
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-complementary nickel halides enable multifaceted comparisons of intermolecular halogen bonds: fluoride ligands vs. other halides†‡
Vargini
Thangavadivale§
a,
Pedro M.
Aguiar
 ab,
Naseralla A.
Jasim
a,
Sarah J.
Pike¶
a,
Dan A.
Smith§
a,
Adrian C.
Whitwood
*a,
Lee
Brammer
*c and
Robin N.
Perutz
*a
ab,
Naseralla A.
Jasim
a,
Sarah J.
Pike¶
a,
Dan A.
Smith§
a,
Adrian C.
Whitwood
*a,
Lee
Brammer
*c and
Robin N.
Perutz
*a
aDepartment of Chemistry, University of York, York YO10 5DD, UK. E-mail: adrian.whitwood@york.ac.uk; robin.perutz@york.ac.uk
bDépartement de Chimie, l'Université de Montréal, Montréal, QC H3C 3J7, Canada
cDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: lee.brammer@sheffield.ac.uk
First published on 23rd March 2018
Abstract
The syntheses of three series of complexes designed with self-complementary motifs for formation of halogen bonds between an iodotetrafluorophenyl ligand and a halide ligand at square-planar nickel are reported, allowing structural comparisons of halogen bonding between all four halides C6F4I⋯X–Ni (X = F, Cl, Br, I). In the series trans-[NiX(2,3,5,6-C6F4I)(PEt3)2] 1pX and trans-[NiX(2,3,4,5-C6F4I)(PEt3)2] (X = F, Cl, Br, I) 1oX, the iodine substituent on the benzene ring was positioned para and ortho to the metal, respectively. The phosphine substituents were varied in the series, trans-[NiX(2,3,5,6-C6F4I)(PEt2Ph)2] (X = F, I) 2pX. Crystal structures were obtained for the complete series 1pX, and for 1oF, 1oCl, 1oI and 2pI. All these complexes exhibited halogen bonds in the solid state, of which 1pF exhibited unique characteristics with a linear chain, the shortest halogen bond d(C6F4I⋯F–Ni) = 2.655(5) Å and the greatest reduction in halogen bond distance (I⋯F) compared to the sum of the Bondi van der Waals radii, 23%. The remaining complexes form zig-zag chains of halogen bonds with distances also reduced with respect to the sum of the van der Waals radii. The magnitude of the reductions follow the pattern F > Cl ∼ Br > I, 1pX > 1oX, consistent with the halogen bond strength following the same order. The variation in the I⋯X–Ni angles is consistent with the anisotropic charge distribution of the halide ligand. The temperature dependence of the X-ray structure of 1pF revealed a reduction in halogen bond distance of 0.055(7) Å on cooling from 240 to 111 K. Comparison of three polymorphs of 1oI shows that the halogen bond geometry may be altered significantly by the crystalline environment. The effect of the halogen bond on the 19F NMR chemical shift in the solid state is demonstrated by comparison of the magic-angle spinning NMR spectra of 1pF and 1oF with that of a complex incapable of halogen bond formation, trans-[NiF(C6F5)(PEt3)2] 3F. Halogen bonding causes deshielding of δiso in the component of the tensor perpendicular to the nickel coordination plane. The results demonstrate the potential of fluoride ligands for formation of halogen bonds in supramolecular structures.
Introduction
Metal fluoride complexes should provide the strongest halogen bond acceptors among metal halide complexes. Their halogen bonding ability has been addressed in solution, but supramolecular structures connecting metal fluoride complexes via halogen bonds are unknown according to a current crystallographic database search. This study reveals that they are indeed capable of forming halogen-bonded chains and allows comparisons of their structures to those of corresponding metal chlorides, bromides and iodides.Halogen bonding (XB) interactions between the electropositive region of a covalently-bound halogen and a Lewis base are increasingly recognised as motifs in secondary bonding that bear comparison with hydrogen bonds.1–5 They have been utilized in solution,4,6,7 for example for anion recognition,4,8 in catalysis,9 in medicinal chemistry,10 in biological systems including proteins and nucleic acid junctions,6,7,11–14 in liquid crystals15 and especially in supramolecular chemistry8 and crystal engineering,16,17 where halogen bonding can provide suitable motifs for self-assembly of molecules. The halogen bond is most commonly described as an electrostatically attractive interaction between a σ-hole generated on a covalently-bound halogen, archetypally iodine in an organo-iodine compound containing fluorine substituents, and a Lewis base.18–20 The organo-iodine compound is described as the halogen-bond donor and the base as the halogen-bond acceptor.
Halogen bonding interactions involving metal complexes in the solid state have been demonstrated extensively.16,21–34 Three approaches to investigating such interactions have been adopted, the design of self-complementary molecules,30,34 co-crystallization of the halogen-bond donor with the halogen-bond acceptor31 or formation of halogen bonds between ion-pairs.21,29,32,33 In a self-complementary molecule, the halogen-bond acceptor and the donor are both present in the same molecule. A series of such complexes exhibiting C–X⋯X′–M (M = Pt, Pd, X = Cl, Br, I; X′ = Cl, Br) halogen bonds was demonstrated by use of halopyridine ligands as the XB donors and halide ligands as the XB acceptors.30,34 Among examples of halogen-bonded co-crystals is a series of pincer palladium halides PdX(PCP) (PCP = 2,6-bis[(di-tert-butylphosphino)methyl]phenyl; X = Br, Cl and I) co-crystallized with I2, 1,4-C6F4I2 and I(CF2)4I as XB donors.31 The metal–halide complexes showed the general trend of the strength of the C–I⋯X–Pd halogen-bonding interaction decreasing in the order X = Cl > Br > I, and of the three XB donors used, I2 showed the strongest interaction.31 These halogen-bond interactions emphasize the different roles of the two halogens involved in C–X⋯X′–M interactions. The carbon-bound halogen serves the Lewis acidic role and adopts an approximately linear C–X⋯X′ interaction geometry, whereas the metal-bound halogen serves in a Lewis basic role and adopts a markedly bent X⋯X′–M geometry. These geometries maximize the interaction between the positive potential of the organic halogen and the most negative region of the inorganic halogen. A different class of co-crystals is illustrated by ruthenium halide complexes that form a halogen-bonded network with dihalogens (Br2 or I2).35 In spite of these thorough studies, the behaviour of metal fluorides with respect to halogen bonding remains unknown in the crystalline state.
We have reported the hydrogen- and halogen-bonding properties of group 10 metal fluoride complexes in solution for complexes of the type trans-[MF(ArF)(PR3)2] (M = Ni, Pd, Pt, ArF = tetrafluoropyridyl and related fluoropyridyls, R = Et, Cy).36–38 Measurement of the dependence of the chemical shift of the 19F NMR resonance of the metal fluoride moiety on the concentration of added iodopentafluorobenzene demonstrated the formation of the F5C6–I⋯F–Ni halogen bonds (Scheme 1a) and allowed us to determine their free energies and enthalpies of formation, and in so doing to establish metal–fluorides as among the strongest halogen bond acceptors. This conclusion is supported by related work showing that zinc and magnesium fluoride complexes behave analogously.39,40
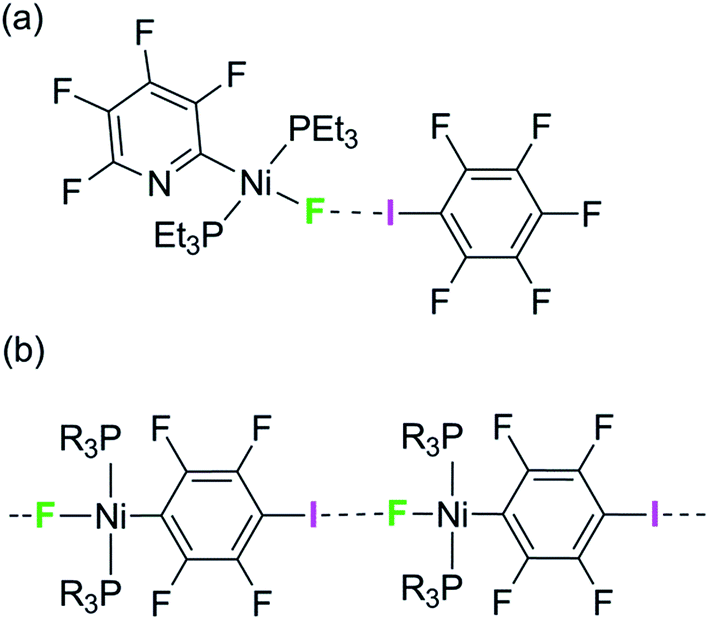 | ||
| Scheme 1 (a) Formation of halogen bonds with nickel fluoride in solution and (b) structural motif with envisaged intermolecular halogen-bonding interaction. | ||
We have attempted to co-crystallize the nickel fluoride complexes with halogen-bond donors in order to determine the geometry of the halogen bond but without success. We turned instead to the alternative strategy, design of self-complementary molecules. The feasibility of this approach is indicated by the crystallization of trans-[NiF{C5NF3(NH2)}(PEt3)2] which shows a hydrogen bond between the amino group on the fluoropyridyl ligand and the nickel fluoride on the adjacent molecule.37 We therefore set about the synthesis of a nickel fluoride complex with an iodine substituent on a fluorophenyl ligand, which would enable formation of an intermolecular halogen-bond network (Scheme 1b) between the iodine as halogen-bond donor and the nickel-fluoride as halogen-bond acceptor. We report the synthesis and structures of a series of Ni–X complexes (X = F, Cl, Br, I) with the halide ligand as halogen-bond acceptors, iodine at the 2- or 4-position on the tetrafluorophenyl ligand as halogen-bond donor and different phosphine groups. Their crystal structures reveal the changes in geometry and strength of the halogen-bonding interaction in the solid state. Geometric trends are determined as a function of halogen, regiochemistry, influence of crystalline environment and temperature. For examples with fluoride ligands (X = F) we also show that the chemical shift tensor in the 19F MAS NMR spectra is highly sensitive to halogen bond formation and strength.
Results
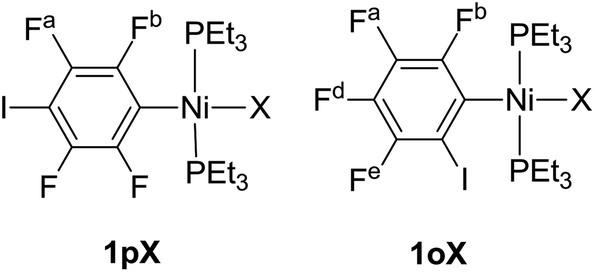
Complexes of the type illustrated in Scheme 1b were accessed by reaction of Ni(COD)2 with 2 equiv. of trialkylphosphine41 to form Ni(COD)(PEt3)2 followed by reaction with 1,4-diiodo-2,3,5,6-tetrafluorobenzene or 1,2-diiodo-3,4,5,6-tetrafluorobenzene. This synthetic method is similar to that used by Bennett with 1,6-dibromo-2,3,4,5-tetrafluorobenzene.42 Oxidative addition of one of the carbon-iodine bonds of diiodotetrafluorobenzene gives rise to a nickel–iodide complex that may then be converted into a nickel fluoride, chloride or bromide. The resulting complexes contain a coordinated C6F4I ligand as a halogen bond donor and the nickel halide as a halogen-bond acceptor. The complexes and their labels are shown in Scheme 2. The labels 1pX and 1oX refer to the position of the iodine substituent on the ring, either para (p) or ortho (o) to nickel, and the halogen (X) bound directly to nickel.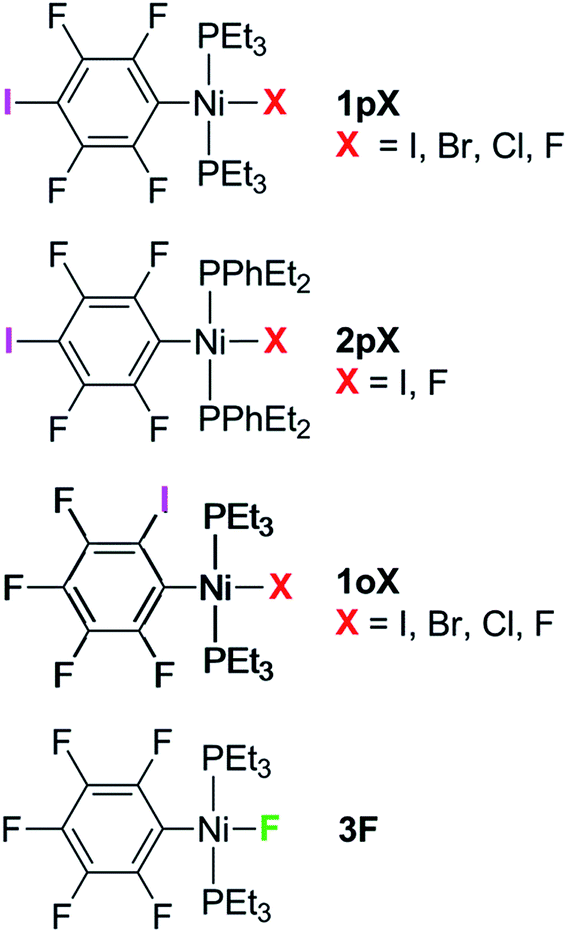 | ||
| Scheme 2 Key to complexes. | ||
Synthesis and characterization of trans-[NiX(2,3,5,6-C6F4I)(PEt3)2], 1pX (X = I, F, Cl, Br)
The synthetic methods for the 1pX series are shown in Scheme 3. The initial reaction with 1,4-diiodo-2,3,5,6-tetrafluorobenzene resulted in red trans-[NiI(2,3,5,6-C6F4I)(PEt3)2] 1pI as the major product in 84% yield. The 31P{1H}-NMR spectrum showed a singlet at δ 15.4 for the two phosphorus atoms in equivalent environments, consistent with a trans geometry. The fluorine atoms of the aromatic ring of 1pI appeared in the 19F-NMR spectrum as two second-order multiplets, AA′XX′ at δ −113.3 and δ −123.6, resulting from the coupling of the neighbouring magnetically inequivalent ortho and para fluorine atoms on the ring. The peak at m/z 695.93 (due to 58Ni with corresponding peaks for other nickel isotopes) in the LIFDI mass spectrum was identified as the molecular ion [M]+ base peak (100%) of trans-[NiI(2,3,5,6-C6F4I)(PEt3)2]. In addition, the 31P{1H}-NMR and 19F-NMR spectra showed singlets at δ 13.5 and δ −117.7, respectively, due to a minor product. The identity of this species as [trans-NiI(PEt3)2]2(μ-2,3,5,6-C6F4) was demonstrated by the peak at 990.17 in the LIFDI mass spectrum which corresponds to the molecular ion [M]+. This complex arises from the oxidative addition of both the iodine atoms on the 1,4-diiodotetrafluorobenzene (Scheme 4).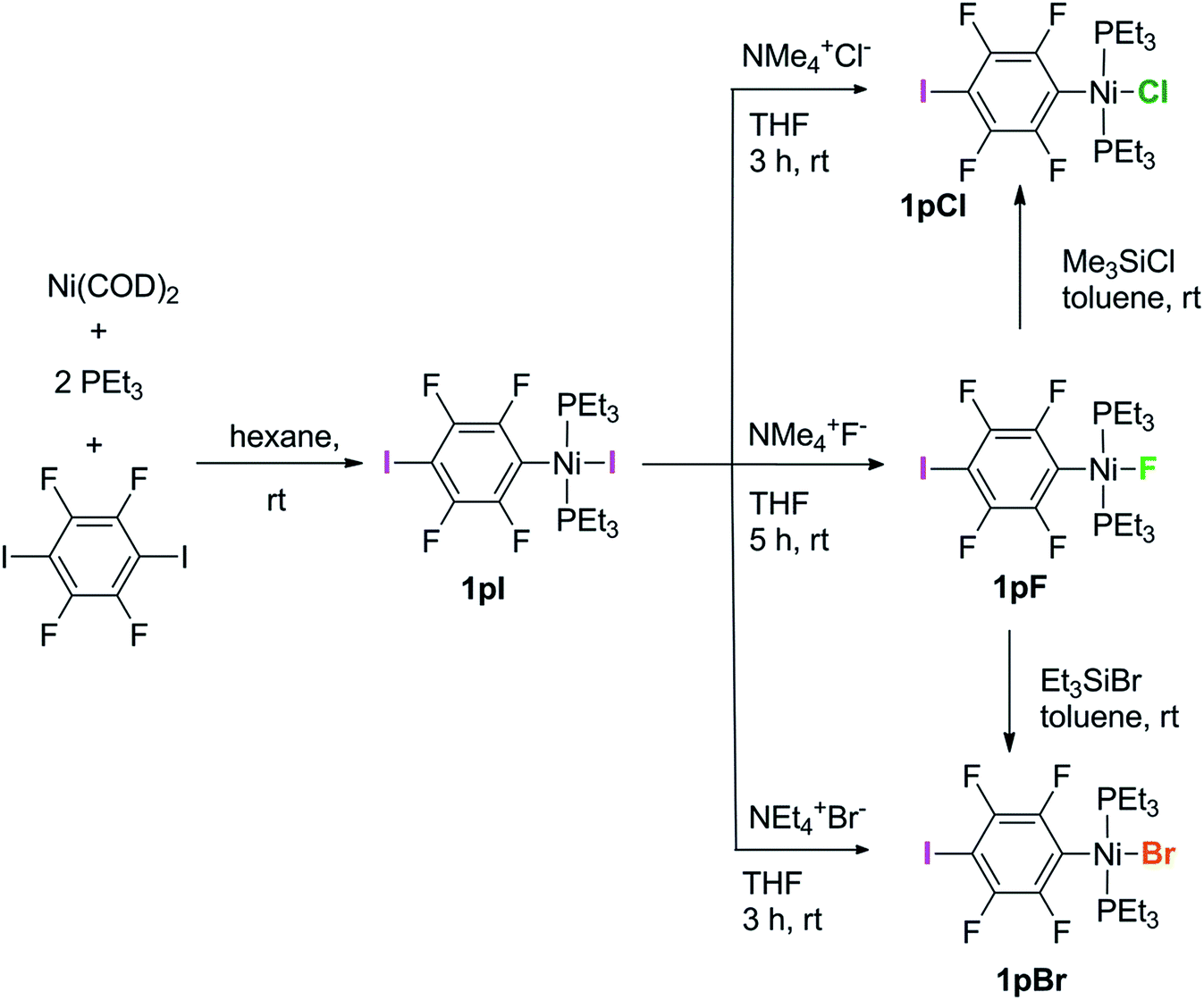 | ||
| Scheme 3 Synthetic routes to complexes 1pI, 1pF, 1pCl and 1pBr. | ||
 | ||
| Scheme 4 Dinuclear product [trans-NiX(PEt3)2]2(μ-2,3,5,6-C6F4). | ||
The reaction of 1pI with [NMe4]F in THF gave a colour change from red to yellow in 5 h at room temperature. After purification and removal of unreacted 1pI, a yellow solid was isolated in 71% yield. The major product of the reaction was identified as trans-[NiF(2,3,5,6-C6F4I)(PEt3)2] 1pF. The 31P{1H}-NMR spectrum showed a resonance at δ 13.1 as a doublet with 2JPF = 46 Hz.41 The 19F-NMR spectrum of the purified product showed two resonances at δ −113.8 and δ −124.9 for the four aromatic fluorine atoms with second-order multiplets AA′XX′ spin system. The nickel fluoride resonance of the complex was observed as a triplet at δ −387.9, JPF = 46 Hz.41 The LIFDI mass spectrum showed a molecular ion [M+] base peak (100%) at 587.99 with appropriate isotopic peaks.
The reaction of 1pI with [NMe4]Cl in THF at room temperature gave rise to a yellow solution of 1pCl. Alternatively, complex 1pCl could be synthesised rapidly and more efficiently by reacting a solution of 1pF in toluene with chlorotrimethylsilane for 15 min at room temperature (Scheme 3). The synthesis of 1pBr paralleled the chloride analogue and used either [NEt4]Br in THF or bromotriethylsilane in toluene.
Synthesis of trans-[NiX(2,3,5,6-C6F4I)(PEt2Ph)2], 2pI (X = I) and 2pF (X = F)
The electronic structure of the spectator ligands on the metal centre may have a significant effect on the halogen-bonding interaction, as demonstrated by earlier work.36 To investigate this effect on the solid-state structures, the triethylphosphine ligands on the complexes were replaced with diethylphenyl phosphine. The complex trans-[NiI(2,3,5,6-C6F4I)(PEt2Ph)2] 2pI was prepared in a similar manner to the PEt3 analogue. The nickel iodide 2pI obtained was converted to the nickel fluoride 2pF by reaction with [NMe4]F (64% yield). NMR data for 2pI and 2pF corresponded closely to those of 1pI and 1pF. The complex 2pF was also characterised by its LIFDI mass spectrum, which showed a molecular ion [M+] base peak (100%) at 684.00.Synthesis of trans-[NiX(2,3,4,5-C6F4I)(PEt3)2], 1oX, X = I, F, Cl and Br
Our next target was to change the position of the halogen-bond donor on the fluoroaromatic ring in order to compare the effect of the electron-withdrawing fluorine atoms ortho to the iodine and to change the relative orientation of the halogen-bond donor and acceptor groups. The synthesis of complexes from 1,2-diiodotetrafluorobenzene proceeded similarly to those from 1,4-diiodotetrafluorobenzene. The reaction of Ni(PEt3)2(COD) with 1,2-diiodotetrafluorobenzene gave rise to trans-[NiI(2,3,4,5-C6F4I)(PEt3)2] 1oI in 67% yield. The 31P{1H}-NMR spectrum of the product exhibited a singlet at δ 10.3 for the two equivalent phosphorus atoms. The 19F-NMR spectrum revealed four resonances for the four aromatic fluorine atoms that were assigned with the help of 19F–19F COSY spectroscopy.The product of the reaction between 1oI and [NMe4]F yielded trans-[NiF(2,3,4,5-C6F4I)(PEt3)2] 1oF. The fluoride resonance was observed in the 19F-NMR spectrum at δ −397.8 as a triplet of doublets as a result of phosphorus coupling (JFP = 46 Hz), and a four-bond coupling to the aromatic fluorine ortho to the metal centre (JFF = 9 Hz). Four further 19F NMR resonances were observed in the aromatic region. The 31P{1H} NMR spectrum exhibited a doublet at δ 9.4 (d, J = 46 Hz). The synthesis of 1oCl and 1oBr followed the same pattern as for 1pCl and 1pBr.
A further minor product of reaction of 1oI with [NMe4]F exhibited resonances at δ −131.4 and at δ −150.2 in the 19F-NMR spectrum and a peak at δ 27.3 in the 31P{1H}-NMR spectrum. Single crystals of this species were obtained from hexane/benzene and allowed us to identify it as the η2-tetrafluorobenzyne complex of Ni(PEt3)2, Ni(PEt3)2(η2-C6F4) (see ESI‡). The spectra and structure are similar to benzyne complexes reported in the literature.42–45
The energetics of halogen bond formation in solution have been reported for complexes such as trans-[NiF(C5NF4)(PEt3)2] with C6F5I.38 We attempted to determine the energetics of the self-complementary interactions of complex 1pF or 1oF by measuring 19F-NMR spectra as a function of concentration. However, the solubility of both complexes 1pF and 1oF proved insufficient to obtain reliable data.
Crystal structures
Single crystals of complexes 1pX were obtained by solvent diffusion. Selected intramolecular bond lengths and angles for X-ray crystal structures of 1pF, 1pCl, 1pBr and 1pI are given in Table 1; intermolecular distances and angles are given in Table 2.| Complex | I1–Caromatic | Ni–P1 | Ni–X | Ni–C1 |
|---|---|---|---|---|
| a Bond I2–C4. b Measurements are for the major component. c Bond I2–C2. d Metal plane Ni–P1–P2–C1–X. e Angle (P1–Ni–P1′). | ||||
| 1pF | 2.096(11) | 2.208(2) | 1.837(5) | 1.873(13) |
| 1pCl | 2.082(2) | 2.2086(5) | 2.2035(5) | 1.881(2) |
| 1pBr | 2.082(2) | 2.2176(6) | 2.3385(3) | 1.881(2) |
| 1pI | 2.092(6)a | 2.224(2) | 2.5221(9) | 1.877(6) |
| 2pI | 2.089(2)a | 2.2200(6) | 2.5348(4) | 1.902(2) |
| 1oF | 2.111(6) | 2.218(2) | 1.841(4) | 1.890(6) |
| 1oCl | 2.095(4) | 2.270(5) | 2.201(5) | 1.899(5) |
| 1oI(α) | 2.096(4)c | 2.225(1) | 2.5332(6) | 1.907(4) |
| 1oI(β) | 2.098(3)c | 2.234(1) | 2.5358(4) | 1.893(3) |
| 1oI(γ) | 2.088(5)c | 2.231(1) | 2.5337(8) | 1.908(5) |
| Complex | X–Ni–C1 | P1–Ni–P2 | P1–Ni–X | P1–Ni–C1–C2 | Angle between metal plane and ring planed |
|---|---|---|---|---|---|
| 1pF | 180.0 | 177.1(1)e | 91.46(6) | 85.2(3) | 85.2(2) |
| 1pCl | 175.82(5) | 175.65(2) | 89.12(2) | 87.9(1) | 82.13(4) |
| 1pBr | 176.61(6) | 176.94(2) | 88.85(2) | 86.2(2) | 85.60(5) |
| 1pI | 178.3(2) | 178.33(7) | 89.55(5) | 84.0(5) | 85.14(16) |
| 2pI | 175.36(2) | 175.36(2) | 89.59(2) | 88.7(2) | 89.07(5) |
| 1oF | 179.3(3) | 173.12(9) | 88.60(16) | 85.3(6) | 84.4(2) |
| 1oCl | 178.1(3) | 171.0(4) | 89.67(16) | 90.1(4) | 85.1(1) |
| 1oI(α) | 173.8(1) | 177.33(5) | 88.87(3) | 89.17(3) | 90.0 |
| 1oI(β) | 179.8(1) | 174.72(3) | 89.67(2) | 89.6(3) | 89.3(1) |
| 1oI(γ) | 178.1(1) | 169.72(6) | 92.67(4) | 84.0(4) | 86.8(1) |
| Compound | Intermolecular I⋯X/Å | R AIX | R BIX | C–I⋯X/° | I⋯X–Ni/° | Angle/° between adjacent metal planesb | Angle/° between adjacent ring planes | Ni⋯Ni⋯Ni/° |
|---|---|---|---|---|---|---|---|---|
| a The normalised distance, R, is defined according to Lommerse et al.48RAIX = d(I⋯X)/(rAI + rAX), where rAI and rAX are the Alvarez van der Waals radii46 of the iodine atom, and halogen (F 1.46, Cl 1.82, Br 1.86 or I 2.04 Å), respectively, in the C–I⋯X–Ni halogen bond. RBIX = d(I⋯X)/(rBI + rBX), where rBI and rBX are the Bondi van der Waals radii47 of the iodine atom, and halogen (F 1.47, Cl 1.75, Br 1.83 or I 1.98 Å), respectively, in the C–I⋯X–Ni halogen bond. b Metal plane Ni–P1–P2–C1–X. | ||||||||
| 1pF | 2.655(5) | 0.76 | 0.77 | 180.0 | 180.00 | 0.0 | 0.0 | 180.00 |
| 1pCl | 3.2414(5) | 0.84 | 0.87 | 167.09(5) | 146.83(2) | 72.69(2) | 88.01(8) | 159.91(1) |
| 1pBr | 3.3320(3) | 0.85 | 0.87 | 167.55(6) | 143.31(1) | 70.91(3) | 87.8(1) | 158.95(1) |
| 1pI | 3.4970(6) | 0.86 | 0.88 | 168.0(2) | 139.18(3) | 67.41(8) | 87.4(3) | 156.79(2) |
| 2pI | 3.6791(4) | 0.90 | 0.93 | 167.39(6) | 158.22(1) | 0.0 | 0.0 | 180.00 |
| 1oF | 2.941(5) | 0.84 | 0.85 | 173.2(2) | 172.4(2) | 72.3(3) | 31.7(3) | 113.55(2) |
| 1oCl major | 3.342(6) | 0.87 | 0.90 | 169.4(2) | 158.1(3) | 54.5(2) | 26.1(2) | 112.1(1) |
| 1oCl minor | 3.26(2) | 0.84 | 0.87 | 178.7(4) | 171.8(8) | 72.1(7) | 26.1(2) | 108.8(2) |
| 1oI(α) | 3.5961(4) | 0.88 | 0.91 | 178.5(1) | 144.16(2)) | 77.1(1) | 0.0 | 97.00(1) |
| 1oI(β) | 3.7456(4) | 0.92 | 0.95 | 151.7(1) | 133.48(1) | 45.1(1) | 12.4(2) | 124.80(1) |
| 1oI(γ) | 4.0752(7) | 1.00 | 1.03 | 169.2(1) | 161.63(2) | 64.6(1) | 39.5(2) | 101.06(1) |
For 1pF the fluoroaromatic ring is oriented almost perpendicular to the square plane of the metal coordination centre, 85.2(2)°. The crystal structure comprises chains of the nickel complexes linked by short C–I⋯F(Ni) halogen bonds (I⋯F 2.655(5) Å, C–I⋯F 180.0°, I⋯F–Ni 180.0°, Fig. 1a). The parallel sets of linear chains align with the b-axis and form layers in the ab plane. Complexes 1pCl, 1pBr and 1pI (Fig. 1b–d, Table 1) crystallize as antiparallel zig-zag chains with molecules linked by a C–I⋯X–Ni halogen bonds. The Ni⋯Ni⋯Ni angle (Table 2) may be taken as an estimate of the zig-zag angle and indicates a slight zig-zag for X = Cl, Br and I (Ni⋯Ni⋯Ni 159.91(1), 158.95(1), 156.79(2)°, respectively, cf. 180° for X = F). The intermolecular distances I⋯X are substantially less than the sum of van der Waals radii (RAIX 0.84–0.86; RBIX 0.84–0.87; see Table 2 for definitions of distances, RAIX and RBIX, which represent the fraction of the sum of van der Waals radii46,47 of the two interacting atoms I and X), but show smaller reductions than observed for 1pF (RAIF 0.76; RBIF 0.77). The C–I⋯X angles deviate slightly from linear (ca. 167°) while the I⋯X–Ni angles lie in the range 139–147°. The torsional angles between adjacent metal coordination planes in the chains are ca. 70°, while adjacent benzene ring planes are approximately orthogonal (ca. 88°, Table 2). In addition, the crystal structure of the minor dinuclear product, [trans-NiBr(PEt3)2]2(μ-2,3,5,6-C6F4) was determined (see ESI‡ and Scheme 4).
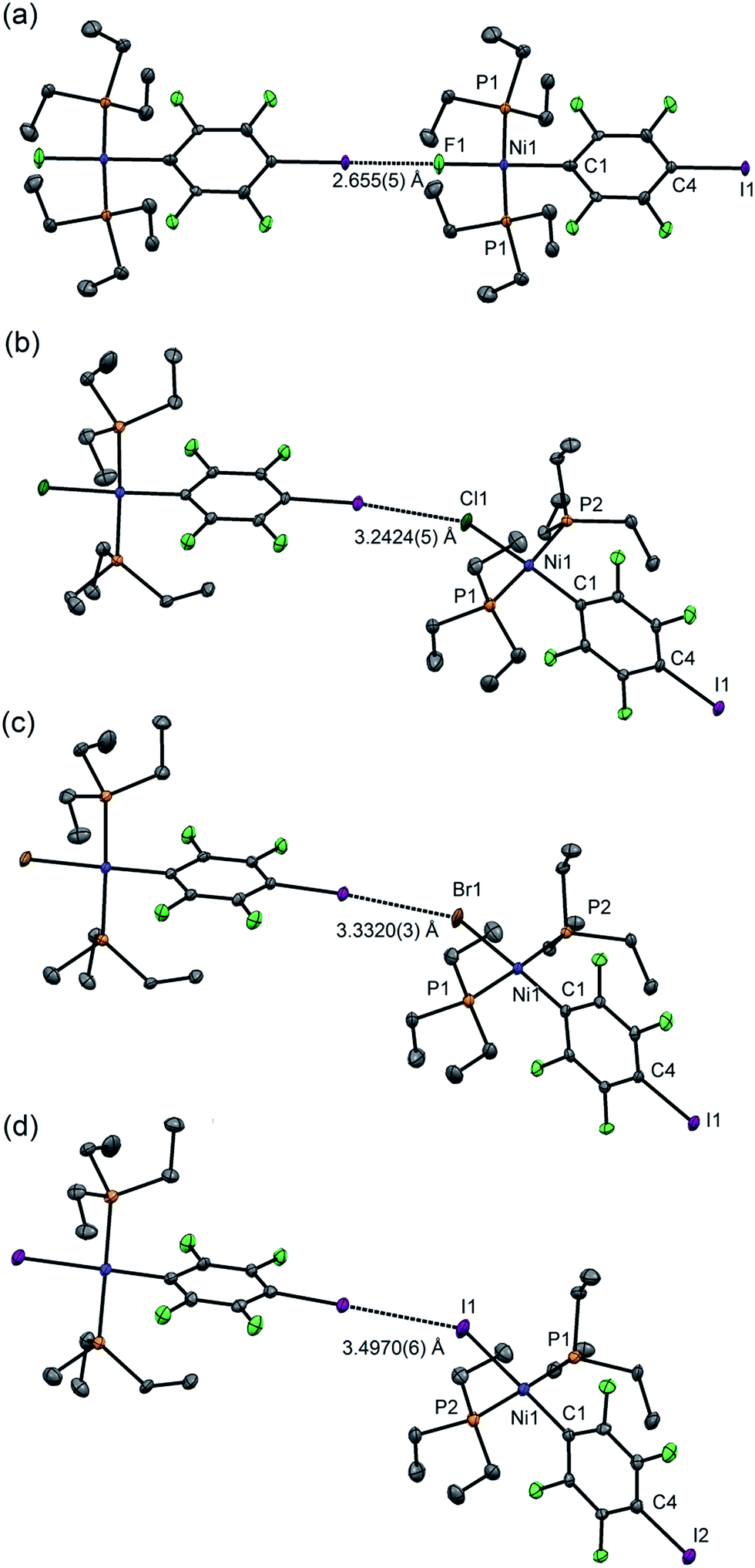 | ||
| Fig. 1 Molecular structures of 1pX (a) X = F, (b) X = Cl, (c) X = Br, (d) X = I, showing intermolecular halogen bonds. Displacement ellipsoids at 50% probability level. Hydrogen atoms not shown. Three-molecule chains are illustrated in the ESI.† | ||
Single crystals of 2pI suitable for X-ray crystallography were grown as dark yellow blocks by solvent diffusion (Fig. 2). The three aromatic rings present in the structure orient themselves away from each other and there are neither inter- nor intramolecular π–π interactions. Unlike 1pI, the Ni coordination planes and the tetrafluoroiodophenyl rings are parallel to one another. The complexes pack with zig-zag halogen-bonded chains, which run parallel (and anti-parallel) to the b-axis. The C–I⋯I–Ni halogen bonds (I⋯I 3.6791(4) Å, C–I⋯I 167.39(6)°, I⋯I–Ni 158.22(1)°) are markedly longer than those of 1pI (Table 2). Attempts to obtain single crystals of 2pF suitable for X-ray crystallography were fruitless.
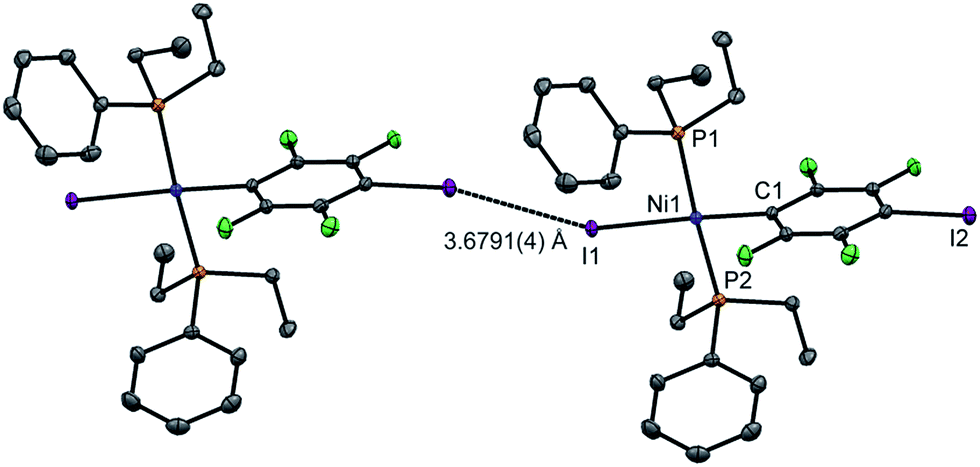 | ||
| Fig. 2 Molecular structure of 2pI showing intermolecular C–I⋯I–Ni halogen bond. Displacement ellipsoids at 50% probability level. Hydrogen atoms not shown. A three-molecule chain is illustrated in the ESI.‡ | ||
Single crystals of 1oF, 1oCl and 1oI were obtained that were suitable for X-ray crystallography.|| Selected intramolecular distances and angles are given in Table 1 and intermolecular distances and angle in Table 2. The crystal structures are illustrated in Fig. 3. It might be expected that the Ni–C distances in the 1pX series would be shorter than those in the 1oX series because of the ortho-fluorine effect.49 Although the Ni–C distances are consistently shorter for 1pX than for their 1oX analogues, the differences are of marginal significance. The differences in the C–I distances are insignificant. In all of the 1oX halogen-bonded complexes, the iodine on the ring in the ortho position causes a pronounced zig-zag of the complexes, as described by the Ni⋯Ni⋯Ni angle (96 ≤ Ni⋯Ni⋯Ni ≤ 125°, Table 2). The normalised distances of the I⋯X contacts RBIX are 0.85 for 1oF, 0.90 for 1oCl (major) and 0.91–1.03 for 1oI, much larger than for 1pX analogues.
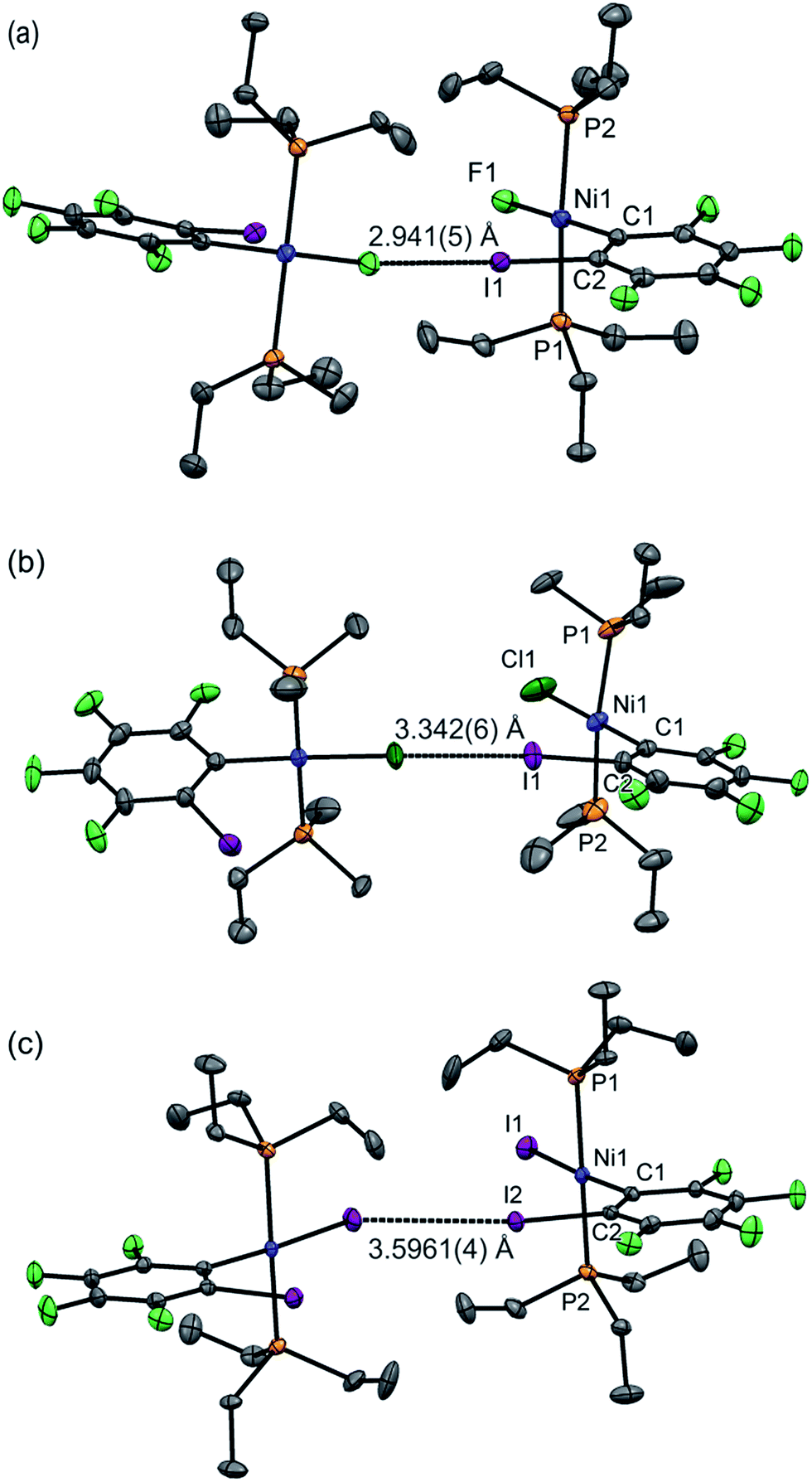 | ||
| Fig. 3 Molecular structures of 1oX (a) X = F, (b) X = Cl, (c) X = I, showing intermolecular C–I⋯X–Ni halogen bonds. The structure of 1oCl is disordered (see Table 2). Only the major component is shown. Hydrogen atoms and some methyl groups omitted for clarity. Displacement ellipsoids at 50% probability level. Three-molecule chains are illustrated in the ESI.‡ | ||
Single crystals of 1oI were obtained as three polymorphs, 1oI(α) crystalized from hexane, and the other two (1oI(β) and 1oI(γ)) from CHCl3 layered with hexane. Polymorph 1oI(α) shows the typical C–I⋯I–Ni halogen-bonding contacts (3.6100(3) Å). The polymorphs from chloroform exhibited the same patterns of halogen bonding but with progressively longer contacts. The C–I⋯I–Ni contact is 3.7456(4) Å for 1oI(β), (major component), while 1oI(γ) has two independent molecules in the unit cell, one of which has a C–I⋯I–Ni distance of 4.0752(7) Å and the other shows no halogen bonding at all. The changes in halogen-bonding geometry accompany changes in orientation of molecules along the halogen-bonded chains, exemplified by the twisting of the ring planes of adjacent molecules with respect to one another for 1oI(β) and 1oI(γ), but coplanar arrangement in 1oI(α).
Variable-temperature X-ray crystallographic studies of 1pF′ and 1pF
The effects of temperature and pressure on C–Cl⋯X–M halogen bonds (X = Cl, Br) have been studied by Mínguez Espallargas et al., illustrating the compressibility of these non-covalent interactions.16,50 In order to quantify the temperature dependence of our system, crystals of 1pF were selected as they showed the strongest X-bonding interaction of the series of complexes prepared. A crystal of 1pF was cooled to 111 K and the structure determined, warmed to 200 K and analysed again, and finally warmed to 240 K and the structure obtained a third time. At 111 K and 200 K, the space group was I2 whereas at 240 K the space group was Cc. The space group change was accompanied by a change in the unit cell, suggesting a phase change between 200 and 240 K. Overlaying the structures at 200 and 240 K revealed that the major difference in structures is the position of one of the triethylphosphine ligands (Fig. S41‡). At the higher temperature, the triethylphosphines are no longer related by a two-fold rotation and one is disordered in two positions (see ESI‡). The variable temperature X-ray crystallographic data of 1pF show that a 4.5% reduction in the volume per molecule (V/Z) is observed when the temperature is changed from 240 K to 200 K and ca. 2% reduction in the volume is observed from 200 K to 111 K (Table 3). The change in space group from I2 to Cc removes the linearity of the halogen-bonded chain. Nevertheless, the key angles remain close to 180°: the C–I⋯F angle is 177.8(8), I⋯F–Ni 164.8(7) and the Ni⋯Ni⋯Ni angle is 171.5(3)°. Although changes in bond lengths to nickel were insignificant, the intermolecular distance between the fluoride on the metal centre and the iodine on the fluoroaromatic ring decreased with temperature by 0.055(7) Å from 240 to 111 K. The normalised halogen-bonded distance RBIF remains below 0.80 throughout the temperature range (Table 2).| Space group/temperature, K | |||
|---|---|---|---|
| I2/111a | I2/200 | Cc/240 | |
| a Independent determination from 110 K study reported in Tables 1 and 2. b Metal plane Ni–P1–P2–C1–X. | |||
| V/Z/Å3 | 577.09(5) | 588.17(4) | 614.7(1) |
| (C)I⋯F(Ni)/Å | 2.652(3) | 2.683(5) | 2.707(7) |
| Ni–F/Å | 1.839(4) | 1.832(5) | 1.855(10) |
| Ni–C/Å | 1.872(10) | 1.879(10) | 1.88(2) |
| Ni–P/Å | 2.211(1) | 2.214(1) | 2.209(5) |
| C–I⋯X/° | 180.0 | 180.0 | 177.8(7) |
| I⋯X–Ni/° | 180.0 | 180.0 | 164.8(7) |
| Angle/° between adjacent metal planesb | 0.0 | 0.0 | 26.3(7) |
| Angle/° between adjacent ring planes | 0.0 | 0.0 | 4.9(10) |
| Ni⋯Ni⋯Ni/° | 180.0 | 180.0 | 171.50(3) |
Solid-state NMR spectroscopic studies
Magic-angle spinning solid-state NMR (MAS SSNMR) spectroscopy offers an opportunity to learn more about the halogen-bonding interactions.51–53 We have found one other example of a 19F-MAS SSNMR spectrum for a nickel fluoride complex54 and one example of calculated 19F tensors for some cobalt fluorides.55 The fluoride bound to the nickel centre can act as a 19F-MAS SSNMR spectroscopic handle because the chemical shift is very sensitive to the fluoride environment36 and appears at high field with no overlap from other resonances. 19F-MAS SSNMR spectroscopy was used to analyse the effect on the fluoride resonance of halogen-bonding interactions in complexes 1pF and 1oF. The complex trans-[NiF(C6F5)(PEt3)2] 3F, which has no halogen-bond donor atom on its backbone was also analysed by 19F-MAS SSNMR spectroscopy for comparison.41 The spectrum of 1pF (Fig. 4) shows the isotropic chemical shift of the nickel fluoride at δ −359.8 with numerous spinning side bands (black dots in Fig. 4). The other peaks in the range of δ −100 to −160 arise from the resonances of the fluoroaromatic ring. Measurement at various spinning speeds allowed simulation of spectrum and determination of the components of the chemical shift anisotropy (CSA) tensor and identification of δiso of the fluoride resonance. Expansions of the isotropic region of the spectra are shown in Fig. 5 and the trends in the CSA components in Fig. 6.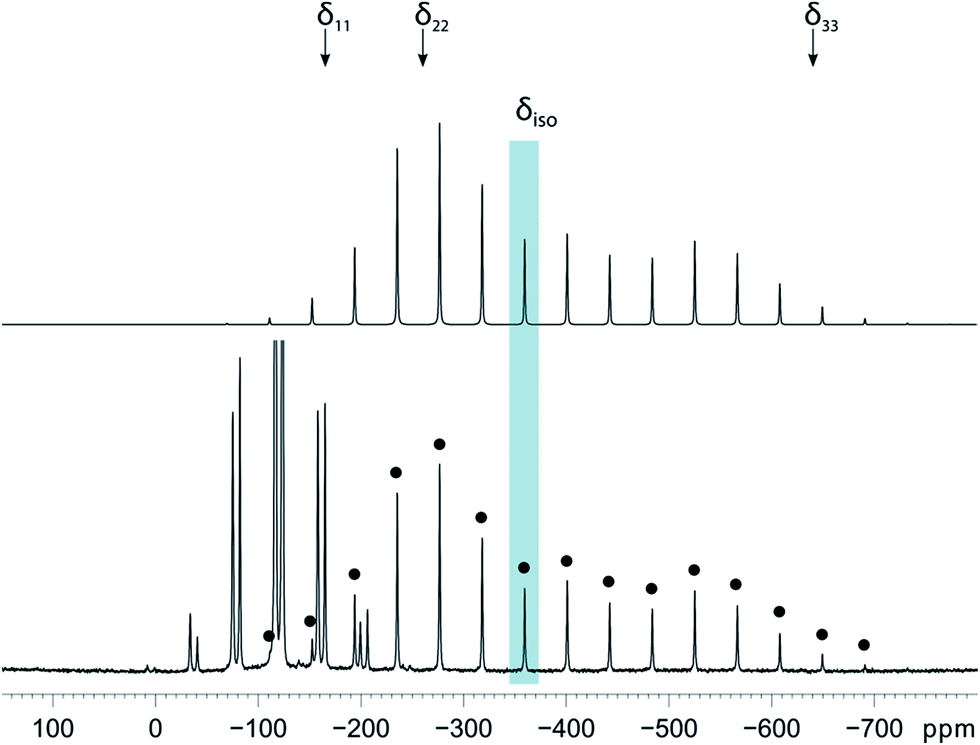 | ||
| Fig. 4 19F-MAS SSNMR spectrum of 1pF. Top: best fit simulated spectrum for the Ni–F resonance with the positions of the shielding tensor marked. Bottom: experimental spectrum. Spinning speed 15.6 kHz. Signals arising from the fluorinated benzene ligand (−110 to −160 ppm) are truncated. The black dots indicate the NiF peak and its spinning side bands. The blue bar indicates δiso. | ||
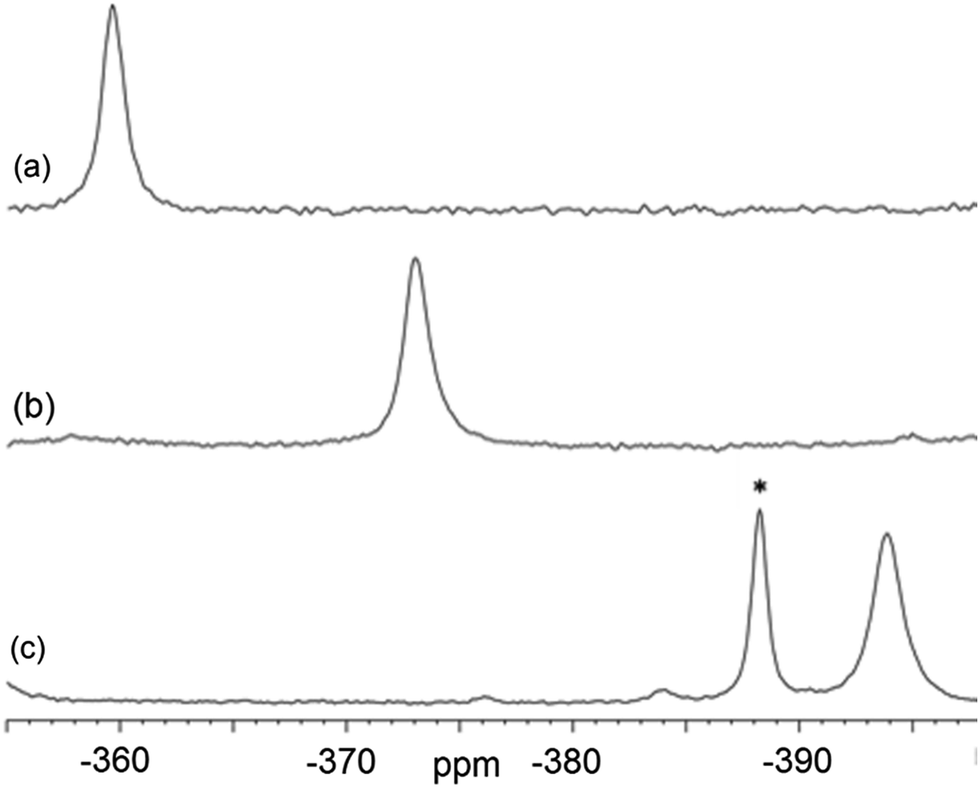 | ||
| Fig. 5 Expansion of isotropic region of 19F-MAS SSNMR spectrum of (a) 1pF, (b) 1oF and (c) trans-[NiF(C6F5)(PEt3)2] 3F with no halogen bond, (asterisk solvated complex impurity <4%). | ||
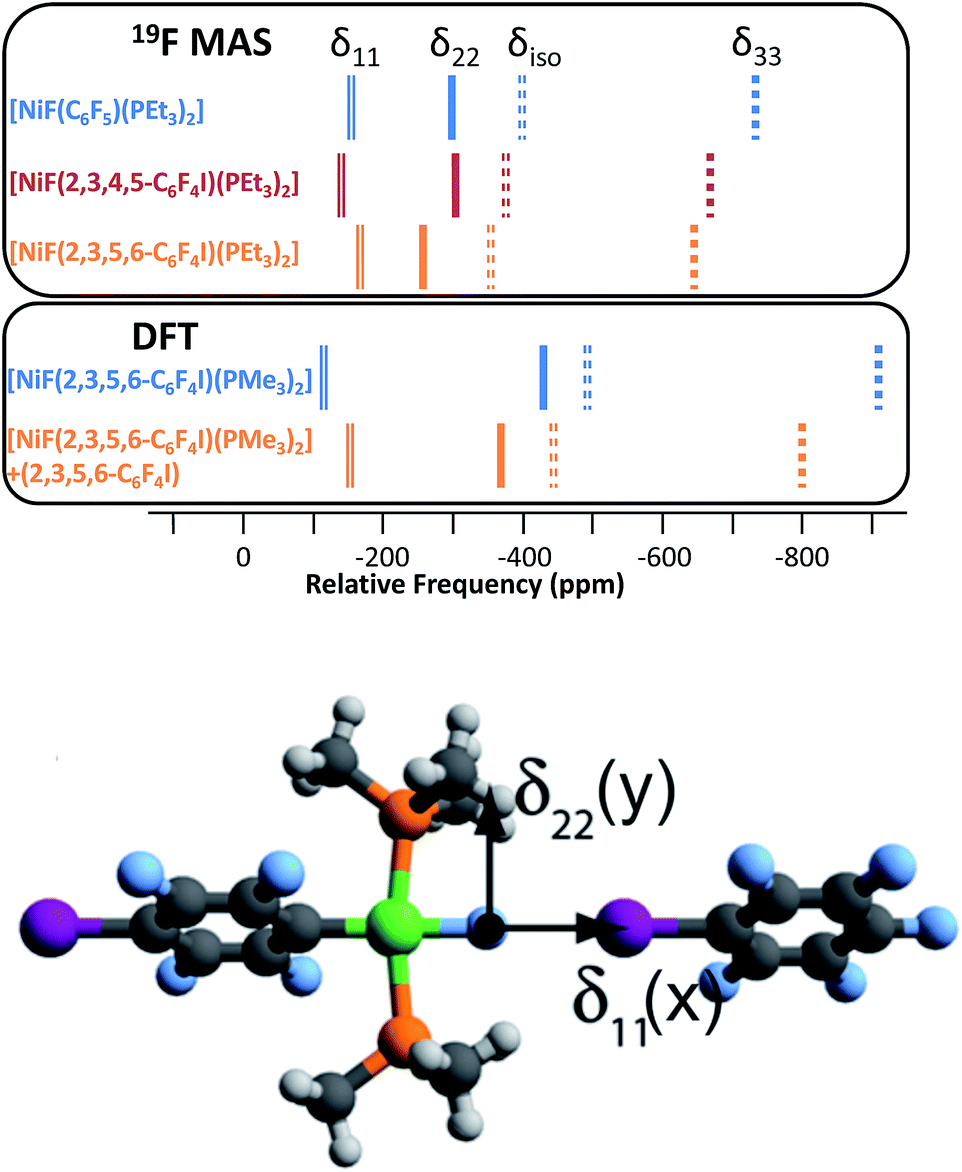 | ||
| Fig. 6 Top: positions of the observed (19F-MAS SSNMR) principal tensor components (δ11, δ22, δ33) and isotropic shift (δiso) for 3F, 1oF and 1pF. Middle: values of δ11, δ22, δ33, and δiso determined by DFT calculations for trans-[NiF(2,3,5,6-C6F4I)(PMe3)2] and its halogen-bonded complex with C6F5I. Bottom: calculated structure of C6F5I⋯FNi(2,3,5,6-C6F4I)(PMe3)2 showing orientation of CSA tensor (δ33 component is perpendicular to the plane of the page). | ||
The components of the anisotropic chemical shift tensor are labelled as δ11, δ22 and δ33, where δ11, is the least shielded component and δ33 is the most shielded component (δ11 ≥ δ22 ≥ δ33).56 For 1pF, the simulations show δ11 at δ −165, δ22 at δ −266 and δ33 at δ −645. The 31P{1H} spectrum of 1pF (see ESI‡) reveals a coupling 2JPF = 42 ± 3 Hz, similar to the value observed in solution (46 Hz). We were unable to obtain the JPF coupling in the 19F-MAS SSNMR spectra for 1oF and 3F.
In addition, the structure of the gas-phase model trans-[NiF(2,3,5,6-C6F4I)(PMe3)2] and its halogen-bonded complex with C6F5I were determined by DFT methods showing a (C)I⋯F(Ni) distance of 2.749 Å. DFT simulation of the 19F-MAS SSNMR spectra of both these species showed that δ11 lies along the Ni–F bond, δ22 lies parallel to the Ni–P bonds and δ33 lies perpendicular to the coordination plane of nickel (Fig. 6, lower pane). Moreover, the CSA calculated by DFT methods for trans-[NiF(2,3,5,6-C6F4I)(PMe3)2] and trans-[NiF(2,3,5,6-C6F4I)(PMe3)2]⋯IC6F5 show that the changes in the shielding of the δ11 and δ22 components are small and of opposite sign, thus minimizing their effect on the isotropic shift.
The isotropic chemical shift δiso of the fluoride resonance of 3F was observed at δ −393.9 in the 19F-MAS SSNMR spectrum (Table 4, Fig. 5), in good agreement with the solution-state fluoride resonance at δ −394.3 (in C6D6) for the same complex. In contrast, the solution-state fluoride resonance of 1pF appeared ∼29 ppm to higher field at δ −388.3 than its solid state isotropic chemical shift (δ −359.8). Similarly, the solution-state fluoride resonance of 1oF was observed ∼25 ppm to higher field at δ −397.9 than its solid state counterpart. Thus the solid-state NMR spectra provide strong evidence of deshielding of δiso with increasing halogen-bond interaction. Table 4 gives values of the SSNMR parameters. The δ33 component is systematically deshielded upon increasing halogen bond strength, mirroring what is observed in the isotropic chemical shift, and there is also a reduction in the span, Ω. Since the DFT calculations indicate a minimal contribution of δ11 and δ22 to the changes in δiso, we conclude that the changes in δ33 are dominated by the effect of halogen bonding. The deshielding of the NiF resonance is also consistent with the effect of addition of C6F5I to trans-[NiF(C5NF4)(PEt3)2] in solution.36,38 A very large chemical shift anisotropy was also reported for another nickel fluoride complex,54 and very strong paramagnetic shielding was observed perpendicular to the metal phosphorus bond for terminal phosphido complexes in their 31P-MAS SSNMR spectra.57 Large chemical shift anisotropies have recently been measured and calculated for metal hydrides.58,59
| Sample | Solution δ | δ iso | Ω | κ | δ 11 | δ 22 | δ 33 |
|---|---|---|---|---|---|---|---|
| a Chemical shift tensor parameters,56δiso Isotropic chemical shift, δiso = (δ11 + δ22 + δ33)/3, Ω Span of the CSA powder pattern, Ω = δ11 – δ33, κ Skew, measures the asymmetry of the powder pattern, κ = (δ22 − δiso)/Ω. | |||||||
| 1pF | −388.3 | −359.8(2) | 480(10) | 0.58(3) | −165 | −266 | −645 |
| 1oF | −397.9 | −373.0(2) | 530(10) | 0.40(3) | −143 | −302 | −673 |
| 3F | −394.3 | −393.9(2) | 575(10) | 0.50(3) | −154 | −298 | −729 |
The paramagnetic contribution to the components of the chemical shift tensor are understood via second order perturbation theory as being affected by the angular momentum operator ![[L with combining circumflex]](https://www.rsc.org/images/entities/i_char_004c_0302.gif) i (i = x, y, z) which couples occupied orbitals to vacant orbitals after a 90° rotation about the direction i.60,61 In this case δ33 lies in the out-of-plane direction z and is rotated by y into the x direction that coincides with the vacant M–F σ* orbital. It is this orbital that is affected by the halogen bonding both through electrostatic effects and through interaction with the C–I σ and σ* orbitals.52 As the δ33 component is deshielded upon formation of the halogen bond, the energy gap between these two orbitals must decrease.
i (i = x, y, z) which couples occupied orbitals to vacant orbitals after a 90° rotation about the direction i.60,61 In this case δ33 lies in the out-of-plane direction z and is rotated by y into the x direction that coincides with the vacant M–F σ* orbital. It is this orbital that is affected by the halogen bonding both through electrostatic effects and through interaction with the C–I σ and σ* orbitals.52 As the δ33 component is deshielded upon formation of the halogen bond, the energy gap between these two orbitals must decrease.
Discussion of trends in halogen bonding
This study establishes that nickel halide complexes trans-[NiX(C6F4I)(PR3)2] form intermolecular C–I⋯X–M halogen bonds by self-recognition in the solid state, leading to halogen-bonded chain motifs. Their crystal structures provide extensive geometric data for these halogen bonds that demonstrate a marked difference between the fluoride complexes and those of the other halides. The strengths of halogen bonds may be compared by the normalised distances (here RAIX or RBIX, see Table 2).16,48 As for other strong halogen bonds, C–X⋯X′–M halogen bonds adopt a characteristic angle C–X⋯X′ close to 180°; the X⋯X′–M angle, however, is typically in the 110–140° range for X′ = Cl, Br, I,30,31,62 but can vary more widely23,63 as the halide ligand exhibits a negative electrostatic potential attractive to halogen bond donors and other Lewis acidic moieties at all angles of approach.64 Despite their established greater strength,36–39 there have been no systematic crystallographic studies prior to this study that establish C–X⋯F–M halogen bond geometries and, in particular typical X⋯F–M angles.In this study we examined the class of C–I⋯X–M halogen bonds through a series of systematic changes and perturbations, by monitoring the geometry as a function of: (i) halide ligand, X, (ii) regiochemistry of halogen-bond donor, I (1pXvs.1oX) (iii) influence of ligand sphere of the metal on halogen-bond acceptor, X, by changing PEt3 for PPhEt2, (iv) the influence of crystalline environment, by comparison of three polymorphs tnqh_x2026;of 1oI, and (v) the effect of temperature, in a study of 1pF.
Common features of halogen-bonded chain structures
1pF forms linear chains, whereas the structures of 1pX (X = Cl, Br, I) and 1oX (all X) form zig-zag chains. The zig-zag angles of the chains of 1pCl, 1pBr and 1pI are characterised by Ni⋯Ni⋯Ni angles in the 155–160° range (see figures in ESI‡). The angles between adjacent metal coordination planes and the angles between adjacent benzene ring planes also show marked differences between 1pF on the one hand and 1pCl, 1pBr and 1pI on the other (Table 2). The zig-zag in the ortho series is dominated by the 60° angle imposed by the ortho substitution, which causes a major reduction in the Ni⋯Ni⋯Ni angle compared to the para analogues (1pF 180° and 1oFca. 114°, 1pClca. 160° and 1oClca. 112°, 1pIca. 157° and 1oI(α)ca. 97°). Both the angle between adjacent ring planes and the Ni⋯Ni⋯Ni angle are significantly smaller for 1oI(α) than for the fluorine and chlorine analogues.Trend with halide ligand for 1pX and 1oX: distance
The normalised distances (Table 2) range from 0.76 for 1pF to 0.90 for 2pI (Alvarez radii, RAIX) or 0.77 to 0.93 (Bondi radii, RBIX).46,47 The normalised distance is markedly smaller for 1pF than for 1pCl, 1pBr and 1pI. Nevertheless, there is a small increase from Cl to Br and from Br to I. Thus the large negative electrostatic potential of fluorine is the dominant effect38,64 and the changes between the other halogens are smaller in this series. The trend is consistent with the observations for hydrogen-bond acceptor behaviour of halide ligands, e.g. D–H⋯X–M, where the H-bond donor D = N, O (or even C). Normalised hydrogen bond distances follow the trend RBHF ≪ RBHCl ≤ RBHBr < RBHI.64 Overall, the geometric results are consistent with fluoride ligands being much stronger halogen-bond and hydrogen-bond acceptors than their heavier congeners. Our results on 1pCl and 1pBr are also comparable to those recorded for the co-crystals of 1,4-(C6F4I2) with PdX(PCP) (X = Cl, Br), but the Pd–F complexes were not synthesised.31The normalised distances for the 1pX series are considerably smaller than for the 1oX series, especially for X = F (Table 2). Considering that the regiochemistry has a very minor effect on the Ni–C and the C–I distances, there is no evidence that the changes in the halogen-bond distance have an electronic origin. On the other hand, they can be rationalised on steric grounds since the σ-hole of the 1pX halogen-bond donor atom is more accessible than that in the 1oX series. The trend in normalised distances with halide ligand for the 1oX series, however, resembles that for 1pX.
Trend with halide ligand for 1pX and 1oX: angle
Halogen bonding interactions are directional forces because of the anisotropic charge distribution around halogen atoms. The charge distribution is compressed along the C–I axis, creating the positive potential trans to the C–I bond, leading to C–I⋯X angles that are close to linear.16,18,48 Halide ligands (M–X) exhibit a similar anisotropy, albeit with a negative potential in all directions at separations appropriate for intermolecular interactions. Brammer et al.30,64 reported the calculated electrostatic potential around the halogen in palladium halide complexes, trans-[PdX(CH3)(PH3)2]. They showed that the most negative value in the molecular plane is consistent with I⋯X–M angles at 155° with a range of 130–180° range when X = F, whereas the other halogens exhibited minima at ca. 124° and a range of ca. 110–140°.64 In the 1pX series of complexes, C–I⋯X angles are close to linear, consistent with typical halogen-bond behaviour (1pF 180°; 1pCl, 1pBr, 1pI all close to 167°, Scheme 5). The I⋯X–Ni angle of 1pF is 180°, exceeding the I⋯X–Ni angles for the other halides, which are in the range 139–147° (Table 2, Scheme 5). This result is in keeping with the analysis of angular variation of the halide negative electrostatic potential mentioned above, and is consistent with more restricted steric access to the halide ligand at lower I⋯X–Ni angles, since Ni–F and I⋯F separations are markedly shorter than for other Ni–X and I⋯X.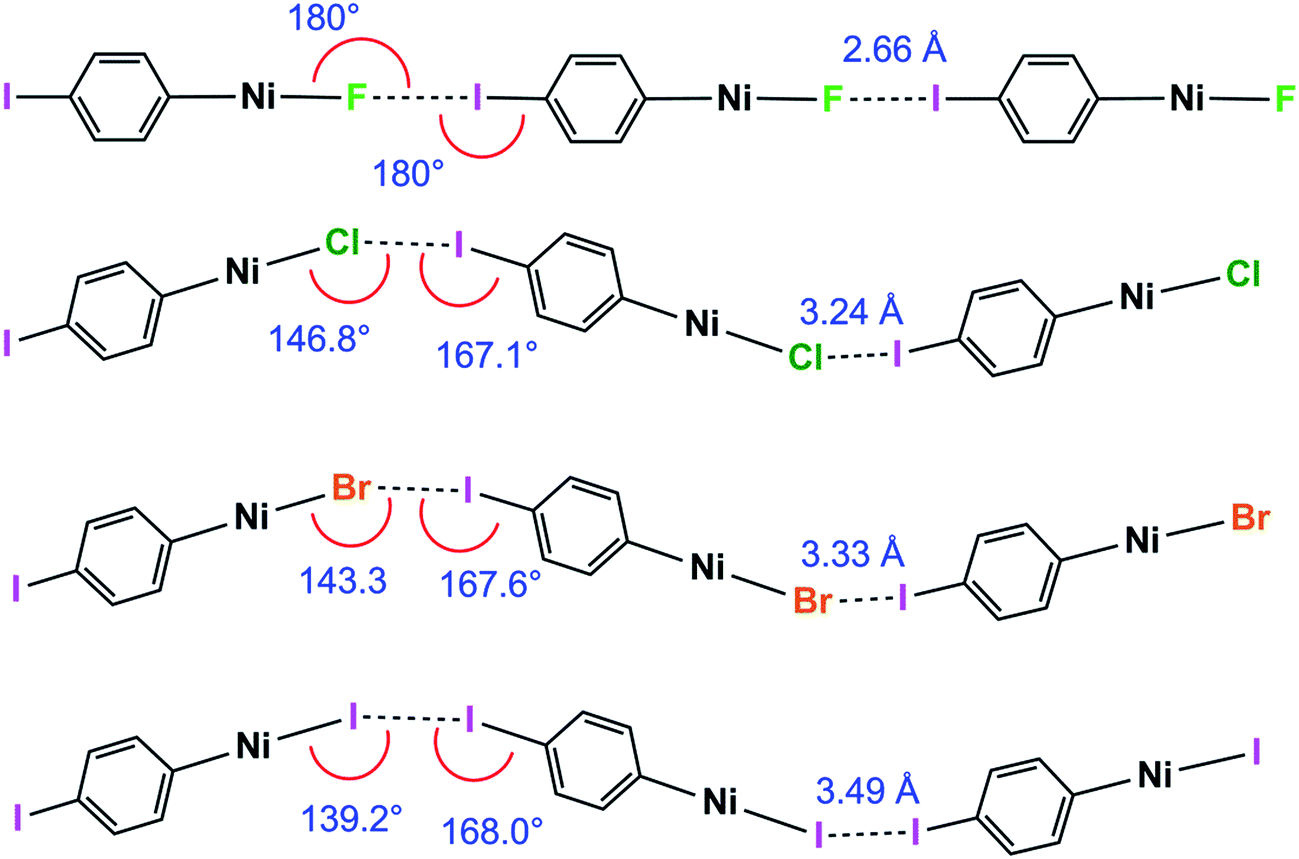 | ||
| Scheme 5 Halogen bond length and C–I⋯Ni and I⋯X–Ni angles for the 1pX series (phosphines and aromatic fluorines omitted, for esd's see Table 2). | ||
The C–I⋯X angles are again similar and close to linear across the 1oX series. The variation in the I⋯X–Ni angle is more marked, moving from 172.4(2)° to 158.1(3)° and 143.66(1)° along the series 1oF, 1oCl, 1oI(α) (Scheme 6, Table 2), which is qualitatively consistent with the trend in location of electrostatic potential minima at the halide ligand.
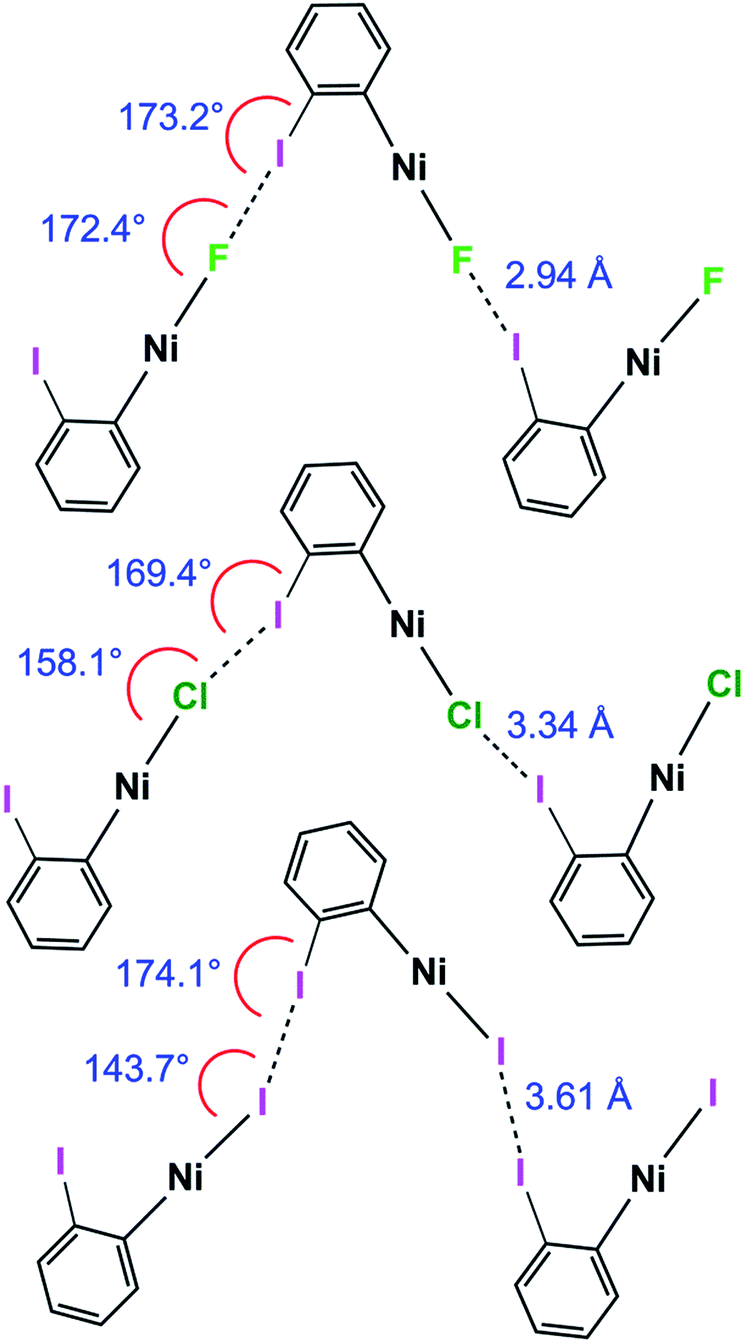 | ||
| Scheme 6 Halogen bond length and C–I⋯Ni and I⋯X–Ni angles for 1oF, 1oCl and 1oI(α) (phosphines and aromatic fluorines omitted, for esd's see Table 2). | ||
Effect of phosphine substituents
The change in phosphine from 1pI with PEt3 to 2pI with PEt2Ph causes a marked increase in the halogen-bond distance, indicating the sensitivity of the halogen bond to the ligand sphere of the metal. There are no solution-phase measurements on PEt2Ph complexes for comparison although an increase in enthalpy of C–I⋯F–Ni halogen bonding when replacing PEt3 ligands by PCy3 has been reported.38Trend with polymorph for 1oI
The crystallisation of three solvent-free polymorphs of 1oI allowed the effect of crystalline environment on formation and geometry of the C–I⋯I–Ni halogen bond to be examined. The three polymorphs provide four different crystalline environments as there are two crystallographically independent molecules in 1oI(γ). The I⋯I distances vary from 3.5961(4) Å in 1oI(α) to 3.7456(4) in 1oI(β) to 4.0752(7) Å 1oI(γ), whereas the second independent molecule exhibits no halogen-bonding interaction. The different crystalline environments also lead to changes in halogen-bond angles C–I⋯I and I⋯I–Ni, and to changes in relative orientation of neighbouring molecules in the halogen-bonded chains (Table 2).Trend with temperature for 1pF
The study of the structure of 1pF using one single crystal at three temperatures** enabled the perturbing effect of temperature on the geometry of the C–I⋯F–Ni halogen bond to be examined. The halogen-bonded distance remains well over 20% shorter than the sum of the van der Waals radii (RAIF, RBIF < 0.8), and C–I⋯F and I⋯F–Ni angles remain close to linearity, across the temperature range studied (111–240 K). Two previous studies of halogen-bonding as a function of temperature also reveal only very small changes in halogen bond angles. Forni et al. reported changes in halogen-bond distance of 0.052(2) Å (C–I⋯N), 0.030(2) Å (C–I⋯O) and 0.059(2) Å (C–Br⋯N) over the temperature range 90–292 K.65 Brammer et al. reported changes of 0.070(2) Å (C–Cl⋯Cl(M)) and 0.079(1) Å (C–Cl⋯Br(M)) over the temperature range 30–300 K.16,50 These values are consistent with the changes observed here, viz. 0.055(7) Å. Such changes are greater than observed for intramolecular distances, but smaller than those for other intermolecular separations, which reflects the strength of these halogen bonds and, in conjunction with their directionality, underlines their utility in supramolecular assembly.Conclusions
Our design strategy for a full range of structures of the type trans-[NiX(C6F4I)(PR3)2], for all halogens X, reveals supramolecular chain structures containing self-complementary halogen-bonding motifs linked by C–I⋯X–Ni halogen bonds. This methodology fulfils the objectives for thorough geometric comparisons and provides a platform for development of supramolecular assemblies with metal–fluoride complexes, which form the strongest halogen bonds. Moreover, the methods offer an entry into solid-state NMR studies of halogen bonding by use of the 19F nucleus of the fluoride ligand as a spectroscopic probe that is not complicated by nuclear quadrupole effects. Such NMR studies will also enable improved comparison between halogen bonds in the solid state and solution phase. This type of comparison is rarely accessible, but is important because the solid state provides the most accurate geometric information through crystallography, while the solution phase provides the most direct experimental means of determining interaction energies through spectroscopic titrations.The strongest halogen bonds are formed by trans-[NiF(p-C6F4I)(PEt3)2], judged both by the reduction in I⋯X distance compared to the sum of the van der Waals distances and by the 19F SSNMR chemical shift. This system contains a linear chain. Other complexes of the type trans-[NiX(p-C6F4I)(PEt3)2] (1pX, X = Cl, Br, I) exhibit zig-zag chain structures with angles C–X⋯I and I⋯X–Ni, and angles between Ni coordination planes and between ring planes that are similar for X = Cl, Br and I. The corresponding complexes trans-[NiX(o-C6F4I)(PEt3)2] (1oX, X = F, Cl, Br, I) all form chains in the solid state with more pronounced zig-zags and weaker halogen bonds than their p-C6F4I counterparts (1pX). The relative strengths of the halogen bonds, as judged by smaller normalised C–I⋯X halogen-bond distances (RAIX, RBIX), follow the pattern 1pX > 1oX and F > Cl ∼ Br > I, consistent with observations of hydrogen bond acceptor capabilities of halide ligands, X. The variation in the I⋯X–Ni angles is consistent with the location of the electrostatic potential minima associated with anisotropic charge distribution of the halide ligand, X, which have a directing effect on the halogen bond.
We have shown by comparison of three polymorphic forms of 1oI that the crystalline environment can have a marked effect on halogen bond geometry and indeed on whether a (weaker) halogen bond is formed at all. These results emphasise the importance of examining trends in intermolecular interaction geometries in the solid state and avoiding overinterpretation of the link, for example, between intermolecular distances and interaction strength based on individual solid-state observations. The study of the crystal structure of 1pF over the temperature range 111–240 K reveals only a small change in halogen bond length, consistent with the C–I⋯F–Ni halogen bond being a strong intermolecular interaction.
Finally, we have shown that it is possible to demonstrate the existence of halogen bonding in the fluoride complexes with 19F-MAS SSNMR spectroscopy. With a suitable reference, trans-[NiF(C6F5)(PEt3)2] 3F, the presence of the halogen bonds is manifest through downfield shifts in the resonance of the Ni–F as has previously been reported in solution. The 19F SSNMR isotropic chemical shifts follow the order 3F < 1oF < 1pF in agreement with the crystal structures, where 1pF shows the strongest halogen-bonding interactions (smallest RIX values). Comparisons between solution and solid-state NMR indicate that C–I⋯F–Ni halogen bonding causes deshielding of δiso by 25–30 ppm; examination of the chemical shift tensor indicates that by far the biggest contributor to the deshielding is δ33, the component perpendicular to the metal coordination plane. This shift is consistent with coupling of the occupied F(2pz) orbital with the vacant M–F σ* orbital; it is this orbital that is influenced by the halogen bonding.
Experimental
All experiments involving oxygen- and water-sensitive materials were performed under an argon or nitrogen atmosphere, in an argon-filled glove box or standard Schlenk (10−2 mbar) techniques or high vacuum lines (10−4 mbar). Solvents (AR grade) for general use were dried over sodium, distilled and stored under argon. Solvents such as hexane and THF were collected from the Innovative Technology solvent purification system and were further dried and distilled. Deuterated solvents were dried over potassium and distilled prior to use.All standard NMR spectra were recorded on Bruker AV I 500 or AV III 500 spectrometers, unless otherwise stated, in tubes fitted with Young's PTFE stopcocks. All 1H and 13C chemical shifts are reported in ppm (δ) relative to tetramethylsilane and referenced using the chemical shifts of residual protio solvent resonances (benzene, δ 7.16), unless otherwise stated. The 31P{1H} NMR spectra were referenced to external H3PO4. 19F NMR spectra were referenced to external CFCl3.
LIFDI mass spectra were recorded on a Waters Micromass GCT Premier orthogonal time-of-flight instrument set to 1 scan per s with resolution power of 6000 FWHM and equipped with a LIFDI probe from LINDEN GmbH. Toluene was used for tuning the instrument.66–68 The polyethylene glycol probe was kept at ambient temperature with the emitter potential at 12 kV. Activated tungsten wire LIFDI emitters (13 μm W from LINDEN) were ramped manually up to 100 mA for the emitter heating current during the experiment. The m/z values are accurate to 0.01 Da. Masses are quoted for 35Cl, 58Ni, 81Br.
X-ray diffraction data were collected on an Oxford Diffraction SuperNova diffractometer with MoKα radiation (λ = 0.71073 Å) at 110 K unless otherwise noted. Data collection, unit cell determination and frame integration were carried out with the program CrysAlisPro.69 Absorption corrections were applied using crystal face-indexing and the ABSPACK absorption correction software within CrysAlisPro. Structures were solved and refined using Olex2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 70 implementing SHELX algorithms. Structures were solved by either Patterson or direct methods using SHELXS-97 and refined by full-matrix least squares using SHELXL-97.71 All non-hydrogen atoms were refined anisotropically. Carbon-bound hydrogen atoms were placed at calculated positions with fixed isotropic displacement parameters and refined using a “riding model”. Detailed crystallographic data are provided in the ESI.‡
70 implementing SHELX algorithms. Structures were solved by either Patterson or direct methods using SHELXS-97 and refined by full-matrix least squares using SHELXL-97.71 All non-hydrogen atoms were refined anisotropically. Carbon-bound hydrogen atoms were placed at calculated positions with fixed isotropic displacement parameters and refined using a “riding model”. Detailed crystallographic data are provided in the ESI.‡
The 19F-MAS NMR spectra were acquired at 9.4 T (376.48 MHz) using a Bruker Avance III HD console and a Bruker 2.5 mm H(F)/X double-resonance probe. The MAS rotors and caps were pumped under high vacuum for 24 hours then stored in the glove box where they remained for at least a week before use. A 30° tip-angle pulse was used to acquire the spectra to ensure sufficient radio frequency bandwidth to yield undistorted spinning sideband intensities. Relaxation delays of 4–7 s were used. Spectra were collected at 295 K unless stated otherwise. All shifts are reported relative to CFCl3 and were calibrated using solid sodium fluoride (δiso = −224.2) as a secondary external reference. Isotropic chemical shifts were determined by comparison of three or more spectra acquired over a range of rotation frequencies (9–30 kHz). Simulation of the shielding tensors was performed using the CSA tensor module72 within the WSolids NMR Simulation Package (Ver. 1.20.18).
Syntheses
Ni(COD)2 was purchased from Sigma-Aldrich. 1,4-C6F4I2, 1,2-C6F4I2 (Fluorochem), Me3SiCl and Et3SiBr (Sigma-Aldrich) were distilled and were stored over molecular sieves. Triethylphosphine was purchased from Fluorochem. Commercially available tetramethylammonium fluoride tetrahydrate (Sigma-Aldrich) was dried following the literature method.73,74 Complex 3F was synthesized as previously described.41
19F NMR aromatic resonances were assigned based on their chemical shifts and coupling constants, and 19F–19F COSY spectra. Note that 19F nuclei ortho to the metal always resonate to lower field than the meta or para19F nuclei. Fluorine atoms are labelled as below:
NMR (C6D6, 298 K). 1H NMR δ 0.97 (apparent quin, J 7.7 Hz, 18H, CH3), 1.06–1.14 (m, 12H, CH2). 31P{1H} NMR: δ 13.1 (d, JPF 46 Hz). 19F NMR: δ −113.8 (AA′XX′, 2F, Fb), −123.6 (AA′XX′, 2F, Fa), −387.9 (t, JPF 46 Hz, 1F, Ni–F). Mass spectrum (LIFDI, m/z+) 587.99, (100% M+). Anal. calcd for C18H30F5INiP2 C, for 36.71; H, 5.13. Found: C, 36.59; H, 5.06.
(b) From 1oF and Et3SiBr. Bromotriethylsilane (17 μL, 0.1 mmol) was added into a solution of THF (10 mL) containing 1oF, (50 mg, 0.08 mmol) and stirred for 1 h. The solvent and excess reagents and by-products were removed under vacuum. The product was washed with pentane and dried. Yield 53 mg, 96%. NMR (C6D6, 298 K). 1H NMR δ 0.92 (apparent quin J 7.7 Hz, 18H, CH3), 1.30 (m, 6H, CH2), 1.38 (m, 6H, CH2). 31P{1H} NMR: δ 9.52 (s). 19F NMR: δ −114.5 (dd, JFF 31.2, 12.2 Hz, 1F, Fb), −115.0 (dd, JFF 21.8, 11.9 Hz, 1F, Fe), −157.0 (dd, JFF 32.0, 19.3 Hz, 1F, Fa), −159.3 (t of m, JFF 20.8 Hz, 1F, Fd). Mass spectrum (LIFDI, m/z+) 649.91, (100% M+). Anal. calcd for C18H30BrF4INiP2 C, 33.27; H, 4.65. Found: C, 33.36; H, 4.54.
DFT calculations
Calculations were performed using Gaussian 09 Revision B.01.75 Geometries optimisations were performed using BH and HLYP hybrid functional38,76,77 with 6-31+G(d,p) for H, C, P, and F atoms and SDD effective core potentials for Ni and I. Calculation of the shielding tensors was done using BH and HLYP functional with 6-311+G(d,p) for H, C, P, F and Ni and SDD effective core potential for I. The computed tensors from Gaussian were symmetrised, diagonalised and the direction cosines visualised using either home-written Matlab scripts or the program EFGShield.78Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Iain Oswald (University of Strathclyde) for determining the crystal structure of [trans-NiBr(PEt3)2]2(μ-2,3,5,6-C6F4) and thank Professors Christopher Hunter (University of Cambridge) and Odile Eisenstein University of Montpellier for useful discussions. We acknowledge an Overseas Research Scholarship from the University of York to VT. We also acknowledge support from EPSRC (grants EP/J012955/1 and EP/J012998/1).References
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- L. Brammer, Faraday Discuss., 2017, 203, 485–507 RSC.
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- M. Erdelyi, Biochemistry, 2017, 56, 2759–2761 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- A. R. Voth, P. Khuu, K. Oishi and P. S. Ho, Nat. Chem., 2009, 1, 74–79 CrossRef CAS PubMed.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J. M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 314–318 CrossRef CAS PubMed.
- D. W. Bruce, Struct. Bonding, 2008, 126, 161–180 CrossRef CAS.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- M. H. Kolar and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef CAS PubMed.
- H. Wang, W. Z. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- R. D. Willett, F. Awwadi, R. Butcher, S. Haddad and B. Twamley, Cryst. Growth Des., 2003, 3, 301–311 CAS.
- R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef CAS.
- F. F. Awwadi, D. Taher, S. F. Haddad and M. M. Turnbull, Cryst. Growth Des., 2014, 14, 1961–1971 CAS.
- D. M. Ivanov, A. S. Novikov, I. V. Ananyev, Y. V. Kirina and V. Y. Kukushkin, Chem. Commun., 2016, 52, 5565–5568 RSC.
- G. Lapadula, N. Judas, T. Friščić and W. Jones, Chem.–Eur. J., 2010, 16, 7400–7403 CrossRef CAS PubMed.
- R. W. Troff, T. Makela, F. Topic, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617–1637 CrossRef CAS.
- J. M. Clemente-Juan, E. Coronado, G. Mínguez Espallargas, H. Adams and L. Brammer, CrystEngComm, 2010, 12, 2339–2342 RSC.
- P. Smart, Á. Bajarano-Villafuerte and L. Brammer, CrystEngComm, 2013, 15, 3151–3159 RSC.
- J. E. Ormond-Prout, P. Smart and L. Brammer, Cryst. Growth Des., 2012, 12, 205–216 CAS.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- M. T. Johnson, Z. Džolić, M. Cetina, O. F. Wendt, L. Öhrström and K. Rissanen, Cryst. Growth Des., 2011, 12, 362–368 Search PubMed.
- L. Brammer, G. Mínguez Espallargas and H. Adams, CrystEngComm, 2003, 5, 343–345 RSC.
- G. Mínguez Espallargas, L. Brammer and P. Sherwood, Angew. Chem., Int. Ed., 2006, 45, 435–440 CrossRef PubMed.
- F. Zordan and L. Brammer, Cryst. Growth Des., 2006, 6, 1374–1379 CAS.
- M. E. G. Mosquera, I. Egido, C. Hortelano, M. López-López and P. Gómez-Sal, Faraday Discuss., 2017, 203, 257–283 RSC.
- S. Libri, N. A. Jasim, R. N. Perutz and L. Brammer, J. Am. Chem. Soc., 2008, 130, 7842–7844 CrossRef CAS PubMed.
- D. A. Smith, T. Beweries, C. Blasius, N. Jasim, R. Nazir, S. Nazir, C. C. Robertson, A. C. Whitwood, C. A. Hunter, L. Brammer and R. N. Perutz, J. Am. Chem. Soc., 2015, 137, 11820–11831 CrossRef CAS PubMed.
- T. Beweries, L. Brammer, N. A. Jasim, J. E. McGrady, R. N. Perutz and A. C. Whitwood, J. Am. Chem. Soc., 2011, 133, 14338–14348 CrossRef CAS PubMed.
- W. Sattler, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2013, 135, 18714–18717 CrossRef CAS PubMed.
- M. Rauch, S. Ruccolo, J. P. Mester, Y. Rong and G. Parkin, Chem. Sci., 2016, 7, 142–149 RSC.
- L. Cronin, C. L. Higgitt, R. Karch and R. N. Perutz, Organometallics, 1997, 16, 4920–4928 CrossRef CAS.
- M. A. Bennett and E. Wenger, Organometallics, 1995, 14, 1267–1277 CrossRef CAS.
- A. J. Edwards, A. C. Willis and E. Wenger, Organometallics, 2002, 21, 1654–1661 CrossRef CAS.
- M. A. Bennett, T. W. Hambley, N. K. Roberts and G. B. Robertson, Organometallics, 1985, 4, 1992–2000 CrossRef CAS.
- A. L. Keen, M. Doster and S. A. Johnson, J. Am. Chem. Soc., 2007, 129, 810–819 CrossRef CAS PubMed.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2009, 131, 7817–7827 CrossRef CAS PubMed.
- G. Mínguez Espallargas, L. Brammer, D. R. Allan, C. R. Pulham, N. Robertson and J. E. Warren, J. Am. Chem. Soc., 2008, 130, 9058–9071 CrossRef PubMed.
- M. Weingarth, N. Raouafi, B. Jouvelet, L. Duma, G. Bodenhausen, K. Boujlel, B. Schollhorn and P. Tekely, Chem. Commun., 2008, 5981–5983 RSC.
- J. Viger-Gravel, S. Leclerc, I. Korobkov and D. L. Bryce, J. Am. Chem. Soc., 2014, 136, 6929–6942 CrossRef CAS PubMed.
- P. Cerreia Vioglio, M. R. Chierotti and R. Gobetto, CrystEngComm, 2016, 18, 9173–9184 RSC.
- M. R. Chierotti, A. Rossin, R. Gobetto and M. Peruzzini, Inorg. Chem., 2013, 52, 12616–12623 CrossRef CAS PubMed.
- M. C. Leclerc, J. M. Bayne, G. M. Lee, S. I. Gorelsky, M. Vasiliu, I. Korobkov, D. J. Harrison, D. A. Dixon and R. T. Baker, J. Am. Chem. Soc., 2015, 137, 16064–16073 CrossRef CAS PubMed.
- H. Saito, I. Ando and A. Ramamoorthy, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 181–228 CrossRef CAS PubMed.
- G. Wu, D. Rovnyak, M. J. A. Johnson, N. C. Zanetti, D. G. Musaev, K. Morokuma, R. R. Schrock, R. G. Griffin and C. C. Cummins, J. Am. Chem. Soc., 1996, 118, 10654–10655 CrossRef CAS.
- P. Garbacz, V. V. Terskikh, M. J. Ferguson, G. M. Bernard, M. Kedziorek and R. E. Wasylishen, J. Phys. Chem. A, 2014, 118, 1203–1212 CrossRef CAS PubMed.
- L. J. L. Haller, E. Mas-Marza, M. K. Cybulski, R. A. Sanguramath, S. A. Macgregor, M. F. Mahon, C. Raynaud, C. A. Russell and M. K. Whittlesey, Dalton Trans., 2017, 46, 2861–2873 RSC.
- C. M. Widdifield and R. W. Schurko, Concepts Magn. Reson., Part A, 2009, 34, 91–123 CrossRef.
- S. Halbert, C. Copéret, C. Raynaud and O. Eisenstein, J. Am. Chem. Soc., 2016, 138, 2261–2272 CrossRef CAS PubMed.
- F. F. Awwadi, R. D. Willett, S. F. Haddad and B. Twamley, Cryst. Growth Des., 2006, 6, 1833–1838 CAS.
- G. Mínguez Espallargas, F. Zordan, L. Arroyo Marín, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Chem.–Eur. J., 2009, 15, 7554–7568 CrossRef PubMed.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CAS.
- A. Forni, P. Metrangolo, T. Pilati and G. Resnati, Cryst. Growth Des., 2004, 4, 291–295 CAS.
- T. A. Dransfield, R. Nazir, R. N. Perutz and A. C. Whitwood, J. Fluorine Chem., 2010, 131, 1213–1217 CrossRef CAS.
- J. H. Gross, N. Nieth, H. B. Linden, U. Blumbach, F. J. Richter, M. E. Tauchert, R. Tompers and P. Hofmann, Anal. Bioanal. Chem., 2006, 386, 52–58 CrossRef CAS PubMed.
- J. H. Gross, J. Am. Soc. Mass Spectrom., 2007, 18, 2254–2262 CrossRef CAS PubMed.
- CrysAlisPro Version 1.171.38.46, Rigaku Oxford Diffraction Ltd Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. Eichele, R. E. Wasylishen, J. F. Corrigan, N. J. Taylor and A. J. Carty, J. Am. Chem. Soc., 1995, 117, 6961–6969 CrossRef CAS.
- K. O. Christe, W. W. Wilson, R. D. Wilson, R. Bau and J. A. Feng, J. Am. Chem. Soc., 1990, 112, 7619–7625 CrossRef CAS.
- N. A. Jasim and R. N. Perutz, J. Am. Chem. Soc., 2000, 122, 8685–8693 CrossRef CAS.
- R. B., Gaussian 09, Rev. B.01, see ESI.‡.
- J. Csontos, N. Y. Palermo, R. F. Murphy and S. Lovas, J. Comput. Chem., 2008, 29, 1344–1352 CrossRef CAS PubMed.
- E. Ruiz, D. R. Salahub and A. Vela, J. Phys. Chem., 1996, 100, 12265–12276 CrossRef CAS.
- S. Adiga, D. Aebi and D. L. Bryce, Can. J. Chem., 2007, 85, 496–505 CrossRef CAS.
Footnotes |
| † Experimental datasets associated with this paper were deposited with the University of York library. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1825404, 1825439–1825452. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00890f |
| § Present addresses: CatScI Ltd, CBTC2, Capital Business Park, Wentloog, Cardiff, CF3 2PX, UK. |
| ¶ Present addresses: School of Chemistry and Bioscience, Richmond Road, University of Bradford, Bradford, BD1 1DP, UK. |
| || The crystal structure of 1oBr exhibited unresolved twinning and poorly resolved disorder and the geometric information was considered not of high enough quality for inclusion. Nevertheless, the major component showed the zig-zag halogen-bonded motif present in the other 1oX structures. |
| ** The separate crystal structure determination at 110 K (Tables 1, 2 and S6) is consistent with that presented here, determined at 111 K (Tables 3 and S15). |
| This journal is © The Royal Society of Chemistry 2018 |