of tetracene or pentacene. These constructs relied on singlet fission7,8 to generate triplet excitons in these molecular films that ultimately transferred to the NC triplet acceptors.9,10 Subsequent to these reports, our laboratory discovered triplet–triplet energy transfer occurring in the reverse direction in toluene solutions featuring selectively photoexcited CdSe NCs with surface-anchored anthryl or pyrenyl molecules.11 This investigation provided clear cut experimental evidence for direct Dexter-type energy transfer occurring across the CdSe quantum dot–molecule interface.12However, PbS quantum dots, while unequivocally generating molecular triplet excitons following direct excitation of the NC,13–16 have proven more mechanistically complex with respect to CdSe. Of particular importance was the identification of an unusual photogenerated reaction intermediate that appeared when 6,13-bis(triisopropylsilylethynyl)pentacene-2-carboxylic acid (TPn),13 a closely related structural derivative of TIPS-pentacene, was anchored to the surface of PbS NCs. This observation was attributed to a sluggish hole trapping process that produced a charge separated state between PbS(e−) and TPn˙+ and upon recombination generated the triplet excited state of TPn. A more recent account offered for PbS quantum dots' spectroscopically-observed deviation from traditional Dexter-type direct TTET relied on quantum chemical calculations to implicate a theoretical NC surface-localized excited state as the kinetic intermediate.16
To continue the mechanistic analysis of this hybrid system, four sizes of PbS NC were synthesized. Upon post-synthetic modification of the PbS NC surfaces with TPn, the sub-picosecond to supra-nanosecond dynamics of these systems were studied using transient absorption spectroscopy. Kinetic analysis of the experimental data indicates the conservation of a stepwise energy transfer mechanism from PbS NCs to TPn, irrespective to the size or band-gap energy of the NC. The rates of the second step in the stepwise mechanism suggest Marcus free energy dependence in the normal region. These results will be rationalized within the context of surface-trapped holes mediating triplet energy transfer.
Experimental
Nanomaterial synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 X-type ligand exchange15 with the oleate ligands. The NC-TPn hybrids were then purified by successive precipitation-centrifugation-resuspension and filtration steps. The average NC:TPn ratio was determined via UV-vis absorption spectroscopy. These values were found to be 1:20.7 for PbS-805-TPn, 1:33.2 for PbS-970-TPn, 1:42.3 for PbS-1000-TPn, and 1:53.9 for PbS-1075-TPn for time resolved spectroscopy samples.
:1 X-type ligand exchange15 with the oleate ligands. The NC-TPn hybrids were then purified by successive precipitation-centrifugation-resuspension and filtration steps. The average NC:TPn ratio was determined via UV-vis absorption spectroscopy. These values were found to be 1:20.7 for PbS-805-TPn, 1:33.2 for PbS-970-TPn, 1:42.3 for PbS-1000-TPn, and 1:53.9 for PbS-1075-TPn for time resolved spectroscopy samples.
Time-resolved spectroscopic methods
Three DADS and an additional fitting parameter could model the complete ultrafast TA spectral datasets for all PbS-TPn materials studied. The additional fitting parameter, resembling a fourth DADS, is included in the model to account for a nonlinear response feature from the toluene occurring coincidentally with the impulse response function (IRF). The spectra produced by the model appear in Fig. 3a–d. Additionally, the fits obtained from the model are compared to the experimental data at the end of the ESI (Fig. S12–S19).† The PbS-TPn TA data was fit separately for the individual white light continua in the visible and NIR, respectively, and the corresponding rate constants were found to be in excellent agreement. The kinetic rate constants extracted from the model have been used to interpret the experimental data (Table S3†).
Results and discussion
The four sizes of PbS NC that were synthesized are displayed in Fig. 1a and b. Consistent with quantum-confined semiconductor nanomaterials, the lowest energy PbS NC exciton absorbance maximum shifts to higher energies with decreasing PbS NC average diameter,24–27 supported by analysis of TEM images (Fig. S1†). The wavelength of maximum photoluminescence intensity shifts in concert with the maximum absorbance wavelength in the first exciton band. The PbS NCs possess photoluminescence intensity maxima near 805, 970, 1000, and 1075 nm, and will be referred to as PbS-805, PbS-970, PbS-1000, and PbS-1075.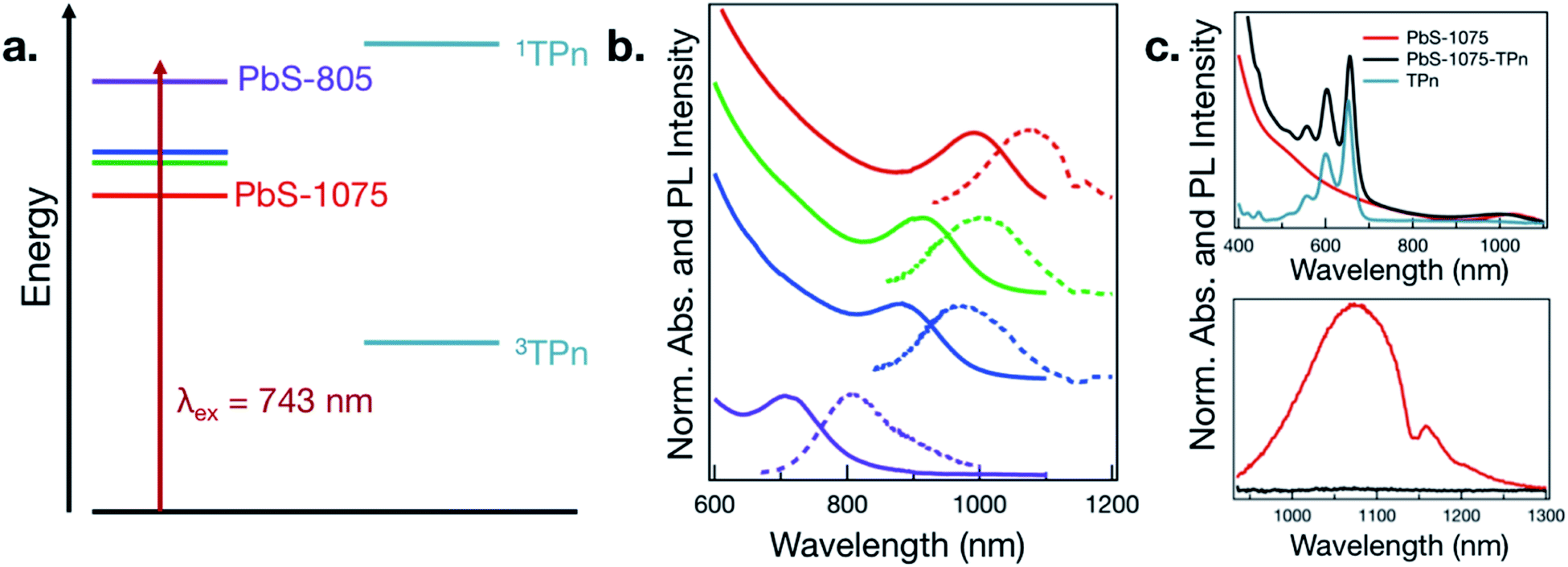 | ||
| Fig. 1 (a) Illustration of PbS nanocrystal first exciton and TPn-centered energy levels. (b) Electronic absorption (solid lines) and emission spectra (dashed lines) for PbS NCs ranging in diameter from 3.3 nm (top, red) to 2.2 nm (bottom, violet). (c) Electronic absorption spectra (top) in toluene of PbS-1075 nanocrystals before and after ligand exchange by TPn with free TPn shown, and photoluminescence spectra in toluene (bottom) of PbS-1075 and PbS-1075-TPn. The depression in the photoluminescence near 1150 nm results from toluene absorption. | ||
As a representative example of the change in electronic effects upon surface-functionalization, the absorption and photoluminescence spectra for PbS-1075 before and after ligand exchange of oleate to TPn and subsequent purification are given in Fig. 1c. The absorption spectrum of free TPn is included for reference. The corresponding spectra for all other hybrid materials subjected to TA spectroscopy are presented in Fig. S2.† While the colloidal NC ligand exchange process and its specific effects in the context of time-resolved optical spectroscopy has been discussed at length elsewhere,11,13–16,19–21 a subtle spectral feature is noteworthy here. In all four discrete materials investigated, a red shift of the TPn absorption bands was consistently observed, often accompanied by a blue shift of the PbS first exciton band. TEM images taken after ligand exchange (Fig. S3†) showed no change in PbS NC average particle size. In the PbS-805 hybrid materials, spectral congestion from the energetic similarity between the PbS first exciton absorbance band and the TPn absorbance band prohibits definitive assignment of the concomitant PbS absorption blue shift. These shifts are reminiscent of NC-molecule electronic wavefunction interactions that occur in strongly quantum-confined materials.13,28
TA spectroscopy was utilized to interrogate the excited state dynamics occurring in the oleate-capped PbS NCs. Fig. 2a displays time dependent TA difference spectra for PbS-805 suspended in toluene using a visible light probe (430–780 nm) following 743 nm pulsed laser excitation (0.15 μJ per pulse, 100 fs fwhm, 0.8 excitations per NC). This excitation wavelength was chosen because it is absorbed by all four NC species but not by TPn, therefore allowing all hybrid materials to achieve selective PbS excitation at a single wavelength. Pumping a solution of TPn without any PbS NCs at 743 nm results in no discernible TA signals (Fig. S4†), indicating that the PbS NCs are selectively excited at this wavelength. Visible and near-IR (830–1380 nm) TA difference spectra for solutions of PbS-1075, PbS-1000, PbS-970, PbS-805 following 743 nm pulsed laser excitation are presented in Fig. S5,† All four PbS species display a prompt, characteristic negative polarity absorption feature, associated with bleaching of their first exciton absorption band. PbS-805 is instructive to illustrate the general excited-state spectral signatures of PbS NCs. This is because the negative polarity TA signal for the PbS-805 first exciton bleach appears within the same visible region of the electromagnetic spectrum as the other notable PbS-TPn excited state features. These PbS-TPn features consist of the broad induced absorption band characteristic of interband transitions in the excited PbS NCs29–32 and some notable TPn transient spectral features. Over the maximum probe delay of the ultrafast TA measurements (6.2 ns), all four PbS TA difference spectra remain mostly unchanged. Nanosecond TA experiments (Fig. S6 and S7†) feature symmetric decay of PbS excited state spectral features on a 1–2 μs time scale. These results are consistent with extant knowledge of the excited state dynamics of carboxylate-capped PbS NCs.13,15,16,32–45
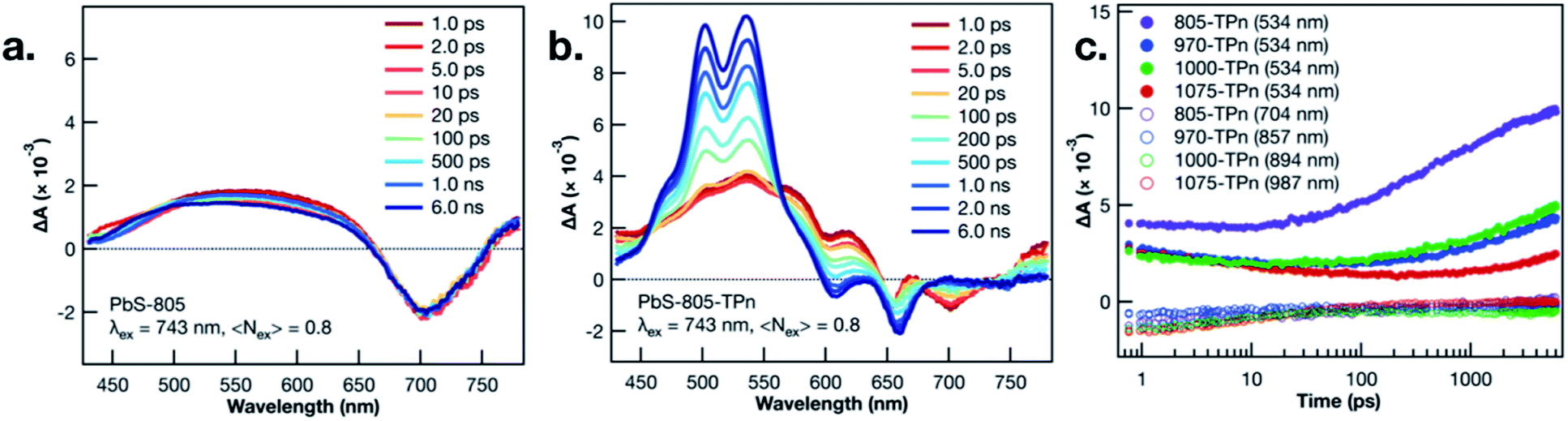 | ||
| Fig. 2 Ultrafast TA difference spectra of PbS-805 (a) and PbS-805-TPn (b) suspensions in toluene following 743 nm pulsed laser excitation (0.15 μJ per pulse, 100 fs fwhm). Time delays range from 1.0 ps (red) to 6.0 ns (blue). Ultrafast TA kinetics (c) probed at 534 nm (closed circles) and different PbS first exciton bleach wavelengths (open circles) for PbS-805-TPn (purple), PbS-970-TPn (blue), PbS-1000-TPn (green), and PbS-1075-TPn (red) suspensions in toluene following 743 nm pulsed laser excitation (0.15 μJ per pulse, 100 fs fwhm). | ||
Following PbS NC surface functionalization with TPn molecules, the TA difference spectra and kinetics following selective excitation of the NCs are changed significantly (Fig. 2b and S8†). Upon pulsed laser excitation at 743 nm, the initially formed excited state resembles the excited state of the oleate-capped PbS NCs with the added presence of three negative absorption features at 550, 600, and 650 nm superimposed on the broad PbS excited state. The location of the negative features implies a relationship to the ground state absorption spectrum of TPn. These negative features have been previously assigned13 and independently reproduced in a closely related study16 as a transient dipole-induced shift of the S0 to S1 electronic transitions in TPn.46–50 In PbS-1075-TPn, the material most analogous to those studied previously,13,16 the first exciton absorption band bleach disappears over the first 250 ps after excitation. Concomitantly, the broad PbS excited state absorbance was observed to symmetrically attenuate. This was followed by the growth of a band centered at 530 nm, previously assigned to the PbS-bound molecular triplet 3TPn.13,16 The molecular triplet develops with a time constant of roughly 8 ns, in stark contrast to the ps-regime decays of the spectral features associated with the PbS NC excited state, but in universal agreement with previous related studies.13,16 The assignments and temporal ordering of the time-resolved spectral features for all PbS NC hybrid materials, regardless of size, are consistent with the same mechanism. This mechanism consists of a decay of the PbS NC excited state on ps time scales to a kinetic intermediate13,16 followed by a nanosecond time scale growth of the TPn excited triplet state.
In all four TPn-modified materials, the time constants for the observed decay of the NIR bleach features were coterminous with the decays of the broad visible absorption features (Table S1†). PbS-805-TPn (Fig. S8a†) is of particular interest to this study because the first exciton bleach is now contained in the region probed by the same visible white light generating crystal. This enables comparison of the dominant 3TPn signal at 534 nm with the PbS NC first exciton bleach at 710 nm. Furthermore, the shift of the bleach wavelength decongests the NIR white light continuum for observation of the 3TPn signal at 994 nm and a characteristic TPn˙+ signal expected near 876 nm.21 The 3TPn signal at 994 nm appears in TA difference spectra for PbS-1000-TPn, PbS-970-TPn, and PbS-805-TPn following 743 nm pulsed laser excitation on time scales matching the growth of the 3TPn feature around 530 nm (Fig. S8b, d and f†). However, the TPn˙+ signal expected near 876 nm is not observed in PbS-1000-TPn or PbS-970-TPn at any time point. There is a feature observed at 876 nm in PbS-805-TPn on the same time scale as the 3TPn features at 530 nm and 994 nm. This kinetic behaviour is indicative that the TPn˙+ formed is unlikely to be acting as the primary kinetic intermediate to 3TPn formation. The decay rates of the characteristic PbS NC excited state features all increased with increasing PbS NC first exciton energy, as did the growth rates of 3TPn (Fig. 2c). The TA difference spectra and kinetics for the PbS-TPn materials on supra-nanosecond time scales (Fig. S9 and S10†) illustrate symmetric decay of the NC-appended 3TPn spectral features on microsecond time scales (Table S2†). The time constants associated with this excited state relaxation process approach the time constant for the relaxation of freely diffusing 3TPn as the PbS NCs decrease in diameter.13 The observed trend in 3TPn lifetime as a function of PbS NC size is consistent with a concentration effect15 induced by the greater number of TPn molecules that can be accommodated by the larger NCs. Temperature-dependent decay kinetics of the 3TPn TA signals (Fig. S11†) indicate the reduction in lifetime is unlikely to be a thermally activated excited state process, as previously observed in other NC-molecule materials featuring reverse triplet–triplet energy transfer.19,20 The lack of experimental evidence supporting any reverse TTET occurring at these interfaces remains inconsistent with a concerted Dexter-like process.
In the interest of assigning consistent rate constants to the three excited state processes occurring in each material, we used a spectral decomposition model23 (vide supra) to fit the time-resolved TA difference spectra. The three decay-associated difference spectra (DADS) fit to the model of the PbS-1075-TPn data collected over the first 6.2 ns after pulsed 743 nm excitation are displayed in Fig. 3a. The first DADS (dark red) is reminiscent of the spectrum observed immediately upon selective excitation of the PbS NC. Specifically, it contains a PbS first exciton bleach in the NIR combined with a broadband PbS induced absorption in the visible with the three negative absorption features from the transient dipole-induced TPn absorption shift. Meanwhile, the third DADS (Fig. 3a, pink) is a strong match to the spectrum of the PbS-appended 3TPn species from the final ultrafast TA time delay and the prompt nanosecond TA difference spectra (Fig. S8g, h and S9d†). The first DADS evolves into the second DADS (Fig. 3a, red) with a time constant of 12.6 ps. Subsequently, the second DADS evolves into the third with a time constant of 8.0 ns. Both time constants are in good agreement with single-wavelength kinetic analyses of the experimental data and previously published results.13,16 Consistent with the single wavelength kinetic analyses, three sequential DADS model the TA difference spectra in every PbS-TPn species investigated here. The sub-microsecond time constants for the PbS-TPn materials extracted from the model are summarized in Table S3.† The final step to obtain consistent rate constants for the two sub-microsecond excited state processes was to average the rate constants on an average TPn per PbS NC basis, which are also included in Table S3.†
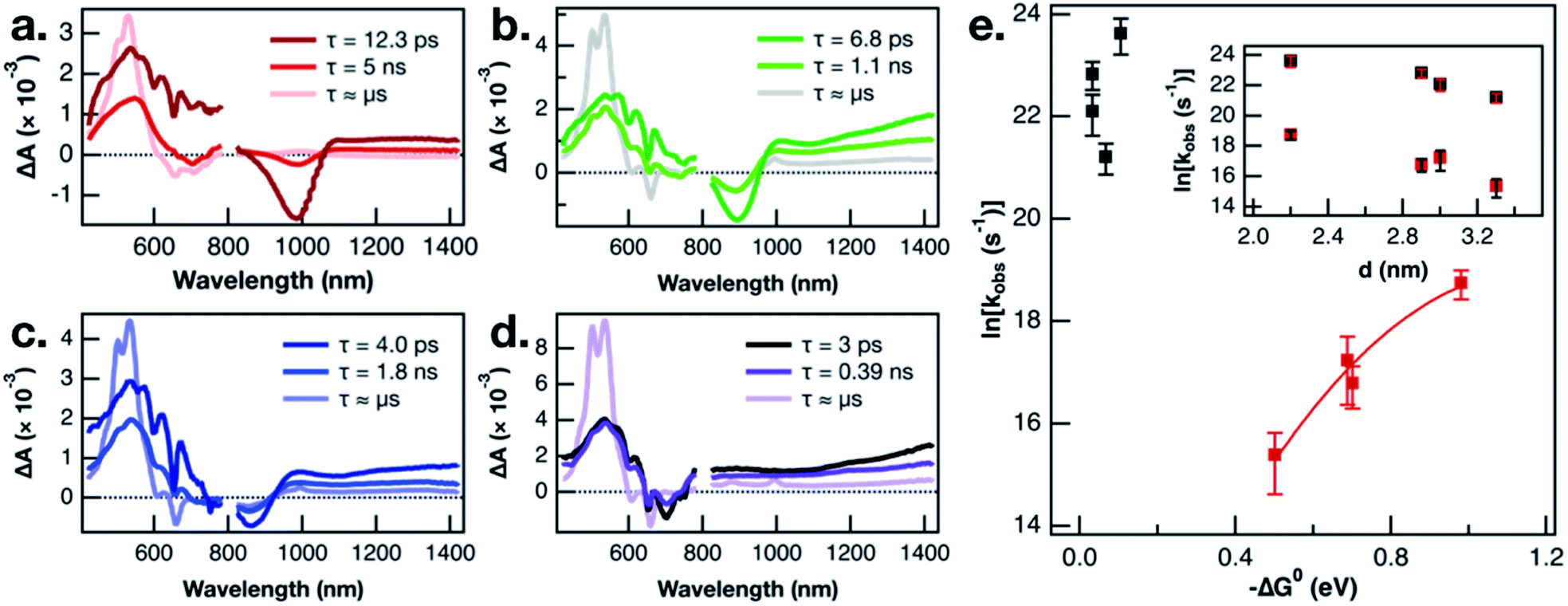 | ||
| Fig. 3 Basis spectra modeled on TA spectra of (a) PbS-1075-TPn, (b) PbS-1000-TPn, (c) PbS-970-TPn, and (d) PbS-805-TPn materials. The ultrafast TA spectra initially resemble the 1st (darkest hues) spectra and eventually come to resemble the 3rd spectra (palest hues) by the end of the 6.2 ns delay stage. The 2nd spectra (intermediate hues) are necessary to accurately model the experimental data. Data from 425 to 775 nm and data from 825 to 1325 nm were fit with matching rate constants. Logarithm of extracted rate constants plotted as a function of thermodynamic driving force (e) and approximate PbS NC diameter ((e), inset). Bars in (e) and ((e), inset) reflect the standard error calculated from three independent measurements. The solid red line in (e) is a Marcus theory fit to the data intended as a phenomenological guide for the eye. | ||
Recent work on CuInS2 NCs has magnified the existence of charge-carrier trapping phenomena in semiconductor NCs, specifically with respect to energy transfer across the NC-molecule interface.51 In the related PbS-acene study by Bender, Raulerson et al., electronic structure calculations provide a rationale for the existence of surface-localized excited states on the PbS NC.16 A direct comparison of the triplet sensitization of two different acene molecules by CsPbBr3 NCs suggested that electrons and holes could possess vastly different trapping kinetics.52 The calculated PbS NC surface states were previously tentatively assigned to hole-trapping phenomena at the NC surface.16 A plausible next step toward validating that mechanistic assignment would be to study the NC size-dependence of triplet–triplet energy transfer from multiple sizes of PbS NC to pentacene. This is the first such PbS NC to acene triplet–triplet energy transfer NC-size-dependence study to employ either NC-selective excitation or a pentacene derivative as the acceptor. If the kinetic intermediate, which is observed comprehensively across all PbS NC sizes investigated, is a surface hole-trapping phenomenon, that could explain the absence of TPn˙+ TA signals discussed earlier. Furthermore, we propose this mechanism provides a suitable explanation for the size-dependent behaviour of the 3TPn TA signal growth rates displayed in Fig. 3e. The rate constants for formation of the 3TPn state increase with increasing PbS conduction band position, featuring a parabolic Marcus free energy dependence in the normal region. As mentioned previously, the rate constants on the vertical axis in Fig. 3e have been adjusted to reflect the differing surface concentrations of TPn on the different sizes of the PbS NC. However, they do not account for any changes in NC-TPn electronic coupling as a function of PbS NC size. Taking these factors into account, the fit applied to the data herein was intended primarily as a phenomenological guide to the eye. However, the values extracted from the fit in this study are broadly consistent with recently reported data from a PbS QD and tetracene study which also employed a fit to Marcus theory.53 Meanwhile, the rate constants associated with the attenuation of the initial PbS NC excited state are not well-predicted by Marcus theory. As shown in the inset of Fig. 3e, the PbS NC size-dependence of the rate constants for decay of the PbS excited state appears to be directly related to the NC diameter. We suggest the rate constants for this first step deviate from Marcus theory in part because they are intrinsic to the PbS NCs or otherwise independent from the energetics of TPn. The combination of the current size and band gap kinetic dependence data, the recent work on charge carrier trapping,51,52 and the theoretical evidence for surface state mediation,16 supports a hole-trapping mechanism for 3TPn generation from excited PbS NCs. On picosecond time scales, surface state trapping of photogenerated holes quenches the PbS band edge exciton, while 3TPn is directly generated from the hole-trapped PbS NC much more slowly.
Conclusions
We have functionalized a series of PbS NCs with TPn, a derivative of TIPS-pentacene, to generate a suite of hybrid inorganic–organic nanomaterials. We have investigated these nanomaterials from sub-picosecond to supra-nanosecond time scales using transient absorption spectroscopy in the visible and near-IR portions of the electromagnetic spectrum. Selective photoexcitation of PbS NCs produces a kinetic intermediate over picosecond time scales, which decays to yield TPn-localized triplet excitons on nanosecond time scales. This stepwise triplet sensitization process is comprehensive across all PbS NC sizes and band-edge exciton energies investigated. The evidence is demonstrative of stepwise (indirect) sensitization and inconsistent with a concerted Dexter-like process. This work demonstrates the inherent complexity in the interactions between direct energy transfer processes, semiconductor trap states, and stepwise electron transfers. Understanding these interactions is fundamental to successfully govern directional triplet energy transfer or sensitization from semiconductor NCs to surface-appended molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support for this work from the Organic Materials Chemistry program in the Air Force Office of Scientific Research (FA9550-18-1-0331). Electron microscopy was performed at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (Award Number ECCS-1542015). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI).References
- Z. Huang and M. Lee Tang, J. Phys. Chem. Lett., 2018, 9, 6198–6206 CrossRef CAS PubMed
.
- S. Garakyaraghi and F. N. Castellano, Inorg. Chem., 2018, 57, 2351–2359 CrossRef CAS PubMed
- H. Lu, G. M. Carroll, N. R. Neale and M. C. Beard, ACS Nano, 2019, 13, 939–953 CAS
-
N. J. Turro, Modern Molecular Photochemistry, 1991 Search PubMed
- A. Olaya-Castro and G. D. Scholes, Int. Rev. Phys. Chem., 2011, 30, 49–77 Search PubMed
- G. D. Scholes, Adv. Funct. Mater., 2008, 18, 1157–1172 CrossRef CAS
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed
- M. Tabachnyk, B. Ehrler, S. Gelinas, M. L. Boehm, B. J. Walker, K. P. Musselman, N. C. Greenham, R. H. Friend and A. Rao, Nat. Mater., 2014, 13, 1033–1038 CrossRef CAS PubMed
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. Wu, N. Geva, M. Welborn, T. Van Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2014, 13, 1039–1043 CrossRef CAS PubMed
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS
- S. Garakyaraghi, C. Mongin, D. B. Granger, J. E. Anthony and F. N. Castellano, J. Phys. Chem. Lett., 2017, 8, 1458–1463 CrossRef CAS PubMed
- Z. Huang, Z. Xu, M. Mahboub, X. Li, J. W. Taylor, W. H. Harman, T. Lian and M. L. Tang, Angew. Chem., Int. Ed., 2017, 56, 16583–16587 CrossRef CAS PubMed
- D. M. Kroupa, D. H. Arias, J. L. Blackburn, G. M. Carroll, D. B. Granger, J. E. Anthony, M. C. Beard and J. C. Johnson, Nano Lett., 2018, 18, 865–873 CrossRef CAS PubMed
- J. A. Bender, E. K. Raulerson, X. Li, T. Goldzak, P. Xia, T. Van Voorhis, M. L. Tang and S. T. Roberts, J. Am. Chem. Soc., 2018, 140, 7543–7553 CrossRef CAS PubMed
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS
- Z. Li, K. Shankar, G. K. Mor, R. A. Grimminger, C.-M. Lin, J. E. Anthony and C. A. Grimes, J. Photonics Energy, 2011, 1, 011106 CrossRef
- C. Mongin, P. Moroz, M. Zamkov and F. N. Castellano, Nat. Chem., 2018, 10, 225–230 CrossRef CAS PubMed
- M. La Rosa, S. A. Denisov, G. Jonusauskas, N. D. McClenaghan and A. Credi, Angew. Chem., Int. Ed., 2018, 57, 3104–3107 CrossRef CAS PubMed
- H. P. Lu, X. H. Chen, J. E. Anthony, J. C. Johnson and M. C. Beard, J. Am. Chem. Soc., 2019, 141, 4919–4927 CrossRef CAS PubMed
- S. Garakyaraghi, E. O. Danilov, C. E. McCusker and F. N. Castellano, J. Phys. Chem. A, 2015, 119, 3181–3193 CrossRef CAS PubMed
- L. J. G. W. van Wilderen, C. N. Lincoln and J. J. van Thor, PLoS One, 2011, 6, e17373 CrossRef CAS PubMed
- P. Kambhampati, Acc. Chem. Res., 2011, 44, 1–13 CrossRef CAS PubMed
- D. B. Turner, Y. Hassan and G. D. Scholes, Nano Lett., 2012, 12, 880–886 CrossRef CAS PubMed
- K. J. Schnitzenbaumer, T. Labrador and G. Dukovic, J. Phys. Chem. C, 2015, 119, 13314–13324 CrossRef
- M. S. Azzaro, M. C. Babin, S. K. Stauffer, G. Henkelman and S. T. Roberts, J. Phys. Chem. C, 2016, 120, 28224–28234 CrossRef CAS
- H. Zhu, Y. Yang, K. Hyeon-Deuk, M. Califano, N. Song, Y. Wang, W. Zhang, O. V. Prezhdo and T. Lian, Nano Lett., 2014, 14, 1263–1269 CrossRef CAS PubMed
- P. Kambhampati, Chem. Phys., 2015, 446, 92–107 CrossRef CAS
-
V. I. Klimov, in Annual Review of Physical Chemistry, 2007, vol. 58, pp. 635–673 Search PubMed
- B. Cho, W. K. Peters, R. J. Hill, T. L. Courtney and D. M. Jonas, Nano Lett., 2010, 10, 2498–2505 CrossRef CAS PubMed
- Y. Yang, W. Rodriguez-Cordoba and T. Lian, J. Am. Chem. Soc., 2011, 133, 9246–9249 CrossRef CAS PubMed
- H. Du, C. L. Chen, R. Krishnan, T. D. Krauss, J. M. Harbold, F. W. Wise, M. G. Thomas and J. Silcox, Nano Lett., 2002, 2, 1321–1324 CrossRef CAS
- B. L. Wehrenberg, C. J. Wang and P. Guyot-Sionnest, J. Phys. Chem. B, 2002, 106, 10634–10640 CrossRef CAS
- J. H. Warner, E. Thomsen, A. R. Watt, N. R. Heckenberg and H. Rubinsztein-Dunlop, Nanotechnology, 2005, 16, 175–179 CrossRef CAS PubMed
- S. W. Clark, J. M. Harbold and F. W. Wise, J. Phys. Chem. C, 2007, 111, 7302–7305 CrossRef CAS
- Y. Chen, D. Yu, B. Li, X. Chen, Y. Dong and M. Zhang, Appl. Phys. B: Lasers Opt., 2009, 95, 173–177 CrossRef CAS
- M. S. Gaponenko, A. A. Lutich, N. A. Tolstik, A. A. Onushchenko, A. M. Malyarevich, E. P. Petrov and K. V. Yumashev, Phys. Rev. B, 2010, 82, 125320 CrossRef
- K. E. Knowles, M. Malicki and E. A. Weiss, J. Am. Chem. Soc., 2012, 134, 12470–12473 CrossRef CAS PubMed
- Y. Yang, W. Rodriguez-Cordoba and T. Lian, Nano Lett., 2012, 12, 4235–4241 CrossRef CAS PubMed
- Y. Yang, W. Rodriguez-Cordoba, X. Xiang and T. Lian, Nano Lett., 2012, 12, 303–309 CrossRef CAS PubMed
- Y. Yang and T. Lian, Coord. Chem. Rev., 2014, 263, 229–238 CrossRef
- K. O. Aruda, M. B. Kunz, M. Tagliazucchi and E. A. Weiss, J. Phys. Chem. Lett., 2015, 6, 2841–2846 CrossRef CAS PubMed
- H. Chung, H. Choi, D. Kim, S. Jeong and J. Kim, J. Phys. Chem. C, 2015, 119, 7517–7524 CrossRef CAS
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef CAS
- L. Sebastian, G. Weiser and H. Bassler, Chem. Phys., 1981, 61, 125–135 CrossRef CAS
- D. J. Norris, A. Sacra, C. B. Murray and M. G. Bawendi, Phys. Rev. Lett., 1994, 72, 2612–2615 CrossRef CAS PubMed
- G. U. Bublitz and S. G. Boxer, Annu. Rev. Phys. Chem., 1997, 48, 213–242 CrossRef CAS PubMed
- S. Ardo, Y. Sun, F. N. Castellano and G. J. Meyer, J. Phys. Chem. B, 2010, 114, 14596–14604 CrossRef CAS PubMed
- S. Gelinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. van der Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512–516 CrossRef CAS PubMed
- Y. Han, S. He, X. Luo, Y. Li, Z. Chen, W. Kang, X. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 13033–13037 CrossRef CAS PubMed
- X. Luo, Y. Y. Han, Z. W. Chen, Y. L. Li, G. J. Liang, X. Liu, T. Ding, C. M. Nie, M. Wang, F. N. Castellano and K. F. Wu, Nat. Commun., 2020, 11, 28 CrossRef CAS PubMed
- V. Gray, J. R. Allardice, Z. Zhang, S. Dowland, J. Xiao, A. J. Petty, J. E. Anthony, N. C. Greenham and A. Rao, ACS Nano, 2020, 14, 4224–4234 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Structural characterization of the PbS and PbS-TPn nanomaterials and additional transient absorption spectra and kinetic data and analyses. See DOI: 10.1039/d0sc00310g |
| This journal is © The Royal Society of Chemistry 2020 |