
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe molecular mechanisms underlying mussel adhesion
Yiran
Li
 ab and
Yi
Cao
*abc
ab and
Yi
Cao
*abc
aShenzhen Research Institute of Nanjing University, Shenzhen, 518057, China. E-mail: Caoyi@nju.edu.cn
bDepartment of Physics, Collaborative Innovation Center of Advanced Microstructures, National Laboratory of Soli State Microstructure, Nanjing University, Nanjing, 210093, China
cChemistry and Biomedicine Innovation Center, Nanjing University, Nanjing, 210093, China
First published on 10th October 2019
Abstract
Marine mussels are able to firmly affix on various wet surfaces by the overproduction of special mussel foot proteins (mfps). Abundant fundamental studies have been conducted to understand the molecular basis of mussel adhesion, where the catecholic amino acid, L-3,4-dihydroxyphenylalanine (DOPA) has been found to play the major role. These studies continue to inspire the engineering of novel adhesives and coatings with improved underwater performances. Despite the fact that the recent advances of adhesives and coatings inspired by mussel adhesive proteins have been intensively reviewed in literature, the fundamental biochemical and biophysical studies on the origin of the strong and versatile wet adhesion have not been fully covered. In this review, we show how the force measurements at the molecular level by surface force apparatus (SFA) and single molecule atomic force microscopy (AFM) can be used to reveal the direct link between DOPA and the wet adhesion strength of mussel proteins. We highlight a few important technical details that are critical to the successful experimental design. We also summarize many new insights going beyond DOPA adhesion, such as the surface environment and protein sequence dependent synergistic and cooperative binding. We also provide a perspective on a few uncharted but outstanding questions for future studies. A comprehensive understanding on mussel adhesion will be beneficial to the design of novel synthetic wet adhesives for various biomedical applications.
 Yiran Li | Yiran Li received his Ph.D. degree from the Physics department of Nanjing University. He joined the University of California, Berkeley as a postdoctoral fellow and then the Nanjing University as a research associate. His research focuses on AFM based single-molecule force spectroscopy and nanoscale mechanical measurement. |
 Yi Cao | Yi Cao received his bachelor's degree in 2001 and Master's degree in 2004 from Nanjing University. He then obtained his Ph.D. in 2009 from the University of British Columbia. He started his independent career at the Department of Physics, Nanjing University as a full professor in 2010. He was the recipient of the 2014 IUPAP Young Scientist Prize in Biological Physics and the 2018 Young Innovator Award by Nano Research. His current research combines single molecule force spectroscopy with synthetic biology and polymer chemistry to address fundamental questions as well as applied challenges in soft materials. |
Introduction
Bioinspired design has enjoyed great success in materials science for the production of novel materials with functions similar or even superior to naturally occurring ones.1–3 The heart of bioinspired design is to understand the structure–function relationship of natural biomaterials.4–6 In the past few decades, considerable efforts have been devoted to the fabrication of mussel-inspired underwater adhesives and surface coating technologies,7–10 owing to the great advances in the understanding of mussel adhesion mechanism. Marine mussels produce a bundle of byssus to firmly fix themselves on a wet rocky seashore to avoid being taken by fierce sea waves. Biochemical studies have revealed that the mussel secretes special proteins to form the major adhesion apparatus, byssus.11 These mussel foot proteins, particularly the ones at the very tip of the byssus typically contain a high percentage of a post-translationally modified amino acid, L-3,4-dihydroxyphenylalanine (DOPA).11 Biophysical characterizations by surface force apparatus (SFA) and atomic force microscopy (AFM)-based single-molecule force spectroscopy (SMFS) has uncovered the direct correlation of the adhesion strength with the contents of DOPA.12–14 Moreover, it was discovered that DOPA can adaptively bind to different surfaces using different binding modes.13,15 The surface chemical properties can greatly modulate the DOPA adhesion.16 It was also revealed that the proximate lysine residues can promote DOPA binding in a sequence dependent manner.17,18 In this perspective, we have first summarized the sequence and biochemical properties of different mussel foot proteins. Then, we have briefly introduced the SFA studies of mussel foot proteins and the synthetic mimetics. Next, we have mainly focused on the AFM-based single molecule force spectroscopy studies of DOPA adhesion, from the basic experimental design to data interpretation and the major scientific discoveries. Furthermore, we have also introduced the recent applications of SMFS in the study of DOPA–Fe3+ coordination bonds and the real-time polydopamine polymerization. Finally, we have provided our perspective on the potential challenges and opportunities in SMFS studies of DOPA adhesion.Biochemical properties of mussel foot proteins
Mussel byssus consists of a bundle of collagenous threads and a flattened adhesive plaque at the end (Fig. 1). The threads can be extended to more than 30% and dissipate a considerable mechanical energy upon stretching through the rupture of reversible metal chelation bonds (i.e. DOPA–Fe3+ and histidine–Zn2+ bonds) and function as shock-absorbers.11,19 The plaques provide strong adhesion strength to various wet substrates. A set of sticky and highly basic proteins were extracted and purified from the phenol gland of mussel byssus.20–22 Further analysis showed that this protein contains large amounts of DOPA, lysine and 3- and 4-hydroxyproline, and is considered to be responsible for the wet adhesion. Later, at least 15 different proteins were characterized from mussel byssus. Eight of these proteins are only present in the adhesive plaques and the rest are also found in other parts of the byssus.20,22,23 For example, collagens dominate the fibrous core of each thread and extend to the plaque. Mfp-1, a major constituent of the protective cuticle, was found covering all the exposed byssus, including the plaque.12,22 Mfp-2 is a cysteine-rich structural element of the plaque matrix, which is in the shape of a structural foam.21,24 Mfp-3 and mfp-5 are found in plaques with the lowest mass and the highest DOPA contents among all the mussel foot proteins.12,23 Mfp-6 is also located at the plaque but surprisingly contains low levels of DOPA and high levels of cysteine.25 It forms cysteinyl-DOPA cross-links with other proteins to stabilize the structure of the plaque and maintains a reducing environment to prevent the oxidation of DOPA.26,27 Mfp-4 is found in the plaque–thread junction and mediates the contact between the fibrous collagen and the foam protein. It has a mass of 93 kDa and has an abundance of histidine, lysine, arginine and aspartate.25 Given that all these proteins contain considerable amounts of DOPA, and the DOPA levels are the highest in the proteins at the adhesive plaque, it is natural to attribute the adhesion properties to DOPA. It was proposed that DOPA can form bidentate H-bonding,28–30 metal coordination14,28 or covalent oxidative cross-linking,31 hydrophobic, π–π and cation–π interactions with different surfaces.13,32 Later on, the hypothesis was confirmed by various biophysical studies, including SFA and AFM.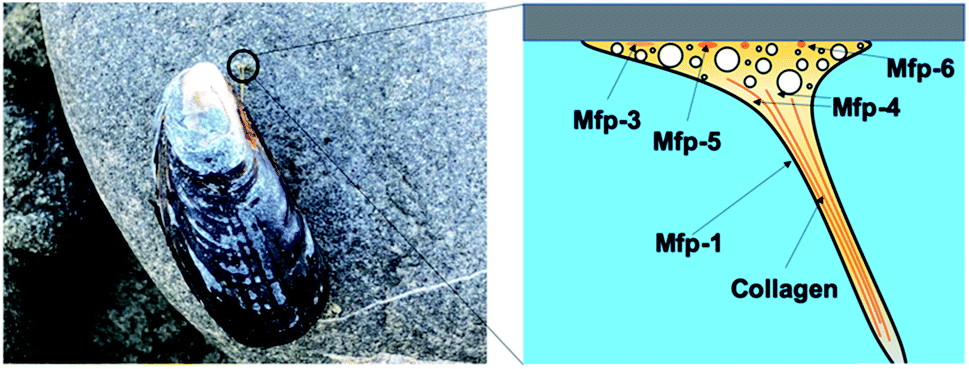 | ||
| Fig. 1 Mussel adhesion and schematic structure of mussel byssus. | ||
The mechanical properties of mfp adhesion can be measured by different techniques, including optical tweezers, bio-membrane force probe, AFM-based SMFS, and bulk tensile test. The ranges of loading rates and forces of these methods are summarized in Fig. 2. As SFA and AFM-based SMFS are two major methods used to study the binding mechanism of mfps, the technical details and the major findings by these methods are summarized below.
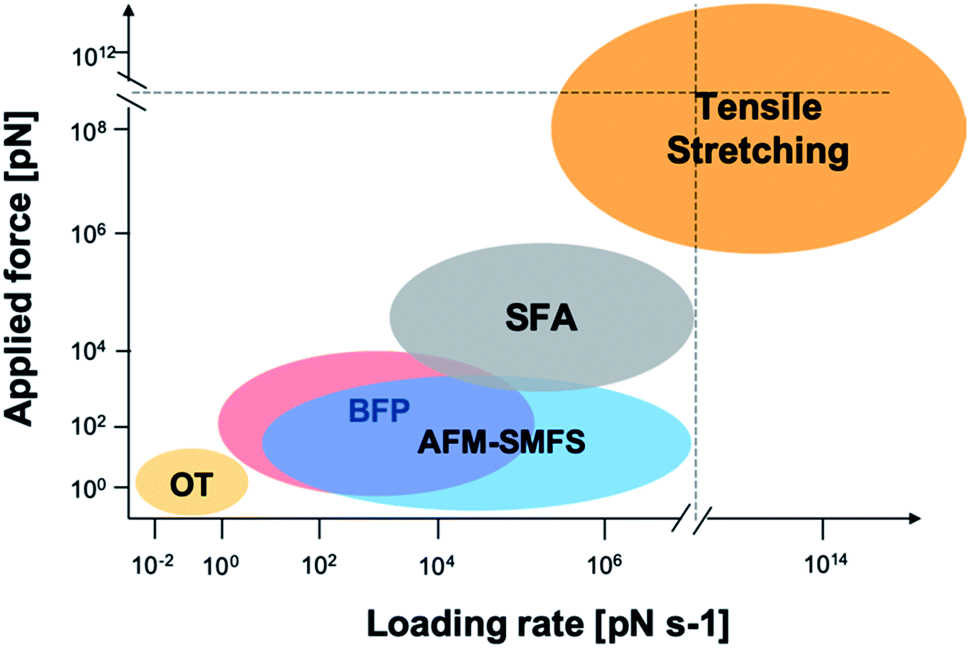 | ||
| Fig. 2 Force and loading rate ranges of different force measurement approaches. OT represents optical tweezers and its range is estimated from ref. 42–44. Bio-membrane force probe (BFP) is estimated from ref. 45. AFM-based SMFS data are from ref. 46–48 and are estimated by general AFM technical details and typical cantilever mechanical properties. SFA data are from ref. 33, 49 and 50. Tensile stretching data are estimated from ref. 47 and 51–54. | ||
SFA studies
SFA is a precise and powerful tool to measure the interactions between two surfaces. It uses multiple beam interferometry to monitor surface separation and the deformation of the contact area. Owing to the sensitive elements, the device can resolve to within 0.1 nm in the z direction and a force to 10−8 N.33 One of the two surfaces of SFA is held by the cantilevered spring, and it measures the normal forces. The other is linked to a piezoelectric positioner to accurately control the separation of the two surfaces. Usually, in the SFA measurement, two orthogonal cylinders coated with atomically smoothed mica are made to approach each other in a direction normal to the axes. The samples are placed between two cylinders. In a typical experiment, a large compressive load force (∼1–10 MPa) is applied to make the formation of sample-surface interaction. Then, the two surfaces are separated, and the separation force is calculated by a spring system based on Hooke's law (Fig. 3a). SFA can measure varieties of interactions, such as van der Waals forces, electrostatic forces and even hydrogen bonding.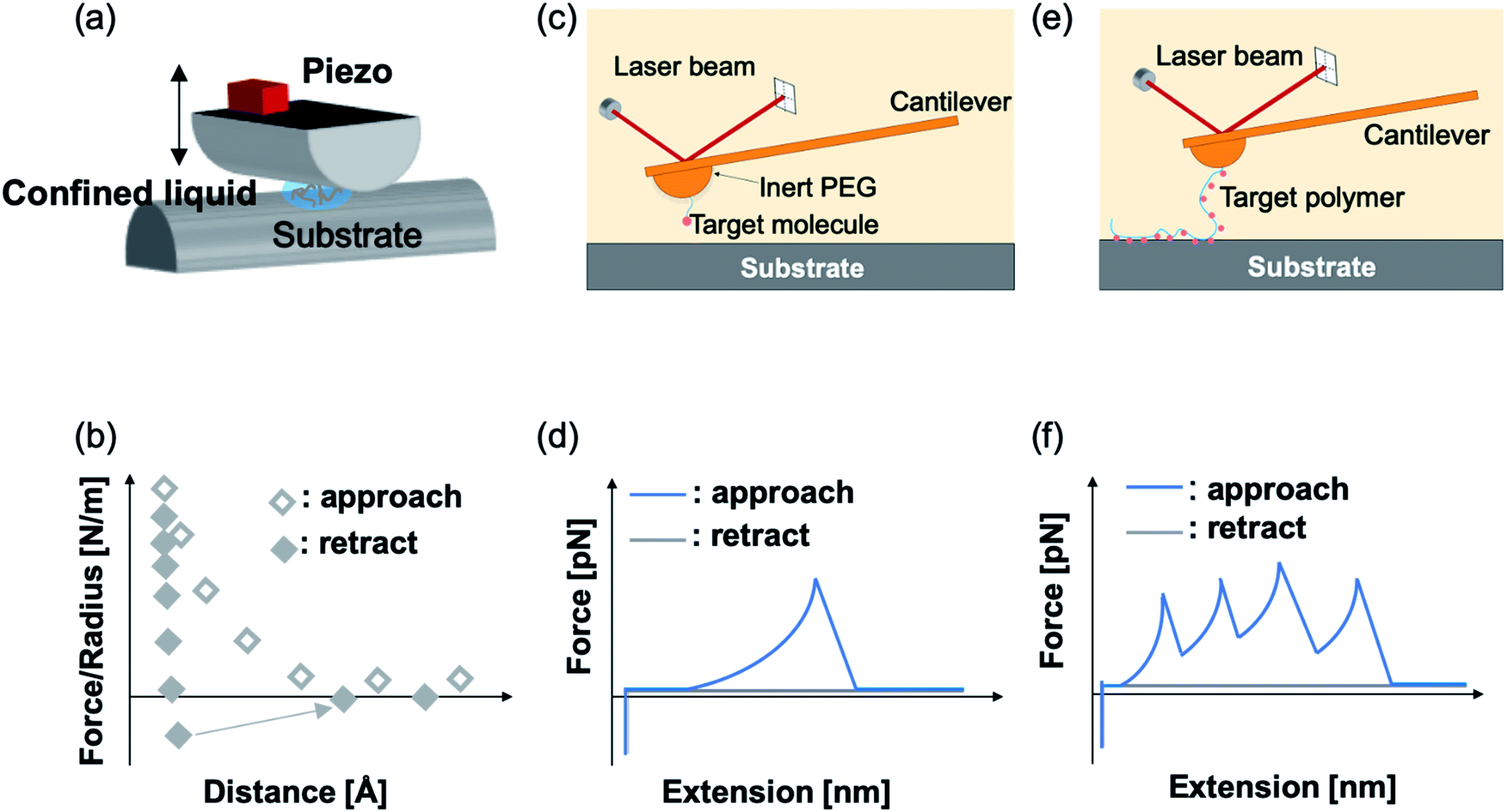 | ||
| Fig. 3 Illustrative scheme of SFA (a) and two types of SMFS (c) and (e) measurements. (b) is the representative force–distance relationship of SFA measurement, (d) is the representative force–extension curve of single-ligand recognition and (f) is the polymer-based fishing measurement. | ||
Using SFA, Jacob N. Israelachvili et al. have greatly advanced our understanding of mussel adhesion in the past decades. They have directly quantified the surface binding strength of various mfps with diverse DOPA contents under different environmental conditions. It was found that mfp-3 and mfp-5, the two major adhesive proteins at the plaque, could achieve ∼3 × 10−4 and ∼1.4 × 10−3 J m−2 adhesion energies to the mica surfaces, respectively.12,15 The adhesion strength of mfp-5 is even higher than that of a highly oriented biotin and streptavidin monolayer.34 Their adhesion energies are directly correlated with their DOPA contents. Interestingly, despite the fact that mfp-1 has a similar DOPA content as mfp-3, its adhesion performance is distinct.12 No adhesion was ever observed for mfp-1 coated single layers in SFA experiments presumably due to the absence of long-range bridging forces. However, a subsequent shearing-separation test observed a finite adhesion between mica and mfp-1. A possible explanation is that most of the binding sites of the mfp-1 were initially attached to a one surface only and were rearranged to the opposite surface upon shearing.12 This case indicated that in a SFA measurement, successful adhesion on both surfaces is the prerequisite for detecting interactions. Therefore, both adhesion between surfaces and the cohesion among the proteins are important for the strong surface adhesion energies measured in SFA.
The effect of the oxidation of DOPA on the adhesion properties of mfps was studied. Oxidation could abolish many important types of interactions between DOPA and the surfaces (e.g. hydrogen bonding and metal chelation). Oxidizing DOPA to dopaquinone by periodate led to the abolition of the adhesion between both asymmetrically and symmetrically deposited mfp-5 films.35 Mussels have evolved an intriguing mechanism to limit DOPA oxidation by secreting an acidic and thiol-rich foot protein, mfp-6, which can revert the oxidized dopaquinone to DOPA using thiol as the reductant.36 Furthermore, the oxidation of DOPA can also promote cohesion if the oxidized surface could bind strongly to the unoxidized surface due to the strong hydrogen bonding between DOPA and dopaquinone.37 Moreover, as oxidized DOPA can crosslink with thiol or amino group via Michael addition,38 some reports also utilized the periodate treatment to enhance DOPA adhesion or cohesion.39–41
The interactions among the adhesive mfp family (mfp-1, mfp-3 and mfp-5) and other surfaces with different chemical properties have also been studied by SFA.15 The results showed that all three mfps could adhere to the four tested substrates (mica, TiO2, silica and polystyrene), yet the adhesion strength was exquisitely dependent on the mfp species, the substrate surface chemistry and the contact time. It was proposed that several different binding mechanisms are involved in the binding, including electrostatic, hydrogen bonding, hydrophobic interactions, cation–π interactions and π–π stacking.15
The contribution of other residues in mfps to the adhesion energies was also explored. Using cyclic model compounds as an example, Butler, Israelachvili, and Waite et al. showed that lysine and DOPA can synergistically enhance the binding strength.18,55 The synergy was interpreted as that the positively charged amine could help break the hydrated salt layer on the mineral surface and serve as vanguards to keep a stable bidentate catechol–surface interaction.18 It remains elusive whether other mechanisms may also contribute to their cooperative surface binding. Moreover, lysine and DOPA interactions can also enhance the intermolecular cohesion of mfp mimicking peptides. Based on the solid state NMR spectroscopy characterization, cation–π interaction was clearly observed.32,56 However, it was quite surprising to discover that the peptides containing lysine–phenylalanine showed a much stronger adhesion energy than those containing lysine–DOPA. It is plausible that lysine–phenylalanine has stronger cation–π interactions. However, it cannot be fully excluded that the formation of certain molecular structures of the peptides may also significantly enhance the cohesion strength. It was noticed that the peptides being studied contained a diphenylalanine motif, which showed a strong self-assembling tendency.57,58
Although SFA has been widely used to measure the adhesion of mfps to different surfaces, we should be aware that these measurements are done at the ensemble level. The adhesion energy does not necessarily correlate with the surface binding strength of individual mfps as the cohesive interactions of proteins and the cooperativity of different molecules also play significant roles. Moreover, it is difficult to prepare a single protein layer for the SFA test in real experiments, even though most SFA measurements declared that single layer adhesives were tested. Besides, adhesion strength is not only determined by the individual bond strength but also by the bond density, which is difficult to control. In general, the bond density can vary dramatically when different proteins or synthetic adhesives are used. Recent studies showed that the distribution of proteins on the two surfaces can greatly affect the SFA observation.35
Single-molecule force spectroscopy experiments by AFM
AFM-based SMFS has been widely used to study the mechanical strength of chemical bonds, ligand–receptor interactions, protein–protein interactions and adhesive molecule–surface interactions.59–74 AFM measurement is complementary to SFA measurement and can directly quantify the strength of single adhesion bonds with an extremely high resolution of 10−9 N on force and 10−10 m on separation. Representative interaction strengths measured by SMFS are summarized in Table 1.| Interaction type | Bond carrier | Bond strength [pN] (at loading rate [nN s−1]) | Dissociation rate [s−1] | Distance to transition state [nm] | Ref. |
|---|---|---|---|---|---|
| Hydrogen bonding | DOPA–Ti [aqueous] | 77 (50) | 20.02 | 0.13 | 13 |
| UPy tandem [toluene] | ∼120 (30) | 0.88 | 0.20 | 86 | |
| P(BTA) stacking [mesitylene] | ∼70 (6.7) | 0.30 | 0.45 | 87 | |
| Hydrophobic interaction | DOPA–PS [aqueous] | 78 (50) | 14.94 | 0.17 | 13 |
| Hexadecane–hexadecane [aqueous] | ∼75 (5.0) | 1.1 | 0.24 | 88 | |
| Unravel PS [aqueous] | ∼70 (—) | — | — | 89 | |
| Unravel PS [aqueous] | ∼80 (—) | 90 | |||
| Hydrophobin | ∼110 (20) | 0.59 | 0.35 | 91 | |
| Covalent bond | SiOx–amylose–SiOx [aqueous] | 2000 (10) | — | — | 92 |
| Cycloreversion of cyclobutene [DMSO] | >1700 (100–500) | — | — | 93 | |
| Thiol-maleimide adduct | 900 (30) | 3.5 × 10−4 (open ring) | 0.05 | 94 | |
| Thioester [in protein] | ∼100 (6) | — | — | 95 | |
| Au–S | 600–1000 (30) | — | — | 112 | |
| Charge interaction | Histidine–histidine | 115 (16) | 0.12 (pH7) | 0.23 (simulated fitting) | 96 |
| Polyanion and positively charged surface [aqueous] | 120 (60) | — | — | 97 | |
| Polyvinylamine–SiO2 [aqueous] | 45–81 (—) | — | — | 98 | |
| Coordinate bonding | His tag/NTA–CO2+, Cu2+, Ni2+, Zn2+ [aqueous] | 22–58 (30) | — | — | 99 |
| Catechol–Fe3+ [aqueous] | 100–200 (50) | — | 0.14 (bis), 0.27 (tris) | 100 | |
| Fe2+–S bond [aqueous] | 146–242 (16) | 3 × 10−6 to 0.7 | 0.11–0.30 (pulling direction dependent) | 101 | |
| Au–S interaction | 500–2900 (30–50) | — | — | 92 and 102–104 | |
| Cu–S [in protein] | 147 (40) | — | — | 105 | |
| Zn–S [in protein] | 90–170 (—) | — | — | 106 and 107 |
Using AFM-based SMFS, the adhesion of mfps has been widely studied. In early AFM experiments, mfps were directly placed between the cantilever tip and the substrate, similar to the design of SFA experiments.75,76 Therefore, it remained difficult to identify the single molecule pulling events. Later on, experiments were carefully designed to minimize the nonspecific interactions and the elastic properties of the polymer were used as the fingerprint of single molecule pulling events. To minimize the interference from other amino acids, SMFS studies mainly focused on DOPA instead of the whole natural mfps. In general, there are two ways to unambiguously study the DOPA adhesion via SMFS (Fig. 3b and c). The first one is to covalently connect DOPA to the AFM cantilever via a bifunctional polymer linker and directly measure DOPA–surface interactions.77–82 In this strategy, inert polyethylene glycol (PEG) is usually used as the linker to minimize nonspecific interactions between the cantilever and the substrates. The other method is to incorporate DOPA into a polymer. By stretching the surface-immobilized polymer chain, DOPA–surface interactions are ruptured one by one. Both methods are widely used and each has its own advantages and disadvantages. The former one allows the direct observation of DOPA–surface interactions without the interference from other components. However, it is not applicable if the interactions involve more than two ligands (e.g. the tris-complex formed between Fe3+ and DOPA). The latter approach is a high-volume method, which is akin to the unfolding of polyproteins.81,83–85 Since it relies on nonspecific interactions to pick up the polymers, it is important to make sure that the sawtooth-like peaks are indeed from the rupture of individual DOPA adhesion bonds. A common criterion is that the persistence length of the force–extension relationship of each rupture event is consistent and matches that of the polymer backbone. The last peak can either be the rupture of a surface adhesion bond or the detachment of the polymer from the cantilever tip, and therefore should be excluded in data analysis. The nonspecific adhesion of the polymer to the cantilever tip is not always high enough to ensure the rupture events in all force regions to be unbiasedly sampled. Nevertheless, with carefully designed experiments and data analysis procedures, this high-volume method can readily probe the rupture of single adhesion bonds. Another aspect that should be emphasized is that SMFS experiments are typically performed at a non-equilibrium condition. The rupture forces largely depend on the force loading rates (Fig. 4). The faster the loading rate is, the higher the rupture forces are. As such, it is not meaningful to compare the binding forces measured at different loading rates. The force–loading rate relationship can be understood by adding a force-dependent term to the Arrhenius model as first introduced by Bell and later by Evans.108–110 The force-dependent term is directly determined by the height of the activation barrier and the width of the potential at the pulling direction. Therefore, based on the force–loading rate relationships, one can extract the kinetic parameters underlying the free energy landscape of the bond dissociation. The measurement of rupture forces at different loading rates is typically named as dynamic force spectroscopy, which provides a unique way to understand the molecular mechanism of the adhesion bonds. In these experiments, the cantilevers were moved away from the substrates at different separation speeds. The force loading rates depend on both the separation speeds and the spring constants of the cantilevers, which can be experimentally determined from the force–time relationships. For some interactions, loading rate-independent rupture forces were also observed at the low loading rate region. This can be attributed to the high rebinding rates of the system and the binding and rebinding were in equilibrium.111
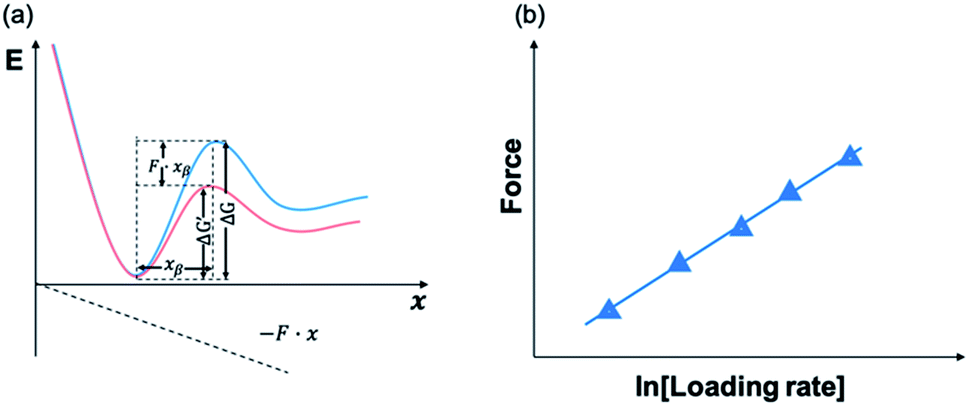 | ||
| Fig. 4 Schematic figures of energy landscape (a) and force–loading rate dependency (b). Applied force can tilt the energy landscape and lower the energy barrier. By fitting force–loading rate relationship, dissociation rate and distance to transition state can be extracted. | ||
In 2006, Messersmith et al. used AFM-based SMFS to study the interactions between individual DOPA and titanium surfaces, which shed new light on the DOPA adhesion mechanism.14 Without the interference from the other components in mfps, the contribution of DOPA to the wet adhesion was first studied. In this test, N-(tert-butoxycarbonyl)-DOPA (N-Boc-DOPA) was end-tethered to a functional PEG linker and the Boc group prevented potential interference from the N-terminal positive charge. Their results showed that the adhesion force is ∼800 pN for the DOPA–Ti surface in deionized water at a loading rate of 60.0 nN s−1, which is a strong noncovalent interaction (Fig. 5a and b). They also performed dynamic force spectroscopy and found that the rupture forces depend on the logarithm of loading rates. Then, they studied the effect of DOPA oxidation to the adhesion strength. At a basic pH of 8.3 or 9.7, they observed a bimodal distribution with a lower value peak located at ∼150 pN and a higher value at 800 pN. The lower value peak was attributed to the detachment of oxidized DOPA from the surface. They also analyzed the relationship between rupture forces and the experimental duration. The results showed that the events of large rupture force values occurred mainly in the first 30 min and after that, most of the rupture events were at low forces. They further studied the binding of oxidized DOPA to an amine-functionalized surface (mimicking the amino groups of organic surfaces). In this test, they observed extremely large rupture forces (∼2 nN), which were similar to the rupture forces of most covalent bonds. After a few pulling cycles, no force signal was observed, suggesting that the bond rupture was irreversible, which was in sharp contrast with DOPA–Ti interactions. The large rupture forces suggested that DOPA may form covalent bonds with the amino groups on the surface via Michael addition. Their results greatly enriched our understandings of DOPA adhesion at the molecular level.
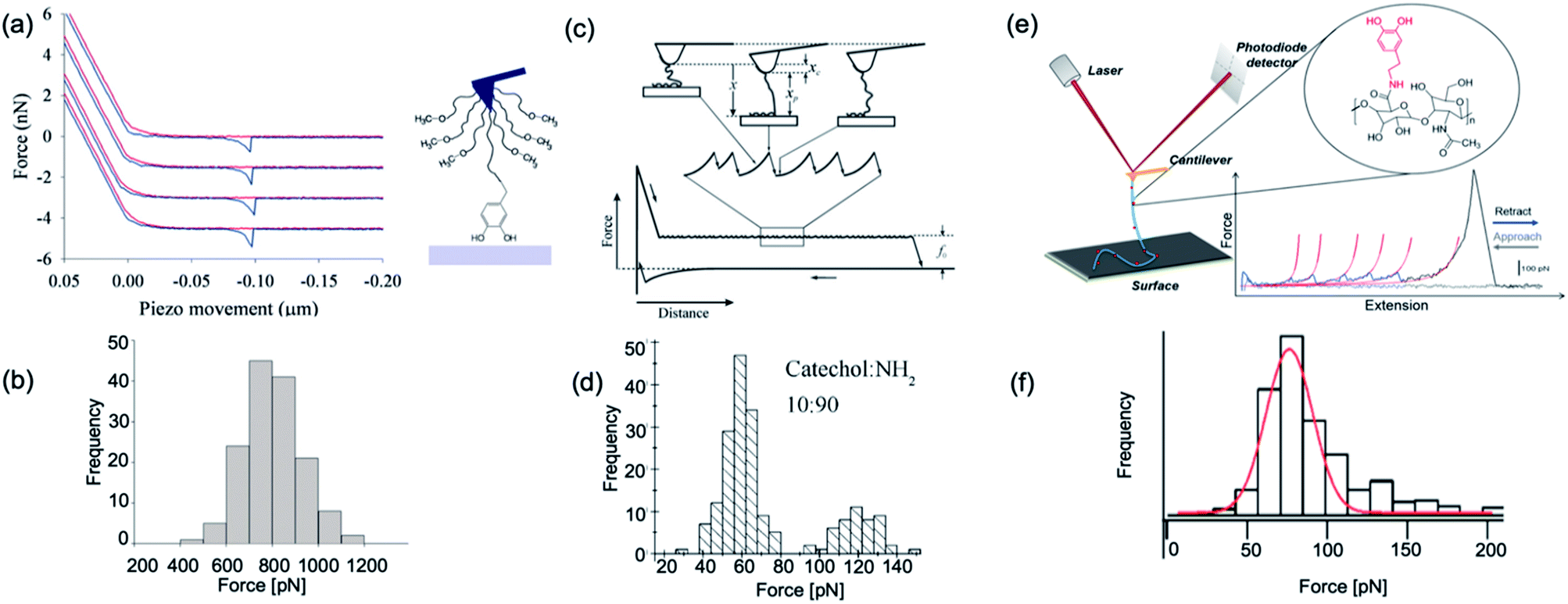 | ||
| Fig. 5 Representative force–extension curve and rupture force histogram from different DOPA surface interaction studies. (a and b) are reproduced from Lee et al.14 Copyright (2006) National Academy of Sciences, U.S.A. (c and d) are reprinted from Wang et al. Copyright (2008) Wiley. Used with permission from ref. 112 and (e and f) are reprinted with permission from ref. 13. Copyright (2014) American Chemical Society. | ||
Later in 2008, Wang et al. used single molecule force spectroscopy to study the surface adsorbed poly[(dopamine methacrylamide)-co-(butylamine methacrylamide)] (p(DMA-co-BMA)). They tuned the ratios of amino and catechol groups in the polymer to reveal the influence of the DOPA density on wet adhesion. Their experiments were performed on titanium-coated silicon wafers in a KNO3 solution at pH 6.8. They relied on nonspecific interactions between the cantilever tip and the polymer to stretch up the polymer and to desorb the catechol groups from the surface. Since the density of DOPA in the polymer was very high, instead of resolving the rupture of each single DOPA–surface bond, they observed plateau-shaped force–extension curves. Their results showed that the interaction strength between catechol and the titanium surface is ∼60–140 pN (Fig. 5c and d). As the force loading rate at the plateau region was close to zero, they assumed that the dissociation of catechol–titanium interactions was under quasi-equilibrium conditions. This was further corroborated by the observation that the adhesion did not change when the retraction speed was modified.112 They have developed a multiple binding model to understand this behavior (Fig. 5c).
In 2014, Li et al. used dopamine-modified hyaluronan acid (HA) to study the interactions between DOPA and different surfaces.13 They set catechol content at ∼10% of total HA carboxyl units, and performed SMFS on nine substrates with different chemical properties. They found that the HA–DOPA polymer has versatile adhesion ability to these substrates with adhesion strengths varying from 60–200 pN. Their force–extension curves are sawtooth-liked and each rupture peak corresponded to an individual DOPA–surface detachment. Worm-like chain model was used to fit each rupture peak, and the obtained persistence length served as the criterion to determine whether the rupture events were from single molecule detachments or not. They also set two important control experiments to support that the observed force events were from DOPA surface detachments. When using unmodified HA, no sawtooth-like force–extension curves were observed, indicating that the sawtooth-like peaks were from DOPA surface interactions. In the other control experiment, they used a similar experimental design as Lee et al.14 Instead of Boc-protected DOPA, dopamine was linked to the cantilever via a PEG linker. The rupture forces were similar to those measured using the HA–DOPA adsorption method. Based on these control experiments, it was safe to conclude that DOPA–surface interactions were indeed measured in this experiment. Their experiments provide a direct evidence that DOPA can indeed strongly and versatilely bind to many kinds of surfaces with different chemical properties. They also inferred that DOPA can form numerous different types of interactions with these surfaces based on the experimentally determined free energy profiles. Their results indicated that DOPA forms coordinate bonds with Ti and Si surfaces via the catechol group while forming mixed hydrogen bonds and coordination bonds with SiO2. For hydrophilic mica and Al2O3 surfaces, hydrogen bonding plays pivotal roles. For hydrophobic surfaces, such as gold, polytetrafluoroethene, and high-density polyethylene, hydrophobic interactions are dominant. For the polystyrene surface, besides hydrophobic interactions, π–π stacking between the catechol group of DOPA and the phenyl group of polystyrene is also involved.
Clearly, there are big discrepancies among the rupture forces measured by the three groups (Fig. 5). It is natural to ask what could be the possible origin of such differences. First, the chemical conditions could affect the SMFS results. In Lee et al.'s experiments, a bulky and hydrophobic Boc group was placed in close proximity to the catechol group, which may provide additional binding to the surfaces.14 On the contrary, in Li et al.'s experiments, the catechol groups were close to negatively charged carboxyl groups,13 while in Wang et al.'s experiments, the polymer backbone also contained positive charges.112 Moreover, the ionic strength and pH may also affect the DOPA adhesion.14 The buffer conditions were different in the three papers. It is also worth mentioning that the loading rates in these papers were also different. The experiments by Wang et al. were under equilibrium conditions, whereas the experiments by Lee et al. and Li et al. were at higher loading rates. The forces logarithmically depended on the loading rates. The experimental condition-dependent rupture forces were further exemplified by Utzig et al.,113 Kinugawa et al.,114 and Das et al.115
The effect of surface properties on DOPA adhesion
Recently, using scanning tunnel microscopy (STM), Li et al. found that DOPA showed different binding status and movement modes on the crystal rutile 〈110〉 surface.116 The surface hydroxyl group greatly enhanced the diffusion ability of absorbed catechols. The capture and release of a proton caused the individual adsorbed catechol molecule to switch between mobile and immobile states. Employing density functional theory calculations, they revealed the energetics between different status. In most previous SMFS measurements, the experiments were performed on amorphous TiO2 and the atoms on the surface were not orderly arranged. Besides, the influence of surface structures was neglected. Inspired by the STM results, the effect of surface properties on DOPA adhesion was also studied. Li et al. covalently attached dopamine onto a cantilever via a bifunctional PEG, and performed SMFS measurements on four TiO2 crystal surfaces (rutile 〈100〉, 〈110〉, 〈111〉, and 〈011〉) with well-defined atomic structures (Fig. 6).16 Their results showed that the adhesion forces of DOPA varied markedly on the four rutile surfaces. The 〈111〉 surface only showed a single distribution at ∼80 pN, which was consistent with the previous observation on amorphous TiO2. On the contrary, the rupture force distributions on other surfaces contained an additional peak located at ∼300 pN. As the chemical components of the rutile surfaces are the same, the only difference is their atomic arrangements. They found that the distances of Ti–O, Ti–Ti and O–O vary on each surface based on the crystal structures. They proposed that the atom distance on the surfaces should match the distance between two phenol oxygen atoms of DOPA in order to form bidentate stable adhesion bonds. Later on, periodic density functional theory (DFT) calculations further revealed the adhesion details of DOPA on a mineral surface.117 These results indicate that the surface structures should be carefully considered when designing new adhesives.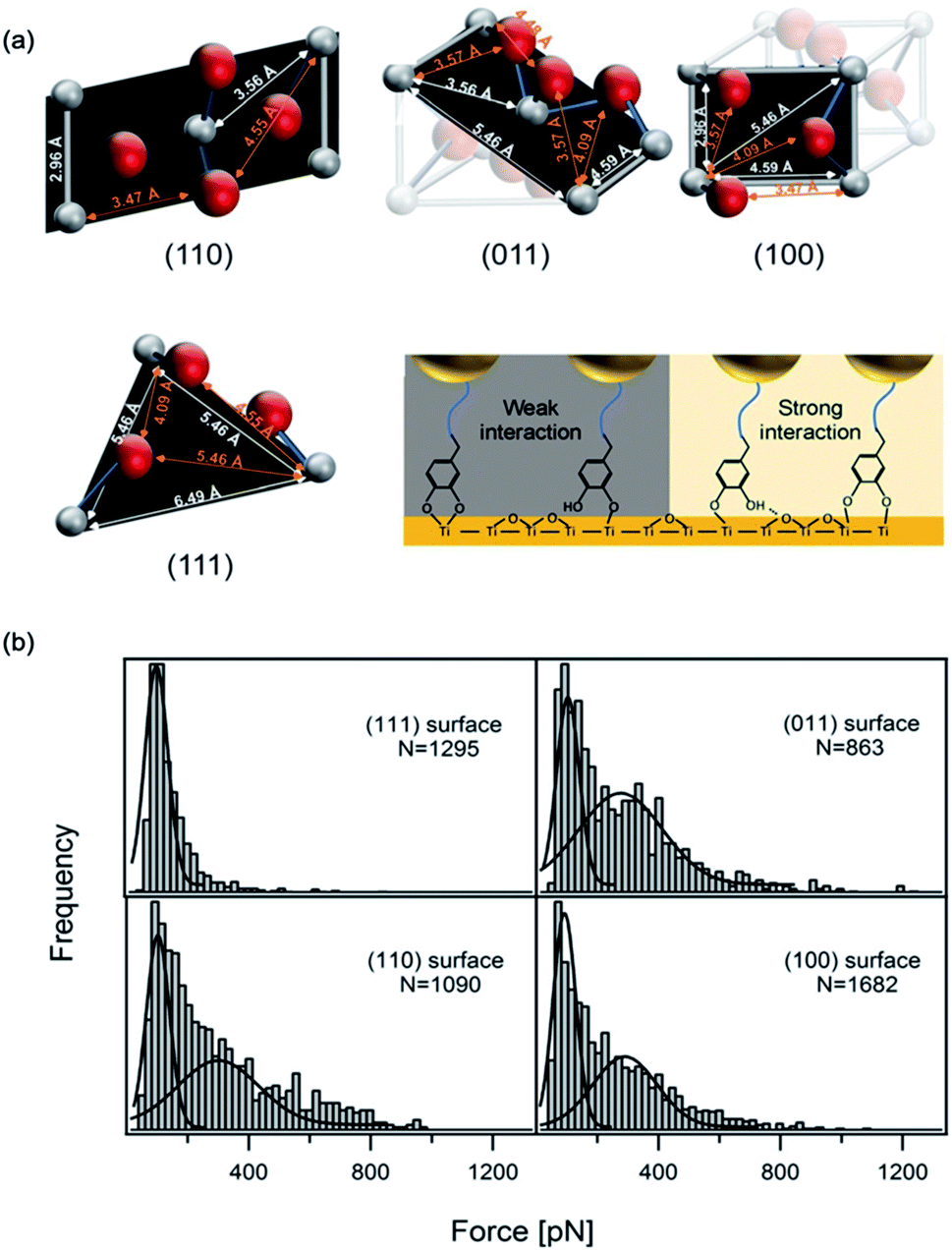 | ||
| Fig. 6 Influence of surface atom arrangement on DOPA adhesion. Schematics of different rutile surface atom arrangements and proposed binding modes of DOPA–surface interactions (a). Rupture force histograms observed from DOPA and different rutile surfaces (b). Copyright (2017) Wiley. Used with permission from ref. 16. | ||
Synergy between lysine and DOPA for surface adhesion
SMFS was also used to study the mechanism of lysine–DOPA synergistic adhesion.17 In 2017, Li et al. synthesized two dipeptides composed of lysine and DOPA but with inverted sequences, and utilized NHS–EDC coupling reaction to connect the dipeptide to cantilever via the natural amine on lysine. A dibenzosuberyl (dde) group was introduced to protect the free amine on lysine. The SMFS results showed that when the amino group was protected by dde, the rupture forces of the two peptides were similar to a single peak located at ∼100 pN. This value is slightly larger than that of DOPA only, which indicates that the hydrophobic dde group could contribute to peptide adhesion. After the cleavage of the dde group, the averaged rupture force of lysine–DOPA increased by almost two times, whereas that of DOPA–lysine remained unchanged (Fig. 7). After analyzing the structures of the two dipeptides, an interpretation based on the lysine and DOPA dipeptide chemical structures was given. Their results showed that the synergy between DOPA and lysine binding is sensitive to the structures of the compounds, and the synergy could be observed only when the external force is evenly distributed to lysine and DOPA. Therefore, the neighboring positive charge can not only displace the hydration layer as well as the surface salt to facilitate DOPA adhesion but can also directly participate in surface binding by forming ionic bonds with the surfaces. Apart from DOPA, hydrophobic amino acid (for example Phe, Leu and Orn) was also found to contribute to wet adhesion. Recently, Leader et al. used SMFS to measure the interactions between various amino acids and the TiO2 surface.56 Their results showed that aromatic interactions dominate over aliphatic interactions. In addition, the affinity of positively charged amino acids to the titanium dioxide surface is higher than that of uncharged, and can be further increased at elevated pH above the pKa of basic residues.56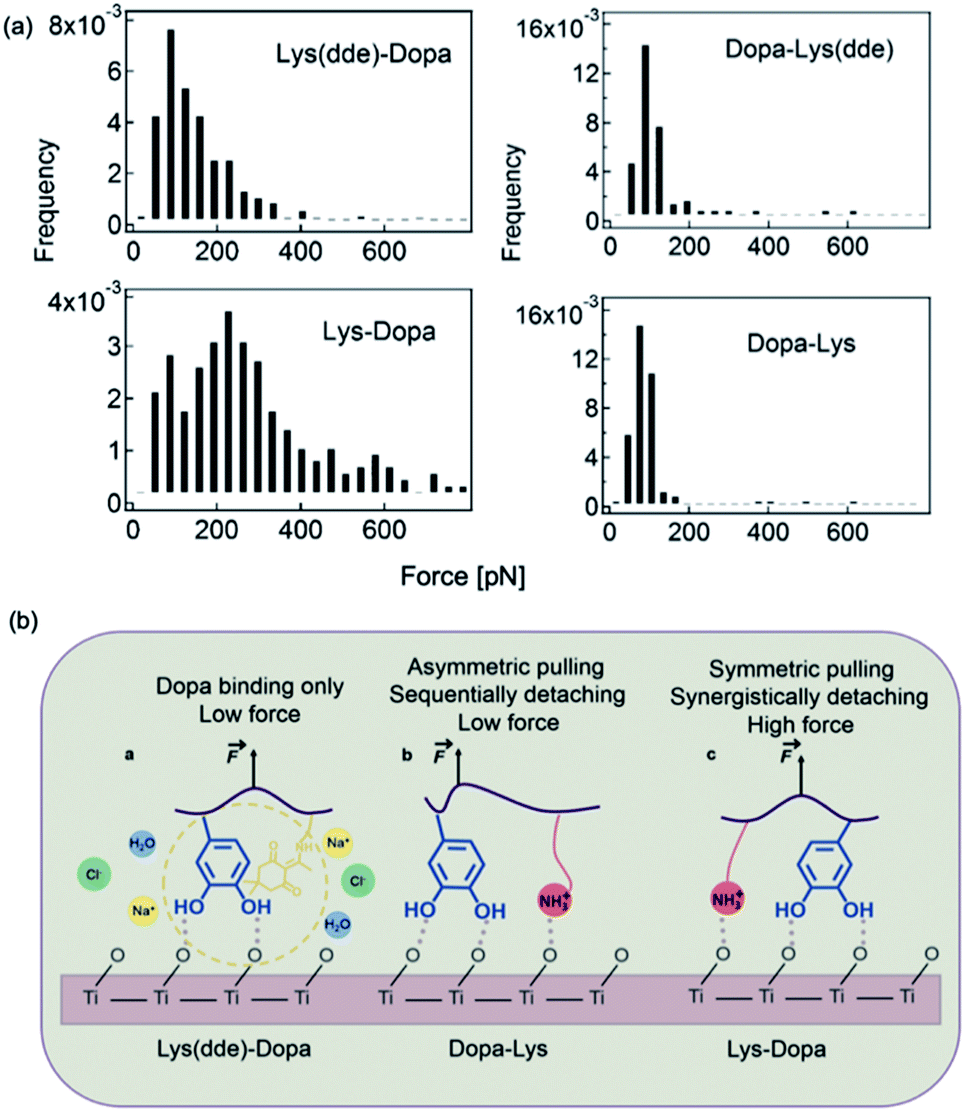 | ||
| Fig. 7 Force distribution of lysine–DOPA and DOPA lysine dipeptides and the TiO2 surface (a), and the schematic of the unbinding process of Dopa containing dipeptides (b). Reproduced from ref. 17 with permission from the Royal Society of Chemistry. | ||
Mechanics of DOPA–Fe3+ coordination bonds
A large amount of DOPA–Fe3+ coordination moieties are found in mfp-1. These coordination bonds act as sacrificial bonds to dissipate shock energy and protect the collagen core in the thread from rupture.19 In 2010, Zeng et al. used SFA to study the DOPA–Fe3+ interactions.118 They added a Fe3+ solution between two mfp-1 coated surfaces and tested the cohesion strength bridged by Fe3+. Their results showed that this Fe3+ mediated cohesion is fully reversible in the presence of 10 μM Fe3+. However, when the Fe3+ concentration was increased to 100 μM, the cohesion abolished. They proposed a stoichiometric change in the catechol–Fe3+ complex from a triple complex at a low Fe3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catechol ratio to a mono complex at a high Fe3+:catechol ratio. The subsequent SMFS study by Li et al. focused on DOPA–Fe3+ interactions on a single molecule level (Fig. 8).100 They conjugated DOPA to HA to form the HA–DOPA polymer, and then mixed the polymer with Fe3+ to form single-polymer micelles.100 The experiments were performed in different solution pH and Fe3+ concentrations to tune the stoichiometry of catechol–Fe3+ complexes. In some conditions, the bis-catechol–Fe3+ complexes were dominant and in the others, the tris-complexes were favored.
:catechol ratio to a mono complex at a high Fe3+:catechol ratio. The subsequent SMFS study by Li et al. focused on DOPA–Fe3+ interactions on a single molecule level (Fig. 8).100 They conjugated DOPA to HA to form the HA–DOPA polymer, and then mixed the polymer with Fe3+ to form single-polymer micelles.100 The experiments were performed in different solution pH and Fe3+ concentrations to tune the stoichiometry of catechol–Fe3+ complexes. In some conditions, the bis-catechol–Fe3+ complexes were dominant and in the others, the tris-complexes were favored.
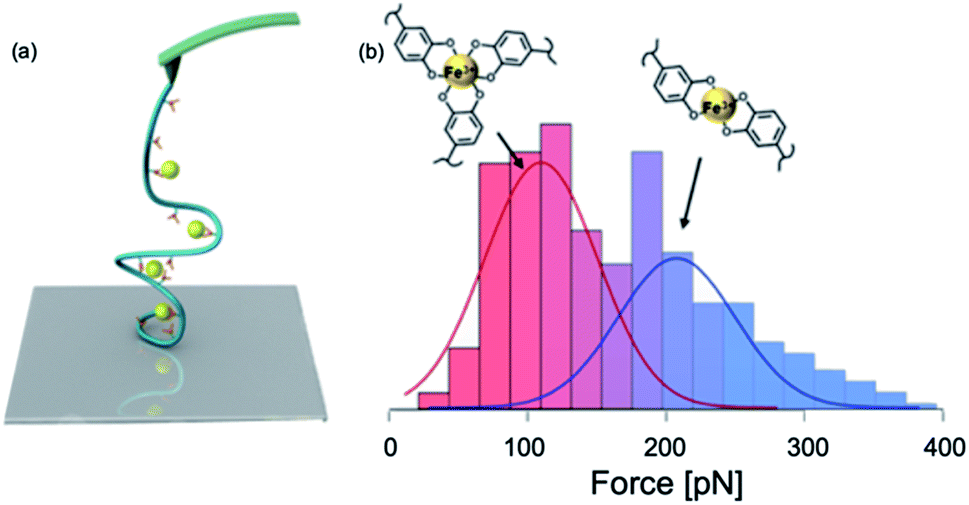 | ||
| Fig. 8 Schematics of SMFS study on catechol–Fe3+ complex (a) and force spectroscopy (b). Reprinted with permission from ref. 100. Copyright (2017) American Chemical Society. | ||
In this experiment, the single molecule micelles cross-linked by DOPA–Fe3+ complexes were deposited on the glass substrates and they were picked up by non-specific interactions between the polymer with the cantilever and the surface to rupture catechol–Fe3+ bonds. The force–extension curves showed sawtooth-like patterns, and each peak corresponded to a rupture of catechol–Fe3+ interaction. They found that tris- and bis-DOPA–Fe3+ complexes had different mechanical strengths. The averaged rupture force of bis-complex was ∼200 pN, whereas the rupture force of tris-complex was ∼100 pN at a pulling speed of 1000 nm s−1. By further performing dynamic force spectroscopy and fitting with Bell–Evans model, they extracted kinetic parameters for the rupture of tris- and bis-catechol–Fe3+ bonds. Combining with first principle calculations, they found that the different mechanical strengths of catechol–Fe3+ bonds originated from their different mechanical rupture pathways. The thermodynamically more stable tris-complexes exhibited lower mechanical stability because of the relatively larger deformation of the complexes at the transition state. They also revealed the fast rebinding kinetics of catechol–Fe3+ complexes. The single-molecule results provided a nanoscale mechanical understanding of cohesive interactions in mfps mediated via DOPA–Fe3+ coordination.
From DOPA to poly-dopamine
SMFS was also applied to study the in situ polymerization of poly-dopamine (pDA).119 Dopamine could auto-oxidize giving rise to o-dopamine-quinone, which cyclized to form a precursor of polydopamine, dihydroxyindole. Although the preparation of pDA coatings is simple and easy, and the pDA coating has been widely employed in surface functionalization, some fundamental facts of pDA remain elusive. This experiment provided the first evidence that pDA is a complex polymer. They performed SMFS experiments on pDA films directly deposited on silicon nitride AFM cantilevers. Plateau pattern force–extension curves with ∼200 nm of contour length in the traces were constantly observed in their experiments, which might have resulted from the sequential desorbing of catechol containing polymers from the surface. The plateau forces were ∼90 pN. Time-dependent force spectroscopy during the early stages of pDA formation revealed that the pDA chain growth occurred at the solid–liquid interface. The formation of films likely started with the adsorption of small oligomeric species, which then underwent further polymerization and maturation to form higher-molecular-weight pDA chains. Their findings clarified that pDA is indeed a polymer instead of the aggregates of oligomers. This study may also inspire the use of SMFS to resolve complex molecule forms in surface coatings and adhesives.120Outlook
Despite the fact that great advances have been made towards the understanding of molecular mechanism of mussel adhesion using SFA and single molecule AFM, there are still many uncharted outstanding questions deserving further efforts. First, it remains unknown whether the other residues in mfps also contribute significantly to mussel adhesion. It was found that the foot protein, pvfp-1 from Asian green mussels, Perna viridis, does not contain DOPA residues but another type of unusual amino acid, C(2)-mannosyl-7-hydroxytryptophan (Man7OHTrp).121 Halogenated DOPA (2-chloro-DOPA) was also found in a large quantity in the adhesive proteins from the sandcastle worm Phragmatopoma californica.122 It would be interesting to reveal the roles of these special residues in underwater adhesion. Second, the components of hydrophobic and charged residues vary dramatically among different mussel foot proteins and even in the same protein from different species. Even though the effects of redox potential,36 hydrophobic environment,123 charge–charge interactions,124 and cation–π interactions on the adhesion have been revealed,55 these effects have not been quantitatively evaluated using single molecule AFM. Third, most adhesive proteins form coacervates via liquid–liquid phase separation. Although this phenomenon is now well appreciated in biological systems,125–127 the mechanism underlying the coacervation of mussel proteins is less explored. Obviously, SFA and single-molecule AFM provide unique platforms to study liquid–liquid phase separation at the molecular level. Fourth, mussel foot proteins are not just random coiled structures and may exhibit unique secondary structures. For example, pvfp-1 from Asian green mussels shows a trimeric structure through the formation of coiled-coil structure of the C-terminus collagen sequence.121 Molecular dynamics simulations revealed that Pvfp-5β adopts a conformation with the aromatic rings of peripheral tyrosine residues facing the solvent.128 How these special conformations affect surface adhesion is eluded in the current studies. Fifth, it was also found that mussel foot proteins rapidly self-assembled into complex architectures during the biofabrication of byssus.25 The barnacle adhesive proteins could preassemble at a low pH value before being secreted out to enhance their underwater adhesion in basic sea water.46 It will also be interesting to study how the special processing and also the surface-induced conformational change affect the adhesion behaviors. Some strong adhesion behaviors were observed in DOPA and TiO2 or silica surface interactions. A reversible dynamic covalent bond could be formed in such a process, particularly in the presence of a large pressure. It would be interesting to figure out the effect of the applied pressure on DOPA adhesion. Last but not the least, how different mussel foot proteins synergistically enhance underwater adhesion by forming protein complexes also remains uncharted but is definitely critical for the understanding of the full picture of mussel adhesion mechanisms. We anticipate that these important aspects will be studied in the near future. These studies are extremely helpful for the design of mussel mimicking peptides and proteins with similar adhesion strengths but with much simplified sequences. These studies can also inspire the rational design of chemically synthesized underwater adhesives.18,129,130Conclusion
Mussel adhesion is a very important, yet complicated system. Despite numerous studies and applications on mussel-inspired adhesion, some of the fundamental mechanisms still remain elusive. In this article, we emphasize the importance of the biophysical studies of mussel adhesion using SFA and AFM-based single-molecule force spectroscopy to reveal the molecular mechanism underlying mussel adhesion. We show that these two methods are complementary, yet different. We explain why some seemly conflicting results may just originate from a slightly different experimental design. As DOPA-mediated adhesion is versatile and adaptive, it will be extremely important to precisely control the surface chemistry in these measurements. It will be helpful to combine these measurements with in situ chemical characterization techniques, such as infrared spectroscopy and Raman spectroscopy, in the future to provide a more comprehensive understanding of mussel adhesion. We believe that with the advances of the characterization techniques, more complicated molecular mechanisms about mussel adhesion will be revealed. Such information is invaluable for the design of the next generation underwater adhesives and surface coating techniques.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work is financially supported by the National Natural Science Foundation of China (11674153), the Basic Research Project of Science and Technology Plan of Shenzhen (JCYJ20170818110643669) and Natural Science Foundation of Jiangsu Province (BK20180335).Notes and references
- B. D. B. Tiu, P. Delparastan, M. R. Ney, M. Gerst and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2019, 11, 28296–28306 CrossRef CAS PubMed.
- C. J. Higginson, K. G. Malollari, Y. Xu, A. V. Kelleghan, N. G. Ricapito and P. B. Messersmith, Angew. Chem., Int. Ed., 2019, 58, 12271–12279 CrossRef CAS PubMed.
- G. M. Whitesides, Interface Focus, 2015, 5, 20150031 CrossRef PubMed.
- J. H. Waite and C. C. Broomell, J. Exp. Biol., 2012, 215, 873 CrossRef CAS PubMed.
- H. Li and Y. Cao, Acc. Chem. Res., 2010, 43, 1331–1341 CrossRef CAS PubMed.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338 CrossRef CAS PubMed.
- Z. Huang, P. Delparastan, P. Burch, J. Cheng, Y. Cao and P. B. Messersmith, Biomater. Sci., 2018, 6, 2487–2495 RSC.
- Y. Li, C. Liang, L. Gao, S. Li, Y. Zhang, J. Zhang and Y. Cao, Mater. Chem. Front., 2017, 1, 2664–2668 RSC.
- H. G. Silverman and F. F. Roberto, Mar. Biotechnol., 2007, 9, 661–681 CrossRef CAS PubMed.
- Q. Lin, D. Gourdon, C. Sun, N. Holten-Andersen, T. H. Anderson, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3782 CrossRef CAS PubMed.
- Y. Li, M. Qin, Y. Li, Y. Cao and W. Wang, Langmuir, 2014, 30, 4358–4366 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999 CrossRef CAS PubMed.
- Q. Lu, E. Danner, J. H. Waite, N. Israelachvili Jacob, H. Zeng and S. Hwang Dong, J. R. Soc., Interface, 2013, 10, 20120759 CrossRef PubMed.
- Y. Li, H. Liu, T. Wang, M. Qin, Y. Cao and W. Wang, ChemPhysChem, 2017, 18, 1466–1469 CrossRef CAS PubMed.
- Y. Li, T. Wang, L. Xia, L. Wang, M. Qin, Y. Li, W. Wang and Y. Cao, J. Mater. Chem. B, 2017, 5, 4416–4420 RSC.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651 CrossRef CAS PubMed.
- J. H. Waite and M. L. Tanzer, Science, 1981, 212, 1038 CrossRef CAS PubMed.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- J. H. Waite, T. J. Housley and M. L. Tanzer, Biochemistry, 1985, 24, 5010–5014 CrossRef CAS PubMed.
- V. V. Papov, T. V. Diamond, K. Biemann and J. H. Waite, J. Biol. Chem., 1995, 270, 20183–20192 CrossRef CAS PubMed.
- L. M. Rzepecki, K. M. Hansen and J. H. Waite, Biol. Bull., 1992, 183, 123–137 CrossRef CAS PubMed.
- H. Zhao and J. H. Waite, Biochemistry, 2006, 45, 14223–14231 CrossRef CAS PubMed.
- J. Yu, in Adhesive Interactions of Mussel Foot Proteins, ed. J. Yu, Springer International Publishing, Cham, 2014, DOI:10.1007/978-3-319-06031-6_4, pp. 31–41.
- H. Zhao and J. H. Waite, J. Biol. Chem., 2006, 281, 26150–26158 CrossRef CAS PubMed.
- T. H. Anderson, J. Yu, A. Estrada, M. U. Hammer, J. H. Waite and J. N. Israelachvili, Adv. Funct. Mater., 2010, 20, 4196–4205 CrossRef CAS PubMed.
- J. Yu, W. Wei, E. Danner, J. N. Israelachvili and J. H. Waite, Adv. Mater., 2011, 23, 2362–2366 CrossRef CAS PubMed.
- S. Bahri, C. M. Jonsson, C. L. Jonsson, D. Azzolini, D. A. Sverjensky and R. M. Hazen, Environ. Sci. Technol., 2011, 45, 3959–3966 CrossRef CAS PubMed.
- J. J. Wilker, Angew. Chem., Int. Ed., 2010, 49, 8076–8078 CrossRef CAS PubMed.
- M. A. Gebbie, W. Wei, A. M. Schrader, T. R. Cristiani, H. A. Dobbs, M. Idso, B. F. Chmelka, J. H. Waite and J. N. Israelachvili, Nat. Chem., 2017, 9, 473 CrossRef CAS PubMed.
- J. Israelachvili, Y. Min, M. Akbulut, A. Alig, G. Carver, W. Greene, K. Kristiansen, E. Meyer, N. Pesika, K. Rosenberg and H. Zeng, Rep. Prog. Phys., 2010, 73, 036601 CrossRef.
- R. Blankenburg, P. Meller, H. Ringsdorf and C. Salesse, Biochemistry, 1989, 28, 8214–8221 CrossRef CAS PubMed.
- E. W. Danner, Y. Kan, M. U. Hammer, J. N. Israelachvili and J. H. Waite, Biochemistry, 2012, 51, 6511–6518 CrossRef CAS PubMed.
- J. Yu, W. Wei, E. Danner, R. K. Ashley, J. N. Israelachvili and J. H. Waite, Nat. Chem. Biol., 2011, 7, 588 CrossRef CAS PubMed.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867 CrossRef CAS PubMed.
- Y. Cao, X. Zhang, L. Tao, K. Li, Z. Xue, L. Feng and Y. Wei, ACS Appl. Mater. Interfaces, 2013, 5, 4438–4442 CrossRef CAS PubMed.
- L. Burdine, T. G. Gillette, H.-J. Lin and T. Kodadek, J. Am. Chem. Soc., 2004, 126, 11442–11443 CrossRef CAS PubMed.
- B. Liu, L. Burdine and T. Kodadek, J. Am. Chem. Soc., 2006, 128, 15228–15235 CrossRef CAS PubMed.
- L. A. Burzio and J. H. Waite, Biochemistry, 2000, 39, 11147–11153 CrossRef CAS PubMed.
- L. P. Ghislain and W. W. Webb, Opt. Lett., 1993, 18, 1678–1680 CrossRef CAS PubMed.
- C. Bradac, Adv. Opt. Mater., 2018, 6, 1800005 CrossRef.
- M. D. Newton, B. J. Taylor, R. P. C. Driessen, L. Roos, N. Cvetesic, S. Allyjaun, B. Lenhard, M. E. Cuomo and D. S. Rueda, Nat. Struct. Mol. Biol., 2019, 26, 185–192 CrossRef CAS PubMed.
- C. Gourier, A. Jegou, J. Husson and F. Pincet, Cell. Mol. Bioeng., 2008, 1, 263 CrossRef.
- C. Liang, Z. Ye, B. Xue, L. Zeng, W. Wu, C. Zhong, Y. Cao, B. Hu and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2018, 10, 25017–25025 CrossRef CAS PubMed.
- J. Wu, P. Li, C. Dong, H. Jiang, X. Bin, X. Gao, M. Qin, W. Wang, C. Bin and Y. Cao, Nat. Commun., 2018, 9, 620 CrossRef PubMed.
- Y. Sun, W. Di, Y. Li, W. Huang, X. Wang, M. Qin, W. Wang and Y. Cao, Angew. Chem., Int. Ed., 2017, 56, 9376–9380 CrossRef CAS PubMed.
- A. M. Schrader, J. I. Monroe, R. Sheil, H. A. Dobbs, T. J. Keller, Y. Li, S. Jain, M. S. Shell, J. N. Israelachvili and S. Han, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2890 CrossRef CAS PubMed.
- J. Yu, N. E. Jackson, X. Xu, B. K. Brettmann, M. Ruths, J. J. de Pablo and M. Tirrell, Sci. Adv., 2017, 3, eaao1497 CrossRef PubMed.
- K. K. Mahato, M. Biswal, D. K. Rathore, R. K. Prusty, K. Dutta and B. C. Ray, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 115, 012017 Search PubMed.
- W. Sun, H. Jiang, X. Wu, Z. Xu, C. Yao, J. Wang, M. Qin, Q. Jiang, W. Wang, D. Shi and Y. Cao, Nano Res., 2019, 12, 115–119 CrossRef CAS.
- L. Zeng, M. Song, J. Gu, Z. Xu, B. Xue, Y. Li and Y. Cao, Biomimetics, 2019, 4, 36 CrossRef PubMed.
- Q. Fan, B. Chen and Y. Cao, J. Mech. Phys. Solids, 2019, 125, 653–665 CrossRef CAS.
- M. V. Rapp, G. P. Maier, H. A. Dobbs, N. J. Higdon, J. H. Waite, A. Butler and J. N. Israelachvili, J. Am. Chem. Soc., 2016, 138, 9013–9016 CrossRef CAS PubMed.
- A. Leader, D. Mandler and M. Reches, Phys. Chem. Chem. Phys., 2018, 20, 29811–29816 RSC.
- M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS PubMed.
- Y. Li and Y. Cao, Chin. J. Polym. Sci., 2018, 36, 366–378 CrossRef CAS.
- M. Mathelié-Guinlet, F. Viela, A. Vijloen, J. Dehullu and Y. F. Dufrêne, Curr. Opin. Biomed. Eng., 2019, 12, 1–7 CrossRef.
- M. Krieg, G. Fläschner, D. Alsteens, B. M. Gaub, W. H. Roos, G. J. L. Wuite, H. E. Gaub, C. Gerber, Y. F. Dufrêne and D. J. Müller, Nat. Rev. Phys., 2019, 1, 41–57 CrossRef.
- C. Feuillie, C. Formosa-Dague, L. M. C. Hays, O. Vervaeck, S. Derclaye, M. P. Brennan, T. J. Foster, J. A. Geoghegan and Y. F. Dufrêne, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3738 CrossRef CAS PubMed.
- T. M. Kennelly, Y. Li, Y. Cao, E. E. Qwarnstrom and M. Geoghegan, Biophys. J., 2019, 117, 688–695 CrossRef CAS PubMed.
- Y. Deng, T. Wu, M. Wang, S. Shi, G. Yuan, X. Li, H. Chong, B. Wu and P. Zheng, Nat. Commun., 2019, 10, 2775 CrossRef PubMed.
- W. Cai, D. Xu, L. Qian, J. Wei, C. Xiao, L. Qian, Z.-y. Lu and S. Cui, J. Am. Chem. Soc., 2019, 141, 9500–9503 CrossRef CAS PubMed.
- D. T. Edwards, J. K. Faulk, M.-A. LeBlanc and T. T. Perkins, Biophys. J., 2017, 113, 2595–2600 CrossRef CAS PubMed.
- J. Li and H. Li, J. Phys. Chem. B, 2018, 122, 9340–9349 CrossRef CAS PubMed.
- B. Li, T. Wang, X. Wang, X. Wu, C. Wang, F. Miao, M. Qin, W. Wang and Y. Cao, Chem.–Eur. J., 2019, 25, 7991–7997 CrossRef CAS PubMed.
- W. Ott, M. A. Jobst, M. S. Bauer, E. Durner, L. F. Milles, M. A. Nash and H. E. Gaub, ACS Nano, 2017, 11, 6346–6354 CrossRef CAS PubMed.
- X. Wang, B. Li, M. Qin, Y. Cao and W. Wang, J. Biomed. Nanotechnol., 2018, 14, 344–353 CrossRef PubMed.
- S. Zhang, H.-j. Qian, Z. Liu, H. Ju, Z.-y. Lu, H. Zhang, L. Chi and S. Cui, Angew. Chem., Int. Ed., 2019, 58, 1659–1663 CrossRef CAS PubMed.
- P. Yang, Y. Song, W. Feng and W. Zhang, Macromolecules, 2018, 51, 7052–7060 CrossRef CAS.
- X. Lyu, Y. Song, W. Feng and W. Zhang, ACS Macro Lett., 2018, 7, 762–766 CrossRef CAS.
- N. O. Alieva, A. K. Efremov, S. Hu, D. Oh, Z. Chen, M. Natarajan, H. T. Ong, A. Jégou, G. Romet-Lemonne, J. T. Groves, M. P. Sheetz, J. Yan and A. D. Bershadsky, Nat. Commun., 2019, 10, 3593 CrossRef CAS PubMed.
- C. Liu, J. Xia, S. Ji, Z. Fan and H. Xu, Chem. Commun., 2019, 55, 2813–2816 RSC.
- B. P. Frank and G. Belfort, Biotechnol. Prog., 2002, 18, 580–586 CrossRef CAS PubMed.
- D. S. Hwang, H. J. Yoo, J. H. Jun, W. K. Moon and H. J. Cha, Appl. Environ. Microbiol., 2004, 70, 3352–3359 CrossRef CAS PubMed.
- P. Hinterdorfer, W. Baumgartner, H. J. Gruber, K. Schilcher and H. Schindler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3477 CrossRef CAS PubMed.
- S. M. Sedlak, L. C. Schendel, M. C. R. Melo, D. A. Pippig, Z. Luthey-Schulten, H. E. Gaub and R. C. Bernardi, Nano Lett., 2019, 19, 3415–3421 CrossRef CAS.
- M. Goktas, C. Luo, R. M. A. Sullan, A. E. Bergues-Pupo, R. Lipowsky, A. Vila Verde and K. G. Blank, Chem. Sci., 2018, 9, 4610–4621 RSC.
- J. L. Zimmermann, T. Nicolaus, G. Neuert and K. Blank, Nat. Protoc., 2010, 5, 975 CrossRef CAS.
- R. C. Bernardi, E. Durner, C. Schoeler, K. H. Malinowska, B. G. Carvalho, E. A. Bayer, Z. Luthey-Schulten, H. E. Gaub and M. A. Nash, J. Am. Chem. Soc., 2019, 37, 14752–14763 CrossRef.
- R. Tapia-Rojo, E. C. Eckels and J. M. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7873 CrossRef CAS.
- J. Valle-Orero, J. A. Rivas-Pardo and I. Popa, Nanotechnology, 2017, 28, 174003 CrossRef PubMed.
- A. Duran, F. J. González-Sánchez, M. J. Fernández, R. Sirera, Í. Navarro-Blasco and I. J. Alvarez, Polymers, 2018, 10, 824 CrossRef PubMed.
- E. Infante, A. Stannard, S. J. Board, P. Rico-Lastres, E. Rostkova, A. E. M. Beedle, A. Lezamiz, Y. J. Wang, S. Gulaidi Breen, F. Panagaki, V. Sundar Rajan, C. Shanahan, P. Roca-Cusachs and S. Garcia-Manyes, Nat. Phys., 2019, 15, 973–981 Search PubMed.
- J. Chung, A. M. Kushner, A. C. Weisman and Z. Guan, Nat. Mater., 2014, 13, 1055 CrossRef CAS.
- N. Hosono, A. M. Kushner, J. Chung, A. R. A. Palmans, Z. Guan and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 6880–6888 CrossRef CAS.
- C. Ray, J. R. Brown and B. B. Akhremitchev, J. Phys. Chem. B, 2006, 110, 17578–17583 CrossRef CAS PubMed.
- I. T. S. Li and G. C. Walker, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16527 CrossRef CAS PubMed.
- W. Di, X. Gao, W. Huang, Y. Sun, H. Lei, Y. Liu, W. Li, Y. Li, X. Wang, M. Qin, Z. Zhu, Y. Cao and W. Wang, Phys. Rev. Lett., 2019, 122, 047801 CrossRef.
- B. Li, X. Wang, Y. Li, A. Paananen, G. R. Szilvay, M. Qin, W. Wang and Y. Cao, Chem.–Eur. J., 2018, 24, 9224–9228 CrossRef CAS PubMed.
- M. Grandbois, M. Beyer, M. Rief, H. Clausen-Schaumann and H. E. Gaub, Science, 1999, 283, 1727 CrossRef CAS PubMed.
- M. F. Pill, K. Holz, N. Preußke, F. Berger, H. Clausen-Schaumann, U. Lüning and M. K. Beyer, Chem.–Eur. J., 2016, 22, 12034–12039 CrossRef CAS PubMed.
- W. Huang, X. Wu, X. Gao, Y. Yu, H. Lei, Z. Zhu, Y. Shi, Y. Chen, M. Qin, W. Wang and Y. Cao, Nat. Chem., 2019, 11, 310–319 CrossRef CAS PubMed.
- D. J. Echelman, A. Q. Lee and J. M. Fernández, J. Biol. Chem., 2017, 292, 8988–8997 CrossRef CAS.
- P. Zheng, Y. Cao, T. Bu, S. K. Straus and H. Li, Biophys. J., 2011, 100, 1534–1541 CrossRef CAS PubMed.
- S. Cui, C. Liu, Z. Wang, X. Zhang, S. Strandman and H. Tenhu, Macromolecules, 2004, 37, 946–953 CrossRef CAS.
- T. Hugel, M. Grosholz, H. Clausen-Schaumann, A. Pfau, H. Gaub and M. Seitz, Macromolecules, 2001, 34, 1039–1047 CrossRef CAS.
- L. Schmitt, M. Ludwig, H. E. Gaub and R. Tampé, Biophys. J., 2000, 78, 3275–3285 CrossRef CAS.
- Y. Li, J. Wen, M. Qin, Y. Cao, H. Ma and W. Wang, ACS Biomater. Sci. Eng., 2017, 3, 979–989 CrossRef CAS.
- P. Zheng, C.-C. Chou, Y. Guo, Y. Wang and H. Li, J. Am. Chem. Soc., 2013, 135, 17783–17792 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- M. A. Hollinger, Crit. Rev. Toxicol., 1996, 26, 255–260 CrossRef CAS PubMed.
- W. Xiang, Z. Li, C.-Q. Xu, J. Li, W. Zhang and H. Xu, Chem.–Asian J., 2019, 14, 1481–1486 CrossRef CAS.
- W. Wei, Y. Sun, M. Zhu, X. Liu, P. Sun, F. Wang, Q. Gui, W. Meng, Y. Cao and J. Zhao, J. Am. Chem. Soc., 2015, 137, 15358–15361 CrossRef CAS.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS.
- S. R. K. Ainavarapu, J. Brujić, H. H. Huang, A. P. Wiita, H. Lu, L. Li, K. A. Walther, M. Carrion-Vazquez, H. Li and J. M. Fernandez, Biophys. J., 2007, 92, 225–233 CrossRef CAS PubMed.
- G. I. Bell, Science, 1978, 200, 618 CrossRef CAS PubMed.
- E. Evans and K. Ritchie, Biophys. J., 1999, 76, 2439–2447 CrossRef CAS.
- Y. Wang, J. Yan and B. T. Goult, Biochemistry, 2019 DOI:10.1021/acs.biochem.9b00453.
- R. W. Friddle, A. Noy and J. J. De Yoreo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13573 CrossRef CAS.
- J. Wang, M. N. Tahir, M. Kappl, W. Tremel, N. Metz, M. Barz, P. Theato and H.-J. Butt, Adv. Mater., 2008, 20, 3872–3876 CrossRef CAS.
- T. Utzig, P. Stock and M. Valtiner, Angew. Chem., Int. Ed., 2016, 55, 9524–9528 CrossRef CAS.
- S. Kinugawa, S. Wang, S. Taira, A. Tsuge and D. Kaneko, Polym. J., 2016, 48, 715 CrossRef CAS.
- P. Das and M. Reches, Nanoscale, 2016, 8, 15309–15316 RSC.
- S.-C. Li, L.-N. Chu, X.-Q. Gong and U. Diebold, Science, 2010, 328, 882 CrossRef CAS PubMed.
- S. A. Mian and Y. Khan, Am. J. Chem., 2017, 2017, 6 Search PubMed.
- H. Zeng, D. S. Hwang, J. N. Israelachvili and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12850 CrossRef CAS PubMed.
- P. Delparastan, K. G. Malollari, H. Lee and P. B. Messersmith, Angew. Chem., Int. Ed., 2019, 58, 1077–1082 CrossRef CAS PubMed.
- X. Kang, W. Cai, S. Zhang and S. Cui, Polym. Chem., 2017, 8, 860–864 RSC.
- D. S. Hwang, H. Zeng, Q. Lu, J. Israelachvili and J. H. Waite, Soft Matter, 2012, 8, 5640–5648 RSC.
- C. J. Sun, A. Srivastava, J. R. Reifert and J. H. Waite, J. Adhes., 2009, 85, 126 CrossRef CAS.
- W. Wei, J. Yu, C. Broomell, J. N. Israelachvili and J. H. Waite, J. Am. Chem. Soc., 2013, 135, 377–383 CrossRef CAS PubMed.
- J. Yu, Y. Kan, M. Rapp, E. Danner, W. Wei, S. Das, D. R. Miller, Y. Chen, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15680 CrossRef CAS PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285 CrossRef CAS PubMed.
- J. B. Woodruff, A. A. Hyman and E. Boke, Trends Biochem. Sci., 2018, 43, 81–94 CrossRef CAS PubMed.
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, eaaf4382 CrossRef PubMed.
- L. Petrone, A. Kumar, C. N. Sutanto, N. J. Patil, S. Kannan, A. Palaniappan, S. Amini, B. Zappone, C. Verma and A. Miserez, Nat. Commun., 2015, 6, 8737 CrossRef CAS PubMed.
- B. K. Ahn, S. Das, R. Linstadt, Y. Kaufman, N. R. Martinez-Rodriguez, R. Mirshafian, E. Kesselman, Y. Talmon, B. H. Lipshutz, J. N. Israelachvili and J. H. Waite, Nat. Commun., 2015, 6, 8663 CrossRef PubMed.
- R. Santonocito, F. Venturella, F. Dal Piaz, M. A. Morando, A. Provenzano, E. Rao, M. A. Costa, D. Bulone, P. L. San Biagio, D. Giacomazza, A. Sicorello, C. Alfano, R. Passantino and A. Pastore, J. Biol. Chem., 2019, 34, 12826–12835 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |