
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defying strain in the synthesis of an electroactive bilayer helicene†
Margarita
Milton‡
a,
Nathaniel J.
Schuster‡
*a,
Daniel W.
Paley
b,
Raúl
Hernández Sánchez
 c,
Fay
Ng
a,
Michael L.
Steigerwald
*a and
Colin
Nuckolls
*a
c,
Fay
Ng
a,
Michael L.
Steigerwald
*a and
Colin
Nuckolls
*a
aDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: njs2154@columbia.edu; mls2064@columbia.edu; cn37@columbia.edu
bColumbia Nano Initiative, Columbia University, New York, New York 10027, USA
cDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA
First published on 20th November 2018
Abstract
We report the synthesis of a bilayer chiral nanographene incorporating a [7]helicene scaffold and two perylene-diimide (PDI) subunits. Twofold visible-light-induced oxidative cyclization of a phenanthrene framework selects for the desired PDI-helicene, despite the immense strain that distinguishes this helicene from two other accessible isomers. This strain arises from the extensive intramolecular overlap of the PDI subunits, which precludes racemization, even at elevated temperatures. Relative to a smaller homologue, this PDI-helicene exhibits amplified electronic circular dichroism. It also readily and reversibly accepts four electrons electrochemically. Modifications to the core phenanthrene subunit change the fluorescence and electrochemistry of the PDI-helicene without significantly impacting its electronic circular dichroism or UV-visible absorbance.
Introduction
This study concerns the regioselective synthesis and characterization of the newest and most highly strained perylene-3,4,9,10-tetracarboxylic-diimide (PDI) helicene. In general, interactions between π-surfaces account for a variety of interesting electronic, optical, and magnetic phenomena. For instance, π-to-π overlap promotes charge transport within organic semiconductors,1,2 a key consideration in the design of organic electronic and optoelectronic devices.3,4 π-Extension of the termini of helicenes – helices of fused aromatic subunits surrounding a nonintersecting stereogenic axis – provides chiral nanographenes with considerable intramolecular π-to-π overlap (Fig. 1a). Such chiral nanographenes may exhibit very large circular dichroism,5–7 circularly polarized luminescence,8,9 non-linear optical properties,10,11 and chiral-induced spin selectivity.12 | ||
| Fig. 1 (a) The fusion of two armchair nanographenes with phenanthrene provides a π-extended bilayer helicene. (b) Fusion with two PDI subunits via oxidative photocyclization at elevated temperatures overwhelmingly favors the formation of PPDH over its isomers, 5PPD and PPPD. | ||
The synthesis of helicenes, especially those with extensively eclipsed π-surfaces, hinges on the strategic accumulation of strain.13–15 Some synthetic complications can be avoided by attaching bulky polyaromatics after forming the helicene core.16 For other approaches, the possibility of unwanted cyclizations and attenuated reactivity must be considered.17–24 Clear synthetic guidelines exist for the preparation of helicenes via the oxidative photocyclization of stilbene-like scaffolds incorporating phenylene and naphthylene subunits.25 However, ensuring the exclusive formation of helicenes from the oxidative photocyclization of phenanthrene-based substrates remains difficult,26,27 especially when both twisted and planar (or nearly planar) polyaromatic products can be formed.28
Here we disclose the synthesis of a π-extended [7]helicene via the regioselective visible-light-induced oxidative fusion of phenanthrene with two PDI subunits. This phenanthryl-linked PDI-dimer helicene (PPDH, Fig. 1b) belongs to an emerging class of chiral, shape-persistent, PDI-based materials prepared by intramolecular oxidative photocyclizations.29–31 Extensive intramolecular overlap of the π-surface distinguishes PPDH from its PDI-dimer helicene predecessors and the vast majority of π-extended helicenes.32–41 The strain attendant on this overlap disfavors the exclusive synthesis of PPDH near room temperature: the undesired [5]helicene isomer (5PPD, Fig. 1b) also forms. Simply raising the reaction temperature gives near-exclusive production of PPDH. This regioselectivity belies the disparity in strain between PPDH, 5PPD, and their planar isomer (PPPD, Fig. 1b). Single-crystal X-ray diffraction (SCXRD) of PPDH confirmed extensive intramolecular overlap of the π-surface, which contorts to mitigate intramolecular PDI-to-PDI contact. PPDH exhibits large electronic circular dichroism (ECD) in the visible range, especially relative to a smaller helical homologue, and readily accepts up to four electrons electrochemically. Finally, the fluorescence and electrochemistry of PPDH can be tuned by changing the substituents on its phenanthrene core.
Results and discussion
Scheme 1 depicts our synthesis of PPDH. Intramolecular oxidative photocyclizations have been used to synthesize several helicenes from aryl-linked PDI-oligomers.42,43 Acenes, in particular, fuse preferentially at the more sterically hindered peri-position under these reaction conditions,29–31,44 whereas oxidative photocyclizations onto 3,6-disubstituted phenanthrene (such as PPD in Scheme 1) may occur at any combination of the adjacent positions. We found that visible-light-induced oxidative cyclization of PPD at 110 °C formed PPDH almost exclusively (entry 3, Table 1). Repeating the oxidative photocyclization of PPD at 70 °C lowered the regioselectivity for PPDH: a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of PPDH and 5PPD, respectively, resulted. A reaction temperature of 30 °C further decreased this ratio of PPDH-to-5PPD to 7:3. We never detected PPPD, no matter the reaction temperature. This absence of sterically unencumbered PPPD and the modest yields of 5PPD underscore a strong electronic preference for PPDH under these reaction conditions. Substituting two pentoxy substituents onto the phenanthrene linker does not alter this preference: these pentoxy substituents slowed the oxidative photocyclization of PPD (entry 4, Table 1), but did not dramatically alter the yield.
:1 mixture of PPDH and 5PPD, respectively, resulted. A reaction temperature of 30 °C further decreased this ratio of PPDH-to-5PPD to 7:3. We never detected PPPD, no matter the reaction temperature. This absence of sterically unencumbered PPPD and the modest yields of 5PPD underscore a strong electronic preference for PPDH under these reaction conditions. Substituting two pentoxy substituents onto the phenanthrene linker does not alter this preference: these pentoxy substituents slowed the oxidative photocyclization of PPD (entry 4, Table 1), but did not dramatically alter the yield.
 | ||
| Scheme 1 Two-step synthesis of PPDH from brominated PDI [R = CH(C5H11)2]. | ||
| Entry | R1 | T (°C) | Solvent | Time (h) |
PPDH:5PPDb |
Isolated yield of PPDH (%) |
|---|---|---|---|---|---|---|
| a The solutions of PPD (0.2 mM) and iodine (1.3 mM) were irradiated with two 55 W compact fluorescent lamps. Only a silica plug was necessary to remove 5PPD from PPDH. See the ESI for additional details. b Determined by 1H NMR of the product mixture. c 5PPD was not observed, perhaps due to decomposition. d Reaction did not go to completion; the remainder consisted of mono-cyclized intermediate and decomposition byproduct. | ||||||
| 1 | H | 30 | PhH | 24 | 70:30 |
63 |
| 2 | H | 70 | PhH | 24 | 91:9 |
83 |
| 3 | H | 110 | PhCl | 24 | >95:<5 |
91 |
| 4 | OC5H11 | 70 | PhH | 76 | —c | 88d |
The strain energies of 5PPD, PPDH, and PPPD have no bearing on the regioselectivity observed for the twofold oxidative photocyclization of PPD. The DFT-optimized (B3LYP/6-31G**) structures of 5PPD and PPDH are more strained (by 11 and 21 kcal mol−1, respectively) than PPPD (see Section VII of the ESI† for the calculation details). Despite this strain, PPDH does not rearrange or decompose otherwise under ambient conditions, enabling the resolution of its left- (M) and right-handed (P) helices via chiral HPLC (Fig. S1†). The obvious rigidity of this PDI-helicene precludes inversion: optically pure PPDH heated in diphenyl ether at 250 °C under air for one hour showed neither racemization nor decomposition by HPLC (Fig. S2†). This stability contrasts with the relative lability of [7]helicene alone, which racemizes completely in solution in just over 3 h at ∼257 °C.45
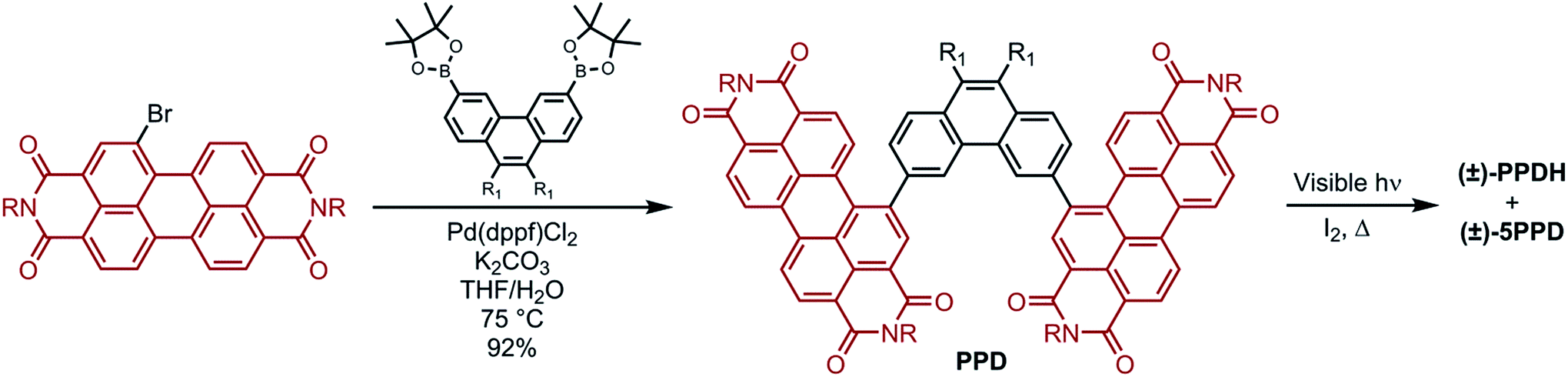
Fig. 2a displays the ECD spectra of the resolved enantiomers of PPDH and its bis(pentoxy) analogue, PPDH-OPe (entry 4, Table 1). They exhibit many Cotton effects across the UV-visible range. The largest transitions of PPDH manifest as a bisignate pair centered at ∼360 nm: |Δε| of 134 M−1 cm−1 at 344 nm and 214 M−1 cm−1 at 396 nm. Naphthyl-linked PDI-dimer helicene (NPDH; see the inset of Fig. 2a) – the [6]helicene homologue of PPDH – shares this feature, albeit its corresponding Cotton effects are diminished: |Δε| of 41 M−1 cm−1 at 355 nm and 56 M−1 cm−1 at 401 nm (Fig. 2a). This strong enhancement in ECD from NPDH to PPDH deviates from the trend observed for the most intense bisignate pair shared by the carbohelicenes. Specifically, lengthening the carbohelicenes (e.g., [6]helicene to [7]helicene) has little effect on the intensity of the longer wavelength Cotton effect of this pair, which plateaus at ∼260 M−1 cm−1.46
 | ||
| Fig. 2 (a) ECD, (b) UV-visible absorbance, and (c) g-factors of PPDH, PPDH-OPe, and NPDH in THF (10 μM, 1 cm path length). (Inset of b) Normalized fluorescence intensities of PPDH and PPDH-OPe in cyclohexane (3 μM, λex = 410 nm; Φf = 41% and 15% for PPDH and PPDH-OPe, respectively). For NPDH, R = CH(C5H11)2. | ||
Chiral oligomers often exhibit ECD whose intensity scales linearly (or nearly linearly) with UV-visible absorbance;23,47 however, the large increase in ECD from NPDH to PPDH in the 340–410 nm regime cannot be attributed to an increase in absorbance of unpolarized light. Relative to the Cotton effects of NPDH at 355 and 401 nm, the Cotton effects of PPDH at 344 and 396 nm show a greater than threefold increase in Δε. The same increases are not observed between the UV-visible absorbance spectra of these PDI-helicenes (Fig. 2b). In fact, the absorbance of NPDH and PPDH essentially match at 401 and 396 nm, respectively, resulting in a significant disparity in g-factor (|Δε|/ε) at these wavelengths: 1.5 × 10−3 for NPDH and 6.8 × 10−3 for PPDH (Fig. 2c).
In addition to amplifying the ECD of PPDH compared to NPDH, the phenanthrene bridge also tunes photoluminescence. The pentoxy chains of PPDH-OPe lower the fluorescence quantum yield (Φf) to 15%, compared to 41% for unsubstituted PPDH. We attribute this decrease to intramolecular donor–acceptor charge transfer following excitation of the PDI-helicene. Excited-state charge transfer also accounts for fluorescence quenching in PDI-based compounds with π-donor substituents.48–50 On the other hand, the UV-visible and ECD spectra of PPDH and PPDH-OPe are nearly identical.
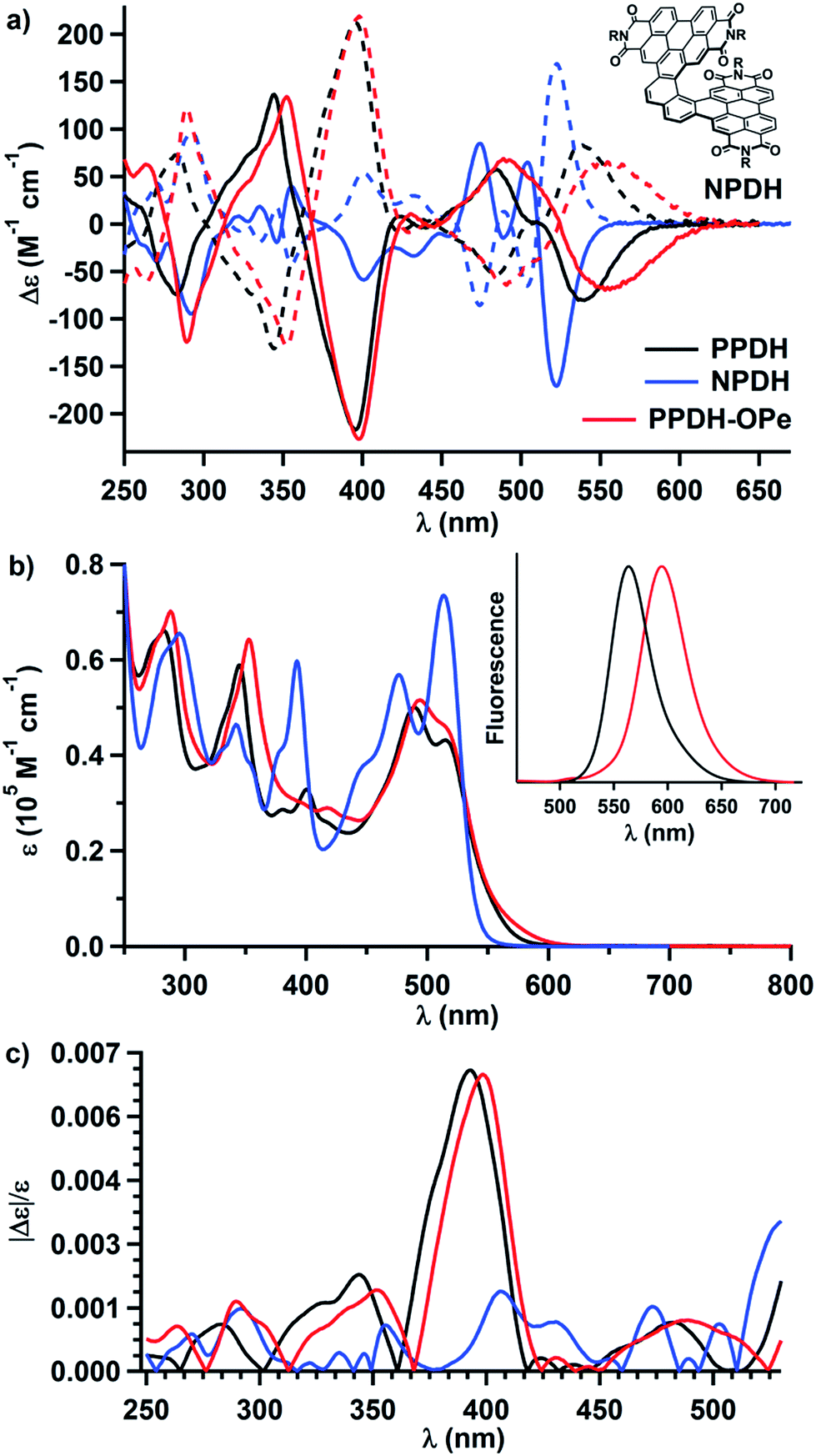
SCXRD revealed that the racemate of PPDH assembles into columns of alternating M- and P-helices (Fig. 3a and S3†). The intermolecular junction in these columns consists of 24 pairs of eclipsing π-bonded carbon atoms (see Fig. S4† for clarification), four of which approach to within 3.4 Å (i.e., twice the van der Waals radius of the carbon atom). This heterochiral columnar packing arrangement resembles that observed previously for a π-helix-of-PDI-helicenes31 but differs from the crystal structures of other helical bilayer nanographenes.16,51 For these latter species, C–H⋯π interactions predominate, stymying intermolecular π-to-π overlap.52
 | ||
| Fig. 3 Structure of PPDH from SCXRD. Free solvent, the CH(C5H11)2 chains, and hydrogen atoms have been hidden to provide an unobstructed view of the aryl surface. Thermal ellipsoids are set at 30% probability. (a) The racemate assembles as heterochiral columns in the solid state. Extensive intramolecular overlap between the PDI subunits (b) distorts their perylene cores into shallow bowls (c). | ||
SCXRD also revealed extensive intramolecular overlap in PPDH (Fig. 3b); this contorts the π-surface. Each PDI subunit warps along its imide-to-imide axis, resulting in bend angles of 9° and 11° between the naphthalene fragments in each perylene (Fig. S5†). For reference, this angle in crystalline PDI-helicenes incorporating [5] and [6]helicene scaffolds measures 3° and 6°, respectively.31,43 The [7]helicene scaffold in PPDH also splays farther from planarity than [7]helicene alone: the dihedral angle defined by the terminal rings of the [7]helicene within PPDH measures 40°, whereas the same angle in crystalline [7]helicene measures 32°.53,54 Moreover, the centroid-to-centroid distance between these same rings in PPDH equals 4.0 Å (Fig. 3c); the difference is 3.8 Å in carbo[7]helicene. The resultant gap between the PDI-subunits can host guests: for instance, part of a molecule of α,α,α-trifluorotoluene fills this π-cavity in the crystal structure of PPDH-OPe (Fig. S6†).
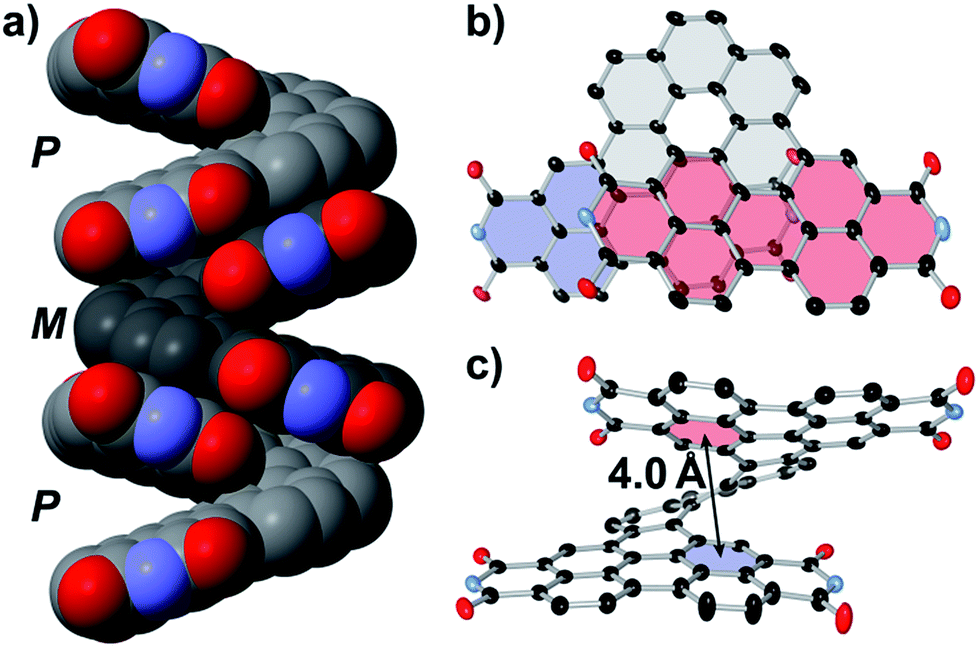
Cyclic voltammetry (CV) of PPDH in dichloromethane revealed three well-resolved reduction events (Fig. 4), all of which are chemically reversible. The two initial events (−1.10 and −1.27 V vs. Fc/Fc+) are one electron processes, whereas the greater intensity of the third (−1.54 vs. Fc/Fc+) affirms the near-simultaneous addition of the third and fourth electrons; thus, PPDH accepts one electron per imide group. Similar three-event CV profiles have been observed for non-conjugated PDI-dimers with extensive intramolecular π-to-π overlap;55–57 therefore, one might assume the fused phenanthrene linker in PPDH has a limited impact on the electrochemistry of the molecule. In fact, the bis(pentoxy)phenanthrene in PPDH-OPe cathodically shifts each reduction event by 200 mV (−1.30, −1.47, and −1.74 V vs. Fc/Fc+). This LUMO elevation exceeds the −180 mV shift observed upon connecting two dodecoxy substituents directly to the bay regions of monomeric PDI.58 As such, the phenanthrene linker can be modified to significantly tune the onset of reduction in PPDH, without altering the overall electrochemical profile.
 | ||
| Fig. 4 Cyclic voltammograms of PPDH, PPDH-OPe, 5PPD, and monomeric PDI (1 mM, 50 mV s−1 scan rate) in Ar-sparged DCM with 0.1 M [Bu4N][PF6] as the supporting electrolyte. For all species, R = CH(C5H11)2. | ||
Conclusions
Visible-light-induced oxidative cyclization of phenanthryl-linked PDI-dimer PPD yields the bilayer [7]helicene PPDH. Increasing the reaction temperature favors the near-exclusive formation of PPDH over its less strained isomers, 5PPD and PPPD. Intramolecular overlap of the π-surface of PPDH precludes racemization, even in solution at 250 °C, and results in a voltammetric profile consistent with those of dimeric PDI molecules with similarly eclipsed surfaces. Substitutions onto the phenanthrene linker change the electrochemistry and some of the spectroscopic properties of PPDH. For instance, bis(pentoxy)phenanthrene quenches fluorescence and cathodically shifts the onset of reduction; however, it negligibly alters the UV-visible absorbance and ECD of PPDH. Racemic PPDH self-assembles into heterochiral columns in the solid state. The extensive intermolecular π-to-π overlap within these columns could facilitate charge transport in organic electronic devices. Retention of the same crystalline morphology for optically pure PPDH would enable the preparation of transistors that respond preferentially to circularly polarized light,59 as well as potentially enhance chiral-induced spin selectivity.60Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. N. thanks Sheldon and Dorothea Buckler for their generous support. Primary support for this project was provided by the U.S. Department of Energy (DOE) under award no. DESC0014563. Partial support was provided by the Office of Naval Research under award no. N00014-16-1-2921. Instruments in the Nakanishi and Owen laboratories at Columbia University were used for this research. We thank both laboratories, as well as Dr Nina Berova and Dr Steffen Jockusch, for their generosity. R. H. S. acknowledges the support from the Columbia Nano Initiative Postdoctoral Fellowship. SCXRD was performed at the Shared Materials Characterization Laboratory (SMCL) at Columbia University. Use of the SMCL was made possible by funding from Columbia University.Notes and references
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603–4625 RSC.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- B. Gu, C. Zhao, A. Baev, K.-T. Yong, S. Wen and P. N. Prasad, Adv. Opt. Photonics, 2016, 8, 328–369 CrossRef.
- D. Dini, M. J. F. Calvete and M. Hanack, Chem. Rev., 2016, 116, 13043–13233 CrossRef CAS PubMed.
- R. Naaman and D. H. Waldeck, J. Phys. Chem. Lett., 2012, 3, 2178–2187 CrossRef CAS PubMed.
- W. H. Laarhoven and W. J. C. Prinsen, Top. Curr. Chem., 1984, 125, 63–130 CrossRef CAS.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- P. J. Evans, J. Ouyang, L. Favereau, J. Crassous, I. Fernández, J. Perles and N. Martín, Angew. Chem., Int. Ed., 2018, 57, 6774–6779 CrossRef CAS PubMed.
- M. Scholz, M. Mühlstädt and F. Dietz, Tetrahedron Lett., 1967, 8, 665–668 CrossRef.
- R. H. Martin, M. Flammang-Barbieux, J. P. Cosyn and M. Gelbcke, Tetrahedron Lett., 1968, 9, 3507–3510 CrossRef.
- R. H. Martin, M.-J. Marchant and M. Baes, Helv. Chim. Acta, 1971, 54, 358–360 CrossRef CAS.
- F. B. Mallory and C. W. Mallory, J. Org. Chem., 1983, 48, 526–532 CrossRef CAS.
- A. Sudhakar and T. J. Katz, Tetrahedron Lett., 1986, 27, 2231–2234 CrossRef CAS.
- E. Murguly, R. McDonald and N. R. Branda, Org. Lett., 2000, 2, 3169–3172 CrossRef CAS PubMed.
- J. Roose, S. Achermann, O. Dumele and F. Diederich, Eur. J. Org. Chem., 2013, 3223–3231 CrossRef CAS.
- Y. Nakakuki, T. Hirose, H. Sotome, H. Miyasaka and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 4317–4326 CrossRef CAS PubMed.
- K. Mori, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 6847–6851 CrossRef CAS PubMed.
- R. El Abed, B. Ben Hassine, J.-P. Genêt, M. Gorsane and A. Marinetti, Eur. J. Org. Chem., 2004, 1517–1522 CrossRef CAS.
- T. Kogiso, K. Yamamoto, H. Suemune and K. Usui, Org. Biomol. Chem., 2012, 10, 2934–2936 RSC.
- W. H. Laarhoven, T. H. J. H. M. Cuppen and R. J. F. Nivard, Tetrahedron, 1970, 26, 4865–4881 CrossRef CAS.
- N. J. Schuster, D. W. Paley, S. Jockusch, F. Ng, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2016, 55, 13519–13523 CrossRef CAS PubMed.
- K. Khokhlov, N. J. Schuster, F. Ng and C. Nuckolls, Org. Lett., 2018, 20, 1991–1994 CrossRef CAS PubMed.
- N. J. Schuster, R. Hernández Sánchez, D. Bukharina, N. A. Kotov, N. Berova, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2018, 140, 6235–6239 CrossRef CAS PubMed.
- C. Lütke Eversloh, Z. Liu, B. Müller, M. Stangl, C. Li and K. Müllen, Org. Lett., 2011, 13, 5528–5531 CrossRef PubMed.
- S. Xiao, S. J. Kang, Y. Wu, S. Ahn, J. B. Kim, Y.-L. Loo, T. Siegrist, M. L. Steigerwald, H. Li and C. Nuckolls, Chem. Sci., 2013, 4, 2018–2023 RSC.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763–7768 CrossRef CAS PubMed.
- T. Fujikawa, Y. Segawa and K. Itami, J. Org. Chem., 2017, 82, 7745–7749 CrossRef CAS PubMed.
- Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef CAS PubMed.
- C. M. Cruz, I. R. Márquez, I. F. A. Mariz, V. Blanco, C. Sánchez-Sánchez, J. M. Sobrado, J. A. Martín-Gago, J. M. Cuerva, E. Maçôas and A. G. Campaña, Chem. Sci., 2018, 9, 3917–3924 RSC.
- D. Reger, P. Haines, F. W. Heinemann, D. M. Guldi and N. Jux, Angew. Chem., Int. Ed., 2018, 57, 5938–5942 CrossRef CAS PubMed.
- Y. Zhu, Z. Xia, Z. Cai, Z. Yuan, N. Jiang, T. Li, Y. Wang, X. Guo, Z. Li, S. Ma, D. Zhong, Y. Li and J. Wang, J. Am. Chem. Soc., 2018, 140, 4222–4226 CrossRef CAS PubMed.
- K. Kato, Y. Segawa, L. T. Scott and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 1337–1341 CrossRef CAS PubMed.
- C. M. Cruz, S. Castro-Fernández, E. Maçôas, J. M. Cuerva and A. G. Campaña, Angew. Chem., Int. Ed., 2018, 57, 14782–14786 CrossRef CAS PubMed.
- P. E. Hartnett, H. S. S. R. Matte, N. D. Eastham, N. E. Jackson, Y. Wu, L. X. Chen, M. A. Ratner, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, Chem. Sci., 2016, 7, 3543–3555 RSC.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS PubMed.
- Y. Li, L. Xu, T. Liu, Y. Yu, H. Liu, Y. Li and D. Zhu, Org. Lett., 2011, 13, 5692–5695 CrossRef CAS PubMed.
- R. H. Martin and M. J. Marchant, Tetrahedron, 1974, 30, 347–349 CrossRef CAS.
- Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS PubMed.
- For example: C. Schaack, E. Sidler, N. Trapp and F. Diederich, Chem.–Eur. J., 2017, 23, 14153–14157 CrossRef CAS PubMed.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem.–Eur. J., 2001, 7, 2245–2253 CrossRef.
- Á. J. Jiménez, F. Spänig, M. S. Rodríguez-Morgade, K. Ohkubo, S. Fukuzumi, D. M. Guldi and T. Torres, Org. Lett., 2007, 9, 2481–2484 CrossRef PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- M. Buchta, J. Rybáček, A. Jančařík, A. A. Kudale, M. Buděšínský, J. V. Chocholoušová, J. Vacek, L. Bednárová, I. Císařová, G. J. Bodwell, I. Starý and I. G. Stará, Chem.–Eur. J., 2015, 21, 8910–8917 CrossRef CAS PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- M. J. Fuchter, M. Weimar, X. Yang, D. K. Judge and A. J. P. White, Tetrahedron Lett., 2012, 53, 1108–1111 CrossRef CAS.
- S. Fujino, M. Yamaji, H. Okamoto, T. Mutai, I. Yoshikawa, H. Houjou and F. Tani, Photochem. Photobiol. Sci., 2017, 16, 925–934 RSC.
- J. Feng, Y. Zhang, C. Zhao, R. Li, W. Xu, X. Li and J. Jiang, Chem.–Eur. J., 2008, 14, 7000–7010 CrossRef CAS PubMed.
- F. Schlosser, M. Moos, C. Lambert and F. Würthner, Adv. Mater., 2013, 25, 410–414 CrossRef CAS PubMed.
- K. M. Lefler, K. E. Brown, W. A. Salamant, S. M. Dyar, K. E. Knowles and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 10333–10345 CrossRef CAS PubMed.
- C. Zhao, Y. Zhang, R. Li, X. Li and J. Jiang, J. Org. Chem., 2007, 72, 2402–2410 CrossRef CAS PubMed.
- Y. Yang, R. Correa Da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1864289 and 1864290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04216k |
| ‡ M. M. and N. J. S. contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |