
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailored homo- and hetero- lanthanide porphyrin dimers: a synthetic strategy for integrating multiple spintronic functionalities into a single molecule†
Jennifer J.
Le Roy‡
*a,
Jonathan
Cremers‡
b,
Isabel A.
Thomlinson
 b,
Michael
Slota
a,
William K.
Myers
c,
Peter H.
Horton
d,
Simon J.
Coles
d,
Harry L.
Anderson
*b and
Lapo
Bogani
*a
b,
Michael
Slota
a,
William K.
Myers
c,
Peter H.
Horton
d,
Simon J.
Coles
d,
Harry L.
Anderson
*b and
Lapo
Bogani
*a
aDepartment of Materials, University of Oxford, 16 Parks Rd, OX1 3PH, Oxford, UK. E-mail: lapo.bogani@materials.ox.ac.uk
bDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford OX1 3TA, UK
cCentre for Advanced ESR, Department of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK
dNational Crystallography Service, School of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
First published on 19th October 2018
Abstract
We present the design, synthesis and magnetic properties of molecular magnetic systems that contain all elements necessary for spin-valve control in molecular spintronic devices in a single molecule. We investigate the static and dynamic magnetic properties and quantum spin properties of butadiyne-linked homo- and hetero-nuclear lanthanide-porphyrin dimers. A heterometallated porphyrin dimer containing both TbIII and DyIII centres is created rationally by the stepwise oxidative homocoupling of distinct lanthanide-porphyrin monomers. TbIII and DyIII mononuclear porphyrin complexes, homodimers and heterodimers all exhibit slow magnetic relaxation below 10 kelvin under a static magnetic field. The coherence times for GdIII porphyrin monomers and dimers are found to be in excess of 3.0 μs at 2 K, allowing distinct magnetic manipulations in low temperature transport experiments.
Introduction
Covalently linked porphyrin oligomers constitute a thriving area of research, particularly as model systems for light harvesting, molecular electronics and catalysis.1 Hetero-metallic porphyrin systems have been studied as donor–acceptor dyads.2 They would also be extremely appealing for the creation of molecular spintronic devices,3 which are typically composed of a spin polariser and a spin analyser, setting and reading out the current, respectively. The ultimate miniaturisation limit of these technology lies in the atomic and molecular level, and there is considerable research effort aimed at reducing spintronic elements down to the single-molecule level.4 The magnetic complexes integrated into molecular spintronic devices have mainly been single-molecule magnets (SMMs), with a particular role played by bis(phthalocyaninato) rare earth sandwich complexes.5 At sufficiently low temperatures, SMMs exhibit molecular hysteresis and behave as magnetic switches that can assume two classical states with a long relaxation time from one to the other.6 The observed slow relaxation could thus allow integrating the spintronic analyser and polariser inside a single molecule.The development of a molecular spin valve requires the synthesis of rationally designed ad hoc systems that contain two different lanthanide-based SMMs. Recent advances in the synthesis of lanthanide hetero-dinuclear complexes have yielded prototypes for two-qubit molecular spin processors, which require coupled lanthanide centres.7 In contrast, for the aims of a molecular spin-valve, the spin centres must be magnetically shielded from one another, so as to operate independently. Moreover, each of the magnetic centres needs to be in contact with an electronic quantum dot, in order to optimise the spintronic transport signal. For these purposes the highly developed chemistry of porphyrin systems8 including homo-lanthanide dimers,8f–h may offer the degree of flexibility and reliability that is necessary for the rational tailoring of the magnetic molecules.
However, the use of metalloporphyrin oligomers in molecular magnetism9 is only scarcely investigated. Moreover, the creation of reliably hetero-dinuclear lanthanide complexes, requires the layout and implementation of a new methodology, in order to produce an original and interesting architecture.10 In particular, it is necessary to build a synthetic route that includes the introduction of protecting groups of very different polarity onto the mononuclear building blocks. Only in this way the subsequent separation by chromatography of the hetero-dinuclear assemblies becomes possible, in a reliable procedure.
Here we present the first synthetic approach to the rational creation of such molecular systems, concentrating on hetero-metallic rare-earth based porphyrin dimers. We analyse the magnetic properties of the independent building blocks and then concentrate on Dy–Tb dimers, where each lanthanide component acts as an individual magnetic source. The synthetic strategy is optimised to retain the presence of two electronic quantum dots in series, in the overall linear arrangement necessary to bridge electronic nanogaps. We investigate the quantum coherent properties of these molecular systems, paving the way to the creation of coherent spintronic devices. By pulsed electron paramagnetic resonance (EPR) we quantify the spin coherence times of Gd-based monomers and dimers, the magnetic interactions and the quantum features relevant for coherent manipulation.
Results and discussion
Molecular design and synthesis
Molecules for molecular spin valves need to possess two magnetic elements that can be tuned by an external magnetic field and can retain their magnetisation state for times as long as the measurement. Different conductivities can be obtained when the spins of the metal centres adopt a parallel or antiparallel configuration. This would constitute a fundamental, conceptual step forward in miniaturising devices to a truly molecular level. In addition, this strategy has the advantage of reducing the number of costly nanofabrication steps, by removing the need of magnetic electrodes.The main requirements that new systems need to fulfil are rather stringent: the magnetic centres need to be reliably slow-relaxing, i.e. they must both have spin-flip times (T1) comparable to the experimental timescale (i.e.>10 s) in the temperature range used for single-molecule transport measurements (sub-K, for our purposes); high magnetic anisotropy barriers; the molecule must allow electrical conductance via single-electron processes, so that the molecular electronic quantum dots must be easy to reduce or oxidise; on the contrary, the spin centres must not easily undergo oxidation–reduction reactions, in order to preserve the magnetic state while in operation; the spin centres must be different, and show different coercive fields; the quantum dots must be connected to the spin centres; the system must be stable enough to permit deposition on different surfaces; it is desirable that a coherent counterpart can also be created, with dephasing times (T2) as long as possible.
We use porphyrin ligands in order to exploit their rich chemistry,8 for their high chemical stability and tendency to crystallise (Fig. 1). Lanthanide insertion can be achieved, as previously reported,9 by heating the free-base porphyrin in the presence of an excess LnIIICl3 (Ln = Y, Gd, Tb, Dy) in a melt of diphenylether and imidazole. While many reports in the literature8 use molten imidazole as a solvent, diphenylether was an essential co-solvent for dissolving our porphyrins. Lanthanides are most commonly found in the +3 oxidation state and tend to adopt high coordination numbers both in the solid state and in solution due to their large ionic radii. Since porphyrins form di-anionic ligands which coordinate to metal ions through four nitrogen atoms, the LnIII ions are not satisfied by a single porphyrin ligand, and one or more supplementary ligands are required in the design of the molecule to balance the charge and account for the remainder of the coordination sphere.
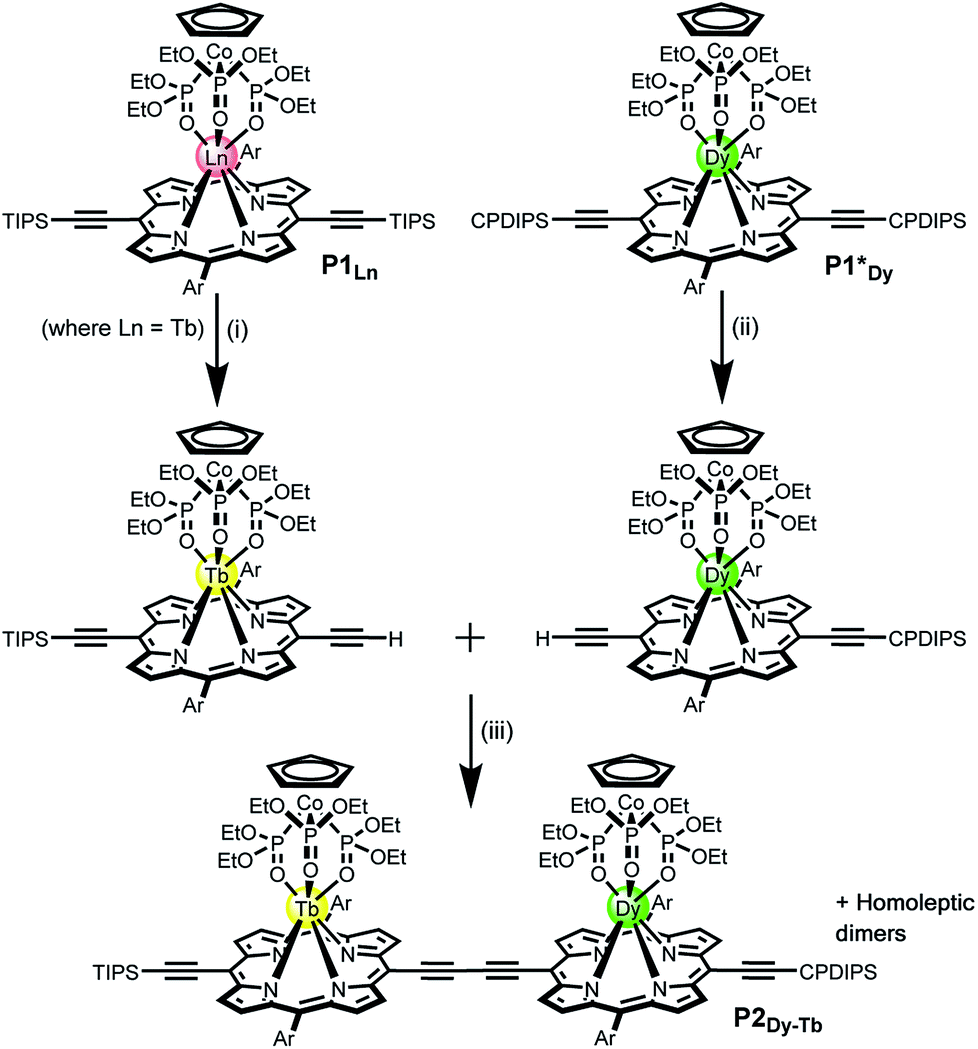 | ||
| Fig. 1 General structure of P1Ln and the synthetic route of the heterometallated dimer P2Dy-Tb (Ln = Tb). Reaction conditions: (i) TBAF, CHCl3/CH2Cl2, 50%; (ii) TBAF, CHCl3/CH2Cl2, 44%; (iii) Pd(PPh3)2Cl2, CuI, 1,4-benzoquinone, toluene/iPr2NH, 48%. Ln = Y, Gd, Tb, Dy; TIPS = triisopropylsilane; CPDIPS = cyanopropyldiisopropylsilane; Ar = 3,5-bis(tert-butyl)phenyl. | ||
For this work we used Kläui's tripodal ligand [CpCo{P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (OEt)2]3]− (Cp = cyclopentadienyl) (Fig. 1) as a redox-stable capping group for the porphyrins.9c,d The tridentate ligand has three coordinating oxygen atoms and a single negative charge, yielding a neutral 7-coordinate lanthanide complex. This choice of ligands is fundamental to the creation of a reliable system that can operate in the non-crystalline state and in molecular electronic devices on surfaces. The tripodal shape of the Kläui ligand effectively shields the LnIII from the environment. Moreover it confers solubility in most organic solvents, and its redox state and binding to Co(III) are resilient to a broad range of reaction and purification conditions. All these characteristics are required if this complex were to be grafted into a devices. Moreover the ligand has an encumbered geometry that is likely to favour grafting onto surfaces via the porphyrin system, thus facilitating the presence of a single geometry in the devices.
O) (OEt)2]3]− (Cp = cyclopentadienyl) (Fig. 1) as a redox-stable capping group for the porphyrins.9c,d The tridentate ligand has three coordinating oxygen atoms and a single negative charge, yielding a neutral 7-coordinate lanthanide complex. This choice of ligands is fundamental to the creation of a reliable system that can operate in the non-crystalline state and in molecular electronic devices on surfaces. The tripodal shape of the Kläui ligand effectively shields the LnIII from the environment. Moreover it confers solubility in most organic solvents, and its redox state and binding to Co(III) are resilient to a broad range of reaction and purification conditions. All these characteristics are required if this complex were to be grafted into a devices. Moreover the ligand has an encumbered geometry that is likely to favour grafting onto surfaces via the porphyrin system, thus facilitating the presence of a single geometry in the devices.
Statistical de-protection of the porphyrin monomers (P1Ln), followed by oxidative homo-coupling, was used for the formation of the homoleptic dimers (P2Ln2).11 We prepared homoleptic dimers containing two Gd, Tb and Dy centres. The hetero-metallated dimer containing both terbium and dysprosium centres was prepared via an alternative route. An analogue of the dysprosium monomer (P1*Dy) was prepared containing polar CPDIPS protected acetylenes. Oxidative coupling of a mixture of mono de-protected P1Tb and P1*Dy, resulted in a statistical mixture of three dimers which could be separated on silica based on the different polarity of the protecting groups. Successful separation was confirmed using MALDI (Fig. 2) where the hetero-dinuclear peak is clearly present with the expected isotopic pattern, and homo-dinuclear species are not present. This indicates that no metal switching occurs during the oxidative coupling of monomer units.
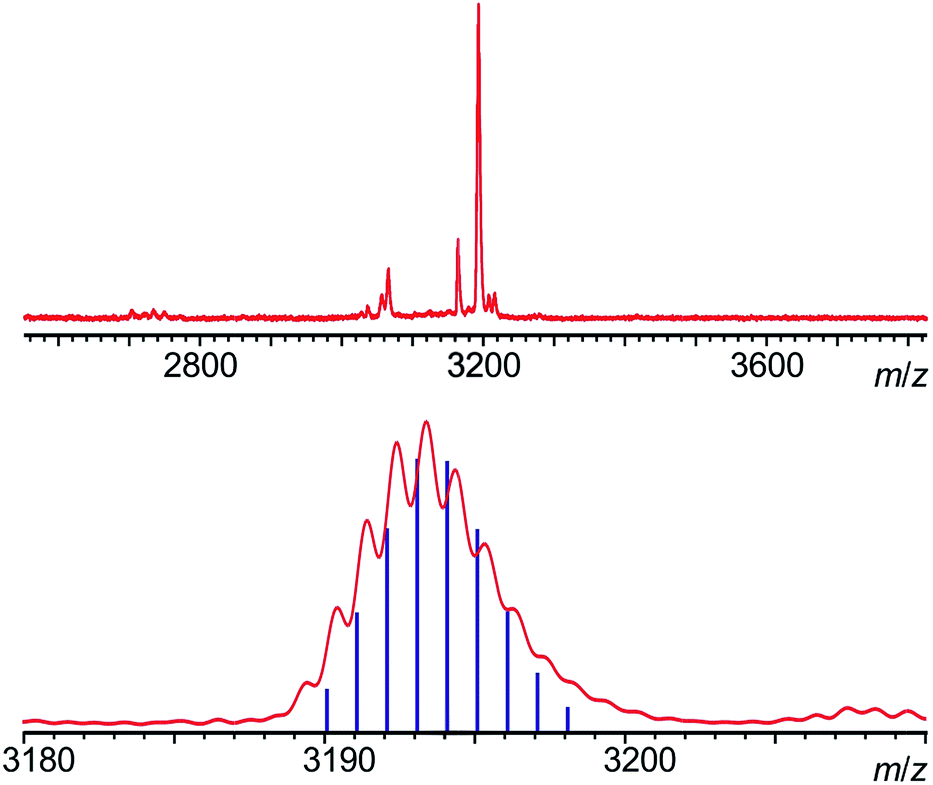 | ||
| Fig. 2 MALDI-TOF mass spectrum of P2Dy-Tb, acquired with a dithranol matrix. The absence of homodinuclear species (which would fall at 3188 and 3198 amu for homo-nuclear TbIII and DyIII complexes) is clearly observable (top), and the agreement of the isotopic pattern with the one expected for P2Dy-Tb is shown in the bottom. | ||
The presence of the paramagnetic lanthanide ions renders the 1H-NMR spectra of the complexes rather uninformative. A diamagnetic yttrium analogue (P1Y) was prepared in order to confirm the general structure by NMR spectroscopy (ESI†).
Structural characterisation
Single-crystals of the porphyrin monomers were grown by slow liquid diffusion of methanol into a CH2Cl2 solution of P1Ln. The homoleptic dimers (P2Ln2) yielded single crystals by slow evaporation of a solution of P2Ln2 in a mixture of ethanol/CH2Cl2.Single-crystals of each P1Y, P1Gd, P1Tb, P1Dy, P2Gd2, P2Dy2 and P2Tb2 were obtained and select crystallographic information is provided in Table 1. P1Gd, P1Tb, P1Dy and P1Y monomers display the same overall molecular connectivity but are not isostructural, with differing packing between the monomers resulting in the adoption of different space groups. P1Gd crystallizes in monoclinic Cc, P1Tb and P1Y crystallise in monoclinic P21/c and P1Dy crystallises in a triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Of these complexes, only the packing structure of P1Tb contains solvent molecules: one dichloromethane molecule per Tb complex. It is surprising that these monomer complexes crystallise differently because they were all crystallised under identical conditions. Additionally, for P1Dy there are two molecules in the asymmetric unit whereas all other monomers have one molecule per asymmetric unit. In related lanthanide structures9e the GdIII, TbIII and DyIII complexes all crystallise in a monoclinic P21/c space group. The discrepancy in packing arrangement is a manifestation of crystal polymorphism.12
space group. Of these complexes, only the packing structure of P1Tb contains solvent molecules: one dichloromethane molecule per Tb complex. It is surprising that these monomer complexes crystallise differently because they were all crystallised under identical conditions. Additionally, for P1Dy there are two molecules in the asymmetric unit whereas all other monomers have one molecule per asymmetric unit. In related lanthanide structures9e the GdIII, TbIII and DyIII complexes all crystallise in a monoclinic P21/c space group. The discrepancy in packing arrangement is a manifestation of crystal polymorphism.12
| Space group | Nearest intramolecular LnIII–LnIII (Å) | Nearest intermolecular LnIII–LnIII (Å) | PorphyrinCentroid–LnIII–CoIII (°) | PorphyrinCentroid–LnIII (Å) | LnIII–P or 1–P or 2–LnIII torsion (°) | CoIII–LnIII (Å) | |
|---|---|---|---|---|---|---|---|
|
a Data from ref. 9e: LnP LOEt− = [(LOEt)Ln-(TPP)]·0.25H2O where TPP = 5,10,15,20-tetraphenylporphyrinate, LOEt− = [(η5-C5H5)Co{P(O) (OEt)2}3]−.
|
|||||||
| P1Y | P21/c | — | 11.1407(3) | 177.83(4) | 1.1958(9) | — | 4.2699(4) |
| P1Gd | Cc | — | 12.10900(9) | 176.97(5) | 1.2360(11) | — | 4.3042(4) |
| P1Tb | P21/c | — | 9.87989(19) | 174.50(13) | 1.233(3) | — | 4.2467(14) |
| P1Dy(A) |
P |
— | 9.8418(3) | 170.48(9) | 1.2084(19) | — | 4.2457(8) |
| P1Dy(B) |
P |
9.8418(3) | 175.49(9) | 1.212(2) | 4.2410(8) | ||
| P2Gd2 |
P |
13.9202(7) | 10.3106(2) | 174.15(10) | 1.266(2) | 180.0 | 4.272(3) |
| P2Dy2 |
P |
13.8726(6) | 10.25340(18) | 174.63(7) | 1.2239(16) | 180.0 | 4.248(2) |
| P2Tb2 |
P |
13.9299(5) | 10.3082(1) | 174.47(9) | 1.2519(18) | 180.0 | 4.270(3) |
| GdP LOEt−a8e | C2/c | — | 10.14 | 177 | 1.23 | — | 4.30 |
| TbP LOEt−a8e | C2/c | — | 10.11 | 177 | 1.23 | — | 4.29 |
| DyP LOEt−a8e | C2/c | — | 10.10 | 177 | 1.22 | — | 4.28 |
| HoP LOEt−a8e | C2/c | — | 10.14 | 177 | 1.23 | — | 4.30 |
For simplicity only one monomer of the Dy-analogue will be described in detail, complete crystallographic data for all the compounds are available in the ESI.† For P1Dy the DyIII ion is sandwiched between four N atoms of the porphyrin ligand and three O atoms of the capping ligand resulting in a 7-coordinate complex (Fig. 3a). The two aryl pendants on the porphyrin are not parallel to the plane of the porphyrin, but are tilted at an angle of 67.42(16)° and 87.74(14)°. The DyIII ion sits 1.212(2) Å out of the plane of the porphyrin and 4.2410(8) Å from the CoIII ion. The Cp ring and porphyrin appendage are near parallel to each other with a tilt angle of 4.4(3)°. The porphyrin, DyIII and CoIII ions also have a near-linear arrangement with a porphyrin-centroid (CPor)–Dy–Co angle of 175.49(9)°, illustrated in Fig. 3a and b.
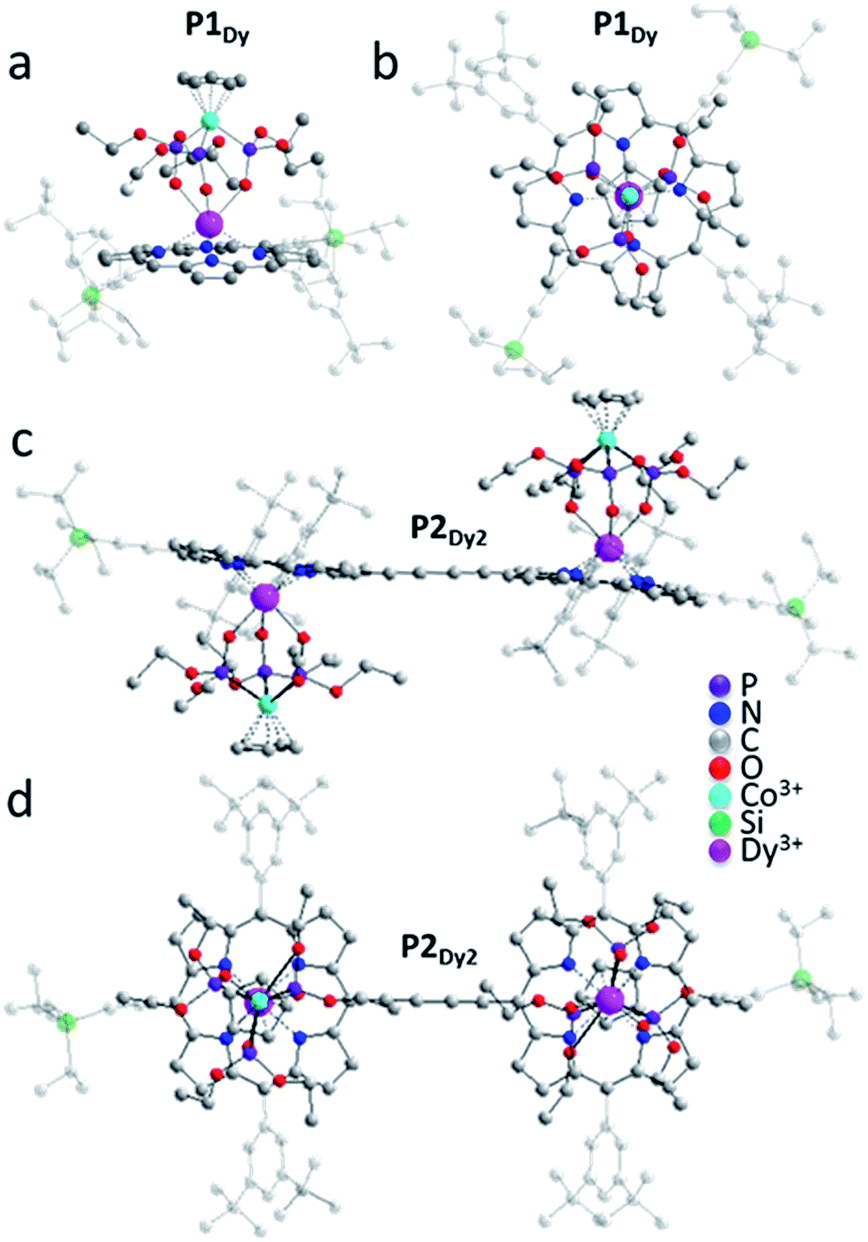 | ||
| Fig. 3 Crystal structures of representative examples of the investigated compounds, as acquired at room temperature. (a) Side view of P1Dy; (b) top view of P1Dy, along the Co–Dy vector; (c) side view of P2Dy2; (d) top view of P2Dy2. H atoms have been omitted for clarity. Triisopropylsilylethynyl and aryl groups are faded for clarity and the colour-scale is: carbon = light grey, oxygen = red, nitrogen = dark blue, phosphorous = purple, cobalt = light blue and dysprosium = pink. | ||
Examination of the four lanthanide monomers reveals that the CPor–Ln distance for P1Gd, P1Tb, P1Dy and P1Y decreased roughly in proportion to the atomic radius from P1Gd through P1Y. The different packing arrangement may explain why we do not observe any periodic trends related to intermolecular Ln–Ln (Å) distance or linearity of the complexes measured through the CPor–Ln–Co tilt angle (Table 1).
We next examined the solid state structures of three homoleptic lanthanide dimers. In contrast to the monomers, each of P2Gd2, P2Tb2 and P2Dy2 complexes crystallise in a triclinic P space group with no solvent molecules in the crystal lattice and all structures contain a centre of inversion. The nearest intramolecular Ln–Ln distance decreases moving from left to right across the lanthanide series P1Gd −13.9202(7) Å, P1Tb −13.9299(5) Å and P1Dy −13.8726(6) Å. There is a centre of inversion at the centre of each porphyrin dimer. This is reflected in the Ln–CPor–CPor–Ln torsion angle (180° in each dimer). We expect that the centre of inversion would not be maintained in solution or on a surface as the barrier to torsional rotation in a butadiyne-linked porphyrin dimer is quite low (ΔH = 5.27 kJ mol−1).13 There is very little deviation of the coordination environment surrounding the lanthanide ions in the monomer and dimer structures. SHAPE14 software was used to compare the polyhedral of P1Dy, P2Dy2, P1Tb, P2Tb2, P1Gd, P2Gd2 with other possible 7-vertex polyhedral. All 6 complexes exhibited predominately either capped octahedron or a capped trigonal prism (see full discussion in the ESI†).
The main objective of creating the P2Dy-Tb is to obtain hetero-dinuclear systems where every molecule in the crystal contains one DyIII centre and one TbIII centre. We demonstrated the purity of this hetero-dimer using Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) mass spectrometry, which is an excellent way of identifying perfectly hetero-dinuclear compounds, as they provide a starkly different mass value and isotopic pattern for hetero and homo-dinuclear molecules (Fig. 2).
Magnetic properties
Each of P1Gd, P1Tb, P1Dy, P2Gd2, P2Dy2, P2Tb2 and P2Dy-Tb have an overall charge of zero. The CoIII ion is diamagnetic and the magnetic properties discussed below originate solely from the localized spin of the LnIII ion(s). We shall investigate both the static and dynamic properties of the systems, relevant to their suitability as spin-valve components.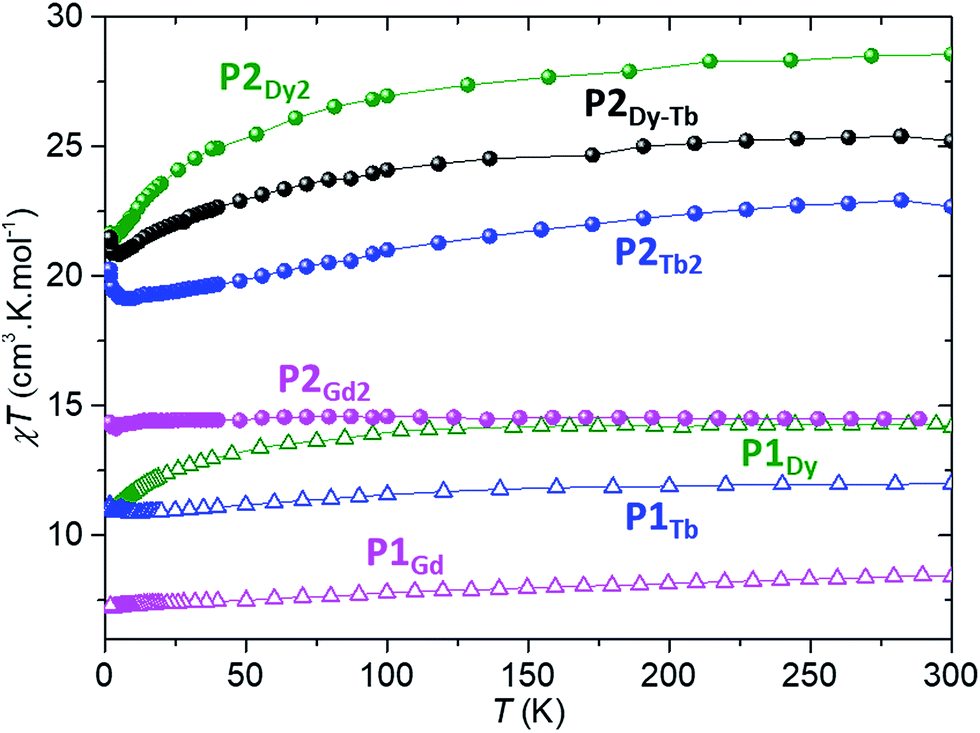 | ||
| Fig. 4 Temperature dependence of the static magnetic susceptibility times the temperature, for all lanthanide-porphyrin complexes described in the text. Spheres indicate dinuclear compounds, open triangles mononuclear ones; Gd compounds are in pink, Dy in green, Tb in blue and the hetero-dinuclear one in black. All measurements were performed on crystalline powder samples under an external static magnetic field of 1 kOe. | ||
The room temperature χT values for each P2Gd2, P2Tb2, P2Dy2 and P2Dy-Tb dimers are 14.50, 22.69, 28.56 and 25.22 cm3 kmol−1 and are in good agreement with twice the theoretical values of monomers 15.76, 23.64, 28.34 and 25.99 cm3 kmol−1 respectively. For P2Gd2 the χT product remains basically unchanged from room temperature to 5 K, with only a very slight decrease below 5 K. This agrees well with an S = 7/2 centre with no anisotropy and with very small anti-ferromagnetic interactions. For P2Tb2 and P2Dy2 the χT product decreases steadily from 300 K down to 5 K, only to eventually increase again below 5 K. In this case, the marked decrease below 50 K is attributed primarily to the large inherent magnetic anisotropy of DyIII and TbIII ions, while the slight low temperature increase may indicate weak ferromagnetic coupling between intramolecular LnIII ions. Such magnetic interactions can be dipolar or exchange-coupling in nature. The distances between Ln ions are presented in Table 1.
The two monomeric systems, P1Tb and P1Dy, show very similar slow magnetic relaxation dynamics, with frequency dependent peaks that shift to higher frequencies on increasing T (Fig. 5a and c). Using the χ′′ peak maxima determined using a Lorentz fit, and the Arrhenius law (τ = τ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(Ueff/kBT) where τ = 1/(2πυ) and υ is the frequency corresponding to the maxima of each χ′′ peak), the effective energy barriers were obtained; P1Tb, Ueff = 7(2) K with a τ0 = 3.4 × 10−5 s and P1Dy, Ueff = 14(4) K with a τ0 = 2.5 × 10−6 s. The relaxation barrier and τ0 for P1Dy is very close to that reported for the similar 10,15,20-tetraphenylporphyrin dysprosium complex.9e
exp(Ueff/kBT) where τ = 1/(2πυ) and υ is the frequency corresponding to the maxima of each χ′′ peak), the effective energy barriers were obtained; P1Tb, Ueff = 7(2) K with a τ0 = 3.4 × 10−5 s and P1Dy, Ueff = 14(4) K with a τ0 = 2.5 × 10−6 s. The relaxation barrier and τ0 for P1Dy is very close to that reported for the similar 10,15,20-tetraphenylporphyrin dysprosium complex.9e
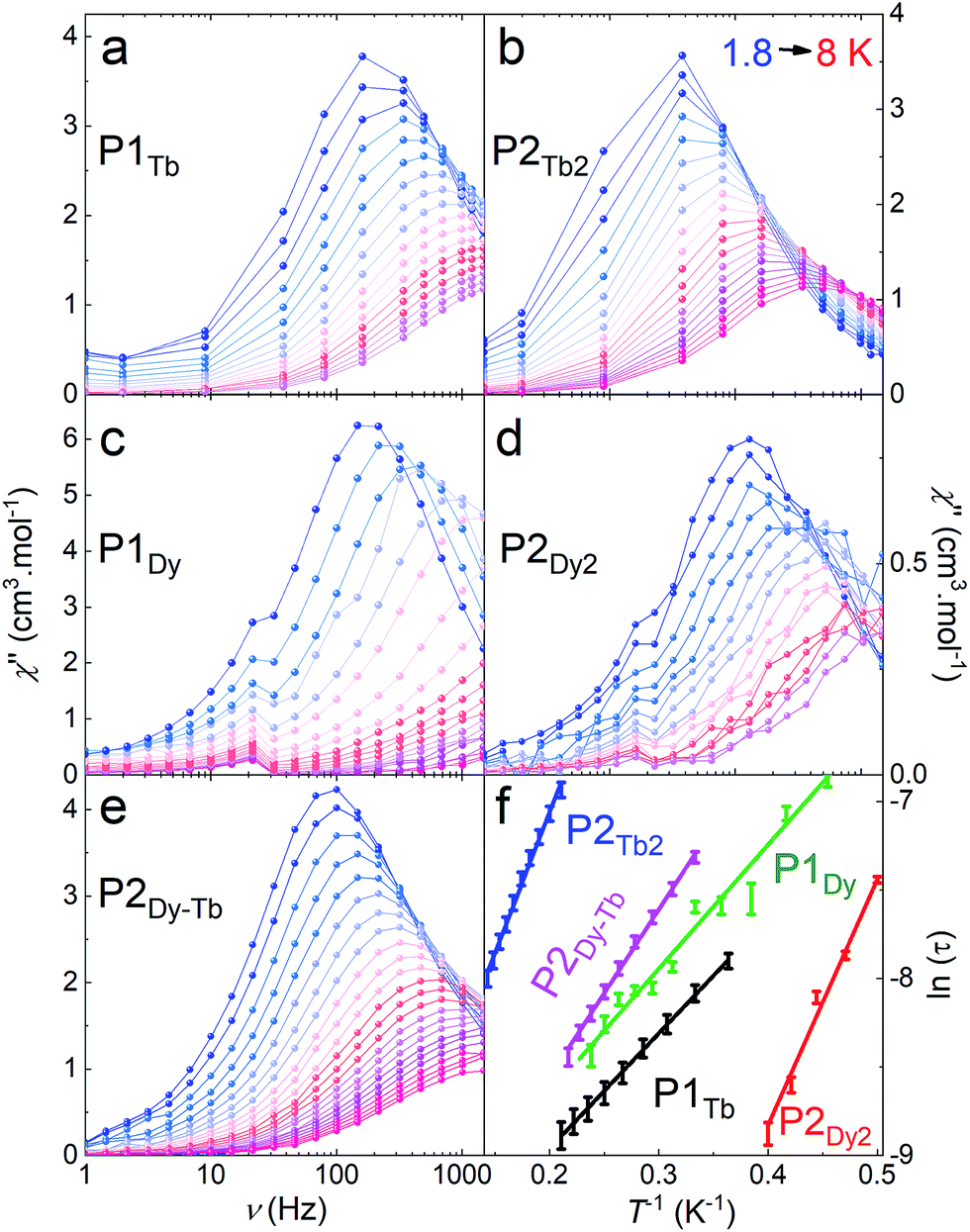 | ||
| Fig. 5 Dynamic magnetic susceptibility of crystalline samples of P1Tb (a), P2Tb2 (b), P1Dy (c), P2Dy2 (d) and P2Dy-Tb (e) for different temperatures (colour scale). All measurements were performed under a 2000 Oe constant magnetic field, in a 1–1500 Hz frequency range. (f) A plot of ln(τ) versus 1/T for all compounds, showing the Arrhenius behaviour. Data points are represented with error bars obtained from the fitting of the peak maxima of the frequency dependence in a–e with Lorentzian curves. Solid lines represent linear regression to the data. | ||
Homoleptic dimers P2Tb2 and P2Dy2 displayed slightly different magnetic properties to their corresponding monomer complexes: P2Tb2, Ueff = 15(2) K with τ0 = 3.6 × 10−5 s; P2Dy2, Ueff = 7(2) K with a τ0 = 4.5 × 10−5 s. The slight differences in the Ueff between mono- and bimetallic complexes is most likely a result of the slight change in the second coordination sphere where the TIPS pendent is replaced by an acetylene resulting in a less symmetric second coordination sphere. Changes to the second-coordination sphere can often influence the magnetic properties of lanthanide ions.15P2Dy-Tb also showed similar behaviour to the above with Ueff = 9(2) K with τ0 = 2.9 × 10−5 s confirming slow-magnetic relaxation at low temperatures.
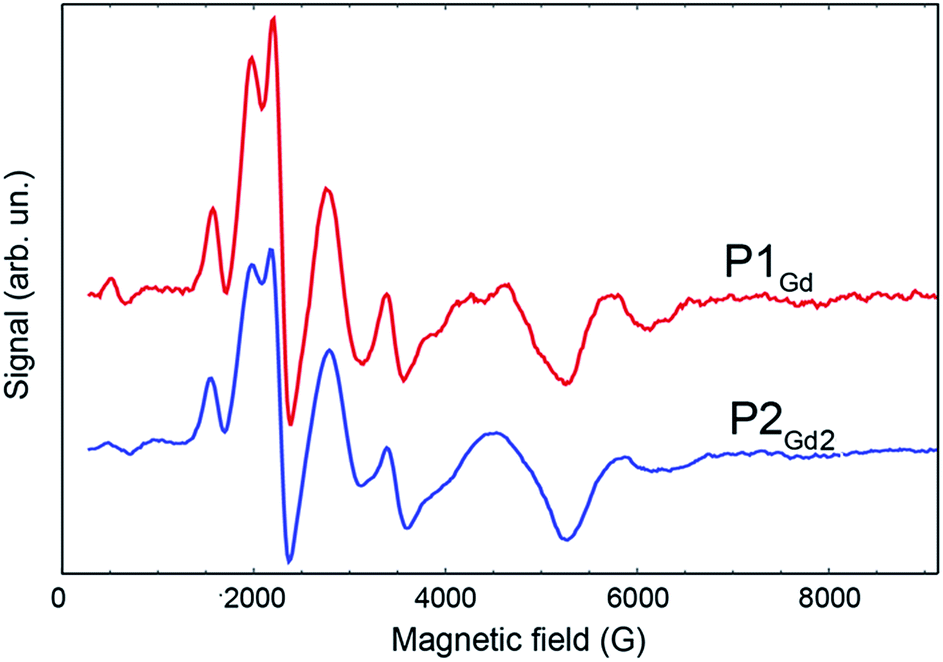 | ||
| Fig. 6 EPR spectra comparing the derivative of the free-induction-decay-detected absorption of P1Gd (red line) and P2Gd2 (blue line). The spectra were acquired at X-band frequency (9.4 GHz) at 3 K in a 1:1 mixture of dichloromethane and toluene. | ||
The phase-memory times, TM, were determined using a variation of the Hahn-echo sequence (Fig. 7, ESI for details†). TM is found to increase steadily on lowering the temperature, with an almost linear trend from 10 to 2 K, reaching 3.6 μs and 3.0 μs for P1Gd and P2Gd2, respectively. This indicates only a marginal decoherence time introduced by the dimerisation, and provides times that amply meet the requirements for possible quantum operations. The echo signal decays mono-exponentially versus the free evolution time τ of the Hahn echo (ESI†), indicating no spectral diffusion. Analogously, no detectable dependence of the T2 times on the magnetic field is observed (ESI†). This means that the dinuclear system will indeed appear composed of two distinct magnetic centres in low temperature transport experiments. Measurements of the single molecule transport and magnetic properties are currently underway.
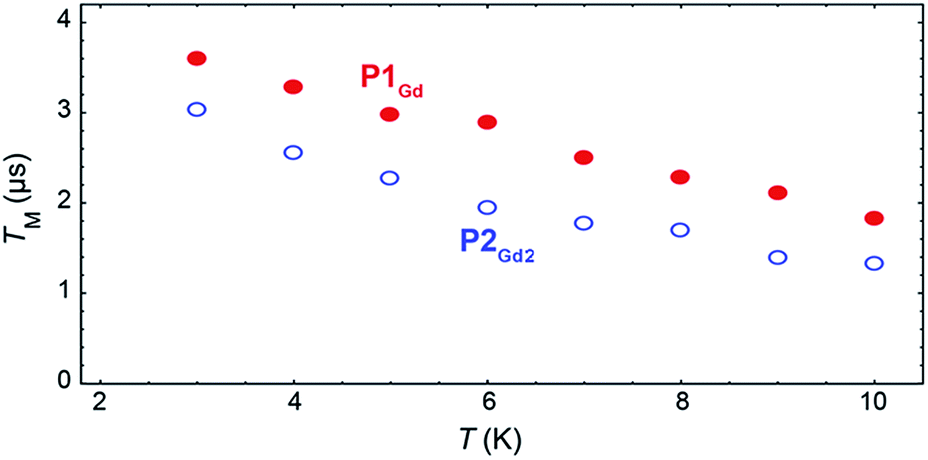 | ||
| Fig. 7 Temperature dependence of the quantum coherence time TM for P1Gd (red full dots) and P2Gd2 (blue empty dots), as measured at 344 mT at 9.37 GHz in a 1:1 mixture of dichloromethane and toluene. | ||
Conclusions
In conclusion, we have created hetero-dinuclear single molecule magnets based on porphyrin ligands, using a rational stepwise approach. We demonstrated the use of protecting groups of very different polarity for the selective creation of hetero-metallic porphyrin complexes. The protecting group methodology can now be used as a tool for the tuning of functional poly-porphyrin molecules, providing a route to hetero-dinuclear molecular magnets.7,16 From this perspective, the dimers here created are the first that are sufficiently stable to undergo purification steps such as chromatographic separations.An extension of these novel synthetic approach would be to rationally assemble poly-heteronuclear lanthanide chains. For example, tetra- and hexa-heteronuclear chains with a pre-defined order of the magnetic centres are planned via stepwise repetition of the oxidative homo-coupling step. Including the optical properties of the porphyrin ligands themselves, future work will include addition of functional groups such as luminescent appendages, light antennae or anchoring groups.
The magnetic properties of the compounds created show the desired slow magnetic relaxation and the coherent properties indicate μs coherence times at low temperatures for GdIII compounds. These compounds offer a testbed to probe magnetic interactions in molecular systems, and indicate a possible applicative direction for ring-shaped poly-porphyrins17 in spintronics and in single-molecule EPR investigations.18 All stringent requirements for the creation of spin-valves are simultaneously met and the new molecules show slow relaxation, magnetic anisotropy, the presence of molecular electronic quantum dots, different spin centres and stability that permits deposition on different surfaces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr F. Tuna and Prof. R. Winpenny for SQUID time at Manchester University during a downtime in Oxford. We acknowledge financial support by the European Union (ERC-StG-338258 OptoQMol, ERC-AdG-320969) and Marie Skłodowska-Curie Research Fellowship MC-SpinReMag-707252, the Violette & Samuel Glasstone Research Fellowship, the Royal Society (URF and Grant funds), EPSRC (QuEEN grant), EPSRC (Centre for Advanced ESR spectroscopy (CAESR) at the University of Oxford (EP/L011972/1)), German DV scholarship and Corpus Christi College. We thank the UK National Mass Spectrometry Facility at Swansea University for recording mass spectra.Notes and references
- (a) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC; (b) D. Holten, D. F. Bocian and J. S. Lindsey, Acc. Chem. Res. 2002, 35, 1, 57 Search PubMed; (c) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS PubMed; (d) G. Sedghi, V. M. García-Suárez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martin, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517 CrossRef CAS PubMed; (e) M. D. Peeks, C. E. Tait, P. Neuhaus, G. M. Fischer, M. Hoffmann, R. Haver, A. Cnossen, J. R. Harmer, C. R. Timmel and H. L. Anderson, J. Am. Chem. Soc., 2017, 139, 10461 CrossRef CAS PubMed.
- (a) A. Harriman, F. Odobel and J. P. Sauvage, J. Am. Chem. Soc., 1995, 117, 9461 CrossRef CAS; (b) J. Fortage, J. Boixel, E. Blart, L. Hammarstrçm, H. C. Becker and F. Odobel, Chem.–Eur. J., 2008, 14, 3467 CrossRef CAS PubMed; (c) J. Cremers, S. Richert, D. Kondratuk, T. D. W. Claridge, C. Timmel and H. L. Anderson, Chem. Sci., 2016, 7, 6961 RSC.
- (a) S. A. Wolf, D. D. Awschalom, R. A. Buhrman, J. M. Daughton, S. von Molnár, M. L. Roukes, A. Y. Chtchelkanova and D. M. Treger, Science, 2001, 294(5546), 1488 CrossRef CAS PubMed; (b) M. Slota, A. Keerthi, W. K. Myers, E. Tretyakov, M. Baumgarten, A. Ardavan, H. Sadeghi, C. J. Lambert, A. Narita, K. Müllen and L. Bogani, Nature, 2018, 557, 691 CrossRef CAS PubMed.
- (a) X. Sun, S. Vélez, A. Atxabal, A. Bedoya-Pinto, S. Parui, X. Zhu, R. Llopis, F. Casanova and L. E. Hueso, Science, 2017, 357(6352), 677 CrossRef CAS PubMed; (b) S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336 RSC; (c) C. Cervetti, A. Rettori, M. G. Pini, A. Cornia, A. Repollés, F. Luis, M. Dressel, S. Rauschenbach, K. Kern, M. Burghard and L. Bogani, Nat. Mater., 2016, 15, 164 CrossRef CAS PubMed; (d) E. Ö. Karabulut, F. Malet, A. L. Fetter, G. M. Kavoulakis and S. M. Reimann, New J. Phys., 2016, 18, 015013 CrossRef; (e) D. D. Awschalom, L. C. Bassett, A. S. Dzurak, E. L. Hu and J. R. Petta, Science, 2013, 339, 1174 CrossRef CAS PubMed; (f) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed; (g) S. D. Jiang, K. Goß, C. Cervetti and L. Bogani, Sci. China: Chem., 2012, 55(6), 867 CrossRef CAS; (h) L. Bogani, Jpn. J. Appl. Phys., 2011, 109, 07B115 CrossRef; (i) L. Bogani, R. Maurand, L. Marty, C. Sangregorio, C. Altavilla and W. Wernsdorfer, J. Mater. Chem., 2010, 20, 2099–2107 RSC; (j) C. Cervetti, E. Heintze and L. Bogani, Dalton Trans., 2014, 43, 4220–4232 RSC; (k) E. Heintze, F. El Hallak, C. Clauß, A. Rettori, M. G. Pini, F. Totti, M. Dressel and L. Bogani, Nat. Mater., 2013, 12, 202 CrossRef CAS PubMed.
- (a) S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135 CrossRef CAS PubMed; (b) F. Troiani, C. Godfrin, S. Thiele, F. Balestro, W. Wernsdorfer, S. Klyatskaya, M. Ruben and M. Affronte, Phys. Rev. Lett., 2017, 118, 257701 CrossRef CAS PubMed; (c) A. Candini, S. Klyatskaya, M. Ruben, W. Wernsdorfer and M. Affronte, Nano Lett., 2011, 11, 2634 CrossRef CAS PubMed; (d) M. Urdampilleta, S. Klayatskaya, M. Ruben and W. Wernsdorfer, ACS Nano, 2015, 9(4), 4458 CrossRef CAS PubMed; (e) M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 165 CrossRef CAS PubMed; (f) M. Misiorny, E. Burzurì, R. Gaudenzi, K. Park, M. Leijnse, M. R. Wegewijs, J. Paaske, A. Cornia and H. S. J. van der Zant, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 035442 CrossRef; (g) E. Burzurì, Y. Yamamoto, M. Warnock, X. Zhong, K. Park, A. Cornia and H. S. J. van der Zant, Nano Lett., 2014, 14, 3191 CrossRef PubMed; (h) R. Frisenda, G. D. Harzmann, J. A. Celis Gil, J. M. Thijssen, M. Mayor and H. S. J. van der Zant, Nano Lett., 2016, 16, 4733 CrossRef CAS PubMed; (i) J. Ponce, C. R. Arroyo, S. Tatay, R. Frisenda, P. Gaviña, D. Aravena, E. Ruiz, H. S. J. van der Zant and E. Coronado, J. Am. Chem. Soc., 2014, 136, 8314 CrossRef CAS PubMed; (j) D. Schmid-Lorch, T. Häberle, F. Reinhard, A. Zappe, M. Slota, L. Bogani, A. Finkler and J. Wrachtrup, Nano Lett., 2015, 15, 4942–4947 CrossRef CAS PubMed.
- (a) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 66 CrossRef CAS; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS; (c) J. J. Le Roy, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2014, 136, 8003 CrossRef CAS PubMed; (d) S. Chen, V. Mereacre, C. E. Anson and A. K. Powell, Dalton Trans., 2016, 45, 98 RSC; (e) M. Gysler, F. El Hallak, L. Ungur, R. Marx, M. Hakl, P. Neugebauer, Y. Rechkemmer, Y. Lan, I. Sheikin, M. Orlita, C. E. Anson, A. K. Powell, R. Sessoli, L. F. Chibotaru and J. van Slageren, Chem. Sci., 2016, 7, 4347 RSC; (f) T. Lacelle, G. Brunet, R. J. Holmberg, B. Gabidullin and M. Murugesu, Cryst. Growth Des., 2017, 17(10), 5044 CrossRef CAS; (g) J. J. Baldoví, Y. Duan, C. Bustos, S. Cardona-Serra, P. Gouzerh, R. Villanneau, G. Gontard, J. M. Clemente-Juan, A. Gaita-Ariño, C. Giménez-Saiz, A. Proust and E. Coronado, Dalton Trans., 2016, 45, 16653 RSC; (h) M. D. Jenkins, Y. Duan, B. Diosdado, J. J. García-Ripoll, A. Gaita-Ariño, C. Giménez-Saiz, P. J. Alonso, E. Coronado and F. Luis, Phys. Rev. B, 2017, 95, 064423 CrossRef; (i) J. D. Hilgar, B. S. Flores and J. D. Rinehart, Chem. Commun., 2017, 53, 7322 RSC; (j) J. Xiong, H.-Y. Ding, Y.-S. Meng, C. Gao, X.-J. Zhang, Z.-S. Meng, Y.-Q. Zhang, W. Shi, B.-W. Wang and S. Gao, Chem. Sci., 2017, 8, 1288 RSC; (k) C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, Angew. Chem., Int. Ed., 2017, 56, 10103 CrossRef CAS PubMed; (l) M. Ding, G. E. Cutsail, D. Aravena, M. Amoza, M. Rouzières, P. Dechambenoit, Y. Losovyj, M. Pink, E. Ruiz, R. Clérac and J. M. Smith, Chem. Sci., 2016, 7, 6132 RSC; (m) E. Kiefl, M. Mannini, K. Bernot, X. Yi, A. Amato, T. Leviant, A. Magnani, T. Prokscha, A. Suter, R. Sessoli and Z. Salman, ACS Nano, 2016, 10, 5663 CrossRef CAS PubMed; (n) F. Pointillart, K. Bernot, B. Le Guennic, S. Golhen, O. Cador and L. Ouahab, Angew. Chem., Int. Ed., 2015, 54, 1504 CrossRef CAS PubMed; (o) J.-K. Ou-Yang, N. Saleh, G. Fernandez Garcia, L. Norel, F. Pointillart, T. Guizouarn, O. Cador, F. Totti, L. Ouahab, J. Crassous and B. Le Guennic, Chem. Commun., 2016, 52, 14474 RSC; (p) L. Bogani, Jpn. J. Appl. Phys., 2011, 109, 07B115 CrossRef.
- D. Aguilà, L. A. Barrios, V. Velasco, O. Roubeau, A. Repollés, P. J. Alonso, J. Sesé, S. J. Teat, F. Luis and G. Aromí, J. Am. Chem. Soc., 2014, 136, 14215 CrossRef PubMed.
- (a) K. Ueta, K. Naoda, S. Ooi, T. Tanaka and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 7223 CrossRef CAS PubMed; (b) C. Stähler, D. Shimizu, K. Yoshida, K. Furukawa, R. Herges and A. Osuka, Chem.–Eur. J., 2017, 23, 7217 CrossRef PubMed; (c) R. Wang, A. M. Brugh, J. Rawson, M. J. Therien and M. D. E. Forbes, J. Am. Chem. Soc., 2017, 139(29), 9759 CrossRef CAS PubMed; (d) Y. Bai, J.-H. Olivier, H. Yoo, N. F. Polizzi, J. Park, J. Rawson and M. J. Therien, J. Am. Chem. Soc., 2017, 139(46), 16946 CrossRef CAS PubMed; (e) B. Basic, O. B. Locos, L. Rintoul, J. Kan, J. Jiang and D. P. Arnold, ChemPlusChem, 2014, 79, 813 CrossRef CAS; (f) W.-K. Wong, L. Zhang, F. Xue and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1999, 0, 3053 RSC; (g) C.-M. Che and S.-Y. Wong, US pat., US20100069345A1, 2005; (h) Y. V. Korovina, N. V. Rusakova, Z. I. Zhilina, Y. V. Ishkovb, S. V. Vodzinsky and V. P. Dotsenko, Mendeleev Commun., 2002, 12(4), 151 CrossRef.
- (a) J. H. Shi, Z. Li, W. Y. Liu, M. Yu, Z. H. Li and G. F. Liu, Inorg. Nano-Met. Chem., 2012, 43, 316 Search PubMed; (b) A. G. Coutsolelos and G. A. Spyroulias, Polyhedron, 1994, 13, 647 CrossRef CAS; (c) W. Kläui, H. Neukomm, H. Werner and G. Huttner, Chem. Ber., 1977, 110, 2283 CrossRef; (d) W. Kläui, Angew. Chem., Int. Ed., 1990, 29, 627 CrossRef; (e) F. Gao, M.-X. Yao, Y.-Y. Li, Y.-Z. Li, Y. Song and J.-L. Zuo, Inorg. Chem., 2013, 52, 6407 CrossRef CAS PubMed.
- (a) Y. Lan, S. Klyatskaya, M. Ruben, O. Fuhr, W. Wernsdorfer, A. Candini, V. Corradini, A. L. Rizzini, U. del Pennino, F. Troiani, L. Joly, D. Klar, H. Wendeh and M. Affronte, J. Mater. Chem. C, 2015, 3, 9794 RSC; (b) D. J. Lewis, P. B. Glover, M. C. Solomons and Z. Pikramenou, J. Am. Chem. Soc., 2011, 133, 1033 CrossRef CAS PubMed; (c) R. Sato, K. Suzuki, M. Sugawa and N. Mizuno, Chem.–Eur. J., 2013, 19, 12982 CrossRef CAS PubMed; (d) J. Gónzalez-Fabra, N. A. G. Bandeira, V. Velasco, L. A. Barrios, D. Aguilà, S. J. Teat, O. Roubeau, C. Bo and G. Aromí, Chem.–Eur. J., 2017, 23, 5117 CrossRef PubMed; (e) S. Haas, E. Heintze, S. Zapf, B. Gorshunov, M. Dressel and L. Bogani, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 174409 CrossRef.
- P. N. Taylor, J. Huuskonen, G. Rumbles, R. T. Aplin, E. Williams and H. L. Anderson, Chem. Commun., 1998, 909 RSC.
- (a) N. Meundaeng, A. Rujiwatra and T. J. Prior, Transition Met. Chem., 2016, 41(7), 783 CrossRef CAS; (b) R. J. Davey, Cryst. Growth Des., 2002, 2(6), 675 CrossRef CAS; (c) L. Borka and J. K. Haleblain, Acta Pharm. Jugosl., 1990, 40, 71 CAS; (d) H. G. Brittain, Polymorphism in Pharmaceutical Solids, CRC Press, 2016 Search PubMed.
- M. D. Peeks, P. Neuhaus and H. L. Anderson, Phys. Chem. Chem. Phys., 2016, 18, 5264 RSC.
- M. Llunell, D. Casanova, J. Girera, P. Alemany and S. Alvarez, SHAPE, Continuous Shape Measures Calculation, version 2.0, Universitat de Barcelona, Barcelona, Spain, 2010 Search PubMed.
- (a) J. J. Le Roy, M. Jeletic, S. I. Gorelsky, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502 CrossRef CAS PubMed; (b) G. Cucinotta, M. Perfetti, J. Luzon, M. Etienne, P.-E. Car, A. Caneschi, G. Calvez, K. Bernot and R. Sessoli, Angew. Chem., Int. Ed., 2012, 51, 1606 CrossRef CAS PubMed; (c) R. Liu, C. Zhang, L. Li, D. Liao and J.-P. Sutter, Dalton Trans., 2012, 41, 12139 RSC; (d) M. Dörfel, M. Kern, H. Bamberger, P. Neugebauer, K. Bader, R. Marx, A. Cornia, T. Mitra, A. Müller, M. Dressel, L. Bogani and J. van Slageren, Magnetochemistry, 2016, 2, 25 CrossRef; (e) K. Bernot, J. Luzon, L. Bogani, M. Etienne, C. Sangregorio, M. Shanmugam, A. Caneschi, R. Sessoli and D. Gattesch, J. Am. Chem. Soc., 2009, 131, 5573–5579 CrossRef CAS PubMed.
- (a) N. Ishikawa, T. Iino and Y. Kaizu, J. Am. Chem. Soc., 2002, 124, 11440 CrossRef CAS PubMed; (b) J. P. Costes, F. Dahan and F. Nicodéme, Inorg. Chem., 2003, 42, 6556 CrossRef CAS PubMed; (c) X.-Y. Chen, Y. Bretonniére, J. Pécaut, D. Imbert, J.-C. Bünzli and M. Mazzanti, Inorg. Chem., 2007, 46, 625 CrossRef CAS PubMed.
- S. Richert, I. Kuprov, M. D. Peeks, E. A. Suturina, J. Cremers, H. L. Anderson and C. R. Timmel, Phys. Chem. Chem. Phys., 2017, 19, 16057 RSC.
- S. Baumann, W. Paul, T. Choi, C. P. Lutz, A. Ardavan and A. J. Heinrich, Science, 2015, 350(6259), 417 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds. CCDC 1565176–1565188. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03762k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |