
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new family of sesterterpenoids isolated around the Pacific Rim
Harry J.
Shirley
a,
Megan L.
Jamieson
 b,
Margaret A.
Brimble
*b and
Christopher D.
Bray
*c
b,
Margaret A.
Brimble
*b and
Christopher D.
Bray
*c
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK
bSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz
cDepartment of Chemistry, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: c.bray@qmul.ac.uk
First published on 16th March 2018
Abstract
Covering: 2009 up to the end of 2017
There has been a recent eruption in the number of known marine sesterterpenoids which have been isolated from Pacific Rim marine organisms. These compounds have novel and unusual structures that exhibit incredibly potent and varied bioactivities. This review details the isolation, biological testing and prospects for this exciting new family with discussion of their potential biogenetic origins.
 Harry J. Shirley | Harry Shirley graduated from The University of Nottingham with MSci (Hons) in 2011, before undertaking PhD research with Dr Christopher Bray at Queen Mary University of London. In 2015 he was awarded a PhD for his work on the total synthesis of sesterterpenoid natural products phorbaketal A and phorones A/B. In the same year he moved to New Zealand, becoming Postdoctoral Research Fellow with Distinguished Professor Margaret Brimble and Dr Daniel Furkert at The University of Auckland. He is currently researching at University of Oxford in Professor Timothy Donohoe's research group. Areas of interest include terpenoid natural products, spiro compounds and new cycloaddition reactions. |
 Megan L. Jamieson | Megan Jamieson completed her BSc Hons at the University of Auckland in 2015, and is currently undertaking a PhD with the supervision of Distinguished Professor Margaret Brimble and Dr Daniel Furkert. Megan's PhD research includes work towards the total synthesis of marine derived sesterterpenoids, and mechanistic elucidation of a recently discovered cobalt catalysed olefination reaction. |
 Margaret A. Brimble | Margaret Brimble is a Distinguished Professor at the University of Auckland where her research program focuses on the synthesis of bioactive natural products and peptides. She has published >450 papers, 65 reviews, holds 30 patents, won the 2016 Marsden Medal, 2012 Rutherford Medal (NZ top science award), the 2010 RSC Natural Products Award, named an IUPAC Distinguished Women in Chemistry (2015) and L'Oreal-UNESCO Women in Science laureate for Asia-Pacific, and conferred the Queen's Honour CNZM. She is President of IUPAC Organic and Biomolecular Division III, co-Founder cancer vaccine company SapVax, Associate Editor for Organic and Biomolecular Chemistry, and Past-President of the International Society of Heterocyclic Chemistry. |
 Christopher D. Bray | Christopher Bray was a doctoral student at the University of Oxford before undertaking post-doctoral studies with Prof Gerry Pattenden at the University of Nottingham on the biogenetically patterned synthesis of the furanocembrance family of natural products. He is currently a Lecturer in Synthetic Organic Chemistry in the Department of Chemistry at Queen Mary, University of London. His research interests include the development of new synthetic methodology to access small rings and the total synthesis of bioactive natural products. |
1. Introduction
Isoprenoids have given the world some of the most important classes of drugs and bioactive molecules including taxanes, steroids, tocopherols, artemisinins, ingenanes and cannabinoids (Fig. 1).1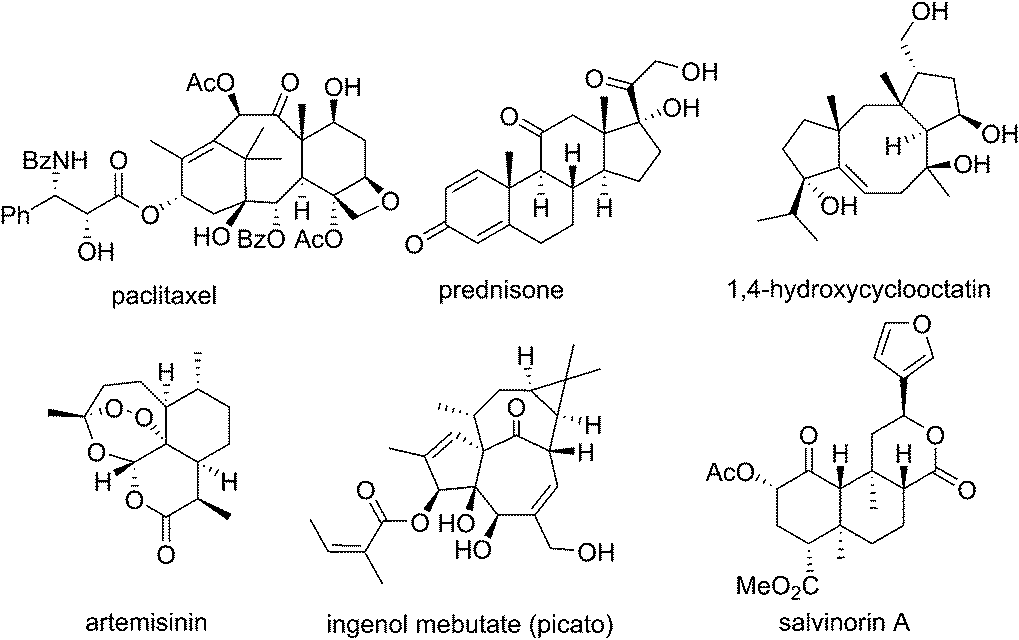 | ||
| Fig. 1 Isoprenoid drugs and bioactive molecules. | ||
This is hardly surprising given that isoprenoids are the largest class of natural products, with estimates of >70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 distinct compounds providing a vast pool of complexity that can interact with biological targets in a huge variety of different ways.2 More generally, natural products play a key role in the drug discovery/pharmaceutical industries. More than half of small molecule drugs can trace their roots back to natural products since they occupy the vastness of chemical space far better than the more focussed areas that traditional medicinal chemistry is able to populate.1a The ongoing discovery of architecturally complex and beguiling natural products also inspires the development of new chemical methodologies which feeds back into the pharmaceutical chemistry arena. The importance of natural products to the pharmaceutical industry and in turn to the wider public has therefore never been more apparent.
000 distinct compounds providing a vast pool of complexity that can interact with biological targets in a huge variety of different ways.2 More generally, natural products play a key role in the drug discovery/pharmaceutical industries. More than half of small molecule drugs can trace their roots back to natural products since they occupy the vastness of chemical space far better than the more focussed areas that traditional medicinal chemistry is able to populate.1a The ongoing discovery of architecturally complex and beguiling natural products also inspires the development of new chemical methodologies which feeds back into the pharmaceutical chemistry arena. The importance of natural products to the pharmaceutical industry and in turn to the wider public has therefore never been more apparent.
2. Pacific Rim sesterterpenoids
Of all the isoprenoids, sesterterpenoids are amongst the rarest with little more than 1000 compounds known.1 Nevertheless, even this small family provides sufficient diversity to serve as a useful mine for bioprospecting.3 Manoalide is a potent inhibitor of phospholipase A2 that reached phase II clinical trials in the 1980's as a topical anti-inflammatory. Although it failed to reach market due to supply issues, it is still used as a standard biochemical tool to block calcium channels. More recently marine sesterterpenoids have shown promise as exciting new anti-cancer drug leads.4,5Biogenetically, sesterterpenoids are derived from five dimethylallyl pyrophosphate (DMAPP) monomers that form the C25 geranylfarnesyl pyrophosphate (GFPP) backbone (Scheme 1). Cyclisation of GFPP is catalysed by sesterterpene synthases (STSs), the dearth of which explains the overall scarcity of sesterterpenoids.6 This landscape has changed significantly over the past 8 years as an entire new subset of sesterterpenoids has been isolated from marine organisms located around the Pacific Rim swelling their numbers considerably. Eight novel skeletal classes: alotane (1), ansellane (2), secoansellane (3), phorbasane (4), isophorbasane (5), phorane (6), gombane (7) and anvilane (8) have been isolated from marine organisms collected in South Korean, Papua New Guinean and Canadian waters (Fig. 2). These alotane and alotane-derived natural products have attracted considerable attention from biologists and synthetic chemists due to their diverse and often potent biological activities, in addition to their elaborate organic frameworks. This review compiles details of the isolation and biological testing of this new family of sesterterpenoids. We highlight previously suggested biogeneses and also introduce a few of our own proposals.
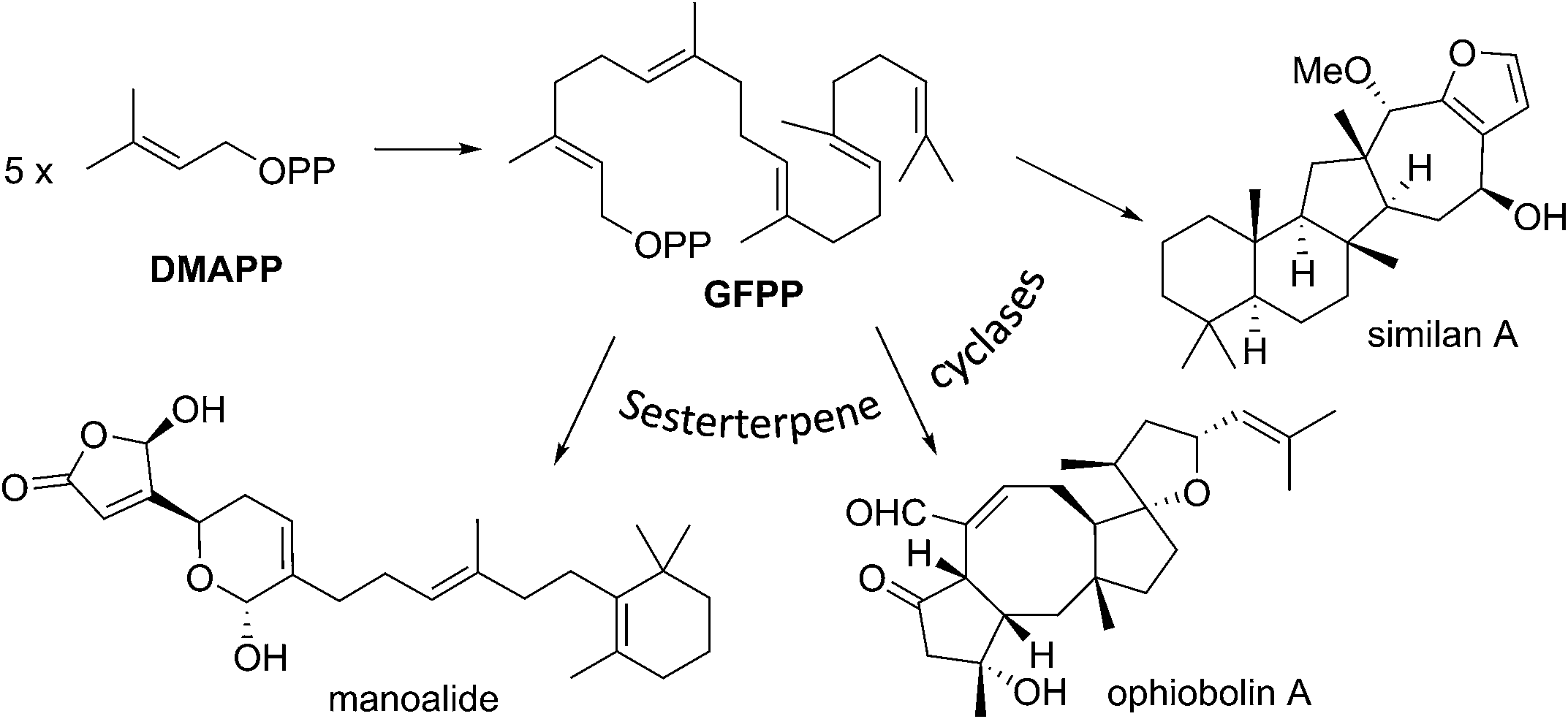 | ||
| Scheme 1 Biogenesis of sesterterpenoids. | ||
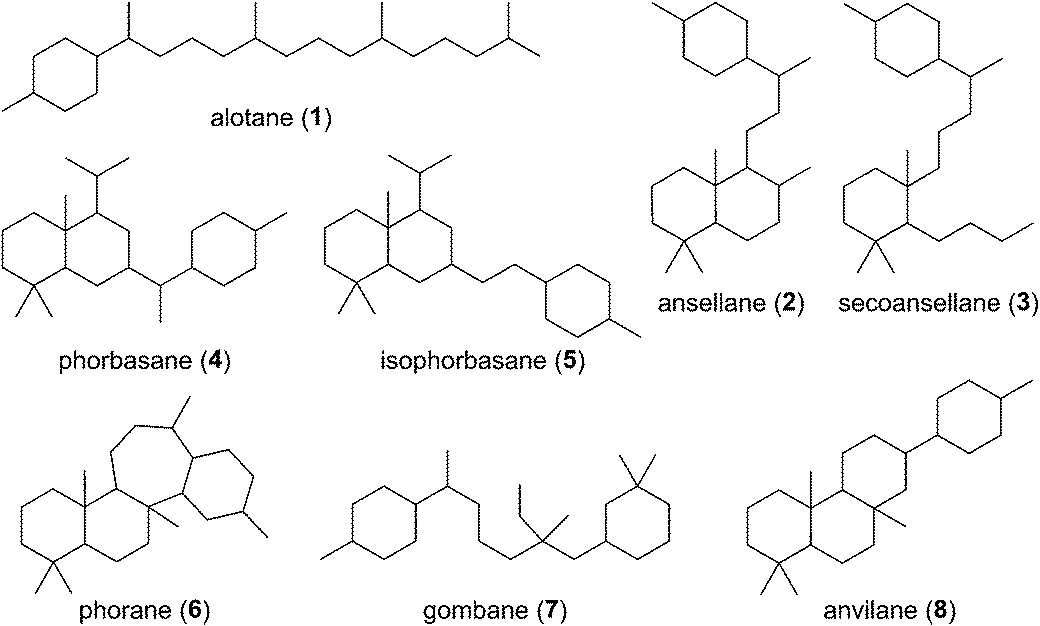 | ||
| Fig. 2 Alotane (1) and its proposed sesterterpenoid skeletal derivatives. | ||
2.1 Alotane sesterterpenoids
This field began in 2009 when Andersen and co-workers isolated alotaketals A and B (9 and 10) from a sample of the marine sponge Hamigera sp. collected off the coast of Papua New Guinea (Fig. 3).7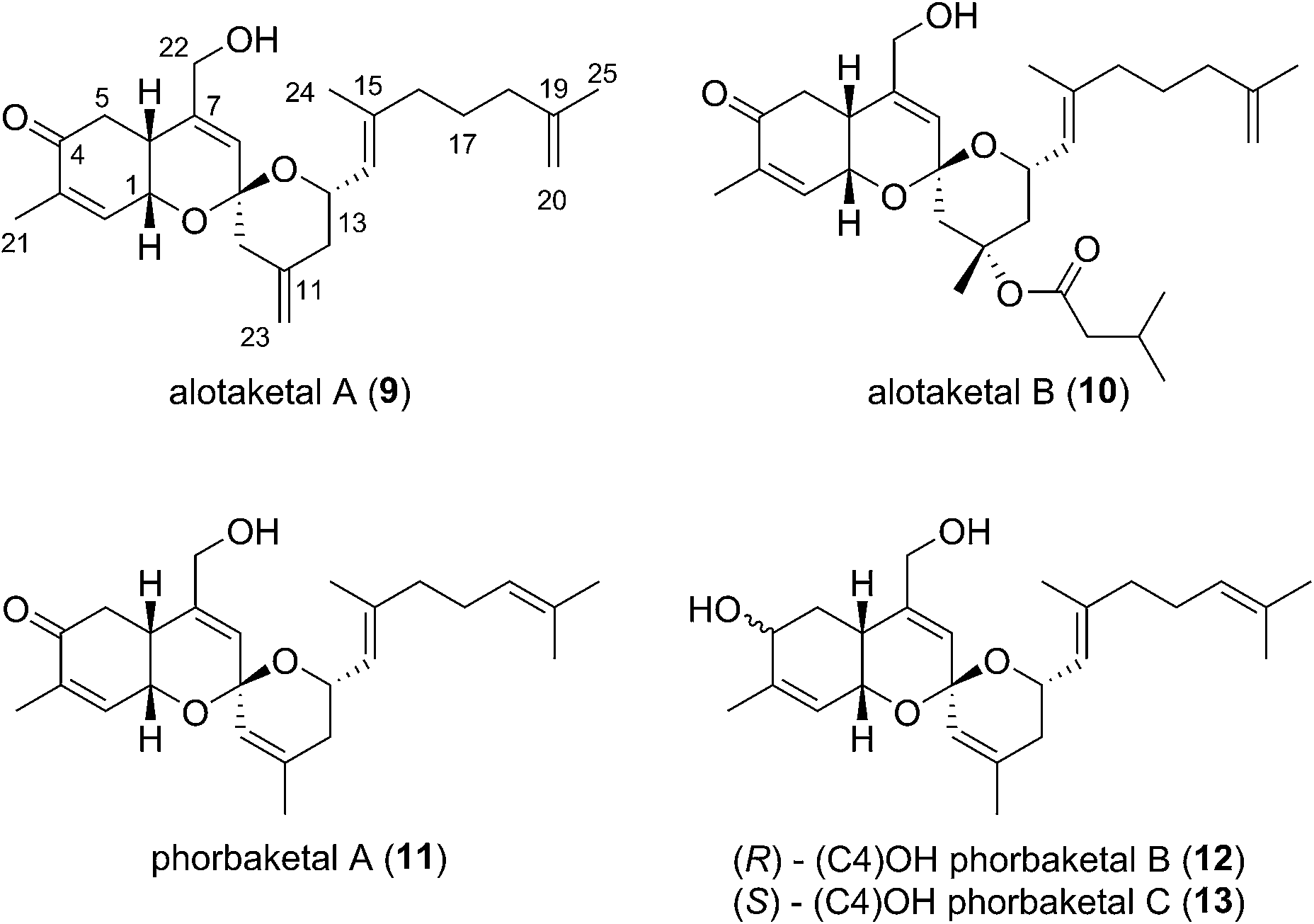 | ||
| Fig. 3 First alotane natural products. | ||
The alotane framework contains novel tricyclic spiroketal and hydrobenzopyran moieties, unprecedented at the time among known natural products. Alotaketals A and B (9 and 10) were found to be potent activators of the secondary messenger cyclic adenosine monophosphate (cAMP) signalling pathway. Treatment of HEK293 cells with alotaketals A or B (9 or 10) resulted in activation of the signalling pathway, and the concentration of cAMP in the cells was greatly increased. Most strikingly, alotaketal A (9) promoted cAMP activation at nanomolar concentrations (EC50 18 nM). In fact, alotaketal A is one of the most potent activators of the cAMP pathway ever to be discovered, being 167 times more potent than forskolin (EC50 3 μM), the standard small molecule adjuvant relied upon for biological investigations of cAMP activation. This startling activity prompted a flurry of efforts from the synthetic community which resulted in a number of impressive total syntheses.8 Soon after the original discovery of the alotaketals, the closely related sesterterpenoids phorbaketals A–C (11–13) (Fig. 3), were isolated from a specimen of the marine sponge Phorbas sp. collected near Gageo Island, South Korea by Rho et al.9 The similarity between these new natural products is particularly striking given they were isolated over 5000 kilometres apart. Rho et al. reported that phorbaketal A (11) could be extracted from a cultivated microbial specimen collected from a homogenized sponge specimen, which might suggest that 11–13 are originated from symbionts inhabiting the sponge, however this is yet to be confirmed and further work is required to identify the true origin of these metabolites. Alotaketal A (9) and phorbaketal A (11) are regioisomers differentiated by alkenes positioned at C11 and C19. The epimeric phorbaketals B and C (12 and 13) were proposed to originate from 11 by reduction at C4. The discovery of numerous other alotaketals and phorbaketals followed between the years 2013–16. Phorbaketals D–K (17–26) were isolated by Kang et al. off the coast of South Korea from the marine sponge Monanchora sp. (Fig. 4).10 This was followed by isolation of phorbaketals L–N (27–29) again from the marine sponge Phorbas sp. from South Korean waters.11 Andersen and co-workers further reported the isolation of alotaketal C (16)12 and later D and E (17 and 18).13 These recently discovered alotanes differ by oxidative modification of the tricyclic core or the geranyl side chain, with phorbaketal N (29) notably being brominated at C14.
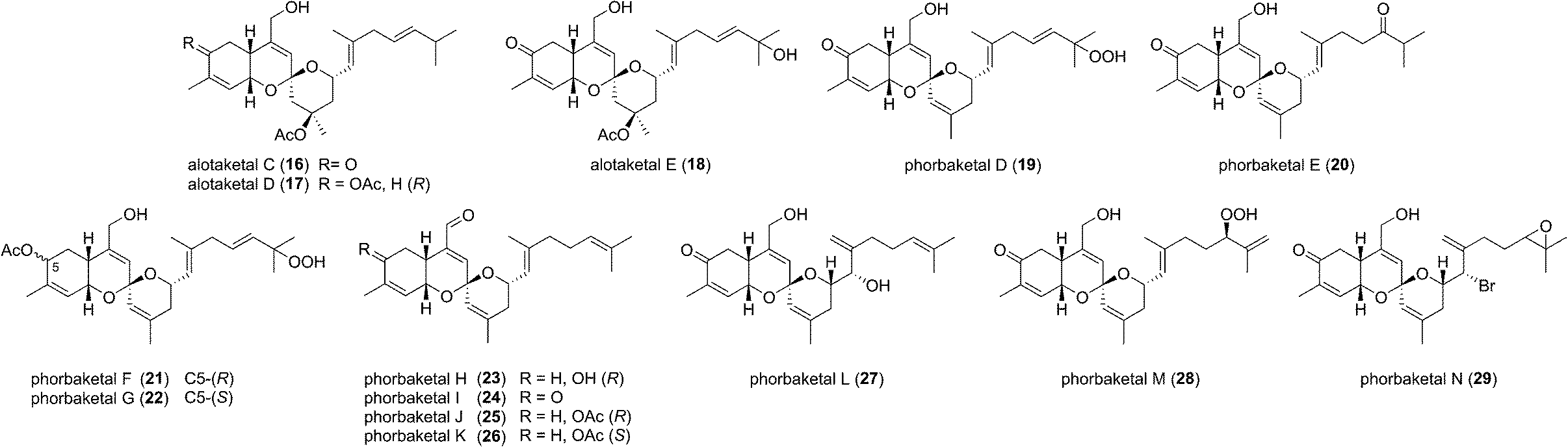 | ||
| Fig. 4 The alotane family of natural products. | ||
Biogenesis of the alotane carbon skeleton is proposed to proceed by partial cyclization of geranylfarnesyl pyrophosphate (GFPP) (Scheme 2).14 An allylic cation would be generated by dissociation of pyrophosphate, prior to attack from the proximal alkene to form a new 6-membered ring. This generates the alotane skeleton which is proposed to be the progenitor to the alotaketals and phorbaketals. Oxidation of alotane (1) at C1, C4 and C9 gives phorbin A (14). This natural product was isolated alongside phorbaketals D–K in 201310 and is currently the least elaborated and hence biogenetically the earliest of the known alotane sesterterpenoids. A possible pathway to the common spiroketal-hydrobenzopyran framework may involve epoxidation/rearrangement about C7 and further oxidation of the unsaturated skeleton to form hydroxy ketones (e.g.15 or similar) prior to spiroketalization. Further modification through alkene migrations and/or oxidation of the geranyl side chain or the tricyclic core would lead to other phorbaketal and alotaketal natural products (Fig. 4).
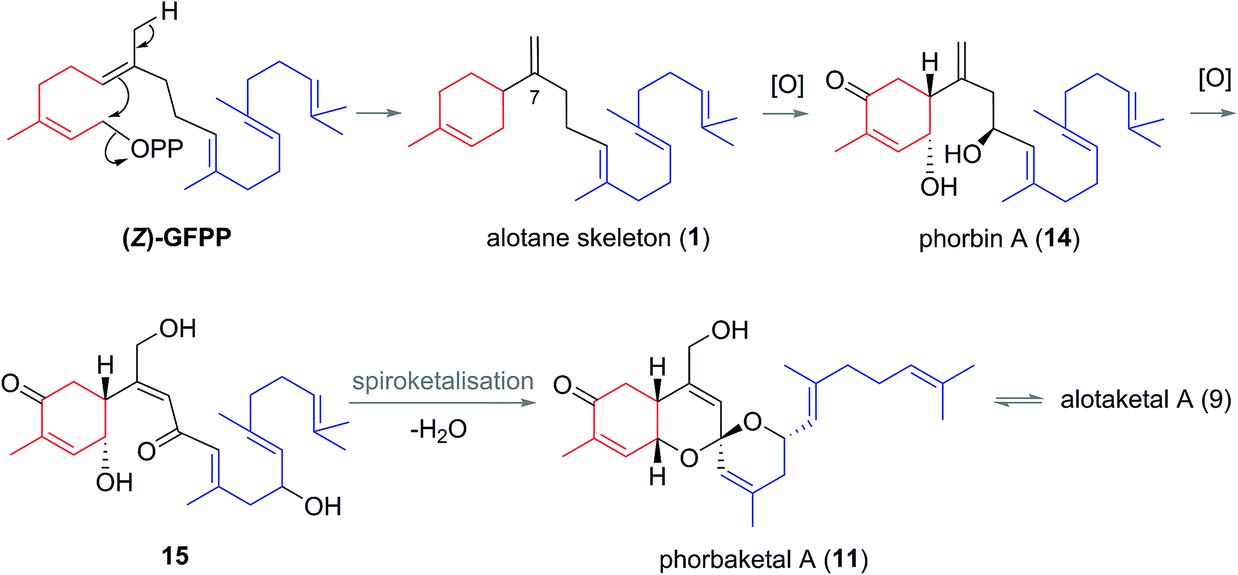 | ||
| Scheme 2 Proposed biogenesis of phorbin A and other alotane natural products. | ||
2.2 Ansellanes and secoansellane
The first of the ansellanes was reported in 2010 by Andersen et al.15 Ansellone A (30) was isolated from the marine sponge Phorbas sp. and from the nudibranch Cadlina luteomarginata observed feeding on the sponge at the time of collection in British Columbia, Canada (Fig. 5). This was the first example of the novel tricyclic ansellane carbon skeleton (2) in a natural product. A few years later Kang and co-workers reported the isolation of ansellone B (37) from a specimen of Phorbas sp. collected from south-western Korean waters. Ansellone B (37) bears an atypical aromatized C ring, absent in all other ansellones isolated subsequently, which instead possess a common benzohydropyran moiety (Fig. 5).14 A number of further ansellones have since been identified from Phorbas sp. including ansellone B1 (38),12,16 phorbadione (35),12 and ansellone C (40).18 All these additional natural products share the hallmark hydrobenzopyran motifs and are differentiated only by the varied oxidation state of the drimane-type trans-decalin skeleton. Secoepoxyansellone A (36) is the first example of the degraded secoansellane carbon skeleton (3), being devoid of the parent ansellane C10–C11 junction (Fig. 5).12 Of the known ansellane natural products these can be split into three sub-categories: pyran-type, aromatic and oxocane-type. Variations within the individual ansellane sub-categories then involve oxidative modification of the drimane-type trans-decalin skeleton (Fig. 5).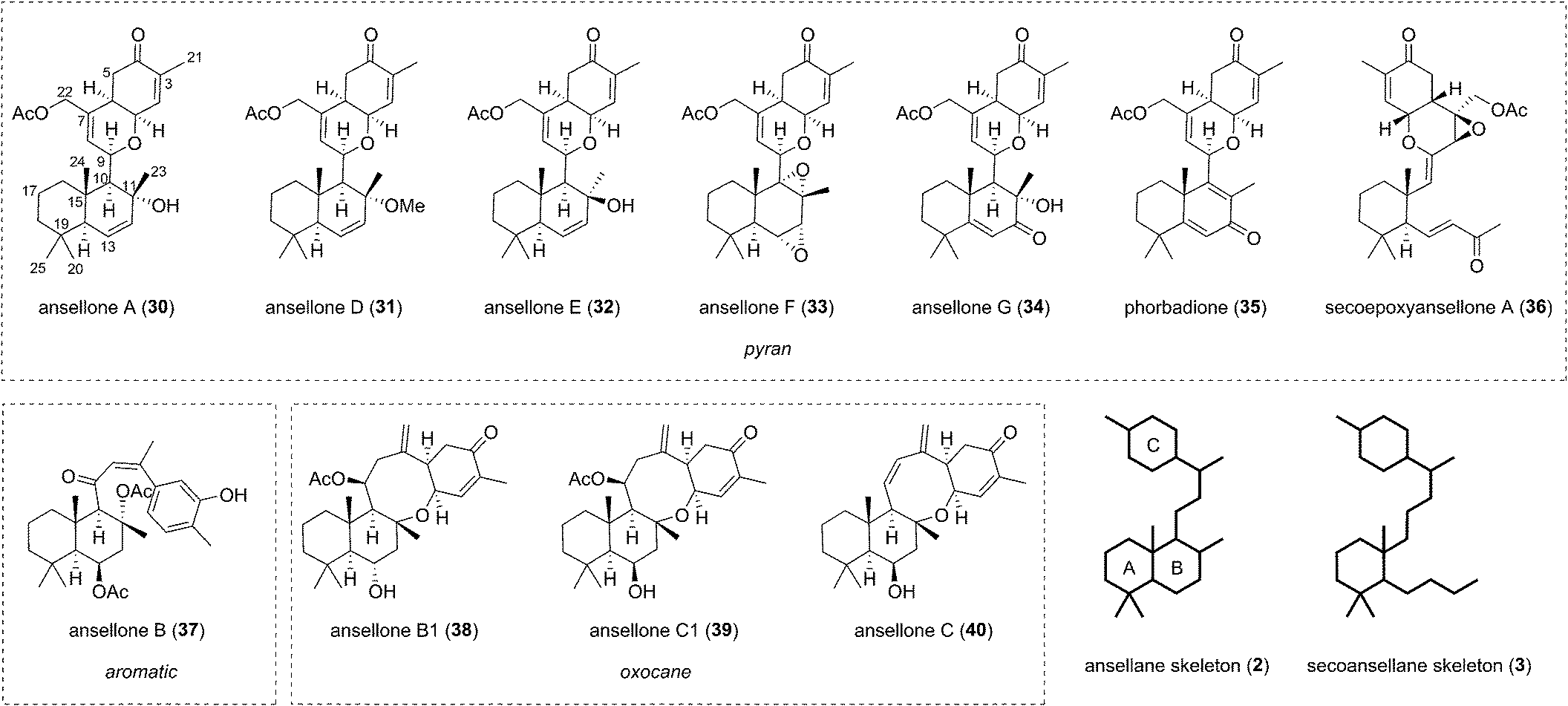 | ||
| Fig. 5 The ansellane natural products. | ||
The ansellane carbon skeleton may be biogenetically derived from cyclization of the pendant oxygenated farnesyl chain of the alotane skeleton starting from 13-hydroxyphorbin 41 to form the drimane-type trans-decalin ring system (in accord with the Stork–Eschenmoser hypothesis) (Scheme 3).19 Oxocane-type ansellones B1 (38), C, and C1 (39 and 40) and pyran-type ansellones A (30) and D–G (31–34) may be derived from common intermediate 42, where the C4 alcohol may substitute alcohols (or similar) at either C11 or C9, respectively. Alternatively, aromatization of ring C in intermediate 42 would lead to ansellone B (37). We now propose that degradation of the ansellane skeleton to the secoansellane skeleton possessed by secoepoxyansellone A (36) may occur via lactolisation of an intermediate such as 43 at C9, before oxocarbenium generation and a retro-Aldol process (Scheme 3).
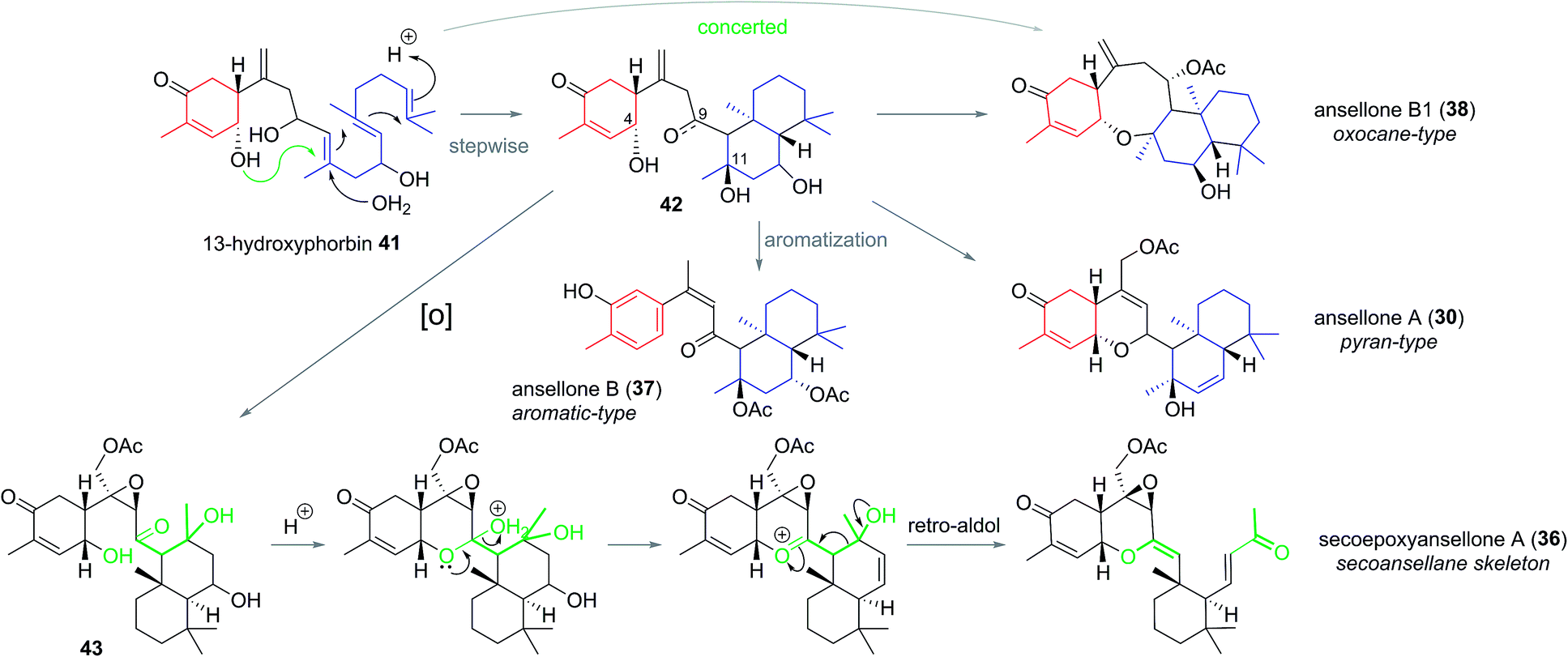 | ||
| Scheme 3 Proposed biogenesis of the ansellane and secoansellane family of natural product. | ||
In 2017, the total syntheses of ansellones A (30), B1 (38) and phorbadione (33) were reported by Tong et al. which they suggest will enable future structure–activity relationship studies of ansellane analogues to be carried out.20
2.3 Phorbasanes and isophorbasane
Phorbasones A (44) and B (45) were reported by Rho and co-workers from a Phorbas sp. Sponge found in South Korean waters.21 The following year Kang et al. isolated isophorbasone A (46) also from a Korean specimen of Phorbas sp. (Fig. 6).14 The phorbasane (4) and isophorbasane (5) skeletons are tricyclic and are related through isomerisation around C11. They are thought to be derived from the alotane skeleton through a series of sequential cyclisations starting from the alotane 47 (Scheme 4), which bears remarkable similarity to phorbin A (14) and 13-hydroxyphorbin 41, the respective progenitors to the alotaketals, phorbaketals and ansellanes.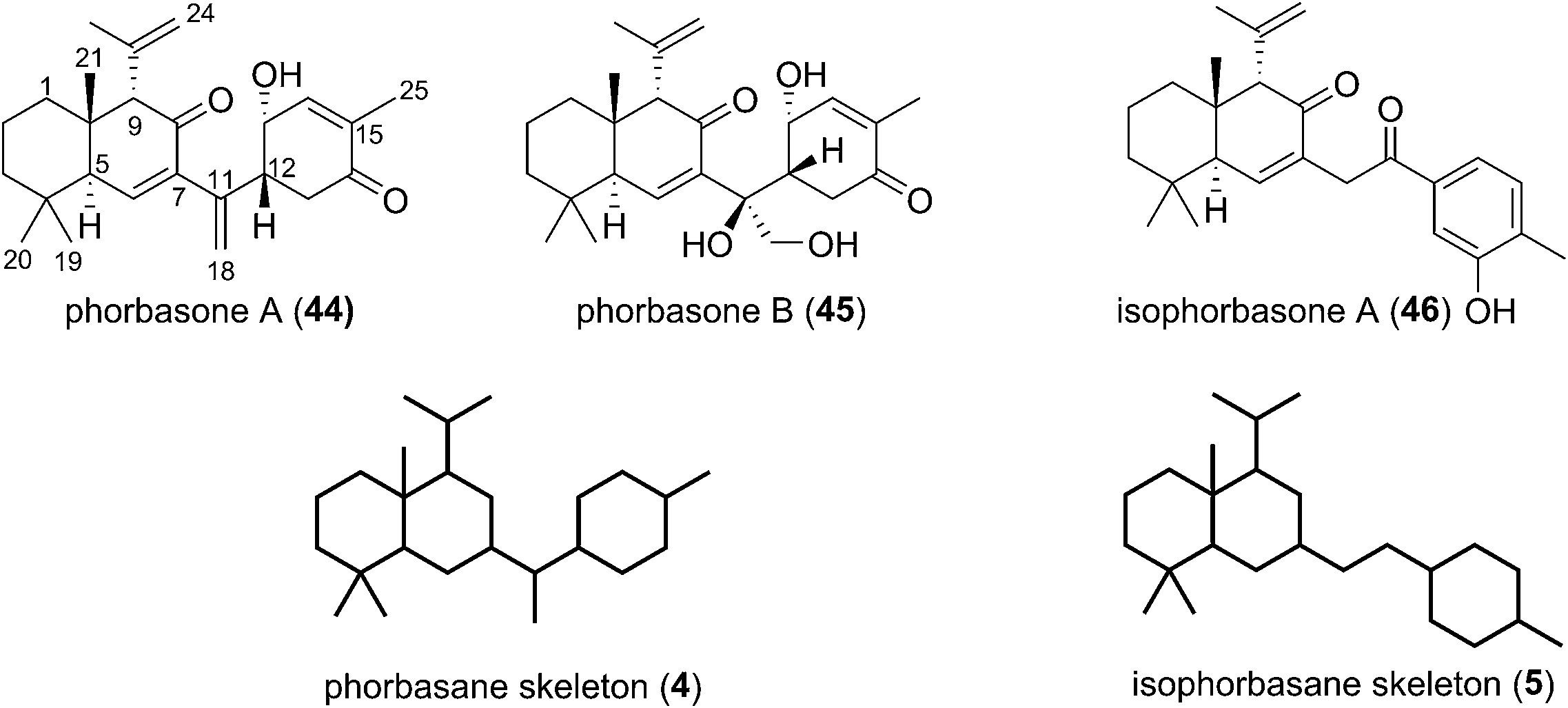 | ||
| Fig. 6 The phorbasane and isophorbasane natural products. | ||
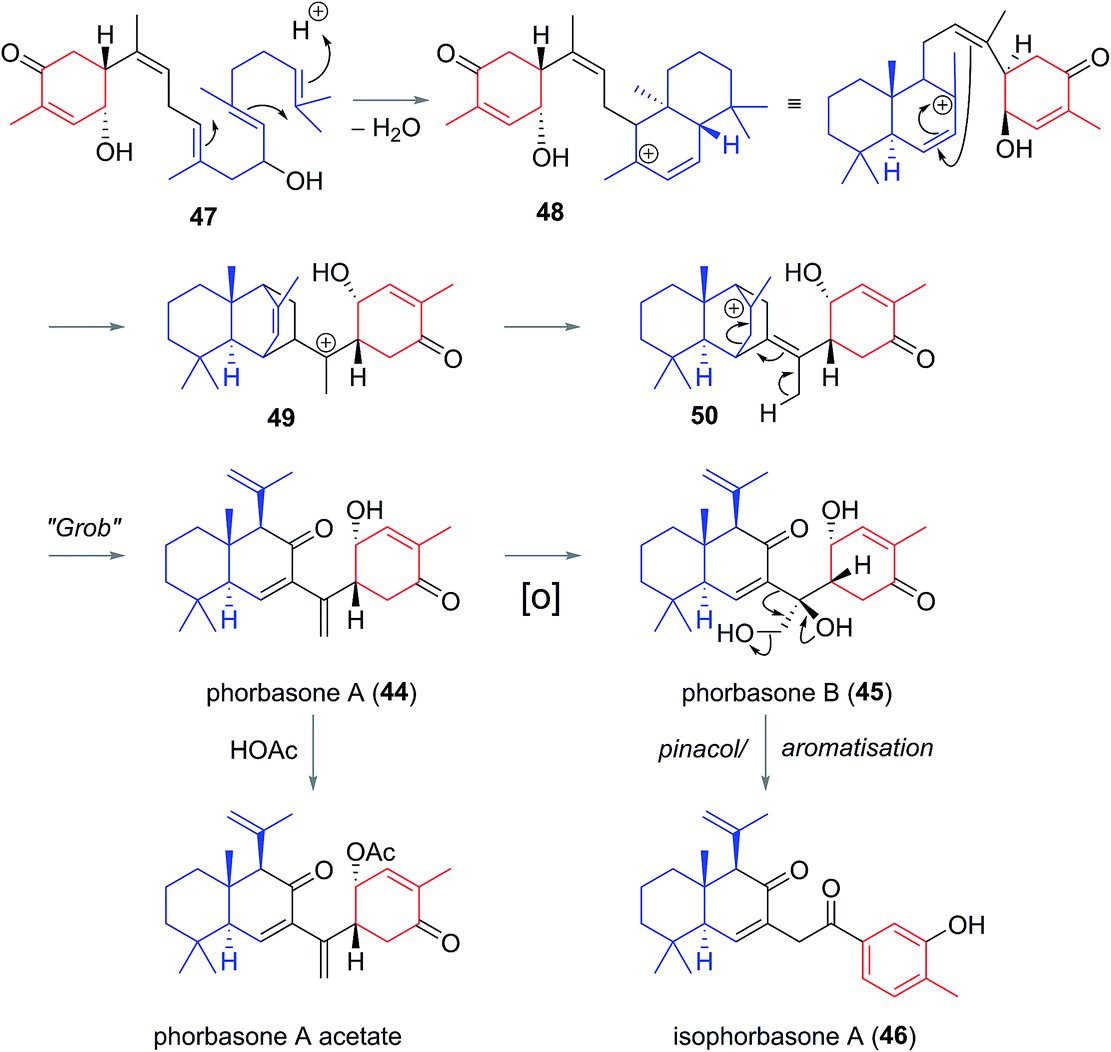 | ||
| Scheme 4 Proposed biogenesis of the phorbasanes and isophorbasanes. | ||
The phorbasane skeleton (4) could be generated by polyene cyclization to give an ansellane type cationic intermediate (48) (Scheme 4)14,21 which closes to an intermediate [2.2.2] fused bicyclic system (49). Proton migration would then give 50, Grob-like elimination of which gives phorbasone A (44). Dihydroxylation of 44 gives phorbasone B (46) before pinacol rearrangement/aromatization leads to isophorbasone A (46).14
2.4 Phoranes
Phorone A (51) was isolated in 2012 in South Korea by Kang et al. from a specimen of the marine sponge Phorbas sp. (Fig. 7).14 More recently in 2015, the isolation of phorone B (52) in the same country was reported by Shin et al. from the marine sponge Clathria gombawuiensis.18 Phorones A and B (51 and 52) are regioisomers due to their aromatic oxidation pattern and are the only known natural products possessing the tetracyclic phorane skeleton (6), which is related to the ansellane skeleton by way of an additional C8–C19 junction.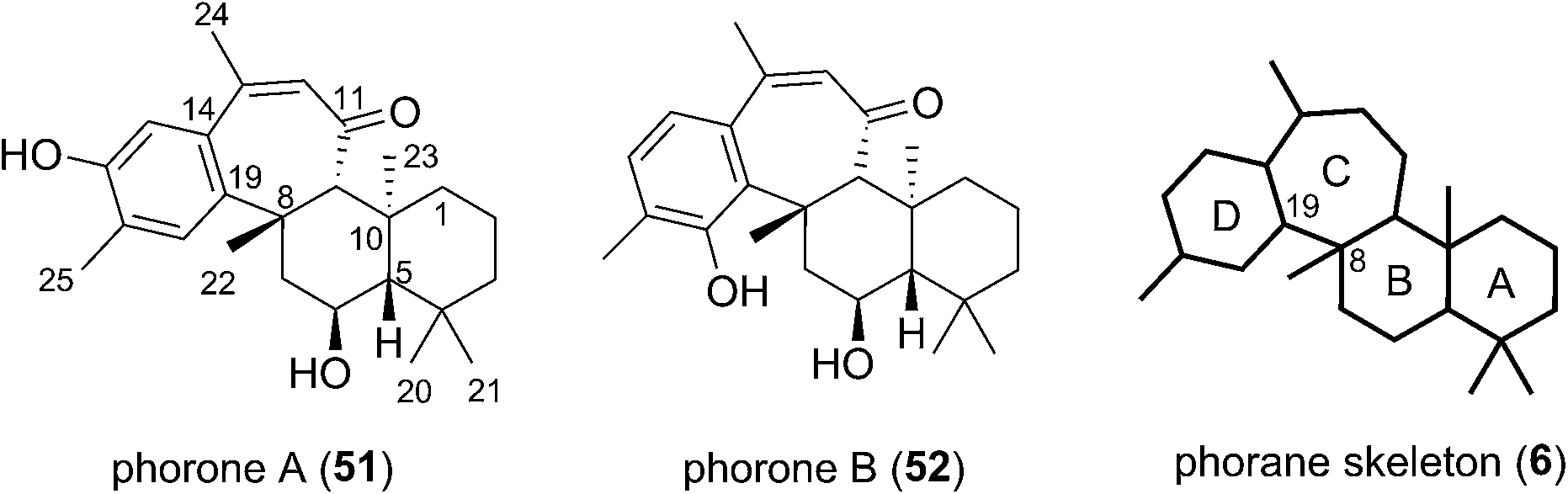 | ||
| Fig. 7 The phorane duo of natural products. | ||
The proposed biogenesis of the phorones suggests a key electrophilic aromatic substitution process to join the parent ansellane ring B to ring C (Scheme 5). Aromatisation of an alotane derivative would give phenol 53. The phorones then likely share a biogenesis similar to that seen with the ansellanes. Cyclisation would give drimane like cation 54 trapping of which by exogenous acetate leads to ansellone B (37). Alternatively the cation could be intramolecularly trapped via electrophilic aromatic substitution at either C19 or C15 leading to phorone A (52) and B (53) respectively. An alternative, and synthetically much more enticing pathway to the phorones might involve a polyene cyclisation22 directly from 55 thereby avoiding intermediates such as 54.
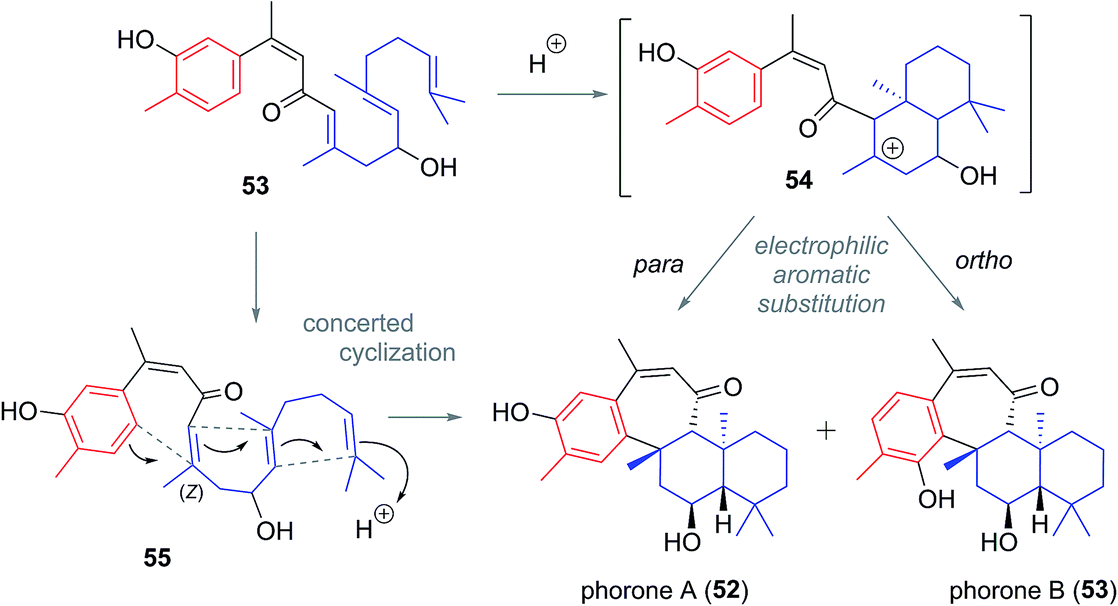 | ||
| Scheme 5 Proposed biogenesis of the phorane natural products. | ||
2.5 Gombanes
In 2014 Shin et al. isolated gombaspiroketals A–C (56–58) from the marine sponge Clathria gombawuiensis (Fig. 8). These are the first and only examples of the bicyclic gombane carbon skeleton (7).23 Being structurally analogous to the phorbaketals and alotaketals, gombaspiroketals A–C (56–58) share their common spiroketal and hydrobenzopyran moieties, but feature a novel, rearranged cyclohexenyl substituent attached at the C13 quaternary centre. Gombaspiroketals A and B (56 and 57) feature an unusual spiroketal-methoxy acetal, and are epimers at C13. Gombaspiroketal C (58) is the analogous spiroketal–hemiacetal of gombaspiroketal A. It is possible that the true natural products possess spiroketal–hemiacetal motifs, since methanolysis may have plausibly occurred during the methanolic sponge extraction procedure.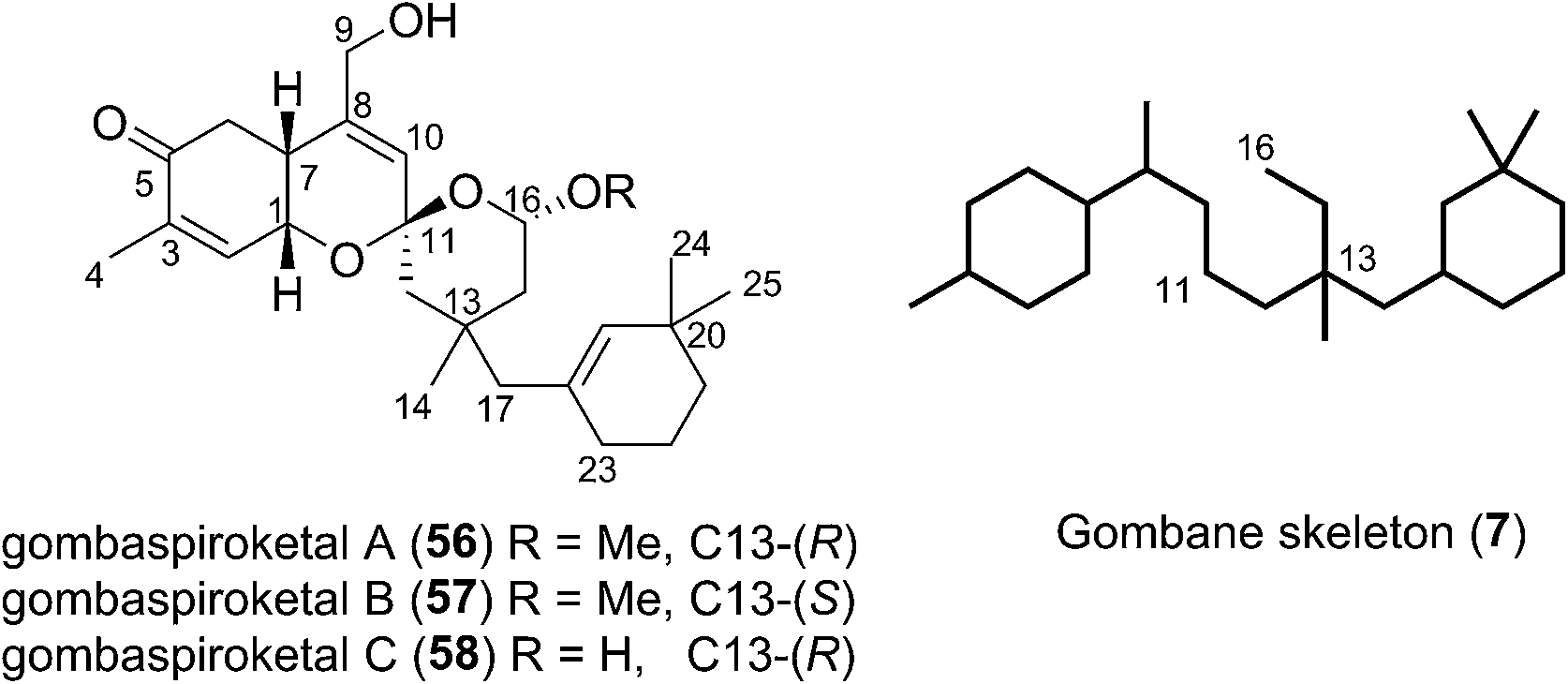 | ||
| Fig. 8 The gombane natural products. | ||
The gombaspiroketals likely share a very close biogenesis to the phorbaketals and alotaketals (whose biogenetic proposal has previously been discussed). The novel dimethylcyclohexenyl methylene motif is proposed to arise from cyclization and migration of the geranyl side chain of phorbaketal A (11) or similar, from C16 to C13 (Scheme 6).18 The resulting carbocation 59 would facilitate formation of the spiroketal-methoxy acetal via oxocarbenium ion 60. This rearrangement could plausibly occur at an earlier stage in their biogenesis i.e. before spiroketalization. The C16 acetal or hemiacetal group resides in the equatorial position, presumably to avoid 1,3-diaxial interactions with groups on the pyran ring.
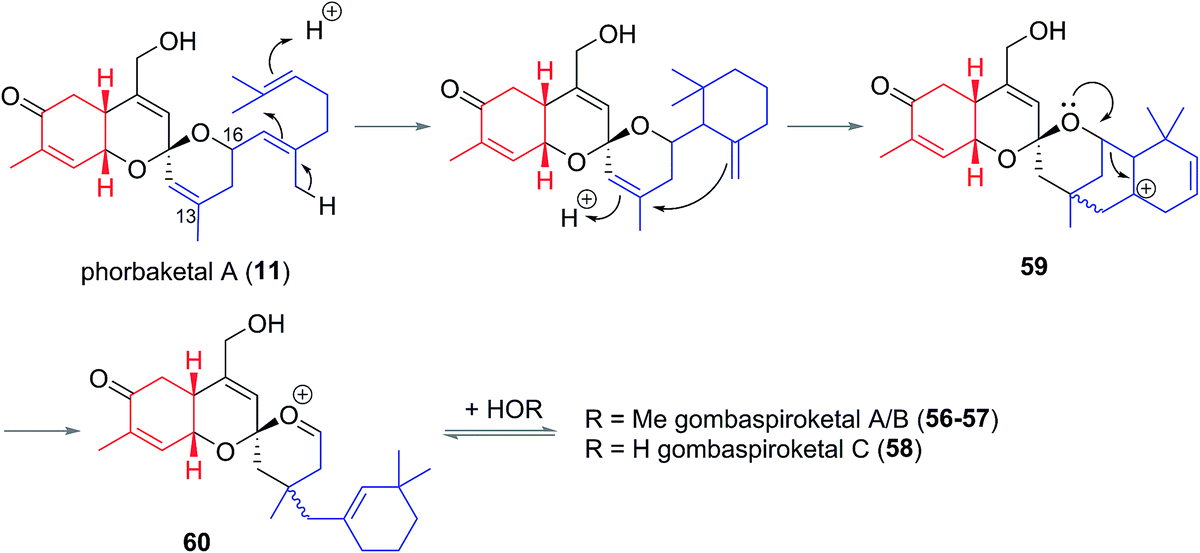 | ||
| Scheme 6 Proposed biogenesis of the gombane natural products. | ||
2.6 Anvilanes
The Phorbas sp. of sponge continues to be a rich source of new sesterterpenoids. Andersen and co-workers isolated anvilones A (61) and B (62) in 2016, from a sample collected in British Columbian waters (Fig. 9). They were isolated alongside eight previously identified natural products of the alotane, ansellane, and secoansellane skeletons. These anvilones are the only isolated natural products of the anvilane skeleton, which features a fused trans,trans-6,6,6-tricycle. Anvilone B (61) is distinguished from anvilone A (62) by oxidation and acetylation at C1.13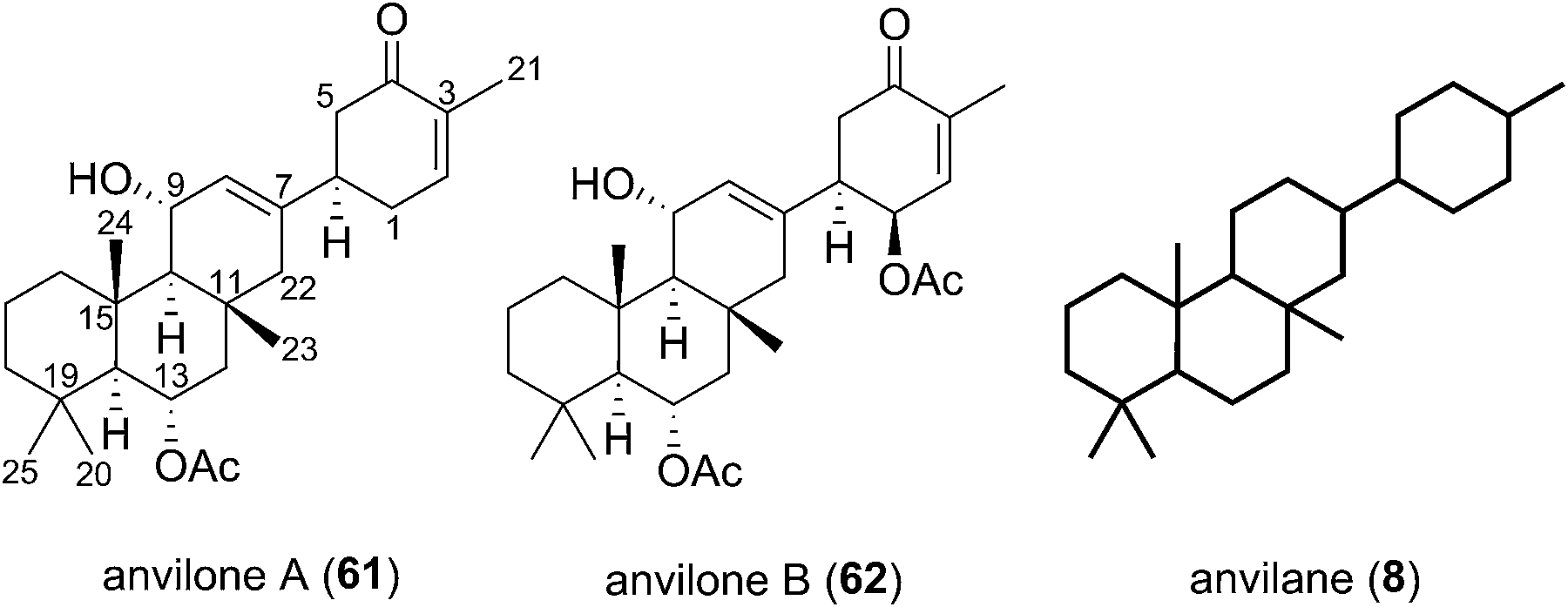 | ||
| Fig. 9 The anvilane natural products. | ||
The anvilane skeleton likely originates from polyene cyclization of an alotane (63). This cascade would install the tricyclic core of the anvilones with further modification through oxidations and/or acetalisations leading to anvilones A or B (61 or 62) (Scheme 7).
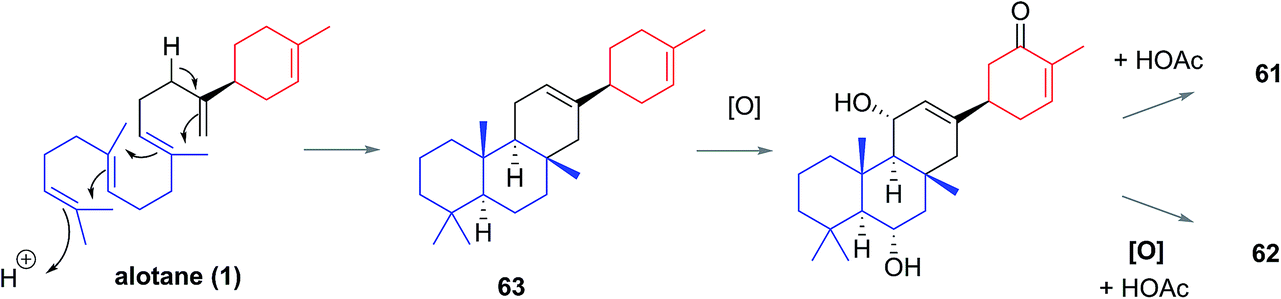 | ||
| Scheme 7 Proposed biogenesis of the anvilane natural products. | ||
3. Biological testing
Various biological investigations have revealed the remarkably diverse and potent bioactivities exhibited by the alotane family of sesterterpenoids. Successful syntheses of alotaketal A (9),8 phorbin A (14),24 ansellones A (30), B1 (38), phorbadione (35)20 and phorbaketal A (11)8d,25 (as well as approaches to its framework26) open up the possibility of future structure–activity relationship studies to explore the properties of alotane analogues.3.1 cAMP activation
In addition to the cAMP activating ability of alotaketal A (9) and B (10), Andersen et al. also found that ansellone A (30) displayed comparable cAMP activation (EC50 14 μM) to that of forskolin in a HEK293 cell-based assay, however alotaketal A (9) remains the most potent modulator to be identified.15 Modulation of the cAMP signalling pathway is used in stem cell techniques and is important for the development of treatments for several diseases including heart failure, cancer, and neurodegenerative diseases. As such, the development of new modulators of this pathway is potentially of high medicinal value (Scheme 2).273.2 Cytotoxicity
Cytotoxicity screening of phorbaketals A–C (11–13) by Rho et al. against human colorectal cancer HT-29, hepatoma cancer HepG2, and lung cancer A549 cell lines showed phorbaketal A (11) to have mild activity (IC50 11–12 μg mL−1) towards all three cell lines, while phorbaketals B and C (12 and 13) were moderately active (IC50 12–460 μg mL−1).9During screening of phorbaketals D–K (17–26) against four cancer cell lines, phorbaketals H and I (23 and 24) were active against the A498 human renal cancer cell line (IC50 9.8 and 9.0 μM respectively).10 When considering structure–activity relationships Kang et al. noted that a hydroperoxy group on the side chain may be detrimental to cytotoxicity, as 21, 22, and 28 were inactive. This is supported by the observation that phorbaketals A–C (11–13) were cytotoxic towards all three human cancer cell lines.9
In a similar fashion, phorbaketals L–N (27–29) were screened for cytotoxicity and phorbaketal N (29) exhibited potent activity.11 Phorbaketal N (29) displayed cytotoxicity against human pancreatic cancer cell line PANC-1 (IC50 11.4 μM), and the renal cancer cell lines A498 (IC50 18.7 μM) and ACHN (LC50 24.4 μM). It should be noted that phorbaketal N (29) was more cytotoxic than the FDA approved anti-cancer drug fluorouracil28 against PANC-1 and thus could provide a useful novel drug lead for the treatment of human pancreatic cancers.
Despite its relatively simple framework, phorbin A (14) displayed micromolar cytotoxicity against MIA-paca (IC50 5.2 μM) and PANC-1 (IC50 7.8 μM) human pancreatic cancer cell lines as well as renal cancer cell lines A498 (IC50 4.9 μM) and ACHN (IC50 10.0 μM).10,29 In terms of structure–activity relationship, this observed potency implies that the spiroketal moiety possessed by the phorbaketals may not be an important feature for anti-cancer activity.
Gombaspiroketals A–C (56–58) also exhibited moderate cytotoxicity in the AS549 and K562 cell lines (LC50 0.85–2.02 μM and LC50 0.77–4.65 μM), comparable to that of doxorubicin (LC50 0.79 and 0.70 μM respectively).23 Phorone B (53) and ansellone C (40) were also both found to be moderately cytotoxic towards AK549 and K562 cancer cell lines.18
3.3 Effects on stem cell differentiation
Biological studies by Hong et al. have revealed that phorbaketal A (11) can promote osteogenic differentiation of human mesenchymal stem cells. In the presence of phorbaketal A (11), mesenchymal stem cells exhibited increased levels of differentiation markers such as osteocalcin, Dlx5, ALP, Runx2, and TAZ. These markers are associated with differentiation into the osteoblast lineage, which is important for bone reformation processes. Thus phorbaketal A (11) is potentially useful for research into anabolic therapeutics in bone diseases such as osteoporosis.30Similarly to phorbaketal A (11), phorbasone A (44) exhibited a distinct effect on bone reformation processes in a study by Rho et al.21 In mesenchymal stem cells phorbasone A (44) increased calcium deposition activity via upregulated gene expression of a range of osteoblast differentiation markers. Phorbasone B (45) showed comparably low activity in these assays, indicating that the 1,2-diol motif disrupts interaction with the target.
Phorbaketal A (11) was also shown by Hong et al. to inhibit differentiation of mesenchymal stem cells into adipocytes. Similar to its effect on osteogenic differentiation, this activity arises from upregulation of markers which inhibit gene transcription required for differentiation into the adipogenic lineage. This activity indicates phorbaketal A (11) may be a useful lead for research into therapeutics to treat obesity.31
3.4 Anti-HIV activity
In 2016 Andersen et al. investigated Phorbas sp. metabolites as therapeutic leads towards the promising ‘shock and kill’ approach to a sterilizing HIV-1 cure.13 The study found that alotaketals C and D (16 and 17), ansellone A (30) and anvilone A (61) activate latent proviral HIV-1 gene expression. Alotaketal C (25) showed latency reversal activity (LRA) (ca. 14% at 30 μM) approximately 2.1-fold more potent than the control compound prostratin at the same concentration. Prostratin is now entering phase I trials as a potential HIV cure. It was also found that alotaketal D (26), ansellone A (30), and anvilone A (61) have LRA profiles comparable to that of the control prostratin. The alotaketals, ansellones, and anvilones, represent a new set of scaffolds that are promising leads for structure activity relationship development of LRA biological tools and drug candidates.4. Summary and outlook
In this review we have presented a remarkable new family of biogenetically related sesterterpenoids isolated from a variety of marine sponges around the Pacific Rim. These have proven to be a rich source of structurally, biogenetically and biologically intriguing new compounds that are based on previously unseen carbon skeletons.This new family of sesterterpenoids have novel scaffolds which exhibit diverse and potent bioactivities. Notably, alotaketal A (9) has multiple biological activities including potent activation of the cAMP signalling pathway. Other important biological activity discovered includes cytotoxicity, various effects on stem cell differentiation and latent HIV-1 reactivation. Therefore the alotane and alotane derived natural products have potential use as drug leads in the treatment of multiple diseases including cancer, heart disease, obesity, osteoporosis and HIV.
The alotane skeleton is thought to be biogenetically derived from geranylfarnesyl pyrophosphate (GFPP) and we have summarized its proposed biogenetic derivations to related sesterterpenoid skeletons as well as introduced our own biogenetic proposals. Identification of the organism(s) generating these secondary metabolites is yet to be conclusively determined, so far with suggestions by Rho et al. of a microbiological origin. We anticipate that the biogenetic proposals presented will inspire future total synthesis endeavours towards these natural products.
5. Conflicts of interest
There are no conflicts to declare.6. Acknowledgements
We thank the University of Auckland for the award of a Doctoral Scholarship (MJ), Queen Mary University of London and the Engineering & Physical Sciences Research Council (EP/J50029X/1) for a DTA studentship and the Royal Society of New Zealand Marsden Fund (both to HS).7. References
- (a) T. J. Maimone and P. S. Baran, Nat. Chem. Biol., 2007, 3, 396–407 CrossRef CAS PubMed; (b) J. Clardy and C. Walsh, Nature, 2004, 432, 829–837 CrossRef CAS PubMed; (c) J. W. H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed; (d) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed; (e) D. Zheng, L. Han, X. Qu, X. Chen, J. Zhong, X. Bi, J. Liu, Y. Jiang, C. Jiang and X. Huang, J. Nat. Prod., 2017, 80, 837–844 CrossRef CAS PubMed; (f) A. Ortega, J. F. Blount and P. S. Manchand, J. Chem. Soc., Perkin Trans. 1, 1982, 2505–2508 RSC.
- S. R. Hare, J. M. Farnham and D. J. Tantillo, Tetrahedron, 2017, 73, 4227–4232 CrossRef CAS.
- (a) S. Nozoe, M. Morisaki, K. Tsuda, Y. Litaka, N. Takahashi, S. Tamura, K. Ishibashi and M. Shirasaka, J. Am. Chem. Soc., 1965, 87, 4968–4970 CrossRef CAS PubMed; (b) H. Fujiwara, K. Matsunaga, H. Kumagai, M. Ishizuka and Y. Ohizumi, J. Pharm. Pharmacol., 2000, 6, 427–431 CAS; (c) E. D. de Silva and P. J. Scheuer, Tetrahedron Lett., 1980, 21, 1611–1614 CrossRef CAS; (d) C. Mahidol, H. Prawat, S. Sangpetsiripan and S. Ruchirawat, J. Nat. Prod., 2009, 72, 1870–1874 CrossRef CAS PubMed.
- D. T. Hog, R. Webster and D. Trauner, Nat. Prod. Rep., 2012, 29, 752–779 RSC.
- L. Wang, B. Yang, X. Ling, Z. Zhou and Y. Liu, Nat. Prod. Rep., 2013, 30, 455–463 RSC.
- H. Chai, R. Yin, Y. Liu, H. Meng, X. Zhou, G. Zhou, X. Bi, X. Yang, T. Zhu, W. Zhu, Z. Deng and K. Hong, Sci. Rep., 2016, 6, 27181 CrossRef CAS PubMed.
- R. Forestieri, C. E. Merchant, N. J. De Voogd, T. Matainaho, T. J. Kieffer and R. J. Andersen, Org. Lett., 2009, 11, 5166–5169 CrossRef CAS PubMed.
- (a) J. Huang, J. Yang, J. Zhang and J. Yang, J. Am. Chem. Soc., 2012, 134, 8806–8809 CrossRef CAS PubMed; (b) M. Xuan, I. Paterson and S. Dalby, Org. Lett., 2012, 14, 5492–5495 CrossRef CAS PubMed; (c) J. Huang, J. Yang, J. Zhang and J. Yang, Org. Biomol. Chem., 2013, 11, 3212–3222 RSC; (d) H. Cheng, Z. Zhang, H. Yao, W. Zhang, J. Yu and R. Tong, Angew. Chem., Int. Ed., 2017, 56, 9096–9100 CrossRef CAS PubMed.
- J.-R. Rho, B. S. Hwang, C. J. Sim, S. Joung, H.-Y. Lee and H.-J. Kim, Org. Lett., 2009, 11, 5590–5593 CrossRef CAS PubMed.
- W. Wang, Y. Lee, R. M. Venkat, Y. Park, J. Lee, H. Kim, D. Hahn, J. Chin, M. Ekins, S. J. Nam and H. Kang, J. Nat. Prod., 2013, 76, 170–177 CrossRef CAS PubMed.
- Y. Lee, W. Wang, H. Kim, A. G. Giri, D. H. Won, D. Hahn, K. R. Baek, J. Lee, I. Yang, H. Choi, S. J. Nam and H. Kang, Bioorg. Med. Chem. Lett., 2014, 24, 4095–4098 CrossRef CAS PubMed.
- J. Daoust, M. Chen, M. Wang, D. E. Williams, M. A. G. Chavez, Y. A. Wang, C. E. Merchant, A. Fontana, T. J. Kieffer and R. J. Andersen, J. Org. Chem., 2013, 78, 8267–8273 CrossRef CAS PubMed.
- M. Wang, I. Tietjen, M. Chen, D. E. Williams, J. Daoust, M. A. Brockman and R. J. Andersen, J. Org. Chem., 2016, 81, 11324–11334 CrossRef CAS PubMed.
- For variants of this proposal see: W. Wang, Y. Lee, T. G. Lee, B. Mun, A. G. Giri, J. Lee, H. Kim, D. Hahn, I. Yang, J. Chin, H. Choi, S. J. Nam and H. Kang, Org. Lett., 2012, 14, 4486–4489 CrossRef CAS PubMed.
- J. Daoust, A. Fontana, C. E. Merchant, N. J. De Voogd, B. O. Patrick, T. J. Kieffer and R. J. Andersen, Org. Lett., 2010, 12, 3208–3211 CrossRef CAS PubMed.
- Unfortunately there has been a lack of continuity in the naming of the ansellones in the literature. The name Ansellone B was first coined in 2012 by Kang et al. (ref. 14) but was later used for a different structure in 2013 by Anderson, et al. (ref. 12). We therefore propose the renaming of Andersons 2013 compound as Ansellone B1(38). The name Ansellone C was first coined in 2015 by Shin et al. (ref. 18) but a different structure (for which we can find no data in the literature) was later referred to as Ansellone C in 2016 by Anderson et al. (ref. 13). We therefore propose the renaming of Andersons 2016 compound as Ansellone C1 (39).
- J. Daoust, M. Chen, M. Wang, D. E. Williams, M. A. G. Chavez, A. Wang, C. E. Merchant, A. Fontana, T. J. Kieffer and R. J. Andersen, J. Org. Chem., 2013, 78, 8267–8273 CrossRef CAS PubMed.
- J.-K. Woo, C.-K. Kim, C.-H. Ahn, D.-C. Oh, K.-B. Oh and J. Shin, J. Nat. Prod., 2015, 78, 4730–4756 CrossRef PubMed.
- (a) G. Stork and A. Burgstahler, J. Am. Chem. Soc., 1955, 77, 5068–5077 CrossRef CAS; (b) R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS PubMed.
- W. Zhang, H. Yao, J. Yu, Z. Zhang and R. Tong, Angew. Chem., Int. Ed., 2017, 56, 4787–4791 CrossRef CAS PubMed.
- J.-R. Rho, B. S. Hwang, S. Joung, M. R. Byun, J.-H. Hong and H.-Y. Lee, Org. Lett., 2011, 13, 884–887 CrossRef CAS PubMed.
- R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC.
- J.-K. Woo, C.-K. Kim, S.-H. Kim, H. Kim, D.-C. Oh, K.-B. Oh and J. Shin, Org. Lett., 2014, 16, 2826–2829 CrossRef CAS PubMed.
- J. G. Hubert, D. P. Furkert and M. A. Brimble, J. Org. Chem., 2015, 80, 2231–2239 CrossRef CAS PubMed.
- S. Joung, R. Kim and H.-Y. Lee, Org. Lett., 2017, 19, 3903–3906 CrossRef CAS PubMed.
- (a) J. G. Hubert, D. P. Furkert and M. A. Brimble, J. Org. Chem., 2015, 80, 2715–2723 CrossRef CAS PubMed; (b) H. J. Shirley and C. D. Bray, Eur. J. Org. Chem., 2016, 8, 1504–1507 CrossRef.
- (a) B. Pavan, C. Biondi and A. Dalpiaz, Drug Discovery Today, 2009, 14, 982–991 CrossRef CAS PubMed; (b) J. A. Beavo and L. L. Brunton, Nat. Rev. Mol. Cell Biol., 2002, 3, 710–718 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- S. E. Bates and T. Fojo, Nat. Rev. Clin. Oncol., 2016, 13, 205–206 CrossRef CAS PubMed.
- M. R. Byun, A. R. Kim, J.-H. Hwang, M. K. Sung, Y. K. Lee, B. S. Hwang, J.-R. Rho, E. S. Hwang and J.-H. Hong, FEBS Lett., 2012, 586, 1086–1092 CrossRef CAS PubMed.
- (a) H. Kang, J.-R. Rho, J.-H. Hong, S.-B. Park, C.-S. Shin, J.-H. Lee, J.-Y. Hong, E.-O. Kim, J.-A. Kim and S.-M. Oh. WO 2010/050783 A2, 2010; (b) H. Kang, D.-H. Won, I. Yang, E. O. Kim, J. A. Kim, A. G. Giri and V. A. Mallepally, WO 2012/033353 A2, 2012; (c) H. Kang, D. H. Won, I. H. Yang, E. O. Kim, J. A. Kim, A. G. Giri and V. R. Mallepally, US 2013/0274212, 2013; (d) Y.-J. Seo, K.-T. Lee, J.-R. Rho and J.-H. Choi, Mar. Drugs, 2015, 13, 7005–7019 CrossRef CAS PubMed; (e) K. Senthilkumar, J. Venkatesan and S.-K. Kim, Biomedicine & Preventive Nutrition, 2014, 4, 1–7 Search PubMed; (f) M. R. Byun, C. H. Lee, J. H. Hwang, A. R. Kim, S. A. Moon, M. K. Sung, J.-R. Roh, E. S. Hwang and J.-H. Hong, Eur. J. Pharmacol., 2013, 718, 181–187 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |