
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zero electrical power pump for portable high-performance liquid chromatography
Stelios
Chatzimichail
a,
Duncan
Casey
 bc and
Ali
Salehi-Reyhani
*ac
bc and
Ali
Salehi-Reyhani
*ac
aDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London, SE1 1DB, UK. E-mail: ali.salehi-reyhani@kcl.ac.uk
bBristol Centre for Functional Nanomaterials, HH Wills Physics Laboratory, Tyndall Avenue, Bristol, BS8 1TL, UK
cDepartment of Chemistry, Imperial College London, SW7 2AZ, UK. E-mail: ali.salehi-reyhani@imperial.ac.uk
First published on 1st October 2019
Abstract
A major trend in analytical chemistry is the miniaturization of laboratory instrumentation. We report a pump requiring no power to operate based on the controlled expansion of a pre-pressurised gas for use in portable applications of high-performance liquid chromatography. The performance of the gas pump is characterised and integrated into a compact liquid chromatography system capable of isocratic separations integrating an LED-based UV-absorption detector. The system weighed 6.7 kg when the mobile phase reservoir was fully charged with 150 mL solvent and included an on-board computer to control the system and analyse data. We characterise the flow-rate through chromatography columns with a variety of geometries and packing materials for a range of pressures up to 150 bar. The maximum variation in flow rate was measured to be 6.5 nL min−1, limited by the resolution of the flow detector. All tests were made on battery power and results are a mixture of those made in the laboratory and in the field. Additionally, we performed a series of 1 m drop tests on the device and show the system's high tolerance to mechanical shocks during operation in the field.
Introduction
High performance liquid chromatography (HPLC) is a gold standard analytical technique crucial to many industries worldwide in its ability to separate and identify chemicals in a complex mixture. LC systems that are portable are becoming increasingly attractive in their ability to perform measurements at the point of sampling. This has potential in field-based applications relating to water and food security; however, conventional LC systems are not suited to point-of-sampling analyses primarily due to their size and lack of robustness outside controlled laboratory conditions. Furthermore, there are significant pressures to minimising the cost of implementing field-based solutions in resource-poor settings. There are four core components of any LC system – 1, an eluent pump to drive the sample and mobile phase; 2, an injector assembly to introduce the crude sample onto the separation phase; 3, a column which separates analytes within the sample; and 4, a detector to record and ideally quantify the sample's individual components as they leave the column. In this work, we focus on the development of a pump system that enables miniaturization of the overall platform.The pursuit of field-based chemical analysis has a long history and is exemplified by early attempts to develop portable chromatography systems. The OB-4 chromatograph reported by Baram et al. in 1983 had dimensions of 70 cm × 30 cm × 50 cm (width × depth × height) and weighed 42 kg.1 In a follow-up report in 1996, they used an iteration of their field chromatograph that weighed 14 kg.2 Similar efforts were reported by Tulchinsky and St Angelo in 1998 with the a battery-operated portable which weighed 9.5 kg, not including the requisite solvent reservoirs or computer for acquisitions.3 In a similar vein, conventional HPLC systems have been installed in mobile vehicle-based laboratories in order to move the point of testing closer to the site of sampling. Each of these approaches has major limitations and has typically added to rather than reduced the complexity of the system in order to provide electrical power and improve the instruments’ robustness to shocks in the field.
Demonstrations of miniaturized LC of varying capability have been made over the last few years and represent a step-change in size and capability of portable solutions. Ishida et al. presented a compact electroosmotic pumping system used in conjunction with a microfluidic chip based column and electrochemical detector.4 The system was not able to achieve high pressure and fell short of LC pressures (0.75 MPa) restricting the choice of chromatographic columns and resulting in long analysis times, even for exemplar separations. Importantly it was the first demonstration of a completely integrated system incorporating a microfluidic chip being lightweight with an overall weight of 2 kg, not including the requisite computer system. Gu et al. have reported an electroosmotic pump capable of generating pressures exceeding 1200 bar (120 MPa) that show incredible potential in supporting miniaturised LC.5,6 However, electroosmotic pumps require high-voltages which necessitate bulky external power supplies, currently defeating the portability afforded by a microfluidic LC. Sharma et al. have characterized isocratic and gradient-based systems incorporating a compact nano-flow pump in a setup with potential for portability that could generate pressures in excess of 50 MPa. They demonstrated the reverse-phase isocratic separation of a series of alkyl benzenes and gradient separation of a three-component mixture of pesticides.7,8 The detector was initially lamp based which limited the potential for miniaturization; however, in a follow up report, an UV-LED based absorption detector was built and helped achieve sub-parts-per-million detection limits for adenosine-5′-monophosphate (AMP), a monomer in the production of RNA.9 UV-LEDs have been an enabling technology for miniaturised LC, with emissions as far down as 235 nm having been reported alongside methods to adequately stabilise their emission.10 Commercial systems based on the works cited here have been developed.
In general, however, groups seeking to miniaturize LC systems have done so by shrinking the size of conventional components. This introduces increased cost and complexity in manufacturing and has potential impacts upon reliability and consistency. The growing need for field-based or point-of-care measurements is best supported by miniaturized systems that not only maintain or exceed the typical performance of their lab-based counterparts but to do so in an affordable and therefore accessible manner. In this paper we report important steps toward achieving portable, high-performance and affordable LC through an approach to miniaturization of the high-pressure pump. Examples of low-pressure pneumatic pumping (up to approx. 6 bar) have been previously reported in open tubular ion chromatography systems; however, field-portable instruments have yet to be reported.11 The pumping mechanism exploits the latent potential energy of a pressurized gas and so has no electrical power requirements, making it highly amenable in supporting field-portable LC. We demonstrate that solvent may be driven through commercial narrow-bore LC columns by exploiting the controlled expansion of a pre-pressurized gas. Incorporating a gas “pump”, we have engineered an isocratic UV-absorption HPLC system the size of a small briefcase (Fig. 1). It is a stand-alone system, requiring only modest battery (6.5 Ah) power to support all on-board flow rate and pressure sensors, an absorption detector and computing unit with all necessary solvents being stored on-board. In this report, we characterize the pressure-flow behaviour of the device, its separation capability and demonstrate its resistance to mechanical shocks. All results are presented from data acquired on the device free from mains power, predominantly in the field.
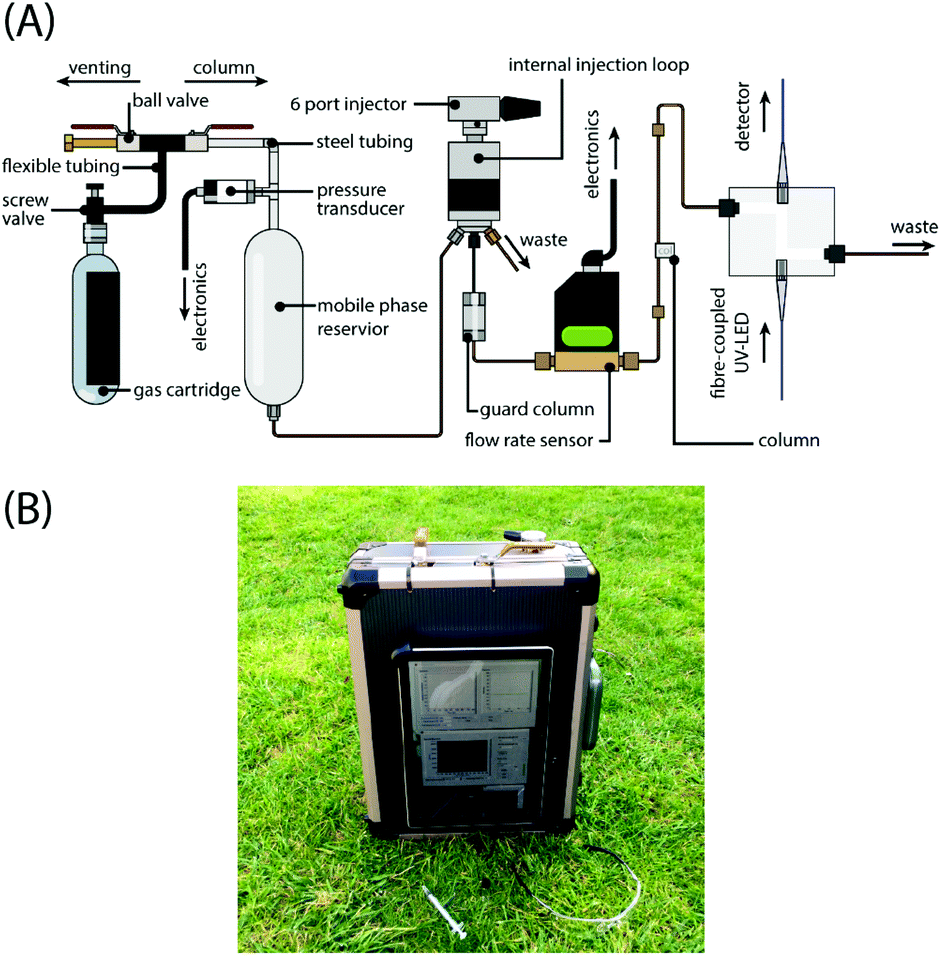 | ||
| Fig. 1 Portable high-performance liquid chromatography. (A) The schematic shows the key system components and their connectivity. The components are depicted for clarity and are not intended to dictate an orientation of operation. (B) Photograph of the ‘chromatography in a suitcase’ system running separations in the field. | ||
Experimental
Reagents
Analytical or HPLC grade solvents and chemicals were used as part of this work. Ultra-pure water was generated on-site (18.2 MΩ; Milli-Q system, Millipore). Isopropanol, L-tryptophan (≥98%) and L-tyrosine (≥98%) were purchased from Sigma-Aldrich. The mobile phase was 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) acetonitrile and water with 0.01% formic acid. Stock solutions of the amino acids were stored in the dark at 4 °C for a maximum of 7 days.
:50 (v/v) acetonitrile and water with 0.01% formic acid. Stock solutions of the amino acids were stored in the dark at 4 °C for a maximum of 7 days.
Instrumentation
The miniaturised LC system incorporates componentry to achieve high-performance liquid chromatography – (1) a pump and injector assembly, (2) a separation column, and (3) a flow cell and detector (Fig. 1). The system has a form factor of 330 mm (width) × 290 mm (depth) × 140 mm (height) and weighs 6.7 kg when fully charged with solvent, including all requisite computer systems and battery modules. The system and its connections are schematically depicted in Fig. 1A. Pump. The pressurised gas source was a nitrogen (oxygen-free) cylinder (BOC, UK) with a maximum fill pressure of 230 bar. This was used to fill a high pressure (3000 psi) 426 mL air tank (Tippmann; BZ Paintball Supplies, UK). The tank was connected via flexible tubing to a ball-valve assembly which allowed head pressure to be vented to atmosphere or transmitted through to the 150 mL mobile phase reservoir (Swagelok, UK). The connections to and upstream of the mobile phase reservoir were a combination of pipe fittings (Swagelok, UK) and stainless-steel tubing (Swagelok, UK). Downstream of the reservoir PEEK tubing and zero dead volume fittings were used to connect components. In the system reported here, the solvent reservoir is in direct contact with the gas pressurising the system head. This may preclude the use of certain combinations of gas and mobile phase owing to outgassing as the pressure drops through the system. This is problematic for the post-column detector where the passage of evolving gas bubbles alters the level of light absorption. Techniques such as enhanced-fluidity LC involves the addition of liquefied CO2 to conventional liquid mobile phases and has been shown to provide enhanced diffusivity, faster solute mass transfer, and reduced system backpressure.12,13 We have not, as yet, explored how this effect might be exploited using our system. We have found the low solubility of nitrogen permits the driving of mobile phase without necessitating a physical barrier or membrane to isolate the phases. To monitor the pressure in the system head up to a pressure of 400 bar a pressure transducer was fitted (Swagelok, UK) and connected to a tablet form-factor PC (Microsoft, UK) via an analog-to-digital converter (USB 6210, National Instruments; USA). Battery operation typically allows for operation times of 13 hours. The most significant limiting factor in the battery lifetime was the tablet computer employed to run laboratory development software. This could be extended significantly with the adoption of low-power single-board microprocessor systems. Injector. A 6-port injector with a 2 μL internal sample injection loop (Rheodyne 7725; IDEX Corporation, USA) was used to inject samples into the flow line. A guard column was fitted immediately downstream of the injector. Chromatography. Isocratic separations of the amino acids were performed using a range of reverse-phase C18 packed capillary, packed standard and monolith columns (length × inner diameter, packing): 30 mm × 150 μm, 3 μm (Agela, China); 70 mm × 150 μm, 5 μm (Agela, China); 80 mm × 150 μm, 5 μm (Agela, China); 150 mm × 1.0 mm, 5 μm (Sigma, UK); and 150 mm × 100 μm, monolith (Chromolith; Merck, UK). Detector. Analytes were detected based on their UV-absorption. The light source was a commercially available 275 nm UV-LED (UVTOP275; Ocean Optics, UK) fibre-coupled to a z-type flow cell with a 2 μL internal volume and 10 mm path length (Ocean Optics, UK). A miniaturised spectrometer (USB2000; Ocean Optics, UK) was fibre-coupled to the output of the flow-cell and used as the detector, which allowed for spectral discrimination without the need for additional filters. Electronics & Data acquisition. Data was acquired using custom-written code in LabView. Absorption was calculated by log10(I0/I) where I and I0 are the intensity of light transmitted through the sample and the initial light intensity measured at the baseline, respectively. For each run, system head pressure and mobile phase flow rate was recorded with each chromatogram. The spectrometer recorded at 10 Hz and the data were sample-averaged every 1 s.Results and discussion
Pump performance
One of the working principles in HPLC is the predictability of solvent flow rate through the system in order to achieve characteristic analyte retention times. If the profile of how flow rate changes as a function of time is known, it is possible in a simplified isocratic system to computationally compensate for this by post-processing chromatograms. Of course, achieving precise flow rates from the outset is desirable. In our system, solvent is driven through microfluidic tubing by the controlled expansion of a pressurised gas. In order to test the gas pump's performance, we attached the system to a lab-based industrial gas cylinder (Nitrogen (Oxygen free), 9.3 m3 tank volume, BOC UK) which allowed us to pressurise the head of the system in a facile manner without the need to detach and fill gas cartridges for each desired pressure; it also allowed us to measure the response of the flow rate to controlled jumps in pressure.In determining the capability of the pump, the flow rate of solvent through a range of packed capillary columns in addition to a monolith column was measured (Fig. 2A). Despite their advantages for portable systems capillary columns have not been adopted to any significant degree. Here we exploit their features of small size, high efficiency and low solvent consumption by incorporating them into the portable LC system. The system head was pressurised to a desired pressure between 0 and 150 bar and isolated from the cylinder using a valve. The 6-port injector valve was subsequently set to connect the pump via the sample loop to the downstream fluidics, and the mobile phase under pressure in the fluid reservoir flowed through the column. The gas pressure in the system head and fluid flow rates through the columns were monitored for a minimum duration of 100 s (typically 300 s) before testing subsequent pressures. A linear increase in flow rate as a function of increasing cylinder pressure was observed spanning ∼50 nL min−1 to ∼1 μL min−1, dependent on the column's geometry and packing type. In the present device, the pressure is directly controlled and, as expected, an increase in pressure leads to a higher flow rate for all the columns tested (Fig. 2A). Predictably, the lowest flow rates were measured for the columns with the highest back pressures. The operational range of flow rates is limited by the back pressure of the system, predominantly dictated by the dimensions of the column. By employing wider bore (>2 mm ID columns), flow rates approaching the mL min−1 range may be achieved. A caveat being the significantly higher consumption of solvent, which can be an issue for portable LC.
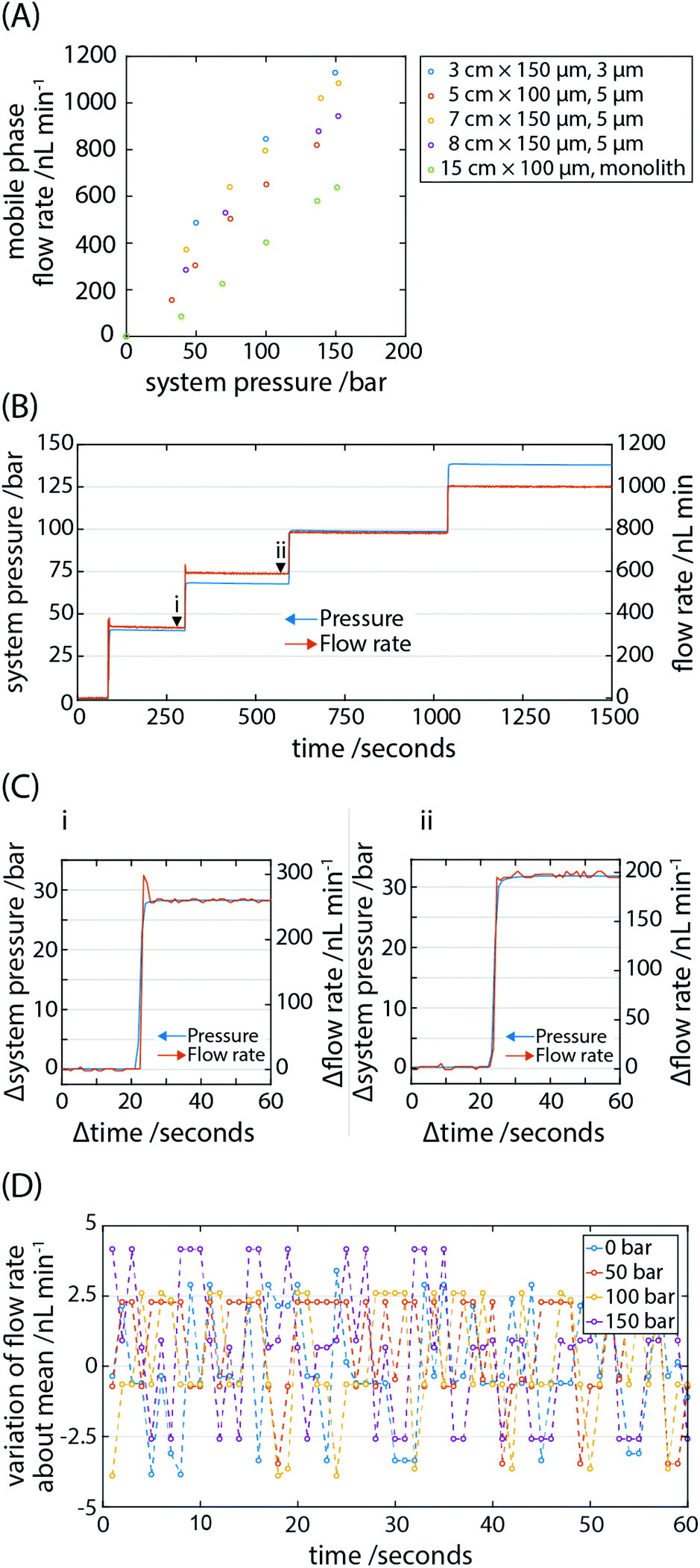 | ||
| Fig. 2 Pump performance. (A) The mobile phase flow rate is measured through a set of capillary columns over a range of system pressures, up to 150 bar. Figure legend specifies the column length, inner diameter and packing type; each column was packed with reverse-phase silica. The error bars lie within the points. (B) An exemplar time-resolved trace of mobile phase flow rate for a series of pressure jumps. (C) Two zoomed in time-resolved trace of a pressure jump where the black arrowheads labelled (i) and (ii) in (B) denote the start of each trace. The change in flow rate tracks the change in system pressure. (D) The variation in flow rate is plotted for a range of system pressures. The maximum peak to peak variation is 6.5 nL min−1 and invariant over the pressure range tested. | ||
We then tested the response of solvent flow rate in the system to both positive and negative jumps in pressure (Fig. 2B). To achieve an increase in pressure, the cylinder valve was opened and the cylinder regulator pressure increased until the desired pressure was indicated by the digital pressure transducer, not the regulator. To decrease pressure, a vent valve was opened briefly, and the flow rate was equally as responsive to drops in pressure. Pressure jumps were achieved by operating manual valves and regulator and were completed in under 5 s. The response in change of flow rate to changes in pressure was immediate (<1 s; Fig. 2C) and expected by Pascal's principle of transmission of fluid-pressure. It is anticipated that the use of digital or electronically operated valves would reduce the time to complete jumps in pressure, if necessary.
A drawback in employing syringe pumps to drive a mobile phase is their pulsatile behaviour at low flow rates and the time required for flow to stabilise. Due to the reciprocal switching behaviour of the driving pistons in a typical commercial HPLC pump, pulsatile flow can develop in the flow rate of the mobile phase. This leads to synchronous baseline fluctuations which can limit the sensitivity of the system. Pulse dampeners are almost universally employed to smooth the pressure-flow profile yet are unable to fully flatten them. We calculated the variation in mobile phase flow rate about the mean for a range of gas pressures up to 150 bar (Fig. 2D). To determine the baseline variation of the pressure transducer and flow sensor, the variation of flow rate about the mean was calculated when the system head was not pressurised i.e. at 0 bar. The peak to peak variation in flow rate was invariant over the pressure range tested. Since we do not measure variations in flow rate above the instrument noise floor even at the highest measured flow rates, we expect the flow rate fluctuation to be well below the absolute peak to peak variation of 6.5 nL min−1. We therefore concluded that we did not observe pulsatile flow in the gas-pumped system and the measured variation in pressure and mobile phase flow rate was limited by the resolution of the pressure transducer and flow rate sensor. For the maximum measured flow rates, the flow precision was ≤0.2% RSD. This compares favourably to current market solutions for research-grade reciprocating pumps, particularly given the low cost and amenability of a gas pump to portable LC.
Chromatographic separations
Although, amino acid analysis is considered to be the gold standard for quantitative peptide and protein analysis, alternative methods are often chosen due to their relatively lower cost, complexity and assay time. Such analyses tend to be performed using colorimetric methods in routine biochemistry or nitrogen determination in food analysis.14,15 For the latter, the Chinese milk scandal of 2008 revealed the vulnerability of standard methods to verify the protein content in milk-based infant formulas based on their inability to discriminate protein from other nitrogen-rich chemicals, or adulterants.16–18 Comprehensive amino acid analysis is necessary in a wide range of biologically facing research but it can be challenging to achieve sufficient sensitivity and selectivity without chemical derivatisation steps.19–21 Reports of derivatisation-free methods have been made22,23 and so the portable system reported here has potential in widening access to LC-based protein quantification methodologies.The system's ability to perform chromatographic separations was tested. The reverse-phase separation of a two-component mixture of the aromatic amino acids tryptophan and tyrosine was performed (Fig. 3A) owing to their favourable absorption at 275 nm, the peak wavelength of the UV-LED which serves as the light source in the UV-absorption detector. These results were measured in a stand-alone fashion whereby the LC system was operated on battery power (6.5 Ah) and the gas pump is supported by a gas cartridge (426 mL fill volume). The gas cartridge was pre-pressurised with nitrogen gas to 100 bar.
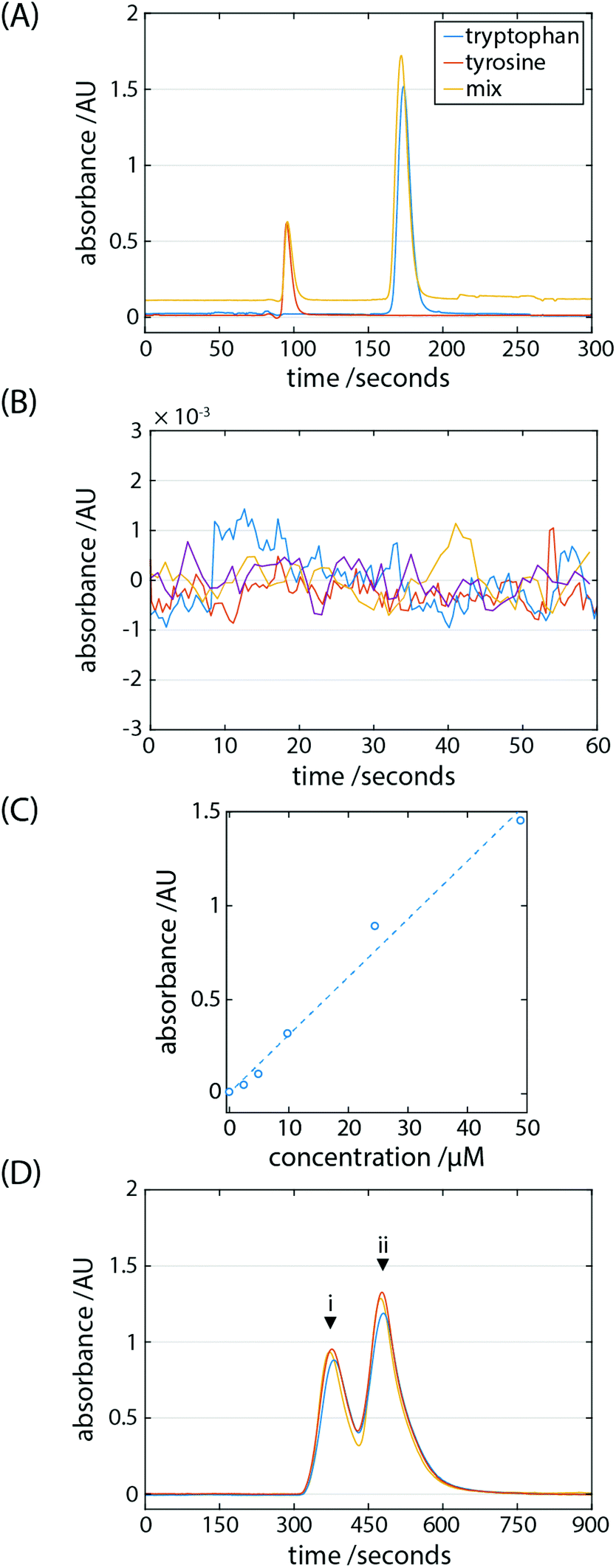 | ||
| Fig. 3 Separations are performed on the system using the pressurised-gas pump. (A) A mix of the amino acids tryptophan and tyrosine are separated and identified using a UV-LED absorption detector arrangement. The mix trace is vertically shifted for clarity. (B) The baseline noise of the detector is shown. (C) A concentration series is established plotting peak heights for tryptophan spanning 2.4 μM (4.8 pmol) to 49.0 μM (98 pmol). The error bars lie within the points. (D) A mix of the pesticides (i) thiabendazole and (ii) pyrimethanil are separated. | ||
The retention times of the analytes were 95.3 ± 0.4 s for tyrosine (n = 3) and 172.9 ± 1.1 s for tryptophan (n = 3). Using the half-peak height method, the column (100 mm) generated 1407 and 1819 theoretical plates for tyrosine and tryptophan, respectively; the peak symmetry factor was found to be 1.87 and 1.31 for tyrosine and tryptophan, respectively. To determine the limit of detection (LOD) of the system the baseline variation was measured (n = 4) to be 3.96 × 10−4 AU by flowing mobile phase only (Fig. 3B). The signal to noise ratio (SNR) can be defined as S/σN, where S is the peak intensity and σN is the standard deviation of the baseline signal; the SNR of 50 μM tyrosine and tryptophan was 1340 and 4100, respectively. A concentration series was established by injecting known concentrations of tryptophan ranging from 2.4 μM to 49.0 μM (Fig. 3C). Defined as 3.3σN/s, where s is the slope of the calibration curve, the LOD of tryptophan of 24.1 nM, or <25 ppb.
Pyrimethanil and thiabendazole are fungicidal pesticides which are applied post-harvest to fruits in order to limit spoilage and increase their shelf-life. Exposure to pesticides is a health concern, therefore, maximum residue levels are set in order to ensure safe and appropriate agricultural practice. Where in use, too little pesticide may lead to resistance in fungal strains thereby limiting their efficacy. Administration of the correct dosage, then, is essential and by extension so are accurate and reliable methods to monitor their levels. There are approximately 300 million central laboratory HPLC tests performed globally per year for the food safety sector. In the case of perishable foodstuffs where there may be a limited time window for testing, field tests are one approach to widening the scope of these tests in monitoring the amount of pesticides entering the supply chain. A separation of a mix of 1.4 mM pyrimethanil and 1.06 mM thiabendazole was performed (Fig. 3D). It is evident that the pesticides are being detected and separated, although with the conditions used here to a resolution short of the baseline. The retention times of the analytes were 381.3 ± 4.2 s for thiabendazole (n = 3) and 482.3 ± 5.6 s for pyrimethanil (n = 3).
Longevity of pump
During the course of a chromatographic run the pre-pressurised gas expands into the mobile phase reservoir in order to drive fluid through the system. As it does so the pressure in the gas reservoir and system head will reduce. A drop in pressure will in turn lead to a commensurate drop in mobile-phase flow rate from run to run. Whether the degree to which this occurs is significant will be in part dictated by the ratio of the volume of the gas cartridge (Vcartridge) and system head (Vhead) to the volume of mobile phase required for each run (Vrun); this is determined by the volume of the downstream fluidics, the mobile phase flow rate and the separation time required. In the embodiment in Fig. 1, the volume of the fluidics downstream of the solvent reservoir is 39.3 μL (300 μm ID tubing) in addition to the full volume of the column; this ranged from 0.4 μL (ID 100 μm, length 50 mm) to 78.5 μL (ID 1 mm, length 100 mm). The volume of the gas cartridge of 426 mL and the system head is 24.8 mL. For the separations in Fig. 3A, it is estimated that a 300 s separation time will require approximately 350 ± 5 μL for the steel column employed of dimensions ID 1 mm and length 100 mm (flow rate 70 ± 1 μL min−1). In this case the ratio of Vrun:(Vcartridge + Vhead) is 7.76 × 10−4. Therefore, such a small ratio ensures a minimal drop in pressure per chromatographic run.
This ratio is helpful to understand the number of runs for which the pumping strategy presented here could support before cumulative drops in pressure preclude the analytical goals of a chromatographic separation. How such constraints manifest will be application specific; however, for our purposes here it is helpful ascertain a ballpark estimate of when this might occur. For example, this might be governed by the absolute change in retention time for specific peaks or perhaps a time limit necessary to complete a chromatographic run. One might arbitrarily impose a constraint that the gas pressure must not drop below 90% of the initial fill-pressure. In this case, one may predict approximately 128 (1 mm ID column) to 13870 (100 μm ID column) supported chromatographic runs for the system described above dependent on the column employed. These estimates are promising for field-based applications. As is typical for lab-based HPLC, employing 4.6 mm ID columns, for instance, flow rates on the order of 1.0 mL min−1 may be achieved due to lower back pressures; however, the solvent required per run results in much fewer supported chromatographic runs, approximately only 10–20.
In terms of longevity, this suggests that the gas pump as reported is better suited to driving solvent through capillary column-based systems. The number of runs over which the pump may be considered useful in the example of the capillary column greatly exceeds the number of runs over which a typical column itself is generally considered to withstand.
To control flow, the system head may be isolated from the downstream fluidics by closing a ball valve. Flow through the system can be fully halted by operating a valve downstream to the column but, to prevent inadvertent damage to optical components, is placed upstream of the flow cell.
Mechanical stability
Due to the environment in which it will inevitably operate, a requirement of a portable LC platform is to be more mechanically stable than its lab-based counterpart. In our portable LC system incorporating a gas pump, curiously, the only moving components are the injector assembly and the mobile phase itself flowing through the fluidics. It is reasonable to expect that this may confer a good measure of mechanical stability to the device overall and result in a relatively low susceptibility to vibrations or physical impacts. We tested this by performing a series of 1 m vertical drop tests during operation (Fig. 4). The baseline noise after the impact was insignificantly different to that prior to impact suggesting that the drops had minimal or no effect on its operation. No published results of mechanical stability or impact analysis on HPLC systems are available and so no comparative assessment of our system is possible. Instrument manufacturers are known to conduct packaging testing to simulate mechanical shocks when being shipped, during which the instruments are not under operation.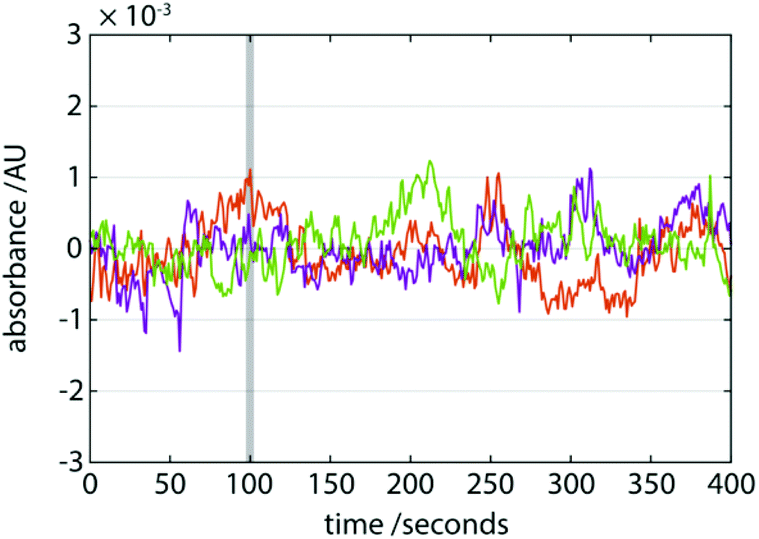 | ||
| Fig. 4 Mechanical stability of the device is tested by repeated 1 m drop tests. Traces (n = 3) show the baseline absorbance before and after impact. The grey vertical bar indicates the approximate time at which the device is released and impacts the floor. No change in baseline absorbance mean or variance is observed as a result of the drop tests. | ||
Conclusions
A major obstacle to miniaturisation of LC is the pump due to the onerous and simultaneous requirements for it to be small-form factor, achieve high-pressure and support stable flow – and achieve this in very challenging environments outside of a lab. We have shown how the controlled expansion of a pre-pressurised gas may be utilised to perform the role of a pump in a high-performance liquid chromatography system. This work characterises the pressure-flow characteristics of a mobile phase driven by the gas pump and demonstrates its use in a miniaturised, lightweight, portable LC system performing reverse-phase separations in the field. In addition to satisfying the requirements above, exploiting the latent energy of a pre-pressurized gas has the major advantage of not requiring power to operate; thereby significantly reducing the requirements of the battery. This in turn can lead to a much smaller class of fully integrated devices. Given the cost of a gas pump is considerably cheaper than conventional pumps, we expect this work to be of interest to those seeking to support or develop field-based applications that are also cost-sensitive.The isocratic separation and detection of analytes is expected to be useful in a number of applications demonstrated here without compromising system performance. Although this work has demonstrated the feasibility of the platform, several key engineering challenges remain. For instance, developing the system to be capable of gradient-based separations would help to broaden the range of applications, particularly to ones involving more complex mixtures. Of course, the operating environment is a very important consideration in portable LC applications and there should ideally be componentry to either control the internal environment of the instrument or active compensation mechanisms to vary pressure and/or flow rate. Given the low power requirements of our system it is possible to accommodate on-column or solvent reservoir heating elements to improve the system's resilience to environmental factors. We predict that the microfluidic volumes of the separation system would help in ensuring a good thermal response to such control mechanisms.
Conflicts of interest
ASR has a financial interest in intellectual property and commercialisation efforts related to the platform described here.Acknowledgements
This work was supported by the Engineering and Physical Science Research Council (EPSRC) through a fellowship (EP/S001603/2) and Impact Assessment Award (EP/R511559/1) to ASR.References
- G. I. Baram, M. A. Grachev, N. I. Komarova, M. P. Perelroyzen, Y. A. Bolvanov, S. V. Kuzmin, V. V. Kargaltsev and E. A. Kuper, J. Chromatogr. A, 1983, 264, 69–90 CrossRef CAS.
- G. I. Baram, J. Chromatogr. A, 1996, 728, 387–399 CrossRef CAS.
- V. M. Tulchinsky and D. E. St Angelo, Field Anal. Chem. Technol., 1998, 2, 281–285 CrossRef CAS.
- A. Ishida, M. Fujii, T. Fujimoto, S. Sasaki, I. Yanagisawa, H. Tani and M. Tokeshi, Anal. Sci., 2015, 31, 1163–1169 CrossRef CAS PubMed.
- C. Gu, Z. Jia, Z. Zhu, C. He, W. Wang, A. Morgan, J. J. Lu and S. Liu, Anal. Chem., 2012, 84, 9609–9614 CrossRef CAS PubMed.
- K. B. Lynch, A. Chen, Y. Yang, J. J. Lu and S. Liu, J. Sep. Sci., 2017, 40, 2752–2758 CrossRef CAS PubMed.
- S. Sharma, A. Plistil, R. S. Simpson, K. Liu, P. B. Farnsworth, S. D. Stearns and M. L. Lee, J. Chromatogr. A, 2014, 1327, 80–89 CrossRef CAS PubMed.
- S. Sharma, L. T. Tolley, H. D. Tolley, A. Plistil, S. D. Stearns and M. L. Lee, J. Chromatogr. A, 2015, 1421, 38–47 CrossRef CAS PubMed.
- S. Sharma, H. D. Tolley, P. B. Farnsworth and M. L. Lee, Anal. Chem., 2015, 87, 1381–1386 CrossRef CAS PubMed.
- Y. Li, P. N. Nesterenko, B. Paull, R. Stanley and M. Macka, Anal. Chem., 2016, 88, 12116–12121 CrossRef CAS PubMed.
- B. Yang, M. Zhang, T. Kanyanee, B. N. Stamos and P. K. Dasgupta, Anal. Chem., 2014, 86, 11554–11561 CrossRef CAS PubMed.
- M. J. Beres and S. V. Olesik, J. Sep. Sci., 2015, 38, 3119–3129 CrossRef CAS PubMed.
- Y. Wang and S. V. Olesik, Anal. Chem., 2019, 91, 935–942 CrossRef CAS PubMed.
- B. J. S. C. Olson and J. Markwell, Curr. Protoc. Pharmacol., 2007, 38, A.3A.1–A.3A.29 CrossRef PubMed.
- H. K. Maehre, L. Dalheim, G. K. Edvinsen, E. O. Elvevoll and I.-J. Jensen, Foods, 2018, 7, 5 CrossRef PubMed.
- J. R. Ingelfinger, N. Engl. J. Med., 2008, 359, 2745–2748 CrossRef CAS PubMed.
- J. W. DeVries, G. W. Greene, A. Payne, S. Zbylut, P. F. Scholl, P. Wehling, J. M. Evers and J. C. Moore, Int. Dairy J., 2017, 68, 46–51 CrossRef CAS.
- J. C. Moore, J. W. DeVries, M. Lipp, J. C. Griffiths and D. R. Abernethy, Compr. Rev. Food Sci. Food Saf., 2010, 9, 330–357 CrossRef CAS.
- M. P. Frank and R. W. Powers, J. Chromatogr. B, 2007, 852, 646–649 CrossRef CAS PubMed.
- T. Teerlink, P. A. M. Van Leeuwen and A. Houdijk, Clin. Chem., 1994, 40, 245–249 CAS.
- R. Schuster, J. Chromatogr. B: Biomed. Sci. Appl., 1988, 431, 271–284 CrossRef CAS.
- A. Hesse and M. G. Weller, J. Amino Acids, 2016, 2016, 1–8 CrossRef PubMed.
- M. Fountoulakis and H.-W. Lahm, J. Chromatogr. A, 1998, 826, 109–134 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |