
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivation of sulfide-protected [FeFe] hydrogenase in a redox-active hydrogel†
Alaa A.
Oughli
 *a,
Steffen
Hardt
a,
Olaf
Rüdiger
b,
James A.
Birrell
*b and
Nicolas
Plumeré
*a
*a,
Steffen
Hardt
a,
Olaf
Rüdiger
b,
James A.
Birrell
*b and
Nicolas
Plumeré
*a
aCentre for Electrochemical Sciences—Molecular Nanostructures, Ruhr-Universität Bochum Universitätsstrasse 150, 44780 Bochum, Germany. E-mail: nicolas.plumere@rub.de; alaa.alsheikhoughli@rub.de
bMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: james.birrell@cec.mpg.de
First published on 13th August 2020
Abstract
[FeFe] hydrogenases are highly active hydrogen conversion catalysts but are notoriously sensitive to oxidative damage. Redox hydrogels have been used for protecting hydrogenases from both high potential inactivation and oxygen inactivation under turnover conditions. However, [FeFe] hydrogenase containing redox hydrogels must be fabricated under strict anoxic conditions. Sulfide coordination at the active center of the [FeFe] hydrogenase from Desulfovibrio desulfuricans protects this enzyme from oxygen in an inactive state, which can be reactivated upon reduction. Here, we show that this oxygen-stable inactive form of the hydrogenase can be reactivated in a redox hydrogel enabling practical use of this highly O2 sensitive enzyme without the need for anoxic conditions.
Hydrogenases are metalloenzymes that catalyze the reversible conversion of H2 into protons and electrons.1 They have been studied extensively as promising candidates for energy conversion applications because of their high catalytic efficiencies.2 However, hydrogenases suffer from oxidative deactivation pathways, which limit their use in devices.3 In the past few years, protective redox-active matrices have been developed in order to tackle these limitations.4–6 The redox active components of theses matrices mediate electron transfer between the entrapped enzyme and the electrode surface, thus, providing a redox buffer that prevents inactivation induced by the high potentials imposed by the electrode.5,7 Additionally, owing to the O2-reducing properties of the redox-active moieties bound to the matrix, O2 is reduced in the outer layers of the film, maintaining anoxic conditions in the inner layers, where catalytic H2 oxidation takes place.5,8 Controlling the film thickness9 enables optimization of the biocatalyst loading and life time,10 reaching time scales of weeks under turnover and constant exposure to oxygen.11
[FeFe] hydrogenases are considered to be the most active and reversible among the hydrogenase classes and, therefore, particularly relevant for biotechnological applications.12 However, the high efficiency of [FeFe] hydrogenases comes with extreme oxygen sensitivity.13 Thus, handling of the enzyme, including for catalytic hydrogel film preparation must be performed under strict anoxic conditions,4 which is impractical, time consuming, and an impediment for upscaling the use of these catalysts. A simple chemical procedure has been reported for the protection of one of the most active [FeFe] hydrogenases, the enzyme from Desulfovibrio desulfuricans (DdHydAB).14 This procedure involves oxidation of the enzyme in the presence of Na2S to produce an air-stable inactive state, named Hinact (Scheme 1). The protected enzyme was stable under air for several days and could be reactivated under reducing conditions.
 | ||
| Scheme 1 Protection of DdHydAB with sulfide under oxidative conditions and its reactivation in a redox film under reductive conditions. | ||
Here, we show that sulfide protected DdHydAB can be embedded in a redox hydrogel film, based on a low potential viologen as the redox mediator, under air, and then reactivated under hydrogen. Hydrogen spontaneously activates some of the enzymes from the catalytically inactive air-stable state Hinact to the active oxidized O2-sensitive Hox state.15 Those active enzymes then reduce the viologen moieties in the hydrogel, which in turn transfers electrons to the rest of the protected enzyme causing them to activate rapidly (Scheme 1). Once activated, the enzyme delivers electrons from H2 oxidation to the viologen modified hydrogel film to block the incoming O2 and thus sustain its electrocatalytic activity in the presence of oxygen (Fig. 1A). Iodide ions added to the electrolyte catalyze the decomposition of reactive oxygen species (generated from the viologen catalyzed O2 reduction) to water preventing damage to the matrix, as reported previously.11
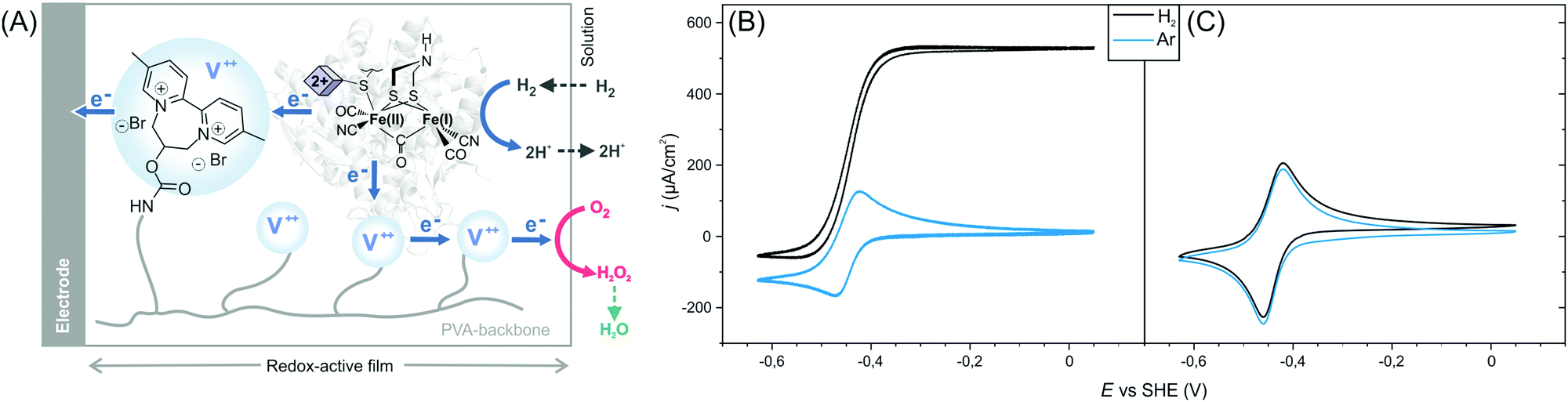 | ||
| Fig. 1 Activation of sulfide-protected [FeFe] hydrogenase DdHydAB in a redox hydrogel. (A) Scheme illustrating the electron transfer routes in the redox-active film. The blue circles represent the viologen (V++) redox mediators. Cyclic voltammograms under Ar (blue trace) and H2 (black trace) of sulfide-protected DdHydAB (B) and standard DdHydAB (C) embedded in the redox polymer P1. Both electrodes were prepared on the lab bench under air. Conditions: Tris-citrate buffer (50 mM, pH 8.8) and KI (100 mM) as supporting electrolyte, room temperature, 2000 rpm, 20 mV s−1. | ||
The efficiency of the protection/reactivation procedure was demonstrated via cyclic voltammetry experiments. Redox hydrogel films were prepared by drop-casting a mixture of DdHydAB and a 2,2′-viologen modified polyvinylalcohol backbone16 (P1) (Scheme S1, ESI†) on a glassy carbon electrode. Electrodes were modified either with the sulfide protected enzyme14 or with the unprotected enzyme (without sulfide treatment)17 and left to dry under aerobic conditions for 16 h. Cyclic voltammograms of the electrodes modified with protected enzymes show catalytic hydrogen oxidation activity (Fig. 1B and Fig. S1, ESI†) with current densities of 720 ± 135 μA cm−2 (standard deviation of n = 5 electrodes). Hence, the reactivation process was successful and the enzyme could sustain its catalytic activity despite exposure to O2 prior to reactivation. In contrast, when similar electrode preparation was performed using unprotected DdHydAB, redox signals only from the viologen of the redox-active polymer can be observed and no catalytic H2 oxidation activity was recorded (Fig. 1C). The same unprotected enzyme exhibited catalytic H2 oxidation activity in the redox-active film when the electrode was prepared and measured under anaerobic conditions (Fig. S2, ESI†). Therefore, the lack of catalytic activity in Fig. 1C can be attributed to O2 damage18 of the unprotected enzyme during the aerobic preparation period.
Evidence for the reactivation mechanism can be obtained from experiments under various H2 concentrations. When cyclic voltammograms of electrodes modified with the sulfide protected enzyme are measured starting from a high potential in H2-saturated buffer, the electrode immediately exhibits maximum catalytic current at the beginning of the scan, even before going to negative potentials (Fig. S3, ESI†). This is attributed to the spontaneous activation of the Hinact state by H2. However, it was reported that the rates of reactivation are faster in the presence of a reducing agent (e.g. dithionite) or an electron mediator.15 In the redox hydrogel matrix, the viologen moieties behave as electron mediators allowing rapid exchange of electrons between the enzymes. Insights on the timescale of the reactivation of the protected enzyme embedded in the redox polymer can be obtained from chronoamperometry experiments at low H2 concentrations and high potentials (Fig. S4, ESI†). The applied high potential under Ar ensures the oxidation of the redox polymer film. After addition of H2 (10%) to the Ar gas flow, the current does not significantly change initially. However, after a delay of 85 ± 4 s (standard deviation for n = 5 electrodes) the catalytic H2 oxidation current then increases rapidly. In contrast, when the same procedure is applied to the activated enzyme, a sharp increase of catalytic activity is observed instantly upon H2 addition indicating that the delay in the case of the protected enzyme is due to an initially slow progression of the reactivation process rather than equilibration of H2 in the electrolyte or in the polymer film. This behavior is attributed to an autocatalyzed reactivation process initiated by spontaneous activation of a small fraction of the enzyme by H2. The initially activated fraction of the hydrogenase then catalyzes H2 oxidation and the resulting electrons are transferred to the viologen moieties, which rapidly activate the rest of the enzyme in the film.
To gain further insight on the reactivation process of the enzyme in the redox-active polymer in the absence of H2, FTIR spectroelectrochemical measurement was performed on protected DdHydAB embedded in the film (Fig. 2 and Fig. S5, ESI†). FTIR assignments of the different states of the DdHydAB active site are well documented in the literature.14,17,19,20 The film was formed on a gold mesh working electrode placed on top of a CaF2 window of a spectroelectrochemical cell based on a previously published design.21 The cell was assembled under air and the applied potential was swept from positive to negative, equilibrating the potential for 30 minutes at each step. At high applied potentials (0 mV vs. SHE) the enzyme is mostly in the air-stable Hinact state (Fig. 2, green peaks). When applying more reducing potentials (−325 mV vs. SHE) approaching the redox potential of the viologen (EV = −440 mV vs. SHE), the FTIR signals from the one-electron reduced, but still inactive, Htrans state14 (Fig. 2, orange peaks) become predominant. This is the first step in the enzyme reactivation in which the enzyme is still protected. The Htrans state is thought to retain the sulfur species bound to the active site. Going to more negative potentials (−425 mV vs. SHE), near EV, leads to the conversion of the Htrans state to the oxidized active state Hox (Fig. 2, red peaks), with H2S release from the enzyme, causing deprotection and activation of catalysis. In parallel to Hox formation, signals from the CO-inhibited state Hox–CO (Fig. 2, gray peaks) are visible. This state is formed when CO coordinates to the active site of a deprotected hydrogenase. The presence of the Hox–CO state suggests that CO has been released from damage of a fraction of the enzyme.20 Within the confined volume of the spectroelectrochemical cell CO cannot escape from the inhibited enzyme. However, when the enzyme is under turnover conditions in the presence of H2 in an open electrochemical cell, this CO is released, giving rise to the large catalytic currents observed in Fig. 1B.
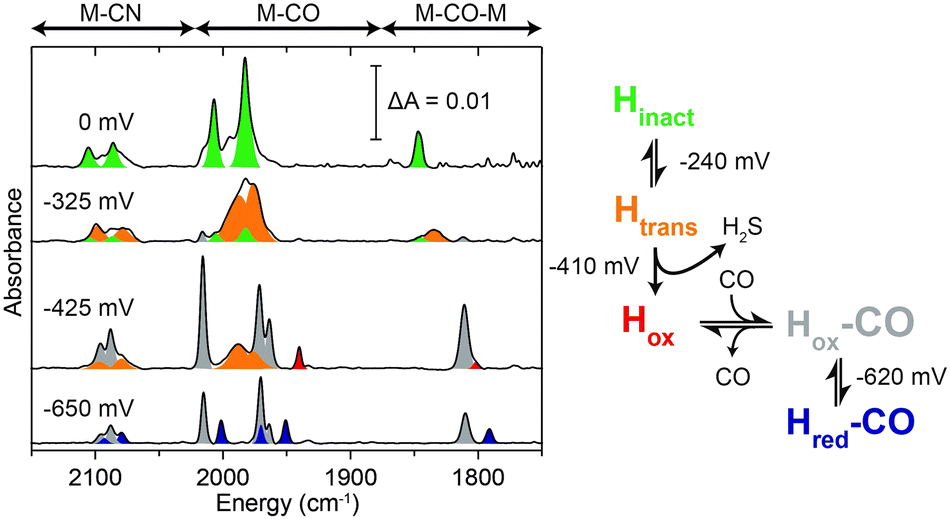 | ||
| Fig. 2 FTIR spectroelectrochemistry of protected DdHydAB embedded in the viologen modified polymer P1 under air. FTIR spectra at selected applied potentials are shown to highlight the main states observed during the redox titration: Hinact (green), Htrans (orange), Hox (red), Hox–CO (gray), Hred–CO (purple). | ||
The reductive deprotection and release of the sulfide ligand from Hinact reactivates the enzyme's ability to oxidize hydrogen but also its sensitivity toward oxygen. This is illustrated in a chronoamperometry experiment of protected DdHydAB after reactivation. When the enzyme is measured in the absence of the hydrogel film as a protection matrix, by simply adsorbing the protein on a graphite electrode surface, catalytic H2 oxidation activity is irreversibly lost upon the addition of 5% O2 to the gas feed (Fig. 3, red trace). The negative current observed under 5% O2 is due to direct O2 reduction at the electrode surface. In contrast, when the protected DdHydAB was embedded in the redox hydrogel, the addition of O2 led only to a slight decrease in the current, which was fully recovered once O2 was removed from the gas feed (Fig. 3, black trace). This decrease of the current in presence of O2 is due to the decrease of the electron flux toward the electrode as some of the electrons generated from the hydrogenase catalyzed H2 oxidation are diverted toward the outer layer of the film to reduce O2 preventing it from reaching the enzyme (Fig. 1A), as reported previously.4,5,8
 | ||
| Fig. 3 Oxygen tolerance under turnover conditions. Chronoamperometry experiments of a glassy carbon electrode modified with a mixture of sulfide-protected DdHydAB and the viologen modified polymer P1 (black trace), and a pyrolytic graphite electrode modified with sulfide-protected DdHydAB in a DET regime (red trace). The concentration of H2 was kept constant at 95% and Ar was used as an inert gas to complete the mixture. The shaded area indicates addition of 5% O2 to the gas feed for 300 s. Chronoamperometry was recorded in Tris–citrate buffer (50 mM, pH 8.8) with KI (100 mM) as supporting electrolyte at −201 mV vs. SHE and 2000 rpm. The DET experiment was performed in an anaerobic glovebox at room temperature. | ||
In conclusion, we have demonstrated that the oxygen-stable sulfide-protected form of DdHydAB can be incorporated into redox-active hydrogel films, under air, and reactivated under reductive conditions giving a catalytically active electrode. The source of electrons for reactivation can either be the electrode poised at reducing potentials or H2. The sulfide protection allowed aerobic preparation of the biocatalytic films, which was not possible with the unprotected enzyme. After sulfide deprotection, the enzyme is intrinsically O2 sensitive but its O2 induced inactivation is prevented in the redox-active hydrogel matrix owing to the local anaerobic conditions established under turnover conditions for H2 oxidation. This combination of matrix-induced reactivation and matrix-induced protection simplifies the integration of highly O2 sensitive catalysts in devices by circumventing the need for anaerobic conditions for both the electrode preparation and for electrocatalytic applications.
This approach may also be advantageous for applying synthetic molecular catalysts that are prone to O2 damage.22 Some recent reports have demonstrated that reductive treatment can reactivate the O2 inactivated states of some artificial systems or that proper design of the coordination sphere can limit the O2 damage.23 While these previous efforts aimed at robust catalysts for H+/H2 conversion under O2, we believe that our demonstration of the reductive reactivation and operation of a highly O2 sensitive enzyme in redox-active matrices opens the possibility to apply synthetic catalysts that show O2 stable inactive and O2 sensitive active states in the same way.
N. P., A. A. O and S. H. acknowledge financial support by the ERC starting grant 715900, by the ANR-DFG project SHIELDS (PL 746/2-1) and by RESOLV, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2033 – Projektnummer 390677874. J. A. B. and O. R. are supported by the Max Planck Society and J. A. B. acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG) Priority Programme “Iron–Sulfur for Life: Cooperative Function of Iron–Sulfur Centers in Assembly, Biosynthesis, Catalysis and Disease” (SPP 1927) Project BI 2198/1-1. The authors thank L. Castaneda-Losada, J. Jaenecke, F. Hiege, L. Dubbert, C. Spula, L. Engelhard and C. Kim for useful discussions and preliminary experiments, Nina Breuer for the preparation of the hydrogenase. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. W. Adams, Biochim. Biophys. Acta, 2015, 1853, 1350–1369 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew. Chem., Int. Ed., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- K. Sakai, Y. Kitazumi, O. Shirai, K. Takagi and K. Kano, ACS Catal., 2017, 7, 5668–5673 CrossRef CAS.
- (a) L. H. Eng, M. Elmgren, P. Komlos, M. Nordling, S.-E. Lindquist and H. Y. Neujahr, J. Phys. Chem., 1994, 98, 7068–7072 CrossRef CAS; (b) S. V. Morozov, E. E. Karyakina, N. A. Zorin, S. D. Varfolomeyev, S. Cosnier and A. A. Karyakin, Bioelectrochemistry, 2002, 55, 169–171 CrossRef CAS.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumeré and C. Léger, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS.
- (a) H. Li, D. Buesen, R. Williams, J. Henig, S. Stapf, K. Mukherjee, E. Freier, W. Lubitz, M. Winkler, T. Happe and N. Plumeré, Chem. Sci., 2018, 9, 7596–7605 RSC; (b) D. Buesen, H. Li and N. Plumeré, Chem. Sci., 2020, 11, 937–946 RSC.
- H. Li, D. Buesen, S. Dementin, C. Léger, V. Fourmond and N. Plumeré, J. Am. Chem. Soc., 2019, 141, 16734–16742 CrossRef CAS PubMed.
- H. Li, U. Münchberg, A. A. Oughli, D. Buesen, W. Lubitz, E. Freier and N. Plumeré, Nat. Commun., 2020, 11, 920 CrossRef CAS.
- B. R. Glick, W. G. Martin and S. M. Martin, Can. J. Microbiol., 1980, 26, 1214–1223 CrossRef CAS PubMed.
- M. W. Adams, Biochim. Biophys. Acta, 1990, 1020, 115–145 CrossRef CAS.
- P. Rodríguez-Maciá, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rüdiger and J. A. Birrell, J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef PubMed.
- E. C. Hatchikian, N. Forget, V. M. Fernandez, R. Williams and R. Cammack, Eur. J. Biochem., 1992, 209, 357–365 CrossRef CAS PubMed.
- S. Hardt, S. Stapf, J. A. Birrell, O. Rüdiger, V. Fourmond, C. Léger and N. Plumeré, Reversible H2 oxidation and evolution by hydrogenase embedded in a redox polymer film, submitted March 2020 Search PubMed.
- J. A. Birrell, K. Wrede, K. Pawlak, P. Rodriguez-Maciá, O. Rüdiger, E. J. Reijerse and W. Lubitz, Isr. J. Chem., 2016, 56, 852–863 CrossRef CAS.
- A. Kubas, C. Orain, D. de Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, J. A. Birrell, W. Lubitz and O. Rüdiger, ChemPlusChem, 2017, 82, 540–545 CrossRef PubMed.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg. Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- D. Moss, E. Nabedryk, J. Breton and W. Mantele, Eur. J. Biochem., 1990, 187, 565–572 CrossRef CAS PubMed.
- (a) A. Dutta, J. A. Roberts and W. J. Shaw, Angew. Chem., 2014, 53, 6487–6491 CrossRef CAS PubMed; (b) P. Rodriguez-Maciá, A. Dutta, W. Lubitz, W. J. Shaw and O. Rüdiger, Angew. Chem., Int. Ed., 2015, 54, 12303–12307 CrossRef PubMed.
- (a) X. Yang, L. C. Elrod, T. Le, V. S. Vega, H. Naumann, Y. Rezenom, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2019, 141, 15338–15347 CrossRef CAS PubMed; (b) X. Yang, L. C. Elrod, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2019, 10, 1368–1373 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc03155k |
| This journal is © The Royal Society of Chemistry 2020 |