
Strapped calix[4]pyrrole as a lithium salts selective receptor through separated ion-pair binding†
Kyeong-Im
Hong
,
Hyeongcheol
Kim
,
Younghun
Kim
,
Moon-Gun
Choi
and
Woo-Dong
Jang
 *
*
Department of Chemistry, Yonsei University, 50 Yonsei-ro, Seodaemun-Gu, Seoul 03722, Korea. E-mail: wdjang@yonsei.ac.kr
First published on 27th July 2020
Abstract
A triazole-bearing strapped calix[4]pyrrole (1) was synthesized as a lithium salt selective ion pair receptor. 1H NMR spectral studies and X-ray crystallography showed that the capture of LiCl by 1 occurs via separated ion pair binding. Furthermore, lithium salts extracted by 1 could be solidified in the form of Li2CO3 through CO2 or K2CO3 treatment.
Lithium is an important element in industrial applications, including lithium-ion batteries, lubricants, and ceramics.1 Currently, most of the available lithium reserves are concentrated in the form of salt flats in a few countries.2 Owing to increasing industrial demand, as well as the limited amount of lithium deposited in salt flats, the price of lithium has greatly increased in recent years. Therefore, finding an alternative lithium source would be of great value. The total amount of Li+ present in seawater is much higher than that in terrestrial reserves. However, the selective extraction of Li+ from seawater is extremely difficult due to the abundance of other cations, such as Na+, K+, and Mg2+.3,4 Recently, ditopic receptors have attracted attention owing to their various applications, including salt solubilization, membrane transport, and as logic gates.4–6 Several types of ion-pair receptors have been designed using calix[4]pyrrole,7–11 calix[4]arene,12,13 and carbazole derivatives14 to capture CsF, KF, LiNO2, and other salts.15 Until now, very few examples of extraction of lithium salts by ion-pair receptors in organic solvents have been reported.16 Most reports of the extraction of lithium salts from aqueous solutions were achieved from aqueous LiNO2 solution.17,18 For the extraction of Li+ from seawater, it would be essential to develop an ion-pair receptor that can extract LiCl, LiBr, and Li2SO4 due to the natural abundance of Cl− (19.35 g L−1), SO42− (2.71 g L−1), and Br− (67 mg L−1).19 However, the development of ion-pair receptors able to extract LiCl or LiBr from aqueous solutions has been challenging. For this reason, Sessler's group reported phenanthroline strapped calix[4]pyrrole as a contact ion-pair receptor for LiCl.20 Recently, we developed triazole-bearing calix[4]pyrroles that showed strong affinities towards halide anions.21 Based on the strong binding affinity of the triazole-bearing calix[4]pyrroles towards halide ions, we designed a new type of ion-pair receptor (1; Scheme 1) with an oligoethylene glycol strap to facilitate the binding of Li+. Through 1H NMR spectral studies, we confirmed the successful binding of LiCl and LiBr to 1 in organic solvent. Although it is unfavorable for LiCl or LiBr extraction through a separated ion-pair binding process due to the high hydration energy of halide ions, we have successfully extracted lithium halide through liquid–liquid extraction using 1. Moreover, lithium salts extracted by 1 could be solidified in the form of Li2CO3 through CO2 or K2CO3 treatment.
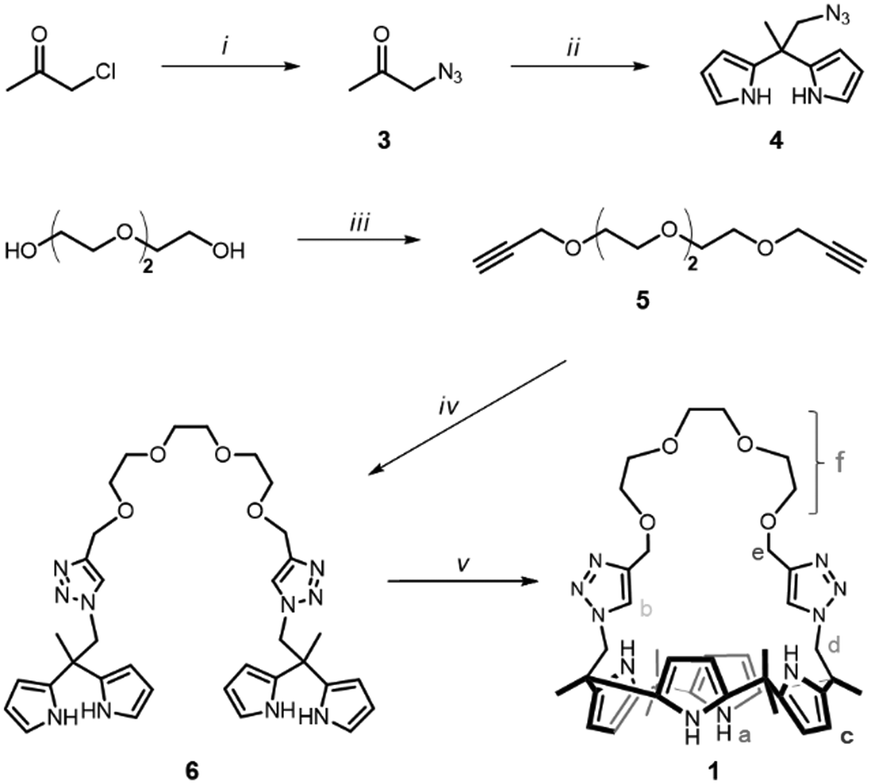 | ||
| Scheme 1 Synthesis procedure of 1. (i) NaN3, acetone; (ii) TFA, pyrrole; (iii) propargyl bromide, NaH, KI, THF; (iv) 4, CuSO4, Na ascorbate, THF/H2O; and (v) BF3·OET2, acetone. | ||
The synthesis of a triazole-bearing oligoether strapped calix[4]pyrrole, 1, is outlined in Scheme 1. 1H NMR observation of 1 in 10% CD3OD/CD2Cl2 was performed upon addition of several salts, such as LiCl, NaCl, and KCl (Fig. 1).
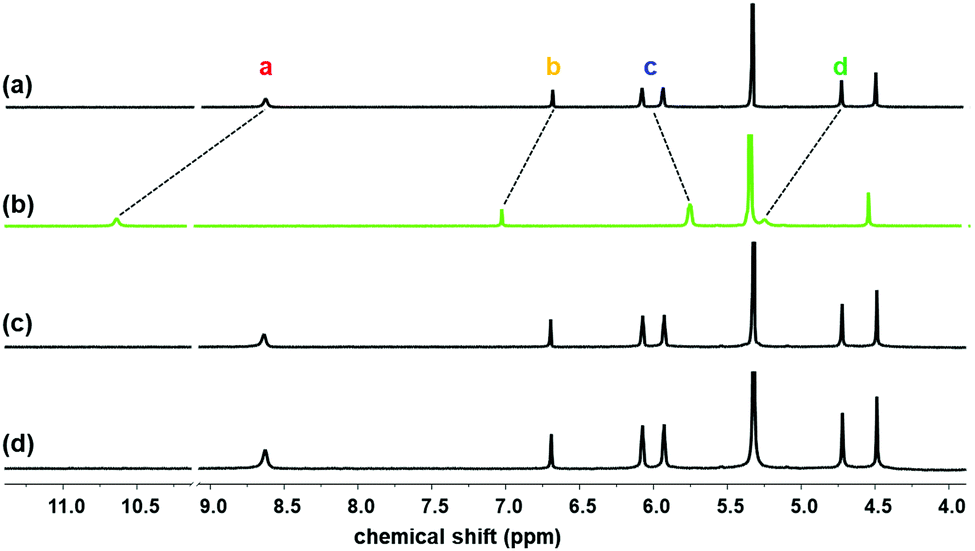 | ||
| Fig. 1 Partial 1H NMR spectra of (a) 1 (2 mM) and 1 with (b) excess LiCl(s), (c) excess NaCl(s), and (d) excess KCl(s) recorded in 10% CD3OD/CD2Cl2. | ||
The 1H NMR spectrum of 1 exhibited large spectral shifts by addition of LiCl, whereas the addition of NaCl or KCl did not cause any spectral changes, indicating the selective binding of LiCl to 1. The pyrrolic N–H (Ha) signal was dramatically downfield shifted from 8.64 to 10.61 (Δδ = −1.97 ppm), indicating the formation of hydrogen bonding with Cl−. The signals of the methylene bridge (Hd) and triazole C–H (Hb) were also downfield shifted from 4.71 and 6.68 to 5.22 and 7.00, respectively (Δδ = −0.51 and −0.32 ppm, respectively). Alternatively, the signals of the pyrrolic beta proton (Hc) were upfield shifted from 6.07–5.93 to 5.73–5.72 due to the structural changes of calix[4]pyrrole to a cone shape.
As 1 exhibited selective binding to LiCl rather than NaCl and KCl, various lithium salts, including LiF, LiCl, LiBr, LiI and LiClO4, were added to 1 to examine the anion binding selectivity (Fig. S1, ESI†). As a result, relatively large 1H NMR spectral changes were observed upon the addition of LiCl and LiBr, indicating that 1 can accommodate both LiCl and LiBr. Therefore, Cl− and Li+ were sequentially added to 1 in the form of tetrabutylammonium (TBA) and ClO4− salts, respectively, and the spectral changes of 1 were monitored to confirm whether they were induced by the binding of LiCl or not. When excess Cl− was added to a solution of 1 in 10% CD3OD/CD2Cl2, the 1H NMR spectral change of 1 differed from the result of LiCl addition (Fig. S2b and d, ESI†). Because Cl− can be tightly bound to calix[4]pyrrole, pyrrolic N–H protons were strongly shifted to 10.94 ppm (Δδ = −2.3 ppm). In the case of the pyrrolic beta protons, the peak appeared as a singlet at 5.67 ppm, indicating the transformation to the cone shaped geometry of the calix[4]pyrrole unit. Subsequently, excess Li+ was added to the solution of 1 with excess Cl−. As a result, the 1H NMR spectrum of 1 with excess Cl− was changed to that with LiCl (Fig. S2c and d, ESI†). Similarly, the sequential addition of Br− and Li+ in the form of tetrabutylammonium (TBA) and ClO4− salts, respectively, to 1, resulted in the same 1H NMR spectrum of 1 with LiBr(s) (Fig. S3, ESI†). These observations again indicated the selective ion-pair binding of 1 for LiCl and LiBr. The association constants of 1 for LiCl and LiBr binding were determined by 1H NMR spectral titrations. From the spectral shift of the 1H NMR signals upon successive addition of LiCl and LiBr, the association constants were estimated to be 60.5 and 19.5 M−1, respectively, as calculated by commercial software HypNMR 2008 (Fig. S4 and S5, ESI†). Although the proton signals of the calix[4]pyrrole unit showed large shifts following addition of LiCl or LiBr, the chemical shifts of the protons in the triethylene glycol units (He and Hf) maintained their initial position while forming 1·LiCl and 1·LiBr complexes. Alternatively, the chemical shifts of He and Hf were relatively large following the addition of Li+ in the form of ClO4− salt, indicating the binding of Li+ to a triethylene glycol unit. From the 1H NMR spectral titration of Li+ to 1 in 10% CD3OD/CD2Cl2, the association constant was estimated to be 4.4 M−1 (Fig. S6, ESI†). As the chemical shifts of He and Hf were not changed by the binding of LiCl or LiBr to 1, we can expect that the binding mode of the ion pairs differs from that of Li+ binding alone. The binding constants of halide ions such as Cl− and Br− were also obtained as 377 and 61 M−1, respectively, through 1H NMR spectral titration (Table S2, ESI†). The binding constants of the anions were greater than that of Li+. Therefore, the binding of Li+ to 1 was enhanced by the participation of halides through conformational changes of 1.
To clarify the binding mode of ion pairs, single crystal X-ray crystallography of 1·LiCl was performed. As shown in Fig. 2, 1 formed a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex with LiCl. Cl− was bound to calix[4]pyrrole units through multiple hydrogen bonds, producing a cone shaped geometry. The C–H proton in the triazole groups also stabilized Cl− binding to the calix[4]pyrrole unit (the distance of C–H⋯Cl was 3.96 Å and 4.01 Å). Interestingly, the crystal structure exhibited binding of Li+ to triazole nitrogen (the distance of Li⋯N was 2.0 Å). Furthermore, three water molecules were coordinated between 1 and LiCl. Within these three water molecules, one was in contact with Li+, another was bound to the triethylene glycol strap (the O⋯H2O distance was 2.4 Å) and the other was bound to Cl− (the Cl⋯H distance was 2.39 Å). The distance between Li+ and Cl− in 1·LiCl was 4.58 Å, which is much larger than the 2.02 Å in LiCl(s). Based on the single crystal structure, computational aided density functional theory (DFT) calculations with Beck's three-parameter hybrid exchange functional and the Lee–Yang–Parr correlation functional (B3LYP), employing the 6-31G(d) basis set in the gas phase were performed for three types of possible binding modes including contact-, separated-, and solvent bridged ion-pairs (Fig. S7, ESI†). The minimized binding energies have been estimated to be −911.06, −815.62, and −1166.70 kJ mol−1 for the three contact-, separated-, and solvent bridged ion-pair models, respectively.
:2 complex with LiCl. Cl− was bound to calix[4]pyrrole units through multiple hydrogen bonds, producing a cone shaped geometry. The C–H proton in the triazole groups also stabilized Cl− binding to the calix[4]pyrrole unit (the distance of C–H⋯Cl was 3.96 Å and 4.01 Å). Interestingly, the crystal structure exhibited binding of Li+ to triazole nitrogen (the distance of Li⋯N was 2.0 Å). Furthermore, three water molecules were coordinated between 1 and LiCl. Within these three water molecules, one was in contact with Li+, another was bound to the triethylene glycol strap (the O⋯H2O distance was 2.4 Å) and the other was bound to Cl− (the Cl⋯H distance was 2.39 Å). The distance between Li+ and Cl− in 1·LiCl was 4.58 Å, which is much larger than the 2.02 Å in LiCl(s). Based on the single crystal structure, computational aided density functional theory (DFT) calculations with Beck's three-parameter hybrid exchange functional and the Lee–Yang–Parr correlation functional (B3LYP), employing the 6-31G(d) basis set in the gas phase were performed for three types of possible binding modes including contact-, separated-, and solvent bridged ion-pairs (Fig. S7, ESI†). The minimized binding energies have been estimated to be −911.06, −815.62, and −1166.70 kJ mol−1 for the three contact-, separated-, and solvent bridged ion-pair models, respectively.
 | ||
| Fig. 2 (a) Side view and (b) top view of the crystal structure of the 2:2:6 complex (1:LiCl:H2O). Minor hydrogen atoms and solvent are omitted for clarity (green: Cl, pink: Li, red: O, grey: C and blue: N). | ||
These theoretical calculations also supported the formation of a water-separated dimeric complex corresponding to the single crystal structure. The result of 2-D NOESY experiments also supported the formation of a dimeric complex (Fig. S8–S10, ESI†). A cross peak between Hd and Hg appeared by the formation of a dimeric complex.
Some ion pair receptors have been explored for the liquid–liquid extraction of lithium salts such as LiNO2 and LiNO3. From a practical perspective, the extraction of halide salts would have great value. To confirm whether the extraction of LiCl is possible or not using 1 from an aqueous to an organic phase through liquid–liquid extraction, 1H NMR spectral studies were conducted with 5.0 mM of 1 diluted in CD2Cl2.
LiCl solution in D2O was added to the solution of 1 and stirred for 1 h. Subsequently, the 1H NMR spectrum of the organic phase was recorded to observe whether spectral changes appeared. The 1H NMR spectrum of 1 exhibited spectral shifts by formation of a 1·LiCl complex, possibly indicating the successful extraction of LiCl from the aqueous phase (Fig. 3b). Several experiments were also performed using 25%, 50%, 80%, and 95% saturated LiCl solutions to determine the minimum amount of LiCl in the aqueous phase for liquid–liquid extraction (Fig. S11, ESI†). 1 succeeded in extracting LiCl with 80% or more saturated LiCl solution. Additionally, the 1H NMR spectra rarely changed when the solution of 1 was exposed to NaCl(aq) and KCl(aq) (Fig. S12b and c, ESI†), indicating the high selectivity of 1 to lithium salt.
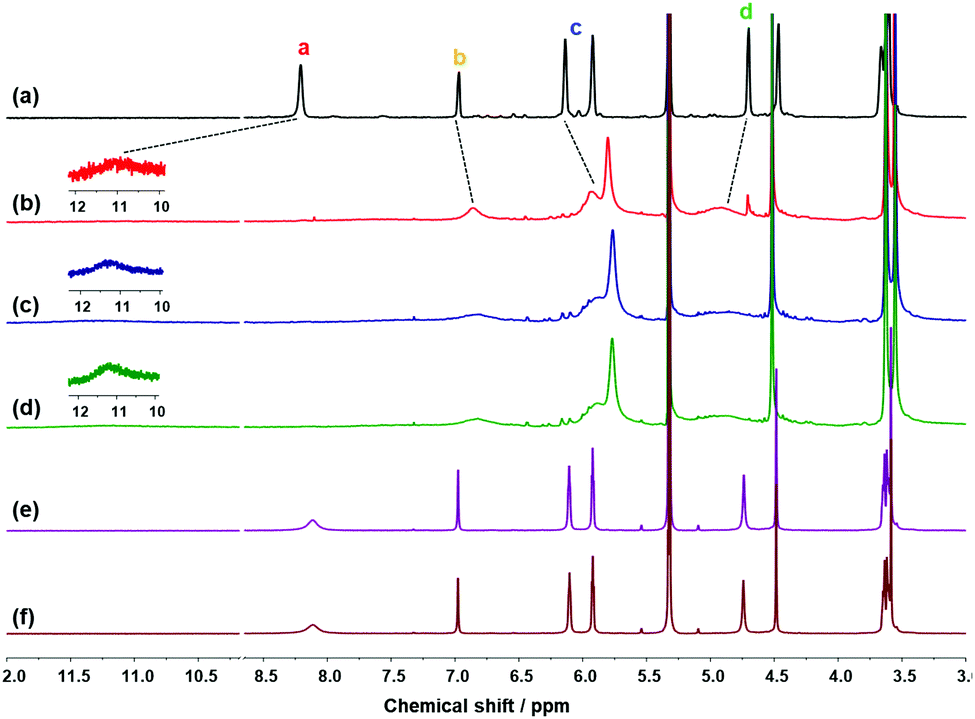 | ||
| Fig. 3 1H NMR spectra of (a) 1 (5 mM) and 1 after extraction with (b) LiCl(aq), (c) LiCl(s), (d) a mixture of LiCl(s), NaCl(s) and KCl(s), (e) excess NaCl(s), and (f) excess KCl(s). | ||
When LiCl(s) was added to the solution of 1 in CD2Cl2 and stirred for 1 h for solid–liquid extraction, the 1H NMR spectrum was consistent with that of 1·LiCl from liquid–liquid extraction (Fig. 3c). For the competition experiments, a mixture of alkali halide(s) containing LiCl(s), NaCl(s), and KCl(s) was added to a solution of 1. As a result, 1 exhibited selective extraction of LiCl(s) (Fig. 3d). The 1H NMR spectra obtained after solid–liquid extraction with NaCl(s) or KCl(s) (Fig. 3e and f, respectively) were identical with those of 1. The extraction experiments with LiBr were also examined in a similar way to the LiCl extraction experiments.
All spectral changes exhibited the successful liquid–liquid and solid–liquid extraction of LiBr (Fig. S13, ESI†). Competition experiments using mixtures of LiBr(s), NaBr(s), and KBr(s) also resulted in the selective formation of host–guest complexes between 1 and LiBr (Fig. S13, ESI†).
For practical applications, the precipitation of lithium salt was attempted after extraction of LiCl by 1. As Li2CO3 has relatively poor solubility in both organic solvents and water, CO32− was added to the host–guest complexes between 1 and LiCl to obtain a Li2CO3 precipitate. For example, the organic phase was collected after the extraction of LiCl, and a small amount of deionized water was added to the solution. Then, CO2(g) was bubbled through the solution to obtain a carbonate solution. As a result, the organic layer became turbid due to the formation of Li2CO3 (Fig. 4). Through modification of the Warder titration method which is known to be a way to calculate the amount of sodium carbonate in a NaOH solution, the amount of Li2CO3 precipitated by 1 was measured through acid–base titration using methyl orange as a pH indicator (Fig. S14 and S15, ESI†). Compared with deionized water, the volume of 0.05 M HCl needed more than deionized water. The formation of Li2CO3 was also confirmed by direct addition of K2CO3. After the liquid–liquid extraction of LiCl(aq) using 100 mg mL−1 of 1 diluted in CH2Cl2, the organic layer was separated from the aqueous layer. Upon addition of K2CO3, the organic layer containing the 1·LiCl complex, became turbid (Fig. S16, ESI†). The extracted Li2CO3 was also confirmed by scanning electron microscopy energy dispersive X-ray spectroscopy (SEM/EDXS) mapping experiments (Fig. S17, ESI†).
 | ||
| Fig. 4 (a) Turbidity and (b) the Tyndall effect of a solution containing the 1·LiCl complex after CO2(g) bubbling with deionized water. | ||
In summary, we synthesized a triazole-bearing oligoether strapped calix[4]pyrrole 1 as a lithium selective ion pair receptor. Through 1H NMR spectral studies, 1 demonstrated selective binding of both LiCl and LiBr in 10% MeOD/CD2Cl2. The binding mode between 1 and LiCl was confirmed by using single crystal X-ray crystallography, which was further supported by theoretical calculations. LiCl was captured by 1 through separated ion-pair binding assisted by three water molecules. Furthermore, we performed liquid–liquid extraction of LiCl and LiBr using 1 to bring the lithium salts into the organic phase. The 1H NMR study indicated successful extraction of LiCl and LiBr into the organic phase through ion pair binding to 1. Moreover, lithium salts extracted by 1 could be isolated in the form of Li2CO3. From a practical point of view, the precipitation of Li2CO3 using K2CO3 and CO2(g) could be very important.
This work was supported by the Mid-Career Researcher Program (2017R1A2A1A17069537 and 2020R1A2C3004520) funded by the National Research Foundation of Republic of Korea, and the authors acknowledge the Pohang Accelerator Laboratory (PAL) for beamline use (2018-1st-2D-007).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Martin, L. Rentsch, M. Höck and M. Bertau, Energy Storage Mater., 2017, 6, 171–179 CrossRef.
- H. Vikström, S. Davidsson and M. Höök, Appl. Energy, 2013, 110, 252–266 CrossRef.
- U. Bardi, Sustainability, 2010, 2, 980–992 CrossRef CAS.
- J. W. An, D. J. Kang, K. T. Tran, M. J. Kim, T. Lim and T. Tran, Hydrometallurgy, 2012, 117, 64–70 CrossRef.
- M. D. Lankshear, A. R. Cowley and P. D. Beer, Chem. Commun., 2006, 612–614 RSC.
- Y.-H. Kim and J.-I. Hong, Chem. Commun., 2002, 512–513 RSC.
- S. K. Kim and J. L. Sessler, Acc. Chem. Res., 2014, 47, 2525–2536 CrossRef CAS PubMed.
- I. W. Park, J. Yoo, S. Adhikari, J. S. Park, J. L. Sessler and C. H. Lee, Chem. – Eur. J., 2012, 18, 15073–15078 CrossRef CAS PubMed.
- M. P. Wintergerst, T. G. Levitskaia, B. A. Moyer, J. L. Sessler and L. H. Delmau, J. Am. Chem. Soc., 2008, 130, 4129–4139 CrossRef CAS PubMed.
- S. K. Kim, V. M. Lynch, N. J. Young, B. P. Hay, C.-H. Lee, J. S. Kim, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 20837–20843 CrossRef CAS PubMed.
- S. Peng, Q. He, G. I. Vargas-Zúñiga, L. Qin, I. Hwang, S. K. Kim, N. J. Heo, C.-H. Lee, R. Dutta and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 865–907 RSC.
- B. M. Rambo, S. K. Kim, J. S. Kim, C. W. Bielawski and J. L. Sessler, Chem. Sci., 2010, 1, 716–722 RSC.
- D. E. Gross, F. P. Schmidtchen, W. Antonius, P. A. Gale, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 2008, 130, 13162–13166 CrossRef PubMed.
- M. K. Chae, J.-I. Lee, N.-K. Kim and K.-S. Jeong, Tetrahedron Lett., 2007, 48, 6624–6627 CrossRef CAS.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 5847–5848 CrossRef CAS PubMed.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, Inorg. Chem., 2004, 43, 7617–7621 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed.
- J. M. Mahoney, K. A. Stucker, H. Jiang, I. Carmichael, N. R. Brinkmann, A. M. Beatty, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 2922–2928 CrossRef CAS PubMed.
- F. J. Millero, in Chemistry of Marine Water and Sediments, ed. A. Gianguzza, E. Pelizzetti and S. Sammartano, Springer Berlin Heidelberg, Berlin, Heidelberg, 2002, pp. 3–34 Search PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- H. Kim, K.-I. Hong, J. H. Lee, P. Kang, M.-G. Choi and W.-D. Jang, Chem. Commun., 2018, 54, 10863–10865 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all compounds, 1H NMR spectral titration for 1·LiCl, 1·LiBr, and 1·LiClO4, 1H NMR spectral experiments for testifying anion selectivity, and computational data. Detailed information for X-ray crystallographic data, and other supporting figures. CCDC of 1·LiCl is 1990576. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04809g |
| This journal is © The Royal Society of Chemistry 2020 |