
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isothermal titration calorimetry as a complementary method for investigating nanoparticle–protein interactions
Domenik
Prozeller
 ,
Svenja
Morsbach
and
Katharina
Landfester
*
,
Svenja
Morsbach
and
Katharina
Landfester
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de
First published on 24th September 2019
Abstract
Isothermal titration calorimetry (ITC) is a complementary technique that can be used for investigations of protein adsorption on nanomaterials, as it quantifies the thermodynamic parameters of intermolecular interactions in situ. As soon as nanomaterials enter biological media, a corona of proteins forms around the nanomaterials, which influences the surface properties and therefore the behavior of nanomaterials tremendously. ITC enhances our understanding of nanoparticle–protein interactions, as it provides information on binding affinity (in form of association constant Ka), interaction mechanism (in form of binding enthalpy ΔH, binding entropy ΔS and Gibbs free energy ΔG) and binding stoichiometry n. Therefore, as a complementary method, ITC enhances our mechanistic understanding of the protein corona. In this minireview, the information obtained from a multitude of ITC studies regarding different nanomaterials and proteins are gathered and relations between nanomaterials’ properties and their resulting interactions undergone with proteins are deduced. Nanomaterials formed of a hydrophilic material without strongly charged surface and steric stabilization experience the weakest interactions with proteins. As a result, such nanomaterials undergo the least unspecific protein-interactions and are most promising for allowing an engineering of the protein corona.
1. Introduction
Resulting from the rapid progress in nanotechnology of the past decades, nanomaterials have been developed with a broad array of applications. Colloidal nanomaterials such as nanoparticles (NPs) in particular hold a high potential for the field of precision medicine.1 In biological media such as blood, NPs undergo highly complex interactions with the different biomolecules in their environment under the formation of a biomolecule corona. Particularly interactions with proteins have a striking impact on the properties and behavior of NPs in biological media.2 Hence, the term ‘protein corona’ – which describes the shell of adsorbed proteins around NPs – is omnipresent in the research of nanomedicine. In order to achieve a controlled behavior of NPs in medicine, gaining control over NP–protein interactions (and therefore the protein corona) is obligatorily required. As a consequence, the physical mechanism of the interactions at the nano–bio interface is the focus of current research.Understanding NP–protein interactions is a challenging task to undertake, as most analytical methods deliver the final composition of the protein corona (or rather parts thereof),3 but fail to elucidate the mechanism behind corona formation. Without answering the many questions around the specifics at the nano-bio interface, the future development of nanomedicines will primarily rely on presumptions. Not many techniques can compete with isothermal titration calorimetry (ITC) in addressing this challenge. ITC yields quantified adsorption parameters from heat absorption or release resulting from intermolecular interactions.4 Combining the thermodynamic information provided by ITC experiments with information from other methods gives deep insight into the mechanism of NP–protein interactions and the fate of protein-covered nanomaterials.5
It is not the aim of this short review to reflect the addressed topics in their entirety, but to focus on the fundamental aspects of ITC, which can be important to the field of protein corona research. Furthermore, limitations of the method's state of the art and the use of auxiliary techniques for overcoming those limitations are addressed. For more detailed information, reviews that are more extensive are highlighted in the beginning of the respective chapter.
2. Nanoparticle–protein interactions
NP–protein interactions embody a very complex and dynamic process and extensive research investigating the protein corona formation has been conducted in the past decades. Excellent reviews addressing and focusing on the protein adsorption onto nanoparticles,6–8 and the impact of the protein corona on nanomedicine2 are available in the literature.The process of protein corona formation is a competition between different proteins for interactions with the accessible surface of NPs. Result of this competition is a dynamic ‘cloud’ of proteins around the NPs, which continuously changes over time. In the beginning seconds to minutes of this interaction, the more abundant blood proteins such as albumin will be dominant in the corona.9 Over time, proteins with higher affinity (and lower concentration) will take over the space close to the NP's surface and proteins with lower affinity will be attached more loosely in the outer layers around the NP.9 This kinetically driven evolution of the corona over time is commonly referred to as ‘Vroman-effect’.10 Common nomenclature for the more tightly bound proteins is the so called ‘hard’ protein corona, while loosely bound proteins are considered ‘soft’ corona proteins.11
Properties of proteins and nanoparticles alike influence their intermolecular interactions with each other. NPs’ properties of interest include size,12 surface charge and functionalization,11 stabilizing surfactants,13 and hydrophilicity.14 On the other hand, the proteins’ properties of interest address their amphiphilic character, which leads to screening of hydrophobic parts of the protein from the surrounding water.15 Similarly, smaller hydrophobic molecules can be transported by amphiphilic proteins such as lipids attached to apolipoproteins.16 This amphiphilic nature in turn also leads to preferred interaction with other surfaces such as nanomaterials. The main forces governing the interfacial interactions between nanomaterials and biological systems are listed in Fig. 1 and in Table 1.8
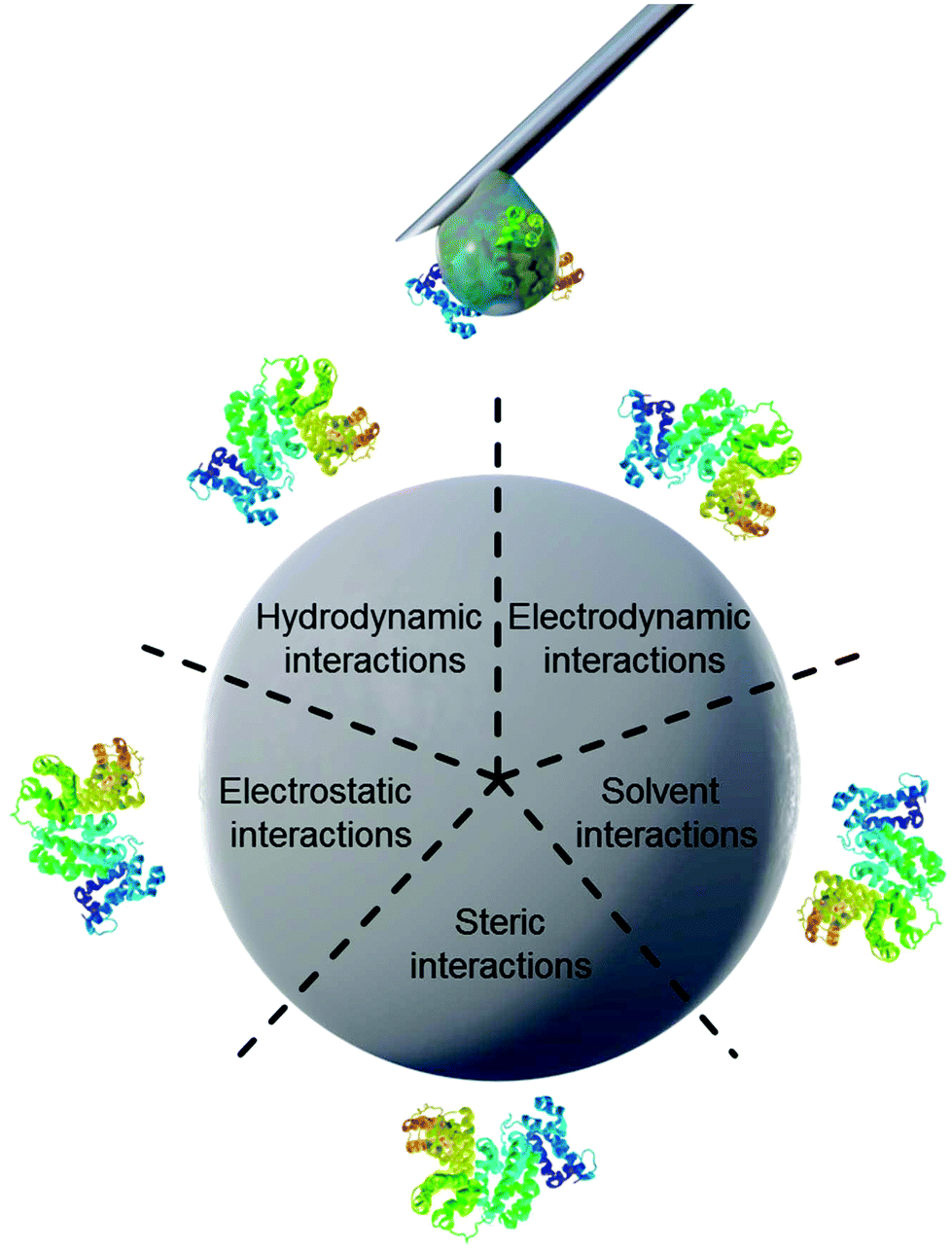 | ||
| Fig. 1 Schematic illustration of main forces governing intermolecular interactions between nanoparticles and proteins. | ||
| Force | Origin and Nature | Range (nm) | Possible impact on the interface |
|---|---|---|---|
| Hydrodynamic interactions | Convective drag, shear, lift and Brownian diffusion are often hindered or enhanced at nanoscale separations between interacting interfaces | 102–106 | Increase the frequency of collisions between nanoparticles and other surfaces responsible for transport |
| Electrodynamic interactions | van der Waals interactions arising from each of the interacting materials in the intervening media | 1–100 | Universally attractive in aqueous media; substantially smaller for biological media and cells owing to high salt content |
| Electrostatic interactions | Charged interfaces attract counter-ions and repel co-ions (coulombic forces), giving rise to the formation of an electrostatic double layer | 1–100 | Overlapping double layers are generally repulsive as most materials acquire negative charge in aqueous media, but can be attractive for oppositely charged materials |
| Solvent interactions | Lyophilic materials interact favourably with solvent molecules | 1–10 | Lyophilic materials are stable (thermodynamically) in the solvent and do not aggregate |
| Lyophobic materials interact unfavourably with solvent molecules | Lyophobic materials are spontaneously expelled from the bulk of the solvent and forced to aggregate or accumulate at an interface | ||
| Steric interactions | Polymeric species adsorbed to particles give rise to spring-like repulsive interactions with other interfaces | 1–100 | Generally increase stability of individual particles but polymers have their own adsorption behavior |
Remaining questions in the field include (but are not limited to) the possibility of protein-multilayers in the corona,17,18 the exact mechanism of the Vroman effect with influence of kinetics on the protein corona,10,19,20 and the thermodynamics of specific NP–protein interactions.21 For these open questions, the thermodynamic information obtainable with ITC can be of significant aid by offering a strong complementary method to the analytical toolbox of protein corona research.
3. Isothermal titration calorimetry
Due to the complexity in the multitude of possible interactions NPs and proteins can undergo, one must design, execute and analyze experiments on these interactions with great caution in order to achieve auxiliary and reliable data. Excellent reviews on the fundamentals of ITC specifically addressing experiment design, execution and analysis in particular are available in literature.22–26In general, ITC is an analytical technique relying on the thermodynamics of binding events. The thermal energy measured stems from binding events resulting from one or multiple titration(s) of one interaction partner to another. These interactions can be of different origins, as previously described for the example of NP–protein interactions in Table 1, and cover the entire spectrum for interactions from the formation of covalent bonds to non-covalent interactions such as hydrogen bonding or electrostatics. The first use of titration calorimetry for analyzing the Gibbs free energy ΔG, the binding enthalpy ΔH and the change of entropy ΔS of the proton ionization from HSO4− and HPO42− in one single titration was published by Christensen et al. in 1966 in form of a method called ‘entropy titration’.27 In the following years, ITC was developed with interactions between different biomolecules in mind, such as enzyme–substrate interactions.28,29 One of the first examples for utilizing calorimetric methods in order to determine the activity of enzymes was published in 1976 by Spink and Wadso.30 However, all processes that lead to the release or absorption of heat during the interaction are accessible with ITC. For example, Chiad et al. determined the thermodynamic parameters and stoichiometry for interactions between silica nanoparticles and surface-active amphiphilic copolymers bearing different types of anchor groups (nonionic, zwitterionic, and acidic) in complex organic–inorganic hybrid systems utilizing ITC.31
During an experiment, a solution of one compound is titrated to the solution of another compound in an isothermal ‘measurement cell’ in equivoluminar injection(s). Because of this isothermal setup implemented by thermal equilibrium of the measurement cell with a ‘reference cell’, the heat increase or decrease resulting from the two compounds’ interaction must be adjusted by controlling the heating rate of the measurement cell. The general setup of an ITC instrument is depicted in Fig. 2.
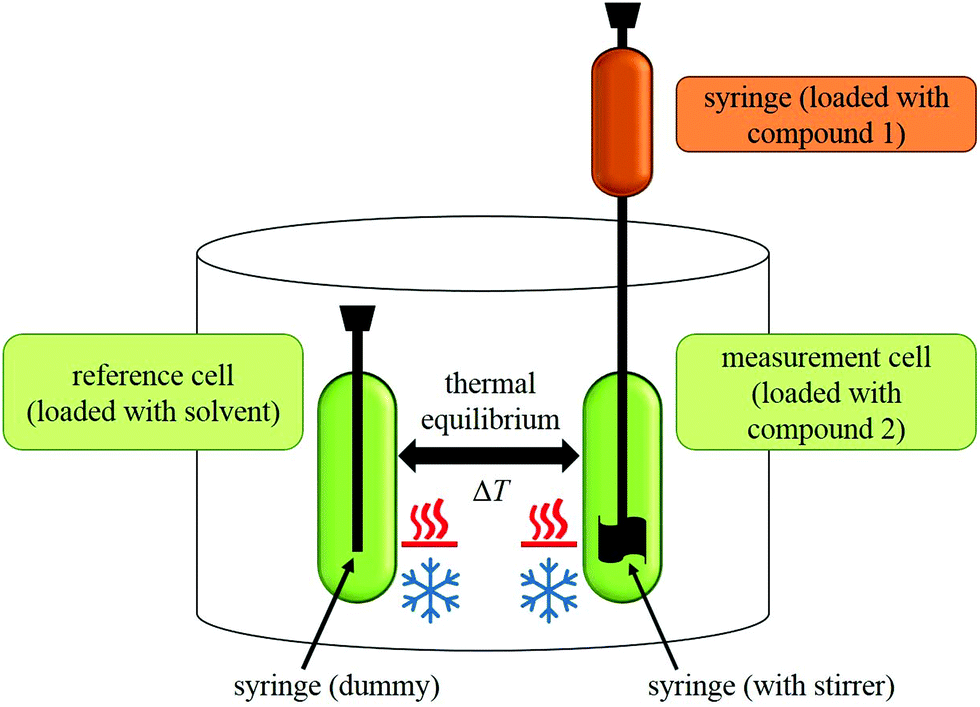 | ||
| Fig. 2 General setup of an ITC instrument. | ||
The heating rate applied in order to maintain a constant temperature within the measurement cell is monitored for the duration of the titration(s) (see Fig. 3, top). Integration of the heat rate over time, leads to the heat of the interaction process taking place during the respective titration step. It is important to consider, that not only interactions between the two compounds lead to heat changes in the measurement cell, but for example titrations of proteins into water result in heat of dilution already. Subtracting the heat of a titration step with the corresponding heat of dilution yields in the ‘corrected heat’ resulting from interactions between the two compounds. Fitting the corrected heat of consecutive titration steps can be achieved with several mathematical models and the resulting fit (called ‘adsorption isotherm’) yields the thermodynamic parameters of the interactions (see Fig. 3, bottom).
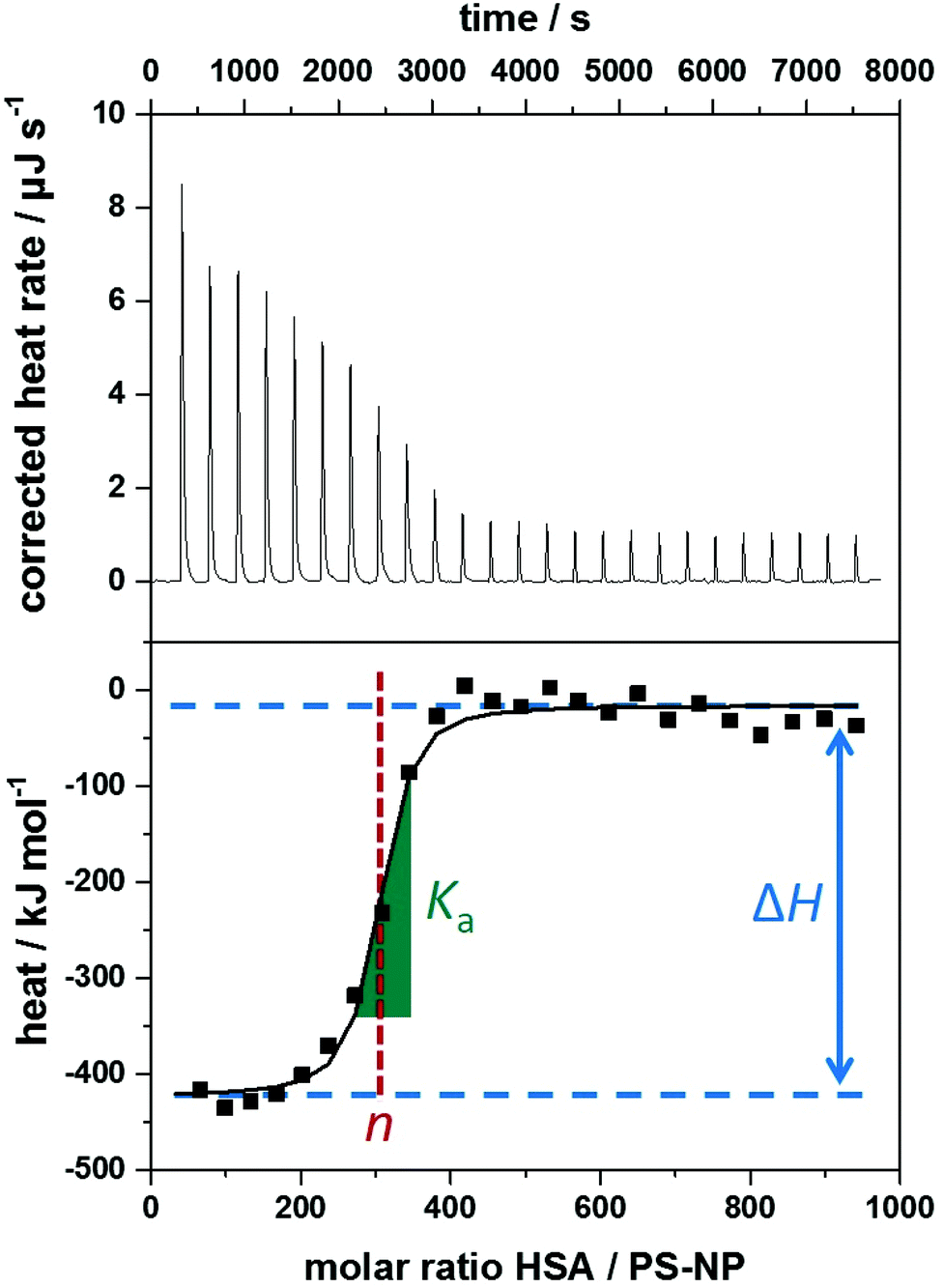 | ||
Fig. 3 Typical data obtained from isothermal titration calorimetry measurements of polystyrene nanoparticles (PS-NPs) titrated with human serum albumin (HSA). Top: Corrected heat rate of the titration. Bottom: Integrated normalized heats from each titration step corrected by the heats of dilution (filled squares) together with a fit corresponding to an independent binding model (straight line). Visual representation of the parameters obtained by ITC experiments within the adsorption isotherm. Ka is derived from the curve's slope in its inflection point (green slope  ), ΔH is represented by the difference between the curve's upper and lower plateaus (blue lines ), ΔH is represented by the difference between the curve's upper and lower plateaus (blue lines  ) and n is the molar ratio of the two components at the curve's inflection point (red lines ) and n is the molar ratio of the two components at the curve's inflection point (red lines  ). ). | ||
Based on the fit resulting from the corrected heat from multiple injections, the association constant Ka, interaction enthalpy ΔH and molar stoichiometry n are obtained. Ka is derived from the curve's slope in its inflection point, ΔH is the difference between the curve's plateaus and n is the molar ratio of the two components at the curve's inflection point (see Fig. 3, bottom). The Gibbs free energy ΔG of the interaction is then calculated using the reaction isotherm equation (see eqn (1)).32 ΔS can then be determined subsequently from the Gibbs–Helmholtz equation (see eqn (2)).
ΔG = RT·ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka Ka | (1) |
| ΔG = ΔH − TΔS. | (2) |
4. Investigating nanoparticle–protein interactions via ITC
In recent years, protein adsorption processes were more and more in the focus of ITC analyses.26 As a result, several protein corona studies employed ITC for characterizing the physicochemical properties of NP–protein interactions specifically.4,11,13,16,33–47 A review on the application of ITC in evaluation of nanoparticle–protein interactions by Omanovic-Miklicanin et al.48 and an excellent review elucidating the thermodynamics of NP–biomolecule interactions in general by Huang and Lau are available in the literature.21One big advantage of ITC studies on the protein corona is the unnecessity to separate the formed NP–protein complexes from the medium for further characterization. This allows analysis of the entire protein corona in situ, including low-affinity soft corona proteins, which might have big influence on the NPs, yet are disregarded in most other analytical approaches. Usually, analysis of the protein corona relies on fractionation steps, such as centrifugation, removing low-affinity proteins from the (soft) protein corona.3 Thus, ITC allows investigation of all NP–protein interactions, limiting the possibility to oversee important adsorption processes. Furthermore, ITC measurements do not require labels on NPs or proteins, allowing investigation of their interaction without modification of any interaction partner. Also, instead of solely yielding information on the binding affinity, ITC studies give information about the complete thermodynamic parameters of the interaction.
In a study by Welsch et al., the thermodynamics of protein adsorption processes onto different nanoparticles were measured with ITC.43 The characterization of nanocarriers’ functionalities and investigations of nanoparticles’ interactions with biosystems via ITC were reviewed by Bouchemal.49 Membrane proteins were studied by Draczkowski et al.4 and Rajarathnam and Rösgen44 utilizing ITC. The stoichiometry and association rates of proteins to nanoparticles was investigated by Cedervall et al.39 Several studies on the empiric, thermodynamic ruleset behind ITC were performed in the past years.50–52
Because of the large amount of possible interactions going on, interpretation of the results that ITC experiments yield about the protein corona in particular is not a simple task. While the significance and meaning of stoichiometry and association constant obtained from ITC studies for single proteins are obvious, interpretation of the thermodynamic parameters concerning the protein adsorption process appears more complex. For their interpretation, the role of hydration water in particular should not be underestimated. Generally, the formation of non-covalent bonds (see Table 1; e.g. van der Waals forces, electrostatic interactions or hydrogen bonds) is an exothermic process (ΔH < 0) dominating for hydrophilic surfaces, while desolvation – the release of hydration water from the surface of NPs and proteins – is an endothermic process (ΔH > 0) and is seen more regularly for hydrophobic surfaces as a result of hydrophobic interactions. In a similarly opposing fashion, the conformational restriction and loss of rotational freedom during protein adsorption yields unfavorable entropy loss (ΔS < 0), while desolvation results in an increase of the system's entropy (ΔS > 0) assuming the protein contains its shape. Therefore, the driving force of the adsorption process strongly depends on the interaction mechanism of the individual NP–protein system and may differ from case to case, requiring cautious analysis. As the heat observed in ITC solely reflects the total energy released or absorbed during the interaction, differentiating the individual contribution of different bonds to the total heat is impossible. For example, relative contributions originating from protein denaturation and aggregation, electrostatic and hydrophobic interactions or desolvation cannot be distinguished and thus cannot be quantified individually.
In order to shed more light onto the trends in correlation between binding mechanism and the properties of NPs and protein, exemplary recent ITC studies on NP–protein interactions are summarized in Table 2.
| NP material | Protein | Parameters of NP–protein interaction | NP properties | Protein chargec | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| K a/105 M−1 | ΔH/kJ mol−1 | ΔS/J mol−1 K−1 | Chargea | Hydrophilicityb | Surfactant | ||||
| a Charge of NPs categorized as positively charged (⊕⊕), mildly positively charged (⊕), neutral (Ø), mildly negatively charged (⊖) or negatively charged (⊖⊖). b Hydrophilicity of NPs base material categorized ranging from very hydrophilic (*****) to very hydrophobic (*). c Charge of protein at physiological pH categorized as overall positively charged (⊕), neutral (Ø), or overall negatively charged (⊖). N/A stands for “not available”. | |||||||||
| Polystyrene (PS) | Apo A-I | 24 | −1438 | −4700 | Ø | * | Lutensol | ⊖ | 53 |
| PS | Clusterin | 371 | −1237 | −4004 | Ø | * | Lutensol | ⊖ | 53 |
| PS | HSA | 0.6 ± 0.2 | −199 ± 54 | −487 ± 74 | Ø | * | Lutensol | ⊖ | 54 |
| PS | HSA | 2.4 ± 0.8 | −192 ± 45 | −540 ± 151 | Ø | * | SDS | ⊖ | 54 |
| hydroxyethyl starch (HES) | HSA | 8 ± 3 | −277 ± 43 | −818 ± 147 | ⊖⊖ | ***** | SDS | ⊖ | 11 |
| HES | Apo A-I | 3330 ± 1240 | −6010 ± 185 | −20000 ± 613 |
⊖⊖ | ***** | SDS | ⊖ | 11 |
| HES (carboxy functionalized) | HSA | 4 ± 2 | −308 ± 30 | −928 ± 103 | ⊖⊖ | ***** | SDS | ⊖ | 11 |
| HES (carboxy functionalized) | Apo A-I | 1880 ± 790 | −5150 ± 787 | −17100 ± 2640 |
⊖⊖ | ***** | SDS | ⊖ | 11 |
| HES (amino functionalized) | HSA | 5 ± 1 | −277 ± 45 | −820 ± 152 | ⊖⊖ | ***** | SDS | ⊖ | 11 |
| HES (amino functionalized) | Apo A-I | 54 ± 1 | 883000 ± 24000 |
2.4 ± 0.9 × 106 | ⊖⊖ | ***** | SDS | ⊖ | 11 |
| Copolymer NIPAM/BAM (50:50) |
HSA | 12.0 ± 0.2 | −595 ± 54 | −1950 ± 230 | Ø | ** | None | ⊖ | 38 |
| Copolymer NIPAM/BAM (85:15) |
HSA | 62.7 ± 0.2 | −104 ± 53 | −350 ± 40 | ⊖ | *** | None | ⊖ | 38 |
| PS (spherical polyelectrolyte brushes) | β-Lactoglobulin | 10 ± 1 | 113 ± 3 | 0.494 ± 0.008 | ⊖ | * | Poly(styrene sulfonate) | ⊖ | 33 |
| Au | HSA | 14 ± 5 | −1960 ± 1290 | −6200 | ⊖ | *** | None | ⊖ | 55 |
| Au (amino acid-functionalized) | α-Chymotrypsin | ≈6 | ≈−45 | ≈−50 | ⊖ | * | 1-Pentanethiol | ⊕ | 40 |
| Au (amino acid-functionalized) | Histone | ≈800 | ≈95 | ≈460 | ⊖ | * | 1-Pentanethiol | ⊕ | 40 |
| Au (amino acid-functionalized) | Cytochrome c | ≈100 | ≈50 | ≈330 | ⊖ | * | 1-Pentanethiol | ⊕ | 40 |
| Au (mannose functionalized) | Concanavalin A | 82 | −10.8 × 104 | N/A | ⊖ | * | None | ⊖ | 56 |
| Au (17% galactose functionalized) | Lecitin PA-IL | 1.7 ± 0.3 | −37 ± 7 | N/A | ⊖ | * | None | ⊖ | 57 |
| Au (100% galactose functionalized) | Lecitin PA-IL | 200 ± 20 | −18 ± 5 | N/A | ⊖ | * | None | ⊖ | 57 |
| Carbon NPs | BSA | 192 | −6477 | N/A | Ø | * | None | ⊖ | 46 |
| Carbon NPs | HSA | 207 | −28024 |
N/A | Ø | * | None | ⊖ | 46 |
| Chitosan (cholesterol modified) | BSA | N/A | −46.1 ± 3.3 | −50 | Ø | * | None | ⊖ | 58 |
| Fe3O4 | BSA | 29.8 | −58.4 | N/A | ⊖ | **** | PEG and oleylamine | ⊖ | 59 |
| Fe3O4 | IgG | 26.1 | −50.2 | N/A | ⊖ | **** | PEG and oleylamine | Ø | 59 |
| CuO | β-Galactosidase | 3.7 ± 0.5 | −67 ± 5.0 | N/A | ⊖ | **** | None | ⊖ | 60 |
| ZnO | ToxR protein | 9 ± 3 | −41.0 ± 3.3 | −21.6 | ⊕ | * | None | ⊖ | 61 |
| ZnO | BSA | 0.26 ± 0.06 | −18.0 ± 2.9 | 25 | ⊕ | * | None | ⊖ | 62 |
| ZnO (polyethyleneimine-functionalized) | BSA | 0.79 ± 0.3 | −26.8 ± 6.7 | 3.14 | ⊕⊕ | * | None | ⊖ | 62 |
It can be seen that most NP–protein interactions result from enthalpy-driven adsorption processes with a loss of entropy (ΔH < 0 & ΔS < 0) corresponding to a general predominance of van der Waals interactions, electrostatics and hydrogen bond formation.21 This is present for example in interactions between human serum albumin (HSA) and N-iso-propylacrylamide/N-tert-butylacrylamide copolymer nanoparticles as reported by Lindman et al. in 2007.38 Similarly, interactions of polymeric polystyrene nanoparticles or hydroxyethyl starch nanocapsules with HSA or different apolipoproteins were dominantly enthalpy-driven under the loss of entropy as reported by Winzen et al.11,54 and Müller et al.53 Notably, many examples can be found where NPs and proteins are of similar charge which also lead to an exothermic interaction that is dominated by van der Waals interactions, electrostatics and hydrogen bond formation, yet appears in weaker forms as nicely seen in the study of Lindman et al. mentioned before.38
In the case of oppositely charged NP and protein, the interactions generally result in an increased entropy gain with relatively high association constants and an endothermic process (ΔH > 0 & ΔS > 0) due to stronger binding and promoted desolvation. As an example, interactions between negatively charged, amino acid-functionalized gold nanoparticles and the positively charged histone and cytochrome c show an entropy-driven adsorption process as reported by De et al.40 However, entropy-driven adsorption processes may also occur in the case of similarly charged NPs and proteins, as reported by Henzler et al. for adsorption of β-lactoglobulin onto spherical polyelectrolyte brushes.33 More highly (positively or negatively) charged NPs generally interact stronger with proteins with higher values for Ka, more negative ΔH and more positive ΔS. This is most likely a result of stronger electrostatic interactions between the charged NP surfaces and oppositely charged protein patches and the resulting desolvation as discussed above. Furthermore, the more hydrophobic the NP system is, the higher is the entropy gain for the same protein, presumably due to higher involvement of hydrophobic interactions in the adsorption process. In many cases, this entropy gain is accompanied additionally by an enthalpy gain, which can also be a result of van der Waals interactions occurring between hydrophobic protein residues and hydrophobic NP surfaces.
During adsorption events, denaturation of proteins may occur, leading to an entropy gain that is accompanied by an enthalpy loss (ΔH < 0 & ΔS > 0). As an example, such constellations were reported by Chakraborti et al.62 for the adsorption of negatively charged bovine serum albumin (BSA) at physiological pH onto (positively) charged zinc oxide nanoparticles. In another example, the adsorption of the protein apolipoprotein A-I (apo A-I) onto hydroxyethyl starch (HES) nanocapsules depends on the surface functionality of the nanomaterial – especially comparing unfunctionalized or carboxy-functionalized with amino-functionalized nanocapsules.11 An often-disregarded factor in the interpretation and analysis trends in NP–protein interactions is the influence of surfactants on the properties of the NPs and therefore the whole adsorption process. This becomes apparent regarding the change in net change and hydrophilicity of NPs with the resulting difference in adsorption observed in multiple studies. HSA showed a significantly higher affinity to PS-NPs stabilized by the more hydrophilic surfactant SDS compared to the more hydrophobic PEG-based Lutensol in a study by Winzen et al.54 In the study by Chakraborti et al.62 discussed above, zinc oxide NPs which were strongly positively charged due to non-covalent polyethyleneimine functionalization interacted with a higher Ka and more enthalpy-driven (more negative ΔH and lower, yet still positive ΔS) than similar NPs lacking the surface functionalization.
Notably, the association constants of different proteins in interactions with different nanoparticle surfaces differ by 3 orders of magnitude. This may be explained by the varying affinities of protein moieties to NPs of different properties and compositions. However, ΔH and ΔS also differ by several orders of magnitudes. This does not seem plausible in all cases and points to limitations of the method, which will be further discussed in chapter 5.
In conclusion, in order to see trends in surface properties of NPs with the interaction mechanisms they undergo with proteins, one must consider the complete surface composition including all of its components instead of solely focusing on the NP-material. Based on the summary of ITC studies on the protein corona discussed above, relations between the properties of NPs and the interactions they undergo with proteins can be concluded. More hydrophobic surfaces of NPs generally result in a higher proportion of hydrophobic interactions with proteins observed by stronger binding and a promotion of desolvation at the NP–protein interface. On the other hand, more hydrophilic surfaces promote van der Waals forces and hydrogen bond formation resulting in enthalpy driven (exothermic) adsorption processes with a loss of entropy. More highly charged (positively or negatively) surfaces lead to stronger interactions compared to surfaces of neutral charge, which is observed by a more positive ΔS due to the release of hydration water from the NP–protein interface. As unspecific NP–protein interactions are desired to be minimal for engineering the protein corona, we deduce that NPs formed of a hydrophilic material with neutral charge and steric (instead of electrostatic) stabilization experience the weakest interactions with proteins. This brief excerpt of the thermodynamic complexity of NP–protein interactions depicts the necessity to take information from other methods into account during analysis of ITC studies on the protein corona (for example on the hydrophilicity or surface charge of NPs) in order to make full use of the technique's potential for the field.
5. Limitations of ITC
While the information obtained from ITC studies on the protein corona nicely complement other methods, ITC also relies on other methods to overcome its own limitations which are discussed in the following. A comprehensive review containing information on the different methods and obtained parameters concerning the protein corona can be found in the literature.5Analyzing ITC data concerning the protein corona of NPs is particularly complex,22–24 as multiple pitfalls arise concerning the essentially required molar concentrations of both, NP dispersion and protein solution. ITC studies of single protein solutions are easily feasible, as calculating the molar concentration of single protein solutions is unproblematic (presupposing knowledge on the molar mass of the respective protein). More complex protein systems (especially in the case of full blood or blood plasma) are more difficult to characterize by their molar protein concentrations. Determining the molar concentration of NPs by their dispersion's solid content is not trivial, as monodispersity and perfectly spherical particles are an often-unavoidable assumption. This gets even more complicated for hollow particles (nanocapsules or self-assembled systems such as polymersomes/liposomes). Furthermore, many ITC instruments rely on relatively high analyte concentrations in order to detect significant heat changes during titration, which is hard to realize for more exotic (and expensive) proteins.
Fits of the adsorption isotherm in all cases require information on the molar concentration of both interacting components. Usually, ITC studies concerning NP–protein interactions apply the so called ‘independent binding model’.23,25 This mathematical model assumes independent protein binding to the nanoparticle according to a standard Langmuir binding. This means that interacting or bound proteins do not influence the binding of other proteins, excluding possible cooperative effects. In reality, the situation might be different and protein–protein interactions also have to be taken into account. Furthermore, entirely reversible adsorption processes are assumed, which is not trivial considering possible structural changes of proteins if denaturation processes occur during adsorption, leading to changing binding affinities over the course of the experiment.63 Addressing the problems arising from the independent binding model, Ballauff et al. developed a binding model including possible affinity changes of proteins during the interactions of proteins with hydrogel NPs.36 Ideally, a new model for each ITC study on the protein corona should be developed, which would hardly be possible and would hinder comparability of different studies. Therefore, new standardized models would enhance the method's ability to address NP–protein interactions dramatically. The limitations of the available binding models are manifested in the binding parameters listed in Table 1. While all interactions of an ITC experiment add up to the overall heat, assuming independent binding between NPs and protein may yield plausible binding parameters in many cases. However, the lacking possibility to differentiate individual heat contributions makes it hard to pick the most appropriate binding model and leaves room for the possibility of misleading binding parameters resulting from false assumptions.
In most studies utilizing ITC, the thermodynamic parameters are calculated from eqn (2). However, this may not always be appropriate, for example in the case for oligomeric proteins, which change their oligomeric state during the interaction. In this case, the protein concentration will change in the course of an ITC experiment and multiple, linked equilibria are needed to describe the interaction. For example, Lin and Lucius studied the phenomenon of linked equilibria extensively in a study of Escherichia coli ClpA and ClpB – two proteins that reside in monomeric, dimeric, tetrameric and hexameric equilibria, which are thermodynamically linked to the binding equilibrium of nucleotides.64
Another pitfall for ITC studies concerning the protein corona is neglecting the possibility of interactions between proteins and molecules that stabilize the NPs (e.g. surfactants). It is known that similar NPs that are stabilized by different surfactants engage in strikingly different interactions with proteins.54 Similarly, the concentration of the surfactant molecules on the surface were shown to be important. Furthermore, the concentration of salts in the different media (protein solution and NP dispersion) should ideally be identical. Significant differences in salt concentrations may result in large heats during injections due to the dilution heat of the salt and resulting noise, which might make it impossible to analyze the recorded data.
6. Conclusion
Summarizing, isothermal titration calorimetry (ITC) delivers important information on nanoparticle–protein interactions by characterizing binding thermodynamics in situ. These thermodynamic information yield insights into the mechanism of all interactions nanoparticles undergo in biological media, which affect the system's surface characteristics and would hardly be obtainable otherwise. Based on the multitude of studies on the protein corona employing ITC, relations between the properties of nanoparticles and the interactions they undergo with proteins can be concluded. While more hydrophobic surfaces of nanoparticles result in a higher proportion of hydrophobic interactions with proteins, more hydrophilic surfaces promote van der Waals forces and hydrogen bond formation. More highly charged surfaces lead to stronger interactions compared to surfaces of neutral charge. As unspecific nanoparticle–protein interactions are desired to be minimal for engineering the protein corona, we deduce that nanoparticles formed of a hydrophilic material with neutral charge and steric stabilization experience the weakest interactions with proteins. The information obtained from ITC studies enhance our understanding of the protein corona formation mechanism as ITC employs and supports the information gained from other methods in a complementary fashion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the DFG/SFB1066 (Nanodimensionale polymere Therapeutika für die Tumortherapie) and thank S. Schuhmacher for graphical assistance. Open Access funding provided by the Max Planck Society.References
- W. Lin, Chem. Rev., 2015, 115, 10407–10409 CrossRef CAS PubMed.
- D. Docter, S. Strieth, D. Westmeier and O. Hayden, Nanomedicine, 2015, 10, 503–519 CrossRef CAS PubMed.
- C. Weber, J. Simon, V. Mailänder, S. Morsbach and K. Landfester, Acta Biomater., 2018, 76, 217–224 CrossRef CAS.
- P. Draczkowski, D. Matosiuk and K. Jozwiak, J. Pharm. Biomed. Anal., 2014, 87, 313–325 CrossRef CAS.
- S. Morsbach, G. Gonella, V. Mailänder, S. Wegner, S. Wu, T. Weidner, R. Berger, K. Koynov, D. Vollmer, N. Encinas, S. L. Kuan, T. Bereau, K. Kremer, T. Weil, M. Bonn, H.-J. Butt and K. Landfester, Angew. Chem., Int. Ed., 2018, 57, 12626–12648 CrossRef CAS PubMed.
- E. Vogler, Biomaterials, 2012, 33, 1201–1237 CrossRef CAS PubMed.
- L. Treuel and G. Nienhaus, Biophys. Rev., 2012, 4, 137–147 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS.
- L. Vroman, Nature, 1962, 196, 476–477 CrossRef CAS PubMed.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailänder, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992–3001 RSC.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- J. Müller, K. N. Bauer, D. Prozeller, J. Simon, V. Mailänder, F. R. Wurm, S. Winzen and K. Landfester, Biomaterials, 2017, 115, 1–8 CrossRef.
- J. Simon, T. Wolf, K. Klein, K. Landfester, F. R. Wurm and V. Mailänder, Angew. Chem., Int. Ed., 2018, 57, 5548–5553 CrossRef CAS PubMed.
- E. A. Vogler, in Wettability, ed. J. Berg, Marcel Dekker, New York, 1993, pp. 184–250 Search PubMed.
- J. Müller, D. Prozeller, A. Ghazaryan, M. Kokkinopoulou, V. Mailänder, S. Morsbach and K. Landfester, Acta Biomater., 2018, 71, 420–431 CrossRef PubMed.
- A. Krishnan, J. Sturgeon, C. A. Siedlecki and E. A. Vogler, J. Biomed. Mater. Res., 2004, 68A, 544–557 CrossRef CAS.
- P. M. Claesson, E. Blomberg, J. C. Fröberg, T. Nylander and T. Arnebrant, Adv. Colloid Interface Sci., 1995, 57, 161–227 CrossRef CAS.
- T. A. Horbett, in Biomaterials: Interfacial Phenomena and Applications, American Chemical Society, 1982, vol. 199, ch. 17, pp. 233–244 Search PubMed.
- D. Prozeller, J. Pereira, J. Simon, V. Mailänder, S. Morsbach and K. Landfester, Adv. Sci., 2019, 6, 1802199 CrossRef.
- R. Huang and B. L. T. Lau, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 945–956 CrossRef CAS PubMed.
- M. W. Freyer and E. A. Lewis, in Methods in Cell Biology, Academic Press, 2008, vol. 84, pp. 79–113 Search PubMed.
-
E. A. Lewis and K. P. Murphy, in Protein-Ligand Interactions: Methods and Applications, ed. G. Ulrich Nienhaus, Humana Press, Totowa, NJ, 2005, pp. 1–15, DOI:10.1385/1-59259-912-5:001.
- A. Velázquez-Campoy, H. Ohtaka, A. Nezami, S. Muzammil and E. Freire, Curr. Protoc. Cell Biol., 2004, 23, 17.18.11–17.18.24 Search PubMed.
- E. Freire, O. L. Mayorga and M. Straume, Anal. Chem., 1990, 62, 950A–959A CrossRef CAS.
- M. Kabiri and L. D. Unsworth, Biomacromolecules, 2014, 15, 3463–3473 CrossRef CAS PubMed.
- J. J. Christensen, R. M. Izatt, L. D. Hansen and J. A. Partridge, J. Phys. Chem., 1966, 70, 2003–2010 CrossRef CAS.
- M. J. Cliff and J. E. Ladbury, J. Mol. Recognit., 2003, 16, 383–391 CrossRef CAS PubMed.
- R. O'Brien and I. Haq, in Biocalorimetry 2, pp. 1–34, DOI:10.1002/0470011122.ch1.
- C. Spink and I. Wadso, Methods Biochem. Anal., 1976, 23, 1–159 CAS.
- K. Chiad, S. H. Stelzig, R. Gropeanu, T. Weil, M. Klapper and K. Müllen, Macromolecules, 2009, 42, 7545–7552 CrossRef CAS.
- P. W. Atkins and J. de Paula, Physikalische Chemie, Wiley-VCH, Weinheim, 2006 Search PubMed.
- K. Henzler, B. Haupt, K. Lauterbach, A. Wittemann, O. Borisov and M. Ballauff, J. Am. Chem. Soc., 2010, 132, 3159–3163 CrossRef CAS PubMed.
- A. L. Becker, N. Welsch, C. Schneider and M. Ballauff, Biomacromolecules, 2011, 12, 3936–3944 CrossRef CAS PubMed.
- N. Welsch, A. L. Becker, J. Dzubiella and M. Ballauff, Soft Matter, 2012, 8, 1428–1436 RSC.
- C. Yigit, N. Welsch, M. Ballauff and J. Dzubiella, Langmuir, 2012, 28, 14373–14385 CrossRef CAS PubMed.
- O. Vilanova, J. J. Mittag, P. M. Kelly, S. Milani, K. A. Dawson, J. O. Rädler and G. Franzese, ACS Nano, 2016, 10, 10842–10850 CrossRef CAS PubMed.
- S. Lindman, I. Lynch, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Nano Lett., 2007, 7, 914–920 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050 CrossRef CAS PubMed.
- M. De, C.-C. You, S. Srivastava and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 10747–10753 CrossRef CAS PubMed.
- K. Chen, Y. Xu, S. Rana, O. R. Miranda, P. L. Dubin, V. M. Rotello, L. Sun and X. Guo, Biomacromolecules, 2011, 12, 2552–2561 CrossRef CAS PubMed.
- J. C. D. Houtman, P. H. Brown, B. Bowden, H. Yamaguchi, E. Appella, L. E. Samelson and P. Schuck, Protein Sci., 2007, 16, 30–42 CrossRef CAS PubMed.
- N. Welsch, Y. Lu, J. Dzubiella and M. Ballauff, Polymer, 2013, 54, 2835–2849 CrossRef CAS.
- K. Rajarathnam and J. Rösgen, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 69–77 CrossRef CAS PubMed.
- M. Pilloni, J. Nicolas, V. Marsaud, K. Bouchemal, F. Frongia, A. Scano, G. Ennas and C. Dubernet, Int. J. Pharm., 2010, 401, 103–112 CrossRef CAS PubMed.
- S. Mandal, M. Hossain, P. S. Devi, G. S. Kumar and K. Chaudhuri, J. Hazard. Mater., 2013, 248–249, 238–245 CrossRef CAS PubMed.
- S. Chakraborty, P. Joshi, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2011, 27, 7722–7731 CrossRef CAS PubMed.
- E. Omanovic-Miklicanin, I. Manfield and T. Wilkins, J. Therm. Anal. Calorim., 2017, 127, 605–613 CrossRef CAS.
- K. Bouchemal, Drug Discovery Today, 2008, 13, 960–972 CrossRef CAS PubMed.
- B. A. Pethica, Anal. Biochem., 2015, 472, 21–29 CrossRef CAS PubMed.
- J.-P. E. Grolier and J. M. del Río, J. Chem. Thermodyn., 2012, 55, 193–202 CrossRef CAS.
- J. C. D. Houtman, P. H. Brown, B. Bowden, H. Yamaguchi, E. Appella, L. E. Samelson and P. Schuck, Protein Sci., 2007, 16, 30–42 CrossRef CAS.
- J. Müller, J. Simon, P. Rohne, C. Koch-Brandt, V. Mailänder, S. Morsbach and K. Landfester, Biomacromolecules, 2018, 19, 2657–2664 CrossRef.
- S. Winzen, J. C. Schwabacher, J. Müller, K. Landfester and K. Mohr, Biomacromolecules, 2016, 17, 3845–3851 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, F. Zhang and S. Yu, Nanoscale, 2017, 9, 4787–4792 RSC.
- X. Wang, E. Matei, A. M. Gronenborn, O. Ramström and M. Yan, Anal. Chem., 2012, 84, 4248–4252 CrossRef CAS PubMed.
- M. Reynolds, M. Marradi, A. Imberty, S. Penadés and S. Pérez, Chem. – Eur. J., 2012, 18, 4264–4273 CrossRef CAS PubMed.
- X. Li, M. Chen, W. Yang, Z. Zhou, L. Liu and Q. Zhang, Colloids Surf., B, 2012, 92, 136–141 CrossRef CAS.
- S. Liu, Y. Han, R. Qiao, J. Zeng, Q. Jia, Y. Wang and M. Gao, J. Phys. Chem. C, 2010, 114, 21270–21276 CrossRef CAS.
- G. Rabbani, M. J. Khan, A. Ahmad, M. Y. Maskat and R. H. Khan, Colloids Surf., B, 2014, 123, 96–105 CrossRef CAS PubMed.
- T. Chatterjee, S. Chakraborti, P. Joshi, S. P. Singh, V. Gupta and P. Chakrabarti, FEBS J., 2010, 277, 4184–4194 CrossRef CAS PubMed.
- S. Chakraborti, P. Joshi, D. Chakravarty, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2012, 28, 11142–11152 CrossRef CAS PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- J. Lin and A. L. Lucius, in Methods in Enzymology, ed. J. L. Cole, Academic Press, 2015, vol. 562, pp. 161–186 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |