
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering multilayer chemical networks†
Maitena
Martinez-Amezaga
,
A. Gastón
Orrillo
and
Ricardo L. E.
Furlan
 *
*
Farmacognosia, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario – CONICET, Suipacha 531, Rosario, S2002SLRK, Argentina. E-mail: rfurlan@fbioyf.unr.edu.ar
First published on 29th July 2019
Abstract
Dynamic multilevel systems emerged in the last few years as new platforms to study thermodynamic systems. In this work, unprecedented fully communicated three-level systems are studied. First, different conditions were screened to selectively activate thiol/dithioacetal, thiol/thioester, and thiol/disulfide exchanges, individually or in pairs. Some of those conditions were applied, sequentially, to build multilayer dynamic systems wherein information, in the form of relative amounts of building blocks, can be directionally transmitted between different exchange pools. As far as we know, this is the first report of one synthetic dynamic chemical system where relationships between layers can be changed through network operations.
Introduction
The implementation of stable covalent bonds that can be broken and re-formed under certain conditions has paved the way for the construction of robust adaptive systems.1,2 Dynamic covalent chemistry has become a valuable tool for different research fields such as materials science,3–7 drug discovery8–10 and delivery,11,12 sensing,13,14 and molecular machines.15–17When dynamic covalent chemistry is applied to the preparation of dynamic combinatorial libraries,18,19 library members are interconnected by reversible reactions and can be represented as a network with interlinked nodes, i.e. a monolayer network (Fig. 1A).20,21 The preparation of complex reaction networks constitutes an important part of systems chemistry, a growing discipline that aims to develop molecular systems of interacting and/or interconverting molecules that show emergent properties.18,22,23
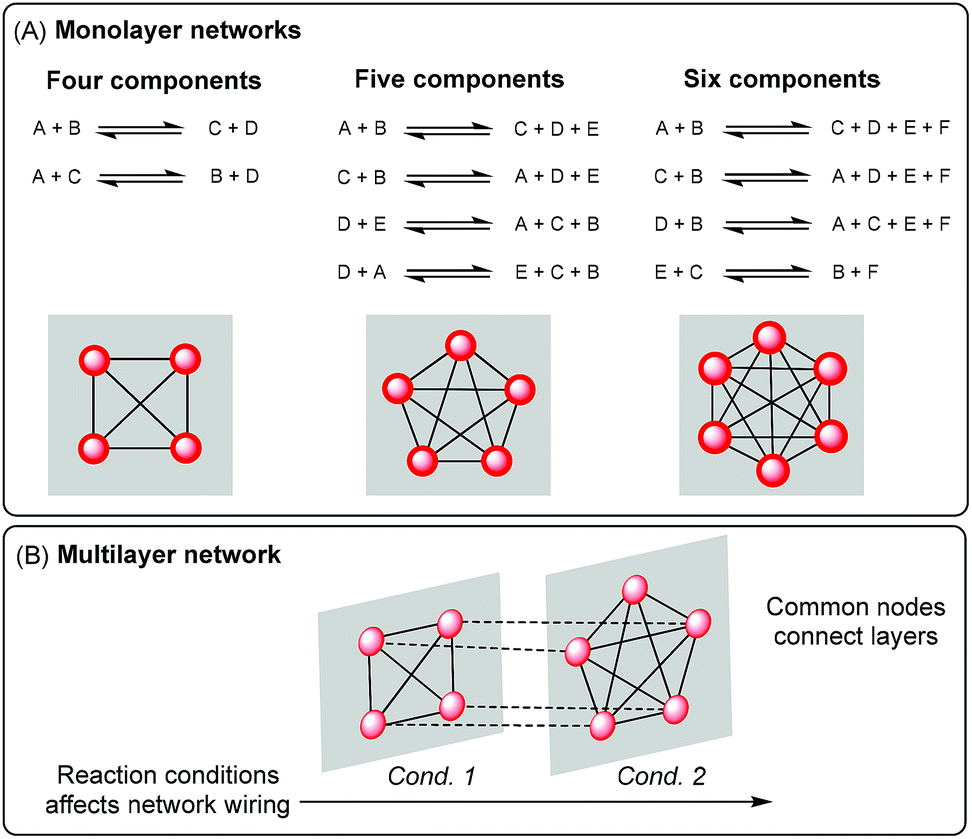 | ||
| Fig. 1 (A) A fully reversible chemical process involving four, five, or six components described as a set of reactions and as a “monolayer” molecular network. (B) A multilayer network is formed from networks that (i) have at least one node in common and (ii) can be sequentially wired under diverse conditions. | ||
In this context, the reversible chemistry used to interconvert library members defines the network wiring and, consequently, affects the system adaptability towards environmental changes.24 One way to exert control on network wiring is through the use of more than one reversible reaction within the same system. When two or more reversible reactions are used in one dynamic system, the relative reactivity of the functional groups involved in each reaction will determine many of the system properties. Most of the multi-reaction dynamic systems reported to date were based on different combinations of two orthogonal reactions,25 and only a handful involved three or more reactions.26–30 The one pot combination of reversible exchange chemistries that can intercommunicate31,32 has been far less explored than their orthogonal counterpart.33 Such a combination generates exchange pools that share one building block type and, consequently, can affect each other. This mutual influence between pools is due to their competence for the building block type they have in common. Control over the composition of such systems has been achieved by controlling the concentration of shared building blocks through irreversible reactions, such as oxidation or reduction,34 or changing the reactivity of those shared building blocks through isomerization.35 In this way, the building blocks can be selectively channelled into different exchange pools.
The combination of communicating reactions that can be sequentially activated represents a special study case since they give rise to multilayer networks.36 In multilayer networks, each molecular network is described as a “layer” and the different layers are connected through common nodes, i.e. connector molecules involved in more than one reaction (Fig. 1B). Such a connectivity could mediate the transference of chemical information between different layers because changes in the molecular composition occurring in one layer (that affect the concentration of connector molecules) will be translated to the following layer. So in principle, an imprint of some events occurring along the process could be recorded in the composition and affect ulterior equilibria.20,28
The only example of a two-layer dynamic chemical network reported is based on the sequential activation of dithioacetal and disulfide exchanges.36 Addition of a third communicating reaction, such as thioester exchange, and the implementation of different reaction conditions to selectively activate the reactions, individually or in pairs, may open the door towards further exploration of multilayer systems.
Here we searched for reagents and conditions to combine three communicating reactions: thiol/disulfide, thiol/thioester, and thiol/dithioacetal exchanges (Fig. 2). Then some selected conditions were applied to build multilayer chemical networks. Although the studied dynamic systems are very simple, they showed some unique properties such as history-dependent information transfer between different compound pools.
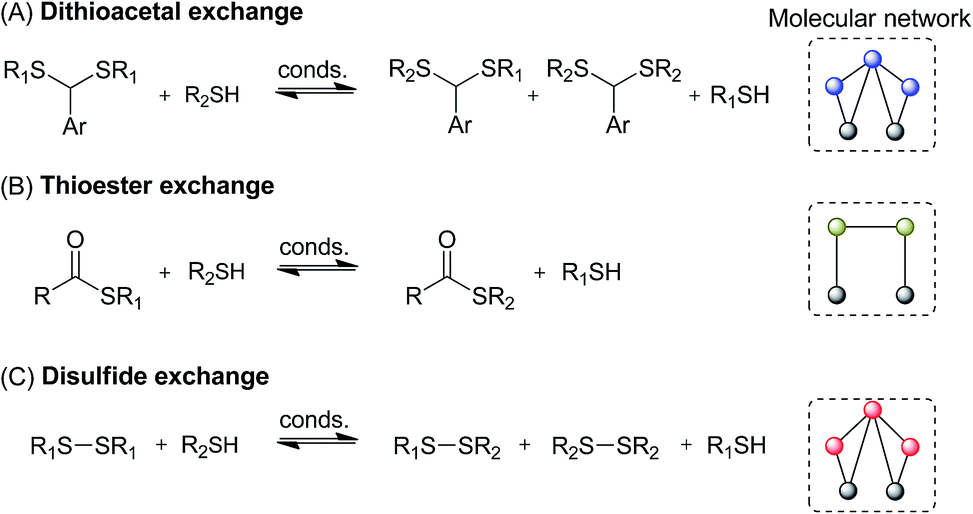 | ||
| Fig. 2 Reversible reactions used in this study. Dashed line squares show molecular networks of each reaction. Color code for nodes: blue, dithioacetal; black, thiol; olive green, thioester; red, disulfide. | ||
Results and discussion
Initially, a catalyst screening was carried out by mixing dithioacetal 1A1, disulfide 11, thioester B1, and thiol 2H (Fig. 3). These compounds can participate in thiol/dithioacetal exchange, thiol/disulfide exchange or thiol/thioester exchange, three communicating reversible reactions that could in principle be simultaneously or sequentially activated.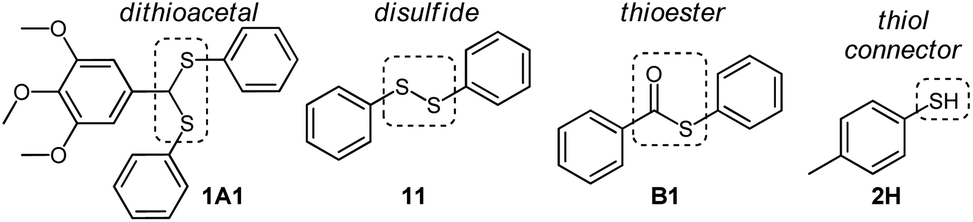 | ||
| Fig. 3 Building blocks used in this study. Dashed line squares highlight functional groups involved in reversible reactions. | ||
Compounds 1A1 (30 mM), 11 (30 mM), B1 (30 mM) and 2H (60 mM) were mixed in acetonitrile at room temperature in the absence or the presence of a catalyst (30 mM), and the composition of the reaction mixture was analyzed after 24 h by HPLC-UV.
In the absence of any catalyst, thioester B1 and dithioacetal 1A1 did not react with thiol 2H, whereas disulfide 11 gave rise to an equilibrated mixture of 11, 12 and 22 (Table 1, entry 1).
| Entry | Catalyst | Dithioacetalb | Thioesterb | Disulfideb |
|---|---|---|---|---|
| a Conditions: 1A1 (15 μmol), 11 (15 μmol), B1 (15 μmol), 2H (30 μmol), and acetonitrile (total volume: 500 μL), r.t., and catalyst (15 μmol). The compositions were measured by HPLC-UV after 24 h of reaction (Abs250). b N.R.: no product could be detected. c Thiol oxidation was observed. d Decreased thiol concentration. e DCA (0.15 μmol) and O2 atmosphere. f Decomposition. g Rose bengal (0.75 μmol) and N2 atmosphere. | ||||
| 1 | None | N.R. | N.R. | Equilibrated |
| 2 | TFA | N.R. | N.R. | N.R. |
| 3 | PTSA | Equilibrated | N.R. | N.R. |
| 4 | H2SO4 | Equilibrated | N.R. | N.R. |
| 5 | Yb(OTf)3 | N.R. | N.R. | N.R. |
| 6 | AlCl3 | N.R. | N.R. | N.R. |
| 7 | FeCl3c | Equilibrated | N.R. | N.R. |
| 8 | Cu(OTf)2d | Equilibrated | N.R. | N.R. |
| 9 | Sn(OTf)2 | Equilibrated | N.R. | N.R. |
| 10 | SnCl2 | Slow exchange | N.R. | N.R. |
| 11 | UV/DCAc,e | Slow exchangef | N.R. | Slow exchangef |
| 12 | UV/rose bengalg | N.R. | N.R. | Slow exchangef |
| 13 | TEAc | N.R. | Equilibrated | Equilibrated |
| 14 | DMAPc | N.R. | Equilibrated | Equilibrated |
| 15 | (Bu2SnCl)2Oc | N.R. | N.R. | N.R. |
| 16 | (Bu3Sn)2Oc | N.R. | N.R. | N.R. |
| 17 | Bu2SnO | N.R. | N.R. | Equilibrated |
| 18 | BuSnO2H | N.R. | Equilibrated | Equilibrated |
It is known that Brønsted acids can stop thioester28,37 and disulfide38,39 exchanges and, at the same time, catalyze dithioacetal exchange.40,41 In the presence of trifluoroacetic acid (TFA), disulfides did not exchange; however, the acid concentration (30 mM) was not enough to activate dithioacetal exchange, so the whole system stayed static (entry 2).
At the same concentration, stronger Brønsted acids such as p-toluenesulfonic acid (PTSA) or sulfuric acid selectively activated dithioacetal exchange to produce equilibrated mixtures of 1A1, 1A2 and 2A2 (entries 3 and 4).
Several Lewis acids have been reported as dithioacetal formation42–46 or hydrolysis catalysts.47,48 In our model system, the addition of ytterbium(III) triflate or aluminium(III) chloride did not activate the exchange of dithioacetals or thioesters; besides, it stopped the disulfide exchange (entries 5 and 6). In the presence of iron(III) chloride, only dithioacetal exchange was observed with concomitant thiol oxidation (entry 7). Copper(II) triflate also favored dithioacetal exchange and deactivated disulfide exchange; however, a significant decrease in thiol concentration was observed after 24 h of reaction (entry 8). Two tin(II) reagents were tested: chloride and triflate (entries 9 and 10). Both mild Lewis acids stopped the disulfide exchange and selectively catalyzed dithioacetal exchange; however, the equilibrium was reached in 24 h only with tin(II) triflate.
The exchange, formation or hydrolysis of dithioacetals has also been achieved using photoirradiation in the presence of photosensitizers such as 9,10-dicyano anthracene (DCA)36 or rose bengal.49,50
When applied to our starting solution of 1A1, B1, 11 and 2H, DCA/UV light induced a very slow exchange of dithioacetals and disulfides along with extensive decomposition (Table 1, entry 11). Similarly, rose bengal/UV light induced disulfide exchange and decomposition, whereas, in this case, dithioacetal and thioester molecules remained unreacted (entry 12).
Thioester exchange in organic solvents is usually achieved with organic bases.37 Addition of TEA and DMAP to our model mixture led to the activation of both disulfide and thioester exchanges (entries 13 and 14). Under these basic conditions, thiol oxidation took place and the free thiols could not be detected after 24 h.
Different organotin(IV) reagents have been used to catalyze carboxylic ester cleavage and/or transesterification;51 however, to the best of our knowledge, their use in transthioesterification has not been reported. Therefore, four organotin(IV) reagents were tested. In the presence of bis(dibutylchlorotin) oxide and bis(tributyltin) oxide the three reactions were inactive (entries 15–16), whereas with dibutyltin oxide only disulfide exchange was active (entry 17). On the other hand, the addition of butylstannonic acid activated thioester and disulfide exchanges, maintaining dithioacetal exchange inactive (entry 18). Despite their differential influence on the thioester exchange, the effect of these reagents on the exchange of disulfides was not expected: two of them inactivated the exchange and the other two did not.
Based on these results, TEA, butylstannonic acid, sulfuric acid, PTSA, tin(II) triflate, and tin(II) chloride were selected for further studies on the isolated reactions as well as binary combinations of simultaneous reactions.
Isolated reactions
Dithioacetal exchange has been recently introduced into the field of dynamic covalent chemistry. Although so far TFA has been the only Brønsted acid used to catalyze the reaction,36,40,41 stronger acids may be more convenient to speed up the exchange (Table 1, entries 3 and 4).When an acetonitrile solution of dithioacetal 1A1 (30 mM) and thiol 2H (60 mM) was stirred in the presence of different Brønsted acids (30 mM), the acid strength determined the reaction kinetics. Sulfuric acid equilibrated the mixture within a few minutes, while a tenfold decrease in the amount of acid caused a dramatic increase in the t1/2 value (Table 2, entries 1 and 2, see full compositions in Fig. S1 and S2†). PTSA led to a t1/2 value of 48 min, whereas the same concentration of TFA was not enough to activate the exchange (entries 3 and 4, Fig. S3 and S4†). A ten times higher TFA loading was required to activate the reaction (entry 5, Fig. S5†).
| Entry | Exchange reaction | Catalyst | Amounte | t 1/2 , (min) |
|---|---|---|---|---|
| a 1A1 (15 μmol), 2H (60 μmol), and acetonitrile (500 μL). b B1 (15 μmol), 2H (30 μmol), and acetonitrile (500 μL). c Argon atmosphere. d 11 (15 μmol), 2H (60 μmol), and acetonitrile (500 μL). e Mol of catalyst per mol of the limiting reagent (1A1, B1 or 11). f The half-life time t1/2 was graphically determined from the kinetics curve of dithioacetal 1A2, thioester B2 or disulfide 12 (monitored by HPLC-UV Abs250). g N.R.: no product was detected. | ||||
| 1 | Dithioacetala | H2SO4 | 1 | 1.2 |
| 2 | Dithioacetala | H2SO4 | 0.1 | 1800 |
| 3 | Dithioacetala | PTSA | 1 | 48 |
| 4 | Dithioacetala | TFA | 1 | N.R. |
| 5 | Dithioacetala | TFA | 10 | 48 |
| 6 | Dithioacetala | Sn(OTf)2 | 1 | 8 |
| 7 | Dithioacetala | Sn(OTf)2 | 0.1 | 8 |
| 8 | Dithioacetala | Sn(OTf)2 | 0.01 | 800 |
| 9 | Thioesterb,c | TEA | 1 | 0.3 |
| 10 | Thioesterb,c | TEA | 0.1 | 0.6 |
| 11 | Thioesterb,c | TEA | 0.01 | 2.5 |
| 12 | Thioesterb | BuSnO2H | 1 | 25 |
| 13 | Thioesterb | BuSnO2H | 0.1 | 420 |
| 14 | Disulfided | None | — | 17 |
| 15 | Disulfidec,d | TEA | 1 | 0.3 |
| 16 | Disulfided | BuSnO2H | 1 | 0.8 |
| 17 | Disulfidec,d | TFA | 1 | N.R. |
In addition to Brønsted acids, tin(II) triflate was the only Lewis acid tested that equilibrated dithioacetals without side reactions in a reasonable time frame (Table 2, entries 6–8, Fig. S6–S8†). Tin(II) triflate appears to be a new promising catalyst for dithioacetal exchange.
Previously, Lewis acid catalysis of dithioacetal exchange had only been attempted by Hirsch et al. using ZnCl2;52 however, the reversibility of the exchange reaction was not confirmed. Interestingly in the DCC context of multilevel systems, tin(II) triflate did not activate thioester exchange and inactivated disulfide exchange.
Transesterification was one of the reactions applied earliest in dynamic combinatorial chemistry.53,54 The thioester exchange was introduced later in the discipline, and it has been used to produce several dynamic systems.55–63 In organic solvents, this exchange has always been carried out under basic conditions.37
Fast exchange between thioester B1 and thiol 2H was catalyzed by stoichiometric or catalytic amounts of TEA (entries 9–11, Fig. S9–S11†).
In addition to the well-known base-catalyzed thioester exchange,37 we studied this reaction with butylstannonic acid (Table 1, entry 18). The reaction with a stoichiometric amount of this organotin compound showed a t1/2 value of 25 min (Table 2, entry 12, Fig. S12†). When a sub-stoichiometric amount of butylstannonic acid was used, the initial exchange was slower (Table 2, entry 13), and the product formation reached a plateau at a concentration lower than the expected value (≅37%, Fig. S13†). At this point, the addition of a second thiol (1-pentanethiol) did not introduce changes in the product distribution, suggesting that the catalyst was not active (Fig. S14a†). In parallel, the addition of more catalyst re-started the exchange and, finally, the mixture reached the expected equilibrium composition (Fig. S14b and c†). Although the thioester exchange was slower with butylstannonic acid (t1/2 = 25 min) than with triethylamine (t1/2 = 0.3 min), the collateral oxidation of thiol 2H was also slower (Fig. S15†).
In the presence of butylstannonic acid, thioester exchange could proceed via an exchange/insertion mechanism similar to that proposed for organotin(IV) catalyzed transesterification reactions.64 Although an efficient exchange can be achieved under base catalysis, alternative conditions for this reversible reaction may be useful for the preparation of base-labile dynamic systems. In that context, the transthioesterification process observed in the presence of butylstannonic acid may represent an interesting starting point for further studies.
Disulfide exchange is one of the most popular reactions in dynamic combinatorial chemistry.65 In aqueous media it is active at nearly neutral pH values, but in organic solvents a base is usually required. In our model, disulfide exchange was achieved in acetonitrile without a catalyst (Table 2, entry 14, Fig. S16†). In this solvent, TEA and butylstannonic acid accelerated the exchange (Table 2, entries 15–16, Fig. S17 and S18†), whereas TFA inactivated it (Table 2, entry 17, Fig. S19†).
Double-level systems based on simultaneous and communicating reversible reactions
In order to study the interplay between different reactions, two binary combinations of simultaneous and communicating reactions were studied, disulfide–thioester and disulfide–dithioacetal exchanges, and the kinetic traces of isolated versus networked reactions were compared.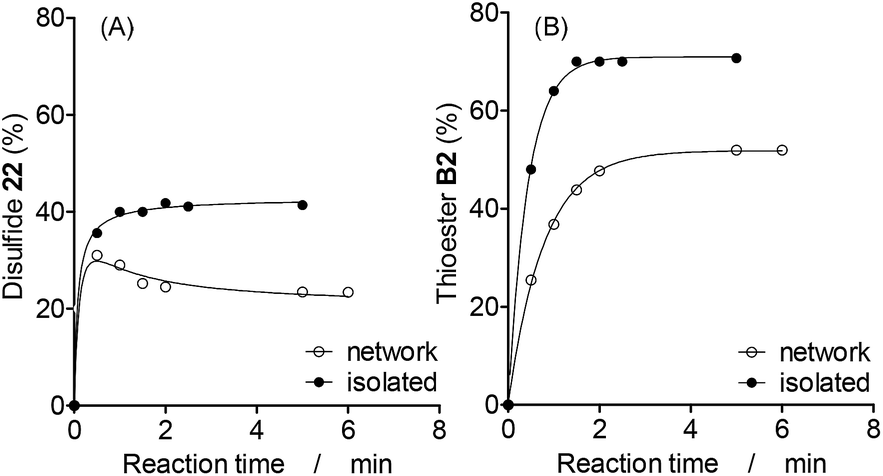 | ||
| Fig. 4 Kinetics profile of the networked and isolated (A) disulfide and (B) thioester reactions. Mixture formed upon addition of thiol 2H (60 mM) in a solution containing TEA (30 mM), disulfide 11 (30 mM) and/or thioester B1 (30 mM) under an argon atmosphere. Solvent: acetonitrile (total volume: 500 μL), r.t. The amount of 22 and B2 at the base of the HPLC peak areas (Abs250) was calculated as follows: 22 (%) = 22 × 100/(11 + 12 + 22) and B2 (%) = B2 × 100/(B1 + B2). | ||
Since the initial concentration of thiol 2H (total concentration of building block 2 in the system) was the same for isolated and networked experiments, the final proportion of disulfide and thioester products containing building block 2 in the networked reactions is lower than that in the isolated reactions (Fig. 4). As expected, this observation is independent of the catalyst used as shown by similar results observed with an alternative catalyst (Fig. S21†).
A more subtle networking effect was observed when the formation kinetics of the networked and isolated disulfide 22 were compared more closely. In the absence of thioesters, the amount of 22 increased until it became constant after 1 min of reaction (Fig. 4A, isolated). In the presence of exchanging thioesters, the amount of disulfide 22 reached a maximum value of 30% after 0.5 min, and then it decreased to reach a constant amount of 24% after 2 min of reaction (Fig. 4A, network). Since disulfides exchange faster than thioesters, 22 is a kinetically preferred product and initially reached a maximum concentration that is higher than its concentration at equilibrium.
The final proportion of product 22 or 2A2 was lower in the networked than in the isolated reactions (Fig. 5, see full composition in Fig. S22†). Under these conditions, dithioacetals exchange faster than disulfides (Fig. 5, isolated). The formation of dithioacetal 2A2 was kinetically favored and reached a transient maximum concentration of 23% after 0.7 h of reaction (Fig. 5B, network). Then, a decrease in 2A2 concentration was observed to reach the final constant concentration of 20% after 2 h.
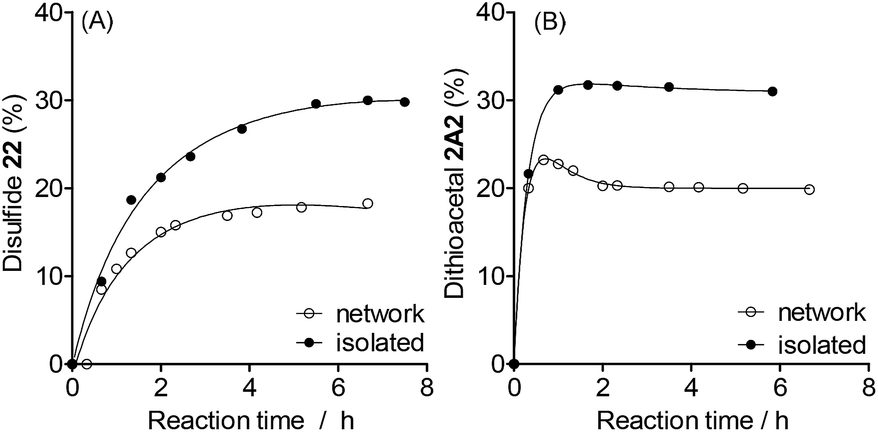 | ||
| Fig. 5 Kinetics profile of the (A) disulfide and (B) dithioacetal layer in the mixture formed from dithioacetal 1A1 (30 mM), disulfide 11 (30 mM), thiol 2H (60 mM), and SnCl2 (1.5 mM). Solvent: acetonitrile (total volume: 500 μL). Temperature: 100 °C under an argon atmosphere. The amount of 22 and 2A2 at the base of the HPLC peak area data (Abs250) was calculated as follows: 22 (%) = 22 × 100/(11 + 12 + 22) and 2A2 (%) = 2A2 × 100/(1A1 + 1A2 + 2A2). | ||
Similar kinetic traces were observed by Lehn et al. in dynamic systems where different nitrogen nucleophiles, such as amines, alkoxy-amines and hydrazides, were competed by one aldehyde.32 As in our examples, a kinetic product is formed first; its concentration reaches a maximum and then decreases. However, the equilibrium was never reached in those systems.
In the future, it would be interesting to study this behaviour further by pushing the system towards more stringent competition for starting materials (changing relative building blocks' concentration).
Triple-level systems based on sequential/simultaneous and communicating reactions: the emergence of multilayer networks
The access to reaction conditions that activate dithioacetal, disulfide and/or thioester exchanges could be a good starting point to build multilayer networks. To this end, the same starting mixture of 1A1, B1, 11 and 2H was used to generate multilevel systems which differ in the number and order of reactions along the sequence.The first sequence started with an acetonitrile solution containing disulfide 11 (30 mM), thioester B1 (30 mM), and dithioacetal 1A1 (30 mM) (Fig. 6A). Initially, disulfide exchange was selectively activated by addition of thiol 2H (60 mM). Under these conditions, thioester and dithioacetal exchanges were inactive (Fig. 6B).
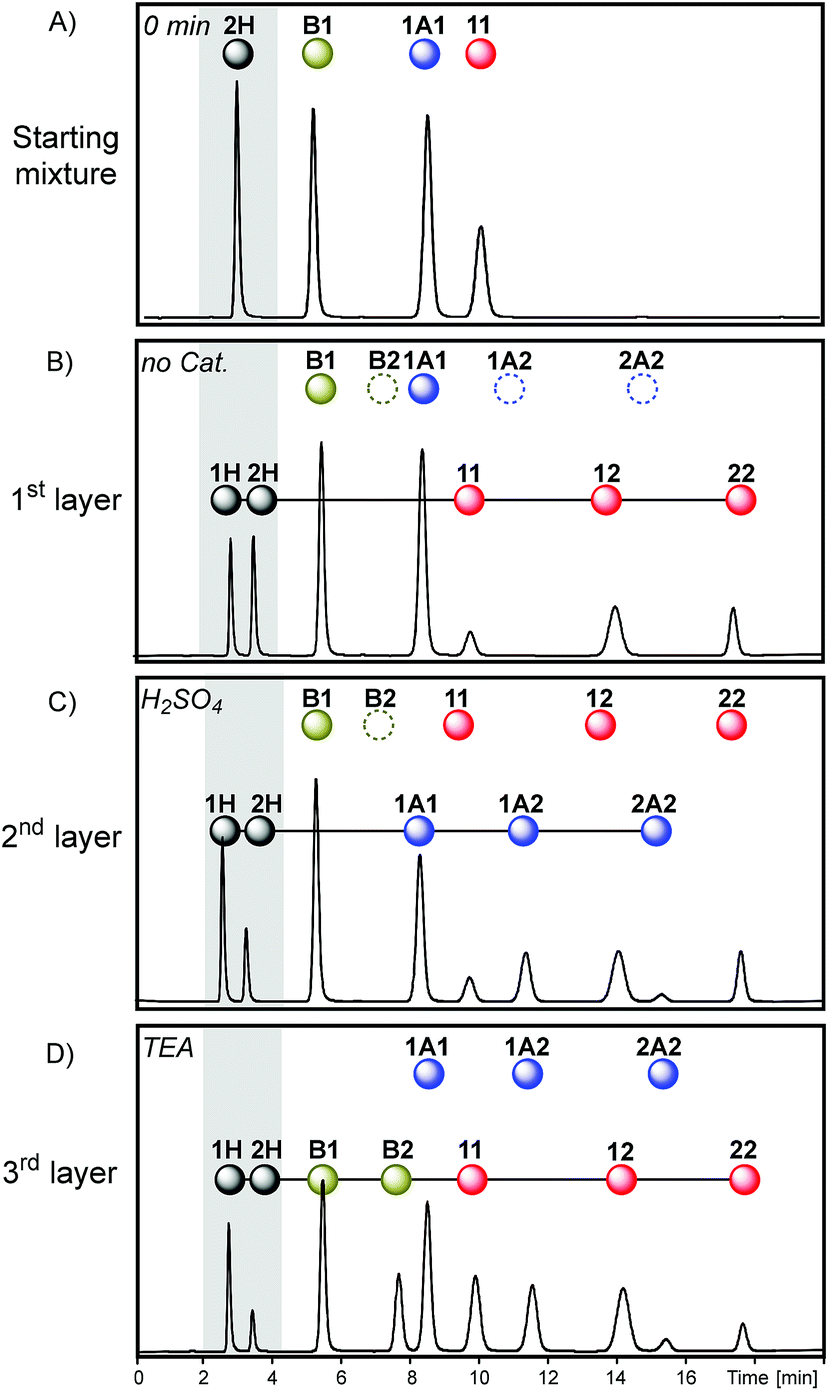 | ||
| Fig. 6 Chromatograms (Abs250) obtained in each step of sequence I. Starting mixture (A), after the equilibrium was reached in the absence of any catalyst (B, first layer), in the presence of H2SO4 (C, second layer) and later in the presence of an excess of TEA (D, third layer). | ||
Next, disulfide exchange was stopped and dithioacetal exchange was activated by adding H2SO4. In this step, a net transference of building block 2 from the thiol pool to the dithioacetal pool was observed generating 1A2 and 2A2 (Fig. 6C). The associated transference of building block 1 in the opposite direction was also noticeable: the 1H/2H peak area proportion increased from 1 (Fig. 6B) to 2 (Fig. 6C). Although at this point disulfides and thioesters stayed disconnected from the network (Fig. 7, 2nd layer of sequence I), the information imprinted in the relative concentration of the free thiols was later transferred to both pools when TEA was added. In the third layer, disulfides and thioesters containing building block 1 were favored. The peak areas of disulfides 11 (42%) and 12 (46%) were higher than that of 22 (12%) (Fig. 6D), an opposite situation when compared to the relative concentration of these disulfides when only disulfide exchange was active (Fig. 6B).
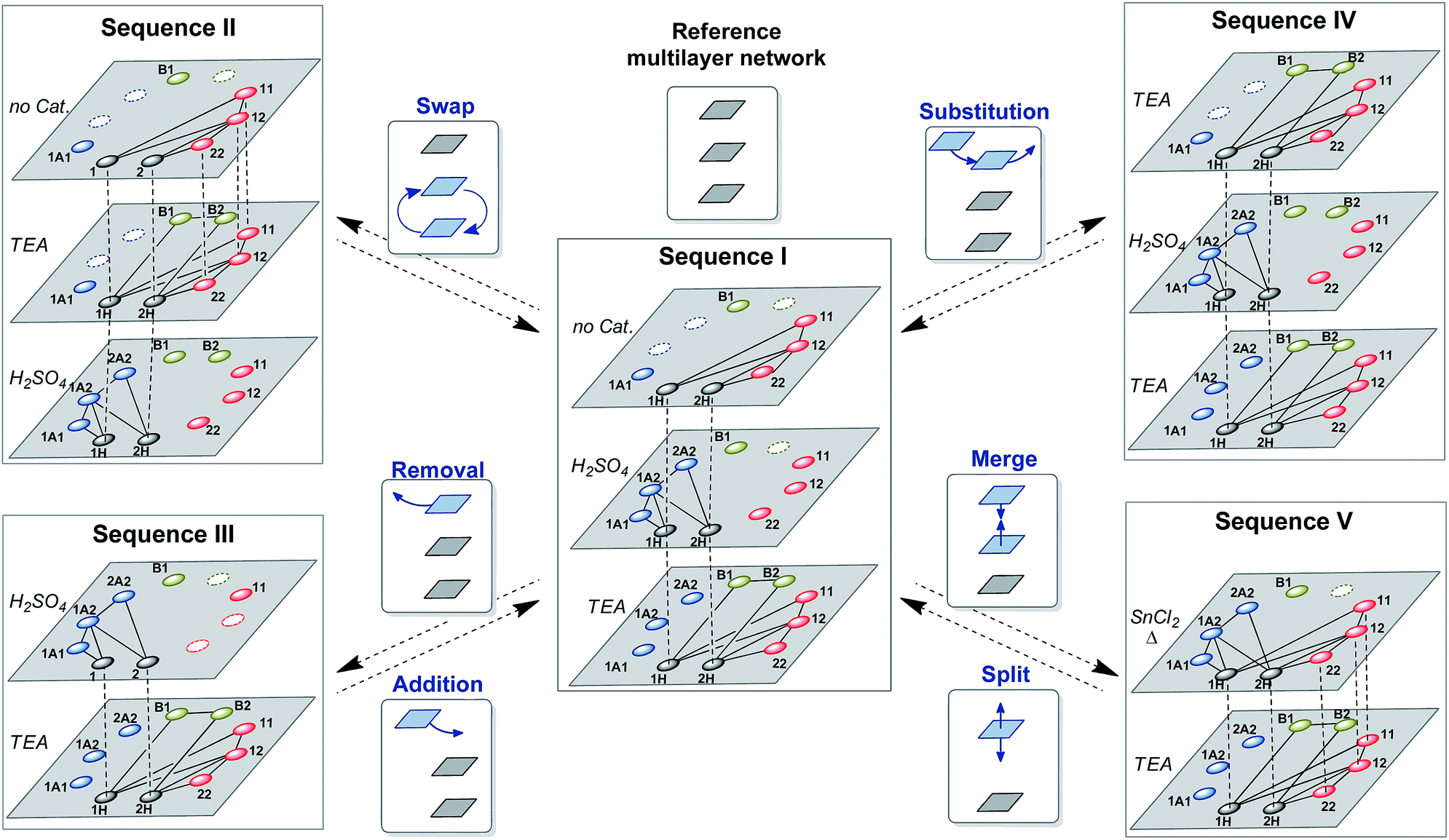 | ||
| Fig. 7 Engineering multilayer chemical networks in dynamic multilevel systems. The introduction of changes into a reference multilayer network gives rise to a variety of networks upon application of operations based on swapping, removal, substitution or merging of layers. | ||
Computer simulations can be useful to analyze the behavior of dynamic combinatorial libraries.66–70 Simulations were carried out in order to predict the composition of the equilibrated libraries constructed with sequential reactions. The experimentally observed equilibrium compositions after each activation step (Fig. 6) were close to those observed in a simulated multilevel system with isoenergetic components (Fig. S23†). This suggests that the library composition was not influenced by non-covalent interactions between library members and/or catalysts. Therefore, the progress of the composition over time (first, second, and third layers of the network) was defined by the covalent network and the relative stability of the components.
Analysis of product distributions along sequence I illustrates how the information stored at the thiol connector level can be transferred between layers. Although dithioacetals and disulfides (or thioesters) were never connected to the network at the same time (Fig. 7, sequence I), the final distribution of disulfides and thioesters was affected by the proportion of dithioacetals in the previous layer (due to its push–pull effect on thiols).
It is expected that the relative amount of free thiols 1H and 2H will change during the sequential process in different manners when different sequences are generated. Since these thiols connect different layers, the relative proportion given by the equilibration in one layer will probably affect the equilibrium composition in the subsequent level.
Network engineering: a new level of design in synthetic chemical systems
Multilevel dynamic systems are usually designed taking in consideration chemical properties such as compatibility between the reversible reactions applied, the effect of the molecular structure on the reactivity, the timing of the reactions, etc. The emergence of multilayer networks gives rise to other design aspects such as the layer order and number. The impact of such features on the final composition was analyzed through some operations at the network level, including layer swapping, removal, substitution and merging.The order in which the layers of the multilayer network are stacked can be altered by changing the activation order of the reactions along the sequence. For instance, the second and third layers of sequence I (Fig. 7) can be swapped by inverting the order of addition of H2SO4 and TEA to produce sequence II (Fig. 7). In sequence II disulfides were initially exchanged as in the first layer of sequence I, and then thioesters were connected to the network through the addition of TEA (2nd layer of sequence II) and, finally, H2SO4 was added to stop the thioester/disulfide exchange and to activate dithioacetal exchange (3rd layer of sequence II). Therefore, once disulfides and thiols were equilibrated (Fig. 8A), thioester exchange was activated with TEA, keeping disulfide exchange active. The connection of thioesters to the network produced the net transference of building block 1 from B1 to the thiol pool and, consequently, to the disulfide pool (Fig. 8B). The proportion of peak areas of disulfides 11/22 increased from 0.4 in the first layer (Fig. 8A) to 1.0 in the second layer (Fig. 8B). In parallel, a concomitant transference of building block 2 from disulfide 22 and thiol 2H to form thioester B2 was observed (Fig. 8B). Next, disulfide and thioester exchanges were stopped and dithioacetal exchange was activated by adding H2SO4. In this step, a net transference of building block 2 from the thiol pool to the dithioacetal pool was observed, generating 1A2 and 2A2 (Fig. 8C). The associated transference of building block 1 in the opposite direction (from the dithioacetal pool to the thiol pool) was also noticeable, and the 1H/2H proportion increased from 1.3 in the second layer to 4.8 in the third layer (Fig. 8B and C, respectively).
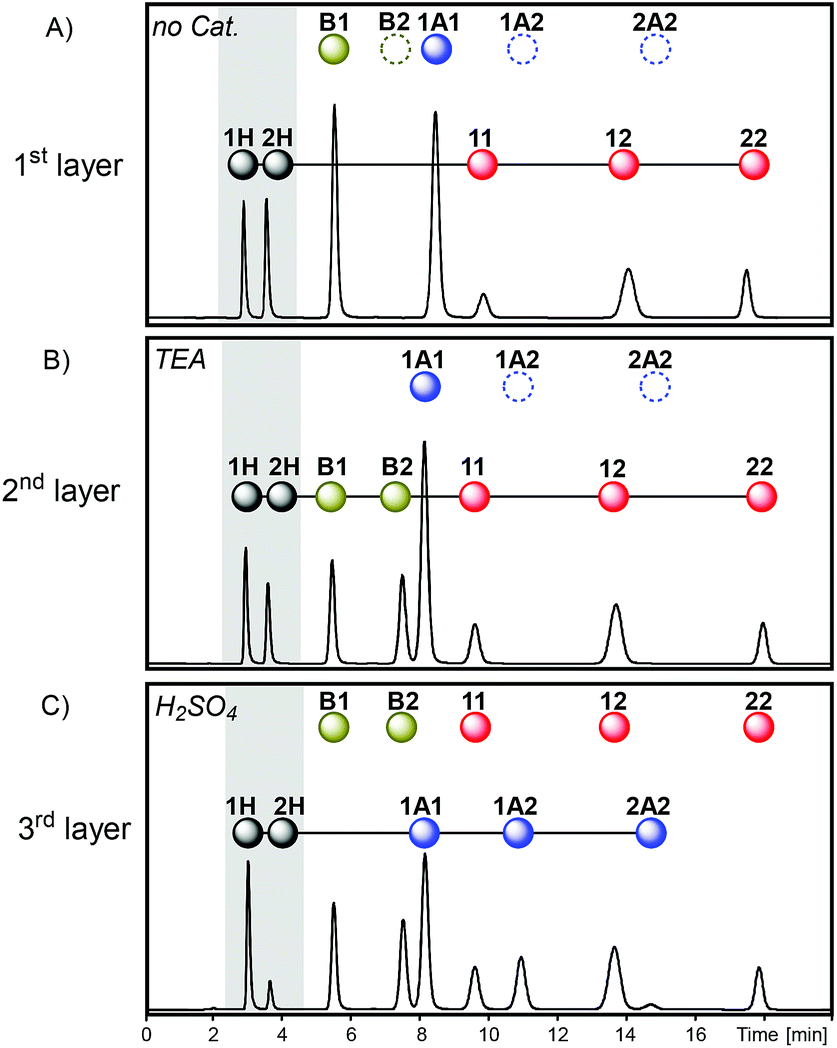 | ||
| Fig. 8 Chromatograms (Abs250) obtained in each step of sequence II after the equilibrium was reached in the absence of any catalyst (A, first layer), in the presence of TEA (B, second layer) and later in the presence of an excess of H2SO4 (C, third layer). | ||
It is worth mentioning, though, that the networks of the last layer of sequences I and II are different (Fig. 7) and this could affect the product distribution. In order to evaluate the effect of the history on the final product distributions of multilayer dynamic systems wherein the last layer is always the same network, three operations that do not involve the third layer of sequence I were studied: layer removal, layer substitution and layer merging (Fig. 7, sequences III–IV).
Thus, sequence III was obtained through the removal of the first layer of sequence I (Fig. 7). In this case, the initial dithioacetal exchange led to an equilibrium composition dominated by products that contain building block 2: the proportion of peak areas 1H/2H was 0.7 while the proportion of 1A1/2A2 was 0.5 (Fig. 9A). This is because building block 1 is mostly trapped in the form of thioester B1 and disulfide 11 so it cannot be exchanged under acid catalysis. Next, the dithioacetal level was blocked while the disulfide-thioester level was activated. At this stage, a net transference of building block 2 was observed from the thiol pool towards the thioester and disulfide pools, to produce B2, 12 and 22 at the expense of 2H (the 1H/2H peak area proportion increases from 0.7 to 4.0, Fig. 9Aversus9B).
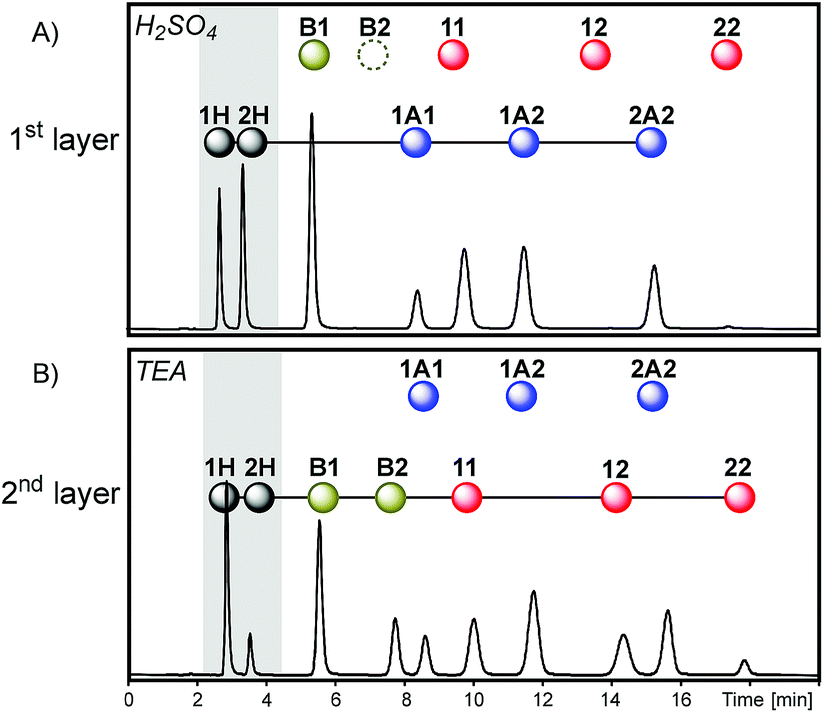 | ||
| Fig. 9 Chromatograms (Abs250) obtained in each step of sequence III after the equilibrium was reached in the presence of H2SO4 (A, first layer) and later in the presence of an excess of TEA (B, second layer). | ||
A comparison of the final compositions for sequences I and III illustrates the influence of history. These two mixtures have the same components and the same network topology (see the last layers of sequences I and III in Fig. 7). However, the relative product concentrations at the equilibrium of these two dynamic mixtures were different. The final layer of sequence III had a proportion of dithioacetals 1A1/2A2 = 0.5 (Fig. 9B), significantly lower than the value of 9.5 observed for sequence I (Fig. 6D).
In sequence I, dithioacetal exchange was activated after equilibration of disulfide exchange and, as a consequence, part of building block 2 was stored in the disulfide pool and was unavailable for exchange in the acidic medium. In sequence III, since the first layer was removed, dithioacetal exchange was the first active reaction and a higher amount of 2 could be processed by the dithioacetal pool.
If the first layer of sequence I remains the same, and the second layer is removed, again, a different final composition is achieved. Since in the reference sequence I, dithioacetal exchange was active only in the second layer, removal of this layer led to a final distribution wherein 1A1 was the only dithioacetal present (Fig. 8B).71 Because building block 2 was never incorporated into dithioacetal products along this sequence IIshort, the final relative concentrations B1/B2 and 11/22 were lower than those in sequence I.
An alternative way of affecting the system history is through the substitution of the first layer of sequence I by a different layer to produce sequence IV (Fig. 7). Initially, disulfide and thioester exchanges were activated in the presence of TEA, and then sulfuric acid was added to stop both exchanges and to activate the exchange of dithioacetal products and, finally, a new addition of TEA (in excess) activated disulfide and thioester exchanges and simultaneously deactivated dithioacetal exchange (Fig. 10).
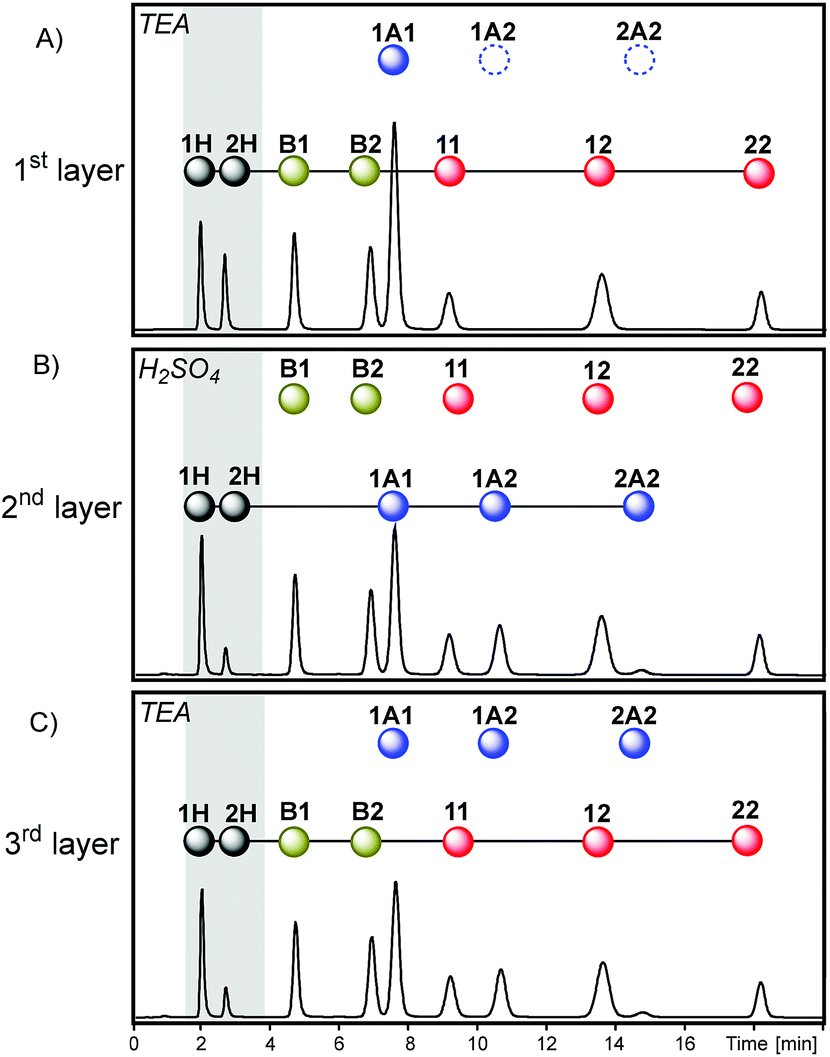 | ||
| Fig. 10 Chromatograms (Abs250) obtained in each step of sequence IV after the equilibrium was reached in the presence of TEA (A, first layer), in the presence of an excess of H2SO4 (B, second layer) and later in the presence of an excess of TEA (C, third layer). The sequence started from a mixture containing dithioacetal 1A1 (30 mM), thioester B1 (30 mM), disulfide 11 (30 mM) and 2H (60 mM). | ||
From the network-wiring point of view, the only difference between sequences I and IV is the connection of thioester exchange to the first layer of sequence IV. This difference was translated into significant differences in the relative concentration of the thioesters B1 and B2 at the end of the sequence (the proportions of peak areas B1/B2 were 1.7 and 0.9 in sequences I and IV respectively, Fig. 6D and 10C). The lower relative concentration of B2 is due to the fact that in sequence I thioester exchange was activated only after some building block 2 had been trapped as dithioacetals, whereas in sequence IV the thioester exchange was activated, for the first time, before dithioacetal exchange.
Finally, a more subtle way to change the system history was attempted by merging the first two layers. With this in mind, the reactions involved in the first and second layers of sequence I were simultaneously activated leading to sequence V (Fig. 7).
Simultaneous dithioacetal and disulfide exchanges (SnCl2/heat) provided a composition where compounds containing building block 1 were present in a higher amount, as indicated by the relative peak areas: 1H > 2H, 11 > 22 and 1A1 > 2A2 (Fig. 11A). Further inactivation of dithioacetal exchange at the time that thioester exchange was activated (along with disulfide exchange) led to a net transference of building block 2 from the disulfide pool to the thioester pool. The most evident change was the appearance of the thioester B2 signal, which is mainly formed with the building block coming from disulfide 22 (Fig. 11B). As previously observed for sequence I, numerical simulations of the equilibrium composition after each activation step of sequences II–V lead to product distributions that resemble those observed experimentally (Fig. S24–S27†). The simulations indicate that the catalysts used did not affect the relative stability of the library members, i.e. the product distribution. Therefore, the differences observed in the final composition of sequences I–IV are defined only by the activation order of the different reversible reactions.
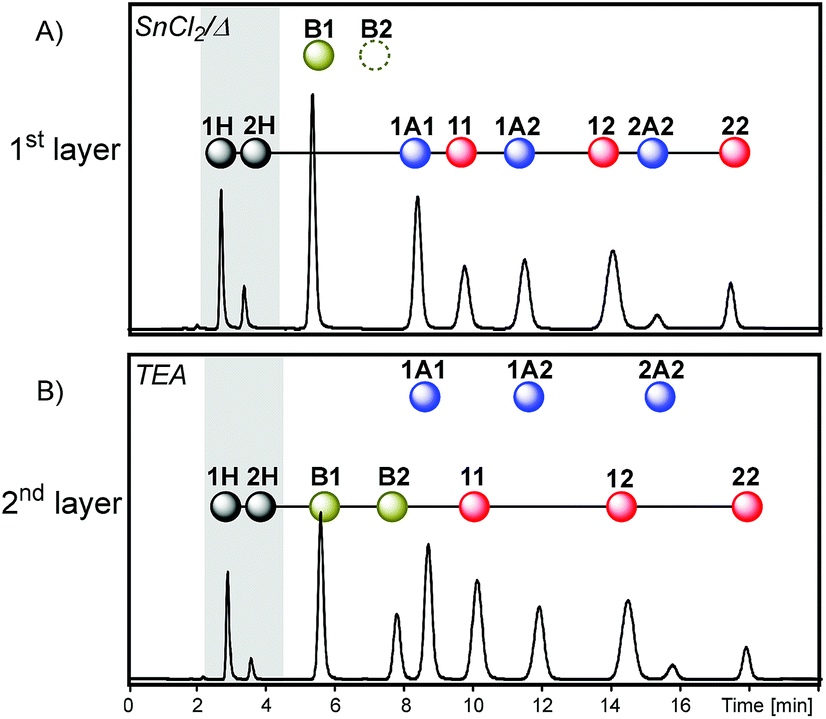 | ||
| Fig. 11 Chromatograms (Abs250) obtained in each step of sequence V starting from a mixture containing dithioacetal 1A1 (30 mM), thioester B1 (30 mM), disulfide 11 (30 mM), 2H (60 mM) and SnCl2 (1.5 mM). Sequential addition of catalysts/conditions is as follows: (A) 960 min after 100 °C heating, Ar atmosphere and (B) 10 min after the addition of TEA (300 mM), Ar atmosphere. Solvent: acetonitrile (total volume: 500 μL). | ||
A comparison of the final product distribution of sequences I–V illustrates how some changes in a layer, early in the sequence, can be read in the last layer (Fig. 12). In this extremely simple multilayer system, the final product distribution depended on the order in which the different reversible reactions had been previously activated/deactivated. Communication between the reversible reactions used allowed spreading of building blocks 1 and 2 over the different pools of compounds (thioesters, disulfides, dithioacetals and thiols). The order of activation/deactivation of the different reactions influenced the proportion in which these building blocks accumulated within each pool.
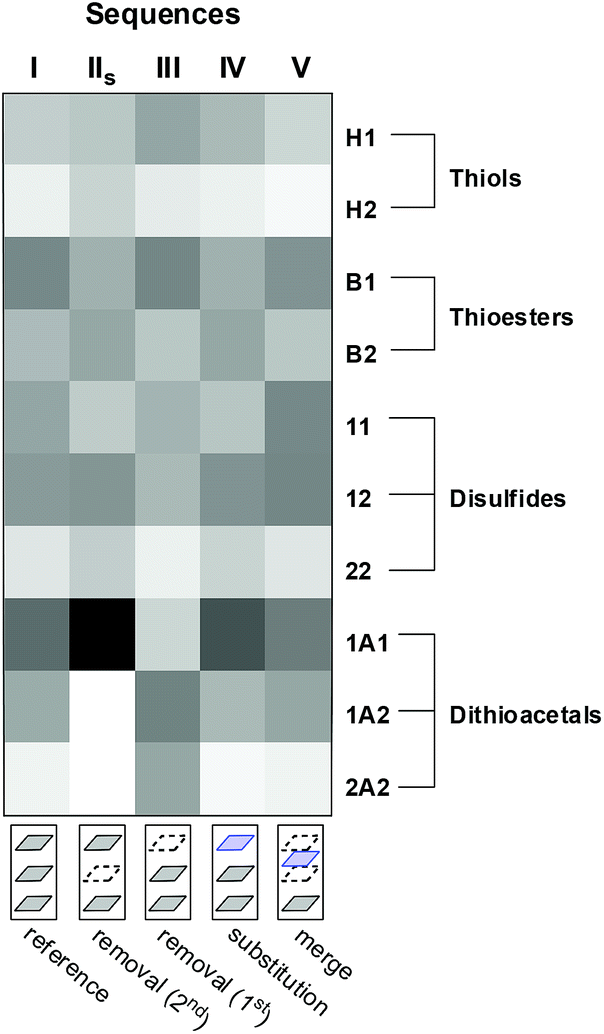 | ||
| Fig. 12 Heat map plot showing the final abundances of library members for the different sequences. Shade represents the peak area normalized to the total peak area. | ||
In the model used, the total amount of 1 is 2.5 times the amount of 2, which introduces a bias towards products containing the former building block. This preference is observed in the final product distribution of sequence I: [B1] > [B2], [11] > [22] and [1A1] > [2A2] (Fig. 12). Some of the operations with the layers of sequence I inverted this preference within the different compound pools. Removal of the second layer (to produce sequence IIshort) or substitution of the first layer (to produce sequence IV) leads to final compositions wherein [B1] < [B2] and [11] ≤ [22] (Fig. 12). Interestingly, in the first case (sequence IIshort) the product preference within the disulfide and thioester pools was reverted when a dithioacetal-related layer was removed. In the second case (sequence IV), the product preferences within the disulfide and the thioester pools were reverted through the rewiring of a layer where these reactions are involved, i.e. the connection of the thioester nodes to the network (compare the 1st layer of sequence I vs. the 1st layer of sequence IV in Fig. 7).
Likewise, removal of the first layer (sequence III) reverted the preference at the dithioacetal level leading to a final composition where [1A1] < [2A2] (Fig. 12). Again, in this case the product preference within the dithioacetal pool was reverted by the removal of a layer that involves only the exchange of disulfides.
Conclusions
In systems with a single reversible reaction, composition changes can be studied under out-of-equilibrium conditions. Once at equilibrium, the situation is rather different. By definition, thermodynamic equilibrium is a state of maximum entropy, so no information about the history of the system can be stored in it.By judicious choice of reactions and conditions, different single-reaction systems can be coupled to produce multilayer networks that include strings of reversible reactions.
Two interesting consequences arise in systems where sequential reversible reactions are communicated through common nodes. First, the activation/deactivation order of the reactions gives the chemical pathway a direction in the time function,72 which configures the history of the system.
Second, given that some compounds (connectors) act as starting materials and as products of different reactions, their concentration can affect the composition of different exchange pools. Thus, thermodynamic systems with sequential reactions can keep track of previous equilibration events in their history. This attribute can be particularly interesting if we consider the intrinsic adaptability of each layer (granted by the use of reversible chemistry). Environmental changes in early layers could be imprinted in final product distributions even if they no longer exist at the time the sequence has finished.
Altogether, these features give rise to a new design level that, in our opinion, deserves further exploration.
Adaptability is a key feature of dynamic covalent chemistry. Such a property has led to the discovery of new functional materials,3–7 drugs8–10 and catalysts.73 Multilayer chemical network engineering can prove useful in the functional screening of dynamic combinatorial libraries (DCLs). By judicious choice of reaction conditions, a library can be biased toward different layers giving access to the modular exploration of different regions of chemical space and connectivity.
DCL adaptation has also been used to sense the presence of analytes.13,14 In multilayer systems, the introduction of the time dimension can be useful to sense analytes added at different times. Such information could be read in the final composition.
Systems chemistry aims to develop molecular systems of interacting and/or interconverting molecules that show emergent properties.22,23 The preparation of complex reaction networks constitutes an integral part of systems chemistry.74–76 In this context, dynamic systems that can respond to environmental changes (light, acidity, metals, etc.) by switching their type of reversible chemistry and connectivity can represent an exciting study case for emergent behaviour.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was funded by the Fondo para la Investigación Científica y Tecnológica (FONCYT) (PICT2015-3574), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), and Universidad Nacional de Rosario. M. M.-A. acknowledges CONICET for a postdoctoral fellowship. A. G. O. and R. L. E. F. are CONICET researchers.Notes and references
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- Y. Zhang and M. Barboiu, Chem. Rev., 2016, 116, 809–834 CrossRef CAS PubMed.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC.
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- T. Maeda, H. Otsuka and A. Takahara, in Dynamic Combinatorial Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2009, pp. 229–260 Search PubMed.
- Dynamic combinatorial chemistry: in drug discovery, bioorganic chemistry, and materials science, ed. B. L. Miller, Wiley, 2009 Search PubMed.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Drug Discovery Today, 2002, 7, 117–125 CrossRef CAS PubMed.
- O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26–36 CrossRef PubMed.
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS PubMed.
- E. Bartolami, C. Bouillon, P. Dumy and S. Ulrich, Chem. Commun., 2016, 52, 4257–4273 RSC.
- A. Herrmann, Chem.–Eur. J., 2012, 18, 8568–8577 CrossRef CAS PubMed.
- S. Otto and K. Severin, in Creative Chemical Sensor Systems, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 267–288 Search PubMed.
- Y. Liu and K. C.-F. Leung, in Dynamic Covalent Chemistry: Principles, Reactions and Applications, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 287–319 Search PubMed.
- D. A. Leigh, U. Lewandowska, B. Lewandowski and M. R. Wilson, Molecular Machines and Motors, in Topics in Current Chemistry, ed. A. Credi, S. Silvi and M. Venturi, 2014, vol. 354, pp. 111–138 Search PubMed.
- M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656–3676 RSC.
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
- R. A. R. Hunt and S. Otto, Chem. Commun., 2011, 47, 847–858 RSC.
- A. G. Orrillo, A. M. Escalante, M. Martinez-Amezaga, I. Cabezudo and R. L. E. Furlan, Chem.–Eur. J., 2019, 25, 1118–1127 CrossRef CAS PubMed.
- S. Otto, Acc. Chem. Res., 2012, 45, 2200–2210 CrossRef CAS PubMed.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- O. Š. Miljanić, Chem, 2017, 2, 502–524 Search PubMed.
- A. S. Y. Wong and W. T. S. Huck, Beilstein J. Org. Chem., 2017, 13, 1486–1497 CrossRef CAS PubMed.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- H. M. Seifert, K. Ramirez Trejo and E. V. Anslyn, J. Am. Chem. Soc., 2016, 138, 10916–10924 CrossRef CAS PubMed.
- S. Lascano, K.-D. Zhang, R. Wehlauch, K. Gademann, N. Sakai and S. Matile, Chem. Sci., 2016, 7, 4720–4724 RSC.
- A. M. Escalante, A. G. Orrillo and R. L. E. Furlan, J. Comb. Chem., 2010, 12, 410–413 CrossRef CAS PubMed.
- L. Rocard, D. Wragg, S. A. Jobbins, L. Luciani, J. Wouters, S. Leoni and D. Bonifazi, Chem.–Eur. J., 2018, 24, 16136–16148 CrossRef CAS PubMed.
- R. C. Lirag and O. Š. Miljanić, Chem. Commun., 2014, 50, 9401–9404 RSC.
- J. Leclaire, L. Vial, S. Otto and J. K. M. Sanders, Chem. Commun., 2005, 1959–1961 RSC.
- S. Kulchat, M. N. Chaur and J.-M. Lehn, Chem.–Eur. J., 2017, 23, 11108–11118 CrossRef CAS PubMed.
- J. F. Reuther, S. D. Dahlhauser and E. V. Anslyn, Angew. Chem., Int. Ed., 2019, 58, 74–85 CrossRef CAS PubMed.
- B. M. Matysiak, P. Nowak, I. Cvrtila, C. G. Pappas, B. Liu, D. Komáromy and S. Otto, J. Am. Chem. Soc., 2017, 139, 6744–6751 CrossRef CAS PubMed.
- Y. Hai, H. Zou, H. Ye and L. You, J. Org. Chem., 2018, 83, 9858–9869 CrossRef CAS PubMed.
- A. G. Orrillo, A. La-Venia, A. M. Escalante and R. L. E. Furlan, Chem.–Eur. J., 2018, 24, 3141–3146 CrossRef CAS PubMed.
- B. T. Worrell, S. Mavila, C. Wang, T. M. Kontour, C.-H. Lim, M. K. McBride, C. B. Musgrave, R. Shoemaker and C. N. Bowman, Polym. Chem., 2018, 9, 4523–4534 RSC.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 12063–12064 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 2002, 41, 898–952 CrossRef PubMed.
- A. G. Orrillo, A. M. Escalante and R. L. E. Furlan, Chem.–Eur. J., 2016, 22, 6746–6749 CrossRef CAS PubMed.
- A. G. Orrillo, A. M. Escalante and R. L. E. Furlan, Org. Lett., 2017, 19, 1446–1449 CrossRef CAS PubMed.
- Y.-C. Wu and J. Zhu, J. Org. Chem., 2008, 73, 9522–9524 CrossRef CAS PubMed.
- S. K. De, Tetrahedron Lett., 2004, 45, 2339–2341 CrossRef CAS.
- B. Tamami and K. P. Borujeny, Synth. Commun., 2003, 33, 4253–4258 CrossRef CAS.
- H. K. Patney, Tetrahedron Lett., 1991, 32, 2259–2260 CrossRef CAS.
- R. V. Anand, P. Saravanan and V. K. Singh, Synlett, 1999, 1999, 415–416 CrossRef.
- B. Cazes and S. Julia, Tetrahedron Lett., 1978, 19, 4065–4068 CrossRef.
- P. Stütz and P. A. Stadler, Org. Synth., 1977, 56, 8 CrossRef.
- B. M. Lamb and C. F. Barbas III, Chem. Commun., 2015, 51, 3196–3199 RSC.
- K. Du, S. Wang, R. S. Basha and C. Lee, Adv. Synth. Catal., 2019, 361, 1597–1605 CrossRef CAS.
- O. A. Mascaretti and R. L. E. Furlan, Aldrichimica Acta, 1997, 30, 55–68 CAS.
- L. R. Sutton, W. A. Donaubauer, F. Hampel and A. Hirsch, Chem. Commun., 2004, 1758–1759 RSC.
- P. A. Brady, R. P. Bonar-Law, S. J. Rowan, C. J. Suckling and J. K. M. Sanders, Chem. Commun., 1996, 319–320 RSC.
- S. J. Rowan, P. A. Brady and J. K. M. Sanders, Angew. Chem., Int. Ed. Engl., 1996, 35, 2143–2145 CrossRef CAS.
- R. Larsson, Z. Pei and O. Ramström, Angew. Chem., Int. Ed., 2004, 43, 3716–3718 CrossRef CAS PubMed.
- R. Larsson and O. Ramström, Eur. J. Org. Chem., 2006, 2006, 285–291 CrossRef.
- Y. Zhang, M. Angelin, R. Larsson, A. Albers, A. Simons and O. Ramström, Chem. Commun., 2010, 46, 8457–8459 RSC.
- S. Ghosh, L. A. Ingerman, A. G. Frye, S. J. Lee, M. R. Gagné and M. L. Waters, Org. Lett., 2010, 12, 1860–1863 CrossRef CAS PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73–77 CrossRef CAS PubMed.
- Y. Ruff, V. Garavini and N. Giuseppone, J. Am. Chem. Soc., 2014, 136, 6333–6339 CrossRef CAS PubMed.
- E. B. Hadley and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 16444–16445 CrossRef CAS PubMed.
- S. A. Berg and B. J. Ravoo, Soft Matter, 2014, 10, 69–74 RSC.
- C. Ghobril, K. Charoen, E. K. Rodriguez, A. Nazarian and M. W. Grinstaff, Angew. Chem., Int. Ed., 2013, 52, 14070–14074 CrossRef CAS PubMed.
- M. R. Meneghetti and S. M. P. Meneghetti, Catal. Sci. Technol., 2015, 5, 765–771 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- P. T. Corbett, S. Otto and J. K. M. Sanders, Chem.–Eur. J., 2004, 10, 3139–3143 CrossRef CAS PubMed.
- J. Atcher, A. Moure and I. Alfonso, Chem. Commun., 2013, 49, 487–489 RSC.
- K. Severin, Chem.–Eur. J., 2004, 10, 2565–2580 CrossRef CAS PubMed.
- A. G. Orrillo and R. L. E. Furlan, J. Org. Chem., 2010, 75, 211–214 CrossRef CAS PubMed.
- M. C. Misuraca, E. Moulin, Y. Ruff and N. Giuseppone, New J. Chem., 2014, 38, 3336–3349 RSC.
- Removal of the second layer of sequence I leads to a sequence that is equivalent to the first two layers of sequence II (sequence II short)..
- O. Penrose, in Chance in physics: foundations and perspectives, ed. J. Bricmont, D. Durr, M. C. Galavotti, G. C. Ghirardi, F. Petruccione and N. Zanghi, Springer, Berlin, 2001, pp. 61–82 Search PubMed.
- G. Gasparini, M. D. Molin and L. J. Prins, Eur. J. Org. Chem., 2010, 13, 2429–2440 CrossRef.
- S. N. Semenov, L. J. Kraft, A. Ainla, M. Zhao, M. Baghbanzadeh, V. E. Campbell, K. Kang, J. M. Fox and G. M. Whitesides, Nature, 2016, 537, 656–660 CrossRef CAS PubMed.
- F. Schaufelberger and O. Ramström, J. Am. Chem. Soc., 2016, 138, 7836–7839 CrossRef CAS PubMed.
- Z. Dadon, N. Wagner, S. Alasibi, M. Samiappan, R. Mukherjee and G. Ashkenasy, Chem.–Eur. J., 2015, 21, 648–654 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of thioester B1, preparation of one-level, two-level and three-level dynamic chemical systems, full kinetics of isolated-networked experiments and numerical simulations for expected equilibrium compositions when the activation sequences I–V are displayed. See DOI: 10.1039/c9sc02166c |
| This journal is © The Royal Society of Chemistry 2019 |