
A cyotosol-selective nitric oxide bomb as a new paradigm of an anticancer drug†
Dongsik
Park
a,
Sooseok
Im
b,
Gurusamy
Saravanakumar
a,
Yeong Mi
Lee
a,
Jinhwan
Kim
a,
Kunho
Kim
b,
Junseok
Lee
 a,
Jihoon
Kim
*a and
Won Jong
Kim
*ab
a,
Jihoon
Kim
*a and
Won Jong
Kim
*ab
aDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), Cheongam-ro 77, Nam-gu, Pohang 37673, Republic of Korea. E-mail: mole@postech.ac.kr; wjkim@postech.ac.kr
bSchool of Interdisciplinary Bioscience and Bioengineering, POSTECH, Jigok-ro 64, Nam-gu, Pohang 37666, Republic of Korea
First published on 14th November 2019
Abstract
We have reported rational design of a polymeric NO delivery micelle as a cytosol-selective NO bomb. Protected NO-donors are released from the micelle under endolysosomal conditions, and then deprotected by cytosolic glutathione. Cytosol-selective NO delivery facilitates significant tumor regression without the aid of other therapeutic modalities even in intravenous administrations.
Nitric oxide (NO) is an endogenous gas molecule that modulates various biological pathways including vasodilation, immune response, angiogenesis, cell proliferation, apoptosis, and metabolism.1 In particular, NO is assumed to provide an alternative to the present clinical anticancer modalities including chemotherapy and radiotherapy as it exerts cytotoxic functions preferentially in cancer cells rather than in the normal cells, as confirmed by in vitro studies, and its endogenous presence and rapid decomposition into innocuous ions after its local actions can minimize the systemic side effects.2 Nevertheless, the clinical setting of NO-based anticancer therapy needs to improve due to the poor stability and inadequate bioavailability of present NO delivery systems.3 Indeed, the present NO delivery systems have mainly been explored as a form of combination therapy with other therapeutic modalities, which is still limited to the conceptual level, such as in vitro cell assays or animal models with intratumoral administrations.4 Therefore, there exists an urgent need for rational design in NO delivery systems with high stability and controlled releasing ability in order to realize the complete clinical potential of NO-based anticancer therapy.
Herein, we design a dual stimuli-responsive polymeric NO delivery micelle that can efficiently accumulate in the tumor and facilitate cytosolic release of NO in cancer cells (Scheme 1). Among various NO-releasing functional moieties (NO-donors), 1-substituted diazen-1-ium-1,2-diolates (N-diazeniumdiolates) were exploited due to their high NO capacity and facile functionalization.1a,5 In particular, the O2-protection of a N-diazeniumdiolate containing a diol group (P-NO) not only makes it possible to prevent the NO release from N-diazeniumdiolates during blood circulation, but also endows glutathione (GSH)-mediated cytosol-selective NO release. We also synthesize diblock copolymers, wherein the phenylboronic acid (PBA) moieties on the polymer chain coordinate a diol group of the P-NO via PBA–diol molecular recognition.6 After the coordination, P-NO provides hydrophobicity to the polymer in order to facilitate the formation of self-assembled micelles (P-NO-PMs). The core–shell micelle not only protects the P-NO during blood circulation, but also allows its high accumulation at a tumor tissue via the enhanced permeation and retention (EPR) effect (Scheme 1). After uptake by cancer cells, the endolysosomal acidic conditions dissociate the interactions between PBA and a diol of P-NO due to the pH-sensitivity of the PBA–diol complex,6 resulting in the release of P-NO into the cytosol. Thereafter, the intracellular GSH at neutral cytosolic pH deprotects the released P-NO to afford N-diazeniumdiolates, which induces the burst release of NO and eventually exerts cytotoxicity (Scheme 1).
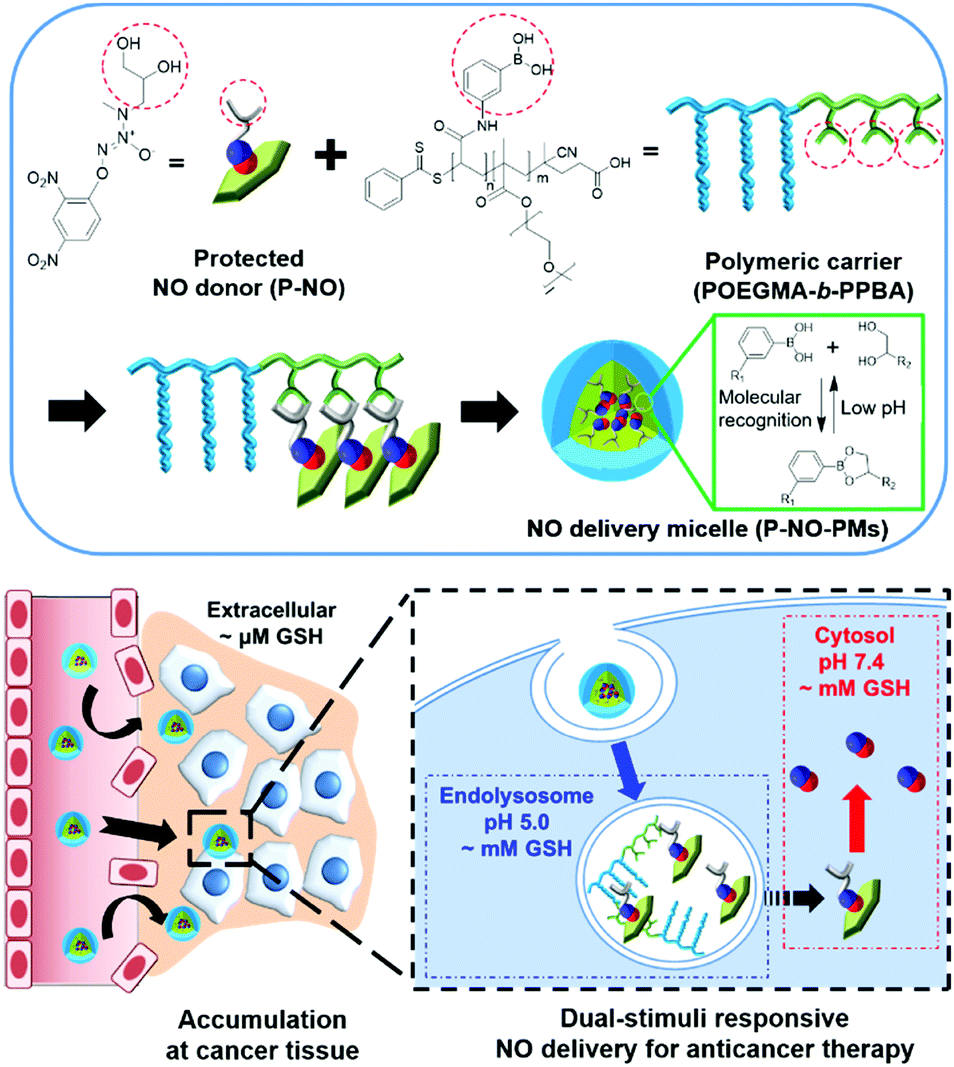 | ||
| Scheme 1 Schematic illustration of P-NO-PMs for anti-cancer therapy. | ||
Initially, O2-(2,4-dinitrophenyl)-1-[N-methyl-(2,3-dihydroxypropyl)amino]-diazenium-1,2-diolate (P-NO) was synthesized by modifying a secondary amine of 3-methylamino-1,2-propanediol (Am-diol) with N-diazeniumdiolate (Am-diol-NO), followed by protecting its O2-position with a 2,4-dinitrophenyl group7 (Fig. S1, ESI†). In 1H-NMR, owing to the down shielding effect of the diazeniumdiolate group, the methyl and methylene peaks attached to the nitrogen atom of Am-diol at 2.31 ppm and 2.51–2.61 ppm were shifted to 2.74 ppm and 2.86–3.07 ppm (Fig. S2, ESI†) in Am-diol-NO, respectively. Am-diol-NO showed spontaneous NO release due to the absence of protecting groups (Fig. S3 and Table S1, ESI†). In addition, the appearance of aromatic peaks associated with 2,4-dinitrobenzene confirmed the successful synthesis of P-NO (Fig. S4, ESI†).
GSH-responsive behavior of P-NO was confirmed by UV-visible absorption spectra and NO releasing profiles. In the absence of GSH, the P-NO exhibited two characteristic absorption peaks corresponding to the N-diazeniumdiolate moiety and 2,4-dinitrophenyl protecting group at 250 and 304 nm, respectively (Fig. 1A). Under intracellular concentration of GSH (2 mM),8 the absorption peak of the protecting group was shifted from 304 to 340 nm and the N-diazeniumdiolate peak was gradually decreased with increasing incubation time. In addition to GSH-responsiveness, P-NO exhibited pH-dependent behavior in the presence of intracellular GSH concentrations (Fig. 1B and Table S1, ESI†). A small amount (0.23 μmol mg−1) of NO was released at pH 5.0 despite the presence of GSH, which could be ascribed to insufficient nucleophilicity of GSH in order to deprotect the 2,4-dinitrophenyl groups from the P-NO under acidic conditions.9 In contrast, a burst release of NO (4.99 μmol mg−1) was observed at pH 7.4 with 2 mM GSH, implying the selective NO release in the cytosol (pH 7.4, with 2 mM GSH) over endolysosomes (pH 5.0, with 2 mM GSH). To the best of our knowledge, different behavior of O2-protected N-diazeniumdiolates between the endolysosomes and cytosol remains unexplored. Considering the potential interference of thiol-containing biomolecules against GSH-responsive NO-donors in intravenous administration, NO release from P-NO was examined at blood concentration of human serum albumin (HSA) (50 g L−1)10 as well as at extracellular GSH concentration (2–20 μM)8 in order to investigate whether the P-NO can prevent nonspecific premature release of NO during blood circulation (Fig. 1C). Negligible NO release was observed in the presence of HSA or extracellular GSH concentrations, implying the stability of P-NO in blood circulations after systemic administration.
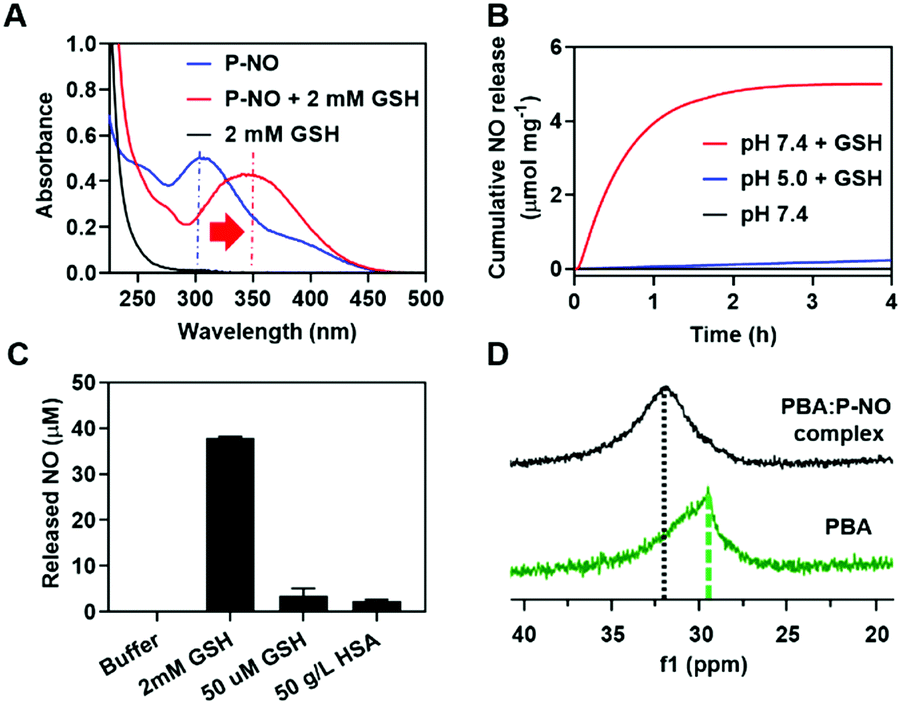 | ||
| Fig. 1 Characteristics of P-NO. (A) UV-Vis spectra of P-NO with or without GSH. (B) GSH- and pH-dependent NO release. (C) HSA-independent NO release at pH 7.4. (D) 11B-NMR of the PBA:P-NO mixture. | ||
The P-NO was designed to coordinate its diol group to the PBA-containing polymer in order to self-assemble the micelles, in which nano-size of the particles has an advantage over the EPR effects. Accordingly, we investigated whether a P-NO with a diol functional group could readily form complexes with a PBA moiety. In the 11B-NMR, the signal of PBA appearing at 29.52 ppm was shifted to 31.91 ppm after complexation with P-NO (Fig. 1D). Similarly in 1H-NMR, the signals corresponding to the adjacent protons from the boronic acid of PBA were shifted from 8.26 to 7.79 ppm and the diol moieties of P-NO were shifted from 4.10, 3.71–3.80, and 3.61–3.65 ppm to 4.92, 4.16–4.53, and 3.72–4.04 ppm, respectively (Fig. S5, ESI†). These results confirmed the successful formation of a boronate ester between a P-NO and a PBA.
An amphiphilic block copolymer comprising oligo(ethylene glycol)methyl ester methacrylate (OEGMA) and (3-acrylamido)phenylboronic acid (PBA) as the block constituents was designed to efficiently deliver P-NO into the tumor. The OEGMA blocks in the polymer take charge of micelle corona, which allows prolonged blood circulation. The PBA block in the polymer can integrate P-NO into the polymer chains via unique PBA–diol coordination, which makes it possible to localize the P-NO in the core of the micelles. This self-assembly was assumed to protect the P-NO from harsh in vivo conditions, accumulate at the tumor via the EPR effect, and facilitate cytosolic release of NO (Scheme 1). The synthesis of POEGMA-b-PPBA was accomplished by a sequential two-step reversible addition–fragmentation chain transfer (RAFT) polymerization (Fig. S6, ESI†). Initially, methacrylated oligoethylene glycol (OEGMA) was polymerized using 4-cyano-4-(phenylcarbonothio)pentanoic acid as a chain transfer agent (CTA) and α,α′-azoisobutyronitrile as a radical initiator (Fig. S7 and S8, ESI†). By employing the as-synthesized POEGMA homopolymer and (3-acrylamidophenyl)boronic acid (AmPBA) as a macro-CTA and monomer, the POEGMA-b-PPBA copolymer with 8 repeating units of OEGMA and 25 repeating units of PBA was obtained (Fig. S9 and S10, ESI†).
In aqueous solution, the amphiphilic POEGMA-b-PPBA copolymer itself formed loose micelles with 150–300 nm diameter, which may be due to the weak hydrophobic interaction among the PBA groups (Fig. S11, ESI†). Owing to the hydrophobic 2,4-dinitrophenyl protecting group, the coordination of P-NO to the POEGMA-b-PPBA copolymer allowed the formation of more compact self-assembled core–shell polymeric micelles (P-NO-PMs) with 80–130 nm diameter known to be of appropriate size for the EPR effect (Fig. 2A and B). The size of P-NO-PMs remained unchanged even in the presence of serum, a general component to induce aggregation or disassembly of various nanoparticles during blood circulation (Fig. 2A and Fig. S12, ESI†). In addition, the P-NO-PMs retained their size even at higher glucose concentration (4.5 g L−1) than that at the normal blood glucose concentrations (1 g L−1) despite the potential interference of a diol-containing glucose against the interaction between PBA and P-NO (Fig. 2A and Fig. S12, ESI†). These results indicate the superior stability of P-NO-PMs in blood circulation.
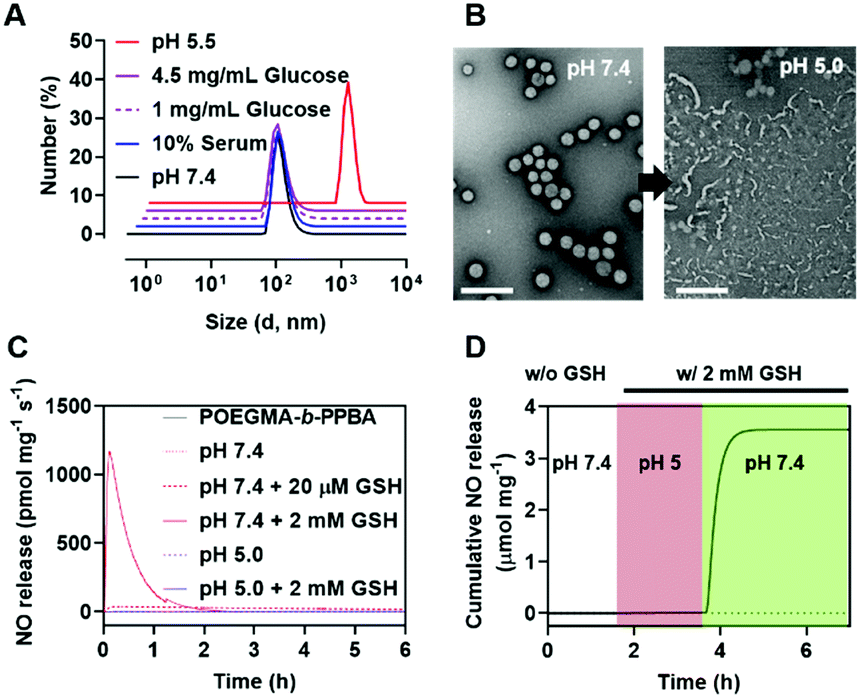 | ||
| Fig. 2 Characteristics of P-NO-PMs. (A) Size distribution of P-NO-PMs dependent on different physiological conditions. (B) TEM image of P-NO-PMs at pH 7.4 and pH 5.0. Scale bar is 100 μm. (C) NO release profiles of P-NO-PMs under various physiological conditions. (D) NO release profile of P-NO-PMs under physiological buffer (white box), endolysosomal (pH 5, 2 mM GSH; red box) and, cytosolic conditions (pH 7.4, 2 mM GSH; green box). | ||
In our design, the P-NO-PMs retain their structures during blood circulation; however, they should be disrupted under intracellular conditions to release NO. Accordingly, intracellular environment-responsive behavior of P-NO-PMs were evaluated under extracellular environments (pH 7.4, with 20 μM GSH), and endolysosomal conditions (pH 5.0, with 2 mM GSH), as well as cytosol conditions (pH 7.4, with 2 mM GSH). In contrast with the insignificant changes under extracellular environments, P-NO-PMs were disrupted under acidic pH due to the pH-sensitive dissociation between PBA and diol in P-NO (Fig. 2B). In particular, P-NO-PMs could also be disrupted under cytosolic conditions, which is ascribed to the deprotection of 2,4-nitrophenyl groups from P-NO. Indeed, a burst NO release was observed in P-NO-PMs under cytosolic conditions, whereas slight or negligible NO release was detected under the extracellular environmental and endolysosomal conditions (Fig. 2C and Fig. S13, Table S2, ESI†). The NO release profiles under dynamic environmental changes clearly demonstrated apparent cytosol selective NO-releasing behavior of P-NO-PMs (Fig. 2D). Overall, while P-NO-PMs protect P-NO during blood circulation (Fig. 2A, C, D and Table S2, ESI†), the micelles are disrupted to release P-NO under endolysosomal conditions (Fig. 2B). The released P-NO exhibits negligible NO release under endolysosomal conditions (Fig. 2C, D and Table S2, ESI†), but in turn releases burst NO under cytosolic conditions (Fig. 2C, D and Table S2, ESI†).
The effects of P-NO-PMs on cancer cells were evaluated in vitro. Diaminofluorescein-2 diacetate (DAF-2-DA)-assisted confocal laser scanning microscopy (CLSM) revealed that the P-NO and P-NO-PMs increase intracellular concentrations of NO (Fig. 3A). In MTT colorimetric assays, insignificant cytotoxicity was observed for the POEGMA-b-PPBA copolymer, Am-diol, and the dinitrobenzene moiety, indicating the biocompatibility of the polymeric carrier (Fig. 3B and Fig. S14, ESI†). The unprotected NO donor (Am-diol-NO) did not exert any anticancer effect even at substantially high concentrations (up to 1 mM; Fig. S14A–C, ESI†), which could be attributed to the rapid transformation of NO into nontoxic ions after unspecific release of NO in the culture media; however, dose-dependent anticancer effects of P-NO and P-NO-PMs were observed in various cancer cells, which demonstrates the anticancer effects of P-NO and the importance of selective intracellular NO release in the NO-based antitumor effect (Fig. 3B and Fig. S14D, E, ESI†). In addition, annexin/propidium iodide (PI)-assisted flow cytometry revealed that P-NO and P-NO-PMs exert anticancer effects via an apoptotic pathway (Fig. 3C and Fig. S15, ESI†). More importantly, P-NO and P-NO-PMs exhibited efficient anticancer effects on MCF-7/adriamycin (ADR; Fig. 3D), a multidrug resistant (MDR) cell line on which doxorubicin (DOX) cannot exert anti-cancer effects (Fig. S16, ESI†). These results imply that P-NO and P-NO-PMs provide an efficient means of overcoming MDR.
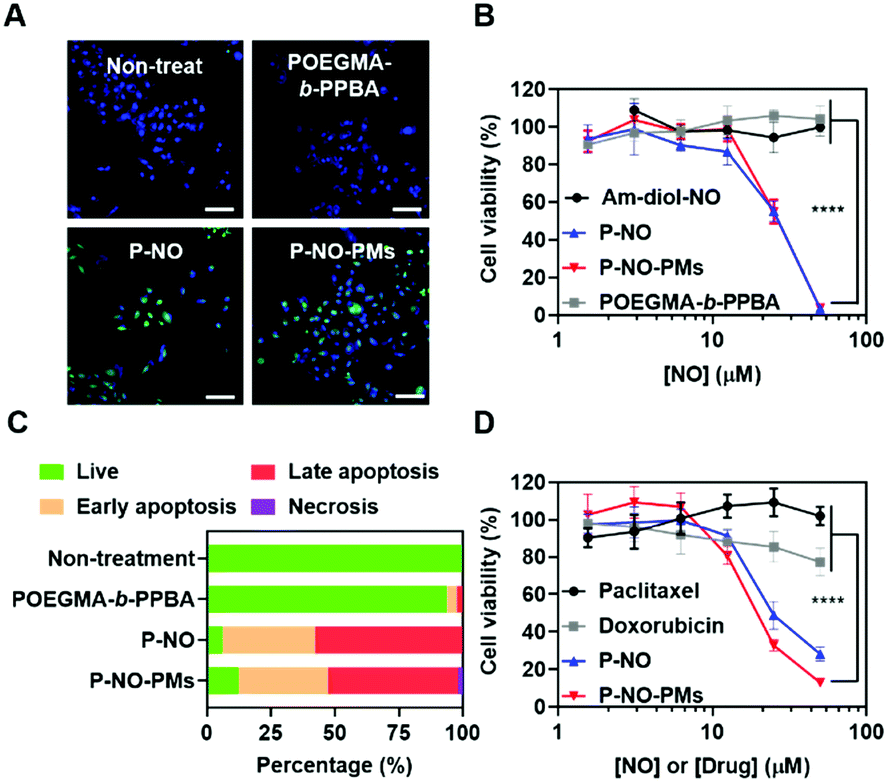 | ||
| Fig. 3 In vitro behavior of P-NO-PMs. (A) Intracellular NO release of P-NO-PMs. Nuclei and intracellular NO were stained with DAPI (blue) and DAF-2-DA (green). Scale bar is 100 μm. (B) Anticancer effects and (C) live/dead assay in MCF-7. (D) Anticancer effects in MCF-7/ADR cell lines. | ||
Eventually, P-NO-PMs were intravenously (i.v.) administered to evaluate the in vivo antitumor effects (Fig. 4). Fluorescent dye (FCR 749)-labelled P-NO-PMs (Fig. S17 and S18, ESI†) demonstrated EPR-mediated rapid accumulations of the micelles as presented in the IVIS spectrum image (Fig. 4A). As a result, only P-NO-PMs exhibited in vivo effects despite the similar anticancer effects between P-NO and P-NO-PMs in vitro (Fig. 4B and Fig S19, ESI†). In addition, body weight change (Fig. S20, ESI†) and hemolytic effect of NO-loaded carriers (Fig. 4C) were not observed. Alanine aminotransferase (ALT) was not significantly changed, and aspartate aminotransferase (AST) activity was increased by 17% after treatment with P-NO-PMs (Fig. S21, ESI†). Considering an increase of more than five times aminotransferase level is known to incur liver damage,11 P-NO-PMs did not induce severe liver toxicity. Overall, the in vivo results indicate the efficient antitumor effects, hemocompatibility, and negligible nonspecific toxicity of P-NO-PMs.
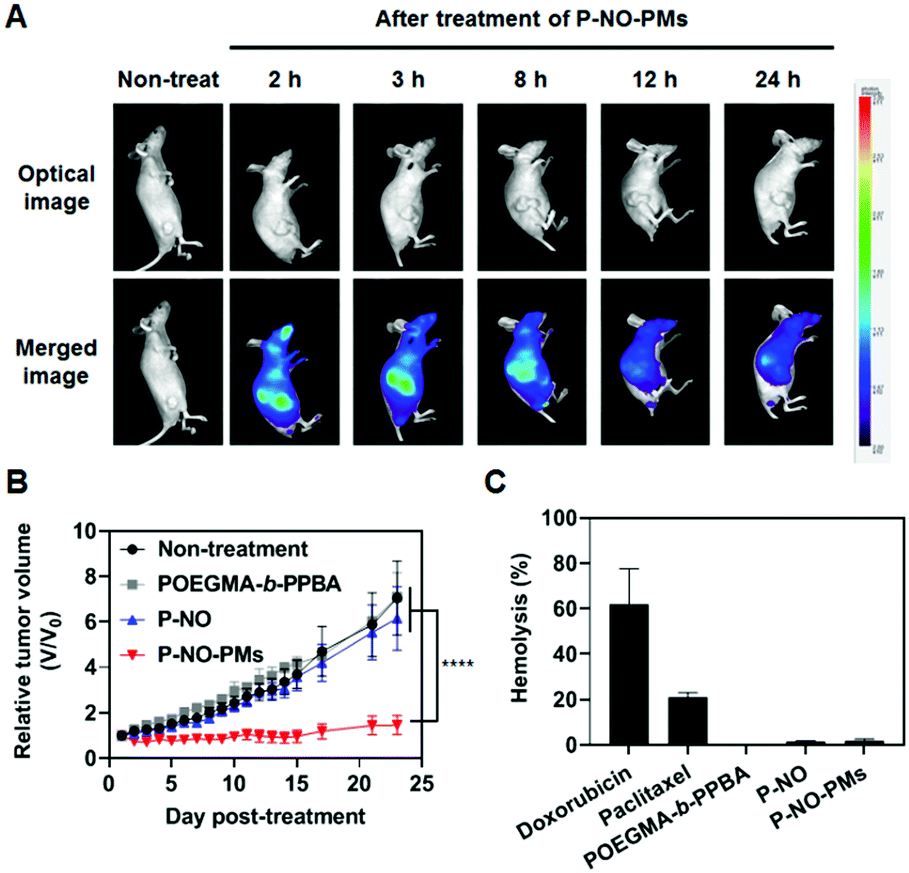 | ||
| Fig. 4 Antitumor effects of i.v. administrated P-NO-PMs in vivo. (A) IVIS image after treatment with P-NO-PMs. (B) Tumor growth of MCF7-bearing mice. Black arrows indicate injections. (C) Hemolysis test of various samples. | ||
In conclusion, we successfully designed and synthesized a cytosol-specific NO delivery system for anticancer therapy. PBA–diol molecular recognition not only allows administration of O2-protected N-diazeniumdiolates into the micelles, but also facilitates the pH-sensitive release of NO-donors. O2-protected N-diazeniumdiolates enable the selective release of NO owing to the GSH-responsive deprotection of the 2,4-dinitrophenyl group at neutral pH. Collectively, the resultant P-NO-PMs were accumulated at the tumor tissue via EPR effects and exhibited a cytosol-selective NO bomb, thus leading to an efficient NO-mediated anticancer effect. We believe that our design imparts knowledge for advancing the development of a NO delivery system and realizing complete clinical potential of NO-based anticancer drugs as a new paradigm for anticancer therapy.
This research was supported by the National Research Foundation of Korea (NRF) grant (NRF-2017R1E1A1A01074088), the Bio & Medical Technology Development Program of the NRF funded by the Korea government (Ministry of Science and ICT) (NRF-2017M3A9F5030930), and the Creative Materials Discovery Program through the NRF funded by Ministry of Science and ICT (NRF-2018M3D1A1058813).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Kim, G. Saravanakumar, H. W. Choi, D. Park and W. J. Kim, J. Mater. Chem. B, 2014, 2, 341–356 RSC; (b) S. Mocellin, V. Bronte and D. Nitti, Med. Res. Rev., 2007, 27, 317–352 CrossRef CAS PubMed.
- (a) J. R. Hickok and D. D. Thomas, Curr. Pharm. Des., 2010, 16, 381–391 CrossRef CAS PubMed; (b) S. Heigold, C. Sers, W. Bechtel, B. Ivanovas, R. Schäfer and G. Bauer, Carcinogenesis, 2002, 23, 929–941 CrossRef CAS PubMed; (c) H. T. T. Duong, Z. M. Kamarudin, R. B. Erlich, Y. Li, M. W. Jones, M. Kavallaris, C. Boyer and T. P. Davis, Chem. Commun., 2013, 49, 4190–4192 RSC.
- (a) J. Kim, B. C. Yung, W. J. Kim and X. Chen, J. Controlled Release, 2017, 263, 223–230 CrossRef CAS PubMed; (b) D. Hirst and T. Robson, J. Pharm. Pharmacol., 2007, 59, 3–13 CrossRef CAS PubMed.
- (a) M. F. Chung, H. Y. Liu, K. J. Lin, W. T. Chia and H. W. Sung, Angew. Chem., Int. Ed., 2015, 54, 9890–9893 CrossRef CAS PubMed; (b) J. Fan, Q. J. He, Y. Liu, F. W. Zhang, X. Y. Yang, Z. Wang, N. Lu, W. P. Fan, L. S. Lin, G. Niu, N. Y. He, J. B. Song and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13804–13811 CrossRef CAS PubMed; (c) S. Pramanick, J. Kim, J. Kim, G. Saravanakumar, D. Park and W. J. Kim, Bioconjugate Chem., 2018, 29, 885–897 CrossRef CAS PubMed.
- (a) J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135–1154 CrossRef CAS PubMed; (b) D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- (a) J. Lee, J. Kim, Y. M. Lee, D. Park, S. Im, E. H. Song, H. Park and W. J. Kim, Acta Pharmacol. Sin., 2017, 38, 848–858 CrossRef CAS PubMed; (b) Y. Li, W. Xiao, K. Xiao, L. Berti, J. Luo, H. P. Tseng, G. Fung and K. S. Lam, Angew. Chem., Int. Ed., 2012, 51, 2864–2869 CrossRef CAS PubMed.
- J. E. Saavedra, A. Srinivasan, C. L. Bonifant, J. Chu, A. P. Shanklin, J. L. Flippen-Anderson, W. G. Rice, J. A. Turpin, K. M. Davies and L. K. Keefer, J. Org. Chem., 2001, 66, 3090–3098 CrossRef CAS PubMed.
- (a) H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS PubMed; (b) D. Buonocore, M. Grosini, S. Giardina, A. Michelotti, M. Carrabetta, A. Seneci, M. Verri, M. Dossena and F. Marzatico, Oxid. Med. Cell. Longevity, 2016, 3286365 Search PubMed.
- (a) H. A. Lindley, Biochem. J., 1960, 74, 577–584 CAS; (b) R. A. Bednar, Biochemistry, 1990, 29, 3684–3690 CrossRef CAS PubMed; (c) F. Sardi, B. Manta, S. Portillo-Ledesma, B. Knoops, M. A. Comini and G. Ferrer-Sueta, Anal. Biochem., 2013, 435, 74–82 CrossRef CAS PubMed.
- (a) M. J. Torres, L. Turell, H. Botti, L. Antmann, S. Carballal, G. Ferrer-Sueta, R. Radi and B. Alvarez, Arch. Biochem. Biophys., 2012, 521, 102–110 CrossRef CAS PubMed; (b) L. Turell, R. Radi and B. Alvarez, Free Radical Biol. Med., 2013, 65, 244–253 CrossRef CAS PubMed; (c) R. E. Wang, L. Tian and Y. H. Chang, J. Pharm. Biomed. Anal., 2012, 63, 165–169 CrossRef CAS PubMed.
- E. G. Gianni, R. Testa and V. Savarino, Can. Med. Assoc. J., 2005, 172, 369–379 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08028g |
| This journal is © The Royal Society of Chemistry 2019 |