
Shaping well-defined noble-metal-based nanostructures for fabricating high-performance electrocatalysts: advances and perspectives
Hai-Jing
Yin
,
Jun-Hao
Zhou
and
Ya-Wen
Zhang
 *
*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: ywzhang@pku.edu.cn; Fax: +86-10-62756787; Tel: +86-10-62756787
First published on 19th July 2019
Abstract
Electrocatalytic reactions have received widespread attention in the recent decades because of their importance in environmental protection and energy storage and utilization, involving fuel cells, electrolysis of water, and electrochemical reduction of carbon dioxide and nitrogen. Among the various nanocatalysts, noble-metal-based ones (containing Ru, Rh, Ir, Pd, Pt, Ag, and Au) exhibit superior performances in most electrocatalytic reactions for their higher catalytic activity and stability as compared to nonnoble-metal-based ones. Currently, the atomic-scale nanoengineering of noble-metal-based electrocatalysts has been demonstrated to be an efficient and robust approach to address specific problems either in catalytic activity/selectivity or durability in a given electrochemical reaction, through understanding the structure–activity relationship of catalysts at the molecular level. In this review, we summarize the progresses made in noble-metal-based nanocatalysts toward some important electrochemical reactions of small-molecule activation (e.g., O2, H2, H2O, CH3OH, CO2, and N2), which involve three aspects: (1) how to synthesize noble-metal-based electrocatalysts with well-defined nanostructures; (2) how to tune the catalytic performance of electrocatalytic reactions; and (3) how to determine the optimal surface structure of catalysts according to the understanding of the structure–activity relationship. Further, we provide prospects for the sustainable development of this cutting-edge field.
 Hai-Jing Yin | Hai-Jing Yin was born in Hebei, China. She received her BSc in Chemistry from the Beijing University of Chemical Technology in 2017. Now, she is working for her PhD under the supervision of Prof. Yawen Zhang in College of Chemistry and Molecular Engineering, Peking University. Her current research focuses on the colloidal synthesis and catalytic property investigations of noble-metal-based nanocrystals. |
 Jun-Hao Zhou | Jun-Hao Zhou was born in Hubei, China. He received his BSc in Material Chemistry from the University of Science and Technology of China in 2016. Currently, he is working for his PhD under the supervision of Prof. Ya-Wen Zhang at the College of Chemistry and Molecular Engineering, Peking University. His current research focuses on the controllable synthesis of noble- and nonnoble-metal-based nanomaterials and their applications in the electrochemical reduction of CO2. |
 Ya-Wen Zhang | Yawen Zhang is a full professor and principle investigator at the College of Chemistry and Molecular Engineering, Peking University. The research interests of his group are rational design, controllable synthesis, ordered assembly, catalytic properties, and structure–function relationships of rare-earth- and noble-metal-based nanostructures. He has published more than 150 papers in peer-reviewed scientific journals and several book chapters; he was also a winner of the National Science Fund for Distinguished Young Scholars in 2010. He obtained his BSc, MSc, and PhD from Peking University in 1988, 1994, and 1997, respectively, and did his postdoctoral research in the State Key Laboratory of Rare Earth Materials Chemistry and Applications of Peking University during 1998–2000. He has been a visiting scholar in the Department of Chemistry of the University of California at Berkeley and Lawrence Berkeley National Laboratory during 2006–2008. |
1. Introduction
Currently, environmental pollution and energy crises are two serious problems that have drawn increased attention. Many efforts have been devoted toward developing clean and renewable energy to replace traditional fossil fuels. Wind, sunlight, and tides have been promising clean and renewable energy sources that can be converted into electrical energy. Although batteries and supercapacitors are often used for storing electricity, both of them suffer from low energy density and high cost. Therefore, it is difficult to store electricity on a large scale, which reduces the utilization efficiency of electricity and causes energy loss. Energy storage in the form of chemical bonds under electrocatalytic reactions is an effective and much cheaper method. A sustainable energy landscape based on electrocatalysis has been proposed, as shown in Fig. 1.1 Renewable energy sources such as sunlight, wind, and tides can be converted into electricity. Then, this electricity can be used via electrochemical reactions toward producing high-value-added chemicals, such as hydrogen, ammonia, alcohols, and hydrocarbons. Thereafter, the obtained fuels (hydrogen and alcohols) can be fed to fuel cells. During the entire cycle, no fossil fuel is involved, and the entire process is clean and environmentally friendly. However, to achieve satisfactory energy efficiency, noble-metal-based (containing Ru, Rh, Ir, Pd, Pt, Ag, Au) and nonnoble-metal-based catalysts, either heterogeneous or homogeneous, should be introduced in these procedures (such as electrolyzing water for hydrogen, carbon dioxide (CO2) reduction reaction (CO2RR) for alcohols or hydrocarbons, or methanol oxidation reaction (MOR) in fuel cells).2–5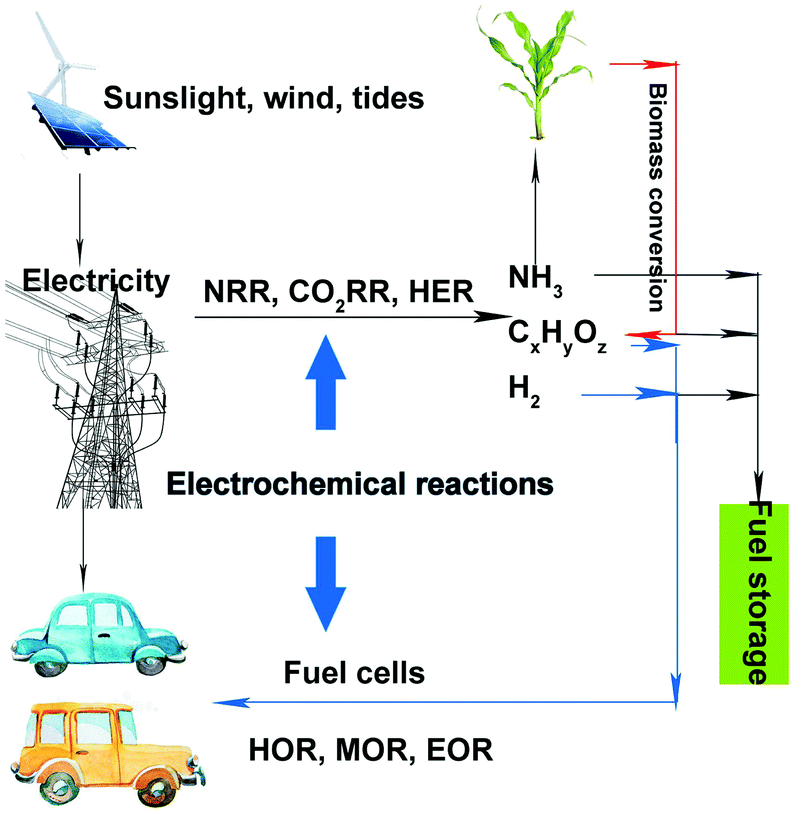 | ||
| Fig. 1 Schematic of a sustainable energy landscape based on electrocatalysis. | ||
Among such electrocatalysts, the heterogeneous noble-metal-based ones exhibit superior performances as compared to the nonnoble-metal-based ones in the above reactions in terms of activity and durability (as the latter are easily corroded in electrolytes). However, there are certain aspects of noble-metal-based catalysts that need to be improved. Firstly, the activity of noble metals needs to be enhanced for practical applications. Secondly, improving the selectivity of noble metals is necessary. For example, we expect that ethanol can be completely oxidized into CO2 for higher energy efficiency in ethanol oxidation reaction (EOR), which requires that the catalysts possess better selectivity. Finally, in complicated reactions (such as MOR or EOR), catalysts are often poisoned by intermediates that are tightly adsorbed on the catalyst surface, which leads to poor durability. Therefore, improving the activity, selectivity, and stability of noble-metal-based electrocatalysts is imperative.
Several studies have been reported to investigate these issues. Most of them have concentrated on two main strategies: adding a second (or even third) metal to the noble metals or tuning the structure of the catalysts. In fact, the size of nanocrystals has a nonnegligible effect. Different particle sizes influence the coordination numbers of the specific atoms in the electrocatalysts. For instance, Calle-Vallejo et al. demonstrated that the coordination numbers of the atoms on the catalyst surface could be regulated by adjusting the size of the nanoparticles (NPs); consequently, the activity of ORR would also change.6 When the particle size is reduced to the subnanometer scale, it may cause significant transitions in the catalytic activity. The extreme case is single-atom catalysts, which may exhibit fantastic catalytic activity for certain reactions.7 For electrocatalytic nanomaterials of a certain size, it has been understood that the addition of a second or third metal can modulate the activity, selectivity, and stability of noble-metal-based electrocatalysts. For example, it has been demonstrated that alloying Pt and Sn can increase the anti-CO poisoning ability of the catalyst, and the addition of a ternary component (say Rh or Ir) can help break the C–C bond in EOR.8–11 In MOR, the addition of Ru can increase the anti-poisoning ability and activity of Pt-based catalysts.12,13 Alloying Pt and Au is beneficial for improving the stability of catalysts used in oxygen reduction reactions (ORRs); a similar effect has also been observed in PtMo and PtRh alloys.14–19
Another factor that affects the catalyst performance is morphology. It is well known that high-index crystal faces with more steps and kinks that possess several unsaturated coordination atoms as the catalytic sites exhibit enhanced activity as compared to terraces.20–22 Lattice strain, which can be found in core–shell nanostructures, also modulates the catalyst activity. For example, Zou et al. synthesized Ru@Pt core–shell NPs via a sequential ethanol reduction method.23 The core–shell structures exhibit superior catalytic performance toward EOR, which was ascribed to the lattice strain of the Pt shell and the downshift in the d-band center of Pt. Moreover, nanostructures such as nanoframes, nanopores, and concave crystals, which expose more active sites, facilitate the catalytic process. However, their inferior structure and poor stability during catalysis are formidable obstacles for their practical applications. Introducing other metals to these nanostructures may enhance their durability and activity.24
Theoretical calculations are also beneficial toward investigating the relationship between structure and activity as compared to the trial-and-error approach. On one hand, we can formulate a suitable model to find a possible activity and/or selectivity descriptor in each reaction; thereafter, high-performance superior catalysts can be screened by calculating the values of specific catalytic descriptors. On the other hand, theoretical calculations may provide an optimal value for the descriptor, which is imperative for designing an optimal catalyst structure.
As mentioned above, the catalytic performances of electrocatalysts can be tuned by altering the composition, shape, and size associated with noble-metal-based nanostructures. Determining the relationship between the surface structure and performance of catalysts facilitates the design of optimal catalysts. In the past few decades, research involving the structure–activity relationship for different reactions has achieved considerable progress. However, an optimal surface structure for most electrochemical reactions is still unclear, and additional efforts should be devoted toward this field. It is obvious that the synthesis of well-defined nanostructures is vital toward investigating the structure–activity relationship. For instance, Liu et al. synthesized a triangular Ag nanoplate by reducing AgNO3 with NaBH4 in the presence of H2O2 and trisodium citrate, which exhibited superior CO selectivity in CO2RR. They attributed this improved selectivity to the presence of dominant Ag(100) facets.25 Zhang et al. fabricated Pt/Ag bimetallic nanostructures with a controlled number of void spaces via a tailored galvanic replacement reaction, which exhibited higher activity in MOR, yielding a higher specific surface area.26 Other nanostructures that play an important role in catalytic reactions, such as core–shell structures, polyhedral nanocrystals, nanoframes, branched structures, etc., have also been produced via different methods. Moreover, when the relationship between the structure and activity is intelligible, the method to formulate the desired catalyst also demands the development of an appropriate synthesis method.
In recent years, several reviews have introduced synthesis methods for fabricating noble-metal-based nanostructures and their applications in catalytic reactions. Some of them have concentrated upon a particular synthesis method in the morphology control of noble-metal-based nanocrystals, namely, seed-mediated growth and oxidative etching.27,28 Others have introduced the synthesis route and the application of a specific nanostructure, such as high-index-facet nanopolyhedrons, concave structures, and icosahedral nanocrystals.29–32 There are several reviews on the routes of well-defined shapes of noble-metal-based nanocrystals, but the introduction of their applications are brief.33–36 However, electrochemical reactions are very important in the area of clean and sustainable energy, as mentioned earlier; therefore, determining the structure–activity relationship between noble-metal-based nanostructures and catalytic performance is beneficial toward the practical applications of electrocatalytic reactions.
In this review, we introduce several common synthesis methods to provide some guidance toward the synthesis of different noble-metal-based nanostructures, particularly facilitating various heterogeneous catalytic reactions. Then, we concentrate on how to modulate the catalytic performance of noble-metal-based catalysts in several representative reactions in the field of electrocatalysis, namely, hydrogen oxidation reaction (HOR), ORR, MOR, EOR, hydrogen evolution reaction (HER), oxygen evolution reaction (OER), CO2RR, and nitrogen reduction reaction (NRR). Moreover, we attempt to clarify the structure–activity relationship in different reactions and formulate better catalysts with higher performance. Finally, we give the challenges and perspectives for noble-metal-based catalysts in electrocatalysis.
2. Method for controlling noble-metal-based nanostructures
It is well known that different catalysts with various surface structures show different performances in certain reactions. Therefore, developing synthesis methods to design well-defined shapes of nanocrystals is essential toward enhancing the activity of catalysts. Moreover, the relationship between activity and structure indicates that there exists an optimal catalyst with a specific composition, structure, and size for each electrochemical reaction. Therefore, it is essential to determine and fabricate a catalyst with an optimal nanostructure. Developing suitable methods to synthesize well-defined shapes of catalysts is conducive toward clarifying the structure–activity relationships and finding an optimal nanostructure. Further, the synthesis of an optimal nanostructure demands a precise synthesis method. Herein, we introduce several common synthesis methods for the formation of different nanostructures, particularly certain specific structures that are usually studied in catalytic processes.The general synthesis methods of noble-metal-based nanostructures include continuous growth, seed-mediated growth, underpotential deposition (UPD), and galvanic replacement. The general strategies used in the shape-controlled synthesis of noble-metal-based nanostructures are shown in Fig. 2. In fact, there are certain similar factors affecting the shape and size of nanocrystals in these synthesis routes: reaction time, temperature, reductant, type of precursors, and facet-specific capping agent. In general, a sufficient reaction time ensures the formation of nanocrystals. The longer the reaction time, the larger is the nanocrystal growth. Temperature, reductant, and precursors can change the reduction rate: high temperature, reductant with higher reduction capacity, and precursors that possess higher redox potential can increase the reduction rate of metal precursors, resulting in the formation of additional nuclei during the nucleation stage and a reduced size of crystals.37–39 Facet-specific capping agents have different binding energies for specific facets and can change the morphology of the final nanocrystals. For example, halide ions are often used as the {100}-facet-specific capping agents that can adsorb onto the (100) facet during the reduction process and prevent the crystal growth on the facet, resulting in cubic NPs exposing the (100) facet.40–42 Citric acid and C2O42− can serve as the {111}-facet-specific capping agents in Pt- and Pd-based nanocrystals.43–45 Further, we will comprehensively discuss the influence of each factor in specific synthesis approaches.
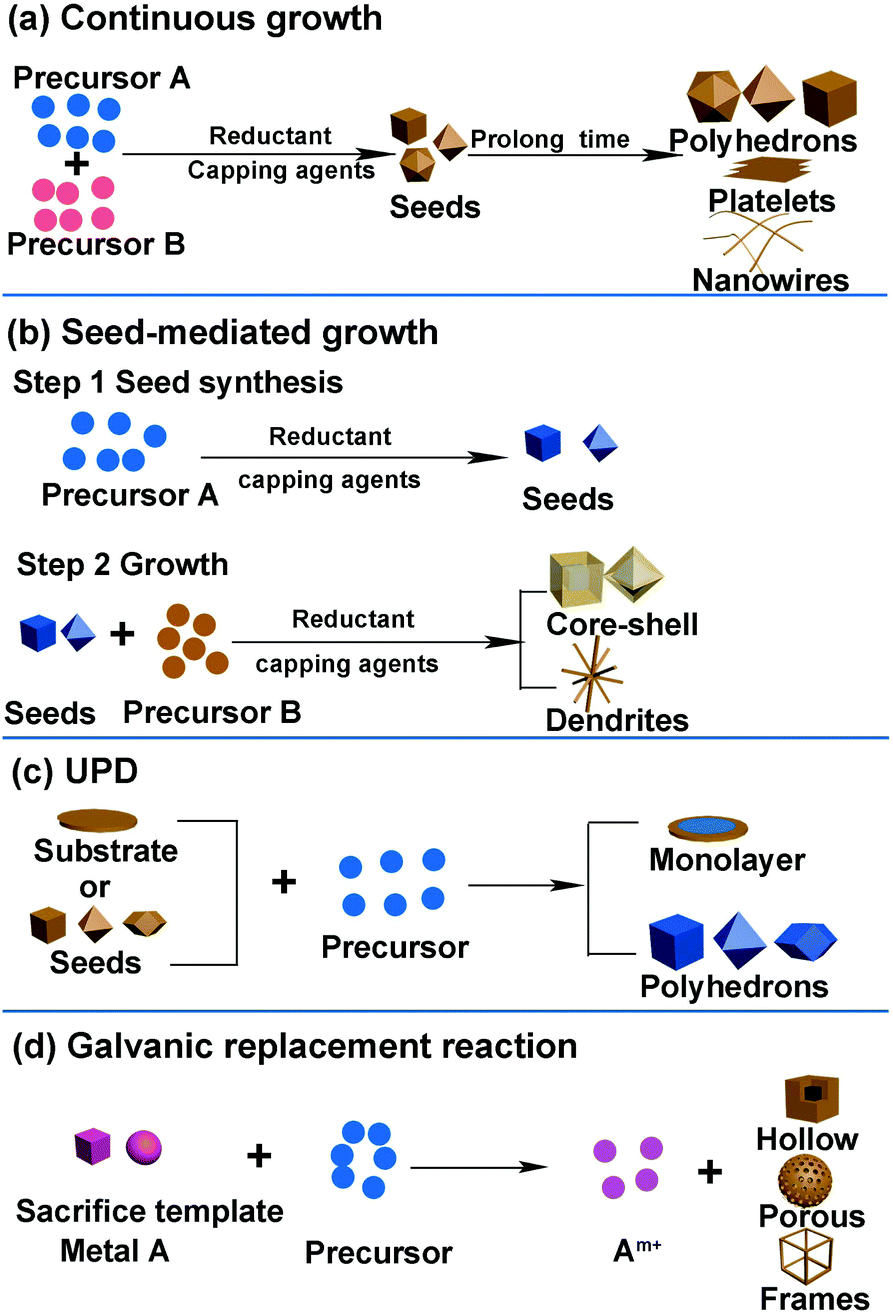 | ||
| Fig. 2 General strategies used in shape-controlled synthesis of noble-metal-based nanostructures. (a) Continuous growth. (b) Seed-mediated growth. (c) UPD. (d) Galvanic replacement reaction. | ||
2.1 Continuous growth
Continuous growth involves two stages. First, the metal precursors form the nuclei in the solution; second, metal ions in the solution are reduced and continuously added to the nuclei (Fig. 2a). Coreduction and thermal decomposition reduction are two common methods used in the continuous growth strategy.Coreduction is the most popular method used for fabricating noble-metal-based nanomaterials as it is facile to operate. In this route, the reaction precursors, solvent, reduction reagent, and facet-specific capping agents can be added together. Both metal alloy and heterogeneous structure can be obtained. When the bond energy between metal A and metal B is stronger than that of A–A and B–B, the alloy is preferentially formed. If one of the metals is preferentially reduced, the other will grow on the interface, which leads to the formation of a core–shell structure. Otherwise, it tends to produce other heterogeneous structures. There are many studies regarding noble-metal-based nanocrystals prepared by the coreduction route, such as nanopolyhedrons (Pt–Pd, Rh–Pd, Pt3M), concave structures enclosed by high-index facets, metallic dendritic nanostructures, nanosheets, nanoplates, and core–shell nanostructures.46–59
As compared to NPs enclosed by low-index facets, concave nanocrystals that expose high-index facets are expected to show enhanced catalytic properties because several atoms and/or kinks with low coordination numbers are distributed on the surface, which possess higher activity. However, during the synthesis process, high-index facets tend to rapidly vanish due to their high surface free energy. Binding organic species, such as surfactants and/or inorganic ions, on the surface could significantly change the surface energy, and hence, stabilize the high-index crystal faces. For example, Jia et al. reported a novel excavated rhombic dodecahedral (ERD) PtCu3 alloy comprising ultrathin nanosheets of high-energy {110} facets, which was prepared by a facile wet chemical route in the presence of n-butylamine and cetyltrimethylammonium chloride (CTAC), as shown in Fig. 3a and b.53 To determine the effect of n-butylamine, its amount was adjusted; the obtained nanocrystals are shown in Fig. 3c–f. Based on the obtained results, they suggested that the adsorption of n-butylamine on the 〈110〉-edge sites of PtCu3 was very important in the synthesis of different nanostructures. Electrochemical characterizations showed that the PtCu3 ERD nanocrystals exhibited high catalytic activity and stability in formic acid oxidation reaction. It is obvious that the capping agent is important for controlling the shape of the nanocrystals.
 | ||
| Fig. 3 (a) SEM images of an individual ERD PtCu3 alloy nanocrystal with different orientations. (b) High-angle annular dark-field scanning TEM (HAADF-STEM) image and HAADF-STEM energy-dispersive X-ray spectroscopy (EDS) elemental maps of a single ERD PtCu3 alloy NP. (c–f) SEM images and (insets) corresponding models of polyhedral PtCu3 alloys synthesized with different amounts of n-butylamine: (c) 1.50 mL, (d) 0.50 mL, (e) 0.30 mL, and (f) 0.05 mL. Adapted with permission from ref. 53. Copyright 2014, American Chemical Society. | ||
In addition to concave structures, many other nanostructures can be synthesized via coreduction because there are many adjustable factors such as precursors, ligands, reductants, capping agents, and reaction time in the synthesis process. Our group demonstrated that Pd–Rh nanocrystals with tunable compositions and morphologies including hollow nanocubes (NCs), nanoicosahedra (NIs), and nanotruncated octahedra (NTOs) could be synthesized by a facile one-pot hydrothermal approach.60 Polyvinylpyrrolidone (PVP) served as both a reductant and a capping agent, and halide anions (Br−/I−) acted as the shape-control agents. Pd–Rh NCs were formed because the higher electrode potential of Pd(II)/Pd than that of Rh(III)/Rh and Br− ions could facilitate selective adsorption on the (100) facets. In the absence of KI, Pd–Rh NIs would be obtained, suggesting that the removal of twinned seeds would occur by the etching effect of halide ions/oxygen pairs.61 When more KBr is added, Pd–Rh NTOs were synthesized because Br coordinated with Pd(II) as well as Rh(III) to form more stable complexes, thereby lowering the reduction rates of metal ions. The as-synthesized catalysts showed different catalytic performances in Suzuki cross-coupling reactions. The nanostructures enclosed only with (100) facets showed better catalytic activity than the other structures. Moreover, we reported a systematic synthesis strategy toward fabricating shape-tunable monodisperse Pt–Ir alloy nanocrystals.62 Both single-crystalline (nanooctahedra (NOs), NTOs, and NCs) and polycrystalline (nanocluster flowers (NCFs), nanowires (NWs), nano-short-chains (NSCs), and nanooctahedral stars (NOSs)) Pt–Ir nanocrystals were synthesized by altering the Br− and I− concentrations during solvothermal processing (Fig. 4a and b). These Pt–Ir alloy catalysts showed enhanced activity than commercial Ir/C catalysts in OER. In particular, Pt–Ir NSCs exhibited higher catalytic activity as that for commercial Pt/C catalysts because of the higher proportion of (110) facets. Therefore, coreduction is a powerful method for performing shape-controlled synthesis.
 | ||
| Fig. 4 Synthesis pathways of (a) single-crystalline Pt–Ir alloy nanocrystals and (b) polycrystalline Pt–Ir alloy nanocrystals. Adapted with permission from ref. 62. Copyright 2017, Royal Society of Chemistry. | ||
In some cases, it is difficult to fabricate noble-metal-based alloys through the coreduction route because certain metal precursors possess relatively low reduction potential, such as Fe, Co, and Ni. In such a case, two metals tend to form a heterogeneous structure rather than a homogeneous alloy. Thermal decomposition reduction is a powerful route to synthesize alloy nanocrystals by the synthesis of metal–surfactant complexes, which can decompose at similar temperatures and be reduced by a reductant, thereby forming an alloy. In this method, the metal precursors need to immediately decompose into Mn+ and M0, respectively, under moderate conditions, such as carbonyls (Mx(CO)y) and acetylacetonates (M(acac)n). For instance, Lanza et al. synthesized Pd–Sn alloys and intermetallic nanocrystals by thermally decomposing the complexes of Pd(II) and Sn(IV) precursors in oleylamine.63 Oleylamine formed coordinating complexes with the two metals, which are stable at room temperature. When heated above the decomposition temperature, the two metal–oleylamine complexes decompose and are simultaneously reduced by oleylamine to form the alloy. Moreover, it is also an efficient method to produce a core–shell structure that necessitates two metals precursors with different decomposition temperatures. Metal precursors with a low decomposition temperature initially form the core and another metal is decomposed that grows on the core to form the shell as the reaction temperature or reaction time increase.64
2.2 Seed-mediated growth
Seed-mediated growth is an effective route to prepare core–shell nanostructures and heterostructures such as dendritic structures.41,65–71 The synthesis process can be divided into two parts: formation of seed crystals and growth of metal atoms that are generated from the reduction and decomposition of precursors (Fig. 2b). During the growth process, the metal precursor added later can undergo homogeneous nucleation and/or heterogeneous nucleation. Heterogeneous nucleation means epitaxial growth on the surface of the seeds, and homogeneous nucleation infers metals that form seeds in solution and then compose with the as-synthesized metal seeds via oriented attachments.72,73 In general, there are three recognized modes during epitaxial growth. Frank–van der Merwe (FM): layered growth; Volmer–Weber (VW): island growth; Stranski–Krastanov (SK): layered growth initially followed by island growth.74 The final morphology of the nanostructures can be influenced by the shape of the added seeds and the subsequent growth process via thermodynamics and kinetics. Atoms reduced from precursors tend to deposit on the sites with high surface energy. The reduction rate of the precursors and the surface diffusion rate of the adatoms can also decide the final structures.Kinetic control over the diffusion and deposition rates of the noble metal can change the shape of the final nanostructures.75–78 For example, Wang et al. reported a facile and convenient approach to synthesize Pt octahedral islands with tunable sizes and densities on Pd NCs by controlling the deposition rate of Pt on Pd seeds (Fig. 5a).79 When the concentration of the reductant (glucose) was low, surface diffusion dominated the process, which induced the formation of flat surfaces on noble metal substrates, such as nanoshells. On the contrary, with an increase in reductant concentration, the deposition rate became higher than the diffusion rate, and Pt would favor the deposition on corners of metal nanocrystals, forming Pt octahedra on a cubic Pd seed. When the deposition rate was further increased, the deposition of Pt octahedra would take place not only on the corners but also on the edge and surface sites of the Pd seeds. Fig. 5b–e show the TEM and high-angle annular dark-field scanning TEM (HAADF-STEM) images of Pd@Pt core–shell nanocrystals prepared at different glucose concentrations. They found that the Pt nanocages with small octahedral islands on the surfaces of Pd cubic seeds exhibited enhanced activity as compared to that in commercial Pt/C in ORR due to the excellent performance of Pt {111} facets toward ORR.
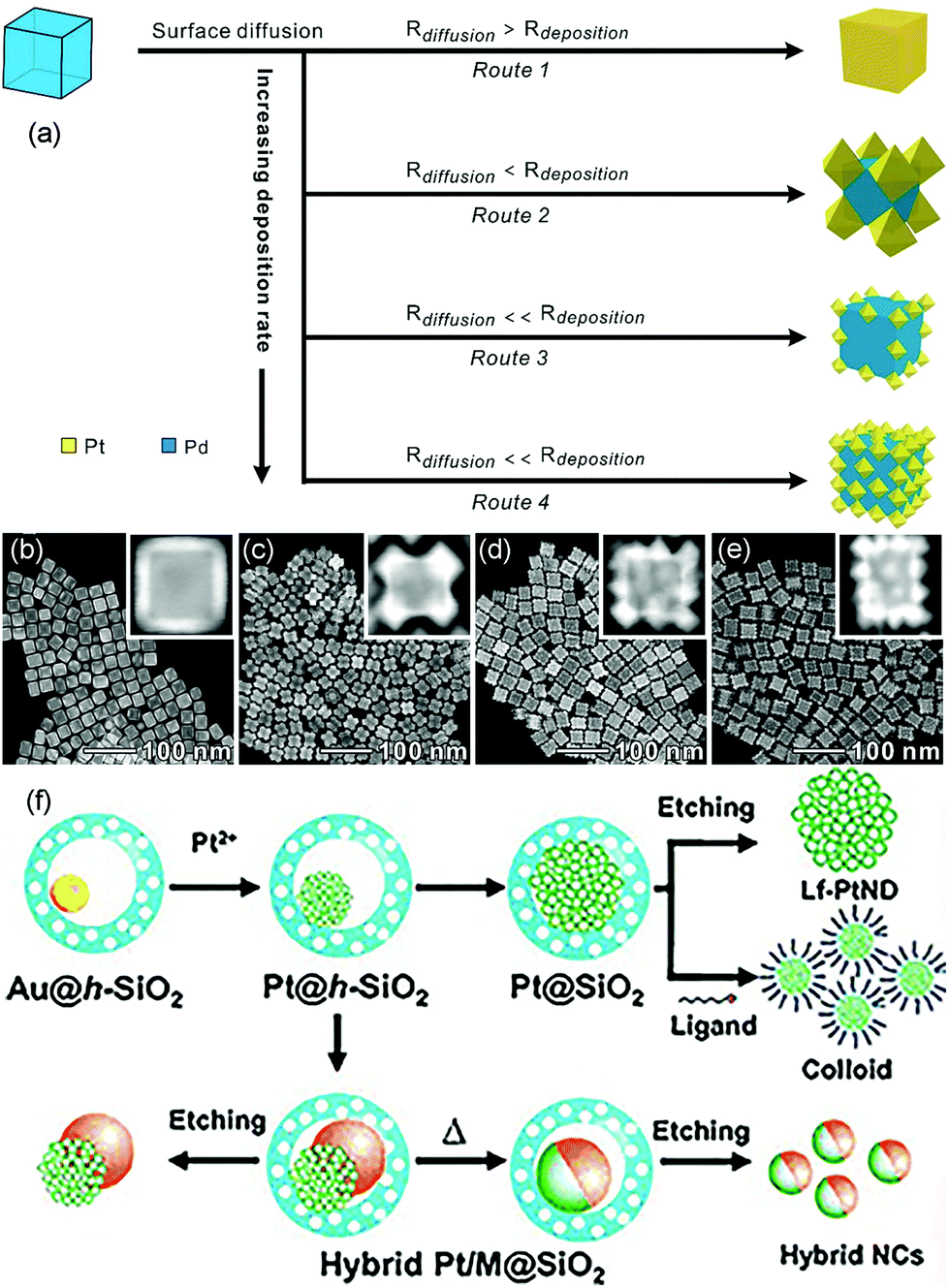 | ||
| Fig. 5 (a) Schematic illustrating the overgrowth of Pt nanoshells on Pd NCs by exercising delicate control over the deposition rate. HAADF-STEM images of Pd@Pt core–shell nanocrystals prepared at different concentrations of glucose: (b) 8.33, (c) 16.67, (d) 20, and (e) 31.67 mg mL−1. Adapted with permission from ref. 79. Copyright 2019, American Chemical Society. (f) Synthesis of Pt-based nanodendrites and hybrid nanocrystals. Adapted with permission from ref. 80. Copyright 2011, Wiley-VCH. | ||
Seed-mediated growth is a common synthesis route to fabricate nanodendrites that have been extensively investigated with regard to various catalytic reactions as the structure yields a relatively large surface area, vast amount of surface atoms, and lattice stress. For instance, Yeo et al. demonstrated a novel synthesis approach for Pt nanodendrites via the Au-seed-mediated growth inside hollow silica nanospheres.80 The hollow silica acted as nanoreactors, providing well-isolated environments for the growth of Pt nanocrystals. The Au nanocrystal in hollow silica nanospheres (Au@h-SiO2) was the seed on which Pt deposited and grew. When an aliquot of Na2PtCl4, Au@h-SiO2, and L-ascorbic acid (H2Asc) were mixed under constant stirring, the Pt nanodendrites on Au crystals would be obtained (Fig. 5f). Then, SiO2 was dissolved with a dilute basic solution at room temperature with no shape change in the Pt-on-Au nanodendrites. The Pt-on-Au nanodendrites exhibited greater ORR activity than that obtained from commercial Pt-black catalysts.
Several factors can be tuned to adjust the morphology of nanocrystals in the seed-mediated growth strategy, which is similar with that in the continuous growth strategy. Moreover, in the former, the structure and surface conditions of the seed have an impact on the ultimately obtained nanostructures. As mentioned before, the metal atoms prefer to deposit on the high-surface-energy sites. When the reduction rate is enhanced by increasing the reaction temperature by using a reductant with a stronger reducing capability, as well as changing the precursors and ligands, kinetic control becomes dominant. Otherwise, thermodynamic control is dominant. All the examples and statements possibly refer to shape-controlled nanocrystals. The appropriate synthesis conditions should be considered according to the desired nanostructures.
2.3 Underpotential deposition (UPD) and galvanic replacement reaction
In this section, we will introduce UPD, galvanic replacement reaction, and a combination of both of the above, which have been used for the fabrication of shape-controlled metal nanostructures. We provide the unique advantages of these three synthesis methods that can be availed in the fabrication of shape-controlled nanostructures. Like the two synthesis methods mentioned earlier, we concentrate on the preferred surface structures in electrocatalytic reactions.For example, Liu et al. successfully controlled the Pt coating to the monolayer thickness on single-crystalline Au (111) surfaces via UPD.81 Li et al. developed a method to fabricate a complete-monolayer Pt coating on a large-surface-area three-dimensional (3D) Ni foam substrate using a buffer layer (Ag or Au).82 They confirmed that the deposition process of the Pt monolayer was indeed self-terminating to a single complete-monolayer Pt, where hydrogen is deposited to protect the Pt surface from further deposition. The HER activity of the as-synthesized Pt monolayers was similar to that of the Pt sheet because both of them yielded large specific surface areas.
Apart from growing a monolayer or a metal film, UPD is an impressive route to obtain a well-defined shape. Ag+ is commonly employed as a cationic additive during the synthesis of Au nanostructures, such as high-index {720}-faceted concave cubic Au nanocrystals, {730}-faceted tetrahexahedron, and {711}-faceted bipyramids. The shape-directing effects of Ag+ are attributed to the UPD of Ag onto the surface of Au particles.83–85 In fact, Ag+ could be reduced and deposited on the surface of Au, just like a kind of capping reagent, thereby controlling the shape of Au NPs. Personick et al. reported the control of the morphologies of Au particles using UPD: octahedra with {111} facets, rhombic dodecahedra with {110} facets, truncated ditetragonal prisms with {310} facets, and concave cubes with {720} facets.86 They first synthesized Au seeds by the rapid reduction of HAuCl4 by NaBH4 in the presence of CTAC. Then, they mixed the seeds with growth solutions consisting of sequentially adding HAuCl4, as well as variable amounts of AgNO3, HCl, and H2Asc. By strategically varying the ratio of Ag+/Au3+ over a range from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 to 1:5, different shaped Au nanocrystals were produced (Fig. 6). The extent of Ag coverage on the surface facets was examined via a combination of X-ray photoelectron spectroscopy (XPS) as well as calculations. They found that a higher Ag coverage was necessary to stabilize the higher-surface-energy facets, which proved to be the most active sites for Au deposition. The phenomenon of UPD occurred because Ag+ could not be reduced by H2Asc when seed-mediated synthesis was used; the presence of Au seeds facilitated the reduction reaction at potential less negative than the Nernst potential.38,87 UPD allows the metals to be more easily reduced and provides more possibilities for the controlled synthesis of metal nanocrystals.
:500 to 1:5, different shaped Au nanocrystals were produced (Fig. 6). The extent of Ag coverage on the surface facets was examined via a combination of X-ray photoelectron spectroscopy (XPS) as well as calculations. They found that a higher Ag coverage was necessary to stabilize the higher-surface-energy facets, which proved to be the most active sites for Au deposition. The phenomenon of UPD occurred because Ag+ could not be reduced by H2Asc when seed-mediated synthesis was used; the presence of Au seeds facilitated the reduction reaction at potential less negative than the Nernst potential.38,87 UPD allows the metals to be more easily reduced and provides more possibilities for the controlled synthesis of metal nanocrystals.
 | ||
| Fig. 6 SEM images of (a) octahedra, (b) rhombic dodecahedra, (c) truncated ditetragonal prisms, and (d) concave cubes of Au–Ag synthesized from reaction solutions containing Ag+/Au3+ ratios of 1:500, 1:50, 1:12.5, and 1:5, respectively. Scale bars: 200 nm. Adapted with permission from ref. 86. Copyright 2018, American Chemical Society. | ||
As the reduction and deposition of one metal atom are accompanied by the dissolution of another metal, the synergy of carving and deposition can offer certain advantages, including the ability to fabricate more significant concaveness on the surface and alter the distribution of elements on the surface. There have been several studies involving the regulation of different shapes of noble-metal-based nanocrystals via the galvanic replacement reaction. For example, the synthesis of Pt–Cu and Pt–Pd concave NCs, cubic Au–Ag nanoframes, Au-based hollow nanostructures, and nanodendritic structures.94–100 Qin et al. conducted a series of studies, and they found that a capping agent can significantly regulate the final nanostructures.101–103 They demonstrated the synthesis of Ag NCs with concave side faces, Au–Ag alloy frames (Ag@Au–Ag concave nanocrystals), and Ag@Au core–shell NCs. As shown in Fig. 7, Ag@Au–Ag concave nanocrystals were obtained using Ag NCs in the presence of H2Asc, NaOH, and CTAC. Ag@Au core–shell NCs were synthesized via PVP as a substitute for CTAC. When CTAC was added to the reaction solution, Cl− ions could selectively bind to the {100} facets of Ag NCs, resulting in the dissolution of Ag atoms, which would occur from the side faces. Simultaneously, AuCl4− and AgCl2− in the solution would be reduced and deposited on the edges and corners of the Ag NCs with high surface energy. However, when PVP served as the capping agent, it would bind to the {111} facets, and therefore, the dissolution of Ag atoms would occur on the facets, resulting in the formation of Ag@Au core–shell NCs.
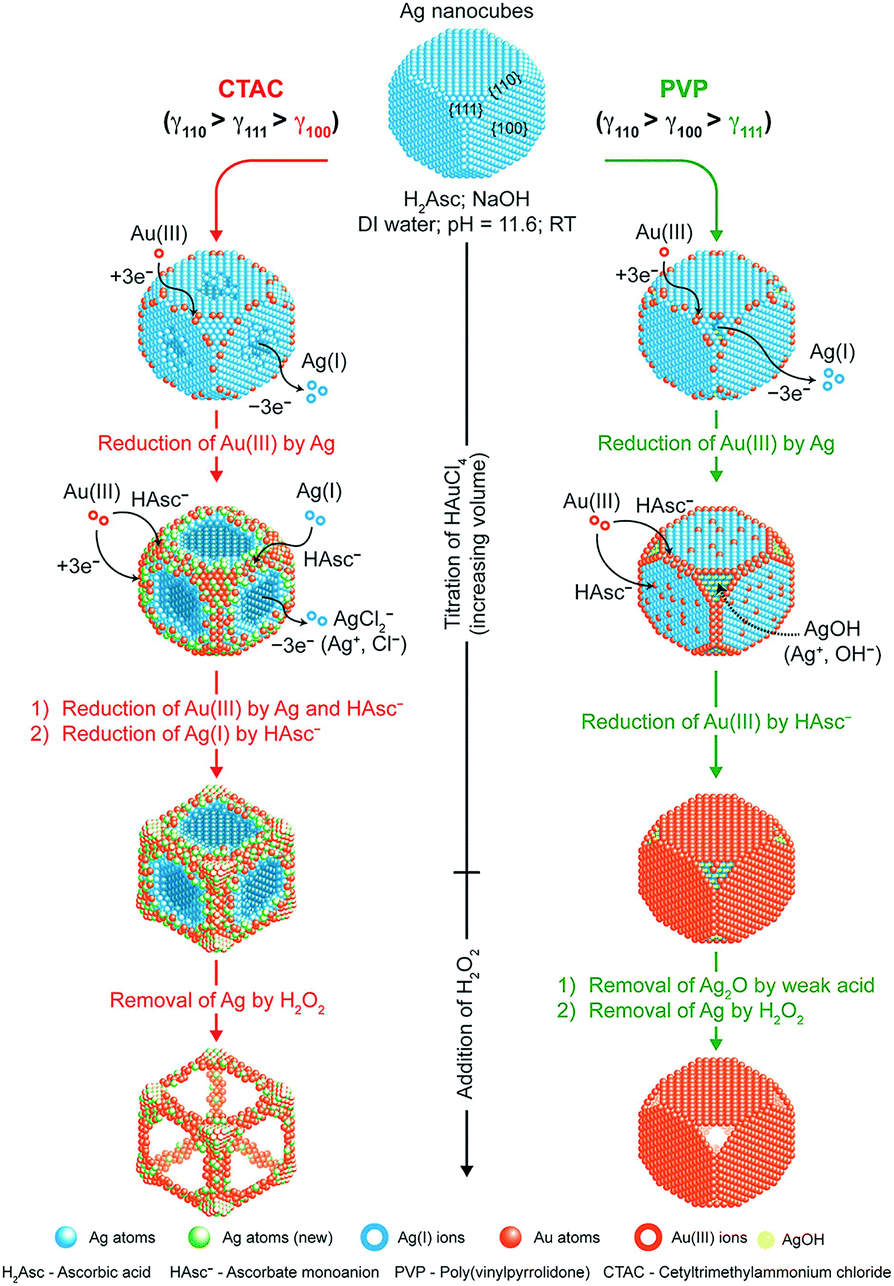 | ||
| Fig. 7 Schematic illustration describing the mechanisms proposed for the deposition of Au and Ag atoms on Ag NCs when HAuCl4 is titrated into an aqueous suspension of Ag NCs in the presence of H2Asc, NaOH, and CTAC (left) or PVP (right) at an initial pH of 11.6. Adapted with permission from ref. 101. Copyright 2017, American Chemical Society. | ||
In brief, the galvanic replacement reaction can be applied to synthesize various shapes (even an amorphous alloy structure), and they are used most commonly to prepare porous or hollow structures.104 The as-synthesized template that can be retained or carved during the synthesis plays a major role in shape control. The dissolved metal ion may or may not be reduced again, depending on the reducing ability of the reductant and the ligands around the metal ions, resulting in changeable components.
000 potential cycles. This implies that the Pd core protected the Pt shell from dissolution. In addition to the core–shell structure, this strategy is a versatile process for engineering the architectural diversity of complex heterogeneous metallic nanocrystals (HMNCs). For instance, Yu et al. fabricated Au/AgPd HMNCs comprising a central Au NC and satellite AgPd NCs by exercising control over the site-selective deposition and shape and size of the satellite nanocrystals.108
2.4 Other synthesis methods
In addition to the methods mentioned above, several other routes such as template-assisted method, etching, and assembly have also been developed to fabricate well-defined nanocrystals. The general strategies of these three methods are shown in Fig. 8.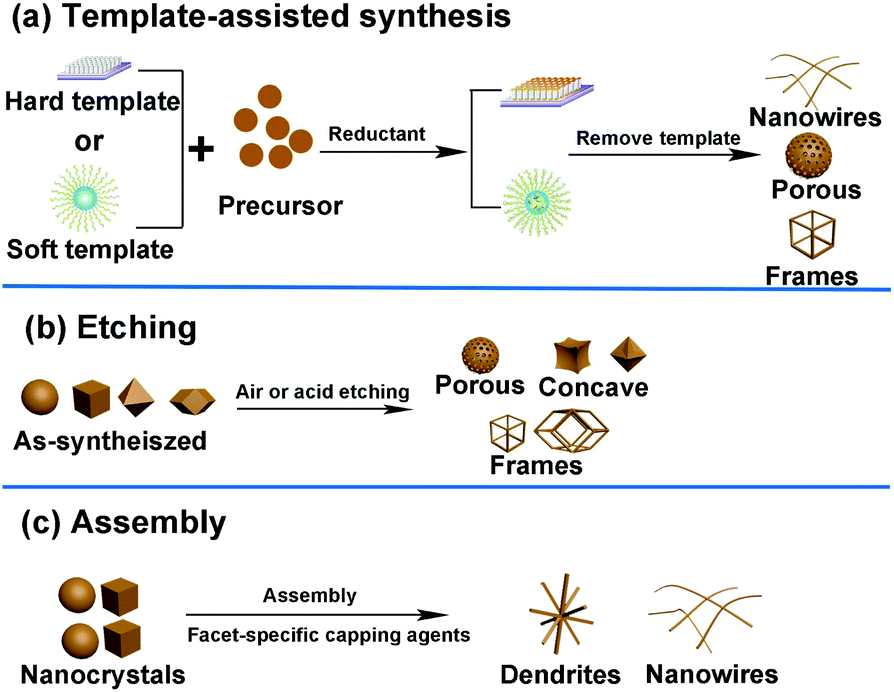 | ||
| Fig. 8 General strategies used in the shape-controlled synthesis of noble-metal-based nanostructures. (a) Template-assisted synthesis. (b) Etching method. (c) Assembly method. | ||
Template-assisted synthesis is an effective way to exercise morphological control over nanomaterials. Nanoframes and nanoporous materials can be synthesized via this route.76,109–111 An intact process based on template-assisted synthesis involves (1) the preparation of a template with the desired shape; (2) growth of the target metal atoms on the template; and (3) removal of the original template without changing the wanted nanocrystals (Fig. 8a). The template includes a hard template (such as mesoporous silica, zeolites, and alumina membranes) or a soft template (emulsion droplets, supramolecular micelles, polymer aggregates/vesicles, gas bubbles, etc.).112–115 By using SBA-15 and KIT-6 as the hard templates, mesoporous Pt–Ru alloy nanostructures and polyhedral and olive-shaped mesoporous Pt nanocrystals were successfully synthesized, respectively.116,117 Liu et al. utilized porous anodic aluminum oxide (AAO) membranes to fabricate nanoporous PtCo alloy NWs (Fig. 9), which exhibited enhanced catalytic activity in MOR as compared to currently used Pt/C.118 Despite the fact that several porous noble-metal-based nanocrystals have been obtained by the hard template method, this method suffers from two intrinsic disadvantages: (1) template fabrication; (2) sometimes, the desired metal crystals are removed along with the templates. Soft templates can avoid these problems as they are formed through self-assembly in the solution and can be easily removed.119,120 For example, polymers as the template can typically aggregate into flexible structures; therefore, the obtained nanocrystals possess different shapes depending on the solvent concentrations. Many porous metal nanomaterials have been synthesized via the soft template method, such as Au–Pt particles, Au–Pd particles, and Pt–Pd films.121,122 Jiang et al. synthesized bimetallic PdPt spheres, trimetallic Au@PdPt spheres, and PdPtCu spheres with large pores by the surfactant-directing method, as shown in Fig. 10.123 F127 not only served as the pore-directing agent but also prevented particle aggregation. Interestingly, they found that by changing the ratio of H2PtCl6/K2PtCl4, the particle size would also be changed: more H2PtCl6 resulted in a larger particle size because the addition of H2PtCl6 slowed the reduction rate of metal species. Trimetallic Au@PdPt and PdPtCu spheres exhibited superior electrochemical activity and durability toward MOR because of the electronic effect of the introduction of a third element.
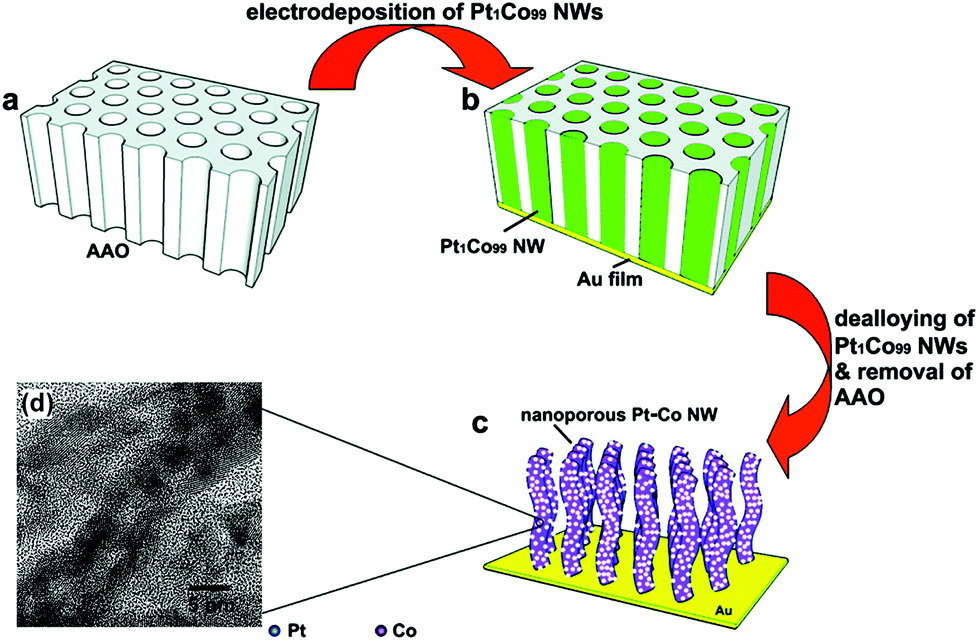 | ||
| Fig. 9 (a–c) Schematic illustrations of the fabrication process of nanoporous Pt–Co alloy NWs. (d) High-resolution TEM micrographs of the as-prepared nanoporous Pt–Co alloy NWs. Adapted with permission from ref. 118. Copyright 2009, American Chemical Society. | ||
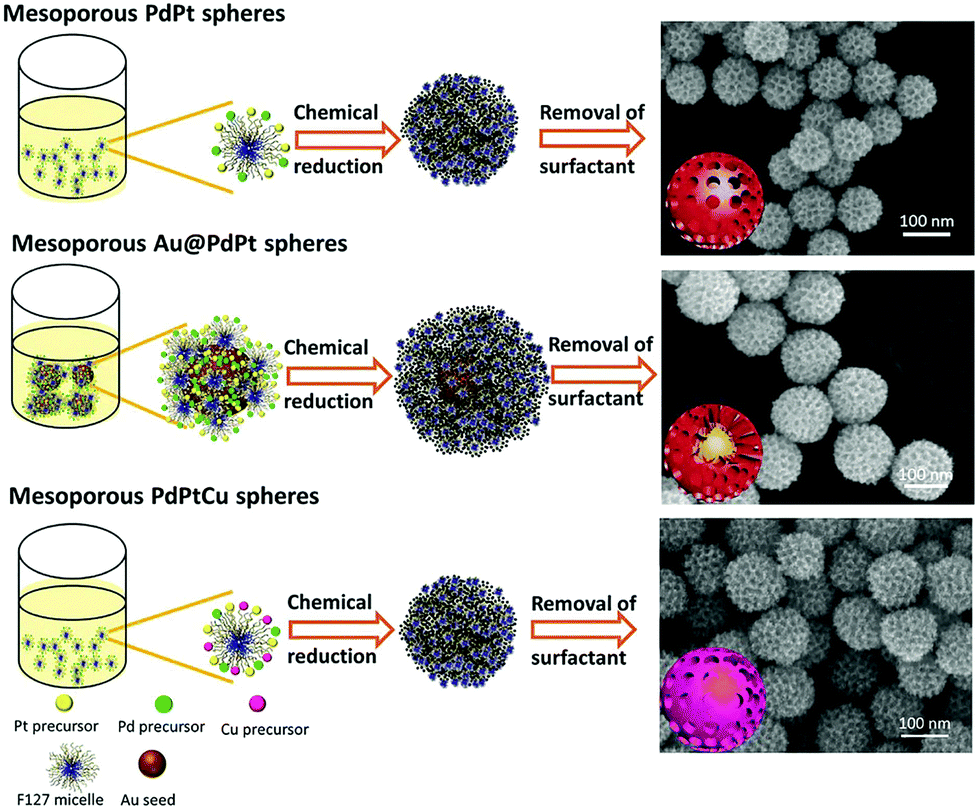 | ||
| Fig. 10 Systematic illustration of mesoporous bimetallic PdPt spheres, mesoporous trimetallic Au@PdPt spheres, and mesoporous trimetallic PdPtCu spheres. Adapted with permission from ref. 123. Copyright 2015, Wiley-VCH. | ||
Selective etching is another powerful approach to fabricate concave nanostructures, nanoframes, and nanopores by using air, acid, and base as the etchants.124–126 Usually, metals that are easily oxidized are also easily etched, such as Cu and Ni.24,126,127 Similar to the galvanic replacement reaction, etching can be controlled at different facets or specific sites to fabricate various morphologies (Fig. 8b). For example, Kwon et al. demonstrated a ternary Co-doped PtCu rhombic dodecahedral nanoframe (Co–PtCu RNF) with a reinforced vertex structure by the acid treatment on a Co–PtCu rhombic dodecahedral nanocrystal (Co–PtCu RNC).24 As shown in Fig. 11a, the doped Co was deposited in the vicinity of the vertices of the PtCu nanoframe (PtCu RNF) and Pt was mainly distributed at the edges and vertices of the nanostructure in Co–PtCu RNF, which can be confirmed from the TEM and HRTEM images (Fig. 11b and c) and the EDS elemental mapping analysis data (Fig. 11d). After acid etching, a portion of Cu would be dissolved, while Pt and Co still remained in the structure; therefore, the Co–PtCu RNF was obtained. Co–PtCu RNF exhibited higher ORR activity and stability as compared to PtCu RNF and Pt/C catalysts due to its ternary composition and vertex-strengthened frame structures.
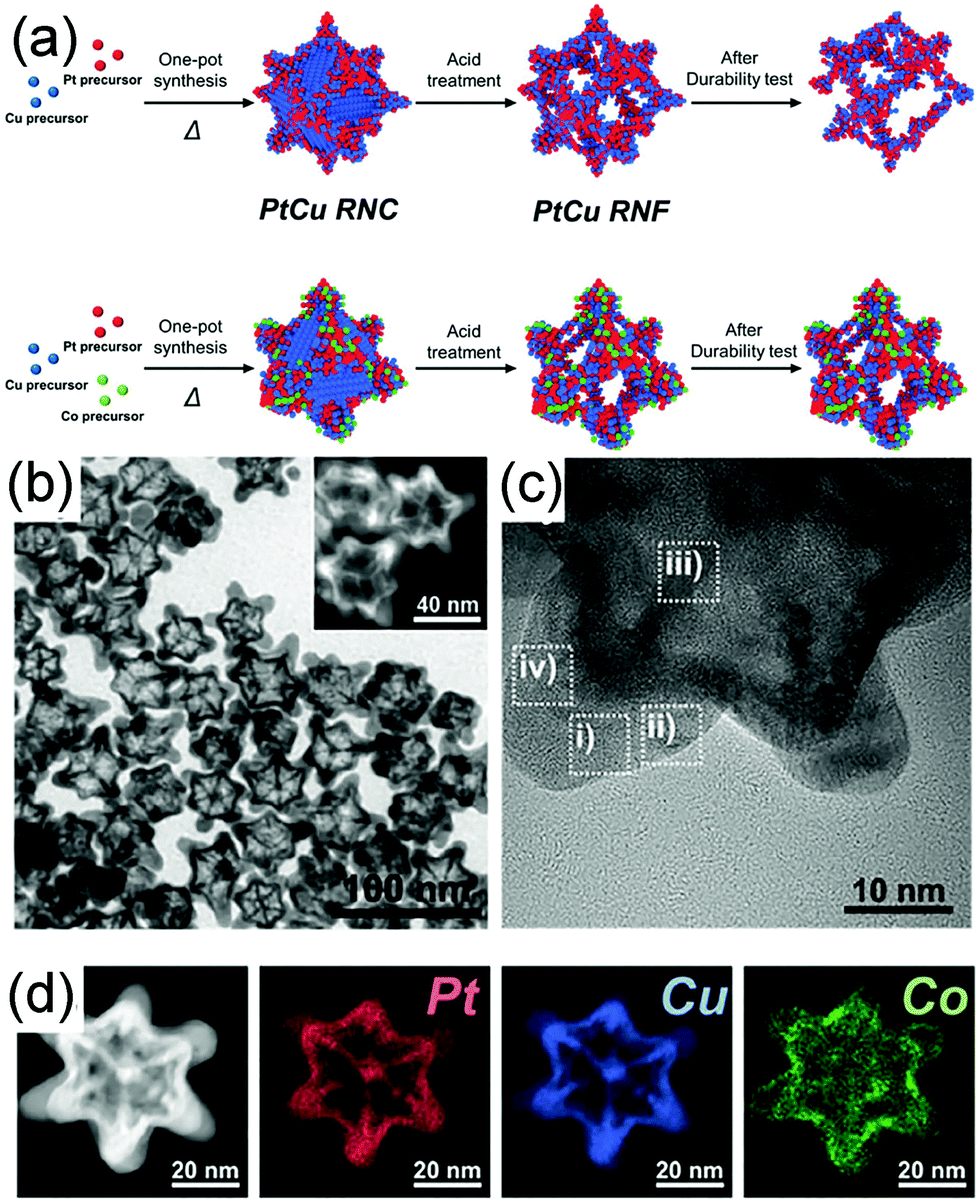 | ||
| Fig. 11 (a) Schematic illustration of the formation process of PtCu RNC, PtCu RNF, Co–PtCu RNC, and Co–PtCu RNF. (b)TEM and (c) HRTEM images and (d) HAADF-STEM image and corresponding EDS elemental mapping analysis of Co–PtCu RNF. Adapted with permission from ref. 24. Copyright 2018, Wiley-VCH. | ||
Assembly is a process in which small building blocks are organized into ordered structures under the effects of thermodynamic and dynamic constraints. Typically, assembly is related to thermodynamic equilibrium powered by minimizing the total free energy of the system. In general, smaller particles are prone to form nanocrystals with highly porous features and 3D channels through a continuous assembly process because a high ratio of the volume to surface area of small particles often yields high surface energy.128 In addition, NWs (such as PtRh, PtCo, PtNi, AuAg, PdBi, etc.) and nanodendrites can be achieved through the self-assembly of small noble-metal-based NPs, forming a one-dimensional (1D) structure (Fig. 8c).129–135 Ortiz et al. reported that the nature of NP nucleation and growth could be manipulated by the local ligand environment of the metal precursor and the selection of extraneous capping agents.136 Pd nanodendrites were synthesized only by using palladium acetylacetonate ([Pd(acac)2]) as the precursor and oleylamine served as both the capping agent and solvent. They demonstrated that precursors with a labile ligand or stable ligand were not favorable for NP aggregation. Only ligand and capping agent with intermediate binding affinity toward Pd could facilitate aggregation-based assembly.
In conclusion, we have presented several common synthesis methods for controlling the surface structure of noble-metal-based nanostructures. There are many factors that can be regulated to change the morphology of the nanocrystals, such as precursors, ligands, capping agents, reductants, reaction time, and temperature. Herein, we focus on the relationship between the surface structure (including composition, shape, and size) of noble metals and their catalytic performances; therefore, we mainly discuss the favorable morphology in catalytic reactions in every synthesis route in this section. Nanopolyhedrons and NPs exposing specific facets can be produced by continuous growth, seed-mediated growth, and a combination of UPD and galvanic replacement. Concave nanostructures are usually synthesized via selective etching and seed-mediated growth. Template-assisted method and oxidative etching serve as a universal method to fabricate nanoframes and nanoporous structures. Assembly can be used to form dendritic nanocrystals and ultrathin NWs. The synthesis methods of representative noble-metal-based nanostructures are summarized in Table 1. Moreover, with regard to catalytic reactions, there is a relationship between surface structure and activity. We can infer that there is an optimal surface structure for each reaction. The synthesis of a well-defined shape can help in understanding the structure–activity relationship, and this can assist in the determination of a possible superior surface structure of nanocrystals yielding high density of the most active sites for the electrocatalysts. Further, once the optimal surface structure is determined, the method of constructing a catalyst with the desired surface structures from the atomic level is still a challenge. Therefore, it is necessary to develop a precise synthesis method to obtain a well-defined nanocrystal shape.
| Nanostructures | Synthetic method | Key factors in synthetic process | Synthetic cases | |
|---|---|---|---|---|
| Nanopolyhedrons |

|
Coreduction | Kinetically control | RhPd48 |
| Coreduction | W(CO)6, oleylamine, oleic acid | Pt–M49 | ||
| Coreduction | Facet-selective agents Br− and I− | PtIr62 | ||

|
Coreduction | Precursors | PtPd46 | |
| Coreduction | Cl−, O2 | Ag61 | ||

|
Coreduction | Precursors halides | PtPd46 | |
| Coreduction | Kinetically control | RhPd48 | ||
| UPD | Ag+ concentration | Au86 | ||

|
Coreduction | Br−, PVP | PdRh60 | |

|
Coreduction | Cl−, O2 | Ag61 | |

|
Coreduction | Br−, PVP | PdRh60 | |
| Coreduction | Br−, I− | PtIr62 | ||
| Nanowires |

|
Oriented attachment | Octadecylamine as surfactant | AuAg133 |
| Hard template | AAO as template | PtCo118 | ||
| Seed displacement and epitaxial growth | PVP, Rh nanocubic seeds | RhPt129 | ||
| Template | Ni seeds, trisodium citrate | PdNi130 | ||
| Coreduction | CTAC as structure-directing agent | PtCo131 | ||
| Nanoplatelets |

|
Coreduction | Precursors, PVP | PtPd47 |
| Coreduction | The amount of n-butylamine | PtCu53 | ||
| Thermal decomposition reduction | NH4Br, oleylamine, | PtBi122 | ||
| Porous structure |

|
Hard template | Mesoporous silica | PtRu116 |
| Soft template | F127 micelle | PdPt123 | ||
| Dealloying | Removing Cu | PtCu124 | ||
| Hollow structure |

|
Coreduction | I−, PVP | PdRh60 |
| Galvanic replacement | Solid template of Ag | Au89 | ||
| Galvanic replacement | Na2S2O3 | CuPt104 | ||
| Concave structure |

|
Coreduction | High temperature, the ratio of oleylamine and oleic acid | Pt3Fe50 |
| Coreduction | Anisotropic overgrowth by control over oleylamine and oleic acid | Pt3Co51 | ||
| Galvanic replacement | PVP, Br−, H+ | PtPdCu94 | ||

|
Etching | Selectively etching Ni | PtNi126 | |
| Nanoframes |

|
Galvanic replacement | Ag nanocubes and AuCl2− | Au96 |
| Galvanic replacement | CTAC, Cl− | AuAg101 | ||
| Etching | Br-Binding on {100}, removal of Pd cores | Rh111 | ||

|
Etching | Cu removed by acid | PtCoCu24 | |
| Interior erosion | Oleylamine, non-polar solvents | Pt3Ni127 | ||
| Dendrites |

|
Coreduction | Pluronic F127 block copolymer, formic acid | PtCu55 |
| Coreduction | Silicon, HF | AgPd56 | ||
| Seed-mediated growth | Truncated octahedral Pd seeds, L-ascorbic acid | PdPt73 | ||
| Galvanic replacement | AgNO3, Cu substrate | AgCu98 | ||
| Template | Gyroid block copolymer as template | Au119 | ||
| Assembly | Oleylamine | Pd136 | ||
| Monolayer |

|
UPD | A buffer layer of Ag (Au) | Pt/3D Ni foam82 |
| UPD and galvanic replacement | Cu monolayer | PtML/Au(111)106 | ||
| PtML/Pd107 | ||||
| Core–shell structure |

|
Coreduction | Microwave radiation | Pd@Pt nanostructures58 |
| Coreduction | PVP as the reductant | Au@Pd nanostructures59 | ||

|
Seed-mediated growth | Au seeds | Au@Ag octahedrons and decahedrons66 | |
| Seed-mediated growth | Incorporation of ammine group | Au@Pt decahedrons67 | ||
| Seed-mediated growth | High temperature, adding Pt at low rate | Pd@Pt nanocubes75 |
3. Applications of well-defined noble-metal-based nanostructures in electrocatalytic reactions
Nowadays, electricity has become cheap, which can be converted from renewable energy (such as solar and wind power); therefore, electrocatalytic reactions have been widely used in several fields to achieve the effective use of energy. Current research has mainly focused on the production of high-value-added products such as hydrogen, alcohols, hydrocarbons, and ammonia via electrochemical reactions. Some of these products (hydrogen, alcohols, etc.) can be used in fuel cells, which convert chemical energy into electricity without being limited by the Carnot cycle and may be a possible strategy that can replace fossil fuel technology. Therefore, there have been several studies directed toward the understanding of catalytic reactions in fuel cells.It has been well known that noble-metal-based nanomaterials are the most active catalysts in most electrocatalytic reactions due to their unique electronic configurations and properties. Moreover, the durability of noble-metal-based catalysts is more superior than that of nonnoble-metal-based catalysts, which can be easily dissolved in acidic or alkaline electrolytes. For many reactions, particularly complicated reactions, the final products are abundant. We prefer a single product in most situations. For example, these products may include CO, HCOOH, CH4, CH3OH, and C2H4 in CO2RR, and the separation of products incurs additional expenditure. Hence, a single product is preferable. In NRR, we expect to obtain ammonia gas with a higher yield. In EOR, it is desirable that ethanol can be completely oxidized to CO2 instead of acetic acid (AA) or acetaldehyde (AAL). It is significant to enhance the selectivity of catalysts. In addition to selectivity, the stability of catalysts should also be improved in the electrocatalytic reactions. Noble metal atoms can get poisoned by intermediates that are generated from such reactions, and therefore, these catalysts can get deactivated. Although we demonstrate that the activity of noble-metal-based catalysts is higher than those of other catalysts, the performance still remains to be improved.
A structure–activity relationship has been found in several electrochemical reactions. Catalytic performance parameters, such as activity, selectivity, and stability, can be regulated by designing the surface structure of the catalyst. In general, the surface structure can be controlled by tuning the composition, morphology, and size of the catalyst. The effects of these three strategies can be summarized as follows: composition can change the activity by tuning the electronic structure of the central atoms or by a synergistic effect; different shapes with different exposed facets, atomic arrangements, lattice strain, and number of unsaturated sites can influence the activity of catalysts; various sizes can be used to adjust the coordination number of metal atoms or specific surface area of the catalysts, which can be used to regulate the catalytic performance.
Here, we focus on several representative electrochemical reactions, and discuss how to resolve the above issues by nanoengineering the surface structure of the catalysts at the atomic scale. Meanwhile, we attempt to clarify the unique structure–activity relationship in each electrochemical reaction. As mentioned before, an optimal nanostructure can exist for a certain catalytic reaction; therefore, we will briefly introduce the current understanding on the catalytic mechanisms of these reactions via theoretical and experimental research, which can provide a reference to formulate the optimal surface structure.
3.1 Fuel cells
Environmental pollution and shortage of fossil fuels are the two major factors that restrict the development of our society. There is an urgent need to find clean, renewable, and efficient energy sources that can replace fossil fuels. Low-temperature fuel cells, such as proton-exchange membrane fuel cells (PEMFCs), direct methanol fuel cells (DMFCs), and direct ethanol fuel cells (DEFCs), are promising renewable energy devices that can convert chemical energy into electric power without the requirement of fossil fuels or environmental pollution. In addition, fuel cells can be used as portable energy devices, as well as form the driving force for vehicles (which have received increased attention in recent years). Although there are many advantages for the use of noble-metal-based nanocrystals in electrocatalysis, there is still a big gap between the current catalytic performance and performance required for practical applications. Here, we introduce several of the main electrochemical reactions occurring in fuel cells and focus on how to improve the catalytic performance of noble-metal-based catalysts with well-defined nanostructures.Pt-Based metals are also the most studied noble-metal-based catalysts toward HOR under alkaline conditions. Alloying Pt with other metals is a valid strategy to achieve higher activity of catalysts. For example, Alia et al. synthesized platinum-coated copper nanowires (Pt/Cu NWs) by the partial galvanic displacement of Cu NWs.141 Pt/Cu NWs outperformed Pt/C in terms of area and mass exchange current densities by 3.5 times and 1.9 times in a hydrogen saturated 0.1 M KOH electrolyte, respectively. They attributed the increased activity to the electronic tuning provided by Cu and the compressive strain on Pt. Both experiments and theoretical calculations demonstrated that the hydrogen binding energy (HBE) is a vital factor that can influence the catalytic activity in HOR. Skúlason et al. constructed a volcano relationship between the exchange current densities of HOR and hydrogen adsorption energies on different metals.142 They suggested that the HBE on a catalyst surface should be neither too strong nor too weak. When the binding energy was strong, the desorption of hydrogen from the catalyst surface would be difficult, and therefore, prevented the further adsorption of other reactants. When adsorption was too weak, the adsorbed species would be hard to activate. Scofield et al. investigated the HOR on a number of crystalline ultrathin PtM alloy NWs (M = Fe, Co, Ru, Cu, and Au) by employing an ambient and facile wet synthesis method. They found that the as-synthesized alloy NW catalysts, particularly PtRu NWs, yielded improved HOR activities as compared to those obtained when using a commercial Pt standard or as-synthesized Pt NWs.143 They attributed this result to the optimized HBE on the PtM surfaces, which was neither too strong nor too weak.
Several studies have shown that the facets and particle sizes of nanostructures also play an important role in determining the catalytic activity under alkaline conditions toward HOR. Sarraf et al. studied three low-index facets of Pt in alkaline solutions toward HOR.144 They found that the (110) facets showed better activity than (100) and (111) facets because of the suitable HBE. Junya et al. conducted the HOR in an alkaline solution on a series of different sizes of Ru NPs in the range from approximately 2 to 7 nm.145 The Ru NPs were synthesized by the liquid-phase reduction of isopropanol and aqueous solution of RuCl3 using NaBH4 in the presence of Vulcan XC-72R. Then, the as-synthesized Ru/C was treated at different temperatures in order to obtain catalysts with different sizes. They found that Ru NPs were an amorphous-like structure when the size of Ru was below 3 nm, whereas the particles possessed well-defined facets when the size of Ru was above 3 nm. The HOR activity normalized by the metal surface area showed volcano-shaped dependence on the particle size, where the maximum activity occurred at approximately 3 nm. They attributed the superior activity of 3 nm Ru NPs in HOR to their unique structure.
Understanding the reason why the same catalyst exhibits different catalytic activities under acidic and alkaline conditions can facilitate the optimization of catalyst performances. Several studies have found that except HBE, the binding energy of OH should be taken into account in alkaline environments.146,147 It can be assumed that the excellent surface structure of the catalyst for HOR should possess not only a suitable HBE value but also optimized adsorption for *OH under alkaline conditions. Qiu et al. fabricated bcc-phase PdCu alloy NPs via a wet chemistry method followed by a critical thermal treatment at various temperatures.148 The result of the HOR implied that the catalyst with a higher ratio of the bcc phase than the fcc phase showed better catalytic performance. The catalyst treated at 500 °C and defined as PdCu/C-500 °C with the highest proportion of the bcc phase, exhibited the highest mass activity of 1.727 mA μgPd−1 and specific activity of 2.922 mA cm−2 at overpotential of 100 mV in 0.1 M KOH electrolyte, which was about 2 times higher that of Pt/C and 4 times higher that of Pd/C, respectively (Fig. 12a and b). They performed DFT calculations to investigate the relationship between enhanced activity and *H and *OH adsorption strengths under alkaline conditions (Fig. 12c). The fcc- and bcc-phase PdCu NPs yielded similar HBE values, but different *OH binding energies. The binding energy of *OH on bcc-phase PdCu was similar to that of Pt (111), while that for fcc-phase PdCu was weaker. From this study, we can also conclude that the binding energy of *OH should also be appropriate, neither too strong (like that in Cu) nor too weak.
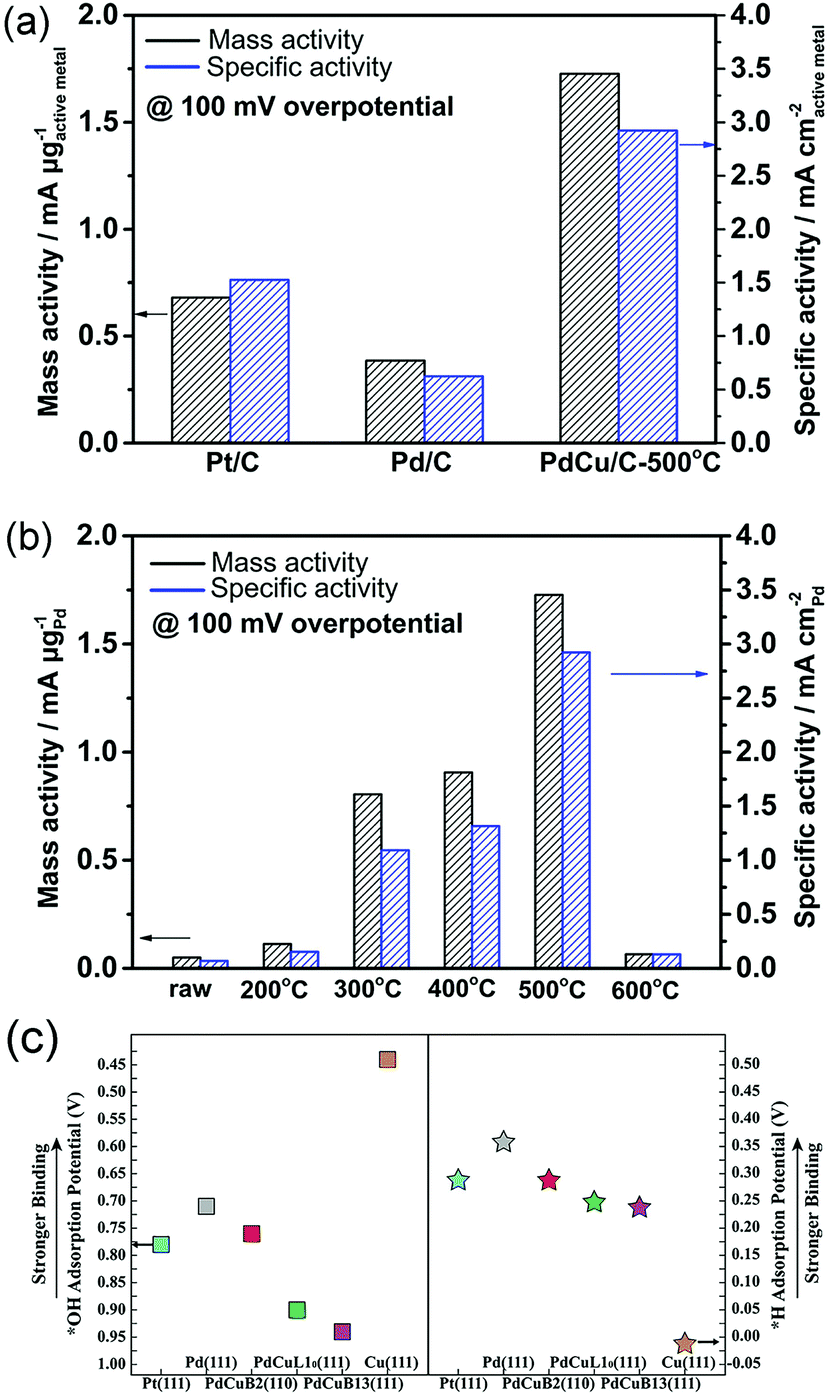 | ||
| Fig. 12 (a) and (b) Comparison of HOR mass and specific activity (MA and SA) of Pt/C, Pd/C, and PdCu/C at overpotential of 100 mV in H2-saturated 0.1 M KOH electrolyte. (c) Equilibrium adsorption potentials (vs. RHE) for *H (1/6 ML) and *OH (1/3 ML) on Pt, Pd, bcc-PdCu (B2 phase), and fcc-PdCu (L10 and B13 phases) terrace surfaces. The three PdCu surfaces shown here are covered by 1 mL Pd. Adapted with permission from ref. 148. Copyright 2018, Royal Society of Chemistry. | ||
As mentioned before, HOR under alkaline conditions yields several advantages, such as reducing the corrosion of noble-metal-based catalysts, ability to use cheaper metals, and increased catalyst stability. However, the catalytic activity in an alkaline solution is not as high as that in acidic media. It is important to enhance the activity under alkaline conditions. As far as we know, studies involving the catalysts used for HOR under alkaline conditions are still relatively few. Additional efforts need to be devoted toward both experiments and calculations to determine better catalysts with outstanding activity.
Several studies have considered improving the catalytic activity by combining Pt with other metals to form a composite structure, such as the deposition of a thin Pt shell over a transition metal core or synthesis of various shapes of catalysts.79,151–155 Research involving a core–shell structure has covered many kinds of transition metals; for example, Ni, Cu, Co, Ti, Cr, and Fe. Interestingly, most of them have exhibited improved activity in comparison to a conventional Pt/C catalyst due to the modification of the Pt electronic structure by transition metals and the lattice strain generated from the lattice mismatch between the core and shell.156–161
The exposed facets of the catalyst can also change the catalytic performance. For example, Duan et al. demonstrated that the catalytic activity for ORR was higher on the (111) surfaces than that on the (100) surfaces by determining the reaction energetics on Pt (100), Pt (111), Pt/Ni (100), and Pt/Ni (111) surfaces.162 Moreover, Pt concave cubes with a higher number of high-index facets exhibited enhanced catalytic activity toward ORR as compared to their low-index counterparts, such as cuboctahedrons, cubes, and commercial Pt/C catalysts because of the existence of several unsaturated coordination sites.163 Xia et al. synthesized concave Pt nanoframes enclosed by {740} facets by a one-pot solvothermal method.164 The Pt nanoframes with highly negative curvature surface had a higher percentage of step, corner, and edge sites, and therefore, showed higher activity.
Several studies have shown that alloying Pt with transition metals, particularly Fe, Co, and Ni, can enhance the catalytic performance. However, transition metals can get easily oxidized and get leached out from nanocrystals during ORR under acidic conditions, resulting in poor catalytic stability. Moreover, when oxygen species intermediates form during the reaction, the segregation of transition metal atoms is accelerated. Adding Au into Pt-based catalysts can significantly enhance the activity and durability by eliminating the oxygen species from the Pt surface or preventing the oxidation of Pt at higher voltages.14,165,166 Except Au, Mo and Rh can also help in retaining the shape and composition of Pt-based alloys by suppressing the migration of surface Pt or the stabilization of undercoordinated Pt sites.16–18 Jia and co-workers synthesized Mo-doped PtNi octahedra via an efficient one-pot approach with N,N-dimethylformamide (DMF) as the solvent as well as the reducing agent and benzoic acid as the structure-directing agent.167 They clarified that Mo atoms were preferentially located on the vertex and edge sites of Mo–PtNi/C in the form of oxides by combining ex situ and in situ spectroscopic techniques, density functional theory (DFT) calculations, and a newly developed kinetic Monte Carlo model. Mo dopants can improve the ORR kinetics by modifying the coordination environments of Pt atoms on the surface. Mo dopants also lead to increased concentrations of subsurface Ni and stabilization of undercoordinated Pt sites such that Mo–PtNi/C octahedral NPs exhibit exceptional activity and durability toward ORR.
In addition to these experiments, some theoretical calculations have been conducted to explore the structure–activity relationship. There are three possible mechanisms (oxygen dissociation, peroxyl dissociation, and hydrogen peroxide dissociation) of ORR that can exist on the surfaces of metal catalysts (Fig. 13).168 Nørskov and co-workers studied the catalytic performance of polycrystalline alloy films Pt3M (M = Ni, Co, Fe, and Ti) in ORR.159 They gave specific activity of Pt and Pt3M electrodes for ORR at 0.9 V in 0.1 M HClO4 and found that Pt3M alloys exhibited higher catalytic activities than pure Pt. Different alloys behaved differently, which meant that the alloy composition was vital for ORR. By using DFT calculations, the activity as a function of the adsorption energy of oxygen is shown in Fig. 14a, where the black dots represent theoretical results and red dots represent the experimental results. Volcano-shaped dependence of the rate of oxygen chemisorption energy explained the reason why PtM alloys showed superior activity: if the binding energy of oxygen was too strong (to the left of the maximum), the rate was limited by the removal of adsorbed O and OH species; on the contrary, if the binding energy was too weak, the dissociation of O2 would be the limiting step. A correlation existed between the d-band center of the metal and the oxygen adsorption energy, where they finally obtained the activity versus experimentally measured d-band center relative to Pt (Fig. 14b). It can be suggested that alloying Pt with other metals can regulate the binding energy of the intermediates, thereby changing the activity. Therefore, tuning the surface structure of the catalyst with a suitable d-band center may be a reasonable technique to enhance the activity. Karan et al. studied the ORR activity of a Pt monolayer shell (PtML) or Pt and Pd bilayer shell (PtML/PdML) on IrRe NP cores with different ratios of Ir and Re, which were synthesized by the galvanic displacement of a Cu monolayer deposited via UPD.169 They found that the molar ratio of Ir to Re influenced the binding strength of the adsorbed OH, and therefore, the O2 reduction activity of the catalysts. PtML/PdML/Ir2Re exhibited the best activity because of the optimal binding energy of the OH species. Bordley et al. synthesized PtAg nanocages for different Pt contents via galvanic replacement reactions.170 PtAg nanocages with the lowest Pt content exhibited the highest activity toward ORR, which was 2–3 times higher than the previously reported Pt catalysts under similar conditions. They attributed the superior catalytic performance to the downshifted d-band position of Pt and advantageous shift in the adsorption energy of oxygen-containing intermediates on the surface of the catalyst.
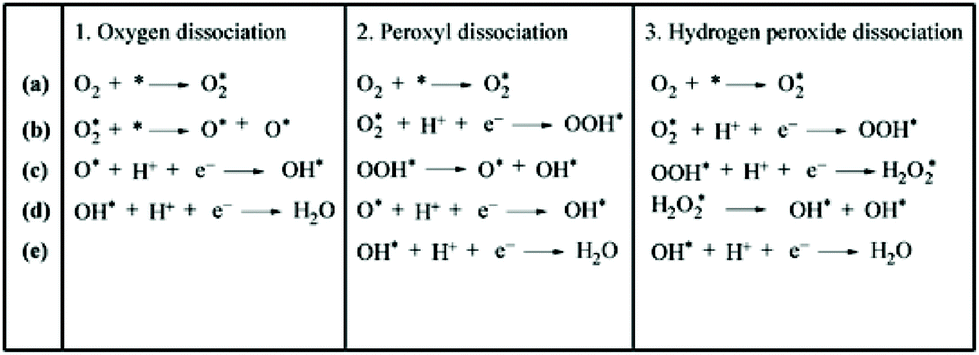 | ||
| Fig. 13 Three possible mechanisms for ORR. Adapted with permission from ref. 168. Copyright 2011, the Owner Societies. | ||
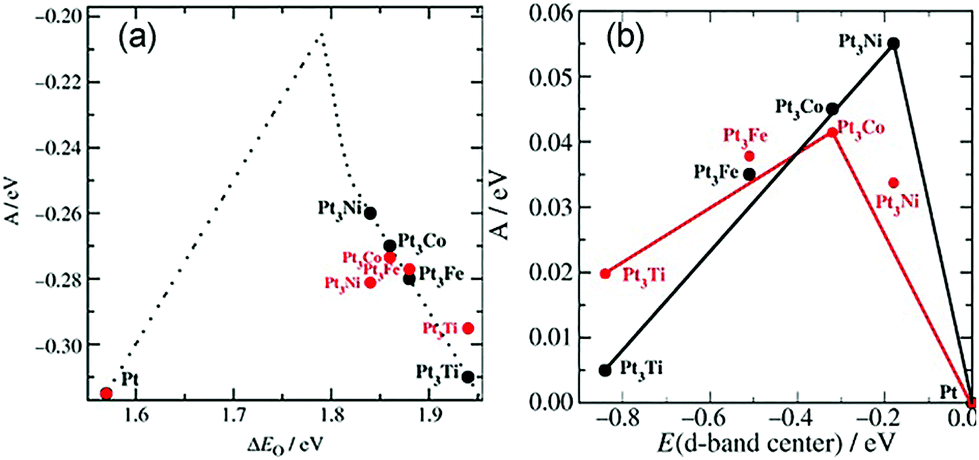 | ||
| Fig. 14 (a) Model of the activity (A = kBTln(r), where r is the rate per surface atom per s) at cell potential of 0.9 V shown as a function of the adsorption energy of oxygen. (b) Activity versus experimentally measured d-band center relative to Pt. The activity predicted from DFT simulations is shown in black, and the measured activity is shown in red. Adapted with permission from ref. 159. Copyright 2006, Wiley-VCH. | ||
Developing high-efficiency electrocatalysts for ORR is a vital step for fuel cell applications. Pt-Based nanomaterials as the most active catalysts have attracted increased attention. Several studies have been devoted toward improving the catalytic activity and durability by modulating the surface structure of the catalyst. Calculations have suggested that the d-band center can serve as the reference for formulating a superior surface structure with high activity. Therefore, we can use DFT calculations to prescreen catalysts with the best activity.
Noble-metal-based nanostructures have been exploited to promote the catalytic activity and increase CO-poisoning resistance. Alloying Pt with other metals or controlling catalyst morphology can change the atomic arrangement and coordination of Pt; therefore, the catalytic activity is also changed.174,175 Currently, Pt-based catalysts, such as Pt–M (M = Pd, Au, Ru, Co, Cu, and Ir) with various morphologies (nanopolyhedra, NWs, nanoframes, core–shell, and nanodendrites), have been widely reported.26,176–181 Our group synthesized sub-10 nm Pt–Pd nanotetrahedra and NCs via one-pot hydrothermal routes with efficient facet-selective agents.182 The (100)-facet-enclosed Pt–Pd NCs demonstrated higher activity, whereas the (111)-facet enclosed Pt–Pd nanotetrahedra exhibited better durability. Wang et al. demonstrated the synthesis of dendritic Pt–Pd nanocages with Pd as the hollow interiors and Pt as the porous dendritic shells via the selective chemical etching of Pd cores from the preformed Pt-on-Pd nanodendrites.183 The dendritic Pt–Pd nanocages exhibited high activity in MOR because of sufficient catalytic active sites present on both their interior and exterior surfaces. Pseudo-Pd–Pt alloy phases were formed at the internal surface of the hollow structures, which was favorable for reducing the electronic binding energy in Pt and facilitating the C–H cleavage reaction in methanol decomposition.
Koenigsmann et al. investigated different chemical components of Pd-based NWs for MOR. They found that all the synthesized NWs exhibited higher activity than commercial Pt/C, and Pt7Pd3 showed the most superior catalytic performance among a series of as-synthesized Pt1−xPdx NWs.184 Both experiments and DFT calculations confirmed that PtRu alloy NPs are suitable for MOR because of their superior activity and CO-poisoning tolerance because of bifunctional and ligand effects. As an oxyphilic metal, Ru can provide adsorbed hydroxyl groups (OHads) at lower potential than that in pristine Pt, which can remove COads by oxidizing it into CO2.179 A series of works have reported that PtRu nanocrystalline particles yield high activity toward MOR. However, studies regarding the structure-dependent electrocatalytic properties of PtRu nanocrystals are scarce because the high reduction potential of Ru and lattice mismatch between Pt and Ru make it a formidable challenge to synthesize shape-controlled PtRu nanocrystals rather than irregular NPs.185,186 Huang et al. synthesized ultrathin PtRu nanocrystals with tunable morphologies (NWs, nanorods, and NCs) through a one-step solvothermal approach (Fig. 15a–c).187 PtRu NWs and PtRu nanorods (PtRu NRs) were enclosed by {111} facets and PtRu NCs were enclosed by {100} facets (Fig. 15d–f). Surprisingly, the {111}-terminated PtRu NWs and PtRu NRs exhibited much higher stability and electrocatalytic mass activity toward MOR. In particular, PtRu NWs showed mass activity that was 2.28 and 4.32 times higher than those of {100}-terminated PtRu NCs and commercial Pt/C, respectively, at 0.70 V vs. RHE in 0.1 M HClO4 aqueous solution with 0.5 M CH3OH at a sweep rate of 50 mV s−1 (Fig. 15g and h). It can be demonstrated that PtRu {111} facets possessed higher tolerance toward CO poisoning as compared to {100} facets.
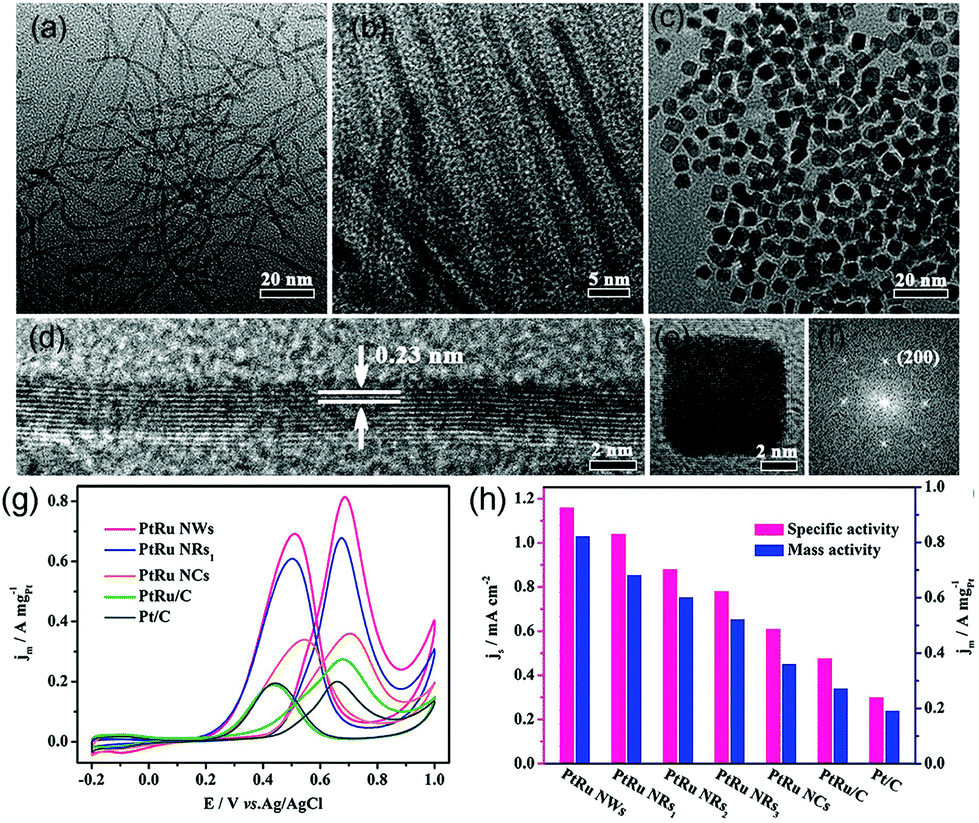 | ||
| Fig. 15 TEM images of (a) PtRu NWs, (b) PtRu NRs1, and (c) PtRu NCs. HRTEM images of an individual (d) PtRu NW and (e) PtRu NC, and the corresponding FFT pattern (f) of (e). (g) Electrocatalytic performances of PtRu nanocrystals and commercial Pt/C catalysts. CVs of (g) PtRu NWs, PtRu NRs1, PtRu NCs, commercial PtRu/C, and commercial Pt/C for MOR in 0.1 M HClO4 + 0.5 M CH3OH solution at a sweep rate of 50 mV s−1. (h) Histograms of the mass and specific activities of different catalysts. Adapted with permission from ref. 187. Copyright 2017, American Chemical Society. | ||
Except for nanocrystalline alloys, amorphous alloys with long-range disordered but short-range ordered atomic arrangements, possessing abundantly unsaturated sites with high surface energy that are crucial for the activation of reactants in catalysis, also perform effectively in this reaction.188,189 Zhao et al. reported amorphous CuPt alloy hollow nanotubes as efficient catalysts for MOR prepared by the Na2S2O3-assisted galvanic replacement reaction.104 The amorphous CuPt alloy exhibited better MOR activity and stability than those exhibited by crystalline CuPt and commercial Pt/C catalysts, which could be ascribed to the enhanced CO tolerance attributable to the strong interaction between the Pt and Cu atoms.
Evidently, tunable composition and catalyst structure are essential factors to increase the activity of catalytic reactions. Obviously, more superior catalysts can be found by designing the surface structure of the catalyst. As compared to the trial-and-error approach, understanding the mechanism of MOR on the catalyst surface may provide guidance toward fabricating the optimal catalyst. Ferrin et al. studied MOR on the (111) and (100) facets of eight transition metals (Au, Ag, Cu, Pt, Pd, Ir, Rh, and Ni) using periodic, self-consistent density functional theory (DFTGGA).190Fig. 16a shows the possible reaction pathways and intermediates in MOR. Intermediates were adsorbed on the catalyst surface through the carbon or oxygen atom. Therefore, the binding energy of the intermediates was linearly correlated with the binding energy of CO* and OH*, respectively. Finally, the potential-determining steps for MOR on the (111) and (100) facets were plotted using the binding energies of CO* and OH* as the descriptors (Fig. 16b). Both potential-determining steps and mechanisms were structurally sensitive; therefore, tuning the composition and structure could be used to adjust the activity and stability. Obviously, the optimal active sites for MOR can be designed according to an optimized binding energy of CO* and (or) OH*. Therefore, we can screen out the possible active surface structures via theoretical calculations. Qi et al. synthesized PtZn intermetallic NPs (3.2 ± 0.4 nm) via a facile and capping-agent-free strategy using a sacrificial mesoporous silica (mSiO2) shell.191 They found that the NPs went through a “non-CO” pathway for MOR because of the stabilization of the OH* intermediate by Zn atoms; therefore, the stability and activity were enhanced than those for a pure Pt system. Wang et al. studied the catalytic performance of a type of ternary Fe1−xPtRux nanocrystals, which were synthesized via a simple cation replacement reaction from binary FePt nanocrystals.192 Fe1−xPtRux exhibited superior catalytic ability to withstand CO poisoning in MOR because the binding energy of CO on Pt was weakened.
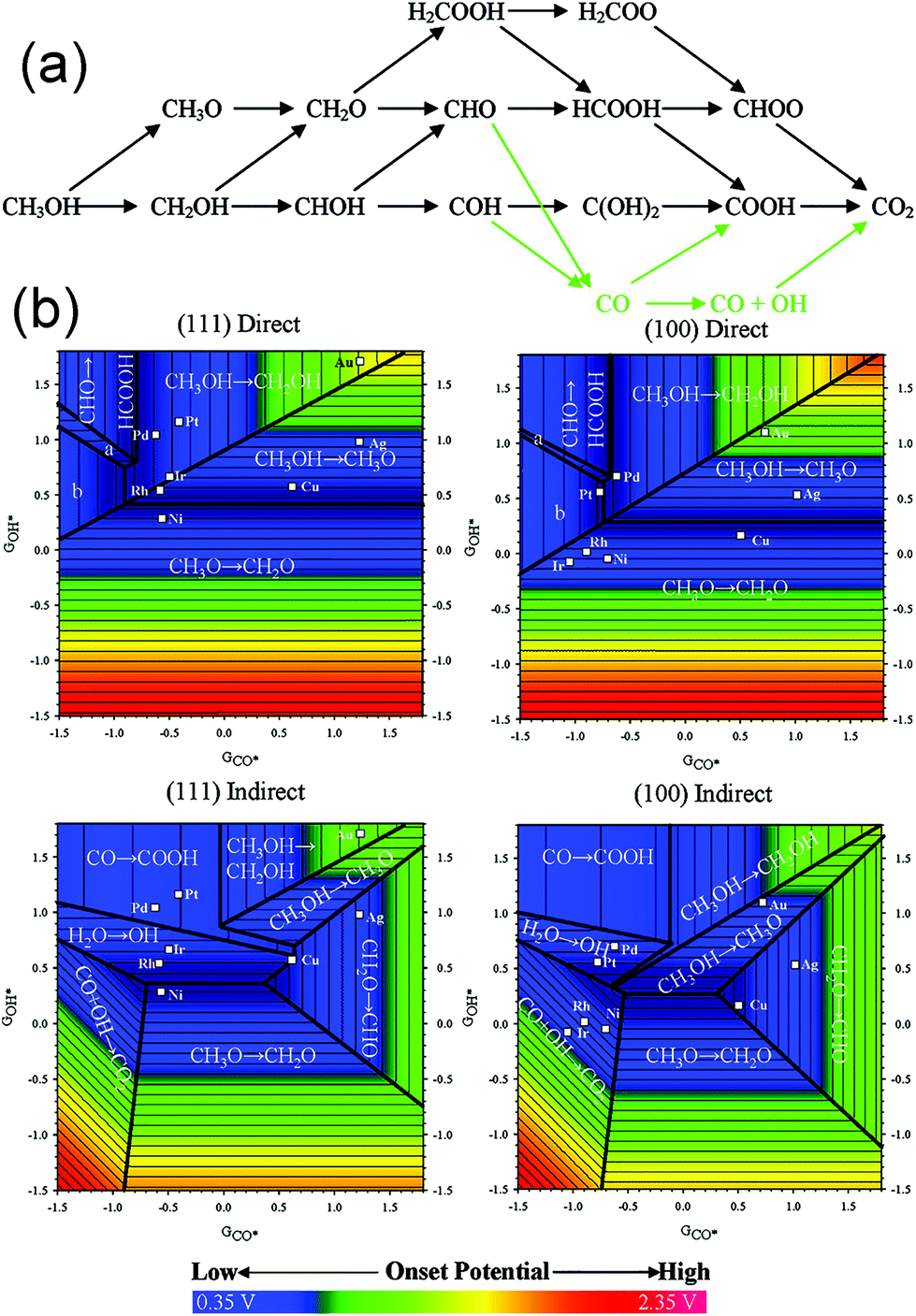 | ||
| Fig. 16 (a) Schematic representation of the reaction pathways and possible intermediates considered here for methanol electrooxidation. Green arrows indicate the indirect mechanism to CO2 formation. (b) Potential-determining steps for methanol electrooxidation on (111) and (100) facets plotted using GCO* and GOH* as the descriptors. Adapted with permission from ref. 190. Copyright 2009, American Chemical Society. | ||
In summary, several groups have contributed toward the development of catalysts with excellent activity and stability for MOR. There is a relationship between the surface structure and activity. The purpose of studying the structure–activity relationship is to formulate a better catalyst with higher activity. We can investigate the structure–activity relationship by using theoretical calculations as well as experiments. As mentioned earlier, the binding energy of the intermediates in the MOR is very important. We can screen out the catalyst with higher activity and stability by calculating the binding energy of *CO and *OH. Moreover, the combination between calculations and experiments is more significant. For example, Pt is easily poisoned by the adsorbed COads, and the addition of oxophilic metals (such as Ru), forming OHads, can oxidize COads.193 DFT calculations indicate that Pt–COads and Ru–OHads species cannot form a transition state (Pt–CO⋯OH–Ru) unless the distance between Pt and Ru is less than (or equal to) 4.0 Å. In this case, alloying is a suitable way to achieve this rather than forming a heterogeneous structure.
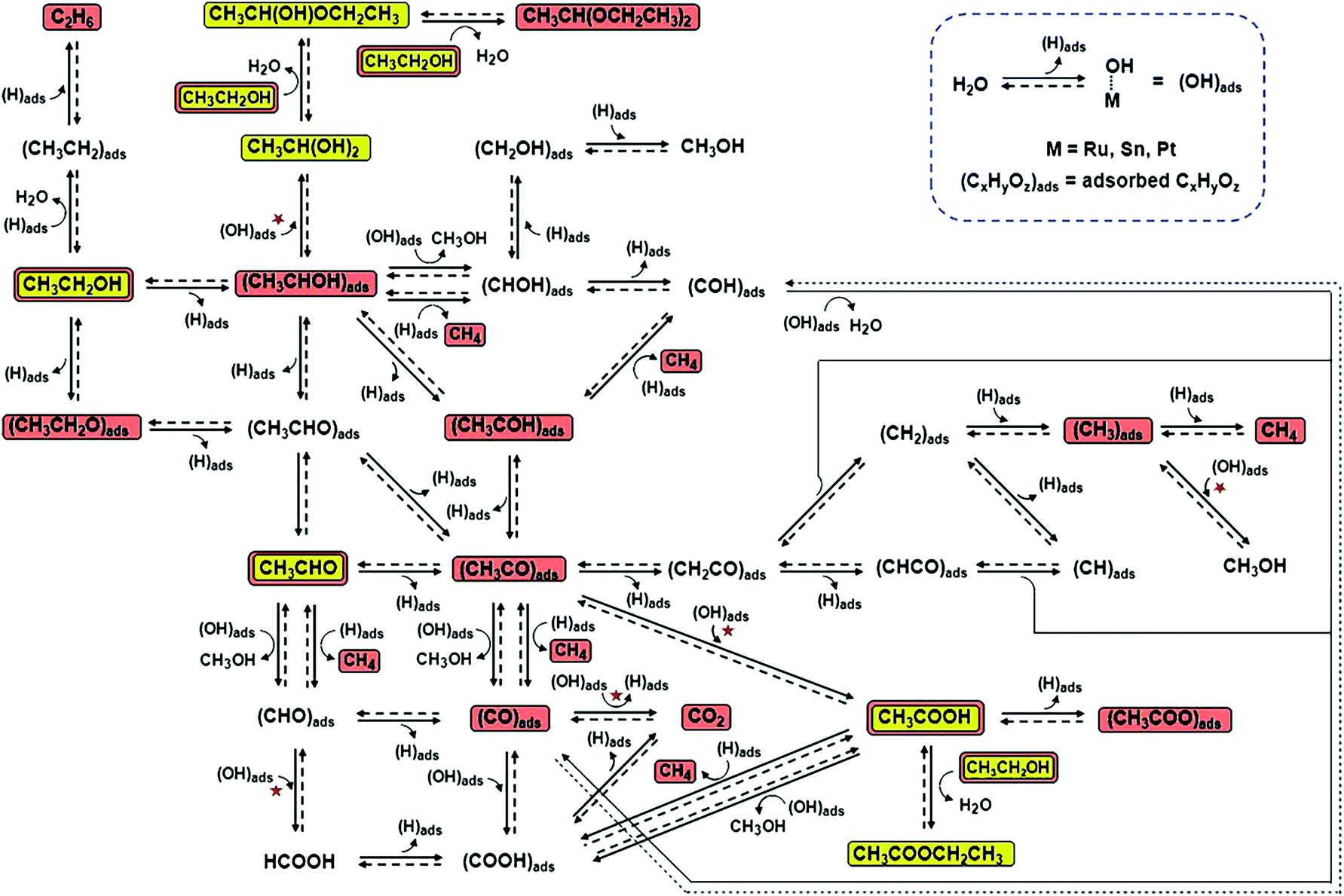 | ||
| Fig. 17 Ethanol electrochemical reaction pathways on Pt-based catalysts. The reaction steps marked with red stars are those in which (OH)ads are involved and catalytic sites are regenerated due to reaction with (OH)ads. The chemical species highlighted in yellow were observed in this study, and those highlighted in brown were observed in earlier studies that used other analytical techniques. Adapted with permission from ref. 194. Copyright 2011, Wiley-VCH. | ||
The two most studied systems are Pt- and Pd-based catalysts in EOR. Pt is the most efficient catalyst in EOR. However, some studies have reported that pure Pd is more active than Pt in EOR in alkaline environments because the kinetics of certain organic molecules can be dramatically enhanced in such environments. Moreover, the in situ FTIR results reveal that the ability of Pd to break the C–C bond of ethanol is slightly better than that of Pt under the same conditions.198,199 Several studies regarding Pd-based catalytic materials for EOR have been reported.20,200–202
The effects of the second or third metals on the catalytic properties have been investigated, such as Sn, Ir, Rh, Ru, Ni, Au, Cu, Ag, and Co.8,203–207 Ir and Rh can help in breaking the C–C bond; therefore, the process of twelve-electron transfer is preferred.8,9,208 Sn is easily oxidized and can adsorb OH, which, in turn, oxidizes the intermediate species (such as *CO) and releases the reaction sites.10,11,209 Other noble or transition metals can regulate the electronic structures of Pt and Pd, and therefore, regulate the reaction activity. Du et al. reported a modified polyol method for the synthesis of PdSn alloy with different Sn contents.10 Some PdSn/C catalysts exhibited specific peak current densities at least two times higher than those of commercial Pd/C (JM) catalysts in 0.5 M ethanol and 0.5 M KOH solution. The activity order of the as-synthesized catalysts in EOR was Pd86Sn14 > Pd75Sn25 ≈ Pd > Pd47Sn53. The promotional effect of Sn in the PdSn alloy could be explained by the bifunctional effect. Sn and/or SnOx had stronger interactions with the hydroxyl group (OHads), and Pd had excellent properties in the adsorption and dissociation of ethanol. Such a synergetic effect yielded an increasingly active binary catalyst than their monometallic counterparts. However, the higher Sn content would also decrease the occupancy of active Pd atoms on the surface, and consequently, impair the overall performance of the dissociation of adsorbed ethanol. Our group reported ternary Pt3RhM (M = Fe, Co, Ni, Cu, Ga, In, Sn, and Pb) nanoalloys with similar geometric structures and sizes prepared via a one-pot solvothermal method.210 We found that Pt3RhSn/C exhibited 67- and 7-fold increases in specific activity and mass activity as compared to those observed in commercial Pt/C at 0.45 V (vs. RHE). We attributed this superior catalytic activity to the synergistic electronic effects of the nanoalloys and moderate adsorption strength of the stable intermediates.
The specific shape of nanocrystals with the designed surface structure can increase the reactivity and stability. Erini et al. reported a shape-controlled PtNiRh octahedral electrocatalyst using a wet chemical approach.205 The octahedral particles with Ni-rich facets, Pt-rich frame, and Rh accumulation at the surfaces exhibited better performance toward EOR than those by the spherical nanocrystal. This strongly suggested that the octahedral {111} facets forming particular sites possessed the ability to break the C–C bond. Our group demonstrated a facile hydrothermal method to synthesize (111)-terminated Pt–Pd–Rh NTOs and (100)-terminated Pt–Pd–Rh NCs with different compositions.211 PtPdRh NTOs exhibited the highest selectivity toward CO2 at 0.5 V vs. RHE. We attributed the capability of the full oxidization of ethanol to the synergistic effect of the three metals.
In addition to the most investigated Pt- or Pd-based catalysts, Zhang et al. synthesized cyclic penta-twinned (CPT) Rh nanobranches (NBs) via a facile solvothermal process.9 The as-prepared Rh NBs possessed a high percentage of open {100} facets with significant CPT-induced lattice strain, as shown in Fig. 18a–c. The as-prepared CPT Rh NBs exhibited outstanding electrocatalytic activity and CO2 selectivity as compared to those of single-crystal tetrahedral nanocrystals, icosahedral nanocrystals, and commercial Rh black (Fig. 18d and e). By combining with DFT calculations, they attributed the high catalytic performance to the open {100} facets. Moreover, the CPT-induced lattice strain further boosted the catalytic activity by enhancing the adsorption strength and lowering the reaction barrier of the dehydrogenation process of ethanol. As compared to the earlier example where the {111} facets were favored, we conclude that this component is an important part of the surface structure of the catalyst. For different catalytic systems, both composition and shape should be taken into account to understand the structure–activity relationship.
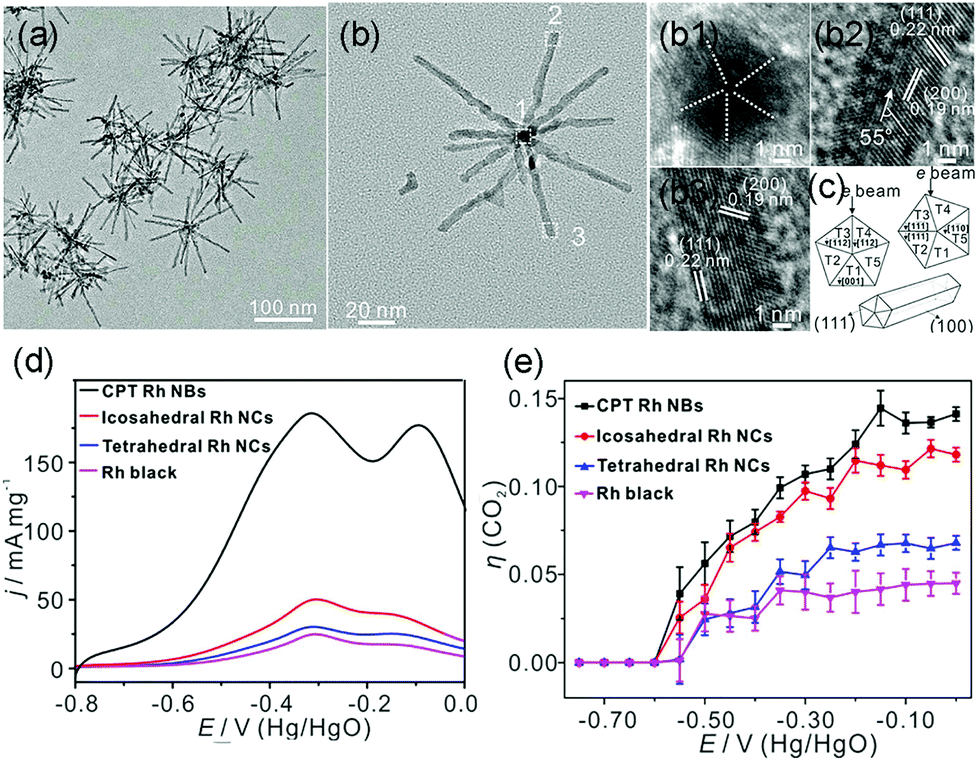 | ||
| Fig. 18 (a) TEM images and (b) high-magnification TEM image of the as-prepared CPT Rh NBs. (b1–b3) HRTEM images taken from different branches. (c) Schematic models of the CPT nanorods and different orientations of the cross-section of CPT nanorods with respect to the electron beam. (d) Positive scan curves of EOR from −0.80 to 0.00 V vs. Hg/HgO (scan rate: 50 mV s−1) in 1.0 M NaOH + 1.0 M ethanol solution. (e) Selectivity (η (CO2)) for complete ethanol oxidation to CO2. Adapted with permission from ref. 9. Copyright 2018, American Chemical Society. | ||
It is well known that for metal particles or clusters with sizes below a few nanometers, strong size effects are expected on the catalytic performance from both geometric and electronic considerations. Von et al. described a series of experiments to investigate the EOR activity on different Ptn clusters supported on indium tin oxide (ITO), which could be prepared and characterized in an ultrahigh vacuum (UHV).212 They conducted the experiment in situ without significant air exposure: an antechamber was attached beneath the main chamber housing a bakeable in situ electrochemical cell (Fig. 19a). The activities of different sizes of Ptn clusters are shown in Fig. 19b, where 0.1 M HClO4 and either 1.0 or 2.0 vol% of ethanol are used. The oscillatory dependence of activity on size was anticorrelated with the binding energy of the Pt 4d core level, which implied that the activity was controlled by the electronic structure of the supported clusters. We can conclude that the size effects are very important in tuning the activity of the nanostructures by regulating the electronic structures of the metal atoms.
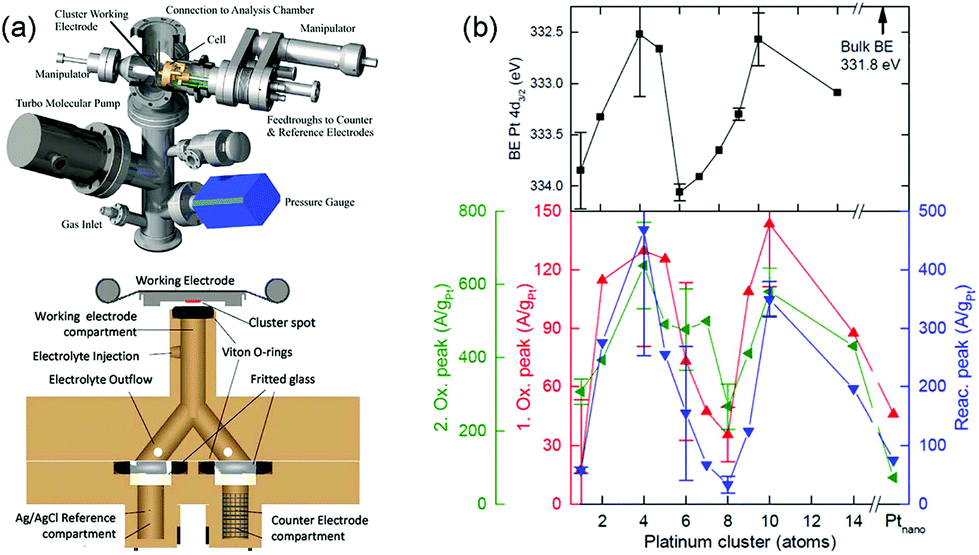 | ||
| Fig. 19 (a) (top) Antechamber with cell. (bottom) Horizontal section of the cell. (b) (top) Pt 4d binding energies. (bottom) Mass activities for each EOR peak as a function of cluster size. Adapted with permission from ref. 212. Copyright 2016, American Chemical Society. | ||
Overall, the current research is mainly focused on the effect of components and facets of the catalyst on the catalytic activity in EOR; however, the effects of nanostructures, such as core–shell structure, hollow, cage–bell, stellated/dendritic, and dimeric, are rarely studied. Novel structures with electronic coupling effects and lateral strain effects may contribute toward improving the performance of noble-metal-based catalysts in EOR. Moreover, due to the complicated reaction process, no specific mechanism has been devised to determine the more active surface structure; therefore, it is urgent to perform theoretical and computational studies to provide insights into the mechanism and recognize the optimal surface structure. Formulating a reasonable theoretical calculation model that is very similar to real reaction conditions can facilitate the design of EOR catalysts.
3.2 Electrolysis of water
As a renewable and environmentally friendly energy source, hydrogen has been considered as an alternative to fossil fuel energy for the future.213–215 Until now, hydrogen has been primarily produced by the reforming of fossil fuels, such as natural gas, in a centralized manner and distributed to user locations in the form of compressed gas. Water electrolysis is a more favorable technique to produce hydrogen because electricity as the main energy supplier can be converted from renewable sources such as windmills and/or solar cells. However, noble-metal-based catalysts that are commonly used in electrolysis (e.g., Pt) prove to be an obstacle in large-scale practical applications. Herein, we focus on research involving HER and OER. In order to decrease the amount of noble metals and maintain or increase the catalytic activity, it is necessary to study the structure–activity relationship to determine the optimum surface structure.Due to the higher activity of the catalyst in acidic media than that in alkaline media, several studies have investigated the activity toward HER under acidic conditions. Three possible reaction steps have been used to explain the HER process with regard to various electrocatalysts in acidic media.216 (1) The first Volmer step is hydrogen adsorption: H3O+ + e− + M → MHad + H2O; (2) Heyrovsky step (electrochemical desorption): MHad + e− + H+ → M + H2; (3) Tafel step (chemical desorption): 2MHad → 2M + H2, where M represents the active site and MHad represents the adsorption of Had on the metal. Studies regarding HER activity versus HBE on various monometallic metals have been reported in bases and acids, and the obtained volcano-shaped curves suggest that catalysts with optimal HBE are the most active (Sabatier's principle).217–219 We can conclude that the catalytic performance can be improved from two aspects: increase in the active sites and optimization of HBE. It is well known that nanostructures (such as nanoframes, nanopores, and nanosheets) can expose more active sites, which improves catalytic performance. Further, alloying noble metals with other compositions or designing specific heterostructures can enhance activity by modulating the surface structure of the catalyst.
There are many investigations on Pt-based catalysts for achieving higher catalytic performance toward HER. The improved catalytic performance of Pt-based multimetallic nanocrystals can be mainly attributed to the strain effect, electronic effect, geometric effect, and surface polarization. Certain studies have demonstrated that interfacial polarization between Pt and other metals may induce the accumulation of negative charges on the Pt surface and facilitate the HER process.220,221 The generated surface strain can affect the surface charge state and consequently change the binding energy of the adsorbates on Pt, which is important for enhancing the electrocatalytic activity of Pt catalysts toward HER. Bao et al. reported that an atomic dodecahedral PtCu alloy shell on Pd nanocrystals achieved 25-fold enhancement of mass activity toward HER as compared to commercial Pt/C in acidic media due to the strain generated from the core–shell structures.222 Wang et al. demonstrated that PtNi3 bimetallic concave octahedrons with a majority of Pt on the framed structure, which were synthesized in ethylene glycol solution, showed substantially enhanced electrocatalytic properties toward HER as compared to commercial Pt/C due to electronic and geometric effects.223 Mahmood et al. fabricated PtAg and PtAgCo nanosheets utilizing an oxidative etching growth strategy for staking faults (Fig. 20a).224 As compared to PtAg nanosheets, the as-prepared PtAgCo nanosheets were largely composed of Pt, particularly at the edges, and the incorporation of Co also took place at the edges. In Fig. 20b and c, PtAgCo nanosheets exhibited higher activity in HER as compared to PtAg nanosheets, which implied the importance of Co. They attributed the reactivity to the polarization effect of doped Co, which resulted in downshifts in the d-band centers of Pt, and therefore, decreased the binding energy of *H.
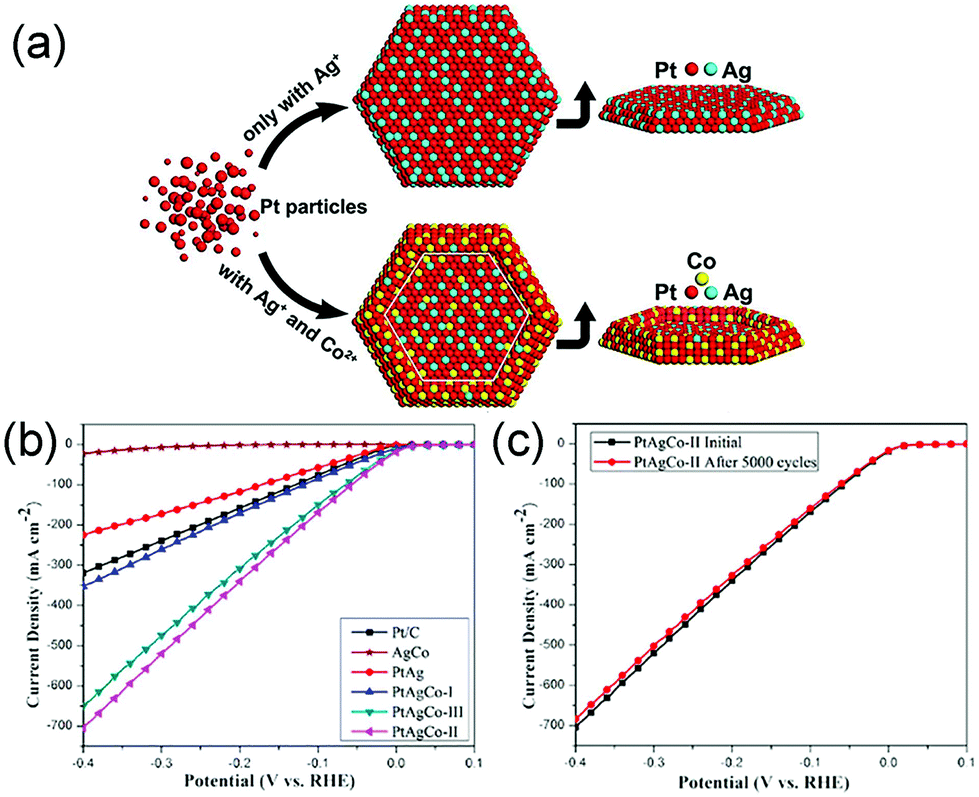 | ||
| Fig. 20 (a) Schematic illustration of PtAg and PtAgCo nanosheets. (b) Polarization curves for PtAg, AgCo, and PtAgCo nanostructures prepared at different molar ratios: (PtAg) 0.01:0.01, (AgCo) 0.01:0.01, (PtAgCo-I) 0.01:0.01:0.005, (PtAgCo-II) 0.01:0.01:0.01, and (PtAgCo-III) 0.01:0.01:0.015 in 0.5 M H2SO4. (c) Durability tests of PtAgCo-II samples. Adapted with permission from ref. 224. Copyright 2017, American Chemical Society. | ||
Other noble metals have been extensively investigated with regard to their design for yielding Pt-like activity and good stability toward HER, such as Ru, Ag, and Pd. Ru is a 4d transition metal, which is also a noble metal, but its price is 10 times lower than that of Pt.225 Ru-Based catalysts have also received increased interest for their similar hydrogen bond strength (∼65 kcal mol−1) as that of Pt. Zhang et al. synthesized ultrathin Ru nanosheets by a facile solvothermal method with the aid of isopropanol and urea.226 The 2D Ru nanosheets exhibited excellent HER activity because nearly all the atoms could be exposed and served as active sites for catalytic reactions, which was beneficial for fast interfacial charge transfer, and therefore, promoted the reaction rate. Huang et al. demonstrated the synthesis of sponge-like highly nanoporous Ag foam via a simple multiple-scan cyclic voltammetry method.227 The nanostructure exhibited superior activity in HER because of the increased number of active sites and the reduced Gibbs free energy of the adsorbed atomic hydrogen (Had) on the catalysts.
In addition to single-component noble metals, several studies have also investigated alloying noble metals with other metals or the synthesis of a heterogeneous structure. Zhang et al. prepared a necklace-like hollow NiRu nanoalloy using the galvanic replacement reaction and hollowing process based on the Kirkendall effect.228 The Ni43Ru57 nanoalloy exhibited the highest catalytic activity with overpotential of 41 mV (vs. RHE) to yield a current density of 10 mA cm−2. This excellent activity could be attributed to the effective electronic coupling of Ni and Ru, causing faster interfacial electron transfer kinetics. Fan et al. reported the synthesis of 4H/fcc trimetallic Au@PdAg core–shell nanoribbons (NRBs) fabricated via the galvanic reaction method under ambient conditions.229 Remarkably, the obtained 4H/fcc Au@PdAg NRBs exhibited excellent electrocatalytic activity toward HER because of the unique geometric effect and synergistic effect among Au, Pd, and Ag. Similar effects were also found in core–shell PdCu@Pd NCs that were synthesized by anodizing PdCu NCs at constant potential in 0.5 M H2SO4 solution.230 These PdCu@Pd NCs show the lowest HER onset potential due to the core–shell strain and ligand effects between Pd and Cu.
Besides the volcano plot correlating with both HER activity and calculated binding energy of *H, the generalized coordination numbers that reflect the location and geometric configuration of the catalyst can also influence the catalytic activity. Pohl et al. constructed various model Pt single-crystal electrodes that possessed different generalized coordination numbers, and they obtained the coordination–activity plots linking the geometry and HER activity of various Pt catalytic centers (Fig. 21a).231 The coordination–activity plots revealed that optimal coordination numbers for catalysts exist that can yield the highest activity. Lv et al. synthesized core–shell NiAu/Au NPs by treating the as-synthesized NiAu NPs in potential cycling between 0.6 and 1.0 V (vs. RHE) in 0.5 M H2SO4.232 The catalyst exhibited enhanced performance toward HER with Pt-like activity and considerably robust durability due to the formation of Au sites with low coordination numbers around the shell, and therefore, a lower reaction barrier (Fig. 21b and c).
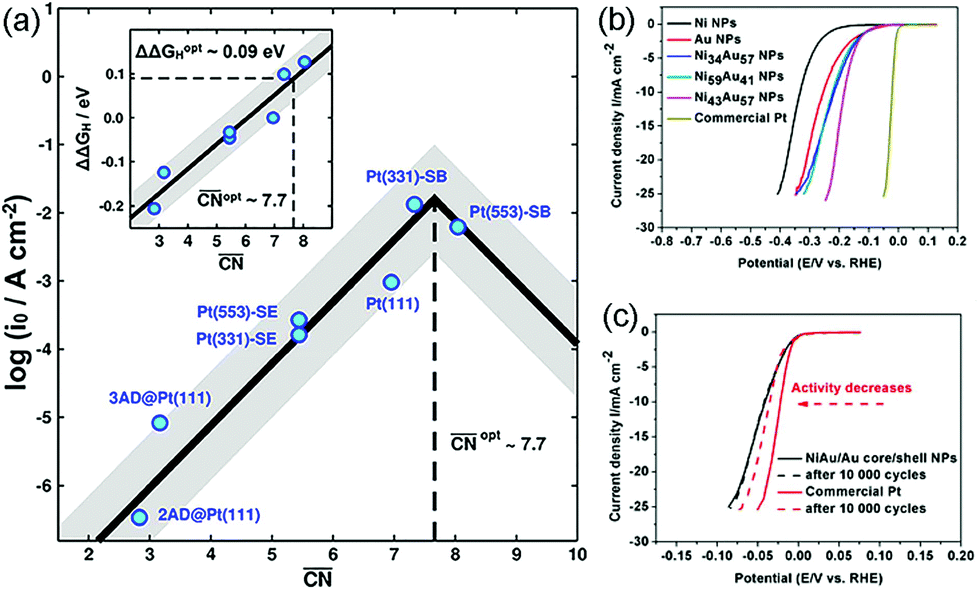 | ||
| Fig. 21 (a) Coordination–activity plot linking the geometry (in terms of generalized coordination numbers) and HER activity of various Pt catalytic centers. Inset: Correlation between differential adsorption energies with respect to Pt (111) and generalized coordination numbers. Adapted with permission from ref. 231. Copyright 2017, American Chemical Society. (b) HER polarization curves of various as-prepared NP catalysts. (c) HER polarization curves of NiAu/Au core–shell NPs and commercial Pt before and after 10000 cycling tests in 0.5 M H2SO4 solution. Adapted with permission from ref. 232. Copyright 2015, American Chemical Society. | ||
As mentioned earlier, the activity toward HER under acidic conditions is much better than that under alkaline conditions. Generally, the reaction rate on most catalysts for HER in alkaline solutions is 2–3 orders of magnitude lower than that in acidic solutions.233 Research involving alkaline solutions is not as comprehensive as those involving acidic solutions. However, electrocatalysts in OER, which is the half-reaction in the electrolysis of water, effectively work in basic or neutral media.234 Therefore, it is crucial to develop catalysts that are suitable for use under alkaline conditions. The reaction pathways in alkaline media comprise three steps, which are similar to those in acidic media.235 (1) The first Volmer step is the water dissociation process: H2O + e− → Had + OH−; (2) Heyrovsky step (electrochemical desorption): Had + e− + H2O → H2 + OH−; (3) Tafel step (chemical desorption): 2Had → H2. Obviously, HER under alkaline conditions depends not only on HBE but also on the splitting ability of water or the binding energy of OH species. Hence, we should consider all the factors when designing catalysts toward HER in alkaline media.
Different noble metals behave differently under alkaline conditions. For example, the kinetics on Pt in alkaline HER is several orders of magnitude lower than that in acidic solutions, but the activity differences for Ru-based electrocatalysts in acidic and alkaline solutions are marginal.236–238 Zheng et al. reported an anomalous fcc Ru catalyst fabricated through reduction and annealing treatment.239 This catalyst showed 2.5 times higher hydrogen generation rate than that of Pt in 0.1 M KOH. They attributed the outstanding catalytic performance to the suitable ability of water dissociation. Catalyst strain also plays an important role in improving the activity of HER under alkaline conditions. Wang et al. compared the HER activity of well-defined Ru@Pt icosahedral nanostructures via thermal reduction and traditional alloy (RuPt) catalysts.240 They found that the Ru@Pt catalyst exhibited better catalytic performance in alkaline HER. The strain between Pt shell and Ru core led to the weak binding of hydrogen and optimal interaction with hydroxyl species, thereby leading to enhanced activity.
Hydrogen, which has the highest energy density (146 kJ g−1), is considered to be a clean energy carrier, facilitating energy storage. Although many nonnoble-metal-based catalysts have been reported for HER (e.g., Mo-based catalysts and various carbon materials), Pt-based materials are still promising catalysts due to their higher activity and stability.241–244 Although many non-Pt noble metals have been investigated in different studies, their catalytic activity is still lower than those of Pt-based catalysts. Therefore, additional efforts need to be devoted toward finding a suitable replacement for Pt. Moreover, alloying Pt-group metals with transition metals or integrating noble metals with nonmetal materials (e.g., MoC2 and MoS2) may increase the activity and stability as the HBE gets optimized via the synergistic effect. Theoretical calculations may also provide high-throughput screening of HER catalysts by calculating the HBE, which facilitates the design of an optimal surface structure.
| * + H2O → *OH + H+ + e− | (1) |
| *OH → O* + H+ + e− | (2) |
| *O + H2O → *OOH + H+ + e− | (3) |
| *OOH → O2 + H+ + e− | (4) |
RuO2- and IrOx-based catalysts are two of the most efficient catalytic systems. However, the incorporation of different metal oxides results in the reduction of conductivity. Moreover, the stability of RuO2 is very poor because it can be easily converted into unstable RuO4 at high anodic potential.250 A considerable amount of research has focused toward Ir- or Ru-based catalysts, as well as compounds of noble metals with enhanced stability and activity.251–254
Studies have suggested that Ru thin films with low-index surfaces show decreased rate of Ru>4+ dissolution during OER and increased stability.255,256 Gloag et al. prepared Pd-core Ru-branch NPs that have nanosized branches with low-index Ru facets via the cubic-core hexagonal-branch mechanism.257 Pd–Ru-branched NPs yielded only 33% geometric current density that decreased after 15 min, which was more stable than Pd–Ru spherical NPs. The enhanced stability was due to the low-index Ru facets. Similar to Ru, Ir can be easily dissolved in acidic media. Designing unique nanostructures can increase the stability of Ir-based catalysts. Park et al. synthesized a robust IrNiCu double-layered nanoframe (DNF) structure via selectively etching CuNi@Ir core–shell structures.258 The IrNiCu DNF exhibited high electrocatalytic activity and stability, which could be attributed to the frame structure that prevented the growth and agglomeration of particles.
In addition to improving catalyst stability, higher activity toward OER is needed. Several studies have suggested that the formation of Ir oxide can enhance the catalytic activity. Pi et al. reported highly porous Ir–Cu (P–IrCu) nanocrystals through a facile chemical dealloying strategy (Fig. 22a).259 Interestingly, as shown in Fig. 22b and c, after chemical dealloying, the content of Ir0 decreased and the content of Ir oxidation state increased due to the formation of surface hydroxyl species (Ir–OH) at low coordinated Ir around the defects. The optimized P–IrCu1.4 NCs exhibited higher activity, which yielded a 1.8-fold improvement in specific activity over that of pristine solid IrCu1.4 NCs (S–IrCu1.4 NCs), as shown in Fig. 22d. This excellent activity was due to the higher oxidation state of Ir, which promoted the optimized intrinsic activity of the catalyst. Our group reported a shape-tunable IrPd alloy nanocrystal, including hollow spheres, NWs, and nanotetrahedra, via a solvothermal method.260 IrPd NWs and nanotetrahedra exhibited more than five times higher mass activity as compared to that of commercial Ir/C catalysts because surface Ir(VI) oxides might be possible key intermediates for OER.
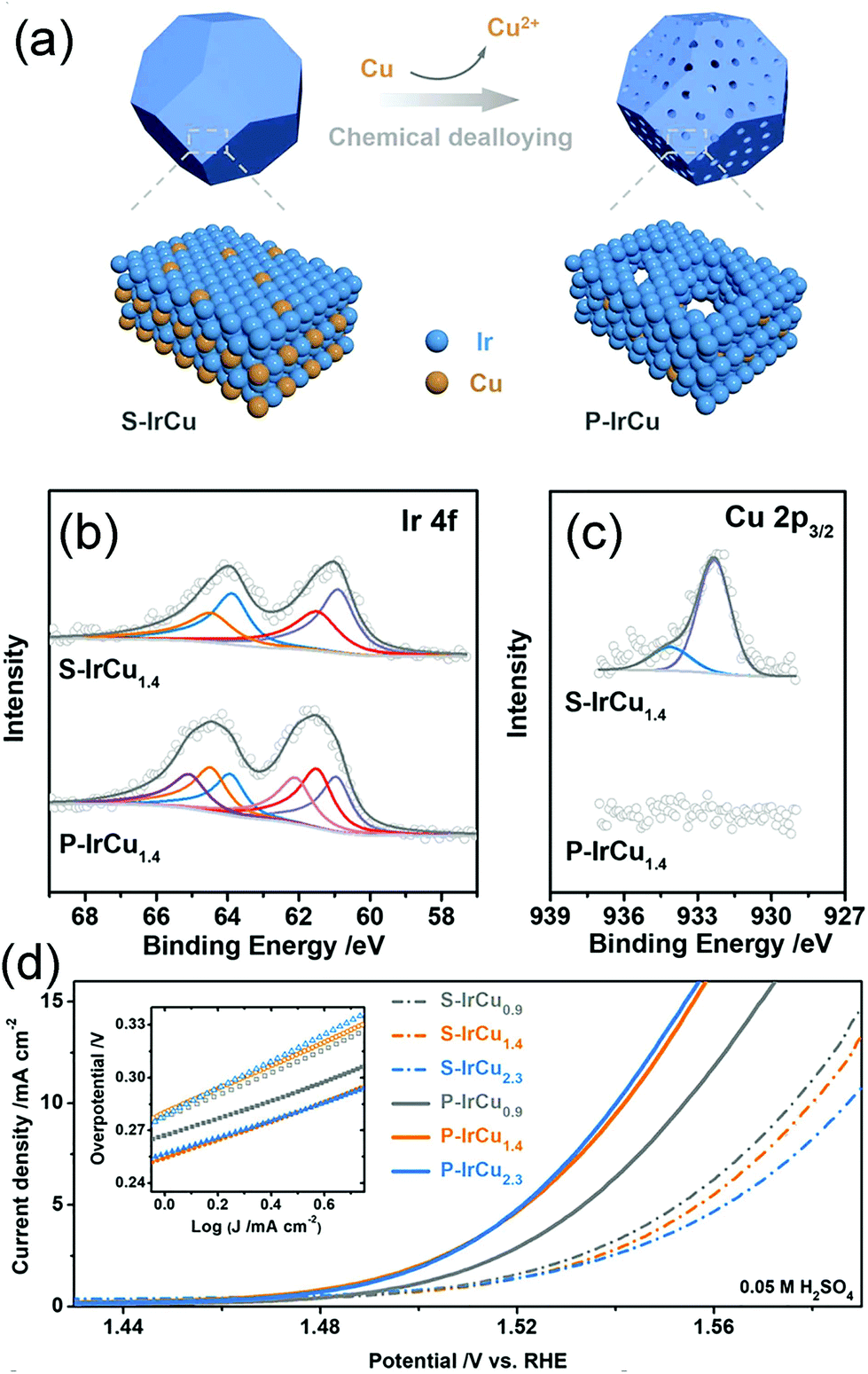 | ||
| Fig. 22 (a) Schematic illustration of the transformation from S-IrCu to P-IrCu NCs via chemical dealloying. Comparison of (b) Ir 4f and (c) Cu 2p XPS spectra of S-IrCu1.4 and P-IrCu1.4 NCs. OER at different potentials on the surface of the sAu/NiFe LDH model. (d) Polarization curves of S-IrCux NCs and P-IrCux NCs. Inset shows the Tafel plots. Scan rate was 5 mV s−1. The electrolyte was 0.05 M H2SO4. Adapted with permission from ref. 259. Copyright 2018, American Chemical Society. | ||
It is well established that elemental doping and synthesis of specific structures are two of the most effective ways to tune the catalytic performance by modulating the electronic and geometric effects, and therefore, improving the electrocatalytic activity. Wang et al. synthesized polycrystalline Ni–Ir nanocages via a galvanic replacement reaction using Ni NPs as the templates.261 The as-synthesized Ni–Ir nanocages yielded overpotential of 302 mV vs. RHE to deliver a current density of 10 mA cm−2 due to the synergetic effect between Ni and Ir. Fan et al. synthesized 4H/fcc-Au@Ir core–shell nanoribbons by a facile epitaxial growth method, and the catalyst showed superior OER activity with mass activity of 693.9 mA mgIr−1 under acidic conditions, which was greater than that obtained from a commercial Ir/C catalyst.262 They attributed this improved catalytic performance to the synergistic effect between Au and Ir. Shan et al. reported doped RuIr alloy nanocrystals with transition metals (M-RuIr; M = Co, Ni, and Fe) via a coreduction polyol method.263 Co-RuIr yielded small overpotential of 235 mV for OER in 0.1 M HClO4 media because of the Co-modified electronic structure and binding strength of reaction intermediates. Moreover, they fabricated a heterostructured OER electrocatalyst (Ru@IrOx) to achieve high activity and stability for OER under acidic conditions.264 They attributed the superior performance to the charge redistribution between Ru and Ir. Zhao et al. synthesized Ru octahedral nanocrystals with an fcc structure enclosed by {111} facets by using 4.5 nm Rh cubes as the seeds (Fig. 23a).265 They compared the OER activity of fcc-Ru octahedra, fcc-Ru truncated cubes, fcc-Ru NPs, and hcp-Ru NPs (Fig. 23b and c). Their results showed that the activity of fcc-Ru was higher than that of hcp-Ru, and Ru {111} facets were more active than Ru {100} facets.
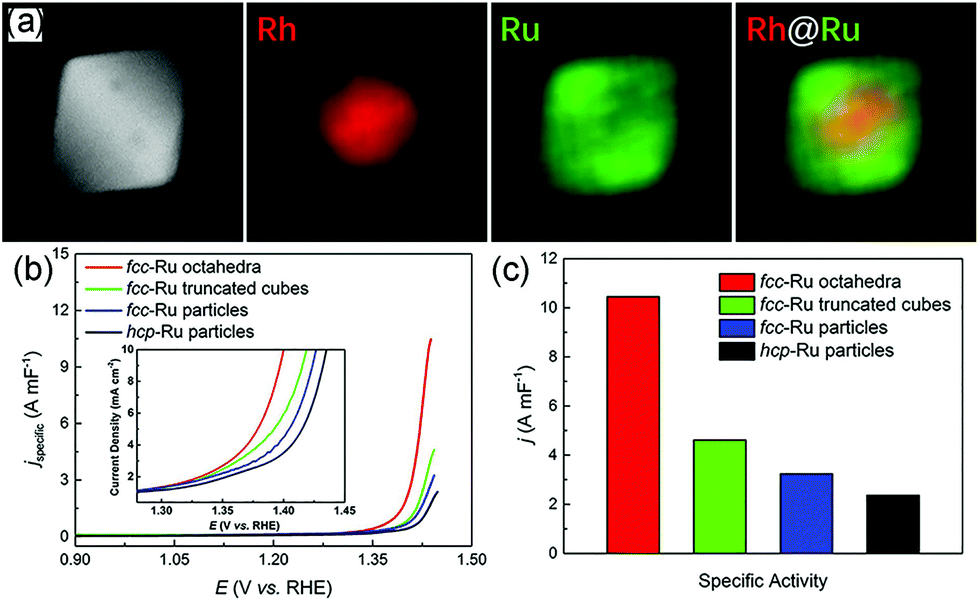 | ||
| Fig. 23 (a) HAADF-STEM image and EDX mapping (Rh: red; Ru: green) of an individual octahedron. (b) Cdl-Normalized polarization curves measured over different Ru catalysts in a 0.5 M H2SO4 solution at a scanning rate of 6 mV s−1. The inset shows the polarization curves of various Ru catalysts in the potential range of 1.28–1.45 V. (c) Summary of the specific activity of different Ru catalysts toward oxygen evolution. Adapted with permission from ref. 265. Copyright 2019, American Chemical Society. | ||
The kinetically sluggish OER process has restricted the use of water splitting for the generation of hydrogen. Although several groups have directed their efforts toward developing superior catalysts and reveal the inherent mechanisms, the studied catalytic systems are limited only to the electrochemical testing systems. Rare practical applications of the catalysts in commercial membrane electrode assemblies used for water electrolysis are carried out. The rational design of catalysts and investigation of their feasibility as OER catalysts in practical application are desirable.
3.3 Electrochemical CO2 reduction reaction (CO2RR) and nitrogen reduction reaction (NRR)
Recently, electrochemical CO2RR and NRR have attracted increased attention due to the increase in environmental problems and the requirement for producing high-value-added chemicals. However, both CO2 and nitrogen are very stable molecules, and a considerable amount of energy must be put into the catalytic system to activate them. The design of catalysts with worthwhile activity that can activate molecules with a low energy input is imperative. Here, we concentrate on research involving noble-metal-based catalysts.| CO2 + e− → CO2˙−E° = −1.9 V | (5) |
| CO2 + 2H+ + 2e− → CO + H2O E° = −0.52 V | (6) |
| CO2 + 2H+ + 2e− → HCOOH E° = −0.61 V | (7) |
| CO2 + 4H+ + 4e− → HCHO + H2O E° = −0.51 V | (8) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O E° = −0.24 V | (9) |
| CO2 + 12H+ + 12e− → C2H4 + 4H2O E° = −0.34 V | (10) |
| CO2 + 6H+ + 6e− → CH3OH + H2O E° = −0.38 V | (11) |
| 2H+ + 2e− → H2E° = −0.42 V. | (12) |
As shown in reaction (5), CO2RR suffers from high overpotential due to the stable nature of CO2 molecules, which is fairly difficult to activate.266,267 Another challenge is competitive HER, because it has similar redox potential as that of reaction (12). Moreover, the CO2RR itself is a multi-electron process, yielding various products (as mentioned earlier); this increases the cost of product separation. Therefore, satisfactory noble-metal-based catalysts in CO2RR should lower the overpotential, possess high activity, and remarkable selectivity and stability. Until now, no desirable catalyst has met these demands. Investigating the structure–activity relationship is necessary to design the desired catalyst.
Early studies on electrochemical CO2RR have mainly focused on polycrystalline bulk transition metal catalysts. The studied catalysts can be divided into four categories in CO2 reduction according to their products. Cu can produce hydrocarbons; Au, Ag, and Zn produce CO; Pb, Sn, and In produce formic acid; Pt, Fe, and Ni lack strong catalytic activity in CO2 reduction and produce H2 instead.268–271 The reaction mechanism of electrochemical CO2RR on metal electrodes in aqueous solutions is shown in Fig. 24.271 Here, we focus on noble metals as the catalysts for CO2RR; the most widely studied are Au, Ag, and Pd because they have high electrical conductivity and good activity and stability under the reaction conditions.
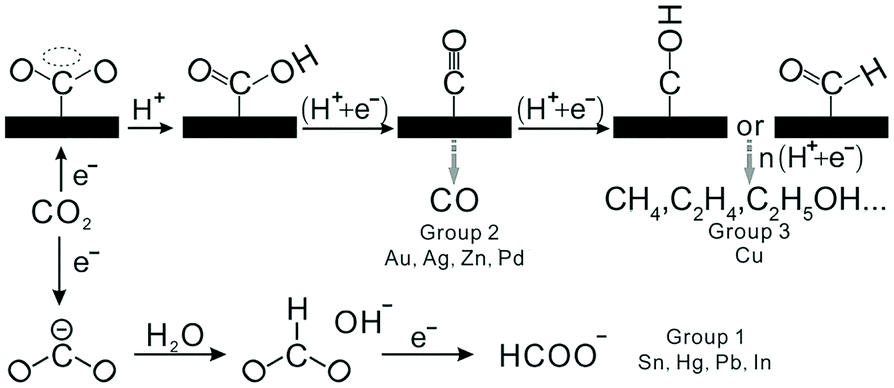 | ||
| Fig. 24 Reaction mechanism of electrochemical CO2RR on metal electrodes in aqueous solutions. Adapted with permission from ref. 271. Copyright 2016, Wiley-VCH. | ||
Different compositions can regulate the binding energy of intermediates via the synergistic effect, and therefore, alter the activity and selectivity of the catalyst. Luc et al. synthesized Ag–Sn electrocatalysts with a core–shell nanostructure that contained a bimetallic core and ultrathin partially oxidized SnOx shell (denoted as AgSn/SnOx) by galvanically displacing Sn with Ag.272 The thickness of SnOx could be changed by altering the Sn content in the core. At an optimal SnOx shell thickness of ∼1.7 nm, the catalyst exhibited high formate Faradaic efficiency of ∼80% and formate partial current density of ∼16 mA cm−2 at −0.8 V vs. RHE. They attributed the catalytic activity and selectivity to the suitable binding energy of the intermediates. Once the Sn concentration increased in the Ag–Sn core, the OCHO* intermediate for the HCOOH formation pathway was further stabilized as compared to the COOH* intermediate that was responsible for CO production, thereby enhancing the Faradaic efficiency toward HCOOH. Enhanced productions of CO, CH4, and CH3OH were observed on CuAu, CuPd, and AgZn, respectively.273–277 It is believed that the neighboring Cu atom ensembles are pivotal to C–C coupling, yielding C ≥ 2 products.278 Based on this principle, Huang et al. fabricated Ag–Cu nanodimers (NDs) including two constituent metals as the segregated domains sharing a tunable interface using the seed-mediated growth strategy.279 Ag acted as the nucleation seeds for the growth of the Cu domain. Fig. 25a–f show the HAADF-STEM images of the synthesized Ag–Cu NDs and Ag and Cu NPs. When Ag NPs and Cu NPs were combined in the form of NDs, CO was significantly suppressed when compared with that in Ag NPs, while the Faradaic efficiency of C2H4 was about 40%, corresponding to a 3.4-fold enhancement over that from Cu NPs. In Fig. 25g, the blue-shift in the surface plasmon resonance (SPR) peak of Ag in the Ag–Cu NDs could be ascribed to the increased electron density in the Ag domain of the dimer. The collected XPS spectra on Cu NPs, Ag NPs, and Ag–Cu NDs confirmed the electron transfer process from Cu to Ag (Fig. 25h). The mechanism of C2H4 promotion in Ag–Cu NDs was explained by coupling the tandem catalysis and electronic effect. Tandem catalysis implies that CO formed on the Ag surface would spillover or sequentially adsorb on the Cu surface, and it could be further reduced. Moreover, the electrons transferred from Cu to Ag could enhance the binding of CO on the catalyst surface, which, in turn, facilitated the coupling of CO into C2H4.280
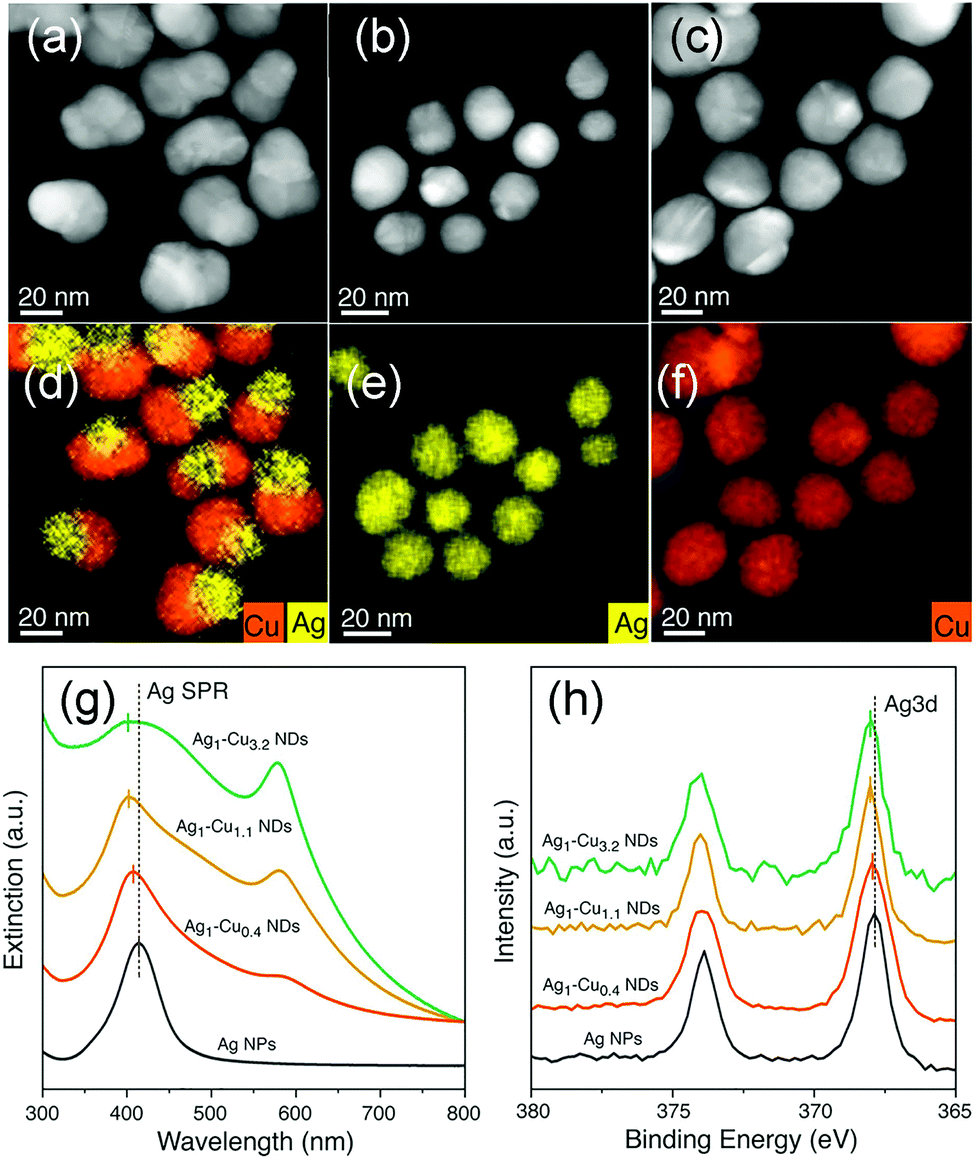 | ||
| Fig. 25 (a–c) HAADF-STEM images of Ag1–Cu1.1 NDs (a), Ag NPs (b), and Cu NPs (c); (d–f) corresponding EDX elemental maps of Cu (orange) and Ag (yellow) of Ag1–Cu1.1 NDs (d), Ag NPs (e), and Cu NPs (f). (g) UV-vis spectra and (h) XPS spectra of Ag/Cu nanocrystals. Adapted with permission from ref. 279. Copyright 2019, American Chemical Society. | ||
With regard to the active and selective CO2 conversion to CO, several studies have investigated the different morphologies of Au catalysts, such as Au NWs, Au NPs, and nanoporous Au thin films.281–283 However, the effects of morphologies on the catalytic performance are unclear. Back et al. investigated the effects of Au morphologies on catalytic activity via DFT calculations by focusing on Au NWs and NPs.284 They found that the edge sites in the NWs were more active than the similar edge sites in NPs, and therefore, NWs showed higher activity than NPs, which was consistent with the experimental results. The difference between the two edge sites was due to the fact that the subtle local environments were clearly distinguishable, which could be described via a generalized coordination number (GCN). GCN may provide a reference to the optimal nanostructure in CO2RR from atomic-level insights, which facilitates in improving the catalytic performance. Several studies have shown that the low-coordinated Au atoms in NPs play an important role in CO2RR.281,285,286 Lee et al. synthesized concave rhombic dodecahedron (RD) Au NPs exposing various high indexes, such as (331), (221), and (553), via seed-mediated growth.286 Concave RDs exhibit 93% CO selectivity at −0.57 V vs. RHE in 0.5 M KHCO3. The current density increased up to 10.6 mA cm−2 at −1.2 V vs. RHE, which was 1.2 times higher than that for Au films. These superior catalytic activity and selectivity of the nanostructure can be attributed to the numerous atomic steps on the surface that serve as active sites. Jiao et al. highlighted the unique ability of low-coordinated surface Ag atoms (active sites) to improve the CO production rates and selectivity both theoretically and experimentally.287,288
In addition to the composition and structure, the size of the nanostructure can also influence the catalytic activity and selectivity. Gao et al. reported a prominent size-dependent activity/selectivity in CO2 electrocatalytic reduction over different sizes of Pd NPs, ranging from 2.4 to 10.3 nm, which were prepared using sodium citrate as the stabilizing agent and NaBH4 as the reductive agent.289 The Faradaic efficiency for CO production varied from 5.8% at −0.89 V (vs. RHE) over 10.3 nm NPs to 91.2% over 3.7 nm NPs, which indicated that CO2 adsorption, COOH* formation, and CO* removal during CO2 reduction can be tuned by varying the size of Pd NPs due to changes in the ratios of corner, edge, and terrace sites.
We mainly introduce noble metal alloying with other main group metals or transition metals for CO2RR. In fact, nonmetal-doped catalysts can also promote catalytic activity and selectivity. Tao et al. demonstrated that doping Pd with a small amount of Te could selectively convert CO2 to CO at low overpotential.290 The PdTe/few-layer graphene (FLG) catalyst, which was synthesized using the ultrasonication-facilitated reduction, showed CO Faradaic efficiency of about 90% at 0.8 V (vs. RHE), which was 3.7 times higher than that of a Pd/FLG catalyst. DFT calculations showed that the higher selectivity could be attributed to the adsorption of Te on the terrace sites of Pd, thereby preventing HER. CO2 adsorption and activation occurred on the high-index sites of Pd to produce CO; therefore, the selectivity toward CO was promoted. Jiang et al. reported a boron-doped Pd catalyst (Pd–B/C) prepared through an aqueous chemical synthesis process, exhibiting formate Faradaic efficiency of 70% in 0.1 M KHCO3 at −0.5 V (vs. RHE), which was 12 times higher than that for Pt.291 They revealed that Pd–B/C preferred to form HCOO*, an intermediate for the formic acid pathway. Zeng et al. reported that silver sulfide (Ag2S) NPs could be loaded on reduced graphene oxide (rGO) via straightforward hydrothermal methods.292 The catalyst showed remarkable selectivity and durable stability for the conversion of CO2 to CO, which could be attributed to the active sites produced by Ag ions in Ag2S along with doped N and S in rGO.
CO2RR is worthy of comprehensive investigations because it can convert CO2 to high-value-added products. Finding superior catalysts with worthwhile activity, selectivity, and stability is meaningful. As mentioned earlier, alloying noble metals with other metals, modulating the size of catalyst NPs, and synthesizing catalysts with different morphologies are powerful ways to enhance the activity and selectivity of catalysts. The binding energy of the reaction intermediates on the catalyst surface is important both for reactive activity and selectivity. Considering that the electrochemical CO2 reduction is a complex reaction that involves multi-electron transfers with various intermediates and products, it is necessary to modulate the adsorption energy of different intermediate species formed during the reaction. Hence, it is reasonable to construct different surface structures to tune the binding energy of intermediates for different target products.
It is well known that owing to the similar theoretical potential values of HER and NRR, HER is a very competitive reaction due to its much faster reaction kinetics than NRR. Moreover, theoretical computation results have revealed that both adsorption of reactants and proton/electron transfer are difficult on most catalyst surfaces in NRR. Therefore, the reaction rate and Faradaic efficiency for ammonia in NRR are very low. Under ambient conditions, the superior reaction rate for the electrochemical synthesis of ammonia is ∼1.14 × 10−9 mol NH3 s−1 cm−2, and the marginally better Coulombic efficiency is about 8.11%. Both rate and selectivity for ammonia are much lower than those in the common ammonia production of commercial systems.297,298 It is important to enhance the activity and selectivity of catalysts toward NRR. Several noble metals have been reported to catalyze nitrogen reduction, such as Pd, Rh, Ru, and Au.299–303 Because of the influence of HER, Au is selected as the most efficient NRR catalyst because of its low HER activity.300,304,305 Research efforts on most of the studied systems are directed toward enhancing the catalytic activity via alloying noble metals with other metals and/or creating larger specific surface areas that can yield the exposure of additional reaction sites.
The fabrication of unique nanostructures, such as porous structures and nanoplates, can increase the exposed active sites, and therefore, increase the activity. Wang et al. fabricated flower-like Au microstructures (denoted as Au flowers) with an average size of approximately 900 nm by the very fast reduction of HAuCl4.300 Au flowers were assembled by using staggered nanoplates as the building blocks, which provided abundant electrocatalytically active sites for NRR. The catalyst achieved high performance (NH3 yield: 25.57 μg h−1 mgcat.−1; Faradaic efficiency: 6.05%) and 100% selectivity (no N2H4 production) due to the increased number of active sites on the surface. Nazemi et al. synthesized pore-size-controlled hollow Au nanocages by the galvanic replacement reaction in solid Ag NCs.305 Among the catalysts with different pore sizes, the highest NH3 yield rates (3.74 μg cm−2 h−1) and Faradaic efficiency (35.9%) were achieved at −0.4 V vs. RHE. Wang et al. fabricated a porous Au film on Ni foam by a micelle-assisted electrodeposition method.306 The catalyst exhibited superior NRR performance at high NH3 yield rate of 9.42 μg h−1 cm−2 and Faradaic efficiency of 13.36% because of its interconnected porous architectonics.
As mentioned earlier, the surface structure of the catalyst is very important in improving the catalytic performance, which can be tuned by changing the composition and synthesizing a specific nanostructure. Wang et al. demonstrated the efficient electrochemical synthesis of ammonia on 3D bimetallic PdRu porous nanostructures, which were synthesized via a rapid reduction process.307 PdRu yielded high activity with NH3 yield of 25.92 μg h−1 mgcat−1 in an acidic solution, which was superior to those obtained from monometallic Pd and Ru nanostructures due to the synergistic effect of the composition. Zhao et al. reported the successful synthesis of Ru icosahedral nanocages by the growth of Ru shells on Pd icosahedral seeds and then performing the selective removal of Pd seeds.308 These Ru nanocages exhibited superior catalytic activities as compared to Pd@Ru core–shell nanocrystals, because the twin boundary regions of these icosahedral nanocages could stabilize the N2 dissociation transition state, reduce the overall reaction barrier, and promote competition with the N2 desorption process. Zhang et al. formulated a quantitative relationship between the GCN and NRR activity.309 The NRR activity linearly increased with a decrease in GCN values of the Au surface atoms. To prove the relationship between the GCN and catalytic activity, the NRR activity of nanoporous Au with high proportion of low-coordinated surface atoms and Au octahedra were investigated. Nanoporous Au exhibited higher NH3 production rate of 30.5 μg h−1 mg−1, which was 5.8 times higher than that of Au octahedra.
Understanding the reaction mechanism of NRR can facilitate the understanding of the structure–activity relationship. However, the specific reaction pathway on a catalyst surface is still indeterminate. Yao et al. studied the intermediates and feasible mechanism during electrochemical NRR by using the powerful surface-enhanced infrared absorption spectroscopy (SEIRAS).310 The inset of Fig. 26a shows a comparison of the CVs of Au films in N2- (red lines) and Ar-saturated (black lines) 0.1 M KOH solutions. The reduction current density below −0.1 V vs. RHE in the former was slightly larger than that in the latter, implying that the NRR could happen on Au. Meanwhile, NH3 could be detected employing the indophenol method, further confirming the formation of ammonia. To identify the possible reaction intermediates during potential cycling, SEIRAS measurements were conducted (Fig. 26b–d). As a comparison, the same test was performed on the Pt surface replacing Au (Fig. 26e); no *N2Hy (1 < y < 4) was detected, which was consistent with the fact that Pt tended to produce H2. Based on the facts obtained in the experiment, they formulated the possible mechanism, as shown in Fig. 26f, which added significant new insights into the reaction mechanisms of NRR and might guide the rational design of more advanced electrocatalysts.
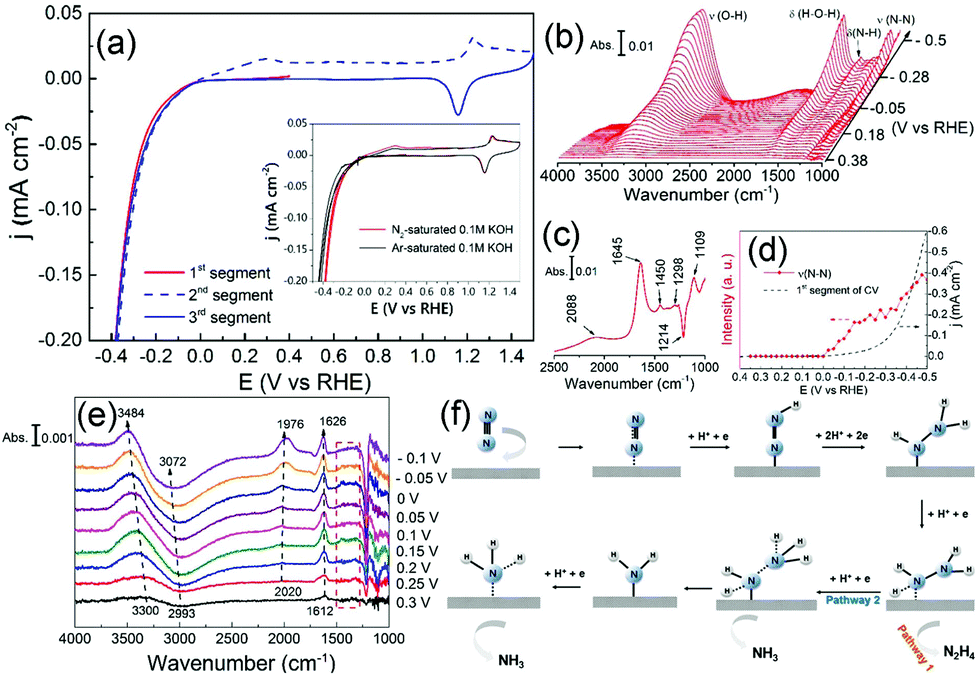 | ||
| Fig. 26 (a) CVs of an Au film electrode deposited on the Si prism in a N2-saturated 0.1 M KOH aqueous solution. (b) FTIR spectra during the first segment from 0.4 to −0.5 V. The reference spectrum was taken at 0.4 V. (c) Single spectrum at −0.5 V in the range of 1000–2500 cm−1. (d) Potential dependence of the IR absorption band of N–N stretching derived from (b). (e) FTIR spectra during the negative scan from 0.4 to −0.1 V on a Pt film electrode in N2-saturated 0.1 M KOH solution. (f) Nitrogen electrochemical reduction reaction pathway on Au surface. Adapted with permission from ref. 310. Copyright 2018, American Chemical Society. | ||
Because studies involving electrochemical NRR are still in their infancy, the specific structure–activity relationship is not clear. To clarify this relationship, in situ detection can be used to monitor the changes in the catalysts and intermediates. However, the yield of ammonia is very low, and therefore, the concentration or quantity of the intermediates formed during NRR is also low, which is difficult to determine. This necessitates more sensitive in situ methods. Besides in situ detection, DFT calculations are another way to determine the structure–activity relationship and to design a better catalyst. For example, Nørskov and coworkers performed DFT calculations combined with the computational standard hydrogen electrode to calculate the free energy profile at each elementary step of the electrochemical NRR on several close-packed and stepped transition metal surfaces at ambient temperature and pressure.311 They found that only the flat metal surfaces of Sc, Y, Ti, and Zr were expected to be covered with N instead of H and could reduce N2 to NH3. Therefore, we can alloy noble metals with Sc, Y, Ti, and/or Zr to prepare NRR catalysts, and there may exist a compositional synergistic effect that enhances the catalytic activity.
4. Summary and perspectives
Electrochemical catalytic reactions are involved in many energy- and environment-related fields, such as fuel cells, electrolysis of water, CO2RR, and NRR. However, sometimes, the catalysts used in these reactions are not sufficiently worthwhile, suffering from low activity, selectivity, and stability. Shaping noble-metal-based nanostructures as efficient catalysts in such reactions is very important, which can enhance the activity, selectivity, and stability of catalysts.312 In this review, we concentrate on the common synthesis methods of noble-metal-based nanostructures, particularly those for shaped nanocrystals, which can yield high catalytic performances. For example, continuous growth, seed-mediated growth, and a combination of UPD and galvanic replacement can be used to produce nanopolyhedrons that possess specific facets or high-index facets with several unsaturated coordination sites. Nanoframes, nanopores, and concave nanostructures exposing large specific surface areas can be synthesized by etching and galvanic replacement. Nanocrystals with significant lattice strain, e.g., dendritic and core–shell nanostructures, are usually formed via seed-mediated growth and assembly techniques. For designing catalysts, in addition to considering the synthesis route, many factors should be taken into account; for example, precursors, ligands, capping agents, reductants, reaction time, and temperature. All these factors can influence the reduction rates, nucleation, growth direction, and thermodynamically or kinetically favored crystal faces, and therefore, change the shape of the nanocrystals.Moreover, we focus on the structure–activity relationship in several electrochemical reactions. The surface structures of nanocrystals play a vital role in determining the catalytic performance. There are mainly two strategies to regulate the surface structure of catalysts: tuning the composition and shape of the nanomaterials. For example, Pt is usually poisoned by COads during MOR. When alloying Pt with Ru (an oxophilic metal), Ru can provide OHads at lower potential that can remove COads by oxidizing it to CO2. EOR can occur by the C1 pathway or C2 pathway; the difference between them is whether the C–C bond is broken or not. Alloying Rh or Ir with Pt can direct the reaction toward the C1 pathway, which is preferred. Nanoframes and nanopores with large specific areas can provide additional reaction sites. Branches and core–shell structures can induce lattice strain to the catalytic system. Concave structures and nanopolyhedrons expose more high-index facets that provide more active facets for catalysis. In addition, modulating the size is another way to improve the catalytic activity. Furthermore, we perform DFT calculations for certain reactions to help clarify the structure–activity relationship and screen out possible catalysts with high performance. For example, the binding energy of *H, and maybe *OH, plays an important role in HOR and HER, and the binding energy of oxygenated species, such as OOH* and O*, is vital for OER. Catalyst activity can be improved by tuning the adsorption of intermediates to obtain the optimal binding energy. Table 2 lists the most representative studies regarding the relationship between structure and catalytic performance in electrocatalysis.
| Catalyst | Synthetic route | Application | Electrolyte | Highlight | Performance | Key factors in catalysis | Ref. |
|---|---|---|---|---|---|---|---|
| MA: mass activity; SA: specific activity; FE: Faradaic efficiency. The peak current ratio of the forward to backward scans (If/Ib) can be used to evaluate the poisoning tolerance of catalysts in DMFCs. Lower If/Ib ratio indicates weaker resistance to the poisoning of carbonaceous species. | |||||||
| Pt-Coated Cu nanowires | Partial galvanic displacement | HOR | 0.1 M KOH | Activity | MA (exchange current densities): 1.9 times than Pt/C, SA (exchange current densities): 3.5 times than Pt/C | Electronic effect | 141 |
| PtM nanowires | Soft template | HOR | 0.1 M KOH | Activity | SA (Pt7Ru3 NW): 2.2 mA cm−2 at 0.05 V vs. RHE | Synergistic ligand and strain effects | 143 |
| bcc-phased PdCu NPs | Wet-chemistry and thermal treatment | HOR | 0.1 M KOH | Activity | MA: 1.727 mA μgPd−1 | bcc surface | 148 |
| SA: 2.922 mA cm−2 at 0.1 V vs. RHE | |||||||
| Au-Modified Pt | Galvanic displacement | ORR | 0.1 M HClO4 | Stability and activity | MA: 5.64 A mPt−2 | Raising Pt oxidation potential | 14 |
| SA: 4.23 mA cm−2 at 0.85 V vs. RHE | |||||||
| Pd@Pt core–shell | Control of the deposition rate | ORR | 0.1 M HClO4 | Activity | MA: 0.68 A mgPt−1 at 0.9 V vs. RHE | {111} facets | 79 |
| Co@Pt NPs | Seed-mediated growth | ORR | 0.1 M HClO4 | Activity | MA: 1.2 A mgPt−1 | Strain effect | 158 |
| SA: 2.24 mA cm−2 at 0.9 V vs. RHE | |||||||
| Pt nanoframes | Solvothermal method | ORR | 0.5 M H2SO4 | Activity | MA: 13.1 A gPt−1 at 0.8 V vs. RHE | {740} facets | 164 |
| Pt/Ag hollow nanostructures | Galvanic replacement | MOR | 1 M CH3OH + 1 M KOH | Activity | SA (Pt/Ag popcorns): 116.1 mA cm−2 | High surface area | 26 |
| Amorphous CuPt hollow nanotubes | Na2S2O3-assisted galvanic replacement reaction | MOR | 0.5 M CH3OH + 0.1 M HClO4 | Activity and stability | MA: 373.7 mA mgPt−1 | Pt 4f binding energies | 104 |
| SA: 7.8 mA cm−2 loss of 9.6% peak current density after 1000 cycles | |||||||
| Porous Pt–Ni–P nanotube arrays | Template-assisted electrodeposition | MOR | 0.5 M CH3OH + 0.5 M H2SO4 | Stability | High If/Ib ratio | Electronic effects and active surface areas | 175 |
| Pd@Pt core–shell hexapods | Seed-mediated growth | MOR | 0.5 M CH3OH + 0.5 M H2SO4 | Activity and stability | MA: 0.52 mA μgPt−1 | Synergistic effect | 178 |
| SA: 1.97 mA cm−2 | |||||||
| Hexapod PtRuCu | Galvanic replacement | MOR | 1 M CH3OH + 0.1 M HClO4 | Activity and stability | MA: 1.35 A mgPt−1 | Synergetic effect | 179 |
| SA: 3.92 mA cm−2 mass activity decreases 27% after 800 cycles | |||||||
| Pt–Pd nanocages | Selective etching | MOR | 1 M CH3OH + 0.5 M H2SO4 | Activity | MA: 0.58 A mgPt−1 | Sufficient active sites | 183 |
| PtRu NWs | Solvothermal approach | MOR | 0.5 M CH3OH + 0.1 M HClO4 | Activity and stability | MA: 0.82 A mgPt−1 | {111} facets | 185 |
| SA: 1.16 mA cm−2 at 0.9 V vs. Ag/AgCl greater If/Ib ratio | |||||||
| PtIr/SnO2 | Seed-mediated growth | EOR | 0.5 M C2H5OH + 0.1 M HClO4 | Selectivity | Improved capability for C–C bond splitting | Ir | 8 |
| Cyclic penta-twinned Rh nanobranches | Solvothermal route | EOR | 1 M C2H5OH + 1 M NaOH | Activity and selectivity | MA: 185.3 mA mgRh−1 | {100} facets and tensile strain | 9 |
| SA: 0.36 mA cm−2 selectivity of CO2: 14.5 ± 1.1% | |||||||
| PdSn NPs | Modified polyol method | EOR | 0.5 M C2H5OH + 0.5 M KOH | Activity | SA: 6.8 mA cm−2 | Synergistic effects | 10 |
| Tetrahexahedral (THH) Pd NPs | Electrodeposition | EOR | 0.1 M C2H5OH + 0.1 M NaOH | Activity | SA: 1.84 mA cm−2 | {730} high-index facets | 20 |
| Au@Pd nanobricks (NBs) | Coreduction | EOR | 0.5 M C2H5OH + 0.1 M KOH | Activity | MA: 783.65 mA mgPd−1 | Exposed stepped surfaces and synergistic effects | 200 |
| SA: 2.87 mA cm−2–0.1 V vs. Ag/AgCl | |||||||
| Pt-Rich core/Sn-rich subsurface/Pt-skin nanocubes | Chemical reduction and etching | EOR | 1.0 M C2H5OH + 0.5 H2SO4 | Activity and stability | Activities six times higher than cubic Pt NPs, lower activities loss after 5000 cycles | (100) facets, lattice strain, and electronic effect | 203 |
| Octahedral PtNiRh NPs | Wet-chemical approach | EOR | 0.5 M C2H5OH + 0.1 M KOH | Activity and selectivity | The lowest onset: 0.1 V vs. RHE, break the C–C bond | Ternary surface site | 205 |
| PtRhCu nanoboxes | Galvanic replacement | EOR | 1 M C2H5OH + 0.1 M KOH | Activity and selectivity | MA: 4090 mA mgPt−1 | Geometric structure and synergistic effects | 208 |
| SA: 14.9 mA cm−2 C1 pathway | |||||||
| Ru nanosheets | Solvothermal method | HER | 0.5 M H2SO4 | Activity | Overpotential 20 mV vs. RHE at a current density of 10 mA cm−2 | Increased active sites | 226 |
| Spongy-like nanoporous Ag foam | Electrochemical method | HER | 0.5 M H2SO4 | Activity | Overpotential 120 mV vs. RHE at a current density of 10 mA cm−2 | Surface active sites and lower the binding energy of H | 227 |
| 4H/fcc Au@PdAg nanoribbons | Galvanic replacement reaction | HER | 0.5 M H2SO4 | Activity | Overpotential 26.2 mV vs. RHE at a current density of 10 mA cm−2 | Electronic and geometric effects | 229 |
| PdCu@Pd nanocubes | Coreduction and electrochemical etching | HER | 0.5 M H2SO4 | Activity | Overpotential 68 mV at a current density of 10 mA cm−2 | The binding energy of hydrogen | 230 |
| Core/shell NiAu/Au | Co-Reduction and cyclic voltammetry procedure | HER | 0.5 M H2SO4 | Activity and stability | Onset potential (∼7 mV) vs. RHE | Geometric effects | 232 |
| Fcc Ru | Reduction and annealing | HER | 0.1 M KOH | Activity | 2.5 times higher H2 generation rate than Pt | Ability of water dissociation | 239 |
| Ru@Pt | Thermal reduction | HER | 0.1 M KOH | Activity | Higher than Pt and PtRu | Interaction with OH species | 240 |
| Pt@Ru cuboctahedrons | Scalable continuous-flow reaction | OER | 0.1 M HClO4 | Activity | Onset potential 1.31 V vs. NHE | Electronic synergy between core and shell | 251 |
| Pd-Core Ru-branch nanoparticles | Hydrothermal synthesis | OER | 0.1 M HClO4 | Activity and stability | Overpotential 225 mV vs. RHE at a current density of 10 mA cm−2 | Low index surfaces of Ru | 257 |
| Ni–Ir nanocages | Galvanic replacement | OER | 0.1 M HClO4 | Activity | Overpotential 302 mV vs. RHE at a current density of 10 mA cm−2 | Synergetic effect | 261 |
| 4H/fcc-Au@Ir nanoribbons | Seed-mediated growth | OER | 0.5 M H2SO4 | Activity | MA: 693.9 mA mgIr−1 at 1.550 V vs. RHE onset potential 1.445 V vs. RHE | Synergistic effect, kinks and steps | 262 |
| Fcc-Ru octahedra | Seed-mediated growth | OER | 0.5 M H2SO4 | Activity | Overpotential 168 mV vs. RHE at a current density of 10 mA cm−2 | Ru {111} facets | 265 |
| AgSn/SnOx | Galvanic replacement | CO2RR | 0.5 M NaHCO3 | Selectivity | Formate Faradaic efficiency of ∼80% at −0.8 V vs. RHE | Binding energy of intermediates | 272 |
| AuCu NPs | Solvent evaporation-mediated self-assembly | CO2RR | 0.1 M KHCO3 | Activity | High CO mass activity at −0.73 V vs. RHE | Geometric and electronic effects | 273 |
| Cu–Pd nanoalloy | Electrodeposition | CO2RR | 0.1 M KHCO3 | Selectivity | FE (CH3OH) 36% at −1.68 V vs. SCE | Synergistic reactivity interplay between Pd–H sites and Cu–CO sites | 275 |
| Zn/Ag foam | Pulse-depositing | CO2RR | 0.1 M KHCO3 | Activity and selectivity | FE (CH3OH) 10.5% SA: −26 mA cm−2 | Strong binding energy of CO | 276 |
| 2.7 mA cm−2 | |||||||
| Ag–Cu nanodimers | Seeded growth | CO2RR | 0.1 M KHCO3 | Activity and selectivity | FE (C2H4) ∼40% at −1.1 V vs. RHE | Tandem catalysis and electronic effects | 279 |
| Concave rhombic dodecahedron Au NPs | Seeded growth | CO2RR | 0.5 M KHCO3 | Selectivity | 93% of CO selectivity at −0.57 V vs. RHE | Numerous atomic steps | 286 |
| Pd NPs | Chemical reduction | CO2RR | 0.1 M KHCO3 | Selectivity | FE (CO) 91.2% at −0.89 V (vs. RHE) | Size, the ratio of corner, edge, terrace sites | 289 |
| Tetrahexahedral gold nanorods | Seeded growth | NRR | 0.1 M KOH | Activity | NH3: 1.648 μg h−1 cm−2, N2H4·H2O: 0.102 μg h−1 cm−2, at −0.2 V vs. RHE | Stepped facets | 298 |
| Rh nanosheet nanoassemblies | Reduction | NRR | 0.1 M KOH | Activity and selectivity | NH3: 23.88 mg h−1 mgcat.−1 at −0.2 V vs. RHE | High specific surface area | 299 |
| Flower-like Au microstructures | Assembly | NRR | 0.1 M HCl | Activity and selectivity | NH3: 25.57 mg h−1 mgcat.−1, FE: 6.05% at −0.2 V vs. RHE | Abundant active sites | 300 |
| Hollow Au nanocages | Galvanic replacement | NRR | 0.5 M LiClO4 | Selectivity and activity | NH3: 3.74 μg cm−2 h−1, FE: 13.36%, at −0.4 V vs. RHE | Increased active sites | 305 |
| Porous Au film on Ni foam | Micelle assisted electrodeposition | NRR | 0.1 M Na2SO4 | Activity and selectivity | NH3: 9.42 μg h−1 cm−2, FE: 13.36% at −0.2 V vs. RHE | Porous architectonics | 306 |
| Au thin film | Chemical deposition | NRR | 0.1 M KOH | Activity | NH3: 3.84 × 10−12 mol cm−2 s−1, FE: 0.12% at −0.5 V vs. RHE | Associative reaction mechanism | 310 |
Although there has been considerable progress in electrocatalysis, the structure–activity relationships in complicated reactions are still unclear, and an optimum surface structure is not obtained. To improve the catalytic performance of catalysts in electrochemical reactions, there are certain approaches for improvement. (1) The well-defined shapes of noble-metal-based nanostructures are important to investigate the relationship between structure and catalytic performance. Therefore, there is an urgent requirement for developing a precise synthesis methodology in formulating a predictable specific nanostructure. Moreover, the synthesis mechanism provides guidance to formulate the desired structure. However, most of the unique structures that have been reported are obtained via trial and error; research has been seldom used to clarify this nanocrystalline formation mechanism. For example, we know that halide ions tend to combine on the {100} facets, but the exact reason for this is unclear. In situ characterization techniques, such as in situ infrared spectroscopy, Raman spectroscopy, and TEM, should be used to study the surface structure evolution during the formation of a nanocrystal. (2) Determining the reaction mechanism necessary to understand the structure–activity relationship: currently, most of the reported possible mechanisms are obtained from DFT calculations. However, the model used in calculations may be different from the real reaction surface. The fabrication of a reasonable model relies on theoretical calculations. Moreover, data on intermediates are important to clarify the mechanism, which can be obtained from in situ detection and operando spectroscopies. In situ characterization should be used to observe changes in the catalyst surface and reactants during the reaction. (3) Determining the optimal surface structure by DFT calculations and experiments: once the reaction mechanism and optimal surface structure are obtained, we need to rationally design the catalyst to fabricate and maximize the optimal surface structure of the catalyst; meanwhile, we need to ensure catalyst stability during the catalytic reaction. The above induces higher requirements on the synthesis methods, characterization techniques, and rational design based on theoretical calculation models.
As mentioned earlier, developing novel synthesis methods to obtain certain nanostructures and determining the synthesis mechanism are very important to fabricate superior catalysts. For example, it is well known that the most stable crystalline structure of Ru is hcp. In general, structural transition can only be achieved under extreme conditions, e.g., temperature and/or pressure. Zheng et al. reported an anomalous fcc Ru-graphitic carbon nitride complex supported on carbon (Ru/C3N4/C catalyst) obtained via a mild synthesis method.239 They clarified that the special kind of carbon-based material (g-C3N4) could induce an anomalous crystalline structure of Ru, which showed better HER activity than that by hcp Ru. This reveals that the support may play a vital role in controlling the crystalline nature and morphology of the noble metal. Zhao et al. added Na2S2O3 in the process of synthesizing CuPt nanotubes, which could turn crystalline CuPt into amorphous nanotubes with superior activity toward MOR.104 With the development of in situ TEM, many synthesis mechanisms can be comprehensively investigated. Zhu et al. investigated the growth of two-dimensional (2D) Pd dendritic nanostructures using in situ liquid-cell TEM.313 They demonstrated that diffusion-limited aggregation and direct atomic deposition were important to form dendritic nanostructures. Therefore, the diffusion and atomic deposition could be controlled in order to synthesize 2D dendritic nanostructures that are beneficial catalysts in most catalytic reactions.
The development of in situ characterization technology provides a powerful means to study the catalytic reaction mechanism. Obviously, understanding this mechanism involving different catalytic reactions is necessary to design optimized catalysts. Dong et al. employed the in situ electrochemical surface-enhanced Raman spectroscopy (SERS) to examine the ORR process at Pt(hkl) surfaces.314 The direct spectroscopic evidence for ORR intermediates indicated that the adsorbed HOO* was stable at the Pt(111) surface, but the adsorbed OH* was stable at the Pt(110) and Pt(100) surfaces in acidic media. Meanwhile, under alkaline conditions, only O2− species were found on the three single-crystal surfaces. According to the different activity on Pt(hkl), we can conclude that the protonation process noticeably affects the ORR activity and its mechanism. González-Quijano et al. investigated the difference in the reaction mechanism in EOR between PtSn/C alloys and Pt/C by in situ FTIR measurements.315 Their results revealed that the addition of Sn to the Pt structure promoted the preferential formation of CO2via a triple parallel pathway: one path involving the conversion of COL (linearly bonded CO) to CO2, while the second path via the reaction of AAL into COL and subsequently into CO2; the third path involved the formation of AA from the adsorbed acetate that might be produced from AAL. It has been acknowledged that the presence of pyridine accelerates the rate of formate production during the CO2RR on Pt catalyst, but the reason for this is unclear. Dunwell et al. proposed that the adsorbed COOHL was a common intermediate in the formation of both formate and CO, and the presence of pyridinium promoted the formate pathway by employing in situ surface-enhanced infrared absorption spectroscopy.316 Therefore, we can speculate that pyridinium can be combined with other noble metals to increase the selectivity of formic acid species.
Moreover, setting up a suitable theoretical calculation model that is similar to the real reaction conditions and developing fast and efficient theoretical calculation methods can facilitate the determination of a descriptor for the reaction as well as ensuring high-throughput screening of the optimized catalyst. As mentioned earlier for the foregoing catalytic reactions, theoretical calculations have indicated that the activity and selectivity of almost all the catalytic reactions can be correlated with the adsorption energy of the intermediate species. Finding superior catalysts with suitable binding energies of the intermediates is important to enhance the catalytic activity. Meanwhile, theoretical calculations can screen out the most promising catalysts.
In summary, electrocatalytic reactions play a pivotal role in renewable energy utilization and environment protection. The development of electrocatalysts still faces numerous challenges; it is necessary to conduct further research in this field. Obviously, in the next few years, additional efforts should be devoted toward the following aspects. (1) Fabricating novel catalysts with unique structures that may possess superior catalytic performances; (2) taking advantage of in situ characterization and DFT calculations to investigate the reaction mechanism on model catalysts for clarifying the structure–activity relationship, thereby facilitating the design of the desired catalysts; (3) with the development of computer science, machine learning and big data analysis can be considered for the design and optimization of catalysts. We are confident that with the development of synthesis methods, characterization techniques, and theoretical calculation approaches, it is highly probable that catalysts with high activity, selectivity, and stability can be developed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledges financial support from the National Natural Science Foundation of China (no. 21832001, 21771009, 21573005 and 21621061), and the National Key Research and Development Program of China (no. 2016YFB0701100).Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 4998 CrossRef PubMed.
- H. Liu, X. Peng and X. Liu, ChemElectroChem, 2018, 5, 2963–2974 CrossRef CAS.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- J. He, N. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- M. Li, D. A. Cullen, K. Sasaki, N. S. Marinkovic, K. More and R. R. Adzic, J. Am. Chem. Soc., 2012, 135, 132–141 CrossRef PubMed.
- J. Zhang, J. Ye, Q. Fan, Y. Jiang, Y. Zhu, H. Li, Z. Cao, Q. Kuang, J. Cheng, J. Zheng and Z. Xie, J. Am. Chem. Soc., 2018, 140, 11232–11240 CrossRef CAS PubMed.
- W. Du, K. E. Mackenzie, D. F. Milano, N. A. Deskins, D. Su and X. Teng, ACS Catal., 2012, 2, 287–297 CrossRef CAS.
- R. Rizo, D. Sebastián, M. J. Lázaro and E. Pastor, Appl. Catal., B, 2017, 200, 246–254 CrossRef CAS.
- T. Frelink, W. Visscher and J. Van Veen, Surf. Sci., 1995, 335, 353–360 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed.
- A. Dorjgotov, Y. Jeon, J. Hwang, B. Ulziidelger, H. S. Kim, B. Han and Y.-G. Shul, Electrochim. Acta, 2017, 228, 389–397 CrossRef CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. F. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- L. Cao and T. Mueller, Nano Lett., 2016, 16, 7748–7754 CrossRef CAS PubMed.
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M.-G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719–1725 CrossRef CAS PubMed.
- Y. Luo, B. Kirchhoff, D. Fantauzzi, L. Calvillo, L. A. Estudillo-Wong, G. Granozzi, T. Jacob and N. Alonso-Vante, ChemSusChem, 2018, 11, 193–201 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, N.-F. Yu, L.-Y. Wang and S.-G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 CrossRef CAS PubMed.
- Z. Y. Zhou, Z. Z. Huang, D. J. Chen, Q. Wang, N. Tian and S. G. Sun, Angew. Chem., Int. Ed., 2010, 49, 411–414 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- J. Zou, M. Wu, S. Ning, L. Huang, X. Kang and S. Chen, ACS Sustainable Chem. Eng., 2019, 7, 9007–9016 CrossRef CAS.
- T. Kwon, M. Jun, H. Y. Kim, A. Oh, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2018, 28, 1706440 CrossRef.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed.
- N. Wenxin, Z. Ling and X. Guobao, Nanoscale, 2013, 5, 3172–3181 RSC.
- Y. Zheng, J. Zeng, A. Ruditskiy, M. Liu and Y. Xia, Chem. Mater., 2013, 26, 22–33 CrossRef.
- Q. Zewei, W. Yuxuan and F. Jiye, Acc. Chem. Res., 2013, 46, 191–202 CrossRef PubMed.
- Q. Chen, Y. Jia, S. Xie and Z. Xie, Chem. Soc. Rev., 2016, 45, 3207–3220 RSC.
- Z. Hui, J. Mingshang and X. Younan, Cheminform, 2012, 51, 7656–7673 Search PubMed.
- H. Wang, Z. Shan, K. D. Gilroy, Z. Cai and Y. Xia, Nano Today, 2017, 15, 121–144 CrossRef CAS.
- Y. Qiang and W. Xun, Nanoscale, 2010, 2, 2328–2335 RSC.
- J. W. Hong, Y. Kim, Y. Kwon and S. W. Han, Chem. – Asian J., 2016, 11, 2224–2239 CrossRef CAS PubMed.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 43, 1442–1442 CrossRef.
- J. Gu, Y. W. Zhang and T. F. Feng, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- Z. Wang, J. Phys. Chem. B, 2000, 104(6), 1153–1175 CrossRef CAS.
- E. Herrero, L. J. Buller and H. D. Abruña, Chem. Rev., 2001, 101, 1897–1930 CrossRef CAS PubMed.
- D. Wang, Q. Peng and Y. Li, Nano Res., 2010, 3, 574–580 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692 CrossRef CAS PubMed.
- X. Huang, H. Zhang, C. Guo, Z. Zhou and N. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808–4812 CrossRef CAS PubMed.
- H. Zhang, M. Jin, J. Wang, M. J. Kim, D. Yang and Y. Xia, J. Am. Chem. Soc., 2011, 133, 10422–10425 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, W. Zhu, H.-S. Wu, Y.-W. Zhang and C.-H. Yan, Chem. Commun., 2012, 48, 543–545 RSC.
- X. Huang, Y. Li, Y. Li, H. Zhou, X. Duan and Y. Huang, Nano Lett., 2012, 12, 4265–4270 CrossRef CAS PubMed.
- B. Lim, J. Wang, P. H. Camargo, C. M. Cobley, M. J. Kim and Y. Xia, Angew. Chem., Int. Ed., 2009, 48, 6304–6308 CrossRef CAS PubMed.
- Y. Yan, F. Zhan, J. Du, Y. Jiang, C. Jin, M. Fu, H. Zhang and D. Yang, Nanoscale, 2015, 7, 301–307 RSC.
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543–18547 CrossRef CAS PubMed.
- C. Wang, C. Lin, B. Zhao, L. Zhang, A. Kumbhar, G. Fan, K. Sun, J. Zhang, S. Chen and J. Fang, ChemNanoMat, 2015, 1, 331–337 CrossRef CAS.
- C. Wang, C. Lin, L. Zhang, Z. Quan, K. Sun, B. Zhao, F. Wang, N. Porter, Y. Wang and J. Fang, Chem. – Eur. J., 2014, 20, 1753–1759 CrossRef CAS PubMed.
- Y. Kang, X. Ye and C. B. Murray, Angew. Chem., Int. Ed., 2010, 49, 6156–6159 CrossRef CAS PubMed.
- Y. Jia, Y. Jiang, J. Zhang, L. Zhang, Q. Chen, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2014, 136, 3748–3751 CrossRef CAS PubMed.
- L.-X. Chen, L.-Y. Jiang, A.-J. Wang, Q.-Y. Chen and J.-J. Feng, Electrochim. Acta, 2016, 190, 872–878 CrossRef CAS.
- J. Zhang, J. Ma, Y. Wan, J. Jiang and X. Zhao, Mater. Chem. Phys., 2012, 132, 244–247 CrossRef CAS.
- L. Chen and Y. Liu, J. Colloid Interface Sci., 2011, 364, 100–106 CrossRef CAS PubMed.
- S. Kang, G. Gao, X. Xie, T. Shibayama, Y. Lei, Y. Wang and L. Cai, Mater. Res. Lett., 2016, 4, 212–218 CrossRef CAS.
- H. Zhang, Y. Yin, Y. Hu, C. Li, P. Wu, S. Wei and C. Cai, J. Phys. Chem. C, 2010, 114, 11861–11867 CrossRef CAS.
- L. Kuai, X. Yu, S. Wang, Y. Sang and B. Geng, Langmuir, 2012, 28, 7168–7173 CrossRef CAS PubMed.
- S.-B. Wang, W. Zhu, J. Ke, M. Lin and Y.-W. Zhang, ACS Catal., 2014, 4, 2298–2306 CrossRef CAS.
- B. Wiley, T. Herricks, Y. Sun and Y. Xia, Nano Lett., 2004, 4, 1733–1739 CrossRef CAS.
- T. Zhang, S.-C. Li, W. Zhu, Z.-P. Zhang, J. Gu and Y.-W. Zhang, Nanoscale, 2017, 9, 1154–1165 RSC.
- R. Lanza, M. Bersani, L. Conte, A. Martucci, P. Canu, M. Guglielmi, G. Mattei, V. Bello, M. Centazzo and R. Rosei, J. Phys. Chem. C, 2014, 118, 25392–25402 CrossRef CAS.
- S. U. Son, Y. Jang, J. Park, H. B. Na, H. M. Park, H. J. Yun, J. Lee and T. Hyeon, J. Am. Chem. Soc., 2004, 126, 5026–5027 CrossRef CAS PubMed.
- X. Xia, S. Xie, M. Liu, H.-C. Peng, N. Lu, J. Wang, M. J. Kim and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6669–6673 CrossRef CAS PubMed.
- M. Tsuji, N. Nakamura, M. Ogino, K. Ikedo and M. Matsunaga, CrystEngComm, 2012, 14, 7639–7647 RSC.
- T. Bian, H. Zhang, Y. Jiang, C. Jin, J. Wu, H. Yang and D. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS PubMed.
- Y. Hu, A.-Q. Zhang, H.-J. Li, D.-J. Qian and M. Chen, Nanoscale Res. Lett., 2016, 11, 209 CrossRef PubMed.
- Q. Li, R. Jiang, T. Ming, C. Fang and J. Wang, Nanoscale, 2012, 4, 7070–7077 RSC.
- Y. Rong, A. Dandapat, Y. Huang, Y. Sasson, L. Zhang, L. Dai, J. Zhang, Z. Guo and T. Chen, RSC Adv., 2016, 6, 10713–10718 RSC.
- J. Satija, J. Tharion and S. Mukherji, RSC Adv., 2015, 5, 69970–69979 RSC.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- B. Lim, M. Jiang, T. Yu, P. H. Camargo and Y. Xia, Nano Res., 2010, 3, 69–80 CrossRef CAS.
- Z. Zhang and M. G. Lagally, Science, 1997, 276, 377–383 CrossRef CAS PubMed.
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z. Xie and Y. Xia, Nano Lett., 2016, 16, 1467–1471 CrossRef CAS PubMed.
- X. Wang, M. Vara, M. Luo, H. Huang, A. Ruditskiy, J. Park, S. Bao, J. Liu, J. Howe, M. Chi, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2015, 137, 15036–15042 CrossRef CAS PubMed.
- W. Wang, X. Li, T. He, Y. Liu and M. Jin, Nano Lett., 2019, 19, 1743–1748 CrossRef CAS PubMed.
- K. M. Yeo, S. Choi, R. M. Anisur, J. Kim and I. S. Lee, Angew. Chem., Int. Ed., 2011, 50, 745–748 CrossRef CAS PubMed.
- Y. Liu, D. Gokcen, U. Bertocci and T. P. Moffat, Science, 2012, 338, 1327–1330 CrossRef CAS PubMed.
- M. Li, Q. Ma, W. Zi, X. Liu, X. Zhu and S. Liu, Sci. Adv., 2015, 1, 1400268–1400268 CrossRef PubMed.
- T. Ming, W. Feng, Q. Tang, F. Wang, L. Sun, J. Wang and C. Yan, J. Am. Chem. Soc., 2009, 131, 16350–16351 CrossRef CAS PubMed.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- J. Zhang, M. R. Langille, M. L. Personick, K. Zhang, S. Li and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 14012–14014 CrossRef CAS PubMed.
- M. L. Personick, M. R. Langille, J. Zhang and C. A. Mirkin, Nano Lett., 2011, 11, 3394–3398 CrossRef CAS PubMed.
- C. Sanchez, M. Del Popolo and E. Leiva, Surf. Sci., 1999, 421, 59–72 CrossRef CAS.
- J. Gu, Y.-W. Zhang and F. F. Tao, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- X. Lu, H.-Y. Tuan, J. Chen, Z.-Y. Li, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2007, 129, 1733–1742 CrossRef CAS PubMed.
- D. Seo and H. Song, J. Am. Chem. Soc., 2009, 131, 18210–18211 CrossRef CAS PubMed.
- M. H. Kim, X. Lu, B. Wiley, E. P. Lee and Y. Xia, J. Phys. Chem. C, 2008, 112, 7872–7876 CrossRef CAS PubMed.
- V. Bansal, H. Jani, J. Du Plessis, P. J. Coloe and S. K. Bhargava, Adv. Mater., 2008, 20, 717–723 CrossRef CAS.
- A. X. Yin, X. Q. Min, W. Zhu, W. C. Liu, Y. W. Zhang and C. H. Yan, Chem. – Eur. J., 2012, 18, 777–782 CrossRef CAS PubMed.
- F. Zhan, T. Bian, W. Zhao, H. Zhang, M. Jin and D. Yang, CrystEngComm, 2014, 16, 2411–2416 RSC.
- L. Au, Y. Chen, F. Zhou, P. H. Camargo, B. Lim, Z.-Y. Li, D. S. Ginger and Y. Xia, Nano Res., 2008, 1, 441–449 CrossRef CAS PubMed.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2009, 10, 30–35 CrossRef PubMed.
- X. Chen, C. H. Cui, Z. Guo, J. H. Liu, X. J. Huang and S. H. Yu, Small, 2011, 7, 858–863 CrossRef CAS PubMed.
- Y. Kang and F. Chen, J. Appl. Electrochem., 2013, 43, 667–677 CrossRef CAS.
- R. Rahmatolahzadeh, M. Ebadi and K. Motevalli, J. Mater. Sci.: Mater. Electron., 2017, 28, 6056–6063 CrossRef CAS.
- J. Ahn, D. Wang, Y. Ding, J. Zhang and D. Qin, ACS Nano, 2017, 12, 298–307 CrossRef PubMed.
- Y. Yang, J. Liu, Z.-W. Fu and D. Qin, J. Am. Chem. Soc., 2014, 136, 8153–8156 CrossRef CAS PubMed.
- X. Sun, Y. Yang, Z. Zhang and D. Qin, Chem. Mater., 2017, 29, 4014–4021 CrossRef CAS.
- Y. Zhao, J. Liu, C. Liu, F. Wang and Y. Song, ACS Catal., 2016, 6, 4127–4134 CrossRef CAS.
- S. Brankovic, J. Wang and R. Adžić, Surf. Sci., 2001, 474, 173–179 CrossRef.
- M. Li, P. Liu and R. R. Adzic, J. Phys. Chem. Lett., 2012, 3, 3480–3485 CrossRef CAS PubMed.
- K. Sasaki, H. Naohara, Y. Cai, Y. M. Choi, P. Liu, M. B. Vukmirovic, J. X. Wang and R. R. Adzic, Angew. Chem., Int. Ed., 2010, 49, 8602–8607 CrossRef CAS PubMed.
- Y. Yu, Q. Zhang, Q. Yao, J. Xie and J. Y. Lee, Chem. Mater., 2013, 25, 4746–4756 CrossRef CAS.
- K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25 CrossRef CAS PubMed.
- M. M. Shahjamali, M. Bosman, S. Cao, X. Huang, X. Cao, H. Zhang, S. S. Pramana and C. Xue, Small, 2013, 9, 2880–2886 CrossRef CAS PubMed.
- S. Xie, N. Lu, Z. Xie, J. Wang, M. J. Kim and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 10266–10270 CrossRef CAS PubMed.
- Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- Y. Yamauchi and K. Kuroda, Chem. – Asian. J., 2008, 3, 664–676 CrossRef CAS PubMed.
- Y. Yamauchi, M. Komatsu, M. Fuziwara, Y. Nemoto, K. Sato, T. Yokoshima, H. Sukegawa, K. Inomata and K. Kuroda, Angew. Chem., Int. Ed., 2009, 48, 7792–7797 CrossRef CAS PubMed.
- A. Takai, Y. Yamauchi and K. Kuroda, J. Am. Chem. Soc., 2009, 132, 208–214 CrossRef PubMed.
- P. Karthika, H. Ataee-Esfahani, H. Wang, M. A. Francis, H. Abe, N. Rajalakshmi, K. S. Dhathathreyan, D. Arivuoli and Y. Yamauchi, Chem. – Asian. J., 2013, 8, 902–907 CrossRef CAS PubMed.
- H. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Terasaki and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 14526–14529 CrossRef CAS PubMed.
- L. Liu, E. Pippel, R. Scholz and U. Gösele, Nano Lett., 2009, 9, 4352–4358 CrossRef CAS PubMed.
- H. Y. Hsueh, H. Y. Chen, Y. C. Hung, Y. C. Ling, S. Gwo and R. M. Ho, Adv. Mater., 2013, 25, 1780–1786 CrossRef CAS PubMed.
- H. Y. Hsueh, Y. C. Huang, R. M. Ho, C. H. Lai, T. Makida and H. Hasegawa, Adv. Mater., 2011, 23, 3041–3046 CrossRef CAS PubMed.
- A. Ma, J. Xu, X. Zhang, B. Zhang, D. Wang and H. Xu, Sci. Rep., 2014, 4, 4849 CrossRef CAS PubMed.
- H. Wang, S. Ishihara, K. Ariga and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 10819–10821 CrossRef CAS PubMed.
- B. Jiang, C. Li, M. Imura, J. Tang and Y. Yamauchi, Adv. Sci., 2015, 2, 1500112 CrossRef PubMed.
- D. Pugh, A. Dursun and S. Corcoran, J. Mater. Res., 2003, 18, 216–221 CrossRef CAS.
- K. Liu, Y. Bai, L. Zhang, Z. Yang, Q. Fan, H. Zheng, Y. Yin and C. Gao, Nano Lett., 2016, 16, 3675–3681 CrossRef CAS PubMed.
- Y. Wu, D. Wang, Z. Niu, P. Chen, G. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 12524–12528 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- Y. Ji, Y. Wu, G. Zhao, D. Wang, L. Liu, W. He and Y. Li, Nano Res., 2015, 8, 2706–2713 CrossRef CAS.
- Q. Yuan, Z. Zhou, J. Zhuang and X. Wang, Chem. Mater., 2010, 22, 2395–2402 CrossRef CAS.
- D. Bin, B. Yang, F. Ren, K. Zhang, P. Yang and Y. Du, J. Mater. Chem. A, 2015, 3, 14001–14006 RSC.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- H. Liao, J. Zhu and Y. Hou, Nanoscale, 2014, 6, 1049–1055 RSC.
- X. Hong, D. Wang, R. Yu, H. Yan, Y. Sun, L. He, Z. Niu, Q. Peng and Y. Li, Chem. Commun., 2011, 47, 5160–5162 RSC.
- B. Lim and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 76–85 CrossRef CAS PubMed.
- H. Zheng, R. K. Smith, Y.-W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- N. Ortiz and S. E. Skrabalak, Angew. Chem., Int. Ed., 2012, 51, 11757–11761 CrossRef CAS PubMed.
- K. Neyerlin, W. Gu, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2006, 153, 1955–1963 CrossRef.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715–6721 CrossRef.
- M. Piana, S. Catanorchi and H. Gasteiger, ECS Trans., 2008, 16, 2045–2055 CAS.
- K. Neyerlin, W. Gu, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2007, 154, 631–635 CrossRef.
- S. M. Alia, B. S. Pivovar and Y. Yan, J. Am. Chem. Soc., 2013, 135, 13473–13478 CrossRef CAS PubMed.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdottir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
- M. E. Scofield, Y. Zhou, S. Yue, L. Wang, D. Su, X. Tong, M. B. Vukmirovic, R. R. Adzic and S. S. Wong, ACS Catal., 2016, 6, 3895–3908 CrossRef CAS.
- N. M. Marković, S. T. Sarraf, H. A. Gasteiger and P. N. Ross, J. Chem. Soc., Faraday Trans., 1996, 92, 3719–3725 RSC.
- O. Junya, S. Takuma, Y. Yuta, A. Shigeo and S. Atsushi, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. Van Der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300 CrossRef CAS PubMed.
- J. Li, S. Ghoshal, M. K. Bates, T. E. Miller, V. Davies, E. Stavitski, K. Attenkofer, S. Mukerjee, Z. F. Ma and Q. Jia, Angew. Chem., Int. Ed., 2017, 56, 15594–15598 CrossRef CAS PubMed.
- Y. Qiu, L. Xin, Y. Li, I. T. McCrum, F. Guo, T. Ma, Y. Ren, Q. Liu, L. Zhou, S. Gu, M. J. Janik and W. Li, J. Am. Chem. Soc., 2018, 140, 16580–16588 CrossRef PubMed.
- H. Xin, A. Holewinski and S. Linic, ACS Catal., 2011, 2, 12–16 CrossRef.
- D. Li, C. Wang, D. S. Strmcnik, D. V. Tripkovic, X. Sun, Y. Kang, M. Chi, J. D. Snyder, D. van der Vliet, Y. Tsai, V. R. Stamenkovic, S. Sunb and N. M. Markovic, Energy Environ. Sci., 2014, 7, 4061–4069 RSC.
- H. Yang, Angew. Chem., Int. Ed., 2011, 50, 2674–2676 CrossRef CAS PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- P. Strasser, Science, 2015, 349, 379–380 CrossRef CAS PubMed.
- A. Fortunelli, W. A. Goddard III, L. Sementa, G. Barcaro, F. R. Negreiros and A. Jaramillo-Botero, Chem. Sci., 2015, 6, 3915–3925 RSC.
- J. Gu, G. Lan, Y. Jiang, Y. Xu, W. Zhu, C. Jin and Y. Zhang, Nano Res., 2015, 8, 1480–1496 CrossRef CAS.
- J. Wu, J. Zhang, Z. Peng, S. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS PubMed.
- J. Zhang, H. Yang, J. Fang and S. Zou, Nano Lett., 2010, 10, 638–644 CrossRef CAS PubMed.
- L. Wang, W. Gao, Z. Liu, Z. Zeng, Y. Liu, M. Giroux, M. Chi, G. Wang, J. Greeley, X. Pan and C. Wang, ACS Catal., 2017, 8, 35–42 CrossRef.
- V. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS PubMed.
- J. Shin, J.-H. Choi, P.-R. Cha, S. K. Kim, I. Kim, S.-C. Lee and D. S. Jeong, Nanoscale, 2015, 7, 15830–15839 RSC.
- Z. Duan and G. Wang, J. Phys. Chem. C, 2013, 117, 6284–6292 CrossRef CAS.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 12337–12340 CrossRef CAS PubMed.
- M. Gatalo, P. Jovanovič, G. Polymeros, J.-P. Grote, A. Pavlišič, F. Ruiz-Zepeda, V. S. Šelih, M. Šala, S. Hočevar, M. Bele, K. J. J. Mayrhofer, N. Hodnik and M. Gaberšček, ACS Catal., 2016, 6, 1630–1634 CrossRef CAS.
- L.-L. Shen, G.-R. Zhang, S. Miao, J. Liu and B.-Q. Xu, ACS Catal., 2016, 6, 1680–1690 CrossRef CAS.
- Q. Jia, Z. Zhao, L. Cao, J. Li, S. Ghoshal, V. Davies, E. Stavitski, K. Attenkofer, Z. Liu, M. Li, X. Duan, S. Mukerjee, T. Mueller and Y. Huang, Nano Lett., 2018, 18, 798–804 CrossRef CAS PubMed.
- Z. Duan and G. Wang, Phys. Chem. Chem. Phys., 2011, 13, 20178–20187 RSC.
- H. I. Karan, K. Sasaki, K. Kuttiyiel, C. A. Farberow, M. Mavrikakis and R. R. Adzic, ACS Catal., 2011, 2, 817 CrossRef.
- J. A. Bordley and M. A. El-Sayed, J. Phys. Chem. C, 2016, 120, 14643–14651 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- T. H. Housmans, A. H. Wonders and M. T. Koper, J. Phys. Chem. B, 2006, 110, 10021–10031 CrossRef CAS PubMed.
- J. Xie, P. Duan, N. Kaylor, K. Yin, B. Huang, K. Schmidt-Rohr and R. J. Davis, ACS Catal., 2017, 7, 6745–6756 CrossRef CAS.
- S. Jones, K. Tedsree, M. Sawangphruk, J. S. Foord, J. Fisher, D. Thompsett and S. C. E. Tsang, ChemCatChem, 2010, 2, 1089–1095 CrossRef CAS.
- L.-X. Ding, A.-L. Wang, G.-R. Li, Z.-Q. Liu, W.-X. Zhao, C.-Y. Su and Y.-X. Tong, J. Am. Chem. Soc., 2012, 134, 5730–5733 CrossRef CAS PubMed.
- Y. Liu, Z. Chen, C. Liu, J. Zhang, X. Han, C. Zhong, D. Rao, Y. Wang, W. Hu and Y. Deng, ACS Appl. Energy Mater., 2019, 2, 1588–1593 CrossRef CAS.
- Q. Wang, Z. Zhao, Y. Jia, M. Wang, W. Qi, Y. Pang, J. Yi, Y. Zhang, Z. Li and Z. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 36817–36827 CrossRef CAS PubMed.
- Y. Xiong, Y. Ma, J. Li, J. Huang, Y. Yan, H. Zhang, J. Wu and D. Yang, Nanoscale, 2017, 9, 11077–11084 RSC.
- S. Xue, W. Deng, F. Yang, J. Yang, I. S. Amiinu, D. He, H. Tang and S. Mu, ACS Catal., 2018, 8, 7578–7584 CrossRef CAS.
- Y. An-Xiang, M. Xiao-Quan, Z. Wei, L. Wen-Chi, Z. Ya-Wen and Y. Chun-Hua, Chem. – Eur. J., 2012, 18, 777–782 CrossRef PubMed.
- Y. An-Xiang, M. Xiao-Quan, Z. Wei, W. Hao-Shuai, Z. Ya-Wen and Y. Chun-Hua, Chem. Commun., 2011, 48, 543–545 Search PubMed.
- Y. An-Xiang, M. Xiao-Quan, Z. Ya-Wen and Y. Chun-Hua, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef PubMed.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 16762–16765 CrossRef CAS PubMed.
- C. Koenigsmann and S. S. Wong, ACS Catal., 2013, 3, 2031–2040 CrossRef CAS.
- W. Zhao, B. Ni, Q. Yuan, Y. Wang, Q. Zhang and X. Wang, Langmuir, 2017, 33, 8070–8075 CrossRef CAS PubMed.
- D. J. Chen and Y. J. Tong, Angew. Chem., Int. Ed., 2015, 54, 9394–9398 CrossRef CAS PubMed.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- Y. Ma, R. Wang, H. Wang, V. Linkov and S. Ji, Phys. Chem. Chem. Phys., 2014, 16, 3593–3602 RSC.
- Y. Pei, G. Zhou, N. Luan, B. Zong, M. Qiao and F. F. Tao, Chem. Soc. Rev., 2012, 41, 8140–8162 RSC.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381–14389 CrossRef CAS PubMed.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. V. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, J. Am. Chem. Soc., 2017, 139, 4762–4768 CrossRef CAS PubMed.
- D. Y. Wang, H. L. Chou, Y. C. Lin, F. J. Lai, C. H. Chen, J. F. Lee, B. J. Hwang and C. C. Chen, Cheminform, 2012, 43, 10011–10020 Search PubMed.
- B.-W. Zhang, T. Sheng, Y.-X. Wang, X.-M. Qu, J.-M. Zhang, Z.-C. Zhang, H.-G. Liao, F.-C. Zhu, S.-X. Dou, Y.-X. Jiang and S.-G. Sun, ACS Catal., 2016, 7, 892–895 CrossRef.
- I. Kim, O. H. Han, S. A. Chae, Y. Paik, S. H. Kwon, K. S. Lee, Y. E. Sung and H. Kim, Angew. Chem., Int. Ed., 2011, 50, 2270–2274 CrossRef CAS PubMed.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J.-M. Léger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- S. Song and P. Tsiakaras, Appl. Catal., B, 2006, 63, 187–193 CrossRef CAS.
- F. Vigier, C. Coutanceau, F. Hahn, E. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81–89 CrossRef CAS.
- C. Bianchini, V. Bambagioni, J. Filippi, A. Marchionni, F. Vizza, P. Bert and A. Tampucci, Electrochem. Commun., 2009, 11, 1077–1080 CrossRef CAS.
- Z.-Y. Zhou, Q. Wang, J.-L. Lin, N. Tian and S.-G. Sun, Electrochim. Acta, 2010, 55, 7995–7999 CrossRef CAS.
- W. Wang, J. Zhang, S. Yang, B. Ding and X. Song, ChemSusChem, 2013, 6, 1945–1951 CrossRef CAS PubMed.
- S. Shen, Y. Guo, L. Luo, F. Li, L. Li, G. Wei, J. Yin, C. Ke and J. Zhang, J. Phys. Chem. C, 2018, 122, 1604–1611 CrossRef CAS.
- S. Shendage, U. Patil and J. Nagarkar, Fuel Cells, 2013, 13, 364–370 CrossRef CAS.
- R. Rizo, R. M. Arán-Ais, E. Padgett, D. A. Muller, M. J. S. Lázaro, J. Solla-Gullón, J. M. Feliu, E. Pastor and H. D. Abruña, J. Am. Chem. Soc., 2018, 140, 3791–3797 CrossRef CAS PubMed.
- E. Antolini, ChemSusChem, 2013, 6, 966–973 CrossRef CAS PubMed.
- N. Erini, V. Beermann, M. Gocyla, M. Gliech, M. Heggen, R. E. Dunin-Borkowski and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 6533–6538 CrossRef CAS PubMed.
- D. Bin, B. Yang, K. Zhang, C. Wang, J. Wang, J. Zhong, Y. Feng, J. Guo and Y. Du, Chem. – Eur. J., 2016, 22, 16642–16647 CrossRef CAS PubMed.
- Z. S. Yang and J. J. Wu, Fuel Cells, 2012, 12, 420–425 CrossRef CAS.
- S. H. Han, H. M. Liu, P. Chen, J. X. Jiang and Y. Chen, Adv. Energy Mater., 2018, 8, 1801326 CrossRef.
- T. Herranz, M. Ibáñez, J. L. G. de la Fuente, F. J. Pérez-Alonso, M. A. Peña, A. Cabot and S. Rojas, ChemElectroChem, 2014, 1, 885–895 CrossRef CAS.
- L. X. Dai, X. Wang, S. S. Yang, T. Zhang, P. Ren, J. Ye, B. Nan, X. Wen, Z. Y. Zhou, R. Si, C. H. Yan and Y. W. Zhang, J. Mater. Chem. A, 2018, 6, 11270–11280 RSC.
- W. Zhu, J. Ke, S. B. Wang, J. Ren, H. H. Wang, Z. Y. Zhou, R. Si, Y. W. Zhang and C. H. Yan, ACS Catal., 2015, 5, 1995–2008 CrossRef CAS.
- W. A. Von and S. L. Anderson, Acc. Chem. Res., 2016, 49, 2632–2639 CrossRef PubMed.
- M. Momirlan and T. N. Veziroglu, Int. J. Hydrogen Energy, 2005, 30, 795–802 CrossRef CAS.
- S. Dunn, Int. J. Hydrogen Energy, 2002, 27, 235–264 CrossRef CAS.
- A. Melis and T. Happe, Plant Physiol., 2001, 127, 740–748 CrossRef CAS PubMed.
- A. R. Zeradjanin, J. P. Grote, G. Polymeros and K. J. J. Mayrhofer, Electroanalysis, 2016, 28, 2256–2269 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, 23–26 CrossRef.
- J. Durst, C. Simon, A. Siebel, P. J. Rheinländer, T. Schuler, M. Hanzlik, J. Herranz, F. Hasché and H. A. Gasteiger, ECS Trans., 2014, 64, 1069–1080 CrossRef CAS.
- B. Song, W. Chengming, D. Mingsen, G. Ming, B. Yu, J. Jun and X. Yujie, Angew. Chem., Int. Ed., 2014, 53, 12120–12124 CrossRef PubMed.
- N. Du, C. Wang, X. Wang, L. Yue, J. Jiang and Y. Xiong, Adv. Mater., 2016, 28, 2077–2084 CrossRef CAS PubMed.
- M. Bao, I. S. Amiinu, P. Tao, W. Li and S. Mu, ACS Energy Lett., 2018, 3, 940–945 CrossRef CAS.
- S. Wang, G. Yang and S. Yang, J. Phys. Chem. C, 2015, 119, 27938–27945 CrossRef CAS.
- A. Mahmood, H. Lin, N. Xie and W. Xun, Chem. Mater., 2017, 29, 6329–6335 CrossRef CAS.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- X. Kong, K. Xu, C. Zhang, J. Dai, S. N. Oliaee, L. Li, X. Zeng, C. Wu and Z. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef CAS.
- J. F. Huang and Y. C. Wu, ACS Sustainable Chem. Eng., 2018, 6, 8285–8290 CrossRef CAS.
- C. Zhang, Y. Liu, Y. Chang, Y. Lu, S. Zhao, D. Xu, Z. Dai, M. Han and J. Bao, ACS Appl. Mater. Interfaces, 2017, 9, 17326–17336 CrossRef CAS PubMed.
- Z. Fan, Z. Luo, X. Huang, B. Li, Y. Chen, J. Wang, Y. Hu and H. Zhang, J. Am. Chem. Soc., 2017, 138, 1414 CrossRef PubMed.
- J. Li, F. Li, S. X. Guo, J. Zhang and J. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 8151–8160 CrossRef CAS PubMed.
- M. D. Pohl, S. Watzele, F. Calle-Vallejo and A. S. Bandarenka, ACS Omega, 2017, 2, 8141–8147 CrossRef CAS.
- H. F. Lv, Z. Xi, Z. Z. Chen, S. J. Guo, Y. S. Yu, W. L. Zhu, Q. Li, X. Zhang, M. Pan, G. Lu, S. C. Mu and S. H. Sun, J. Am. Chem. Soc., 2015, 137, 5859–5862 CrossRef CAS PubMed.
- P. J. Rheinländer, J. Herranz, J. Durst and H. A. Gasteiger, J. Electrochem. Soc., 2014, 161, 1448–1457 CrossRef.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- S. Sun, G. Wang, Y. Zhou, F. Wang and X. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 19176–19182 CrossRef CAS PubMed.
- Y. Luo, X. Luo, G. Wu, Z. Li, G. Wang, B. Jiang, Y. Hu, T. Chao, H. Ju, J. Zhu, Z. Zhuang, Y. Wu, X. Hong and Y. Li, ACS Appl. Mater. Interfaces, 2018, 10, 34147–34152 CrossRef CAS PubMed.
- Y. Li, L. Zhang, Y. Qin, F. Chu, Y. Kong, Y. Tao, Y. Li, Y. Bu, D. Ding and M. Liu, ACS Catal., 2018, 8, 5714–5720 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. Li, Y. Han, Y. Chen, M. Jaroniec and S. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- X. Wang, Y. Zhu, A. Vasileff, Y. Jiao, S. Chen, L. Song, B. Zheng, Y. Zheng and S. Qiao, ACS Energy Lett., 2018, 3, 1198–1204 CrossRef CAS.
- Y. J. Tang, M. R. Gao, C. H. Liu, S. L. Li, H. L. Jiang, Y. Q. Lan, M. Han and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 12928–12932 CrossRef CAS PubMed.
- S. Y. Bae, I. Y. Jeon, J. Mahmood and J. B. Baek, Chem. – Eur. J., 2018, 24, 18158–18179 CrossRef CAS PubMed.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W. C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- M. Ledendecker, J. S. Mondschein, O. Kasian, S. Geiger, D. Göhl, M. Schalenbach, A. Zeradjanin, S. Cherevko, R. E. Schaak and K. Mayrhofer, Angew. Chem., Int. Ed., 2017, 56, 9767–9771 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- J. X. Feng, H. Xu, Y. T. Dong, S. H. Ye, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2016, 55, 3694–3698 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. Subhramannia, B. K. Balan, B. R. Sathe, I. S. Mulla and V. K. Pillai, J. Phys. Chem. C, 2007, 111, 16593–16600 CrossRef CAS.
- N. M. AlYami, A. P. LaGrow, K. S. Joya, J. Hwang, K. Katsiev, D. H. Anjum, Y. Losovyj, L. Sinatra, J. Y. Kim and O. M. Bakr, Phys. Chem. Chem. Phys., 2016, 18, 16169–16178 RSC.
- L. C. Seitz, T. J. Hersbach, D. Nordlund and T. F. Jaramillo, J. Phys. Chem. Lett., 2015, 6, 4178–4183 CrossRef CAS PubMed.
- R. Frydendal, M. Busch, N. B. Halck, E. A. Paoli, P. Krtil, I. Chorkendorff and J. Rossmeisl, ChemCatChem, 2015, 7, 149–154 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- S. H. Chang, N. Danilovic, K. C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 1548 CrossRef CAS PubMed.
- L. Gloag, T. M. Benedetti, S. Cheong, C. E. Marjo, J. J. Gooding and R. D. Tilley, J. Am. Chem. Soc., 2018, 140, 12760–12764 CrossRef CAS PubMed.
- J. Park, Y. J. Sa, H. Baik, T. Kwon, S. H. Joo and K. Lee, ACS Nano, 2017, 11, 6600–5509 Search PubMed.
- Y. Pi, J. Guo, Q. Shao and X. Huang, Chem. Mater., 2018, 30, 8571–8578 CrossRef CAS.
- T. Zhang, S.-A. Liao, L.-X. Dai, J.-W. Yu, W. Zhu and Y.-W. Zhang, Sci. China Mater., 2018, 61, 926–938 CrossRef CAS.
- C. Wang, Y. M. Sui, M. Xu, C. Liu, G. J. Xiao and B. Zou, ACS Sustainable Chem. Eng., 2017, 5, 9787–9792 CrossRef CAS.
- Z. Fan, Z. Luo, Y. Chen, J. Wang, B. Li, Y. Zong and H. Zhang, Small, 2016, 12, 3908–3913 CrossRef CAS PubMed.
- J. Shan, T. Ling, K. Davey, Y. Zheng and S. Qiao, Adv. Mater., 2019, 31, 1900510 CrossRef PubMed.
- J. Shan, C. Guo, Y. Zhu, S. Chen, L. Song, M. Jaroniec, Y. Zheng and S. Qiao, Chem, 2019, 5, 445–459 CAS.
- M. Zhao, Z. Chen, Z. Lyu, Z. D. Hood, M. Xie, M. Vara, M. Chi and Y. Xia, J. Am. Chem. Soc., 2019, 141, 7028–7036 CrossRef CAS PubMed.
- D. T. Whipple and P. J. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Modern Aspects of Electrochemistry, 1975 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- A. V. Rudnev, K. Kiran, A. C. Lopez, A. Dutta, I. Gjuroski, J. Furrer and P. Broekmann, Electrochim. Acta, 2019, 306, 245–253 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- S. Zhang, P. Kang, M. Bakir, A. M. Lapides, C. J. Dares and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15809–15814 CrossRef CAS PubMed.
- Q. H. Low, N. W. X. Loo, F. Calle-Vallejo and B. S. Yeo, Angew. Chem., Int. Ed., 2019, 58, 2256–2260 CrossRef CAS PubMed.
- J. H. Zhou, D. W. Lan, S. S. Yang, Y. Guo, K. Yuan, L. X. Dai and Y. Zhang, Inorg. Chem. Front., 2018, 5, 1524–1532 RSC.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974 CrossRef CAS PubMed.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- J. T. Song, H. Ryoo, M. Cho, J. Kim, J. G. Kim, S. Y. Chung and J. Oh, Adv. Energy Mater., 2017, 7, 1601103 CrossRef.
- S. Back, S. Y. Min and Y. Jung, J. Phys. Chem. C, 2018, 122, 4274–4280 CrossRef CAS.
- B. Hvolbæk, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Nørskov, Nano Today, 2007, 2, 14–18 CrossRef.
- H. E. Lee, K. D. Yang, S. M. Yoon, H. Y. Ahn, Y. Y. Lee, H. Chang, D. H. Jeong, Y. S. Lee, M. Y. Kim and K. T. Nam, ACS Nano, 2015, 9, 8384–8393 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5(7), 4293–4299 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- D. F. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. X. Wang, J. G. Wang and X. H. Bao, J. Am. Chem. Soc., 2015, 137, 4288 CrossRef CAS PubMed.
- H. Tao, X. Sun, S. Back, Z. Han, Q. Zhu, A. W. Robertson, T. Ma, Q. Fan, B. Han, Y. Jung and Z. Sun, Chem. Sci., 2018, 9, 483 RSC.
- B. Jiang, X. Zhang, K. Jiang, D. Wu and W. Cai, J. Am. Chem. Soc., 2018, 140, 2880–2889 CrossRef CAS PubMed.
- L. Zeng, J. Shi, J. Luo and H. Chen, J. Power Sources, 2018, 398, 83–90 CrossRef CAS.
- C. Zamfirescu and I. Dincer, J. Power Sources, 2008, 185, 459–465 CrossRef CAS.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64 CrossRef CAS PubMed.
- R. Lan, J. T. Irvine and S. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- D. Bao, Q. Zhang, F. L. Meng, H. X. Zhong, M. M. Shi, Y. Zhang, J. M. Yan, Q. Jiang and X. B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- H.-M. Liu, S.-H. Han, Y. Zhao, Y.-Y. Zhu, X.-L. Tian, J.-H. Zeng, J.-X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- Z. Wang, Y. Li, H. Yu, Y. Xu, H. Xue, X. Li, H. Wang and L. Wang, ChemSusChem, 2018, 11, 3480–3485 CrossRef CAS PubMed.
- M. M. Shi, D. Bao, S. J. Li, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Energy Mater., 2018, 8, 1800124 CrossRef.
- C. J. van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- Y. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2016, 138, 3970–3973 CrossRef CAS PubMed.
- M. Nazemi, S. R. Panikkanvalappil and M. A. El-Sayed, Nano Energy, 2018, 49, 316–323 CrossRef CAS.
- M. Nazemi and M. A. El-Sayed, J. Phys. Chem. Lett., 2018, 9, 5160–5166 CrossRef CAS PubMed.
- H. Wang, H. Yu, Z. Wang, Y. Li, Y. Xu, X. Li, H. Xue and L. Wang, Small, 2019, 15, 1804769 CrossRef PubMed.
- Z. Wang, C. Li, K. Deng, Y. Xu, H. Xue, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 2400–2405 CrossRef CAS.
- M. Zhao, L. Xu, M. Vara, A. O. Elnabawy, K. D. Gilroy, Z. D. Hood, S. Zhou, L. Figueroa-Cosme, M. Chi, M. Mavrikakis and Y. Xia, ACS Catal., 2018, 8, 6948–6960 CrossRef CAS.
- K. Zhang, R. Guo, F. Pang, J. He and W. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 10214–10220 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- Bimetallic Nanostructures: Shape-Controlled Synthesis for Catalysis, Plasmonics, and Sensing Applications, ed. Y.-W. Zhang, John Wiley & Sons Ltd, London, 2018 Search PubMed.
- G. Zhu, Y. Jiang, F. Lin, H. Zhang, C. Jin, J. Yuan, D. Yanga and Z. Zhang, Chem. Commun., 2014, 50, 9447–9450 RSC.
- J. Dong, X. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z. Yang, D. Wu, J. M. Feliu, C. T. Williams, Z. Tian and J. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- D. González-Quijano, W. J. Pech-Rodríguez, J. A. González-Quijano, J. I. Escalante-García, C. Morais, T. W. Napporn and F. J. Rodríguez-Varela, ChemElectroChem, 2018, 5, 3540–3547 CrossRef.
- M. Dunwell, Y. Yan and B. Xu, ACS Catal., 2017, 7, 5410–5419 CrossRef CAS.
| This journal is © the Partner Organisations 2019 |